Molecular Evolution
Charles F. Aquadro
OUTLINE
1. What is molecular evolution and why does it occur?
2. Origins of molecular evolution, the molecular clock, and the neutral theory
3. Predictions of the neutral theory for variation within and between species
4. The impact of natural selection on molecular variation and evolution
5. Biological insights from the study of molecular evolution
6. Conclusions
The molecules of life (DNA, RNA, and proteins) change over evolutionary time. Much can be learned about evolutionary process and biological function from the rates and patterns of change in these molecules. The study of these changes is the study of molecular evolution. This chapter discusses why these molecules change, what can be learned about pattern and process from these changes, and how the changes in the molecules of life can be used to infer important past evolutionary events.
GLOSSARY
Fixation. The population process in which, either by drift or by natural selection, a new mutation increases in frequency in a population until it replaces all other variants and reaches a frequency of 100 percent.
Molecular Clock. When the time at which organisms last shared a common ancestor is plotted over time (e.g., estimated time from the fossil record), a roughly linear accumulation of genetic changes in DNA and the encoded proteins is frequently observed. In 1965, the rough linearity of this accumulation of change motivated Emile Zuckerkandl and Linus Pauling to propose that these data represent a sort of “molecular clock” by which the amount of molecular divergence could be used to infer the date of a last common ancestor.
Molecular Evolution. Changes in the molecules of life (DNA, RNA, and protein) over generations, for many reasons, including mutation, genetic drift, and natural selection, resulting in different sequences of these molecules in different descendant lineages. The study of molecular evolution is the study of the patterns and process of change that result in these different sequences.
Mutation. Heritable change in genetic material, including base substitutions, insertions, deletions, and rearrangements; the ultimate source of new variation in populations.
Neutral Theory. Short for neutral mutation–random drift theory of molecular evolution, proposing that molecular variation is equivalent in function (selectively neutral), making genetic drift the main driver of molecular genetic change in populations over time.
Positive Selection. New advantageous mutations, or changing environments, can present opportunities for new, or currently existing, variants to now have a reproductive advantage. They thus relentlessly increase in frequency until they fix in the relevant population. The selective pressure that leads to this fixation is termed positive selection.
Purifying Selection. Selection against harmful (i.e., deleterious) mutations “purifies” the population of these harmful variants. Such selection is due to constraint, typically to maintain a specific important biological function.
1. WHAT IS MOLECULAR EVOLUTION AND WHY DOES IT OCCUR?
The molecules of life (DNA, RNA, and proteins) are not static. They change over evolutionary time, hence the term molecular evolution. Some molecules evolve rapidly and some only very, very slowly. Their change is due to the interplay between two fundamental evolutionary processes: mutation and fixation. Before discussing these two processes, it is important to note that mutations come in two basic varieties: heritable mutations that occur in the germ line and are passed on in the genome of progeny, and somatic mutations that can occur in the process of cell division during normal growth and development. For example, if the latter affect the control of certain cellular processes, they can lead to uncontrolled cell growth and cancer. Molecular evolutionary studies have traditionally focused only on heritable genetic changes that accumulate within and between organisms.
Inherited mutations occur primarily during DNA replication in the production of gametes and introduce new genetic variants into the population. For animals and plants, the relevant genetic molecule is DNA. For some viruses, the heritable molecule is RNA. Certain segments of DNA or RNA genomes are translated into proteins by the cells, and some changes lead to changes in the encoded protein sequence, leading to molecular evolution at the amino acid level as well. Heritable mutations are largely considered to occur at random in time and space and location across our genome, and at a relatively constant rate, at least for many organisms (see chapter IV.2).
The second process of molecular evolution is fixation, which is fundamentally the “population” phase leading to molecular evolutionary change. Most new mutations are lost from populations as a result of chance (genetic drift; see chapter IV.1), or because they are harmful (deleterious). Mutations remain in populations because of chance or because they increase the reproductive success of offspring carrying them. Drift can lead to rapid changes in frequencies of mutations in very small populations, but is much less influential in large populations. Ultimately, the outcome of drift alone is always the loss or fixation of every new mutation; in other words, every mutation will eventually reach 0 or 100 percent frequency. Fixation means that the new mutation now replaces all previous variants present (segregating) in the population at a particular site in the genome.
