5.7 MAMMALS
‘Home again’ to mammals, to discuss gene variants with functions that are known in much more detail than in insects and nematodes. Humans and other mammals share a core of cellular, physiological, and genetic characteristics in aging patterns (Section 1.2). Trends for obesity and insulin resistance are also common. A fundamental difference in mammalian aging from fly and worm is the role of abnormal cell growths, which have major roles in arterial disease and cancer. Although aging monkeys accumulate Alzheimer-like β-amyloid (Aβ) deposits, aging mice lack Aβ deposits, unless given human transgenes (Section 1.6.1). The discussion is organized by three functions: growth and metabolism, inflammation, and lipoproteins. The discussion of human genotypes emphasizes common polymorphisms to the neglect of rare or familial mutants that dramatically shorten life span in major diseases.
5.7.1 Growth and Metabolism
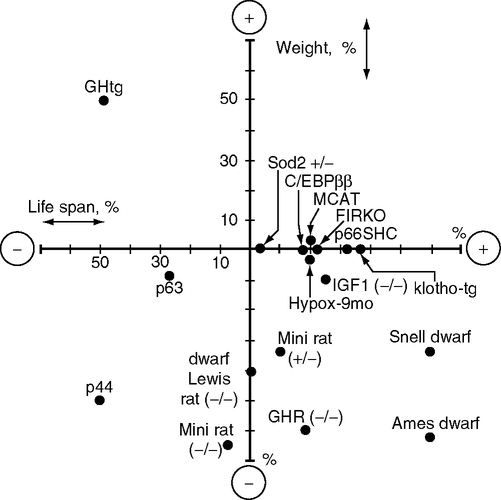
More than 15 mutations influence longevity in association with alterations in growth and metabolism (Fig. 5.8). This summary of manipulations of longevity from Table 5.3 plots the percentage change of weight and life span relative to controls (0,0). The graph also shows transgenic mutants described in Section 1.2.6, mice overexpressing catalase (MCAT) and another wth partial superoxide dismutase 2 deficiency (Sod2+/–) Table 5.3 lists rodent models by order of life span in four categories with detals in footnotes: (A) primary pituitary hormone defects; (B) GH/IGF-1 receptor defects; (C) other growth factor defects (p44/p53, p63, p66shc); (D) adipocyte-targeted defects (C/EBPβ, FIRKO). Table 5.3 and notes give more details and references. Generalizations must be tentative because many studies compare the effects of mutants with diverse genetic backgrounds that include varying combinations of inbred strains.9 The associations of size and longevity vary widely. A provisional conclusion may be drawn that dwarfism is neither necessary nor sufficient for longevity, as was also concluded for the fly insulin-signaling mutants.
TABLE 5.3
Long-Lived Rodent Mutants Ranked in Order of Increased Life Span
TABLE 5.3 A Primary Pituitary Hormone Defects
| Mutant; ref. | Change in Life Span | Body Weight (Young Adult) | Hormonal Defects | Fasting Plasma Hormones | Reproduction | ||
| Glucose | Insulin | IGF-1 | |||||
| Ames dwarf mouse | F, +68% | −67%, | no GH, TSH, | lower | lower | > −99% | F, sterile |
| Pit1dw/dw;a | M, +49% | PRL | lower | M, low fertility | |||
| Snell dwarf mouse | F, +42% | −67% | no GH, TSH, | lower | lower | > −99% | F, sterile |
| Prop1df/df;b,c | M, +26% | PRL | lower | M, low fertility | |||
| mini rat antisense | +10%. tg/– | −35%, tg/– | −90% pituitary | normal | lower | −35%, tg/− | |
| GHtgd | −7.5%,tg/tg | −65%, tg/tg | GH | (tg/−) | (tg/−) | −70%, tg/tg | |
| dwarf Lewis rat | M, 0% | −40%, +/− | −40% plasma | normal | normal | −40%, −/− | low fertility |
| (dw/dw)e | F, 0% | −40%, −/− | GH | ||||
| excess GH (GH-tg)f | −45% | +50% | >10-fold GH elevation; corticosterone elevated | premature loss | |||
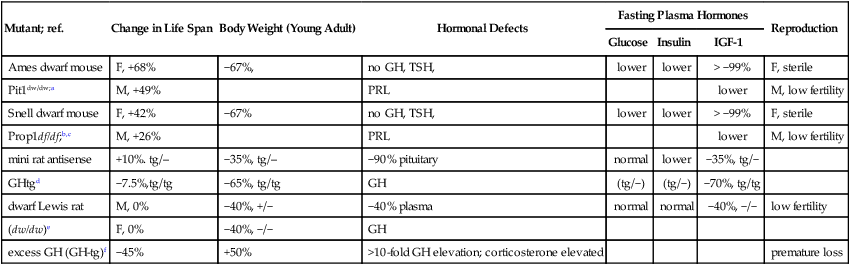
GH, growth hormone (pituitary); GHR/BP, growth hormone receptor/binding protein; the GHR/BP gene encodes the GH receptor (liver) and its truncated form, the GH-receptor binding protein (serum protein, secreted by the liver); Ghrh: growth hormone releasing hormone (hypothalamus); IGF-1, insulin-like growth factor-1 (liver); IGF-1R, the IGF-1 receptor; IRS, insulin receptor; PRL, prolactin; TSH, thyroid stimulating hormone.
