IV.2
Mutation
Charles F. Baer
OUTLINE
1. The meaning of mutation
2. Types of mutations
3. Causes of mutation
4. Mutation and evolution: Basic principles
5. How random is mutation?
6. Variation in mutation rate: Among taxa
7. Variation in mutation rate: Within the genome
8. The mutational spectrum and mutational bias
9. Mutation, genome size, and genomic complexity
10. Mutation and extinction
11. Mutation and evolution: Other long-term consequences
Evolution depends on genetic differences among individuals, and ultimately all genetic variation has its origins in mutation. There are many ways in which DNA can change in a heritable fashion—from changes in single nucleotides, to rearrangements, to wholesale insertion or deletion of new sequences of DNA—and many causes of these mutations. Mutation rates vary among individuals, among species, among regions of the genome, and according to environmental conditions, and these mutation rates themselves can evolve. It may be energetically expensive to minimize mutation rates, and lineages with low mutation rates may slow their rates of evolution. On the other hand, a large fraction of mutations are deleterious, and on average the typical mutation can harm the function of organisms, even to the point where populations cease to survive.
GLOSSARY
Ectopic Recombination. Recombination between two nonhomologous sites in the genome. Recombination requires sequence similarity, but the similar sequences need not represent the same feature in the genome of the common ancestor.
Gene Conversion. Nonreciprocal exchange of genetic information from one homologous nonsister chromatid to the other in a heterozygous individual during meiotic recombination that results from template-directed repair of double-stranded breaks. The effect is to convert one allele in a heterozygote into the other. Gene conversion is a kind of mutation, but the state of the “mutant” allele depends on the state of the other allele present in the individual.
Mutation. A change in the nucleotide sequence of the genome from the parent to the offspring.
Transposable Element (TE). A genetic element that encodes the information necessary for its own replication, independently of the replication of the “host” individual’s genome. The behavior of a TE is analogous to the replication of a parasitic organism; TEs are examples of “selfish” genetic elements.
1. THE MEANING OF MUTATION
Many a textbook chapter, research paper, and grant proposal begins with the phrase “mutation is the ultimate source of genetic variation.” Without mutation, every locus will ultimately fix, the population will be devoid of genetic variation, and evolution will cease. Ultimately, all life everywhere would be (genetically) identical. This logic has a profound implication: all evolutionary innovation (see, e.g., chapter II.6) must ultimately have as its origin a single mutant allele in a single population. The mutant allele must initially increase in frequency in the population by genetic drift when rare, before proceeding to fixation.
The primary focus of this chapter is the ways in which mutation influences evolution; its secondary focus is the mechanisms by which mutation itself evolves (for a fuller exploration of theories on the evolution of mutation rate, see chapter III.9). Before embarking, it is useful to define exactly what is meant by mutation. Prior to the rediscovery of Mendel’s work, “mutation” referred to a heritable, discontinuous change in the phenotype, a so-called sport. Following the rediscovery of Mendel’s work, it was recognized that many, if not most, mutations obeyed Mendel’s laws. The Oxford dictionary defines mutation as “the changing of the structure of a gene, resulting in a variant form that may be transmitted to subsequent generations, caused by the alteration of single base units in DNA, or the deletion, insertion, or rearrangement of larger sections of genes or chromosomes.” From the perspective of evolution, the mutations that matter are heritable mutations. In organisms with a distinct germ line (e.g., animals), somatic mutations can harm the individual (e.g., cause cancer), but since they are not passed to the next generation, they have no evolutionary consequence beyond their effect on an individual bearer.
Because faithful transmission of biological information is necessary for life to continue, living organisms have evolved multiple mechanisms to ensure accurate replication of the genome. It is useful to consider the mutational process in terms of “input,” that is, damage to the DNA or replication errors, and “output” (i.e., “mutation” per se), which is the fraction of the input that makes it into the next generation after DNA repair. The distinction between input and output matters, because the mutational process can evolve in two ways, either by changing the input (e.g., by avoiding environmental mutagens), or by changing output (e.g., by evolving a proofreading polymerase).
