Infections, Inflammogens, and Drugs
Publisher Summary
This chapter examines environmental inflammatory factors in vascular disease and dementia with a focus on infections, environmental inflammogens, and drugs that modulate both vascular disease and dementia. Infections and blood levels of inflammatory proteins are risk factors for future coronary events and possibly for dementia. When early age mortality is high, the survivors carry long-term infections that impair growth and accelerate mortality at later ages (“cohort morbidity phenotype”). Chronic infections, which are endured by most of the world’s human and animal populations, cause energy reallocation for host defense. Infections and inflammation may impair stem cell generation, with consequences to arterial and brain aging. Diet may introduce glycotoxins that stimulate inflammation. Some anti-inflammatory and anti-coagulant drugs may protect against coronary artery disease and certain cancers, and possibly also for Alzheimer disease. These “pharmacopleiotropies” implicate shared mechanisms in diverse diseases of aging.
2.1 INTRODUCTION
Chapter 1 emphasized the role of inflammatory processes in vascular disease, from early beginnings before birth. Alzheimer disease shares many of the same inflammatory changes, although cause and effect are less clear. This chapter follows the pathways of Fig. 1.2 further by examining the role of infections and inflammatory agents in vascular disease and Alzheimer disease and selected cancers. Pharmacologic interventions through NSAIDs and anti-coagulant drugs further establish the inflammatory mechanisms in vascular disease and may extend to Alzheimer disease. These examples are discussed in relation to Query II (Section 1.1) that inflammation causes bystander damage and Query III that environmental pathogens and inflammogens influence chronic diseases with inflammatory processes through bystander damage (Section 1.4).
The environmental role in these diverse, slowly developing diseases remains counter-current to traditional thinking, because in general, Alzheimer disease, cancer, and vascular disease are not ‘infectious,’ by Koch’s postulates. That is, with few exceptions for these diseases, infectious agents cannot be isolated, and the disease cannot be transferred and propagated to a test animal.
2.2 VASCULAR DISEASE
2.2.1 Historical Associations of Infections and Vascular Mortality
The traditional risk factors for vascular disease (hypertension, obesity, elevated LDL cholesterol, smoking) do not explain about 35% of cases (Section 1.5.3.2). From epidemiologic and pathologic studies, chronic infections may be primary causes or co-factors of inflammation in vascular disease. This controversial concept has been discussed for a century or more (Frothingham, 1911). The hypothesis of inflammation as a co-factor is strongest in human arterial disease, because prodromal microscopic foci of oxidized lipids and activated macrophages are present before birth (Section 1.5.3.1). The evidence for the role of infections in arterial disease, while considerable and supported by animal models, is still largely circumstantial for humans.
Rheumatic heart disease is a classic example of infection-caused heart disease, but with a different etiology than most cardiovascular cases. Until about 1950, rheumatic fever from ‘strep’ infections was still an important cause of damage to heart valves. In particular, streptococcal A substrains cause high incidence of endocarditis and mitral valve scarring (Bispo, 2000; Stollerman, 1997; Wilson, 1940). Rheumatic fever with mitral damage is life-shortening (Jones, 1956). In the 1930s, for example, few survivors of childhood infections lived to age 40, and most died within 15 years of infection (Wilson, 1940, p. 272). Rheumatic heart disease has become rarer in developed countries from public health improvements and, then after 1950, the availability of antibiotics. However, heart valves without rheumatic disease often harbor a diverse bacterial flora (see below).
Eileen Crimmins and I hypothesize that historical and modern levels of early infection are major determinants of adult vascular disease (Crimmins and Finch, 2006a,b; Finch and Crimmins, 2004, 2005). More generally, historical and modern populations also show associations of infections with later mortality. In some rural parishes of 17th century Sweden, high early infectious mortality was followed by high late life mortality among survivors (Bengtsson and Lindstrom, 2000; Bengtsson and Lindstrom, 2003). Among U.S. Civil War veterans, infectious disease in early adulthood has been associated with heart and respiratory problems after age 50 (Costa, 2000). Cardiovascular disease was twice as prevalent among older Army veterans born before 1845 compared to veterans born in the early 20th century (Fogel and Costa 1997; Fogel, 2004). In Norway 1896–1925, infant mortality, which is a proxy for exposure to infections, correlated strongly with arteriosclerotic deaths 40–69 years later (Forsdahl, 1977) (Fig. 2.1).
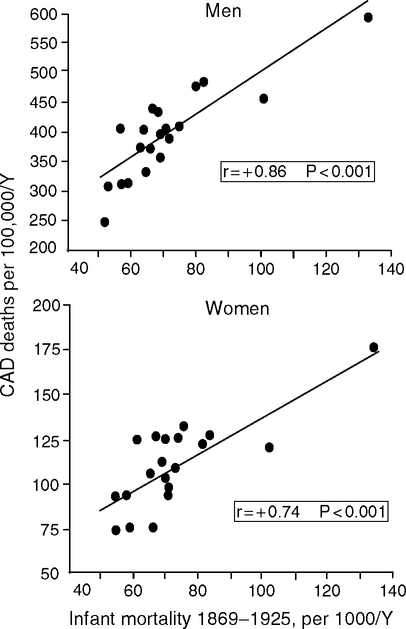
In the United States 1961–1971, adult cardiovascular disease is also associated with birth cohort levels of infant diarrhea and enteritis (Buck and Simpson, 1982). Other examples are discussed in Crimmins and Finch (2006a). Considered over most of the 20th century, the associations of prior infections on later mortality may explain up to nearly 25% of the decline of both morbid and mortal conditions at later ages (Costa, 2000). Relationships of early and later age mortality in birth cohorts are developed further below.
2.2.2 Modern Serologic Associations
Stepping forward, we have access to individual histories of infections through persistent antibodies. Serologic associations with cardiovascular disease were first noted in 1987 for cytomegalovirus (CMV) (Adam et al, 1987), soon followed by Chlamydia1 pneumoniae in 1988 (Saikku et al, 1988). Other associations include the ubiquitous Helicobacter pylori and Mycoplasma pneumoniae; and cytomegalovirus (CMV), hepatitis virus A and -C viruses (HAV, HCV), and herpes simplex virus (HSV-1 and -2) (Belland et al, 2004; Campbell and Kou, 2004; Stassen et al, 2006; Vassalle et al, 2004). Cerebrovascular disease is also associated with C. pneumoniae and H. pylori (CagA strains) (Lindsberg and Grau, 2003). Carotid thickness correlates with antibodies to E. coli endotoxin (LPS) (Xu, 2000), while anti-LPS antibodies correlate with antibodies to oxidized LDL (Mayr et al, 2006). The list grows.
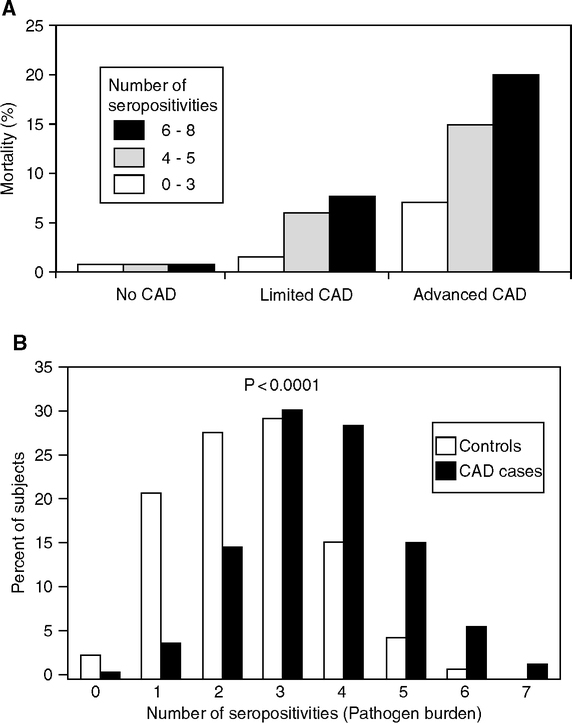
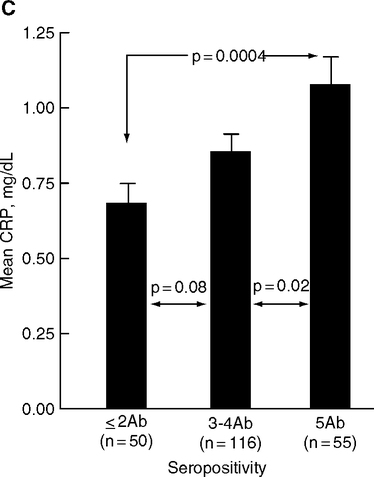
In the AtheroGene Study (Mainz and Paris), cardiovascular mortality and coronary stenosis were 2–3-fold higher in patients seropositive for ≥ 4 pathogens, relative to those with 0 to 3 seropositivities, with the highest odds ratios for C. pneumoniae and M. pneumoniae (Espinola-Klein et al, 2002a, b; Georges, 2003) (Fig. 2.2A, B). Carotid and femoral artery thickening (IMT) are greater in individuals with chronic infections who also carry proinflammatory alleles of IL-6, IL-1 receptors, and the endotoxin receptor CD-14 (Bruneck Study, northern Italy) (Markus et al, 2006). Blood levels of C-reactive protein (CRP), an inflammatory protein and strong risk indicator of coronary artery disease (Section 1.5, Fig. 1.16B), may also correlate with the number of different seropositivities (Georges et al, 2003; Zhu et al, 2000) (Fig. 2.2C). However, others did not find these serological associations with CRP elevations (Epstein et al, 2000; Lindsberg and Grau, 2003). This is not surprising, because seropositivity often persists long after an infection has subsided and transient elevations of CRP have subsided. Over the life span, the majority of adults become seropositive for C. pneumoniae and CMV (Almanzar et al, 2005; Miyashita et al, 2002).
Epidemiological associations of infections and vascular disease are increasingly supported by clinical studies and animal models (Campbell and Kuo, 2003; Coughlin and Camerer, 2003; Libby, 2003; Liuba et al, 2003). C. pneumoniae illustrates several key issues. This gram-negative bacterial pathogen grows only as an intracellular parasite. Infections typically begin in lungs and may propagate systemically to the vasculature by circulating macrophages. Infections are ubiquitous and reinfections very common (Belland et al, 2004; Campbell and Kuo, 2004; Grayston, 2000). C. pneumoniae is notorious for its broad cell targets, including endothelia, macrophages, and smooth muscle cells of atheromas. It resists antibiotics, which can suppress normal replication without eradicating its effects. Dead C. pneumoniae still activate the transcription factor NF-kB in endothelial cells, which could promote atherogenesis without active infection (Baer et al, 2003). C. pneumoniae are detected in the majority of atheromas by immunological, genomic, or ultrastructural criteria, but not in healthy arteries (Muhlestein et al, 1996; Shor et al, 1998; Shor, 2001). Heart valves tend to have more C. pneumoniae and other pathogens, in diseased than normal hearts (Juvonen et al, 1998; Nilsson et al, 2005; Nystrom-Rosander et al, 2003). Live C. pneumoniae was cultured from vascular tissues from some cardiac patients (Belland et al, 2004; Campbell and Kuo, 2004). T cells cultured from atherosclerotic carotids were immunopositive in 40% of 17 patients (Mosorin, 2000). These individual variations may arise from successful elimination of the pathogen by the host. The suppression of pathogen growth by antibiotics or cell stress may also add to these variations (Belland et al, 2004; Campbell and Kuo, 2004). However, assay criteria for C. pneumoniae are not well standardized and detectability varies from 0–100% (Kalayoglu et al, 2002; Peeling et al, 2000). The high genetic diversity of C. pneumoniae (Belland et al, 2004) may also contribute to variability. Some argue that C. pneumoniae and H. pylori in vascular lesions are an epiphenomenon because damaged or necrotic tissues, such as found in vascular plaques, are vulnerable to superinfections (Black, 2003). It is hard to prove the causal role of infections in atherogenesis because of their earthly ubiquity— everyone experiences infections (Belland et al, 2004; Campbell and Kou, 2004).
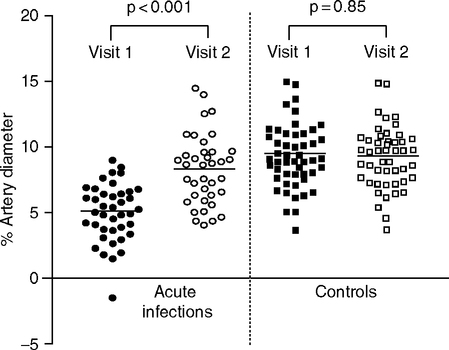
Peripheral arteries also show effects of infections. Children with acute respiratory infections had impaired regulation of the brachial artery endothelium, by the flow-mediated vasodilation test (Avon Longitudinal Study of Parents and Children, or ALSPAC Study, Fig. 2.3). The effects of infection may have persisted for a year in some individuals (the statistical significance was P<0.06) (Charakida et al, 2005). Longitudinal follow-up continues. Studies of children are valuable because seropositivities are less frequent than in adults.
Antibiotics give another test of the infection hypothesis. The large WIZARD trial [weekly intervention with zithromax (azithromycin) for atherosclerosis and its related disorders] is so far inconclusive (Dunne, 2000). Experimental design is difficult, because the treatments may be most effective early during infections (de Kruif et al, 2005). Other long-term studies include the Azithromycin and Coronary Events (ACES) (Belland et al, 2004) and the Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT) (Campbell and Kuo, 2004). The first placebocontrolled, double-blind, randomized clinical trial of antibiotics on C. pneumoniae in vascular tissue was inconclusive (Berg et al, 2005). Although 81% of cardiac bypass patients were seropositive, C. pneumoniae DNA was not present in plaques of patients with advanced CAD; antibiotic treatment did not alter seropositivity.
Animal models show that infections may have greater synergy with arterial disease when lipids are elevated, as is common during infections (Section 1.4). In rodents, rabbits, and pigs, C. pneumoniae accelerated atherogenesis, but only when the models were made hyperlipidemic (Belland et al, 2004, de Kruif et al, 2005; Liuba et al, 2003a,b,c). Chronic endotoxin also required hyperlipidemia to accelerate atherogenesis (Engelmann et al, 2006). The apoE-knockout (–/–) mouse is an important model, with greatly elevated cholesterol on non-atherogenic diets that promote progressive arterial lesions not found in normal mice. ApoE knockouts develop aortic plaques by 4 months, followed by vascular rigidity and aneurysms (Wang, 2005, Wouters et al, 2005). Moreover, the lipidemia-induced lesions depend on pathogen-signaling pathways via Toll-like receptors (TLRs) linked to MyD88, an adaptor that activates kinases (Laberge et al, 2005) (Chapter 5, Fig. 5.4). The double apoE and MyD88 knockout mouse had much small aortic lesions, with fewer macrophages and lower chemokines In apoE knockouts, C. pneumoniae caused rapid vascular endothelial damage (aortic contractility, 2–6 weeks after infection) (Liuba et al, 2000). Subsequently, the arterial wall thickens with increased ROS production. The convergence of hyperlipidemia in infection and arterial disease through pathogen-activated pathways suggests that atherogenesis is bystander outcome of the indispensable host defense mechanisms.
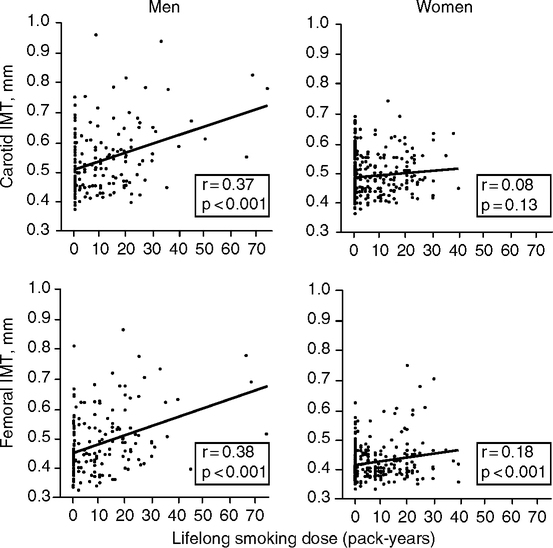
Future case control studies with longitudinal follow-up may be more conclusive. We may learn how to quantify effects of infections on vascular damage by the intensity and duration. There could be a threshold for acute infections of sufficient brevity that do not cause enduring damage. We may anticipate some dose-duration relationships in chronic subclinical infections and arterial disease that are like the ‘pack-years of smoking’ in relation to carotid thickening, as discussed below (Fig. 2.6) and lung cancer. The pathogen burden is indicated by the scaling of vascular event risk to the number of seropositivities, discussed above (Espinola-Klein, 2002; Georges et al, 2003; Zhu et al, 2000). Both research groups use similar terminology, ‘pathologic burden’ (Zhu et al, 2000) and ‘infectious burden’ (Espinola-Klein, 2002), to represent seropositivities, which does not inform on whether infections are active. I suggest the alternative term inflammatory burden to more comprehensively represent these long-term inflammatory influences. Besides infections, the inflammatory burden includes non-infectious inflammogens such as smoke and other aerosols and dietary AGEs produced during cooking (see below). These complexities are well expressed by Stephen Epstein and colleagues:
Given that atherosclerosis is a multifactorial disease, Koch’s postulates to establish causality will never be satisfied. These postulates … assume a single pathogen, require that all patients with the disease must have evidence of being infected with the casual agent and that all the infected develop the disease. In contrast … infectious agents are … neither necessary nor sufficient for [vascular] disease development … proof of causality can be achieved only in terms of probability rather than as certainty. (Epstein et al, 1999, p. e26).