a.Ames dwarf mouse: (Bartke et al, 2001; Brown-Borg et al, 1996; Tatar et al, 2003; Tsuchiya et al, 2004). The mean life span of ad lib fed Ames is 1062 d; current maximum is 1550 d (Ikeno et al, 2003) (Fig. 5.10). The Ames mutant (Prop-1 gene, Ames dwarf) is upstream of the Pit1 transcription factor and impairs differentiation of the anterior pituitary (Andersen et al, 1995). The mutant has been maintained on a genetically’ ‘heterogeneous background’ for 20 y (Ikeno et al, 2003) The Ames dwarf has delayed neoplastic lesions and attenuated kidney pathology. The <1% normal IGF-1 was estimated from the radioimmunoassay sensitivity in consultation with Bartke and Chandrashekar (Chandrashekar and Bartke, 1993). The Ames dwarf shows increased insulin sensitivity but intolerance to glucose; insulin-stimulated phosphorylation of the insulin receptor is enhanced (Dominici et al, 2002).
b.Snell dwarf mouse: (Flurkey et al, 2002). Longevity of Snell dwarfs, relative to non-mutant litter-mates, depends on the genetic background (Brown-Borg et al, 1996; Liang et al, 2003). Life span is about 40% longer than the C3H/HeJ × DW/J)F1 background (Flurkey et al, 2001). Mean survival, dwarf, 1,178 ± 235 d versus background genotype, 832 ± 158 d. When Snell dwarfs are housed with normal males, life span was shorter than the normal; life spans were much longer when females were the cage mates, which is attributed to greater social stress in male company, or the greater access to body warmth in huddling with females (Vergara et al, 2004). Collagen cross-linking (tail-tendon breaking time) increased more slowly with age in the dwarfs. Knee-joint pathology during aging was also slowed (Silberberg, 1972). Spleen hypertrophy during aging is also attenuated (Flurkey and Harrison, 1990).
c.Snell dwarf history. The Snell dwarf was the first mutant dwarf identified (Snell, 1929). In an earlier phase of biogerontology, Fabris and colleagues described the Snell dwarf as a model for accelerated aging because of short life span, < 6 mo (Section 2.5.2) and early pathology (Fabris et al, 1972; Fabris et al, 1988). In a European colony, dwarfs developed cataracts, graying hair, kyphosis, and sagging skin by 4–5 m. Pneumonia or other infections, however, were not noted (Fabris et al, 1971a). These accelerated aging changes were blocked and life span was increased to >12 months by injecting lymphocytes from normal mice or by bovine GH (Fabris, 1972). The thymus gland deficiencies were attributed to deficits in GH (Fabris, 1971; Fabris et al, 1971b; Pierpaoli, 1969). Snell dwarfs were also short-lived in two U.S. breeding colonies. At Cornell U. Medical College (Winick, 1968), dwarfs died before maturing; survival was increased by limiting litters to 1–3 mice, which increased body weight, possibly by more access to milk and maternal care. All mice seemed to lose weight after weaning, implying pathogenic infections. I can add the personal observation that animal facilities were suboptimum at Cornell in 1970–1972, my first academic appointment. Graying is atypical of aging in mice (Finch, 1973b). At the National Cancer Institute (NCI) (Chen et al, 1972), dwarfs were also short-lived, although some survived to 12 mo. This colony had a high incidence of tumors, unlike modern colonies. However, obesity was observed, as in currently colonies. In great contrast to these three colonies, dwarfs in two other colonies from that period were longer lived. Dwarf mice at Washington U. lived up to 41 mo and were studied as models of retarded skeletal aging (Silberberg, 1972; Silberberg, 1973). In Glasgow U., dwarf mice lived at least 8 mo and without cataracts, or other aging markers; potential survival to later ages was not followed (Shire, 1973). We cannot know what conditions differed critically between these short- and long-lived dwarf colonies. The high incidence of lymphomas and leukemia in the NCI colony could have had viral origins (Chen, 1972). Reduced infections and other husbandry improvements are more likely than genetic changes (Section 2.5.2). As discussed in the text, dwarf mice are sensitive to stress-induced immune deficiencies and wasting conditions (Foster, 2000). At the Jackson Laboratory, improved conditions included temperature control and reductions of infections (Section 2.4.2) (Kevin Flurkey, pers. comm.).
d.Mini rat antisense GHtg: (Shimokawa et al, 2002; Yamaza et al, 2004). Transgenic expression of anti-sense GH in pituitary. Heterozygotes (tg/–) lived 10% longer than the (tg/tg) homozygotes. Although plasma GH was not different, despite the major decrease in pituitary GH, detailed sampling might show a dampened pulsatile secretion of GH. Pathology at death of tg/tg—mostly neoplasia, particularly leukemia, which was never observed in –/– rats; lower incidence of pituitary tumors and kidney disease. Natural killer (NK) cell activity was lower, but normal T-cell responses to mitogens; the smaller spleens and thymus were scaled to body size
e.Dwarf Lewis rat (dw/dw):(Sonntag, 2005). Dwarf Lewis rats carry a recessive mutation (not mapped or characterized) that has no effect on other pituitary hormones (Charlton et al, 1988; Ramsey et al, 2002). Augmentation of GH after puberty slightly increased the male life span (14%), with no effect on females. Lewis dwarfs have no abnormalities in other pituitary hormones, corticosterone, insulin, or glucose. Learning was impaired at all adult ages, but without obvious brain abnormalities.