Fixation can also be caused, or assisted, by natural selection. Selection acting to directly increase a variant frequency, as the result of an increased relative reproductive success or survival of individuals carrying it, is termed positive selection, or often simply adaptation. Such selection can cause rapid changes of allele frequencies in populations, over tens of generations, versus millions of generations by genetic drift alone in large populations. While the underlying mutation rate is thought not responsive to selective challenges, the fixation process is strongly influenced by selection.
Harmful or deleterious mutations, or ones that reduce reproductive output or success, often will not be fixed but rather reduced in frequency or eliminated from populations. This is known as purifying selection, and it prevents the otherwise-inexorable, but very slow, march of successive neutral mutation fixations over time in all finite populations.
2. ORIGINS OF MOLECULAR EVOLUTION, THE MOLECULAR CLOCK, AND THE NEUTRAL THEORY
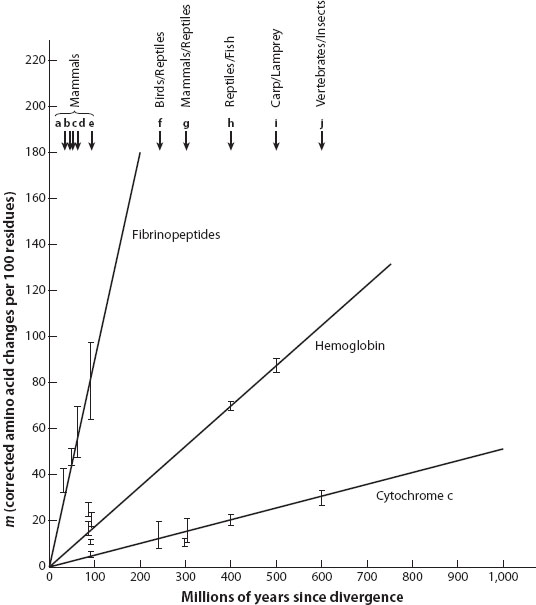
Molecular evolution as a field of study originated sort of accidentally, as biologists discovered how to determine the sequence of proteins and started collecting data from diverse organisms in the 1950s and 1960s. Key early data were for hemoglobin and histones, proteins chosen for their biomedical importance. The study of proteins was emphasized because of their clear functional relevance, and because methods were developed first for sequencing these biological molecules. Comparison of sequence divergence among proteins revealed two important patterns: specifically, fibrinopeptides, hemoglobin, and histones from various vertebrates with well-defined fossil records revealed that (1) different proteins evolved at different rates, but (2) each protein seemed to accumulate changes at a surprisingly consistent rate, a pattern that led to the concept of a “molecular clock” (figure 1).
Figure 1. Molecular clocks. Rates of amino acid substitution in three proteins: fibrinopeptides, hemoglobin, and cytochrome c. The number of amino acid differences (per 100 residues, and corrected for multiple changes at the same residue) is plotted for comparisons between various mammals, birds and reptiles, mammals and reptiles, reptiles and fish, carp and lamprey, and vertebrates and insects, all lineages of organisms for which fossil data provided estimates of the time since divergence. Note that while some comparisons of the three proteins are from the same pair of organisms and time of divergence, the three proteins are evolving at very different rates (i.e., fibrinopeptide the fastest, and cytochrome c the slowest). In addition, the rough linearity of the rate of accumulation of molecular divergence with time illustrates the molecular clock concept. (Modified from Richard E. Dickerson. 1971. The structure of cytochrome c and the rates of molecular evolution. Journal of Molecular Evolution 1: 26–45).
The term molecular clock refers to both a mechanism and a tool for evolutionary studies. As a mechanism, the presence of a roughly constant accumulation of change led to the inference that chance, not local adaptation, is the cause of much of the observed molecular change. As a tool, the presence of a clock representing a molecule also meant that if its rate could be calibrated with organisms of known age (e.g., from the fossil record), then observed sequence differences for organisms without a fossil record could also be “dated.”
