Recombination and Sex
N. H. Barton
OUTLINE
1. Molecular recombination
2. Rates of recombination
3. Linkage disequilibrium
4. What generates linkage disequilibria?
5. Recombination facilitates selection
Sex and recombination are among the most striking features of the living world, and they play a crucial role in allowing the evolution of complex adaptation. The sharing of genomes through the sexual union of different individuals requires elaborate behavioral and physiological adaptations. At the molecular level, the alignment of two DNA double helices, followed by their precise cutting and rejoining, is an extraordinary feat. Sex and recombination have diverse—and often surprising—evolutionary consequences: distinct sexes, elaborate mating displays, selfish genetic elements, and so on. Indeed, a substantial fraction of molecular evolution—as measured by the rate of protein evolution—is driven by sex. For example, the most striking changes along the lineage leading from our common ancestor with chimpanzees are in genes expressed in the testis, presumably influencing sexual selection between sperm. Although sex and its consequences are most obvious among eukaryotes, whose genes regularly pass through meiosis, sex is also important—and perhaps, essential—for bacteria and archaea, which often adapt to new environments (and to antibiotics) by bringing in genes from other lineages. The evolution of sex itself is discussed in another chapter (see chapter III.9). Here, I focus on the molecular mechanism of recombination, its effects on the composition of a population, and its interaction with other evolutionary processes—especially, with selection.
GLOSSARY
Allele. A particular form of a gene.
Centimorgan (cM). A distance on the genetic map that corresponds to a rate of recombination of c = 1%.
Epistasis. A state in which the value of a trait is not equal to the sum of effects of the genes that influence it.
Gene Conversion. During meiosis, a DNA heteroduplex forms; repair of mispaired heterozygous sites leads to an excess of one or the other allele.
Hitchhiking. The increase in a neutral allele that happens to be associated with a selectively favorable allele at another locus.
Introgression. Movement of genes from one genetic background to another, as a result of hybridization between individuals from distinct populations.
Linkage. Genes that are carried on the same chromosome are said to be linked.
Linkage Disequilibrium. Nonrandom associations between alleles at two or more genetic loci.
Meiosis. A cellular division process in eukaryotes in which gametes are produced, each with half the number of copies of each chromosome as the parents.
Recombination. The generation of new combinations of genes.
Sex. Production of offspring that are a mixture between two different parental genotypes.
1. MOLECULAR RECOMBINATION
Soon after the rediscovery of Mendel’s work in 1900, it was found that alleles at different genes sometimes tend to be inherited together. This phenomenon of linkage could be used to identify the linear order of genes on a chromosome. Consider a diploid parent that is heterozygous at two genes, A and B: one genome carries alleles ab, and the other, AB. The fraction of recombinant gametes (Ab and aB) that are passed on can be measured by crossing to a true-breeding stock. If the genes are on different chromosomes, this fraction is cAB = 50%. If they are closely linked on the same chromosome, then cAB is small, and measures the probability of a crossover between the two genes; by measuring rates of recombination between multiple alleles, one can determine their order on the chromosome; for example, if three genes lie close together, in the order ABC, then we expect that cAC = cAB + cBC. This relationship is not exact, because there can be multiple crossovers in an interval; an even number will yield a nonrecombinant gamete, while an odd number will produce a recombinant gamete. If crossovers occur independently, then the chance of observing an “effective” recombination between A and C is the chance of an effective recombination between A and B, but not between B and C, and vice versa: thus, cAC = cAB(1 – cBC) + (1 – cAB)cBC. Thus, genes A, Z that are far apart on the same chromosome may appear to be unlinked (i.e., cAZ ∼ 50%), but can be shown to be linked by mapping the genes between them. Distances on the genetic map are measured in centimorgans, with 1 cM corresponding to a 1 percent probability of crossover; 1 morgan = 100 cM.
The most important finding of classical genetics was that this linear genetic map corresponded to the linear arrangement of the chromosomes. Genetic mapping of model organisms, especially Drosophila, became ever more detailed, ultimately identifying the location of mutations within genes. Once the genetics of bacteria and their viruses was established in the 1950s, it was possible to map large numbers of mutations very precisely; by the mid-1960s, the order of mutations in the genetic map was shown to be the same as their order in the protein sequence, thus identifying the physical basis of the abstract alleles that had been mapped by classical genetics.