fGH transgenic over-producer Giant GH-transgenic mice. Life span: mean, 425 versus 733 d controls; maximum, 550 versus 1150 d controls (Bartke, 2003). Corticosterone is elevated (basal and ether-induced) (Cecim et al, 1996). Pathology includes premature enlargement of seminal vesicles and increased mammary tumors (Bartke, 2003; Steger, 1993). Fertility declines prematurely, by 5–7 mo (Cecim, 1995). Brain aging is accelerated, with elevated GFAP (astrocytic hypertrophy marker, Fig.1.7E, Fig.1.9 Section 1.8.1) and slowed turnover of hypothalamic dopamine, confirming similar age changes in normal C57BL/6J mice after 600 d in my lab (Finch, 1973a; Nichols et al, 1993; Rozovsky et al, 2005).
5.7.1.1 Rodent Mutants with Altered Insulin Signaling and Fat Metabolism
Presaging the findings of long-lived fly and worm mutants, the pioneering work of Andrej Bartke showed that deficits in insulin and IGF-1 increase mouse life span (Bartke et al, 2002; Brown-Borg et al, 1996). The Ames, little, and Snell dwarf mice lack pituitary growth hormone (GH) and live longer than most background strains (Fig. 5.9) under protected conditions (Table 5.3A, note c). The lack of plasma GH consequently greatly attenuates the hepatic secretion of IGF-1 (Chandrashekar and Bartke, 1993), which is required for normal growth. Currently, more than five gene mutants that reduce GH/IGF-1 levels or signaling also increase life span; other mutations have little or no effect on life span (Table 5.3A) (Fig. 5.8).
The Ames dwarf lives about 50% longer than most lab mice and was the first mutation (prop-1) shown to increase longevity in a mammal. The Snell dwarf (pit-1) is just behind in longevity. Both male and female life spans are increased. With favorable husbandry conditions, the GH deficits increase life span more than diet restriction. The Ames and Snell dwarfs have low insulin and glucose. However, other disturbances are less desirable from a human perspective: hypothyroidism, low body temperature, and impaired reproduction. Obesity is prevalent with aging. Brain aging changes may also be slowed. Tumors are decreased, discussed below.
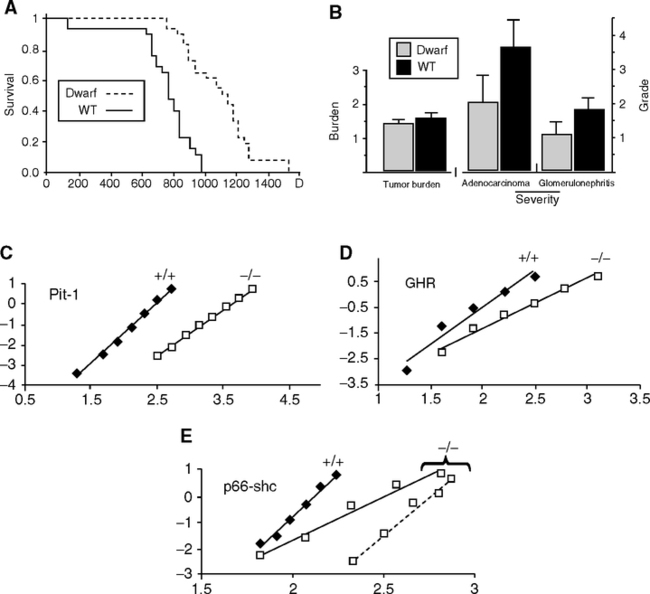
Causes of death during aging in these mutants are not generally reported. The Ames and Snell dwarfs are the best described. In the Ames mutant, neoplasia was the major attributed cause of death (67% vs 95% in the background strain) with delayed occurrence (Fig. 5.9B), corresponding to the increased life span (Fig. 5.9A) (Ikeno et al, 2003). Lung adenocarcinoma was the most common lesion and was less severe in the dwarfs. Glomerulonephritis was also lessened. The 15% decrease in ‘total pathologic burden’ is modest relative to effect of diet restriction in attenuating pathology (Section 3.2.2). Snell and ‘little’ mice have less neoplasia and splenic hypertrophy (Flurkey and Harrison, 1990; Flurkey and Currer, 2004). Snell dwarfs had fewer lymphomas, plus milder kidney pathology and lens opacity (cataract score) (Vergara et al, 2004). The GH-receptor-knockout also had less neoplasia (no details given) (Bartke, 2005). About 25% of old Ames dwarfs and 47% of the Snell dwarfs had no visible gross pathologic lesions at natural death, as seen in about 25% of aging F334 rats on diet restriction (Ikeno et al, 2003).
The Ames and Snell dwarfs have multiple pituitary defects and lack GH, TSH, and PRL. To evaluate the role of growth deficits and interactions with hypothyroidism, Snell dwarfs were injected with both GH and thyroxine (T4) from 4–15 wk; a subgroup continued to receive T4 (Vergara et al, 2004). Adult weight was partially restored, with greatest effect from continuing T4. Survival curves did not differ between untreated dwarfs and those receiving GH/T4 only early in life. However, continuing T4 replacement decreased survival to levels close to the background controls.