2.3 INFECTIONS FROM THE CENTRAL TUBE: METCHNIKOFF REVISITED
A century ago, Metchnikoff suggested that autointoxication by microbial toxins in the intestinal flora causes chronic poisoning of body cells and premature death (Metchnikoff, 1901, Podolsky, 1998). Recent evidence implicates bacterial leakage from periodontal disease in vascular disease. Moreover, I suggest the lower gut should also be considered in bacterial leakage, which could be a factor in elevated circulating acute phase proteins during aging.
2.3.1 Humans: Leakage from Periodontal Disease and Possibly the Lower Intestine
The mouth normally harbors several hundred bacterial species, mostly as very high density biofilms on teeth. About 10 species of gram-negative anaerobes may be the main pathogens in vascular disease, particularly Porphyromonas gingivalis and Actinobacillus actinomycetemcomitans (Asikainen and Alaluusua, 1993). Their subgingival location is less accessible to antibiotics. We are unavoidably exposed to this high-density flora: Even tooth brushing and flossing can cause transient bacteremia (Carroll and Sebor, 1980, Slots, 2003).
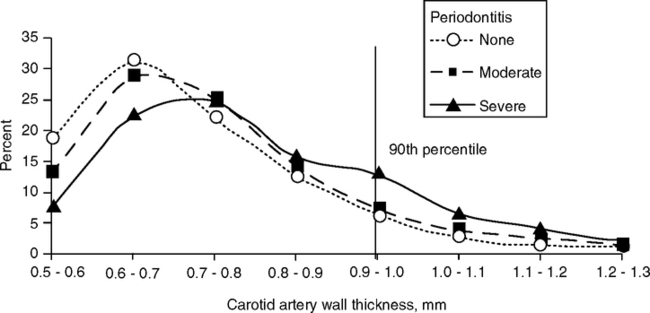
The evidence for oral-vascular disease relationships is controversial. In the Atherosclerosis Risk in Communities Study (ARIC Study), severe periodontal disease was associated with thick carotid walls (Fig. 2.4); the effect was greater in men than women (Odds ratio, OR 1.46, range 1.18–1.81) (Beck et al, 2001, 2005; Beck and Offenbacher, 2001). In ARIC (Slade et al, 2003) and other studies, serum CRP, fibrinogen, and IL-6 tended to be elevated in individuals with periodontitis who were otherwise healthy (Chun et al, 2005; D’Aiuto et al, 2004; Schwahn et al, 2004). In atheromas from vascular surgery, nearly half contained DNA from at least one periodontal pathogen (Haraszthy et al, 2000).
The periodontitis-vascular association is experimentally supported. In rabbits, periodontitis induced by P. gingivalis increased aortic lipid deposits in proportion to the severity of periodontitis (Jain et al, 2003). P. gingivalis generally forms biofilms beneath the gingiva and can invade oral epithelia and vascular endothelial cells. Activation of Toll receptors by P. gingivalis is associated with increased IL-1, TNFa, prostaglandin E2, and leukocyte adhesion molecules (ICAM-1, VCAM-1) (Choi et al, 2005a; Chun et al, 2005; Hajishengallis et al, 2004).
The associations with vascular disease are considered circumstantial (see the analysis of 14 studies) (Kolltveit and Eriksen, 2001), because few studies measured infections by serology or bioassay (Danesh et al, 1999); smoking adds other confounds (Hujoel et al, 2000). In ARIC, 68% were seropositive for one or more of 17 bacterial species associated with periodontal disease (Beck et al, 2005). High antibody titers to >1 oral pathogen were associated with higher prevalence of cardiovascular disease, particularly in never-smokers. However, there were no associations of periodontal disease with prevalent cardiovascular disease after adjusting for covariates. Prospective studies should include the individual histories of oral health, smoking, and other lifestyle covariates; multiple time samples of serology for the spectrum of major pathogens; and screening for genetic polymorphisms in IL-1a and IL-1β, which are associated with risk of periodontal disease (Lopez et al, 2005).
Diverse microbial ‘communities’ reside in the mouth and lower intestine. The intermediate gut is normally quite sterile, stomach through jejunum (Lin, 2004). The gut epithelial cells in the crypts of Lieberkuhn have tight junctions (zona occludens) that maintain characteristic epithelial cell polarity as part of the barrier to the body cavity and prevent leakage of gut contents (Mullin et al, 2005). Cholera and other pathogenic bacteria alter this vital tight junction barrier. In aging rats, tight junctions become leakier, as assayed by transcolonic epithelial permeability; these aging effects were greater on high fat diets (Mullin et al, 2002) (Fig. 2.5A). The aging gut may have increased leakage, allowing entry of endotoxins into the circulation. As a precedent, CRP elevations are common in inflammatory bowel disease (Poullis et al, 2002). At some threshold, leakage of endotoxins could cause elevations of blood CRP and other acute phase proteins (Section 1.7).
The increase of colonic permeability with aging may be due to aberrant crypts (Mullin et al, 2002). Aberrant crypts and villi with cellular dysplasia and loss of epithelial cell polarity increase during aging in the gut of rodents (Mullin et al, 2002) (Fig. 2.5B) and humans (Finch and Kirkwood, 2000, pp. 132–137; Shpitz et al, 1998; Takayama et al, 1998). Stem cell depletion may contribute to these aging changes. Epithelial cells in the crypts of Lieberkuhn proliferate throughout life and are extruded at the tips of the intestinal villi. The crypt stem cells (Potten et al, 2001; Potten et al, 2003) apparently become stochastically depleted during aging (Finch and Kirkwood, 2000, pp. 132–137; Martin, 1998). Radiation and some carcinogens accelerate the loss of stem cells and increase the incidence of abnormal crypts (Magnuson et al, 2000; Martin, 1998). Alternately, TNFa can alter tight-junction permeability via NF-kB activation, as implicated in Crohn’s disease and other chronic intestinal inflammations. Mucosal layer breakdown is not necessary for inflammatory transients to cause gut leakage of potential significance to arterial disease (Lin, 2004).
Obesity and diabetes predispose to chronic low-grade infections, which are discussed with effects of diet restriction in Section 3.2.4. The burden of infections (HSV-1 and -2, enteroviruses) shows correlations with insulin resistance particularly in those with C. pneumoniae seropositivity. Thus, metabolic adaptive responses to low-grade infections could be atherogenic by altering insulin sensitivity (Fernandez-Real, 2006). Moreover, the sensitivity to low-grade infections may be associated with inflammatory gene variants.
2.3.2 Worms and Flies as Models for Human Intestinal Microbial Intrusion
In the worm model of aging, bacterial autotoxicity may be a proximal cause of death during aging (Garigan et al, 2002; Gems and Riddle, 2000; Lithgow, 2003; Mallo et al, 2002) (also discussed in Section 5.2). Wads of bacteria pack the pharynx in aging worms and there is bacterial overgrowth in the pharynx and intestine (Garigan et al, 2002) (Fig. 2.5C)—“… the final coup de grace is bacterial invasion” (Lithgow, 2003, p. 16). The constipation of aging worms may model aspects of human inflammatory bowel syndrome, in which bacterial overgrowth into the normally sterile small intestine causes chronic inflammation (Lin, 2004; Pimentel et al, 2000). Several long-lived worm mutant resist pathogenic bacteria (Garsin et al, 2003) (Section 5.5.2).
In the standard culture conditions, worms are fed on live E. coli strain OP50 (Brenner, 1974). However E. coli OP50 may not be the optimum food because life spans may be longer and constipation lessened on diets of some species of yeast (Mylonakis et al, 2002a) or diets of the soil bacterium Bacillus subtilis, which may be more natural foods (Garsin et al, 2003). The concern that live E. coli OP50 is mildly pathogenic is now 40 years old (Croll and Yarwood, 1977; De Cuyper and Vanfleteren, 1982; Hansen et al, 1964): “The longer life span in the absence of bacteria suggests a possible toxicity of bacterial products in the monoxenic cultures” (Hansen and Yarwood, 1964, p. 629).
Elimination of live bacteria from the diet increases life spans. A diet of UV-killed bacteria delayed the pharyngeal pack-up of bacteria (Garigan et al, 2002) and increased life spans up to 55%, without loss of fecundity (Gems and Riddle, 2000). Moreover, axenic growth on sterile media supplemented with nutrients doubled the life span (Croll et al, 1977; De Cuyper and Vanfleteren, 1982; Houthoofd et al, 2002; Houthoofd et al, 2004). The axenic cultures maintained the rate of pharyngeal pumping to later ages and increased stress resistance, but at the expense of lower fecundity (Croll et al, 1977; Houthoofd et al, 2002). Switching from axenic to bacterial media after larval maturation eliminated the longevity benefit; conversely, raising larvae on bacteria followed by brief antibiotic treatment before transfer to axenic media increased longevity almost as much as growth on sterile media throughout life (De Cuyper and Vanfleteren, 1982). However, these benefits are due not only to the elimination of bacterial toxicity, because this axenic medium was deficient in ubiquinone, a micronutrient obtained from the bacterial diet (Jonassen et al, 2003; Larsen et al, 2002).
These findings raise uncomfortable questions about artifacts from standard lab conditions that are widespread in lab models. I argue that all of our experimental models adapted to the lab should be scrutinized for atypical outcomes of aging. Lab husbandry has eliminated most infections and provides a uniform quality ad lib diet rarely found over the life span in nature. Moreover, our highly inbred lab models were initially selected for early reproduction and high fecundity that may be atypical of the evolutionary background. These concerns will be discussed further in the next chapter in interpreting the obesity common in lab rodents.
Flies also show the importance of enteric microbes to aging. In Drosophila melanogaster antibiotics given later in life increased life span by 30% (Brummel et al, 2004). Cell changes in the aging fly gut are consistent with the leakage of gut bacteria later in life. Aging intestinal epithelial cells accumulate virus-like particles (Anton-Erxleben et al, 1983). In the aging housefly (Musca domestica), intestinal cells accumulate concretions (Sohal et al, 1977) and lipid inclusions (Sohal, 1981). Bacteria are seen in sick-looking flies (the insect body cavity is usually sterile) (Flyg et al, 1988). Recent data document the increased bacterial and fungal load of aging flies (Section 5.6.4). The extensive increase of antimicrobial genes during aging in flies (Section 1.8) is consistent with the increased pathogen load of aging flies, possibly from breakdown of the barriers from the gut and exoskeleton. Chapter 5 discusses these and other genetic influences on longevity through inflammation and stress resistance.
2.4 AEROSOLS AND DIETARY INFLAMMOGENS
Chronic inflammation is stimulated by intake of non-infectious inflammogens by inhalation and ingestion. These sources have received less attention than infectious pathogens in relation to arterial disease. Airborne inflammogens may be of looming importance to future life expectancy with the accelerating global increases of air particulates (Section 6.4).
2.4.1 Aerosols
Aerosols are characterized by size (PM10, <10 μ particle diameter) and composition (mineral, hydrocarbon, sulfur, endotoxin, etc.) and whether the aerosols carry infectious agents (viable vs. non-viable aerosols). Inflammatory responses independently of infectious agents are induced by airborne inflammogens: Among many examples are smoke from tobacco, fossil fuel, and biomass combustion; dust from agriculture; and endotoxins from feces in the many urban locations with poor sanitation and in livestock and poultry. These sources are pertinent to current aging and to historical improvements in public health (Fig. 1.1A).
Smoke is well recognized as a non-infectious (‘non-viable’) aerosol with major consequences to vascular health. Cigarette smoke strongly increases the risk of heart attacks. In the United States in 1990, 20% of deaths from cardiovascular disease are attributable to smoking (Centers for Disease Control and Prevention, 1993). Second-hand smoke is also strongly associated with coronary disease (Zhang and Smith, 2003; Zhu et al, 1997) and lung cancer (Section 2.4.2). Smoking increases the carotid wall thickness in men with dose-dependency (number of pack-years as an estimate of lifetime exposure) (Gariepy et al, 2000) (Fig. 2.6). The mechanisms include proatherogenic increases of oxidized LDL and acute phase proteins. In NHANES III, smokers were twice as likely than non-smokers to have very high CRP (>10 mg/L), with dose responses to the intensity and history of smoking (Bazzano et al, 2003). Second-hand smoke also increased serum CRP into the range of primary smokers and coronary risk in the ATTICA Study (Barnoya and Glantz, 2004; Panagiotakos et al, 2004). Men incur more adverse effects of smoking than women (Fig. 2.6) (Gariepy et al, 2000).
Other types of smoke cause chronic lung damage and inflammatory responses consistent with vascular diseases. Common sources of smoke are open combustion in fireplaces, furnaces, and factories, which diffuse into the breathing environment with adverse effects on the lungs (Singh and Davis, 2002). Until the mid-20th century, exposure to wood and coal smoke was almost unavoidable, and still is in many countries (Zhu et al, 1997). ‘Hut lung,’ or domestically acquired particulate lung disease, is associated with inhaled smoke particulates from burning coal, wood, or other fuels and wastes (Gold et al, 2000). Cardiovascular admissions to hospitals were associated with recent exposure to black smoke PM(10) in some studies, e.g., Edinburgh (Prescott et al, 1998).
Particulate air pollutants induce vascular endothelial damage (Sandhu et al, 2005; Schulz et al, 2005). Mortality gradients in vascular diseases followed indexes of ambient air pollution in residential zones; not surprisingly, higher income zones had the least exposure to pollution (Finkelstein et al, 2005). Animal models support this epidemiology. Particulate inhalants cause chronic lung damage with lung alveolar macrophage hyperplasia, fibrosis, and accelerated atherosclerosis, e.g., rats exposed to wood smoke (Tesfaigzi et al, 2002) or to fly ash (Schreider et al, 1985). In hypercholesterolemic Watanabe rabbits, exposure to particulate aerosol increased the size of coronary atheromas in proportion to the number of lung macrophages that had phagocytosed particles (Suwa et al, 2002).
Dust from corn and grain also induces inflammatory responses (Buchan et al, 2002; Jagielo et al, 1996). Even lab animal bedding materials can contain appreciable bacterial endotoxin and (1- >3)-β-D-glucan from bacteria, molds, and plants. When inhaled, these common aerobiosols induce chronic inflammation (Ewaldsson et al, 2002). Humans also experience varying exposures to bioaerosols according to occupation and income, which can be a factor in the strong socio-demographic gradients in longevity. Bioaerosols are now a major concern of industrial safety (Burrell, 1994; Menetrez et al, 2001), but are still a hazard of agricultural workers. Workers in sewage plants and garbage collectors also suffer from inhalants that cause chronic systemic inflammation and elevated CRP (Rylander, 1977). Farm workers entering a swine confinement building had rapid elevations of blood complement C3 peaking at 1 h, followed by peak CRP at 2 h (Hoffmann et al, 2003). The smokers in this group had greater responses. Besides airborne live bacteria and fungi, non-viable bioaerosols may contain endotoxins of fecal origin. A specific role of LPS inhalation was shown by the induction of plasma CRP and other inflammatory responses with well-defined dose responses (Michel et al, 1997; Thorn, 2001).
The aerosol-vascular disease association is relevant to the historical increase of human longevity during the developments of public sanitation (Section 2.5) and to earlier phases of human evolution as population density increased and encountered increasing exposure to infections, inflammogens, and especially to domestic smoke for cooking and heating (Section 6.2). Genetic risk factors for resistance to domestic smoke and other types of air pollution may have evolved during this time. Curiously, European populations have a high prevalence of a null allele (GSTM1*0) of glutathione-S-transferase M1. GST makes the key antioxidant glutathione (Fig. 1.11) and belongs to a superfamily of xenobiotic detoxifying enzymes with potential importance to human evolution (Section 6.4.2). The M1*0 homozygotes (equivalent to GSTM1 knockout) have impaired lung functions as children and a higher risk of asthma (Peden, 2005).
2.4.2 Food
Cooked foods have inflammogens produced by the chemistry of glyco-oxidation (Sandu et al, 2005). As discussed in Section 1.4.2, advanced glycation endproducts (AGE) and advanced lipid oxidation endproducts (ALEs) are produced endogenously from chemical reactions of glucose and other reducing sugars with peptide lysine and arginine, which are proinflammatory, atherogenic, and carcinogenic (Kikugawa, 2004; Skog et al, 1998; Vlassara et al, 2002). This saga began in 1912 with Louis Maillard’s discovery of chemical reactions between amino acids and glucose that lead to the loss of lysine and the formation of brownish condensation products (Finot, 2005; Maillard, 1912; Nursten, 2005). These reactions are the basis for browning of foods by broiling, or frying, which can increase AGE content 3- to 5-fold (Table 2.1) (Goldberg et al, 2004). AGEs and ALEs are also formed during food processing and storage. AGEs ingested from cooked foods are detected by immunoreactivity for the glycation adduct CML (N-carboxymethyl-lysine) (Table 2.1). In healthy adults, plasma CML strongly correlated with the dietary intake of AGE over a 3-fold range (Urribari et al, 2005).
TABLE 2.1
Advanced Glycation Endproduct (AGE) Content of Common Foods and Effects of Cooking
| Food | CML, kU/g food |
| beef, boiled 1 hr | 22 |
| broiled, 15 min | 60 |
| tofu, raw | 8 |
| broiled | 41 |
| milk (pasteurized) | 0.05 |
| butter | 265 |
CML (N-carboxymethyl-lysine, an AGE formed by heating); radioimmunoassay (Goldberg et al, 2004).
The proinflammatory effects of dietary AGEs were directly shown in diet cross-over studies of diabetics (Vlassara et al, 2002). Nutritionally equivalent diets were prepared by different degrees of heating that yielded 5-fold differences in CML. Six weeks on the high AGE diet elevated inflammatory markers, serum C-reactive protein by 35%, and TNFa by 85% in association with 30% higher CML. Similarly, ingested dietary AGEs correlated with serum CML in renal failure patients (Uribarri et al, 2003).