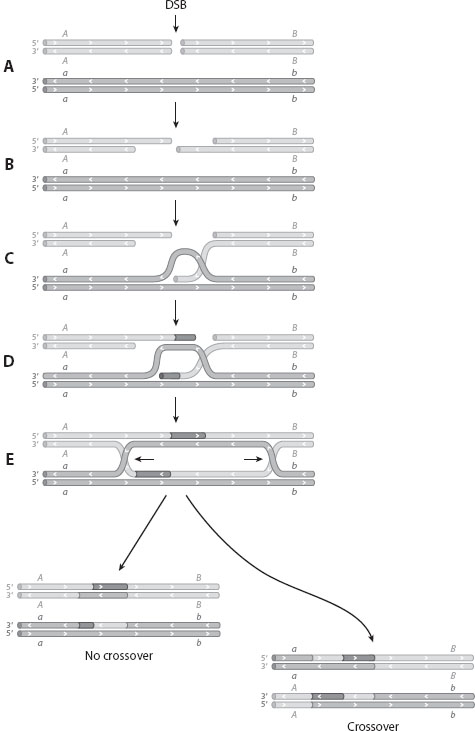
At the molecular level, the primary function of recombination is to repair double-stranded breaks in the DNA. If both strands of the double helix are broken, accurate repair is possible only if the broken strands can be aligned with an intact homologue, and the missing information copied across. An intermediate structure is formed, which can either be resolved into the two original strands, or lead to a crossover (figure 1). In either case, a small segment is copied from one fragment to the other, leading to gene conversion: any heterozygous sites within the segment will be “converted” into a homozygote. Molecular recombination is crucial for the repair of double-stranded breaks, and remarkably efficient: if human cells in tissue culture are irradiated with ultraviolet light, their chromosomes are broken into many separate fragments, yet such extreme damage can be almost perfectly repaired.
Figure 1. Molecular recombination between two homologous DNA strands is initiated by a double-stranded break (DSB). The outcome can be resolved in two ways: with or without generating a crossover between the two loci, A and B. In either case, a region of the DNA is converted to the homologous allele (orange). (A) Two DNA double helices are aligned, and a double-stranded break is made in one of them. (B) DNA is degraded to make two single-stranded tails. (C) One strand invades the intact double-stranded homologue. (D) New DNA is synthesized (orange), homologous to the invading allele. (E) Strands are rejoined, producing two “Holliday junctions” that can migrate along the DNA. These can each be resolved by breaking and rejoining the strands in two ways. (F) shows the outcome with no crossover, while (G) shows the outcome with a crossover. Note that in both cases, there is gene conversion (orange segments), in which heterozygous sites may become homozygous. (After Watson et al. 2004. Molecular Biology of the Gene. New York: Cold Spring Harbor Laboratory Press.)
In this article, I focus not on the process of molecular recombination but on its consequences for the evolution of populations. In this context, the terminology is different: recombination refers to any process that produces different combinations of genes, and includes the segregation of different chromosomes at meiosis, as well as crossing-over between homologous chromosomes, as described above. More broadly, the transfer of DNA from one bacterium to another is a form of recombination—albeit one that is asymmetrical, and involves only a small part of the genome. The term could even refer to the transfer of genes from the mitochondrial to the nuclear genome that followed the symbiotic union of an alphaproteobacterium with the ancestor of modern eukaryotes (see chapter II.12).
Sex has a slightly different meaning, referring to the coming together of genes from different individuals; the term may also be used broadly, applying to both prokaryotes and eukaryotes. If sexual union is followed by segregation of a single chromosome pair, to produce haploid offspring identical to the parents, then there has been sex but no recombination. Such an alternation between haploid and diploid phases is nevertheless important, since deleterious recessive alleles are masked in the diploid stage (see chapter IV.8).