Both Ames and Snell dwarfs have very low blood glucose, insulin, and IGF-1. Both become obese during aging, particularly males, which may reach 40 g, with corresponding increases in serum leptin (Flurkey et al, 2001). The ‘little’ mutation (lit/lit) in the GH-releasing hormone receptor (GHRH) restricts the pituitary hormone deficits to GH; however, the mice still become obese (Godfrey et al, 1993). Concurrent obesity and longevity as in these dwarfs may also co-exist in some human populations (Section 3, Fig. 3.4A) and in the fat accumulated by long-lived mutant flies. The ad lib food intake of Ames and Snell dwarfs is greater normal when calculated per g body weight (BW), but is normalized if calculated by BW0.75 (metabolic BW) (Bartke, pers. comm.).
Ames and Snell dwarfs’ learning ability declined little if at all during aging, unlike controls (Bartke, 2005). The Ames dwarf may have increased adult neurogenesis in the hippocampus, a brain region critical for memory (Sun et al, 2005b). Neuronogenesis in the adult brain is dependent on IGF-1 (Lichtenwalner et al, 2006) produced by local cells not dependent on serum GH; in the hippocampus, IGF-1 was normal (mRNA and protein) for the background strain. The brain also has autogenous GH expression, which is lower in the Ames dwarf (Sun et al, 2005a).
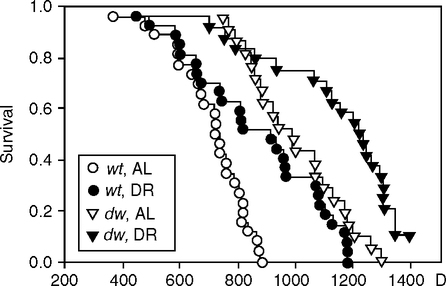
Diet restriction (DR) further increases the Ames life span in both sexes by about 30% (Fig. 5.10), which is relatively more than in the fly dwarf chico mutant (Fig. 5.6). Despite the obesity, the dwarf mouse metabolic mutants share some features of DR with lower blood glucose and insulin, increased resistance to oxidative stress, and trends to slower mortality acceleration (MRDT) (Bartke, 2002, 2005). Skin fibroblasts from the Ames, Snell, and GH-receptor mutants are resistant to H2O2, paraquat, and UV (Salmon et al, 2005). Increased expression of FoxO family transcription factors that regulate stress-resistance genes is hypothesized as downstream of attenuated insulin/IGF signaling in dwarfs, as in DR (Bartke, 2005).
The lit/lit and Snell dwarfs have slower immune maturation (Cross et al, 1992; Foster et al, 2000) and are more vulnerable to infections, stress-induced immune deficiencies, and wasting conditions (Foster et al, 2000). Immune system age changes are slowed in Snell dwarfs (Flurkey et al, 2001), with smaller increases of memory T cells (CD4, CD8) and smaller loss during aging of nHTL and pCTL cells (mitogen-stimulated T cells that produce IL-2 or that differentiate into cytotoxic effectors, respectively). Ames dwarfs may have milder immune aging changes in different markers: B and T cell numbers are normal, as is production of antibodies to tetanus toxoid (Cross et al, 1992; Hall et al, 2002). However, lit/lit mice have 3-fold higher mortality after infection with influenza virus (Alt et al, 2003). This may be a model for the higher mortality of hypopituitary humans from respiratory infections, as shown in a large prospective study (Tomlinson et al, 2001).
The IGF-1 receptor knockout benefits females; males were hyperglycemic in glucose tolerance tests and lived slightly longer. Conversely, the GH-receptor knockout mainly benefited male life span; glucose and insulin were much lower, and female fertility was lower.
In an earlier phase of biogerontology, the Snell dwarf was described very differently as a model for accelerated aging because of its short life span, <12 m and early onset pathology (Fabris et al, 1972; Fabris et al, 1988), including lymphomas that are currently rare (Chen et al, 1972) (Table 5.3A, note c summarizes this important history). Reduced infections and other husbandry improvements are more likely than genetic changes (Section 2.5.2).
Mortality rate analysis of some of these mutants indicates slower aging, but statistics are weakened by the small numbers of mice in most studies (Table 5.4) (de Magalhaes et al, 2005). In healthy rodent colonies, life span is mainly determined by the acceleration rate of mortality (Gompertz slope, or mortality rate doubling time [MRDT] (Section 1.2.1; Table 5.1 notes). However, decrease in the background mortality, or ‘initial mortality rate’ (IMR), will increase life span without slowing aging. Despite the statistical significance of mutant effects on life span, the Gompertz parameters did not differ (Table 5.4). The Ames dwarf (prop-1) had the clearest slowing of aging, with longer MRDT, confirming (Flurkey et al, 2001) (Fig. 5.9 C, D, E). This mortality profile implies a delayed onset of aging, without change in rate of aging, which represents a life history model predicted earlier (Finch, 1990, p. 26). The histopathology was also well characterized and was attenuated relative to background strain (Fig. 5.9B). The MRDT was also longer in the C/EBPβ/β fat storage deficient mutant, discussed below. Table 5.4 also shows the short-lived senescence accelerated mouse (SAM) (Takeda et al, 1981), with faster mortality accelerations (shorter MRDT) and the diet restricted F344 rat (longer MRDT) (Finch, 1990, p. 508). de Magalhaes et al. (2005) emphasize that different survival curves and mean life spans do not resolve whether the mutant actually slowed aging, and I fully agree. The slowing of aging must be determined by assessing mortality rate parameters and pathology. The sample must be sufficient for mortality statistics, usually at least 100 mice. Histopathology is always needed to define the nature of senescent mortality in each experiment. Few other reports in Table 5.3 even described gross pathologic lesions, and none noted the ubiquitous pituitary tumors of aging female rodents (Finch et al, 1984). An exemplary study is the mitochondrial catalase overexpressing mouse (Fig. 4.8, MCAT) (Section 1.2.6) (Schriner et al, 2005), in which life span was increased by right-shifting the Gompertz slope together with delayed and decreased organ pathology.