Rodents show adverse effects of dietary AGEs. In a mouse model of both atherosclerosis and diabetes (apoE–/– genotype, with STZ-induced diabetes), the high-AGE diet increased aortic lesions, whereas a low-AGE diet decreased lesions below the level in the standard chow diet (Lin et al, 2003). The lesions of the high-AGE diet group had more arterial foam cells and receptors for AGE (RAGE). In another mouse model, 6 months on a high-AGE/high-fat diet induced type-2 diabetes, with impaired glucose regulation and insulin insensitivity (Sandu et al, 2005). The low-AGE/high-fat controls had normal glucose regulation despite similar adiposity. Plasma 8-isoprostane, a marker of lipid oxidation, was increased by the high-AGE diet. As a further scary example, a caramel component used for coloring beverages (2-acetyl-4-tetrahydroxybutylimidazole, THI) inhibits lymphocyte egress from the thymus by inhibiting the sphingosine 1-phosphate receptor (Schwab et al, 2005). Dietary AGE content may have unrecognized influences on rodent studies, because lab chows are typically heated during preparation.2
These findings point to an expanding role of dietary AGEs in atherosclerosis and diabetes, and support their designation as glycotoxins’ (Koschinsky et al, 1997; Vlassara, 2005). Vlassara hypothesizes that dietary AGEs, possibly synergizing with tobacco and other environmental inflammogens, sustain oxidative stress and chronic inflammation. The AGE receptors (RAGEs) that activate signaling pathways with PI3K (Section 1.3.3) may link dietary glycotoxins to longevity pathways that involve insulin/IGF-1 signaling (Fig. 1.3A) and that are also implicated in vascular disease (Fig 1.3B).
Besides their color, some Maillard products have definitive tastes and aromas (Schieberle, 2005). Caramel coloring and flavorings have been added to commercial foods and beverages for more than a century (Chappel and Howell, 1992; Nursten, 2005). These preferences may have been important in the development of cooking during the last half million years when early humans learned to control fire (Section 6.2). Cooking could have enhanced health by killing parasites and infectious organisms in animal tissues. Moreover, cooking increases the usability of many plants as foods by increasing digestibility and by inactivating toxins that are widely found, e.g., cassava and potatoes. (De Bry, 1994) suggests that early humans used Maillard products as olfactory cues to indicate when tubers with heat-sensitive toxins were sufficiently cooked. Nonetheless, evolution of the omnivorous human diet would have greatly increased exposure to toxins, implying the importance of detoxification mechanisms to enable these new foraging strategies (Sections 3.7 and 6.2.3).
Future increases of human longevity may come from better knowledge of these interactions and the mechanisms that remove ingested and endogenously produced AGEs. Dietary changes during human evolution may also have selected for genes that detoxified dietary AGEs, e.g., the recently discovered amadoriases (‘AGE-breakers’) (Monnier and Sell, 2006). Our ancestors ate meat increasingly by a million or more years ago, a major departure from the plant-based diets of the great apes, and, it is presumed, that of the shared human-chimpanzee ancestor (Section 6.2) (Finch and Stanford, 2004). The more recent use of fire for broiling or roasting meat would have increased AGE ingestion and selected for meat-adaptive genes.
2.5 INFECTIONS, INFLAMMATION, AND LIFE SPAN
2.5.1 Historical Human Populations
The recent longevity increases (Fig. 1.1A) also implicate the relationship of infection and inflammation to arterial disease (Finch and Crimmins, 2004, 2005; Crimmins and Finch, 2006a,b). Anonymous reviewers of these papers questioned the importance of vascular disease in deaths before the modern era. However, all evidence points to vascular disease as ancient and ubiquitous “… its pattern has always been the same regardless of race, diet, and the stresses of survival” (Magee, 1998, p. 663). The 5,300 year old Tyrolean “iceman” of the Bronze Age evidenced carotid artery calcification (Murphy et al, 2003), which is common in advanced atherosclerosis (Fig. 1.13) (Section 1.5.3.1) and is an independent risk factor of vascular mortality (Doherty et al, 2003; Sangiorgi et al, 1998). Two millennia later, Egyptian mummies of the 18th dynasty preserved calcified arteries and other vascular pathology (Ruffer, 1911). Most large arteries (16/24) in this sample met criteria for atherosclerosis, with half of these specimens showing vascular calcification (9/16). By the European Middle Ages, anatomists were describing arteriosclerosis as “natural to old age” (Long, 1933). Approaching the modern era, the records, scarce as they are, also show cardiovascular disease as a major cause of death in older adult ages. In 19th and mid-20th century England and Sweden, which had low life expectancy, cardiovascular disease was one of the two most important causes of mortality at the oldest ages (Preston et al, 1972; Preston, 1976). For cohorts born after the first decade of the 1800s, the deaths recognized as due to cardiovascular diseases exceed those attributed to infectious conditions. From the earliest date in Sweden, deaths from cardiovascular disease are two times higher than from infectious conditions for those 70–74. For the U.S. Civil War, Fogel and colleagues compared doctors’ reports of heart disease from Union Army veterans aged 65 and over in 1910 versus veterans of the same age in 1983. Heart disease was nearly twice as prevalent in the U.S. Civil War Veterans (76% vs. 40%, age-adjusted) (Fogel, 2004, p. 31). William Osler’s statement from 1892 still holds true today, “Longevity is a vascular question, which has been well expressed in the axiom that ‘a man is as old as his arteries.’ To a majority of men, death comes primarily or secondarily through this portal.” (Osler, 1892, p. 664).
Moreover, early human ancestors were also likely to incur vascular pathology. Chimpanzees, our closest biological relative, also show extensive hypercholesterolemia, even on non-atherogenic diets, and die from heart attacks and strokes in captivity (Finch and Stanford, 2004; Steinetz et al, 1996) (Section 6.2). Crimmins and I provisionally conclude that vascular pathology during aging has been prevalent throughout human history and, quite possibly, throughout human pre-history as well (Crimmins and Finch, 2006a).
Crimmins and I are evaluating the role of infection and inflammation on later mortality in historical cohorts (Finch and Crimmins, 2004; Crimmins and Finch, 2006a, b). According to our ‘cohort morbidity hypothesis,’ exposure to infections early in life causes chronic infections that, in turn, promote vascular disease, leading to earlier mortality. Tuberculosis, Helicobacter pylori, Chlamydia pneumoniae, and other gastro-intestinal pathogens noted above are among many chronic infections that have recently diminished. The human environment in rural and urban areas alike was typically filthy by modern standards, with gross continuing exposure to human and animal feces. Running water was not available for convenient washing and bathing. Conditions gradually improved with national efforts in public hygiene even before the identification of infectious pathogens and development of immunization at the end of the 19th century. Besides the infectious environment, it was difficult to keep clothes clean and free of ectoparasites, especially before the availability of cheap cotton for clothing, which is easier to wash than wool or leather. Improved nutrition was another major factor in resistance to infectious conditions, due to agricultural improvements and the development of national transport systems of canal and rail (Fogel and Costa, 1997; Fogel, 2004; McKeown, 1976).
We chose to consider birth cohorts before the 20th century when infections were very common or rampant, but before tobacco smoking became popular. Smoking is a major inflammatory stimulus, as discussed above. Complete birth and death records are available from Sweden from 1751. Sweden also pioneered a national program of inoculation against small pox, begun in 1756, which attenuated these epidemics by the 1820s, decades ahead of other European countries (Skold, 2000). We also included England (1841–1899), France (1806–1899), and Switzerland (1871–1899). These early cohorts also did not benefit from antibiotics, which were not widely available before 1950. All the old age mortality examined occurred before 1973. In many countries, dramatic declines in mortality after 1970 are explained best by lifestyle and medical factors.
Initial life expectancy was low due to high early mortality characteristic of preindustrial societies, but increased considerably by 1899 (Fig. 2.7A). The historical trends for declining childhood mortality and old age mortality were remarkably parallel in Sweden, England, and France (Fig. 2.7B). As early age mortality declined, so did later age mortality in the survivors, seven decades later. The increased life expectancy at age 70 was clear in Sweden by 1850 (Fig. 2.7C). These findings support my early estimate that the steady historical increase in the adult age when mortality reaches 1% implies a slowing of aging processes (Finch, 1969, p.12).
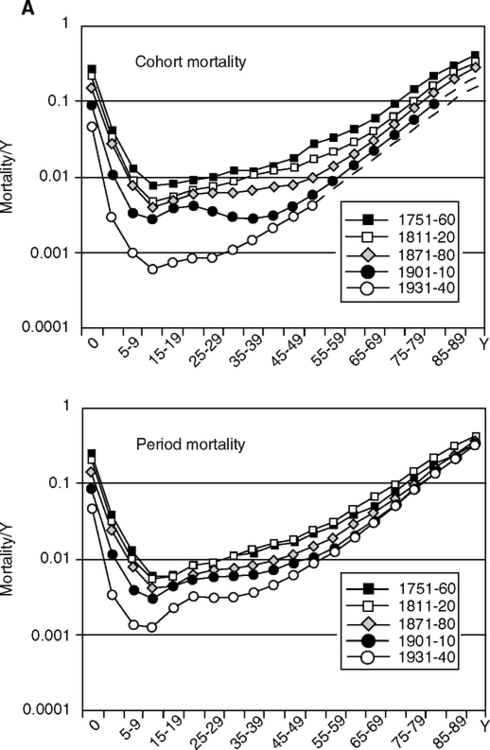
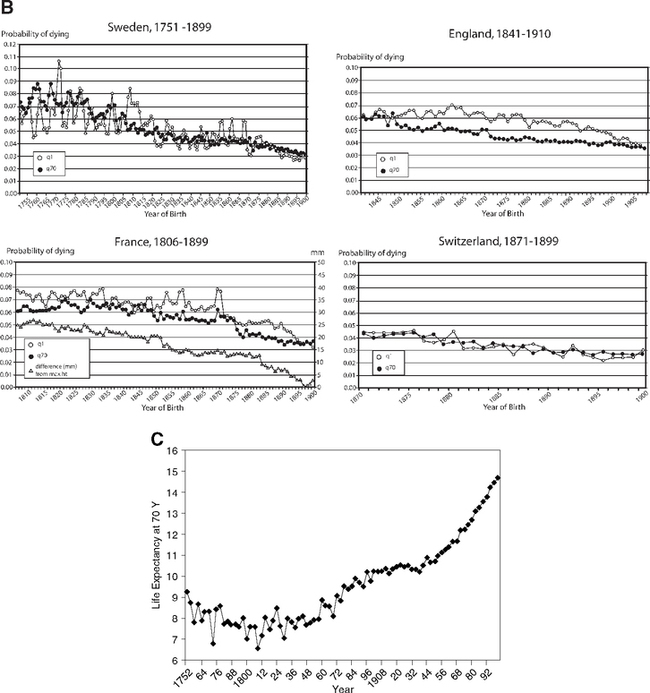
These associations were tested with regression models for the relationships of temporal change in mortality at ages 70–74 with four childhood stages: infancy, <1 year; early childhood, 1–4; later childhood, 5–9; and adolescence, 10–14 years (Crimmins and Finch, 2006a). Infant mortality is largely attributed to infections. A key feature of this analysis is the comparison of birth cohort, followed throughout life, with the same ages in the corresponding periods. The results are consistent across these four countries: Most of the variance in old age mortality is explained by the early mortality in that birth cohort, 87–96%. The early and later mortality association of cohorts was much stronger than for periods. At a given year, the older adults in a population were born seven decades before the children and had experienced different environments that had a stronger effect on their mortality than in the current environment. Mortality of intermediate adult ages also did not predict old age mortality. The overall mortality curves shift downward quite uniformly as early mortality improves (Fig. 2.7A). Separate analysis of males and females also showed consistent associations in cohorts between early and later mortality (Crimmins and Finch, 2006b). Many specific mechanisms can be considered in cohort morbidity through which recurrent exposure to acute infections or continiued chronic infections accelerates atherosclerosis as well as causing direct damage to heart valves and myocardium (focal lesions and diffuse fibrosis). Extensive immune activation through hyperantigenic stimulation of T cells could also have increased T cell participation in atheroma instability (Section 1.5).
Height was also examined because infections slow growth (Section 4.4). The level of early mortality strongly predicted adult height (Fig. 2.7B). In birth cohorts with high early mortality, the survivors were shorter as adults, which we attribute to the greater exposure to infections in childhood. Infections and inflammation cause the reallocation of metabolic resources and energy from growth (Fig. 1.2B), as discussed in detail in Chapter 4. There are also associations of inflammatory genes with fetal growth (TNFa-308 G/G is more prevalent in lower birthweights) (Casano-Sancho et al, 2006) (Section et al 4.10.1). These mechanisms may also account for the progressively decreasing size of adults after 50,000 years ago in human pre-history (Chapter 6, Fig. 6.7).
Our model of inflammation in the pathobiology of aging (Fig. 1.2A) also includes important links between maternal infections and inflammation to fetal and infant growth and inflammation. Influenza, malaria, and tuberculosis were common maternal inflammations until recently (Riley, 2001). Malaria and possibly other maternal infections can retard fetal growth and increase fetal cytokines (Moormann et al, 1999) (Section 4.5). Smaller babies may have lower resistance to environmental pathogens. These possibilities are not included in the Barker hypothesis of fetal origins that focused on maternal malnutrition as the main cause of the fetal retardation effect on later vascular disease (Barker, 2004), discussed at length in Chapter 4. These manifold effects of inflammation and infection on growth during childhood and on later arterial disease point to a potential unifying theory of human development and aging.
2.5.2 Longer Rodent Life Spans with Improved Husbandry
With intriguing parallels to the increasing human longevity discussed above, rodent life spans have nearly doubled in the past 50 y. Elimination of chronic infections through improved husbandry is a major factor. Additionally, arterial and myocardial disease may have been more severe in the early rodent colonies, as indicated for 19th century humans above.
Life span increases are best documented for mice of the C57BL/6J (‘B6’) strain, inbred since 1936 at the Jackson Laboratory (Bar Harbor, ME), a pioneering center of mouse genetics (Staats, 1985). B6 males had mean life spans of 18 m in 1948–1956 (Russell, 1966) that gradually increased to the present range of 26–30 m (Finch, 1972; Kunstyr and Leuenberger, 1975) (Fig. 2.9.A, B). Survival curves became increasingly ‘rectangularized’ and right-shifted: Sporadic deaths before 20 m decreased, while maximum longevity increased from 30 to 44 m (Finch, 1969; Tanaka et al, 2000; Turturro et al, 2002). These right-shifts of mortality indicate reduction of infections and match those of human populations as health improved (Fig. 1.1A). Unfortunately, the pathology of aging was not well documented for B6 mice during this transition. In modern colonies of B6 mice, the cumulative incidence of cardiomyopathy is about 40% by 24 m (Schriner et al, 2005; Turturro et al, 2002) (Section 1.2.2). Rat life spans also increased during the same period: In McCay’s rat colony at Cornell University, where he conducted pioneering studies of nutirion and aging (Chapter 3), mean life span increased from 13 m in 1934 to 20 m in 1943 (McCay et al, 1943). Current lab rat life spans are 26-32 m.

Another striking example is the greatly improved longevity of dwarf mice with growth hormone deficiency. Thirty years ago, the Snell dwarf mouse was considered a model for accelerated aging because of short life span (< 6 m) and wizened appearance with gray hair, cataracts, and early onset tumors (Fabris et al, 1972) (Chapter 5, Table 5.3, footnote c). However, in the past decade with improved husbandry, dwarf mice have ‘switched teams’ to become models of slow aging, with life spans over 4 y and delayed onset of tumors. Gray hair is not common in contemporary dwarf mice and, in any case, is not a general trait of aging in B6 mice or other strains (Finch, 1973b). Husbandry improvements that enabled this remarkable transformation probably include reduced infections (see below) and better vivarium temperature control.
The general improvements in longevity across all genotypes of rodents in the past decades are not well understood. Even in the early days of laboratory rodent husbandry, some colonies were maintained well enough to achieve contemporary longevity. Slonaker’s rats lived up to age 46 m (Slonaker, 1912) (Chapter 3, footnote 5), while Robertson’s white mice averaged 25 m (Robertson and Ray, 1920) and the Berg-Simms colony females averaged 31 m in the 1960s (Chapter 1; footnote 3, this chapter), discussed below. I suggest that these early colonies were less inbred and closer to wild-types that were recently shown to have greater longevity (Harper et al, 2006a) (Chapter 3, Fig. 3.3).
Because the age-related pathology of aging mice at Jackson Labs before 1960 was not reported, we may look to the occasional reports on pathology from other early aging colonies.3 This scattered literature describes conditions in aging rats that may surprise readers. In the 1960s, Wexler and colleagues documented in detail that repetitive mating can accelerate arterial degeneration in males as well as females (Wexler and Miller, 1958, 1960; Wexler and True, 1963; Wexler, 1964; Wexler, 1976). These studies employed standard rat stocks (Holtzman, Long-Evans, Sprague-Dawley, Wistar) fed on low fat (4%) diets. Lesions developed in coronary and carotid arteries in proportion to the breeding experience. Heart valve damage was common. ACTH injections or restraint stress also accelerated spontaneous atherosclerosis. Wexler and colleagues postulated that the repetitive mating caused severe stress. It is impossible to define the conditions in Wexler’s colony that caused this level of stress during reproduction. Myocardial fibrosis was also associated with the severe coronary artery changes. In current clinical practice, myocardial fibrosis is associated with arrhythmias and sudden death (Siwik and Colucci, 2004; Zannad and Radauceanu, 2005).