2. TYPES OF MUTATIONS
Three fundamental types of mutation have been identified, each of which comes in several varieties and has a variety of causes, and consequences. Base substitutions occur when one nucleotide is substituted for another at the same homologous position in the genome. A base substitution may be either a transition (purine → purine or pyrimidine → pyrimidine) or a transversion (purine → pyrimidine, or vice versa). Insertions and deletions (collectively, indels) occur when an additional sequence is inserted into an existing sequence, or part of an existing sequence is deleted. Genome rearrangements, including inversions and translocations, occur when pieces of chromosomes change position in the genome. Inversion occurs when a piece of a chromosome becomes detached and reattaches in the opposite orientation (e.g., gene order goes from ABCDE to ADCBE, following inversion of the segment BCD. Translocation occurs when a piece of a chromosome becomes detached and is reattached either on a different chromosome or in a different place on the same chromosome. In eukaryotes, rearrangement can result in the suppression of recombination, either from mispairing at synapsis or from lethal recombinant genotypes resulting from improper gene dosage.
Two important classes of indels are short tandem repeats (STRs), and copy-number variants (CNVs). STRs are a particular type of CNV consisting of a short motif repeated one after another (“in tandem”) multiple times, and are highly mutable because the DNA polymerase tends to “slip” during replication, with the result that one of the resulting daughter strands contains either one (or more) additional repeat(s) or one (or more) fewer repeat(s). Mutation rates of STRs can be many orders of magnitude greater than base-substitution mutation rates. More generally, CNVs are sequences present in multiple copies in the genome that vary in number between (haploid) genomes; CNVs are generated by any mechanism capable of generating an indel mutation. An important feature of CNVs is their use as substrate for ectopic (nonhomologous) recombination.
An important source of CNVs is transposable elements (TEs), genetic elements that encode their own replication throughout the genome, independent of the host cell’s replication machinery; they are “selfish” elements because natural selection operating at the level of the TE will favor an increase in TE copy number, even to the detriment of the fitness of the host organism.
3. CAUSES OF MUTATION
Mutations may have their ultimate cause in factors either endogenous or exogenous to the organism. Replication errors, TEs, and free-radical by-products of metabolism are examples of the former; environmental mutagens are examples of the latter. Each comes with its own implications for evolution. The strategy of an organism wanting to avoid the deleterious consequences of exogenous mutation is simple: “don’t go there.” For example, to reduce the mutational input from incident UV radiation, an organism might evolve the choice of spending more time in the shade. Similarly, if circumstances favor an increase in metabolic rate, the organism will need to cope with the increased mutational input from metabolic by-products. One way to reduce the mutational output would be to evolve more efficient DNA repair; another possibility might be to increase free-radical “scavenging” mechanisms.
4. MUTATION AND EVOLUTION: BASIC PRINCIPLES
Evolution requires variation. All else equal, the more genetic variation produced by mutation, the more opportunity for evolution, and the faster evolution can proceed; however, that genetic variation comes with a cost: most mutations that are not neutral are deleterious, and only a relatively few mutations are beneficial.
Before turning to the many ways in which deleterious mutation influences evolution, consider neutral mutations, those with no effect on fitness. The simplest case is that of a single, bi-allelic locus (alleles A and a at frequencies p and q = 1 – p, respectively) in an infinite population in which A mutates to a with probability μ and a mutates to A with probability ν. At equilibrium, 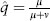 . If μ and ν are equal, at equilibrium the allele frequencies will be equal; if, say, μ is 10 times greater than ν, at equilibrium the population will consist of 10 times as many a alleles as A alleles. Less obvious is the timescale. It can be shown that the magnitude of change in allele frequency in one generation due to mutation is on the order of the mutation rate itself, typically a very small number.
. If μ and ν are equal, at equilibrium the allele frequencies will be equal; if, say, μ is 10 times greater than ν, at equilibrium the population will consist of 10 times as many a alleles as A alleles. Less obvious is the timescale. It can be shown that the magnitude of change in allele frequency in one generation due to mutation is on the order of the mutation rate itself, typically a very small number.
In a closed finite population at genetic equilibrium, the amount of “standing” genetic variation present at a single neutral locus, Ĥ (H represents “heterozygosity”), is proportional to the product of the mutation rate, μ, and the genetic effective population size, Ne (see chapter IV.1); that is, (H)^(N1eμ). This result is intuitive: the higher the mutation rate, the more genetic variation there will be, and similarly, the larger the population, the more genetic variation it can hold. If different groups harbor different amounts of genetic variation, it might be because they differ in Ne (usually the explanation of first resort) or because they differ in mutation rate, or both.