TABLE 5.4
Gompertz Parameters for Mouse Mutants and Diet-Restricted Rat
| Gene/Intervention (Number of Animals) | Genotype | Change in Life Span | Initial Mortality Rate+, IMR/y | Mortality Rate Doubling Time+, MRDT, y |
| C/EBP (60) | CTL | 22%* | 0.52 | 0.27 |
| C/EBP β/β | 0.38 | 0.45** | ||
| Diet restricted F344 rat (230) | ad lib fed | 30%* | 0.031 | 0.17 |
| restricted | 0.034 | 0.37** | ||
| GHR Growth hormone | CTL | ≥ +16%* | 0.52 | 0.36 |
| receptor-KO (26) | KO | 0.59 | 0.46 | |
| GHRHR little mouse (66) | CTL | ≥ +23%* | 0.19 | 0.29 |
| KO | 0.15 | 0.28 | ||
| Igf/1r+/–dwarf (37) | CTL | ≥ +16%* | 0.30 | 0.39 |
| KO | 0.06 | 0.32 | ||
| INSR FIRKO (127) | CTL | 18%* | 0.078 | 0.36 |
| KO | 0.035 | 0.29 | ||
| p66shc–/– (29) | CTL | 30%’ | 1.7 | 0.11 |
| KO | 0.29 | 0.25 | ||
| PIT1, Ames dwarf (59) | CTL | ≥ +49%* | 0.020 | 0.23 |
| KO | 0.0035 | 0.30** | ||
| PROP1, Snell dwarf, F.(29) | CTL | +42%’ | 1.29 | 0.75 |
| KO | 0.74 | 0.35 | ||
| SAM senescence - | CTL | −26%* | 0.63 | 0.37 |
| accelerated mouse (870) | SAM | 0.77 | 0.21** |
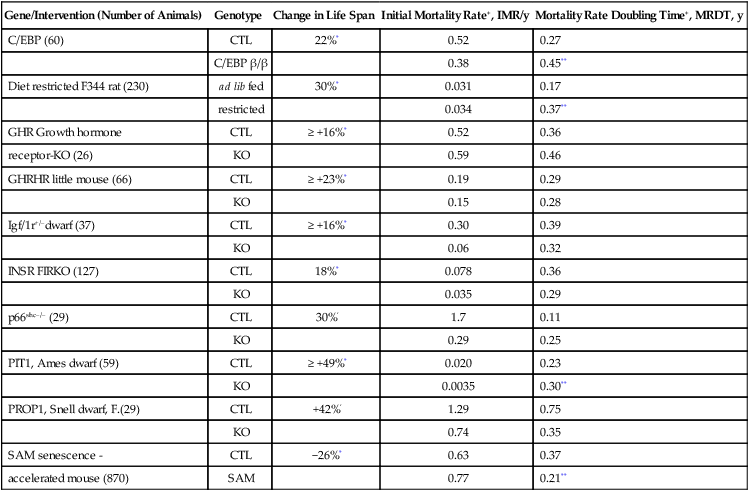
+, for definitions of IMR and MRDT, see TABLE 1.2.
CLT, control strain, see TABLE 5.3.
*significant difference of life span, according to original reports (TABLE 5.3).
**significant difference of parameter in (de Magalhaes et al, 2005).
Hypophysectomy (surgical ablation of the anterior pituitary) gives another approach to pituitary hormones and aging, and can be accomplished with high survival in rodents as in humans. In Arthur Everitt’s pioneering experiments, hypophysectomy at 2 m slowed many aging changes that are also slowed by diet restriction: major decrease in endocrine tumors and kidney lesions; less aortic wall thickening (Everitt et al, 1980). Life spans were increased by 16%, more than in the diet restricted rats of this study. Hypophysectomy at 15 m had less effect. These effects were verified in mice: Hypophysectomy at 1 m increased life span by 15% and at 9 months by 21%, with progressively less effect at later ages (Powers et al, 2006b).
Conversely, life span is shortened by 30% in transgenic mice that overexpress GH with plasma GH ≥ 1000-fold elevations. Excess GH may cause a progeria syndrome, with increased tumors, elevated corticosteroids, and impaired reproduction and immunity (Bartke, 2003; Hall et al, 2002; Steger et al, 1993). These adverse outcomes are at odds with the popularity of GH supplementation in some circles (see below).