Until the mid-1960s, most evolutionary biologists considered natural selection the primary determinant of evolutionary change. Genetic drift was considered important only in small populations; for example, drift played an important role in Wright’s shifting balance theory, but not as a primary driving force in evolution, rather as a source of new combinations of alleles on which selection could act; thus, drift was rarely considered, and most models focused on selection.
Several observations in the mid-1960s and early 1970s challenged the dominance of selection as the driver of evolution. First, high levels of protein polymorphism (i.e., variation within populations) were observed in fruit flies, humans, and bacteria. Could all that variation within populations really be maintained by natural selection? Second, extrapolating from available data, Motoo Kimura estimated that there were as many as two amino acid replacements per generation across the genome in mammals. Again, could selection really drive that many amino acid replacements to fixation without an intolerable reduction in population fitness? Third, Jack King and Thomas Jukes considered the genetic code with its built-in redundancy (meaning some nucleotide changes were “silent” and didn’t change the encoded protein sequence) and the conservative characteristics of many amino acid changes seen between proteins isolated from different organisms. They argued, like Kimura, that much of the variation observed in proteins within and between species did not alter function and therefore accumulated by mutation and genetic drift. Surely adaptation occurred, but Kimura argued that it occurred in only a small proportion of the genome at any one time and that natural selection was unlikely to account for the maintenance of extensive molecular variation observed within species and for the fixation of variation between species. It was also inferred that many mutations were in fact harmful and eliminated from populations, so those regions of genes and the genome that were functionally critical would remain largely invariant. If selection could not reasonably explain these observations, then it followed that there might indeed be a significant role for drift.
This broad concept became known as the neutral allele theory of molecular evolution, and it has formed a conceptual and model framework on which much of the current field has been based. Perhaps most important has been the recognition that all populations of organisms are finite in size, so that the stochastic process of genetic drift forms a background on which all other evolutionary forces act. The term neutral theory (as it is often called) can be misleading, as not all variation is selectively neutral; rather, the theory allows that a significant portion of new mutations are strongly deleterious and nearly immediately removed from the population. And the theory does allow for a limited number of adaptive mutations; however, the remaining mutations are selectively equivalent (neutral), and their dynamics are determined solely by genetic drift. Thus, the majority of variation we see within and between species is assumed to have no effect on fitness of organisms.
In the 1970s, the emergence of methods to directly sequence DNA, which were much less laborious than protein sequencing, began an inexorable shift to the study of DNA sequences in the field of molecular evolution. Not only does DNA sequence data provide an estimation of the frequency of variation at individual nucleotides, but the tight genetic linkage of adjacent nucleotides also means that sequences retain more of their evolutionary history. The availability of these correlated evolutionary histories allowed for the development of new statistical and computational approaches for testing models of molecular evolution, particularly the neutral theory. The field was no longer theory rich and data poor, as now the data began pouring in at an astounding rate. The ability to obtain larger sample sizes of DNA sequences has also increased the statistical power to discriminate models and to infer evolutionary history, demography, and the targets and magnitude of selection acting on the genome. The strict neutral theory was clearly an oversimplification but has provided the field with a valuable reminder of the importance of stochastic processes in all populations and a valuable null hypothesis against which to evaluate data.
3. PREDICTIONS OF THE NEUTRAL THEORY FOR VARIATION WITHIN AND BETWEEN SPECIES
The probability that a neutral allele will eventually become fixed is equal to its frequency in the population. And the rate at which new alleles become fixed in a population (the substitution rate) is essentially equal to the “neutral” mutation rate per generation. Thus, if the neutral mutation rate remains constant, so should the rate of evolution. For new neutral mutations destined to be fixed by drift, average time to fixation (in units of generations) is approximately four times the long-term population size. For mutations destined for loss, average time to loss is quite short. Thus, for large populations, we expect long times to fixation, and thus lots of “transient” genetic variation in populations drifting slowly through them. Together, these processes mean that the level of variation within species is a function of population size and mutation rate.