The role of mutation in determining the standing genetic variance for a quantitative trait (e.g., height) is determined jointly by the per locus mutation rate μ, the number of loci that affect the trait, n, and the phenotypic effects of mutant alleles, a. In the simplest case, that of a haploid organism, the genome-wide mutation rate for the trait is 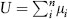 . The genetic variation introduced into the population by mutation each generation (the mutational variance, VM) is equal to the product of U and the expected squared effect of a new mutation, denoted as E(a2), so that VM = UE(a2). The same principles apply to diploids, except that dominance must be taken into consideration.
. The genetic variation introduced into the population by mutation each generation (the mutational variance, VM) is equal to the product of U and the expected squared effect of a new mutation, denoted as E(a2), so that VM = UE(a2). The same principles apply to diploids, except that dominance must be taken into consideration.
For a neutral quantitative trait, the standing genetic variance in a population, VG, is proportional to the product of effective population size (Ne) and the mutational variance, that is, VG ∝ NeVM. The scaling with Ne and mutation rate (μ) is the same as for single-locus variation, but the standing quantitative variance also depends on the number of loci that affect the trait (n, the mutational “target size”) and the average effects of alleles at those loci, E(a2).
Clearly, not all mutations are neutral; for example, many human genetic disorders are caused by mutations of large effect (usually recessive) at individual loci. For loci under directional selection, mutation adds genetic variation to the population and selection removes it; at equilibrium, referred to as mutation-selection balance (MSB), the two forces exactly offset. For a single haploid locus, the equilibrium frequency of the deleterious mutant allele, 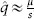 , where μ is the mutation rate from wild type to mutant and s is the selection coefficient against the mutant allele. This result is intuitive: the greater the mutation rate and the weaker the strength of selection, the greater the equilibrium frequency of the mutant allele (see chapter IV.5).
, where μ is the mutation rate from wild type to mutant and s is the selection coefficient against the mutant allele. This result is intuitive: the greater the mutation rate and the weaker the strength of selection, the greater the equilibrium frequency of the mutant allele (see chapter IV.5).
Similar reasoning applies to quantitative traits; many traits are not neutral, in which case natural selection would prefer the most-fit allele be fixed at every locus (balancing selection leads to a similar conclusion). At MSB the standing genetic variance, VG, is established by the counterbalancing effects of the input of genetic variation by mutation (VM) and the removal of deleterious alleles by natural selection; in many cases VG≈VM/S, where S represents the average selection coefficient against a mutant allele.
In finite populations, the effects of selection and Ne become entangled. If the strength of selection (s) acting on an allele is less than (about) 1/2Ne, the evolutionary dynamics are governed by genetic drift rather than selection; that is, the allele is said to be “nearly neutral” (see chapter IV.1). The strength of selection is an inherent property of an allele, whereas the efficiency of selection depends on the population size. The consequences of the relationship between efficiency of selection and Ne are profound; we return to this result throughout the chapter.
5. HOW RANDOM IS MUTATION?
Mutation is almost always assumed to be a “random” process, by which is meant that mutations do not occur based on the potential future effect on fitness. The conclusion that mutation is random in this regard stems from the pioneering work of Luria and Delbrück in the 1940s. It was known that when E. coli sensitive to the bacteriophage T1 were plated in the presence of T1, initially no colonies would grow on the plate, but eventually colonies would begin to appear, and those colonies consisted of resistant bacteria that bred true. The question was, Are the resistant cells derived from slow-growing mutants that existed in the population prior to plating in the presence of phage, or from mutations that occurred subsequent to exposure to phage? The question cut to the heart of evolutionary biology, because if the resistant mutants occurred only (or much more frequently) after exposure to phage, it would mean the environment directly influences the heritable genome in such a way as to increase the fitness of the affected organism—in which case evolution would be “Lamarckian” (although Darwin himself was Lamarckian in this regard, particularly in later editions of On the Origin of Species). Luria and Delbrück and others convincingly demonstrated that preexisting mutants were sufficient to explain the delayed population growth and there was no need to invoke acquired immunity.