Other mutants altering growth have modest effects on longevity (Table 5.3B) (Fig. 5.8). Mutants targeting GH/IGF-1 functions without other pituitary changes live slightly longer than background strains and are small-sized: GH-receptor knockouts (GHR–/–) and IGF-1 receptor knockout heterozygotes (Igf/1r+/–). The GH-receptor KO(-/-) has very low plasma IGF-1 (Kopchick and Laron, 1999); these mice are very insulin-sensitive (slightly lower to normal blood glucose, low insulin); however, GH is elevated (Andrej Bartke, pers. comm.; Coschigano et al, 2000; Coschigano, 2003). Gluconeogenic and stress-resistance genes are more active (Al-Regaiey et al, 2005). Diet restriction had no effect on median life span and increased the maximum of females only (Bonkowski et al, 2006b). This major contrast to Ames dwarf responses to DR (Fig. 5.9) implies the dependence of diet restriction responses on intact GH-receptor signaling. The GHR–/– mutant is a model for Laron syndrome in humans (next section).
TABLE 5.3B
| Change in Life Span | Body Weight (Young Adult) | Hormonal Defects | Fasting plasma Hormones | ||||
| Mutant | Glucose | Insulin | IGF-1 | Reproduction | |||
| IGF-1 receptor | F, +33% | <−8% | normal | normal | +20% | normal | |
| knockout mouse; | M, +16% | higher | |||||
| Igf/1r+/−;g | |||||||
| GH-receptor | F, +33% | −60%; growth | GH, 5–10x | −80% | −20% | ≥−80% | F, low fertility; |
| knockout GHR−/−;h | M, +19% | slows by 4 w | higher; | lower | lower | lower | maturity |
| delayed 4 w | |||||||
| little (lit) mouse, | F, +25% | −33% | >99% lower | ||||
| Ghrhlit/lit;i | M, +23% | GH; corticos- | −75% | ||||
| terone elevated | lower | ||||||
| Klotho over- | F, +20% | normal | impaired | normal | F, +50% | 50% fewer | |
| expressionj | M, +30% | insulin and | M, norm | pups | |||
| IGF-1 recep- | |||||||
| tor activation | lower | ||||||
| midi mouse, m/mk | −35% | low IGF-1 | >−99% lower | F, low fertility | |||
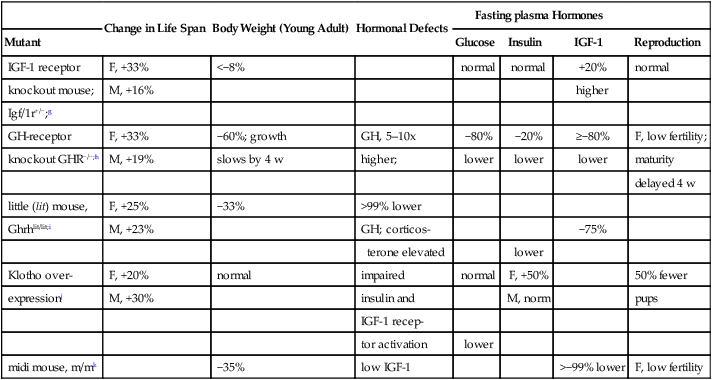
g.IGF-1 receptor knockout mouse (Holzenberger et al, 2003). Life span background strain 129: female mean 568 d, max 810 d; male mean, 585 d, max 1000 d. Life span +/–: female, mean 756 d; female max, ca. 960 d; male, mean 679 d; male max 1020 d (not significant). Fasting glucose, normal; fed glucose, females, +12%; males, −4%. Glucose tolerance, males hyperglycemic (peak about 2x females). Normal body temperature.
h.GH-receptor knockout (GHR–/–): (Coschigano et al, 2003; Kopchick and Laron, 1999).The GHR/BP−/– mutation in the GH binding protein impairs the GH-receptor by blocking dimerization required for GH signal transduction in hepatocytes that secrete IGF-1. Homozygotes Igf/1r−/– are not viable. Life span changes averaged from the two background strains: C57BL/6J, female mean 821 d, female max 1100 d; male mean 765 d, male max 1000 d; GHR –/– female mean 956 d, female max 1250 d; male –/– mean 951 d male max 1200 d. Ola-BALB/cJ, female mean 759 d, female max 1250 d; male mean 656 d, male max 980d. –/– female mean 921 d, female max 1250 d. Male GHR –/– C57/BL/6J gained 5g slowly. Another transgenic introduced a GH antagonist peptide (GHA) that did not alter life span; although weaning weight was the same as the GHR–/–, the GHA gained weight progressively, unlike the GHR—/—, and nearly caught up to controls by 1 y. Food intake in both mutants was normal for body weight or higher.
i.little (lit) mouse (Ghrhlit/lit): (Flurkey and Harrison, 1990; Flurkey et al, 2001). Growth impairments are very modest. The low blood GH is caused by a pituitary receptor defect, which impairs responses to GH-releasing hormone (GHRH) from the hypothalamus. lit/lit males, 1,093 ± 186 d versus background, 886 ± 148 d; females, 1070 ± 127 d versus background, 857 ± 169 d. In lit/lit homozygotes, tail growth ceased after 7–10 mo, while body mass and skeleton continued to grow; postnatal tail growth may be a better bioassay for GH than body weight (Flurkey and Harrison, 1990). Corticosterone is also elevated (Alt, 2006) Spleen hypertrophy during aging is attenuated relative to wild-type. Heterozygotes lit/+ grew normally.