For a stable population, the balance of new mutations and loss or fixation by drift leads to an equilibrium level of variation. It can be shown that this level of variation is such that the probability that an average nucleotide site shows a difference between two randomly chosen chromosomes (or is heterozygous in a randomly chosen diploid sexually reproducing organism) is approximately equal to four times the long-term population size multiplied by the rate of mutation. The amount of divergence between two sequences sampled from two different species will be equal to the mutation rate times twice the time since speciation plus an additional amount equal to the expected number of differences between two randomly chosen chromosomes in the ancestral population. Because variation between species is but an extension of variation within species, and both are ultimately driven by mutation, then strictly neutral variation within species should be positively correlated with strictly neutral variation between species.
4. THE IMPACT OF NATURAL SELECTION ON MOLECULAR VARIATION AND EVOLUTION
Mutations that confer a fitness advantage will increase in frequency in the population because of positive selection. If the variant goes to a frequency of 100 percent, the population has now undergone a substitution of one variant for another (e.g., a new A has replaced the ancestral G nucleotide). Positive selection can lead to very rapid rates of fixation, orders of magnitude faster than the rate of fixation due to genetic drift alone.
Because adjacent nucleotides are tightly linked genetically, selection impacts not only the beneficial mutation but also the region of the genome in which that variant is located. Rapid fixation can therefore fix not only the favored variant but also the surrounding segment of the genome, resulting in a “selective sweep” and consequently a genomic region of initially no or very reduced adjacent neutral variation. Only over time will new mutations introduce variation back into this region. Perhaps surprisingly, the average divergence of linked neutral sites is unchanged from the neutral prediction. Such patterns provide much insight into the frequency and location of adaptive fixations throughout the genome (see chapter V.14).
While the rate of new mutations in the population is unchanged with natural selection, the fixation rate for beneficial mutations is dramatically increased, leading to increased sequence divergence between species for those specific sites under positive selection compared with adjacent neutral variants whose dynamics are determined by stochastic processes of genetic drift alone. The study of protein-coding sequences provides a particularly illustrative, and useful, example of how this contrasting pattern of positively selected, negatively selected, and neutral variation can be used to infer where and how natural selection has acted in the genome. Amino acids are encoded in mRNA in a triplet code of three nucleotides. While there are 61 possible combinations of three nucleotides that encode amino acids (three additional encode protein synthesis “stop” signals), there are only 20 common amino acids. Many amino acids are encoded by more than a single nucleotide triplet. Those triplet codons that encode the same amino acid differ by what is known as synonymous or silent variants. Those that result in a change in the encoded amino acid are termed nonsynonymous or replacement variants.
Since protein function is largely determined by its amino acid sequence, the fitness consequences of nonsynonymous mutations are much greater than those of synonymous mutations. Constraints on protein function result in strong purifying selection on nonsynonymous variants, preventing them from reaching substantial frequencies in populations (polymorphism) or from going to fixation (substitutions). These proteins evolve at very slow rates (e.g., histones in figure 1). Proteins with very relaxed constraints on amino acid sequence (e.g., fibrinopeptide, which are nonessential “spacer peptides” cleaved from fibrinogen in the clotting of blood, figure 1) evolve near the neutral rate of evolution. Some proteins play key roles in adaptation to new enzyme substrates or respond to biotic or abiotic challenges. Here, positive selection favoring new amino acid variants leads to the accelerated fixation of mutations. Some adaptive responses require repeated changes in protein sequence (such as at the antigen-binding sites of some immunity proteins), resulting in successive accelerated replacements. Somewhat counterintuitively, such positive and negative selection has little to no effect on the rates of substitution of adjacent strictly neutral variants. Thus, contrasting levels of variation and/or divergence at nonsynonymous to synonymous sites can provide estimates of the strength of both positive and purifying selection for a protein-coding gene. The ideal neutral benchmark is found in “dead genes” that no longer function (pseudogenes), in which levels of variation and divergence are usually close to those seen at synonymous sites. These contrasts of nonsynonymous and synonymous variation and divergence form the basis of several tests to detect natural selection acting on genomes and uncover the functional targets of that selection (see chapter V.14).