Moreover, myocardial fibrosis with microscopic scarring was also common in several early rat colonies that may have contributed to premature death. “In older rats fibrosis may be so extensive that it is difficult to understand how the animals remain alive” (Fairweather, 1967, p. 227). Other examples include colonies with 60% prevalence of fibrosis (mild to severe fibrosis) by 20 m (Wilens and Sproul, 1938) and 100% by 20 m (Humphreys, 1957). In modern colonies, myocardial fibrosis is apparently uncommon and arises later (Bronson, 1990). Myocardial fibrosis is associated with inflammation and chronic stress (Holloszy and Smith, 1986), e.g., in rodent models, increased by TGF-β1 overexpression and decreased by TGF-β1 deficiency (Brooks et al, 2000; Siwik and Colucci, 2004). Diet-restricted humans have lower myocardial stiffness and plasma TGF-β1 (Section 3.3.2).
Arterial calcification was common in many early colonies but may be rarer today. In the Wilens-Sproul colony, calcification was noted in 46% in pulmonary arteries and 3% in coronary arteries and the aorta (Wilens and Sproul, 1938). In McCay’s colony, aortic calcification occurred in 20% of ad lib fed, but was unexpectedly 3-fold higher (60%) with diet restriction (McCay et al, 1939). Others described ‘bamboo stick aorta’ with disintegration of the elastic layers and secondary calcification resembling Monckeberg’s medial sclerosis (Fairweather, 1967; Mawdesley-Thomas, 1967; Wilgram, 1959). Sporadic arterial and myocardial calcification in aging rats was also reported by (Hummel, 1938; Wilgram, 1959). Moreover, calcification was associated with repetitive breeding in females (Gillman and Hathorn, 1959; Wexler, 1964). Current aging rodents have a low incidence of arterial calcification (<5%) (Bronson, 1990). Arterial calcification is associated with local nodules of Chlamydia pneumoniae in humans (Pierri et al, 2005) and in renal failure (Oh et al, 2002).
Coronary artery disease (CAD) was variable in the early colonies. The first report of spontaneous coronary disease on a normal (not fat-loaded) diet may have been from the Wilens-Sproul rat colony, in which 60% had some degree of coronary sclerosis by 24 m (Wilens and Sproul, 1938). Coronary artery stenosis to varying degrees was concurrent with myocardial fibrosis. In the Edinburgh colony, occlusive CAD with intimal plaques was present in 60% of rats by 17 m on a low-fat diet, causing complete blockage of a coronary vessel in some rats (Humphreys, 1957). In another colony, the incidence of coronary stenosis was about 15% (Wissler et al, 1954). CAD was particularly high in female ‘retired breeders’ (Wilgram and Ingle, 1959). However, in two other contemporary colonies, CAD was rare (Berg, 1967; Fairweather, 1967).
Three factors may be at work in the increased longevity of laboratory mice, ranked in reverse order of likeliness, in my opinion: genetics, diet, and infectious diseases. Improvements at the Jackson Laboratory occurred after 1959 when the Pedigreed Expansion Stock (begun in 1948) was moved to cleaner facilities (Russell, 1966). Longevity increases were not the result of intentional selection for longevity, although routine culling of sickly pups should lower overall mortality by reducing the pool of infections. The lack of correlation between life spans of parents and offspring in these early B6 colonies (Gunther Schlager, in Russell op. cit.) can be considered evidence against genetic drift (crosses of B6 and other strains clearly show inheritance of life span) (Jackson et al, 2002; Finch and Tanzi, 1997). Cardiomyopathies, nonetheless, may arise more frequently in aging rats maintained on diet restriction with exercise (McCarter et al, 1997) (Section 3.4.2). Dietary fat could be a factor because fatty diets can shorten life span (Chapter 3). The fats fed the first longevity group at Jackson are not known: The composition of the commercial diet was then a ‘trade secret’ (Elizabeth Russell, pers. comm.). In 1959, the Jackson Lab switched to Guilford Chow (11% fat, 19% protein), routinely given breeding females to enhance milk production. Chows with 4–5% fat are currently favored for aging studies (Finch et al, 1969; Turturro et al, 2002).
The major change from the 1940s to the 1970s at Jackson and elsewhere was reduction of chronic infections through improved animal and human hygiene. Some reported deaths from epidemic infections as merely ‘accidental’ (Robertson and Ray, 1920). Until the 1970s, laboratory colonies were often infected with microbial infections and skin parasites. Numerous pathogens were gradually minimized or eliminated, including bacteria (Salmonella, Mycoplasma); viruses [coronavirus; ectromelia (mousepox), mouse hepatitis virus, Sendai virus]; and ectoparasites (mites, pinworms) (Bell et al, 1964; Cotchin and Roe, 1967; Flynn et al, 1965; Miller and Nadon, 2000). Early colonies often had chronic respiratory disease (CRD) from endemic Mycoplasma4 recognized by wheezy breathing and crusty noses. While minimizing infections and dietary fat could only increase longevity, we may never know the causes of the extensive myocardial and arterial lesions described above. The Wilens-Sproul colony rats had numerous abscesses (‘suppurative lesions’) in brain, ears, genitourinary tract, and lungs (Wilens and Sproul, 1938).
Besides the Jackson Lab mice, another early benchmark rat colony was founded at Columbia University by Benjamin Berg and Henry Simms, which maintained advanced husbandry and exemplary documentation of age-related pathology (Fig. 1.5) (Berg, 1967; Simms and Berg, 1957; Simms and Berg, 1962). Although the Berg-Simms colony was begun in 1945, there was little respiratory disease (< 5% of rats). These rats were not selectively inbred, except to eliminate an ‘eye anomaly’ (Simms and Berg, 1957). Longevity in the Berg-Simms colony was in the range of modern colonies: females, median life span of 31 m and maximum of 34 m; males, median of 27 m and maximum of 29 m (Berg, 1976). Chronic lesions of aging approximated those of other rat strains in modern colonies (Bronson, 1990) and in the same age ranges: glomerulonephropathy arose before cardiomyopathy and abnormal growths (Simms and Berg, 1957; Simms and Berg, 1962), and arterial calcification was occasional. This health and longevity is remarkable for that time.
The current best practice in rodent husbandry is the ‘specific-pathogen free’ (SPF) colony, in which the pathogen load is regularly monitored with sentinel mice (Lindsey, 1998; Miller and Nadon, 2000). SPF status with minimal mycoplasmas and other pathogens increases fecundity and post-weaning growth and lowers spontaneous mortality (Bell et al, 1964). However, pathogen loads can fluctuate in SPF colonies with agents carried by humans and other adjacent lab animals (Taylor, 1974; Taylor and Doy, 1975). Infections are still embarrassingly common within SPF colonies at major research institutions (Jacoby and Lindsey, 1997). Although stricter barrier facilities can further reduce transmission of external infections, the expense and effort are prohibitive. Germ-free (axenic) animals lacking bacteria are problematic for aging studies: Their adaptive immunity is undeveloped, and their flaccid, grossly enlarged caecums develop fatal constrictions (volvulus) (Gordon et al, 1966).
Recent examples also show the importance of animal husbandry. Age-changes in skeletal muscle composition and function observed in ‘dirty’ colonies are negligible in aging rodents from SPF colonies of the same strains (Florini, 1989). Moreover, age-changes in rat liver protein oxidation (carbonyl content) disappeared in a subsequent cohort of the same strain of rats obtained 10 years later; these differences were confirmed with stored samples (Stadtman and Levine, 2003). Lastly, sporadic hippocampal neuron loss in aging may have been common in early colonies (Landfield et al, 1977; Meaney et al, 1988) but is not obvious currently (Gallagher et al, 1996; Rasmussen et al, 1996). Variable stress and infections may have been involved. Moreover, early reports of neuron loss (reduced density of large neurons) could be interpreted as neuron atrophy (Fig. 1.7A). In many ways, the hygiene of lab animals parallels that of humans in modern health care: We can minimize childhood infections by immunization and hygiene, but we remain vulnerable to sporadic epidemics. “La pest reste ici” (J.P. Sartre, The Plague, 1947).
2.6 ARE INFECTIONS A CAUSE OF OBESITY?
Viral infections as causes of obesity are being discussed because of evidence that four viruses cause obesity in vertebrate models and serological associations of obesity with infections in humans with obesity and glucose intolerance. These findings should be considered provisional.
This story began with the finding that mice developed obesity when infected as weanlings with canine distemper virus (CDV, paramyxovirus closely related to measles) (Lyons et al, 1982; Lyons et al, 2002). CDV causes focal lesions of hypothalamic appetite centers with selective loss of leptin receptors, POMC and catecholaminergic neurons in the arcuate nucleus, and hyperplasia of adipocytes and pancreatic islets. Obesity is also induced in other animal models by infections with scrapie (prion disease), Bornavirus, the retrovirus (RAV-7), and AD-36 (human group D adenovirus) (Atkinson et al, 2005; Lyons et al, 2002). In a recent study impressive for its large subject pools, AD-36 seropositivity was 3-fold more prevalent in obese adults (30% than controls, 11%; 502 Ss); moreover, in 28 twin pairs discordant for AD-36, the seropositive individual was fatter than the co-twin (Atkinson et al, 2005). Curiously, AD-36 seropositive individuals had lower cholesterol and triglycerides. In vitro, AD-36 infections of adipocytes decreased increased glucose uptake and leptin secretion; these effects depend on transient expression of viral mRNA, but not viral DNA replication (Rathood et al, 2007). Unlike CDV, no hypothalamic lesions have been found in AD-36 infected mice (Dhurandhar et al, 2000). Another study of obese men who were otherwise healthy found inverse correlations between insulin sensitivity and seropositivity for common infections (HSV-1 and -2, enterovirus, and C. pneumoniae; AD-36 was not included in this panel) (Fernandez-Real, 2006).
If the 30% prevalence of AD-36 seropositivity in obesity is generally validated, viral infections may contribute as much to obesity as life style behaviors of eating and exercise, and moreover, may be a causal factor in these behaviors by their impact on the hypothalamus. However, responses to viral infections depend on many factors of host defense, including food intake and exercise (Chapter 3). Resolution of cause and effect in these associations is difficult because obesity and diabetes increase vulnerability to infections (Falagas and Kompoti, 2006) (Chapter 3). Nonetheless, these recent findings support the suggestion of (Lyons et al, 1982) that viral infections have roles in sporadic childhood and adult obesity. AD-36 infections that lead to obesity by causing hypothalamic lesions could be another, and milder example, of viruses that propagate by modifying behaviors, although how obesity could particularly favor AD-36 viral propagation is far from obvious.
2.7 INFLAMMATION, DEMENTIA, AND COGNITIVE DECLINE
2.7.1 Alzheimer Disease
Infections may also be causes or promoters of Alzheimer disease (AD). This new possibility is even less settled than associations of infections with vascular disease. As a first example, identical Swedish twins who are discordant for dementia may also show long-term outcomes of infections. The first twin to be affected was 3.6-fold more likely to have had periodontal disease (Gatz et al, 2006). Periodontal disease is also associated with infections that interact with arterial lesions (see above), but links of infections to AD are even more speculative.
The current (and incomplete) evidence on infections and AD centers on herpes viruses and Chlamydia pneumoniae (Mattson, 2004; Ringheim, 2004; Robinson et al, 2004; Wozniak et al, 2005). Causality is elusive because these infections are ubiquitous. Herpes virus infections occur widely. Postmortem, about 2/3 of all brains, normal and Alzheimer, have HSV-1 (Itzhaki, 1997). HSV-1 infections reside in some brain regions affected by AD (frontal lobe and hippocampus), as well as the trigeminal ganglion. At later ages, HSV-1 infections appear to be active by the presence of HSV-1 antibodies in the cerebrospinal fluid of about half AD and normal controls (Wozniak et al, 2005). Clearly, AD can arise in the absence of active HSV-1 infections! However, in one sample, the apoE4 allele and HSV-1 co-occurred 10-fold more frequently in AD brains than normal elderly (PCR assay). ApoE4 carriers may have a greater risk of neurodegeneration from the activation of HSV-1 (Itzhaki et al, 1997). HSV-1 activation in the trigeminal ganglion causes shingles, which afflicts many elderly. A much rarer condition is herpes simplex encephalitis; survivors often have life-long cognitive impairments, possibly from neuronal apoptosis from HSV-1 (Aurelian, 2005). Other candidates are human herpes virus 6 (HHV6) seropositivity in Alzheimer (22% AD vs. 0% controls) (Wozniak et al, 2005) and CMV in vascular dementia (93% VascD vs. 34% controls) (Lin et al, 2001).
Some evidence suggests that vaccination against common infections may be protective. In the Canadian Study of Health and Aging, the risk of developing dementia during 5 years was lowered by vaccination for influenza (OR, 0.75), poliomyelitis (OR, 0.6), or diphtheria (OR, 0.41) (4392 Ss, aged ≥ 65) (Verreault et al, 2001). None of these infections is otherwise implicated in dementia. The protective effects of vaccination could be indirect by reduced vulnerability to other infections.
The case for C. pneumoniae as a casual factor in AD is less developed than for arterial disease. On one hand, C. pneumoniae persistently infects macrophages and can enter the brain, as in HIV (Stratton and Sriram, 2003). In a culture model, C. pneumoniae promoted monocyte migration across brain endothelia (MacIntyre et al, 2003). Infections of cultured human cerebral microvascular endothelial cells altered the levels of proteins associated with entry of this pathogen into the brain (increased β-catenin, N cadherin; decreased occludin) (MacIntyre et al, 2002). However, the evidence is mixed for the postmortem detection of infections. One study detected C. pneumoniae in cerebral vessels and glia in more Alzheimer brains than controls (Balin et al, 1998; MacIntyre et al, 2003). However, subsequent studies could not detect C. pneumoniae DNAs even in these same brains (Gieffers et al, 2000; Nochlin et al, 1999; Ring and Lyons, 2000). Vascular dementia studies were also mixed: positive association (Yamamoto et al, 2005) versus no association (Chan Carusone et al, 2004; Wozniak et al, 2003). Antibiotic treatment of AD patients with doxycycline and rifampin did not alter seropositivity for C. pneumoniae but, unexpectedly, slowed early cognitive declines (Loeb, 2004).
C. pneumoniae isolated from AD brains caused rapid formation of amyloid plaques in a normal mouse (BALB/c) (Little et al, 2004), which, like other ‘wildtype’ mice, never develops plaques during normal aging. Aβ1-42 containing plaques increased during the three months after infection, and a subset of plaques had fibrillar amyloid. Activated astrocytes around the plaques and distant from plaques suggest general inflammatory responses. Neuronal perikarya also had increased Aβ1-42, an early Alzheimer change (Section 1.6.3). The formation of fibrillar Aβ is puzzling because the mouse Aβ1-42 protein differs from the human in three substitutions that reduce aggregation (Section 1.6.). Nonetheless, mice overexpressing TGF-β1 (an inflammatory factor upregulated in Alzheimer brains) slowly developed fibrillar Aβ deposits around cortical microvessels; these deposits were preceded by thickening of the basement membrane. These changes are absent during aging in wild-type mice (Wyss-Coray et al, 2000).
Transgenic AD mice also show acceleration by systemic inflammation from i.p. injections of the endotoxin LPS (Godbout et al, 2005; Konsman et al, 2004; Scott et al, 2004). In the triple transgenic mouse model of AD, which has both amyloid plaques and neurofibrillary degeneration (Section 1.6), LPS caused earlier hyperphosphorylation of neuronal tau but, contrary to expectations, did not alter amyloid deposits (Kitazawa et al, 2005). In other AD transgenics, LPS induces APP and Aβ in neurons of the cerebral cortex and hippocampus, which are AD brain regions (Sheng et al, 2003). Microglial activation correlated strongly with neuronal APP and Aβ.
Aging mice had greater inflammatory responses and more prolonged ‘sickness behaviors’ in response to LPS (Godbout et al, 2005). The effects of aging on responses to LPS were not examined by these transgenic studies, which used relatively young mice. In stroke models, LPS pretreatment increases subsequent brain damage from cerebral artery blockade (Becker et al, 2005). Moreover, blood CRP is also elevated by LPS (Section 1.2) and elevated peripheral CRP increased damage from stroke in rats (Gill et al, 2004). Thus, in populations with high levels of infection and inflammation, stroke may cause greater brain damage and higher mortality. It is unclear if systemic endotoxins cross the blood-brain barrier. In rodents, systemic LPS binds to Toll-like receptors (TLRs) (Section 1.2 and Section 2.2.2 above) on cerebrovascular endothelia, which may increase vascular permeability to some sugars (Singh and Jiana, 2004).
Do environmental inflammogens promote AD? Smoking, while a strong risk factor in vascular disease (above), is not a clear risk factor for AD. While some case control studies indicate that smokers had lower risk of AD, recent cohort studies showed higher risk (Letenneur et al, 2004; Luchsinger and Mayeux, 2004; Sabbagh et al, 2005). Occupation and education are confounding variables. Smokers with AD may be younger at death but had the expected neuropathology (Sabbagh et al, 2005). Lastly, obesity and the metabolic syndrome are associated with chronic, low-grade inflammation (Section 1.7). As discussed in the next chapter, obesity and diabetes increase the risk of infections, while obese mice also have greater neuroinflammatory responses to LPS (Scott et al, 2004).