j.Klotho over-producer: Klotho encodes an uncharacterized membrane protein with an extracellular domain shed into blood and cerebrospinal fluid that regulates insulin signalin (Kurosu et al, 2005). Klotho deficits shortened life span drastically: dramatic involutional, age-like changes arose after weaning and few survived to maturation (2 months) (Kuro-o et al, 1997). Pathology included arterial calcification at a level atypical of current aging mice (Section 2.5.2) and loss of subcutaneous fat. Conversely, transgenic overexpressors lived 20–30% longer (two lines); KL48 female, 830 versus 697 d, background; male: 940 versus 715 d background (Kurosu et al, 2005). The Klotho peptide was elevated 2-fold in plasma. Glucose clamp studies showed resistance to IGF-1 and insulin in male mice, and IGF-1 resistance only in females. Klotho inhibits activation of the IGF-1 and insulin receptor and suppresses insulin-receptor mediated signaling including interactions of the PI3-kinase (p85 subunit with the insulin receptor). Pathology was not reported.
k.IGF-1 hypomorph mouse ‘midi’ (Lorenzini et al, 2004); reported as abstract.
The IGF-1 receptor mutant (Igf/1r+/-) with 33% longer life span is studied as a heterozygote (homozygotes die at birth) (Holzenberger et al, 2003). Body weight is slightly lower (5–8%), with normal intake of food and water. Blood IGF-1 is 32% higher, which may be a compensation for 50% lower density of IGF-receptors (brain, kidney, lung). While glucose-insulin regulation is nearly normal, IGF-1 signaling downstream of the receptor was decreased (50% lower phosphorylation of Akt and p66shc after IGF-1 stimulation). Body temperature is normal, unlike the Ames dwarf (Hunter et al, 1999). A new IGF-1 hypomorph, the midi mouse, has no detectible IGF-1, yet is only 35% smaller; its life span approximates the IGF-1R+/- (Christian Sell, pers. comm.; Lorenzini et al, 2004).
The Klotho gene10 has added another link to insulin signaling. Klotho encodes a highly pleiotropic peptide that influences blood glucose and insulin signaling (Kurosu et al, 2005), enzymes that influence arterial disease (ACE and PAL-1) (Arking et al, 2005), and calcium metabolism (Lewin and Olgaard, 2006). KLOTHO-deficient mice have very short life spans, with dramatic involution by 2 months (Kuro-o et al, 1997). The gross pathology includes extreme arterial calcification atypical of current aging mice (Section 2.5.2) and loss of subcutaneous fat. Conversely, transgenic KLOTHO overexpressors lived 20–30% longer than the background strain (Kurosu et al, 2005). KLOTHO inhibits activation of the IGF-1 and insulin receptor and suppresses insulin-receptor mediated signaling, including interactions of the PI3-kinase (p85 subunit) with the insulin receptor. Body weight was normal with either KLOTHO deficits or elevations. The pathology of the KLOTHO-deficient mice was attenuated by inhibiting insulin signaling with a loss-of-function insulin receptor gene (IRS+/-), which recalls that Ames dwarf mice also show deficits of insulin and IGF-1 signaling (Bartke, 2006). In humans, the klotho polymorphism KL-VS with two coding changes may be associated with mortality risk (Arking et al, 2002; Arking et al, 2003; Arking et al, 2005). KL-VS heterozygotes were more prevalent than homozygotes at advanced ages in Ashkenazi Jews and Czechs, suggesting a survival advantage. There may be associations of KL-VS with blood pressure and HDL cholesterol and stroke.
Returning to rodent models, two dwarf rats show little or no difference in longevity. The Lewis dwarf has a recessive mutation (Charlton et al, 1988), which reduces body size by 40% in association with 40% deficits of plasma GH and IGF-1. However, life span is normal (Sonntag, 2005). Pathology showed counterbalancing changes: dwarfs had fewer fatal neoplastic diseases and milder kidney degeneration, whereas there was a high incidence of intra-cranial hemorrhage (an unusual lesion). Also puzzling is the ‘mini’ rat, which carries a transgene antisense GH (Shimokawa et al, 2002; Shimokawa et al, 2003). By comparison with wild-type (-/-), plasma IGF-1 and adult weight decreased by 35% per transgene copy (tg/, tg/tg). Despite size reductions comparable to the dwarf mice, the homozygotes (tg/tg) had 15% shorter life spans. Transgenics had greatly decreased chronic nephropathy, but much more leukemia, a different balance of pathologies than in the Lewis dwarfs.
A new set of growth factor mutants affecting p44, p63, and p66-shk have altered aging phenotypes. These mutants, described below, alter activities related to the p53 oncogene, which, when not mutated, acts as a tumor suppressor. p53 is in a superfamily (p53/p63/p73) of transcription factors with multiple isoforms that regulate cell cycle arrest, metabolism, and apoptosis (Keyes and Mills, 2006; Levrero et al, 2000). In human tumors, p53 mutations are famous as the most common lesion, whereas p63 and p73 mutations are rare in human malignancy. p53 controls IGF-1 at the IGF-1 receptor as well as PTEN, which modulates IGF-signal transduction through Akt. Moreover, some p53/p63/p73 isoforms regulate transcription of IGFBP-3, which may be a vital link to somatic growth, inflammation, and metabolism. Reciprocally, SIRT1 regulates p53 transcription (Yang et al, 2006), which links p53 to nutritional status through NAD dependence of SIRT1 acetylase activity. As discussed above, flies also show a sirtuin-p53 link to life span and responses to diet restriction.