Not all mutations are simply strictly neutral, lethal, or strongly favored; rather, functional and population genetics studies have demonstrated that many mutations affect function only slightly, most in a slightly negative manner but sometimes improving function a bit. In these instances, whether a mutant acts as a neutral variant can be influenced by the population size. Consideration of the relative strength of natural selection and genetic drift reveals that if the difference in reproductive success (fitness) is less than the reciprocal of the long-term population size, then the mutant will behave as a neutral variant, even if it would have a (slight) selective advantage or disadvantage in an infinitely large population.
Data have also revealed just how much of observed DNA variation segregating in a population is nearly neutral, with much of it being slightly deleterious. One impact of this class of variants is that fluctuations in population size among lineages, or even along lineages, leads to fluctuations in the ability of natural selection to “see” these variants. This phenomenon is most clearly observed in the “generation time effect” observed in the molecular clock for some types of variants. For example, germ line mutations are most often caused by DNA replication; therefore, short-generation mammals have more synonymous mutations per year than do long-generation species; however, short-generation mammals also tend to have very large population sizes, so that selection is more efficient and thus could result in more nearly neutral mutations. As predicted then, rates of protein evolution are slower on a per generation basis in short-generation mammals than rates of substitution in long-generation mammals.
5. BIOLOGICAL INSIGHTS FROM THE STUDY OF MOLECULAR EVOLUTION
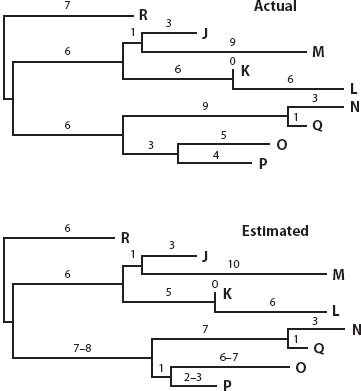
The comparison of sequences from different organisms, and particularly from short-generation organisms such as microbes and viruses, has provided one of the clearest illustrations of a core principle of evolution: descent with modification. For example, these valuable studies have provided real data sets with known phylogenies with which to evaluate the accuracy of statistical methods to estimate the phylogenetic relationships of organisms and their ancestors when we have only sequence data from the extant end points of the evolutionary process for study (figure 2). Experimental microbial and viral evolution studies are also allowing direct tests of evolutionary hypotheses regarding adaptation, including whether recurrent adaptation takes place by the same or novel mechanisms, particularly since samples of every step of the evolutionary process can be saved for future study and even functional reanalysis.
Figure 2. Mutational change in virus cultures demonstrates descent with modification and molecular evolution, and the molecular clock concept. Shown is a comparison of an actual “true” phylogeny of a virus (bacteriophage T7) with an estimated phylogeny constructed using only DNA sequences of the viruses in the experimental population. This population was initiated with one virus and split in binary fashion into several sequential derived lineages, the end points being denoted as the letters on the right side of the phylogeny. Numbers above the branches indicate the actual or estimated number of nucleotide substitutions that occurred along each branch, respectively. Actual substitutions were determined by sequencing 1091 base pairs of the ancestral viruses at each branch point in the tree. (Modified from David M. Hillis, John P. Huelsenbeck, and Clifford W. Cunningham. 1994. Application and accuracy of molecular phylogenetics. Science 264: 671–677).
With the introduction of the polymerase chain reaction (PCR) for amplifying specific genomic regions from very small samples (and even ancient bones, scat, and skins; see chapter V.15), coupled with wave after wave of advances in sequencing technology and automation, the field of molecular evolution has gone from being data limited to data overloaded, and no organism is now technically inaccessible. A striking example is the genomic study of microbes inhabiting our guts, our skin, the soil, and the oceans. Many of these microbes were not even known to science, since they could not be cultured in the lab or identified on the basis of morphology alone. Many are now known and identified only via their DNA sequences, and the relationships of these microbes are being revealed by the extent of sequence divergence from other known microbes.