In 1988, in a paper provocatively titled “The Origin of Mutants,” John Cairns and colleagues reopened the issue of “directed mutation.” Cairns et al. employed a different system, in which the selective agent—the ability to utilize lactose as a carbon source—does not kill sensitive (Lac-) cells but merely prevents their growth. An excess of Lac+ mutations occurring subsequent to the onset of selection, and moreover, the selective environment, apparently did not increase the rate of nonadaptive mutations. The basic feature—the appearance of adaptive mutations after the onset of selection unaccompanied by a simultaneous increase in nonadaptive mutations—was subsequently observed in other systems and precipitated a lively controversy. Cairns et al. initially speculated that starvation induced “highly variable” transcription, which, when coupled with reverse transcription, would result in eventual incorporation of the adaptive mutant into the genome. Although Cairns’s hypothetical mechanism was not borne out, other mechanisms were proposed. Barry Hall proposed that starvation might induce a transient state of hypermutation in a small subset of cells that would then revert to wild-type mutation rate after starvation was alleviated by an adaptive mutation. Alternatively, Roth and coworkers argue that apparently adaptive mutations can be explained by very slow growth in cells carrying additional copies of the critical genes, providing additional targets for beneficial mutations; the apparent high frequency of adaptive mutations is therefore a product of simple Darwinian selection plus increased mutational target size.
The mutation rate in E. coli and other microbes apparently does increase under various physiological stresses, including starvation. In many cases, the mechanism involves recombination induced by double-strand breaks in the DNA, followed by the (mutagenic) action of an inherently error-prone polymerase; however, it remains unclear whether the stress-induced increase in mutation represents an adaptation or is simply a feature of a sick organism functioning at a subpar level; evolutionary orthodoxy suggests the latter.
6. VARIATION IN MUTATION RATE: AMONG TAXA
Mutation rates vary among organisms, among genomic locations, among sequence motifs, and among nucleotides. At the outset, it is important to distinguish exactly what is meant by rate. Mutation rate may be expressed per genome replication, per generation, or per unit time. For unicellular microbes and viruses, mutation rates per replication and per generation are equivalent; for multicellular organisms they are not. Further, mutation rates can be expressed as per site, per gene, or per genome. From the perspective of natural selection, the relevant mutation rate is the rate per genome, per generation (U). Natural selection (usually) acts via individuals, whose fitness is integrated over the entire genome, and is manifested by its contribution to the next generation. All else equal, natural selection favors a reduction in U. There are two basic ways to effect a change in U. First, the per-site mutation rate may remain unchanged and the number of sites necessary to build the organism changes. Second, the number of sites may remain unchanged and the per site mutation rate changes. In organisms that undergo multiple rounds of genome replication per generation, the number of rounds of replication between generations may be changed.
Given that mutation rate varies among taxa, are there underlying regularities? The answer appears to be yes. In the early 1990s, Jan Drake observed that the per nucleotide, per generation mutation rate in DNA-based microbes (viruses and prokaryotes) varied nearly inversely with genome size across four orders of magnitude, leading to a nearly constant per genome mutation rate of about 0.003–0.004, which he and others argued must be due to the existence of a globally optimum mutation rate, presumably due to the existence of a cost of fidelity associated with replication speed.
As data from multicellular eukaryotes accumulated, it became apparent that the per nucleotide, per generation mutation rate varies positively with genome size in cellular organisms (prokaryotes and eukaryotes), although the per nucleotide, per replication mutation rate is similar between microbes and multicellular organisms. What could explain the remarkable difference in scaling within microbes, on the one hand, and among cellular organisms (including multicellular eukaryotes), on the other? Michael Lynch has argued that the difference is related to the relationship between genome size, body size, and Ne. Distilled, the argument is as follows: larger organisms have smaller population sizes, reducing the efficiency of natural selection to reduce mutation rate; at some point a further decrease in mutation rate provides such a small fitness advantage that selection cannot overcome drift. Although the absolute cost of fidelity can be the same among different groups (it need not be), groups differ in how close to the optimum (low) mutation rate they can get, based on population size.