2.7.2 HIV, Dementia, and Amyloid
Dementia with memory loss is common in HIV sufferers with AIDS (acute immunodeficiency syndrome) (Selnes, 2005). The HIV virus enters the brain through macrophages but does not infect neurons with the production of further infectious virions. Diffuse damage and inflammation are found throughout the brain. Cases are often complex because of infections by other pathogens that cause diverse damage, e.g., demyelination (multifocal leukoencephalopathy) from the neurotropic JC virus (Del Valle and Pina-Oviedo, 2006).
Recently, diffuse amyloid Aβ deposits were found in the brains of AIDS patients (Green et al, 2005; Rempel and Pulliam, 2005) in proportion to the duration of infection (Fig. 2.10). The average age at death was 43 years, which is two decades before amyloid deposits become generally common. Nearly 50% of 150 brains from AIDS victims had diffuse Aβ deposits in the frontal cortex, most frequently near arteries (Green et al, 2005). Neuronal Aβ was also common, an early change in Alzheimer disease (Section 1.6.3). So far, AIDS brains have not shown two hallmarks of Alzheimer disease: compact neuritic plaques (Rempel and Pulliam, 2005) or neurofibrillary tangles (Green et al, 2005). These findings with well-characterized monoclonal antibodies confirm smaller studies of AIDS brains, which also included younger ages (Esiri et al, 1998; Izycka-Swieszewska et al, 2000; Rempel and Pulliam, 2005). Two mechanisms are noteworthy because of possible synergy: (I) induction of the amyloid precursor protein (APP) in neurons as an acute phase response during the brain inflammation of AIDS (Section 1.6.4) and (II) decreased degradation of Aβ by neprolysin, an endopeptidase enzymatically inhibited by Tat peptides from the HIV-encoded protein (Daily et al, 2006; Rempel and Pulliam, 2005). Both mechanisms should increase the production of oligomeric and solid Aβ. More information may be anticipated from HIV carriers who did not develop AIDS because of successful anti-viral therapy.
2.7.3 Peripheral Amyloids
Tissue amyloid deposits (amyloidosis) are associated with chronic infections, e.g., tuberculosis (de Beer et al, 1984, Sipe, 1994, Urban et al, 1993). Peripheral tissue amyloid accumulations are also increased by endemic bacterial flora and common endemic pathogens in mice. Specific-pathogen free mice of several genotypes had no tissue amyloids (thioflavin-binding, not otherwise characterized) up through 28 m (mean life span), whereas ‘conventional’ (dirtier) colonies had amyloid deposits (Lipman et al, 1993). In mice transgenic for mutant human transthyretin (TTR), the amyloid deposits of the gut and the penetrance of the mutant TTR-induced peripheral neuropathy (polyneuropathy) were strongly influenced by the microbial flora (Noguchi et al, 2002). TTR amyloidosis was increased by exposure to enterobacteria and yeast, and decreased by anaerobic cocci. Social stress or social deprivation may also influence amyloidosis, e.g., more amyloid accumulated during aging when mice were housed in groups versus solitary (Lipman et al, 1993).
2.7.4 Inflammation and Cognitive Decline During ‘Usual’ Aging
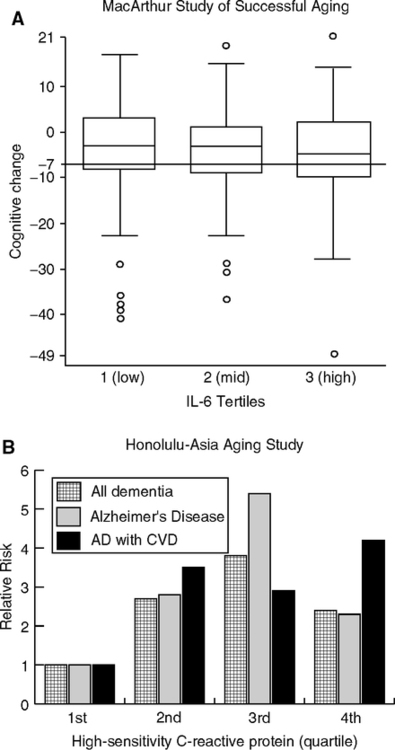
Elevations of CRP and other acute phase proteins are common at later ages in populations (Fig. 1.23) and may be modest risk factors of cognitive declines. The MacArthur Study of Successful Aging followed high performing elderly for 7 years in a carefully controlled analysis (Weaver et al, 2002). Elderly in the highest IL-6 tertile had twice the risk of cognitive decline (Fig. 2.10A). This subgroup may be > 25% of all elderly with elevated IL-6.
Other studies show similar risks. In the Health, Aging, and Body Composition Study of well-functioning elderly (African American and Caucasian) (Yaffe et al, 2003), during 2 years, the top tertile of inflammatory markers showed a risk of cognitive declines in association with levels of CRP (OR 1.41) and IL-6 (OR, 1.34), but not for TNFa. During 5 years of observation, 23% had significant cognitive decline. Subgroups with both the metabolic syndrome (Chapter 1) and high inflammatory markers had 1.66-fold higher risk of cognitive impairment (Yaffe et al, 2004). In the Helsinki Aging Study over 5 years, the risk of cognitive decline increased with CRP < 5 mg/L (OR, 2.32) and diabetes (OR 2.18) (Tilvis et al, 2004).
However, the Longitudinal Aging Study (Amsterdam) found mixed associations: Cognitive decline was associated with elevated α1-antichymotrypsin (ACT), but not with elevations of CRP or IL-6 (Dik et al, 2005). In the Maastricht Aging Study (MAAS) for those over 50 during 6 years, elevated CRP was associated with decline in only one cognitive test (word learning) (Teunissen et al, 2003). The InCHIANTI study of aging in Tuscan communities documents the age-related increase of IL-6 and other acute phase proteins (Cesari et al, 2004) (Fig. 1.21) and may soon report on cognition. Overall, the associations of acute phase elevations and cognitive declines are modest and may not generalize between populations. Some divergences may be due to different tests used. But, a larger question lurks.
Do the cognitive declines in “normal aging” represent incipient dementia from Alzheimer or cerebrovascular disease? Although these studies of normal aging excluded individuals with signs of dementia at entry, none was designed to identify the early, preclinical stages (CDR 0.5 or mild cognitive impairment) (Section 1.6.3). However, the Honolulu-Asia Aging Study of men clearly shows associations of blood CRP at middle age with Alzheimer and vascular dementia 25 years later (Schmidt et al, 2002). Relative to the lowest quartile, the top three quartiles showed 3-fold higher risk of Alzheimer and vascular dementias 25 years later (Fig. 2.10B). The risks did not scale smoothly with CRP levels, but are highly significant for each of the top three quartiles: for Alzheimer, for vascular dementia, and for mixed cases. These associations were independent of cardiovascular disease in this sample, which may represent selective mortality. However, InCHIANTI attributed most of the elevations of CRP and other acute phase proteins to cardiovascular disease (Cesari et al, 2004). Seropositivity to C. pneumoniae was prevalent among the elderly and correlated with IL-6 and TNFa (Blanc et al, 2004). As noted in Section 1.5.4, vascular and Alzheimer disease share many risk factors including elevated acute phase proteins, elevated cholesterol, and hypertension.
2.8 IMMUNOSENESCENCE AND STEM CELLS
Immune system aging is very complex: Innate immunity tends to increase, exemplified by arterial macrophages and local inflammation, whereas adaptive immunity tends to develop restriction of repertoire from oligoclonality and depletion of T cells (Sections 1.2 and 1.5.1). Inflammatory processes may interact with aging in instructive immunity and stem cell generation.
2.8.1 Immunosenescence and Cumulative Exposure
The thymus is highly sensitive to atrophic changes during acute infections, which alter the critical micro-environment and impair thymocyte proliferation (Savino, 2006). During the life span, humans and other mammals show major shifts from naive T cells to memory T cells with progressively restricted repertoire (oligoclonality) (Sections 1.2.2 and 1.4.3). This aspect of immunosenescence can be accelerated by greater exposure to antigens. The ‘hyperstimulation hypothesis’ of naive T cell depletion is most strongly developed for the very common infections by cytomegalovirus (CMV). Deficits of memory cells and the presence of highly differentiated T cells with CMV-specificity are strongly linked to CMV seropositivity. These changes characterize an ‘immune risk phenotype’ with higher mortality in the elderly, observed in two European populations (Akbar and Fletcher, 2005; Huppert et al, 2003; Pawelec et al, 2005; Wikby et al, 2005). Continued antigenic stimulation is hypothesized to drive clonal expansions of memory T cells and is predicted to eventually clonally deplete cells with the highest antigenic affinity. Moreover, the hyperstimulation by one antigen can have bystander effects that activate other T cell specificities due to local secretion of IFNα and TNFa (Fletcher et al, 2005) (Section 1.4.3).
Although CMV is a ubiquitous infection, in 60–100% of adults depending on the population, nonetheless, some individuals reach old age without becoming seropositive. By comparison with CMV-seronegative individuals aged 22–91, healthy CMV-seropositive individuals with latent infections at all ages had 25–50% fewer naive T-cells and an increase of differentiated memory T cells (CD8+ CD28– effector cytotoxic) in all age groups, particularly the older (Almanzar et al, 2005). Moreover, IFγ expression is higher in CD8+ T cells with CMV-seropositivity, which would increase the body load of this powerful cytokine and possibly synergize with inflammatory processes throughout the body.
The hyperstimulation hypothesis of immunosenescence predicts that populations with chronic infectious disease should show premature T cell senescence. HIV is being examined from this perspective. Young HIV carriers have impaired T-cell responses to vacci-nation for preventing childhood infections; e.g., measles, polio, and influenza antigens induce weaker responses (Rubinstein et al, 2000; Setse et al, 2006). HIV-specific T cells have lower proliferation and cytotoxicity, both as found in immunosenescence of the elderly (van Baarle et al, 2005). Moreover, HIV patients show immune impairments to other antigens. Multiple immunizations with a T cell-dependent neoantigen (φ174, non-infectious bacteriophage) caused progressively smaller responses, unlike healthy controls, presumably because of the already limited pool of naive T cells; booster immunizations with φ174 also increased plasma HIV viremia (Rubinstein et al, 2000). These findings support the hyperstimulation hypothesis of immunosenescence.
These effects extend to antigenic stimulation by intestinal nematodes (helminth parasites). In Ethiopians infected with HIV and nematodes, the plasma HIV load was correlated with the helminth load; successful de-worming treatment also decreased HIV viremia in 8/11 patients (Wolday et al, 2002). Ethiopian immigrants to Israel have given unique opportunities to study the reversibility of age-like immunological changes after leaving a highly infectious environment (van Baarle et al, 2005). About 60,000 Ethiopians of all ages arrived in two waves, 1984–1985 and 1991. Immigrants were often infected with worms, but not with HIV, malaria, or TB. The immigrants were characterized by abnormally low proportions of naive CD4+ T cells and excess T memory cells (‘broad T cell activation’) (Borkow et al, 2000; Kalinkovich et al, 1998; Kalinkovich et al, 2001). Nearly half had inverted CD4:CD8 ratios <1, not found in control groups that resemble the ‘immune risk phenotype’ of elderly northern Europeans (Section 1.2). T cell impairments included decreased activation of ERK2 kinases, β-chemokine secretion, and delayed type hypersensitivity (DTH, memory T cell dependent). Effective de-worming restored the DTH (Borkow et al, 2000), normalized the ratios of CD4:CD8 T cells, and almost normalized the T cell activation profile (Kalinkovich et al, 1998). Evidently, elimination of the nematode antigen load partly regenerated a naive T cell pool, as occurs in HIV infections. Even when not infected with HIV or worms, by the age of 5, Ethiopians have abnormally high proportions of memory T cells, indicative of antigenic hyperstimulation (Tsegaye et al, 2003). Nonetheless, their neonatal T cell proportions were normal, suggesting effective placental barriers to maternal infections (see Section 4.6.4). Their short life expectancy (about 40 y, like other pre-industrial populations, Fig. 1.1 legend) and high memory T cell profile suggest that some Ethiopians are exposed to conditions prevailing in 18th century Europe where life span was also about 40 y (Section 2.4). These continuing studies promise unique insights on the rate of reversibility of immunosenescence after the transition to a 21st century environment with greatly improved health care, hygiene, and nutrition.
Another population in transition is the Tsimane of the Bolivian Amazon basin. These forager-horticulturalists have had limited access to modern medicine and manifest a high incidence of parasites and other infections (Gerven et al, 2007; McDade et al, 2005). Their short life expectancy (43 y, about the same as the Ethiopian immigrants above) and age-specific mortality profile resemble mid-19th century Sweden (Fig. 1.1B, Fig. 2.7A). Blood CRP elevations are consistent with their high pathogen load: By age 40, the Tsimane have had as many years with high CRP as those in the United States by age 60. Immune cells have not been characterized. Other populations of interest for accelerated immunosenescence are Gambians, who have seasonal cycles of infections that alter T cell functions (Section 4.6.2) (Moore et al, 2006), and rural Guatemalans, who received nutritional supplements and health care in controlled studies (INCAP, Section 4.2) (Scrimshaw, 2003).
Mice also accumulate memory T cell clonal expansions in response to chronic antigenic stimulation, e.g., by HSV-1, which induces a strong cytotoxic lymphocyte response (Messaoudi et al, 2006). Unexpectedly, these memory T cells were not antigen specific and were induced just as effectively by the immune adjuvant, suggesting important by-stander effects that increase T cell clonal expansion (Section 1.4.3). Moreover, normal intestinal flora also influence memory T cell clonal expansions, as shown with germ-free mice (Kieper et al, 2005). We may conclude that the individual profile of aging in instructive immunity is closely linked to the cumulative antigenic experience that certainly begins at birth with the first full encounter to the germy world and may well begin earlier in the undocumented levels of prenatal infections. Internal sources of antigens also extend far beyond enteric and oral fauna to auto-antigens, possibly including AGEs accumulated on long-lived proteins. Lastly, the trends for increased levels of proinflammatory cytokines may also influence adaptive immunity, because IL-1, IL-6, IL-18, etc., modulate many aspects of immune progenitor cell differentiation, including stem cells.
2.8.2 Immunosenescence and Telomere Loss
Chronic inflammation without infections can cause telomere shortening. Chronic smokers, for example, have shorter telomeres in peripheral blood lymphocytes (Valdes et al, 2005) and less telomerase (hTERT) (Getliffe et al, 2005). The pathway linking smoking to peripheral lymphocytes may be direct because smoke components rapidly activate blood monocytes by redox-dependent pathways (Walters et al, 2005). Chronic skin inflammation is also associated with telomere shortening. Children with atopic dermatitis have activated skin T cells; when cultured, skin T cell cultures had 25% shorter telomeres than normal blood lymphocytes (Bang, 2001). In psoriasis, blood T cells also have shorter telomeres (Wu et al, 2000).
Chronic psychological stress was correlated with telomere shortening in peripheral lymphocytes in women (Epel et al, 2004). Although these premenopausal women were considered ‘healthy,’ this study did not report on recent infections, which would be expected with histories of stress and which could have caused immune cell proliferation independently of perceived psychological stress. Stress is well known to increase the incidence of infections in humans (Marsland et al, 2002) and animals (Sheridan et al, 2000).
Short telomeres may predict greater mortality and shorter longevity. As noted above, CMV-seropositive individuals had shorter telomeres. Although CMV is considered a benign infection in those with healthy immune systems CMV-seropositivity is associated with higher mortality in the elderly (Pawelec et al, 2005). Again, short telomere length in blood lymphocytes was associated with increased mortality at all ages by 2-fold, with 8.5-fold more infections and 3.2-fold more heart disease (Utah blood donors) (Cawthon et al, 2003). Among stroke survivors, those with short telomeres in blood lymphocytes were more likely to develop dementia and die sooner (von Zglinicki and Martin-Ruiz, 2005). The generality of these findings is unclear.
2.8.3 Inflammation and Stem Cells
Inflammation may prove to impair stem cell generation and survival. For example, in the adult rodent brain, neuron stem cell formation (de novo neurogenesis) was strongly inhibited by LPS endotoxin (direct i.c.v. infusion) (Ekdahl et al, 2003). Neurogenesis was inversely correlated with the density of local microglia. Neurogenesis was restored by minocycline, an antibiotic that passes the blood-brain barrier and suppresses microglial reactions in neurodegenerative disease models. These findings give a rationale for evaluating the role of inflammation in neurogenesis during Alzheimer and Parkinson disease, which have chronic local inflammatory responses, as well as in the milder inflammation of normal brain aging (Chapter 1).
Circulating C-reactive protein (CRP) may inhibit generation of vascular endothelial progenitor cells (EPCs), which are implicated in adult vascular repair and regeneration (Section 1.5.3.4). In vitro, clinical levels of exogenous CRP inhibited EPC migration and adhesion; moreover, high CRP also inhibited formation of capillary tubules (angiogenesis) and the expression of proangiogenic factors (VEGF, IL-8; eNOS) (Verma et al, 2004; Suh et al, 2004). Other factors are anticipated besides elevated CRP because angina patients showed very modest correlations of CRP and circulating EPCs (r2 = 0.09) (George et al, 2004).
Responses to bacterial pneumonia imply further roles of EPC. Elderly patients with bacterial pneumonia, but without clinical cardiovascular disease, had several-fold more circulating EPCs during the acute phase, relative to posttreatment (Yamada et al, 2005). Those with low EPCs before treatment had persistent lung fibrosis, which suggests that EPCs contribute to lung repair. Moreover, EPC and total lymphocyte counts were strongly correlated, consistent with the poor prognosis of lymphocytopenic pneumonia patients. This association implies the co-regulation of EPCs with other marrow stem cell populations.