2. RATES OF RECOMBINATION
The amount of recombination depends on the number of chromosomes, and on the length of the genetic map, summed over the chromosomes. In eukaryotes, these both vary widely; for example, Drosophila melanogaster has three chromosomes (plus a tiny nonrecombining chromosome) with a total map length in females of 2.4 morgans, while humans have 23 chromosome pairs, with a map length of about 35 morgans. The number of chromosomes ranges up to many hundreds, while the map length per chromosome is limited by the (usual) requirement that there be at least one crossover per chromosome arm, to ensure proper segregation of the chromosomes at meiosis; however, if the crossover is at the tip of the chromosome, then it may contribute negligible recombination among genes. There are exceptions: one reason Drosophila is a convenient model is that no crossing-over occurs at all in males.
Rates of recombination per base pair vary substantially between species, because both the length of the genetic map and the physical length of the genome vary greatly; however, in both humans and D. melanogaster, the rate of recombination between adjacent base pairs averages about 10-8 per generation (allowing for the absence of crossing-over in males in Drosophila). This average figure masks great heterogeneity across the genome; typically, the rate of recombination per base pair is much lower near the centromere. This broad-scale variation in recombination rate provides an opportunity to see the evolutionary effects of recombination. In Drosophila melanogaster, genetic diversity is strongly correlated with recombination rate—an observation that has stimulated much work on molecular evolution (see below, and chapter V.1).
On a still-smaller scale, variation in recombination rates can be extreme, with most recombination concentrated in “hot spots.” Classical genetics cannot measure such fine-scaled variation in recombination rates; recombination hot spots were first discovered by screening very large numbers of human sperm for recombinants between closely linked genetic markers. They were confirmed by population genetic methods (discussed below) that have allowed detailed maps of hot-spot locations across the human genome. Surveys of single-nucleotide polymorphisms in large pedigrees allow the precise location of recombination events, and they have shown that approximately 60 percent of these occur in hot spots that were estimated by population genetic methods.
The molecular basis of recombination hot spots has recently been determined, and it has interesting evolutionary implications. In mammals, they are initiated by PRDM9, a methyl transferase that marks specific sites on the chromosome; these are then targeted by Spo11, a highly conserved enzyme that initiates double-stranded breaks. The system is puzzling, because any binding site variant that increased the local recombination rate would tend to be eliminated by Spo11, and replaced by the alternative, less active allele—in other words, gene conversion would tend to favor “cold spots.” Hot spots are indeed transient, being polymorphic within the human population, and being almost entirely distinct from the hot spots found in chimpanzees. It is possible that a dynamic equilibrium exists between loss of hot spots by gene conversion, and the generation of new binding sites by mutation of the PRDM9 gene, to recognize different sequences; this latter process could itself be driven by broad-scale selection to maintain the optimal distribution of recombination across the chromosomes. This hypothesis is supported by the conservation of broad recombination patterns across species, despite the rapid evolution of the PRDM9 gene.
In bacteria, recombination appears to be a side effect of other processes: DNA from other bacteria may be acquired through transfer of plasmids, infection by viruses, or feeding on DNA from outside the cell. It occurs very rarely per cell division, but because bacterial populations are so large, the total number of recombination events can be large, and cause significant evolutionary consequences. Because only small fragments are transferred, they can come from very different lineages, and still function; for example, antibiotic resistance can be acquired from bacteria that are more than 20 percent divergent in sequence. Rates of transfer by direct uptake of DNA do decrease with sequence divergence; nevertheless, selectively favored alleles can be picked up from very distant lineages.
3. LINKAGE DISEQUILIBRIUM
This article concentrates on recombination (including both segregation of different chromosomes, and crossing-over within chromosomes). Recombination produces offspring gametes that carry new combinations of the alleles inherited from either parental genome, and so can generate enormous variability: if the parents differ at 40 sites, recombination can generate 240 ∼ 1012 combinations. However, at the level of the whole population, recombination has no effect on average if the alleles are already randomly combined; further shuffling makes no difference. Thus, recombination can alter the composition of the population only when there are nonrandom associations among alleles. Such associations are technically called linkage disequilibria—an unfortunate term, since there can be linkage disequilibria between alleles not physically linked on the same chromosome, and because there may be a steady level of associations at equilibrium. To understand how recombination influences evolution, we must understand how nonrandom associations can be produced.