7. VARIATION IN MUTATION RATE: WITHIN THE GENOME
Not all parts of the genome evolve at the same rate, nor do they harbor the same amount of genetic variation. One obvious possibility is that mutation rate varies consistently with particular features of the genome; another is that natural selection does. Three features stand out: level of transcription, chromatin architecture, and local recombination rate. Transcription per se is believed to influence the probability of mutation in two opposing ways. First, DNA is necessarily single stranded during transcription, and single-stranded DNA is more vulnerable to damage (transcription-induced mutation, TIM); the effect is more pronounced on the nontranscribed (coding) strand. Second, transcription-coupled repair (TCR) mechanisms repair damage to transcribed DNA, but the repair is primarily on the transcribed (noncoding) strand. Both TIM and TCR predict strand asymmetry, in which mutations are more likely on the coding strand; the degree of strand asymmetry has been shown to be positively correlated with transcription level.
Chromatin can be broadly classified as “open” or “closed,” based on the degree of compaction. In the human genome, regions of open chromatin are both gene rich and enriched for broadly expressed genes. Interestingly, mutation rates appear higher in closed than in open chromatin. The cause of the distinction is not known; a possibility is that open chromatin is more accessible to the DNA-repair machinery.
It is overwhelmingly clear that within-species polymorphism correlates positively with local recombination rate. There are two possible (nonexclusive) explanations. First, natural selection is more efficient in regions of high recombination (the Hill-Robertson effect). Second, recombination might be mutagenic. The “mutagenic recombination” hypothesis predicts that both polymorphism within species and genetic divergence between species should be correlated with local recombination rate, whereas selective hypotheses in general do not predict a relationship between recombination and divergence. Early evidence from Drosophila melanogaster found no association between recombination and divergence, which was taken as support for selection underpinning the relationship between polymorphism and recombination. At this writing the evidence must be considered equivocal, although it seems very likely that selection plays the predominant role.
8. THE MUTATIONAL SPECTRUM AND MUTATIONAL BIAS
To say that mutation is a random process with respect to fitness does not mean that all mutations are equally probable: numerous sources of mutational bias are known or suspected. For example, certain kinds of mutations are more likely to occur on the lagging strand during DNA synthesis (others are not), leading to base composition asymmetry over evolutionary time. Base pairs consisting of A:T are more likely to mutate to G:C or C:G base pair than vice versa, and transitions appear more common than transversions. Similarly, gene conversion is often biased such that A:T pairs are more often converted to G:C or C:G than vice versa. Most genomes are too GC rich, given the apparent extent of the mutational bias, suggesting that equilibrium base composition is established by mutation-gene conversion balance.
Genomes, even those of closely related taxa and of the same ploidy level, can vary substantially in size. Changes in genome size have their ultimate cause in mutation; the equilibrium must be established by the balance of insertions and deletions, mediated (maybe) by natural selection. TEs provide an obvious source of (selfish) insertion bias. There appears to be an overall bias toward small deletions in all taxa, although the extent of the bias is stronger in microbes. Natural selection must at some point establish a lower bound on genome size, and presumably an upper bound as well. Understanding the relative influences of mutational bias, genetic drift, and selection on the evolution of genome size is an important unresolved issue.
9. MUTATION, GENOME SIZE, AND GENOMIC COMPLEXITY
In 1971, Manfred Eigen introduced the concept of an error threshold, in which the relationship between mutation rate, genome size, and information content of the genome was first formalized. The basic idea is that the error (= mutation) rate puts an upper bound on the length of a biological sequence (e.g., an RNA virus); for a given mutation rate, natural selection will be unable to maintain a (functional) sequence longer than the critical length in the face of mutational loss of information. This finding, referred to as an “error catastrophe,” led to an apparent paradox, because mutation rates of RNA viruses, which lack proofreading capacity, appeared too high to allow the evolution of a proofreading enzyme in the first place. Importantly, this theory applies in an infinite population and is therefore a deterministic phenomenon. Various solutions to the paradox have been proposed, including the suggestion that there is no paradox, but the general inverse relationship between genome size and information content, on the one hand, and mutation rate, on the other, seems robust.