In summary, these observations suggest that chronic exposure to infection and inflammogens may drive increased mortality during aging from three causes. (I) The diminished pool of naive memory T-cells truncates the primary immune response to new infections, whereas the highly differentiated state of memory cells limits the strength of the secondary responses. (II) The cytokine shift to increased IFNγ production by differentiated T cells may interact with the proinflammatory trend during aging. (III) Atherogenesis may interact with these processes at many levels, from local reactions of CMV-specific T cells in unstable atheromas to reduced generation of stem cells that may be critical in repair of vascular, lung, and neural tissues.
2.9 CANCER, INFECTION, AND INFLAMMATION
Some infections that cause chronic local inflammation also increase the risk of cancer and other abnormal growths. About 15% of all cancers are attributed to infections, directly or indirectly (Herrera et al, 2005). Links between aging, cancer, and endogenous inflammation are also hypothesized (Sarkar and Fisher, 2005). Many cancers and abnormal growths are associated with local inflammatory cell responses that stimulate cell proliferation. In the mutational theory of oncogenesis, the mutagenic progression to malignancy begins with DNA damage. Mutations accumulate during DNA replication, which may be stimulated endogenously, e.g., by sex steroids in reproductive tissues or exogenously, e.g., ultraviolet damage, carcinogens, viruses. Inflammatory processes after initiation are illustrated by the ras pathway. Ras protooncogenes (H-ras, K-ras, N-ras) are often activated by mutations; e.g., 50% of colon cancers have mutations in ras (Bos, 1989). Activated ras protein, in turn, induces IL-1 and IL-8 and other proinflammatory cytokines (Sparmann and Bar-Sagi, 2004; Liu et al, 2004). IL-8 induction involves ERK-MAPK and PI3K pathways, which also influence longevity (Fig. 1.3). IL-8 mediates the recruitment of macrophages and other inflammatory cells that promote tumor vascularization (angiogenesis). Local inflammatory responses may also produce ROS that induce further mutations on the pathway to malignancy. Several examples are discussed in more detail that represent inflammation from infections (next) and from smoking (following section) and that further illustrate the importance of bystander damage during inflammation (Chapter 1.4).
2.9.1 Helicobacter Pylori and Hepatitis B Virus
H. pylori is conclusively linked to inflammatory bowl disease and gastro-intestinal cancers, which rank second among malignancies as cause of death (Herrera et al, 2005; Parkin et al, 2005; Sugiyama and Asaka, 2004). These links are much stronger than associations of H. pylori with vascular disease (Section 2.2). Most humans have submucosal infections of H. pylori: 76% in developing countries, 58% in developed countries (Parkin et al, 2005). Infections are usually acquired early in life, and the risk is several-fold higher if parents are infected (Webb et al, 1994; Rothenbacher et al, 2002). Fortunately, most carriers (85%) are asymptomatic. However, about 15% of carriers develop peptic ulcers, with 1% proceeding to gastro-intestinal cancers. As hygiene and public health improve, the prevalence of infections decreases. In successive birth cohorts of the Bristol Helicobacter project, cancer incidence has decreased correspondingly with H. pylori infections (Harvey et al, 2002; Lane et al, 2002). This progression supports the general relationships of the early-life infectious load to later mortality in successive cohorts (Fig. 2.8) and is a benchmark example of by-stander effects in infections (Queries 1 and 2, Section 1.1).
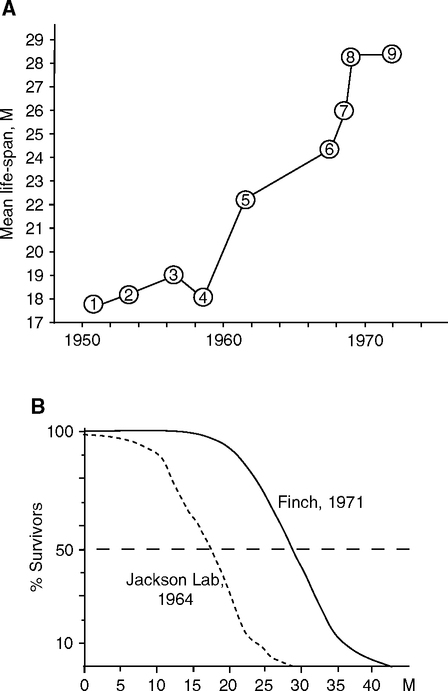
The oncogenicity of H. pylori arises from chronic localized inflammatory responses around the mucosa to which it attaches extracellularly (Penta et al, 2005). Mucosal cell proliferation is stimulated. Infiltrating macrophages and neutrophils produce promutagenic ROS that damage epithelial cells, in association with increased oxidative DNA damage, as assayed by intestinal 8-OHdG (Farinati et al, 2003) . As further proof of the inflammation-oxidative damage link, treatments that eradicated H. pylori also decreased tissue level of 8-OHdG (Farinati et al, 2004) . As expected from the increased intestinal proliferation, intestinal telomere DNA is also shortened in H. pylori infections, even without local metaplasia (Kuniyasu et al, 2003). There are instructive genetic interactions of host cytokine alleles with virulence factors of H. pylori. For example, cancer risk was 10-fold higher by the combined presence of a high-risk H. pylori virulence factor and high-risk IL-1 allele than in the opposite low-risk combination (Figueiredo et al, 2002). Anti-inflammatory NSAIDs, which inhibit COX-2, are protective in H. pylori infections (more on this in the next section). H. pylori infections are strong examples of inflammation-induced oxidative damage (Query II).
Inflammatory bowl disease is among the top three main risk conditions for colorectal cancer. After 7 y of exposure to inflammatory bowl disease, the incidence of colorectal cancer increases by 0.5–1%/y (Itzkowitz and Yio, 2004). The area of the colon surface involved in inflammation also increases the risk of colorectal cancer, giving an example of bystander damage with dose effects from time and target area (Section 1.4). Oxidant stress from inflammation is a leading mechanism in colorectal cancer. Chromosomal instability, mutational loss of function in the APC and p53 genes, and induction of COX-2 and NO synthase are typical. Consistent with the inflammation hypothesis, aspirin and NSAIDs reduce the risk of colorectal cancer (Section 2.10.2).
Virus-induced inflammation is also linked to liver cancer, which ranks sixth in the world and third in the U.S. as causes of death (Parkin et al, 2005). Most liver cancers, 75% worldwide, are caused by hepatitis B and C viruses (HBV, HBC). Each infection increases the risk about 20-fold, while co-infection may increase the risk >100-fold (Donato et al, 1998). A substantial minority (5–20%) of HBV infections progresses from cirrhosis, within 5 y of diagnosis, to hepatocellular carcinoma (Imperial, 1999; Marcellin et al, 2005). HBV-induced cancer is associated with increased inflammatory expression: Although the acute phase responses during HBV were not reported in detail, IL-6 elevation correlates with the clinical severity (Tangkijvanich et al, 2000). Tumorigenesis depends on Hepatitis-Bx (HBx), a virally encoded signaling peptide that stimulates hepatocyte proliferation by activating NF-kB after viral integration. Hbx also facilitates metastasis by inducing matrix remodeling through matrix metalloproteinases and COX-2 (Lara-Pezzi et al, 2002). These latter inflammatory responses are all-too-familiar in atheromas (Section 1.5.3.1). COX-2 inhibitors suppress cell invasion in experimental models (Lara-Pezzi et al, 2002), but their efficacy has not been reported for HBV or HBC. These examples of H. pylori and hepatitis virus in inflammation and cancer merely scratch the surface of a growing literature that links chronic inflammation to cancer and that shows promise for anti-inflammatory drugs (see below).
2.9.2 Smoking and Lung Cancer
Smoking is the most robust example of an external inflammogen (bioaerosol) that is not infectious and that causes acceleration of mortality and premature mortality from multiple causes. Lung cancer was extremely rare before the advent of popular smoking beginning in the early 20th century. In Richard Doll’s classic longitudinal study of British physicians since 1951, smokers have a risk that increases in proportion to the numbers of ‘pack-years.’ In contrast, those avoiding contact with cigarette smoke have negligible risk (Fig. 2.11) (Doll et al, 2004; Doll et al, 2005; Peto R et al, 2000; Vineis et al, 2004). For lifetime use, the risk of death from lung cancer is 5% in men and 10% in women. Smokers who quit retain the same risk for lung cancer indefinitely; e.g., men quitting at 60 had stabilized 10% during the remainder of their lives.
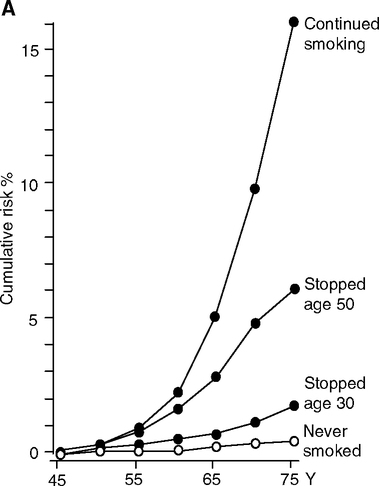
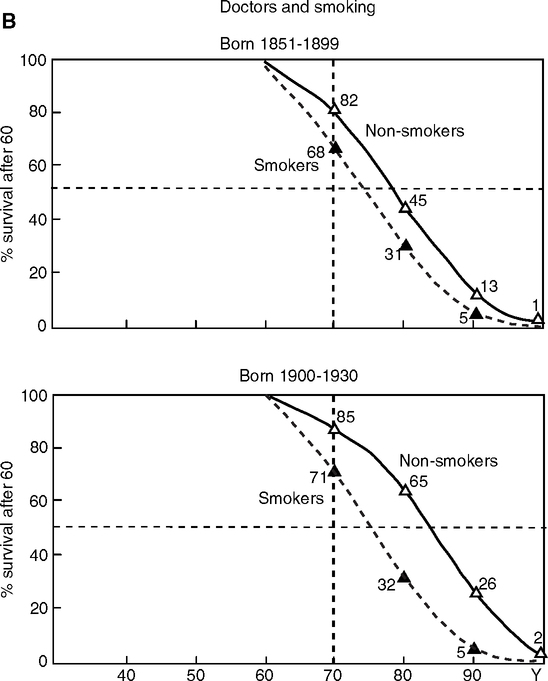
These powerful dose-response relationships in smoking and cancer define a paradigm for environmental exposures that may extend to environmental factors in cancers in general. Besides the lung, smoking also increases the risk of cancer elsewhere in the larynx, stomach, liver, pancreas, and bladder. However, prostate cancer is only weakly, if at all, related to smoking (Vineis et al, 2004). Cancer risk in smokers may be subject to genetic variations in detoxifying enzymes, DNA repair (Wu X. et al, 2004), and apoE4 (Fig. 5.18).
Smoking also increases the risk of vascular disease events, but the dose-response to pack-years is less clear. It is not known how tobacco smoke promotes, directly or indirectly, carcinogenesis in enteric organs that are not in direct contact with the airways.
2.10 PHARMACOPLEIOTROPIES IN VASCULAR DISEASE, DEMENTIA, AND CANCER
I suggest the term ‘pharmacopleiotropy’ to represent multiple domains of drug effects with overlapping specificities. Many drugs influence biochemical and cell systems far beyond the original targets. Negative or adverse effects of drugs are well recognized, as in gastric bleeding from aspirin. However, unexpected positive pharmacopleiotropies are emerging for drugs that protect against heart disease which may also reduce the risk of cancer and of AD (Table 2.2). These cross-over effects are consistent with shared anti-inflammatory effects of diverse drugs in heart disease and with evidence for the pervasiveness of inflammatory processes in “normal” aging and in Alzheimer and vascular disease. Moreover, pharmacopleiotropies are consistent with genetic pleiotropies of apoE4 as a shared risk factor in Alzheimer and vascular disease and with the shared risk factors from the environment and lifestyle (Section 5.7).
TABLE 2.2
Possible Benefits of Adult-Onset Degenerative Diseases by Aspirin, NSAIDs, and Statins
| Aspirin | NSAIDs | Statins | |
| cancer | |||
| colorectal & esophageal | +2 | +2 | +/− |
| bladder, breast, ovary, prostate | +/– | +/– | +/– |
| non-Hodgkin lymphoma | −1 | −1 | |
| neurodegenerative diseases | |||
| Alzheimer disease | + | + | +/– |
| Parkinson | + | + | |
| vascular disease events | |||
| myocardial infarct | +2 | −2 | +2 |
| stroke | +/− | −1 | +2 |
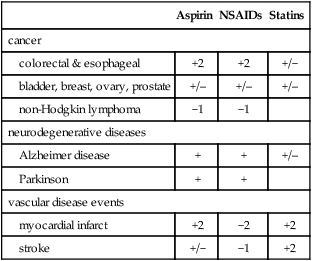
aspirin and NSAIDs (Baron, 2003; Bosetti et al, 2002; Herendeen and Lindley, 2003; Perron et al, 2003;
Meric et al, 2006); non-Hodgkin lymphoma (Cerhan, 2003). statin benefits to cancer are in Phase II trials with various tumors and are being considered for adjunct chemotherapy and chemopreventives (Chan et al, 2003; Etminan et al, 2003).
Alzheimer (Breitner, 2003; Etminan et al, 2003)
Parkinson (Chen et al, 2003)
aspirin (Antithrombotic Trialists’ Collaboration, 2002; Eidelman et al, 2003; Bartolucci and Haward, 2006) statins (Fonarow and Watson, 2003; Heart Protection Collaborative Group, 2002; Law et al, 2003; Mostaghel and Waters, 2003; Wald and Law, 2003) stroke:
aspirin (Bartolucci and Howard, 2006)
NSAIDs (Kearney et al, 2006)
statins (Amarenco et al, 2006; Endres and Laufs, 2006).
+2, consistent benefit across many studies; +1, possible benefits according to initial observations; +/–, possible benefits, but not consistent; −1, possible adverse effects; −2, definite adverse effects. Other references in Section 2.1.
2.10.1 Anti-inflammatory and Anti-coagulant Drugs
The remarkable possibility can be considered that a cluster of major degenerative diseases is attenuated by the widely used drugs that were developed for heart disease. Meta-analyses agree in the shared efficacy of quite different drugs in reducing the risk of vascular events, certain cancers, and possibly Alzheimer disease, with effects in the broad range of 10–60% (Table 2.2). No single study can suffice, and there are many divergences between indications of efficacy from animal models and clinical effects. It is hard to interpret the drug literature on vascular disease and dementia (Baron, 2003): Clinical end-points differ between studies, as do drug doses and durations. There is great heterogeneity in age, health status, ethnicity, socioeconomics, and education. Those taking aspirin and other drugs often modify their lifestyles, which reduces their risks independently of the drug candidate. Thus, the ‘possible benefits’ of the drugs in Table 2.2 should be considered a moving target of continuing critical evaluation for complex drug interactions to identify subgroups by risk and benefit for remaining life expectancy.
The pharmacology of anti-inflammatory drugs distinguishes two broad groups: steroidal anti-inflammatory drugs (SAIDs) and non-steroidal anti-inflammatory drugs (NSAIDs) (Hardman and Limbird, 2001; Vane and Botting, 2003). NSAIDs alter production of prostaglandins and thromboxanes, ‘isoprenoids’ that have diverse and broad roles in normal physiology and in disease, e.g. the thromboxane TXA2 enhances local platelet aggregation, while blockers of TXA2 can be protective against thrombosis. The ubiquitous isoprenoids are also referred to as ‘autocoids’ because they are produced from local membrane lipids. Arachidonic acid is an autocoid substrate of the cyclooxygenase enzymes, COX-1, -2, and -3. COX-1 products mediate normal functions of gastric mucosa and kidney, as well as coagulation: COX-2 is induced during inflammatory reactions, e.g., by LPS and other endotoxins. COX-1 and COX-2 are coded by distinct genes, whereas COX-3 is a new splicing variant of COX-1 with overlapping functions (Botting, 2003; Vane et al, 2003). PI3-K, of the ‘insulin/IGF-1 metabolic longevity’ pathways (Fig. 1.3), mediates many NSAIDs’ actions. For example, PI3-K is required for induction of COX-2 by endotoxin (LPS) in mesangial cells (Sheu et al, 2005).
Steroidal anti-inflammatory drugs, such as dexamethasone and prednisolone (synthetic glucocorticoids), act via specific receptors to modulate subcellular pathways that are in part distinct from those regulated directly by NSAIDs. Glucocorticoids have metabolic effects during chronic treatments that differ from NSAIDs. However, there are notable convergences; e.g., the COX-2 gene promoter has glucocorticoid response elements (GRE), which enable repression by cortisol and synthetic glucocorticoids (Kramer, 2004; Santini et al, 2001).
Of the NSAIDs, aspirin (acetylsalicylic acid) has been long known as an analgesic and antipyretic at low doses and is the mostly widely taken drug worldwide (Vane et al, 2003). Aspirin has anti-coagulant activities that reduce the risk of vascular events. At 160 mg/d, aspirin doubles the clotting time by irreversibly inhibiting platelet COX-1, which makes TXA2 from arachidonic acid, an omega-6 polyunsaturated fatty acid (PUFA). Aspirin effects on clotting last up to 10 days, the life span (t½) of blood platelets. Higher doses of aspirin are used for rheumatic inflammatory diseases, but often cause gastric bleeding due to blockade of COX-1 in the gastric mucosa. However, even after its acetylation by aspirin, COX-2 activity persists for certain PUFA ω-3 and ω-6 substrates, some of which are anti-inflammatory, e.g., 15 epi-lipoxin A4 (Arita et al, 2005). Ibuprofen and naproxen are non-selective for COX-1 and-2 (Cryer and Feldman, 1998). Other NSAIDs were developed to target COX-2 without inhibiting COX-1, effectively reducing inflammatory responses with fewer gastric side effects, e.g., celecoxib, naproxen, and rofecoxib. COX-2 and prostaglandin production are also inhibited by synthetic glucocorticoids, as noted above.