We measure the strength of associations between alleles simply by the difference between the actual frequency of a particular pair of alleles, pAB, and the frequency expected if they combine at random, pApB: DAB = pAB – pApB, where pA and pB are the frequencies of the A and B alleles at the two loci. (An equivalent definition is that DAB = pABpab – paBpAb.) Linkage disequilibria are defined for particular sets of alleles, but it is always true that DAB = Dab = –DaB = –DAb. Describing multiple alleles, and more than two genes, is much more complicated: we can define coefficients of association among sets of alleles, but because so many genotypes are possible, a correspondingly large number of coefficients is needed. This fundamental problem makes the population genetics of multiple recombining loci difficult.
In the simple case of two loci with two alleles, recombination at a rate c simply reduces DAB by a factor (1 – c) in every generation; if the loci are unlinked, c = 1/2. Thus, linkage disequilibria will typically persist for approximately 1/c generations: alleles 10 cM apart will remain associated for about 10 generations while alleles I kb apart on the human genome recombine at about 10-5 per generation, and so stay together for about 100,000 generations—or about halfway back to our common ancestor with chimpanzees.
4. WHAT GENERATES LINKAGE DISEQUILIBRIA?
Recombination changes the composition of a population by breaking up statistical associations between alleles (linkage disequilibria); thus, its effect on evolution depends on what generates these associations. Mutation typically acts independently at different sites, and so breaks down associations.
Selection
Epistatic selection can favor certain combinations of alleles, and so generate linkage disequilibria; recombination will thus reduce fitness by destroying the favorable associations just built by selection (see chapter IV.5). An important example occurs near a sex-determining locus, where alleles that increase fitness in one or other sex will accumulate, leading to strong selection against recombination, and eventually, to sex chromosomes that do not recombine with each other at all (see chapter V.4).
Migration
Migration can also generate strong nonrandom associations. These are seen most strikingly in narrow hybrid zones between genetically distinct populations, in which strong linkage disequilibria are maintained by a balance between mixing and recombination (see chapter VI.6). Because such associations allow selection to act on whole sets of alleles, rather than on each one individually, they can greatly reduce the effective rate of gene exchange by increasing the effectiveness of selection. Migrants bring in sets of alleles that may not be adapted to the local environment or genetic background, and that are eliminated by strong selection. Recombination breaks up these associations, scattering incoming alleles across different native genetic backgrounds and making it harder to eliminate them.
Random Drift
Perhaps the most important, and the most general, cause of linkage disequilibria is random genetic drift (see chapter IV.1). As we look forward in time, there will be random fluctuations in linkage disequilibrium, simply because individuals that carry some combinations of alleles happen by chance to leave more offspring. The variance in linkage disequilibrium between two alleles depends on the number of recombination events between them per generation: var(DAB) is proportional to 1/(1 + 4Nec), where Ne is the effective population size.
Looking back, blocks of genome will share the same genealogical ancestry, to the extent that they have passed intact through meiosis without being broken apart by recombination. This correlation in ancestry is described by an elegant extension to the coalescent process. As we trace the ancestry of a segment of genome back through time, it may encounter a recombination event, such that portions are inherited from different ancestral genomes, and from then on back in time have separate genealogies. Different lineages may coalesce, so the blocks they carry become identical by descent from some ancestral genome—an event in which one parental genome passed a block of genome on to two offspring, both blocks surviving to be found in our present-day sample. In the simplest case of a single well-mixed population, with constant effective size, recombination and coalescence occur at rates that do not change through time. Thus, each present-day genome traces back to many different ancestral genomes, each contributing one or a few small segments.
Typically, blocks of genome of length c ∼ 1/2Ne will have the same ancestry. This can be seen directly in the genome sequence: although the proportion of sites that are heterozygous averages π = 4Neμ, this nucleotide diversity varies greatly along the genome, as the genealogy changes abruptly from one block to the next. In the human genome, the boundaries between such haplotype blocks are sharpened by recombination hot spots, but even if recombination rates were uniform, there would be abrupt changes as discrete recombination events occurred in the ancestry of the sample.