Patterns and rates of molecular evolution can tell us about function in two ways. First, regions of genes and genomes that do not change are likely of critical and unchanging function. This basic principle of molecular evolutionary conservation has pervaded all of biology, and has been a guiding principle underlying the study of function in the exponentially growing number of genome sequences now being completed for virtually every type of plant, animal, microbe, and virus. Conserved regions of genes are what have changed biology from being simply organism focused to drawing on comparative functional data about a gene of interest from all studied forms. Second, regions of molecules that do change have turned out to include both those of little or no function, and those for which rapid change is itself adaptive. Distinguishing between adaptive change and relaxed functional constraint can be challenging; however, numerous statistical and computational methods have been developed over the last two decades allowing discrimination between positively selected change and relaxed constraint, and an area of very active research is that of evaluating the functional and fitness consequences of the adaptive fixations.
Another feature of molecular evolution that has emerged in the last decade is how much of the detected positive selection appears associated with conflict (e.g., “arms races” between hosts and pathogens), and not adaptation in the traditional sense (e.g., to new environments or nutrients). Microbial and viral pathogens clearly have driven, and still drive, much of the rapid molecular evolution in genomes, but additional conflicts between males and females, between host genomes and transposable elements, and even between hosts and endosymbionts have emerged as important drivers of rapid molecular evolution, including the evolution of new genes and new gene functions.
The role of noncoding portions of the genome in adaptation has also become strongly apparent in recent years. Variation in regulatory sequences (including enhancers, splicing machinery, and transcription factors) has been demonstrated as key to certain adaptive evolutionary changes in both molecules and phenotype (see chapters V.11 and V.12). Additionally, new and unanticipated types of functional sequences, such as small and long noncoding RNAs of various types (e.g., micro-RNAs, long noncoding RNAs [lncRNAs], and long intergenic noncoding RNAs [lincRNAs]), have been identified and not only demonstrate remarkable levels of evolutionary conservation and control of key developmental and cellular processes but also could underlie instances of adaptive change. Clearly, much remains to be discovered about genomes and the means and mechanisms by which the molecules of life evolve and adapt.
The study of the patterns and rates of evolution of biological molecules has provided data and results that clarify the relative roles of mutation, genetic drift, and natural selection in populations. The changes in these molecules underlie the evolution of organismal form and function, and the field of molecular evolution is alive with new discoveries about how genomes evolve and how observed molecular changes contribute to the stunning biological diversity of life we observe around us.
FURTHER READING
Graur, D., and W-H. Li. 2000. Fundamentals of Molecular Evolution. 2nd ed. Sunderland, MA: Sinauer. A solid introduction to many of the core principles of molecular evolution, though it was written in the “pre-genome” era and is thus missing many of the recent discoveries.
Kimura, M. 1983. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University Press. The classic and well-written summary of the neutral theory argued by Motoo Kimura, a central figure in its development.
Kumar, S. 2005. Molecular clocks: Four decades of evolution. Nature Reviews Genetics 6: 654–662. A comprehensive review of the development and use of the molecular clock concept from the 1960s through 2004.
Lynch, M. 2007. The Origins of Genome Architecture. Sunderland, MA: Sinauer. A recent summary of the patterns and mechanisms of the molecular evolution of genomes.
Nei, M., and S. Kumar. 2000. Molecular Evolution and Phylogenetics. New York: Oxford University Press. An accessible review of methods of analysis of DNA and protein-sequence data for molecular evolutionary studies, including the powerful and easy-to-use software MEGA, available for download at www.megasoftware.net/.
Nielsen, R. 2005. Molecular signatures of natural selection. Annual Reviews of Genetics 39: 197–218. A clear and concise summary of the statistical inference of natural selection from molecular data from within and between species.
Page, R.D.M., and E. C. Holmes. 1998. Molecular Evolution: A Phylogenetic Approach. Oxford: Blackwell Science. An introduction to molecular evolution and molecular population genetics with particularly clear figures and graphs.
Yang, Z. 2006. Computational Molecular Evolution. New York: Oxford University Press. A mathematically and statistically rigorous review of methods of analysis of DNA and protein-sequence data for molecular evolutionary studies.