The NSAIDs field is in turmoil because of conclusive evidence for increased risk of vascular events (Hippisley-Cox and Coupland, 2005; Juni et al, 2004; Levesque et al, 2005). For example, a population-based case-control study in Finland showed that NSAIDs users had 40% higher risk of a myocardial infarct (OR, 1.40, 95% CI, 1.33–1.48) (Helin-Salmivaara et al, 2006). The various types of COX-2 inhibitors had similar effects. An underlying mechanism may be that inhibiting COX-2 may induce both proinflammatory and prothrombotic homeostatic compensatory responses (Doux et al, 2005). Homeostatic responses would be expected because of the importance of COX-2 to many normal cellular functions, e.g., celecoxib induced COX-2 in rat spinal cord (Hsueh et al, 2004). Moreover, due to their short half-lives in circulation (e.g., t½ 11 h, celecoxob), there could be prothrombotic transients in prostaglandin conversion in the declining phase before another dose is taken. Withdrawal from chronic aspirin in stable cardiac patients, for example, may increase the incidence of thromboses (Senior, 2003). These early observations merit full study.
Statin drugs were designed to lower blood cholesterol by blocking a rate-limiting enzyme of cholesterol synthesis (HGM-CoA reductase). In addition, statins have many effects on inflammation (Balk et al, 2003; Halcox and Deanfield, 2004; Kwak et al, 2003; Schonbeck and Libby, 2004; Stuve et al, 2003), which may account for vascular benefits not directly linked to blood lipids. Cholesterolindependent pleiotropic effects of statins include the lowering of blood CRP (Albert et al, 2001), inflammatory cell infiltration and cell death (Wierzbicki et al, 2003), and impairing of lymphocyte proliferation (Palinski and Tsimikas, 2002b). Some statin effects result directly from the decrease of mevalonate from the inhibition of HMG CoA reductase. Intracellular mevalonate is a precursor to the isoprenoids (farnesyl pyrophosphate, geranylgeranyl) that modulate enzymes with links to inflammation and vascular smooth muscle cell proliferation, e.g., the Ras and Rho kinases (Endres and Laufs, 2006; Kamiyama et al, 2003). PI3K/Akt is also involved in the induction of nitric oxide synthase by statins (Walter et al, 2004). Statins also increase circulating endothelial progenitor cells (EPCs) (Urbich and Dimmeler, 2005; Vasa et al, 2001a, b), which are implicated in vascular repair (Sections 1.5.3.4 and 2.5.2) and may modulate EPC cell senescence by suppressing Chk2, a DNA damage checkpoint kinase induced by telomere dysfunction (Spyridopoulos et al, 2004). Cardioprotective effects of NSAIDs and statins involve neovascularization (Vagnucci and Li, 2003), which may be important to both AD neuritic plaques and in vascular atheromas (Chapter 1, Table 1.3).
2.10.2 Aspirin and Other NSAIDs
Aspirin and NSAIDs have well established benefits to risks of heart attack and probably to stroke. In the huge Anti-thrombotic Trialists’ Collaboration, aspirin and antiplatelet drugs collectively reduced vascular events: 25% risk reduction for myocardial infarction (primary or recurrent) and 11% reduction of stroke; aspirin at a low dose (75–150 mg/day) reduced risk by 32% (Antithrombotic Trialists’ Collaboration, 2002). Low dose aspirin in other studies reduced stroke by 13–16%. This net figure represents a greater reduction in thrombotic strokes than the increase of hemorrhagic stroke in aspirin users (van Gijn and Algra, 2002; Wald and Law, 2003).
Some cancer risks may benefit from aspirin and NSAIDs (Baron, 2003; Meric et al, 2006). Colorectal cancer is the best case, in which aspirin lowered risk by 20–30% in case control and cohort studies (Morgan, 2004; Slattery et al, 2004). Gastric cancer shows similar benefits of aspirin and NSAIDs (Wang et al, 2003b). As discussed above, gastric cancer is strongly associated with Helicobacter pylori infections, which elevate mucosal prostaglandins (Laine et al, 1995). In one mechanism, aspirin directly inhibits H. pylori growth (Wang et al, 2003c). Breast cancer may also benefit by aspirin and NSAIDs (Zhang et al, 2005), particularly for cancers that have both estrogen and progesterone receptors (Terry et al, 2004). The optimum dose of aspirin for cancer prevention may be higher than for coronary disease (Chan et al, 2004). These effects are controversial and may not hold for low-dose aspirin (Cook et al, 2005).
Alzheimer disease risk may also benefit from aspirin and NSAIDs, another controversial possibility that has been considered for nearly two decades. An early indication came from informal clinical observations that rheumatoid arthritis patients seemed to show less Alzheimer disease (Jenkinson et al, 1989); although this study could not fully document the drug use profile, rheumatoid patients are typically heavy users of anti-inflammatory drugs. Another indication came from identical twins discordant for use of anti-inflammatory drugs (all classes): The twin taking anti-inflammatories had about 75% lower relative risk (Breitner et al, 1994). Recall from Section 2.7 that periodontal disease increased the risk of dementia in twins (Gatz et al, 2006).
Larger post hoc studies indicate improvements of 13% for aspirin and 28% for NSAIDs (Akiyama et al, 2000; Breitner, 2003; Etminan et al, 2003; McGeer et al, 1996; Nilsson et al, 2003). In recent longitudinal observational studies, two groups reported less Alzheimer’s in users of NSAIDs, but not aspirin (in t’ Veld et al, 2001; Cornelius et al, 2004). However, others observed less Alzheimer’s and loss of cognition in normal individuals with 75 mg or more aspirin; NSAIDs showed similar trends (not statistically significant) (Nilsson et al, 2003).
Interventional studies of NSAIDs and Alzheimer’s are in an early stage (Akiyama et al, 2000; Davey Smith and Ebrahim, 2002; Etminan et al, 2003). In Joseph Rogers’s pioneering pilot study, Alzheimer patients with mild to moderate impairments had slower decline after 6 months of indomethacin, an inhibitor of both COX-1 and -2 (Rogers et al, 1993). However, subsequent trials with the selective COX-2 inhibitor Rofecoxib (alternate names: Refecoxib or Vioxx) did not show benefit in two large randomized trials lasting 1 y (Aisen et al, 2003; Reines et al, 2004).
Experimental studies give some insights into these divergent results (Imbimbo, 2004). In cell cultures, indomethacin decreases the production of Aβ42 (Fig. 2.12), as does ibuprofen (Weggen et al, 2003; Kukar et al, 2005). These effects are well documented in transgenic Alzheimer mice, in which chronic ibuprofen at clinical levels slowed brain amyloid accumulation (solid and soluble), IL-1β increase, and behavioral changes (Lim et al, 2001b; Morihara et al, 2005).
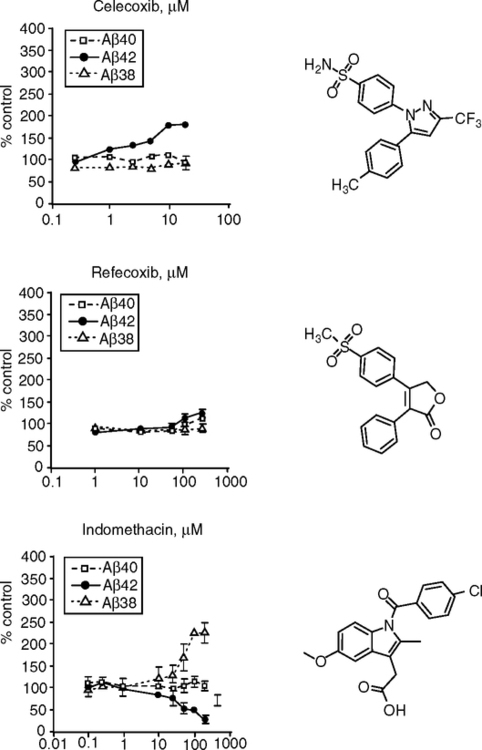
However, contrary to the consistent effects of ibuprofen, targeted COX-2 inhibitors did not give the expected benefits. Rofecoxib increased Aβ42 production in cells (Fig. 2.12) and in the brain (Kukar et al, 2005). Celecoxib (Celebrex) was similar. Contrary to assumptions, these activities do not require COX-1 inhibition and appear to modify amyloid production through direct allosteric effects on γ-secretase, the enzyme complex that cleaves Aβ42 from the amyloid-precursor protein. As noted above, COX-2 drugs are associated with increased risk of myocardial infarctions. Possibly, their withdrawal from general long-term use may also benefit the risk of dementia on two grounds: the shared prothrombotic risk factor of myocardial and cerebrovascular events and the increased production of Aβ42 by these drugs.
In considering further large-scale trials of NSAIDs as preventives for Alzheimer disease, we need more preclinical understanding of the pharmacopleiotropies of these powerful drugs. Genotype may also be important, e.g., apoE4 carriers taking aspirin had a higher incidence of Alzheimer disease (Cornelius et al, 2004). Other inflammatory gene variants could influence drug responses, particularly those associated with vascular disease (Chapter 4). Meanwhile, we may learn more about the benefits of aspirin to Alzheimer and vascular dementia, because aspirin continues to be widely advised and used.
2.10.3 Statins
2.10.3.1 Vascular Disease
Statins are considered separately because of their multiple effects on cholesterol and on inflammation. In a meta-analysis, statins reduced the risk of coronary events by about 60% and stroke by 17%, in association with lower LDL cholesterol (Law et al, 2003). Among secondary prevention medications for cardiovascular disease, statins reduced cardiac mortality by 24% to 42%, i.e., at least as much as aspirin and blood pressure medications (β-adrenergic blockers, and ACE inhibitors) (Fonarow and Watson, 2003; Grobbee and Bots, 2003). Statins also lowered cardiovascular mortality in patients with normal cholesterol in two major studies: the Scandinavian Simvastatin Survival Study (‘4S’) and the Long-Term Intervention with Prevastatin in Ischaemic Disease (LIPID) trial (Fonarow and Watson, 2003).
Statins influence vascular plaque size by mechanisms that may be independent of effects of statins on blood cholesterol levels (Wierzbicki et al, 2003). The FATS trial showed 75% decrease of cardiovascular events, despite the small improvement in stenosis (<1%) (Brown et al, 1993). These authors suggested that statins selectively depleted unstable fatty plaques with large lipid cores (Fig. 1.13, type IV), thereby stabilizing the lesions and reducing thrombosis. Plaque shrinkage during statin use is documented by sonography (Sato et al, 2003) and magnetic imaging (Vigen et al, 2005).
These unexpected benefits of statins point to broad anti-inflammatory actions, possibly through the reduction of isoprenoids from blocking HMG-coA reductase, which modifies many receptor functions in inflammatory pathways. Stain therapy is being considered for a wide range of inflammatory conditions, including multiple sclerosis, rheumatoid arthritis, and sepsis.
2.10.3.2 Dementia
Lower risk of Alzheimer disease was observed in statin users in epidemiological studies (Crisby et al, 2002; Jick et al, 2000; Vagnucci and Li, 2003; Wolozin et al, 2000). Evidence seems definitive that statins lower the risk of a first stroke (Amarenco, 2005; Endres and Laufs, 2006) or recurrent stroke (Amarenco et al, 2006), which should also lower dementia risks (Section 1.6). The slow turnover of brain cholesterol (about 6 months) (Casserly and Topol, 2004) implies that prolonged treatment with statins may be needed.
Animal models raise caveats for statins, as for NSAIDs. On the positive side, in cell culture models, lowering of cell cholesterol by statins decreased Aβ42 production, apparently by directing APP cleavage to the non-amyloidogenic α-secretase pathway of APP cleavage (Cole et al, 2005b). However, the lower levels of isoprenoids had opposing effects by increasing production of APP and Aβ42, which would be pro-amyloidogenic. This may be why lovastatin increased brain amyloid in a transgenic Alzheimer mouse (Tg2676), despite lower plasma cholesterol (Park et al, 2003). Rebound effects are also observed in discontinuation of statins, which are associated with increased risk of stroke and mortality (Endres and Laufs, 2006). Statin clearance after sustained administration suddenly activates rho and Rac-1, leading to oxidative bursts and decreased NO availability. Rebound transients could also arise with the statins that are rapidly cleared from plasma, noted above for NSAIDs.
2.10.4 Sex Steroid Replacement (Hormone Therapy)
Estrogen replacement therapy to compensate for age-related deficits is a possible intervention for vascular and brain dysfunctions and diseases that interact with inflammation at many levels. Major ongoing studies address benefits and risks of hormone therapy.5 Animal models are largely supportive, but clinical findings are mixed. Besides their direct effects on many brain cell and vascular cell functions, sex steroids also modulate adaptive immune and inflammatory functions. My summary of these complex and controversial topics begins with a demographic perspective.
Cardiovascular disease, cerebrovascular disease, and Alzheimer disease show an accelerating incidence during mid-life into later ages. Sex differences in mortality are present throughout life with greater overall male vulnerability (Section 5.2): In developed countries during recent decades, age-specific death rates of adult women lag behind those of men and approximate those of adult men who are 5 y or more younger (Fig. 5.2). After middle age, cardiovascular disease becomes the first or second main cause of death in most populations (Section 1.2.2, Fig. 1.4). Mortality rate accelerations tend to be slightly faster for women than men, for both total and cardiovascular mortality (Horiuchi, 1997), but within the population average mortality rate doubling times of about 7–10 y (Section 1.2.1). The mortality accelerations are concurrent with menopause, which implies a role for the sharp decrease of ovarian steroid levels. However, behavioral and environmental risk factors vary widely between populations, making it is hard to resolve the contributions of age-changes in sex hormones to health and mortality after middle age.
Dementia risks accelerate exponentially after age 60, with a doubling time of about 5 y (Fig. 1.10A) (Kawas and Katzman, 1999; Mayeux, 2003). Sex differences in dementia are variable and, in a meta-analysis of 23 studies, were not statistically significant (Jorm and Jolley, 1998). The age-specific prevalence was also similar in men and women in three studies [Rotterdam Study (Ott et al, 1998), Cardiovascular Health Study (Fitzpatrick et al, 2004), and AHEAD (Assets and Health Dynamics of the Oldest Old, a survey of community living U.S. elderly) (Suthers et al, 2003)]. Nonetheless, relatively more women are demented at later ages, when calculated as a fraction of the surviving population. In developed countries at 55, the female:male ratio is about 1:1; at 85, about 2:1, and climbing (Section 5.2. Fig. 5.2). In populations where women have a greater life expectancy (Section 5.2), women are exposed to a proportionately greater lifetime risk of dementia (the ‘demographic hypothesis of dementia prevalence’) of Suthers, Kim, and Crimmins (2003). As examples, at age 55, Rotterdam women had a 2-fold higher risk of dementia in the remaining life span: women, 0.33; and men, 0.16 (Ott et al, 1998). In the AHEAD sample at age 85, the remaining life expectancy without cognitive impairment was 15% longer for women.
The greater mortality risk of males in cardiovascular deaths has been interpreted as due to protection from cardiovascular disease by ovarian steroids, principally by estrogens, which decrease >90% after menopause. Until recently, estrogen was the most widely prescribed medication in the United States and remains popular because of its effectiveness in suppressing menopausal hot flushes (Hickey et al, 2005). Estrogen therapy is supported by experimental and clinical studies. Many observational studies concluded that estrogen replacement lowered cardiovascular events by 40–60% (Barrett-Connor et al, 2005; Grady et al, 1992). For example, the Nurse’s Health Study recruited women who reported no cardiovascular disease (Grodstein et al, 1997) at entry ages 30–55 in 1976: 18 years later, current hormone users had a lower mortality risk (0.63); those who acquired coronary risk factors since entry had the greatest benefit (relative risk, 0.51); the risk of death from coronary disease was 0.47. In EPAT (Estrogen in the Prevention of Atherosclerosis Trial), estradiol replacement (oral, micronized) of healthy postmenopausal women for 24 months slowed (actually decreased) carotid artery thickening (Hodis et al, 2001; Karim, 2005), a major marker of atherosclerosis (Section 1.5). In the Women’s Health Initiative (WHI)-estrogen (E) Trial, coronary disease outcomes were decreased by about 35% in women who began CEE before 60. Estrogen plus progestin (CEE + medroxyprogesterone) had similar benefits (Women’s Health Initiative Steering Committee, 2004; reviewed in Hodis and Mack, 2007a,b). These findings are supported by other studies and meta-analyses, which also show benefits of estrogen therapy are reduced or reversed if begun after age 60.
Hormone therapy (estrogen replacement) ‘should be’ protective because of consistent improvements in vascular risk markers for (lowering of LDL cholesterol and IL-6, etc.), as shown by several hundred studies (Godsland, 2001). A complication is that oral estrogen may have proinflammatory effects.6 Experimentally, estrogen attenuates the systemic and local inflammatory responses (Amantea et al, 2005; Carlsten, 2005; Thomas et al, 2003) and can attenuate apoptosis, e.g., in endothelial cells by Akt-dependent mechanisms (Koga et al, 2004).
Estrogens also have anti-oxidant effects (Behl et al, 1997; Sugishita et al, 2003). In clinical studies, estradiol decreased plasma antibodies to oxidized LDL (Hoogerbrugge et al, 1998) and the susceptibility of LDL to oxidation (Sack et al, 1994). In vascular smooth muscle cultures, estradiol decreased the production of ROS from hydrogen peroxide or CMV infections; estradiol also inhibited the CMV infection (Speir et al, 2000).