The generation of linkage disequilibria by random sampling is seen most strikingly in the spread of a new mutation. This mutation arises on a particular genome and carries a fragment of that one genome with it as it increases in frequency. If the mutation takes T generations to get to its present frequency, then on average, it will carry with it a block of map length c ∼ 1/T. Thus, the pattern of reduced diversity around such a new allele can give an estimate of the strength of the selection that drove it (see chapter V.14). The same argument applies both to favorable mutations that increase rapidly through selection—the classic process of hitchhiking (see chapter V.14)—and to deleterious mutations that increase through random drift, despite selection against them. These patterns allow us to estimate the age of some alleles. For example, this method has shown that the ΔF508, a deleterious allele that causes cystic fibrosis, arose approximately 3000 years ago.
5. RECOMBINATION FACILITATES SELECTION
In the examples above, of sex chromosomes and of migration with local adaptation, recombination reduced mean fitness. This, together with the obvious costs of sex and recombination, raises the question of why they are so widespread, at least among eukaryotes. At the end of the nineteenth century, August Weissman argued that sexual reproduction provided variation that would allow more efficient adaptation by natural selection, and this intuitive explanation was widely accepted; however, it is not at all easy to show exactly how sex and recombination can generate useful variation, and to show that it gives an advantage that can outweigh the various costs (see chapter III.9).
How might recombination facilitate selection? In principle, selection can be effective on a strictly asexual population, acting simply on the variation generated by mutation. Indeed, if mutations are fixed one at a time, recombination makes no difference, since only two alternative types at most are ever present together; however, if new favorable mutations arise while others are still on their way to fixation, there is strong interference between them—they can be brought together only by recombination. This difficulty can be avoided in very large populations, so large that many copies of every possible mutation arise in every generation. However, in more modestly sized populations (N<1/μ, say), the rate of adaptation may be limited primarily by the rate of recombination. Another way to look at the issue is to see that recombination randomizes alleles across genetic backgrounds of different quality, allowing selection to disentangle the effects of any particular allele from the effects of the random set of alleles with which it happens to find itself in any one individual.
To see the evolutionary role of recombination in a wider perspective, it is helpful to think of it in relation to speciation. The separation of populations into distinct biological species restricts the field of recombination, and so allows each species to specialize in different ecological niches. Hybrids produced by recombination between species are typically less fit, because they contain new combinations of alleles that have not been favored by selection and may be poorly adapted to the niche of either parent; however, speciation also reduces the size of the gene pool, making it more important to bring together the best combinations of mutations, whose supply is limited by the population size. It may be that regular sex and recombination have made it possible for eukaryotes to adapt to specialized niches, involving large body size and slow reproduction, despite the small population size that such specialization implies.
FURTHER READING
Barton, N. H., D.E.G. Briggs, J. A. Eisen, D. B. Goldstein, and N. Patel. 2007. Evolution. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. Chapter 15 describes how recombination interacts with random drift to shape the ancestry of genomes; chapter 23 shows how recombination evolves, and its consequences for other aspects of the genetic system.
Baudat, F., J. Buard, C. Grey, A. Fledel-Alon, C. Ober, M. Przeworski, G. Coop, and B. de Massy. 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327: 836–840. This landmark paper shows how recombination hot spots move over time, even though the overall pattern of recombination is conserved.
Burt, A. 2000. Sex, recombination and the efficacy of selection—Was Weissman right? Evolution 54: 337–351. A review that discusses Weissman’s explanation for the function of recombination in modern terms.
Charlesworth, B., and D. Charlesworth. 2010. Elements of Evolutionary Genetics. Greenwood Village, CO: Roberts & Company. Chapter 8 discusses the evolutionary role of recombination.
Coop, G., and M. Przeworski. 2007. An evolutionary view of human recombination. Nature Reviews Genetics 8: 23–34. A clear and concise review, with a focus on humans.
Whitehouse, H.L.K. 1973. Towards an Understanding of the Mechanism of Heredity. London: E Arnold. A classic text that gives an excellent historical account of the development of our understanding of recombination.