The consequences of the relationship between genome size, genome “complexity” (roughly defined as the number of features), mutational target size, and natural selection have been extensively explored by Lynch. In general, increasing the size and/or number of genomic features increases the probability of deleterious mutation. For natural selection to favor increasing the size of a feature, the selective benefit must outweigh the cost of increasing the mutational target size. Since mutations with selective effects s < 1/2Ne are effectively neutral, the ability to grow the genome is more constrained in organisms with large Ne (e.g., microbes). It seems intuitive that complex organisms must somehow require complex genomes, and the genomes of multicellular eukaryotes are arguably more complex than those of microbes. However, the direction of causality is not certain, and (employing a favorite phrase of evolutionary biologists) “theory predicts” that genome complexity should scale inversely with Ne.
10. MUTATION AND EXTINCTION
In 1964, H. J. Muller observed that finite populations of nonrecombining organisms would accumulate mutations by the combined action of mutation and drift. Once the least-loaded genome in the population is lost by drift, it can never be reconstituted (except by back mutation). Thus, mean fitness of finite asexual populations will steadily decay over time by the mechanism known as Muller’s ratchet. The long-term consequences of the ratchet in an ecological context have been investigated by Lynch, Lande, and others. Once fitness declines below the point at which individuals replace themselves on average, population size begins to decline and the rate of accumulation of deleterious mutations increases as selection becomes progressively less efficient, in a self-reinforcing process culminating in extinction, dubbed a mutational meltdown. Although the effect is more pronounced in asexual populations, sexual populations are not immune from the cumulative long-term effects of very slightly deleterious mutations.
11. MUTATION AND EVOLUTION: OTHER LONG-TERM CONSEQUENCES
Deleterious mutations have been implicated as a cause of or leading contributor to a wide variety of evolutionary phenomena that are difficult to explain, including the evolution of: sexual reproduction and recombination, ploidy, mating systems, sex chromosomes, sexual selection, and senescence, among others. Many of these topics are covered in more detail in other chapters; the reader is also encouraged to delve into the further reading suggested below.
FURTHER READING
Drake, J. W., B. Charlesworth, D. Charlesworth, and J. F. Crow. 1998. Rates of spontaneous mutation. Genetics 148: 1667–1686. Remains the most comprehensive review of the properties of spontaneous mutations across all domains of living organisms, and includes a very readable summary of the theoretical predictions regarding the evolution of mutation rate, circa turn of the century.
Loewe, L., and W. G. Hill, eds. 2010. The population genetics of mutations: Good, bad, and indifferent. Philosophical Transactions of the Royal Society B 365. A theme issue, on the occasion of Brian Charlesworth’s 65th birthday, dedicated to the population genetics of mutations. Filled with fascinating reading.
Luria, S. E., and M. Delbrück. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28: 491–511. In which it was first shown that mutations are random and not “directed” with respect to future fitness. Also an elegant example of the application of mathematical theory in biology.
Lynch, M. 2007. The Origins of Genome Architecture. Sunderland, MA: Sinauer. The grand synthesis of the Lynchian worldview, in which effective population size and slightly deleterious mutation are sufficient to explain many disparate features of gene and genome evolution.
Lynch, M. 2010. Evolution of the mutation rate. Trends in Genetics 26: 345–352. In which it is argued that random genetic drift, not a “cost of fidelity,” imposes the lower bound on the achievable mutation rate.
Prendergast, J.G.D., H. Campbell, N. Gilbert, M. G. Dunlop, W. A. Bickmore, and C.A.M. Semple. 2007. Chromatin structure and evolution in the human genome. BMC Evolutionary Biology 7: doi72 10.1186/1471-2148-7-72. Evidence that open and closed regions of chromatin have characteristically different mutation rates.
Sturtevant, A. F. 1937. Essays on evolution. 1. On the effects of selection on mutation rate. Quarterly Review of Biology 12: 467–477. Perhaps the first paper to clearly lay out the basic issues involved in the evolution of mutation rate, it remains extremely influential.
Wilke, C. O. 2005. Quasispecies theory in the context of population genetics. BMC Evolutionary Biology 5: doi44 10.1186/1471-2148-5-44. Concisely explains the relationship between the body of theoretical work concerning genome evolution derived from Eigen’s pioneering work, on the one hand, and mainstream theoretical population genetics, on the other hand.