Moreover, estrogen enhances endothelial progenitor cell (EPC) production and inhibits EPC senescence (Sections 1.6.2 and 2.5.2). Young women receiving estradiol in a fertility clinic had 3-fold more circulating EPCs, probably due to increased bone marrow production, as was shown in mice (Strehlow et al, 2003). Estrogen attenuates EPC cell senescence, at least in part, by augmenting telomerase (hTERT) through PI3kinase/Akt-dependent mechanisms (Imanishi et al, 2005).
Bone density is another key benefit of estrogen therapy. The consensus holds that estrogen therapy attenuates osteoporosis and spontaneous osteoporotic fractures that are an important cause of mortality in older women (Barrett-Connor et al, 2005; Col et al, 2005; Raisz et al, 2005). Some of the same inflammatory factors that are risk factors in cardiovascular disease and that are improved by estrogen are also regulators of osteoclasts (Chapter 1) (Ginaldi et al, 2005); e.g., estrogen suppressed IL-6 in bone marrow cells and formation of new osteoclasts (Jilka et al, 1992).
But adverse side effects of estrogen are also widely recognized, particularly for endometrial cancer (Amant et al, 2005; Finch and Flurkey, 1977). The continued stimulation of cell proliferation in the endometrium by sex steroids inevitably leads to the accumulation of mutations from errors during DNA replication (bystander effect, type 3, Section 1.4.3). Moreover, breast cancer risk was increased 20% by estrogen plus progestin in the WHI Trial and Heart and Estrogen/Progestin Replacement Study (HERS) (reviewed in Hodis and Mack, 2007a,b). Although venous thromboembolism is also increased by most forms of hormone therapy (Cosman et al, 2005), the effects diminish after the first year of treatment (Hodis and Mack, 2007a). The risk of stroke in WHI-E and WHI-EP was higher for women beginning therapy after 60, while much rarer for those beginning within 5 y of menopause (<1 per 1000).
Similar reversals have occurred for dementia. Just as for cardiovascular disease, hormone therapy has shown benefits to dementia and cognitive aging in many experimental and clinical studies (Birge et al, 2001; Sherwin, 2005). In animal models, estrogen favors neuronal growth and memory, and increases resistance to amyloid and other neurotoxins (Brinton, 2004; Pike, 1999; Quintanilla et al, 2005; Rozovsky et al, 2005; Simpkins et al, 2005). Estrogen also decreases neuronal damage in stroke models (Wise et al, 2005). However, the Women’s Health Initiative Memory Study (WHIMS), an ancillary study of WHI, found that estrogen increased risk of cognitive decline by 1.38-fold for either ‘mild cognitive impairment, MCI’ or ‘probable dementia’ (Shumaker et al, 2004). Adverse effects on cognition were greater among women with lower scores at entry (Espeland et al, 2004).
These recent findings are devastating to millions of women and their physicians (Barrett-Connor et al, 2005), but strong conclusions are premature (Schneider, 2004; Morrison et al, 2006; Hodis and Mack, 2007a,b). The timing of replacement may be particularly important (Clarkson, 2002). In WHI and several other trials, estrogen therapy was initiated at 10 or more years after menopause (age at entry averaged 63 y). This delay is important because early menopause accelerates ‘subclinical’ atherosclerosis, as does surgical ovariectomy (Stampfer et al, 1990). The duration of estrogen deficits also influences carotid artery thickness in correlation with the time since menopause (surgical and natural) (Mack et al, 2004): thickness increased by 0.0032 mm/y after menopause (age-adjusted), which is about 50% of the overall age trend of 0.006 mm/y (author’s calculation).7 In monkeys, estrogen replacement was most effective in suppressing arterial plaque growth if begun at ovariectomy; estrogen replacement was ineffective if delayed 2 y (equivalent human age 52) (Clarkson, 2002).
Consistent with these findings, estradiol replacement did not benefit existing coronary stenosis in women about 10 y after menopause (Hodis et al, 2003a,b; Hodis and Mack, 2007a,b). The timing of estrogen replacement is the focus of ELITE (Early versus Late Intervention Trial) (Howard Hodis, University of Southern California). The use of estradiol rather than conjugated equine estrogens (CEE) may improve trial reproducibility because of variations in equine estrogen components (footnote 5).
Lastly, I return to influences of sex steroids on immune functions, as implied by the anti-inflammatory effects of estrogens. In general, females have higher serum Ig and antibody responses to diverse antigens and incur more autoimmune disease than males (Min et al, 2005; Olsen et al, 1996). During aging, lymphopoiesis differs between immune cell subclasses (Min et al, 2005). Estrogen deficits may derepress regulatory T-cell functions (CD4+) that control self-tolerance and responses to microbial antigens (Sakaguchi, 2004). Benefits of estrogen replacement to osteoporosis depend on T-cell subsets that support bone-resorption by osteoclasts via TNFa and TGF-β1 (Gao et al, 2004; Roggia et al, 2001). These mechanisms may extend to endothelial progenitor cells (EPC) of bone marrow origin that mediate vascular repair. As noted above, estrogen enhances EPC functions and suppresses osteoclasts, which mediate osteoporosis. The importance of EPCs in vascular repair (Section 1.5.3.4) and in attenuating inflammation is a rationale for optimizing hormone therapy for both osteoporosis and EPC production and functioning.
2.10.5 Plant-derived Micronutrients and Neutriceuticals
The powerful benefits of aspirin and other drugs with anti-inflammatory and anti-coagulant activities may be mimics of plant-derived biochemicals present in human diets to varying amounts: polyunsaturated fatty acids (PUFAs), curcumin, and salicylate. These and many other biogenic agents contribute to health through regulatory influences, rather than through their caloric value, and may be broadly considered as micronutrients.
PUFAs are ‘essential’ micronutrients required for normal brain development and adult health (DeMar et al, 2006) and are ultimately derived from plant foods (Crawford et al, 2004; Holub and Holub, 2004; Zamaria, 2004). PUFAs may have been important in human brain evolution, during the transitions to meat-rich diets (Section 6.2.2).
PUFAs bind to PPAR, SREBP, and other transcription factors that directly modulate the genetic control of fat and carbohydrate metabolism in brain, fat, and liver cells (Sampath and Ntambi, 2004).
α-linolenic acid (18:3ω3) is an essential ‘omega-3’ fatty acid that must be obtained from the diet8. The closely related linoleic acid (18:23ω6) is an ‘omega-6’ PUFA. After ingestion, the ‘parental’ PUFAs are enzymatically elongated and desaturated to yield eicosinoids, which have certain opposing activities. Eicosapentanoic acid (20:5ω3) (‘EPA’) and docosahexanoic acid (22:6ω3) (‘DHA’) have anti-inflammatory and anti-thrombotic activities, whereas arachidonic acid (20:4ω6) gives rise to ω-6 autocoids with opposite activities. Cyclooxygenases and lipoxygenases convert arachidonic acid to prostaglandins and leukotrienes (Section 2.6 above).
Fish fat and oils are rich sources of eicosapentanoic acid and docosahexanoic acid (both ω-3). Plant foods lack EPA and DHA, but may contain α-linolenic acid (ω-3). Egg yolk also contains ω-3 PUFAs (Renaud, 2001). Popular vegetable oils (corn, safflower, sunflower) are rich in linolenic acid (ω-6). Meat is rich in arachidonic acid (ω-6): pork and poultry < beef and lamb, in both the lean meat and visible fat (Li et al, 1998). Of these, pork fat was the highest (175 mg arachidonic acid/100 g) and lean beef the lowest (21 mg/100 g). Typical U.S. diets yield 1–3 g of α-linolenic acid (ω-6), but 80 mg DHA (ω-3). ‘Mediterranean’ diets tend to be rich in α-linolenic acid (ω-3). At high dietary ratios, the ω-6 PUFAs compete at the enzymatic level with ω-3 to diminish the yield of eicosapentanoic acid and docosahexanoic acid (ω-3); this shift is considered prothrombotic (see above). The optimum ratios and amounts of dietary PUFAs remain controversial.
Epidemiological studies link the intake of linolenic acid (ω-3) to lower cardiovascular and stroke and improved risk indicators (Crawford et al, 2004; Harris and Levine, 2005; Holub and Holub, 2004). For example, in a rural Japanese community, the carotid artery thickness (IMT) at age 63 varied inversely with ω-3 consumption (Hino et al, 2004). Their high fish consumption yielded about 2.2 g/d of ω-3 PUFAs; this level approximates the ‘Mediterranean’ diets and exceeds the typical U.S. consumption of about 1.3 g/d. Nonetheless, intervention trials and case control studies with fish oils have been inconclusive (Wilkinson et al, 2005).
The dietary proportions of ω-3 versus ω-6 PUFAs influence coagulation through platelet membrane composition. For example, Eskimos with diets rich in fish oils have low cardiovascular disease in association with slow coagulation in some studies (von Schacky and Dyerberg, 2001). ω-3 PUFAs decrease platelet production of the prothrombotic thromboxane TXA2 (Section 2.10.1 above) (Adan et al, 1999; Kramer et al, 1996) and attenuate atherosclerosis in hypercholesterolemic rats (Adan et al, 1999). Dietary PUFA regulation of coagulation is attributed to the partial replacement of arachidonic acid (ω-6) by eicosapentanoic acid (ω-3) in platelet membranes, which lowers production of TXA2; e.g., 3 wk of increased fish consumption lowered TXA2 (Mann et al, 1997). Diet composition may have had a major role in human evolution, in the transitions from the herbivory of great ape ancestors to the high meat intake favored by humans (Chapter 6).
The PUFA framework is being extended to Alzheimer disease (Calon et al, 2005; Mucke and Pitas, 2004). PUFAs are important for normal brain functions and development; e.g., synapses are enriched in docosahexanoic acid. The same transgenic Alzheimer mice that responded to NSAIDs (see above) also show strong effects of dietary PUFAs (Calon et al, 2004; Calon et al, 2005). Depletion of docosahexanoic acid (ω-3) by an ω-6 rich diet (safflower oil) caused huge deficits (>90%) in glutamatergic synapses (NR2A & -B) and in the PI3K subunit, p85a. Oxidative stress (carbonyl content) was increased and learning was impaired. Supplements of docosahexanoic acid partly blocked these deficits. Docosahexanoic acid activates the PI3K/Akt pathway and blocks caspase activation, which may link PUFA functions to insulin-like signaling pathway in longevity (Fig. 1.3A).
Human elderly have not consistently associated dietary ω-3 and supplements with cognitive functions (Maclean et al, 2005). Short-term supplements with EPA did not slow cognitive decline in Alzheimer patients, not surprisingly (Boston et al, 2004). A good model for such studies is EVA (Etude du Vielliessement Arteriel; Nantes, France), which used erythrocyte membrane lipid composition as an index of dietary PUFA (Heude et al, 2003). Over 4 years, higher erythrocyte ω-6 content was associated with higher 1.91-fold risk of cognitive decline, whereas ω-3 content reduced risk by 0.59. If the vascular studies of PUFAs are any guide, the benefits of PUFAs to Alzheimer disease will not be proven soon.
Another type of inflammatory regulator involves ω-3 PUFAs, which are oxidized by COX-2 or cytochrome P450 (CYP gene family) to numerous agents. Among the emerging functions are the ‘resolvin’ RvE1, which inhibits leukocyte infiltration (IC50 of 5 nM) (Arita et al, 2005). These aspirin-independent pathways confound analysis of dietary PUFAs in vascular disease (Arita, 2005), e.g., in self-reports, blood salicylate was detected in 14% of aspirin ‘non-users’ (Smith et al, 1999). This discrepancy could represent benign inaccuracy, or the more interesting possibility of unrecognized dietary salicylates (see below). Many other examples show the difficulty of assessing dietary or other lifestyle factors in health.
Two other plant-originated substances in human diets, curcumin and salicylate, are interesting candidates for benefits to shared risk factors for vascular and Alzheimer disease. Curcumin, a biphenol with a remarkable range of activities, is a major component of turmeric, a traditional Asian spice and preservative from the herb Curcuman longa. In Arurvedic medicine, curcumin is considered antiinflammatory and, in fact, inhibits COX-2 in various cells (Cole et al, 2005a; Lantz et al, 2005; Sharma et al, 2005). Relevant to arthritis, curcumin synergized with celecoxib to inhibit synovial cell growth (Lev-Ari et al, 2006), which is relevant to arthritis. Curcumin induced glutathione biosynthesis and inhibited NF-kappaB activation and interleukin-8 release in alveolar epithelial cells (Belmin et al, 1993; Biswas et al, 2005; Orlandi et al, 2000; Pardio et al, 2005). Curcumin may also be anti-thrombotic. Lastly, and relevant to atherosclerosis in human endothelial cells, curcumin attenuated the repression of thrombomodulin by CRP (Nan et al, 2005). In diet-induced hypercholesterolemia, curcumin increased HDL-cholesterol by 50% and decreased LDL cholesterol by 40% (Arafa, 2005).
Curcumin fed to transgenic Alzheimer mice attenuated oxidative damage and amyloid deposits, whether introduced early before amyloid formed or at later ages (Cole et al, 2005a; Lim, 2001; Yang et al, 2005). Curcumin crosses the blood-brain barrier and binds directly to amyloid deposits. The apparently low incidence of Alzheimer disease in India (Section 1.6.4) (Chandra et al, 2001) is intriguing, in view of the heavy use of turmeric in regional cooking. Clinical trials are evaluating curcumin in Alzheimer disease, vascular disease, cancer, and osteoarthritis. Moreover, there are potential synergies of curcumin with DHA in Western diets that are typically low in DHA and polyphenolic anti-oxidants (Cole and Frautschy, 2006).
Curcumin also has antimicrobial activities; e.g., curcumin attenuated HSV-2 transmission in a mouse model (Bourne et al, 1999). Other useful drugs may be found among ancient traditional spices and food preservatives. Some spices have anti-bacterial, anti-viral, and anti-fungal activities that may have been highly adaptive (“some like it hot”) before the advent of refrigeration and sterilization (Billing and Sherman, 1998; Goff and Klee, 2006).
Dietary salicylates may give some of the benefits identified with aspirin (acetylsalicylate) (Hare et al, 2003; Paterson and Lawrence, 2001; Paterson et al, 2006). Many fruits, vegetables, herbs, and spices are rich in salicylates. Vegetarians had 40% higher serum salicylate, with some overlap in non-vegetarian low dose (75 mg/d) aspirin users (Blacklock et al, 2001). The higher salicylate intake of vegetarians is consistent with dietary plant sources. How fascinating that salicylates are important to plants as host-defense mechanisms and are induced in response to attacks by insects and viruses (Shimoda et al, 2005; Ton et al, 2002; Traw et al, 2003). Fruit and vegetables grown without insecticides may have higher salicylates. The anti-inflammatory activities of salicylates work by different mechanisms than aspirin. About one-third of oral aspirin is rapidly converted by carboxyesterases in the gut and liver to salicylic acid and related dihydroxybenzoic acids (Blacklock et al, 2001). Acetylsalicylate is rapidly cleared (t1/2 20 min), while salicylate clears more slowly (blood t1/2, up to 30 h) (Hare et al, 2003). In contrast to acetylsalicylate, which inhibits COX-1 and COX-2 by acetylating serine at the enzyme active site (see above), salicylate inhibits prostaglandin synthesis by repressing COX transcription (Awtry and Loscalzo, 2000; Hare et al, 2003). There is always more to aspirin.
Two other natural COX inhibitors have come to light. Olive oil contains ‘(–)oleocanthal’, a phenolic adduct that strongly inhibits COX-1 and -2 in rank order: indomethacin < oleocanthol < ibuprofen (Beauchamp et al, 2005). Oleocanthol may be among the benefits imputed to the Mediterranean diet. Ingestion of 50 ml extra-virgin grade olive oil is estimated to yield 9 mg of (–)oleocanthol, which is 10% of the ibuprofen dose recommended for analgesia.
The overlapping risk factors for vascular disease and Alzheimer disease are tantalizing targets for simple nutritional interventions. PUFAs, curcumin, and salicylates are likely to be joined by many other dietary micronutrients in wild plants and cultivars. There may be a common biochemistry in view of the conjugated double bond systems that these three classes of compounds share. Synthetic drugs developed for vascular disease and cancer may interact with micronutrients from animal and plant foods, as indicated for curcumin and celecoxib, and alter therapeutic outcomes.
2.11 SUMMARY
Arterial disease, some cancers, and possibly Alzheimer disease are promoted or accelerated by infections and environmental inflammogens. Drugs with antiinflammatory actions appear to show corresponding benefits. These strong bidirectional effects are consistent with the major role of inflammation in the progression of vascular disease, cancer, and Alzheimer disease. Query III (Section 1.1) is well answered with major evidence for inflammation in bystander damage in aging from infections and inflammogens in aerosols and diet. The acceleration of telomere shortening by inflammation may prove to be a major mechanism in immune dysfunctions during aging (bystander type III damage, Section 1.4.3). The molecular mechanisms may include PI3K/Akt signaling, which is on the insulin signaling pathway of many mutations that increase life span of laboratory animals (Fig. 1.3A and Chapter 5). New drugs that selectively target PI3K isoforms for cancer (Kang et al, 2005; Osaki et al, 2004) and vascular disease (Walter et al, 2004) may have broader influences on longevity. Chapter 3 considers evidence that lower caloric intake and moderate physical activity can attenuate many of these chronic diseases and aging processes, in approximate parallel with the beneficial effects of anti-inflammatory drugs. The evolution of human longevity (Chapter 6) will further consider how humans became increasingly exposed to inflammation and the role of diet-aging-disease interactions.