The p44 mutant mouse with a truncated p53 peptide, grows slowly, reaches only 50% normal size, and has 50% shorter life span (Maier et al, 2004) (Table 5.3D). Tumors were not reported. The IGF regulation is abnormal: Despite the slight elevations of IGF-1, there was notable activation of IGF-1R and Akt phosphorylation. The integration of p53 and IGF-1 points to deep-level controls of development and, possibly, aging.
TABLE 5.3D
| Mutant; ref. | Change in Life Span | Body Weight (Young Adult) | Hormonal Defects | Fasting Plasma Hormones | Reproduction | ||
| Glucose | Insulin | IGF-1 | |||||
| C/EBPβ/β mouse; deficient fat storageo | +22% | normal | low white fat | ||||
| FIRKO mouse, Fat-specific insulin receptor knockoutp | +18% | <25% total wt; –50% body fat | loss of insulin-signaling in fat only; normal food intake | normal | lower | normal | |

oC/EBPβ/β mouse: C/EBP (CCAAT/enhancer-binding proteins) are required for adipocyte differentiation and activates PPARγ (Chiu et al, 2004). β/β mouse is a knockin replacement of C/EBP α by C/EBPβ, thus carrying two copies. Life span β/β 28.9 mo; β/+ 23.7 mo. Fat storage is reduced, but without hyperlipidemia. Food consumption and core temperature are higher than controls. White adipose tissue had increased thermogenic mitochondria. Pathology of aging not known.
pFIRKO mouse, fat-specific insulin receptor knockout: (Bluher et al, 2002; Bluher et al, 2003).
The p63 deletion heterozygotes also had 22% shorter life spans, but were only slightly smaller (Keyes et al, 2005). Tumors were absent as observed in the p44/p53 deletions. Immune functions are impaired, with a much higher prevalence of spontaneous suppurative infections and hemorrhage than in the control mice or other colonies during normal aging.
The p66 ‘shick’ knockout (p66shc–/–) is normal size and lives 30% longer (Migliaccio et al, 1999). The p66-SHC protein has an Src-homology2 (SH2) domain that is phosphorylated by the activation of cell growth factor receptors and mediates p53-dependent response to oxidative stress. The p66SHK protein, in turn, phosphorylates the transcription factor FKHR (DAF-16 orthologue), which regulates stress-resistance genes. IGF-1 is normal; no hormonal deviations were reported. Resistance to oxidative stress was increased and to oxidative kidney damage from diabetes, with possible greater benefit to females (see Table 5.3C, note n). Importantly, there was also increased resistance to atherosclerosis induced by fatty diet (Napoli et al, 2003a,b). Aortic wall aging was also diminished (cholinergic epithelial relaxation and NO release, which decline in aging) (Francia et al, 2004). Human variants of p66shc are rare (Sentinelli et al, 2006).
TABLE 5.3C
| Fasting Plasma Hormones | |||||||
| Mutant | Change in Life Span | Body Weight (Young Adult) | Hormonal Defects | Glucose | Insulin | IGF-1 | Reproduction |
| p44 (p53 isoform)1 | −50% | −50% | activated IGF-receptors | +20% | early M. infertility | ||
| p63+/−;m | −22% | −5% | |||||
| p66shc−/−;n | +30% | normal | normal | normal | normal | ||
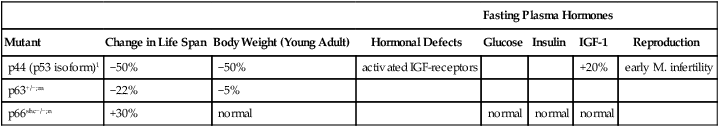
p53 as a deacetylase (Marmorstein, 2004; Vaziri et al 2001). A larger p53 deletion p53+/m (first 6 exons and 20 kb upstream sequences including the Efnb3 gene) did not alter body size but had similar pathological phenotypes with short life span, osteoporosis, and ’generalized organ atrophy,’ which is a mysterious term (Tyner et al, 2002). Age-related tumors were absent in both p53 deletions. m. p63 deletion: p63 is in the gene family of transcription factors with multiple isoforms that regulate cell cycle arrest and apoptosis (Keyes and Mills, 2006; Levrero et al, 2000). Unlike p53, mutations in p63 are rare in human malignancy and p63+/- does not increase tumors in mice. Loss of p63 induces cell cycle arrest with cell senescence phenotypes (Section 1.2.3), e.g., SA-β-galactosidase induction. The p63+/- deletion heterozygote on a F1(129/C57B6) background appeared normal as young adult, but lifespan was shorter: median 95 wk versus 104 wk background; maximum, ca. 110 versus 156 wk (Keyes et al, 2005). Survival curves give the impression of increased IMR and shorter MRDT. Tumors were absent as observed in the p44/p53 deletions (note l). Skin cells had striking induction of two human fibroblast cell senescence markers, SA-β-galactosidase and p16(INK4a), which also increase in human skin during aging (Ressler S et al, 2006). However, neither was detected in aging control mice. Aging mice had ’some lordokyphosis’ (spinal curvature), which is common at later ages in background strains. Chronic infections were unusually common during aging: 52% had skin lesions, or infections (abscesses with bacterial inclusions); 12% had hemorrhage; other sites of infection in subcutaneous tissues, mouth and pharynx, genitourinary tract.
n. p66shc knockout mouse: (Migliaccio et al, 1999). Knockout of p66 (p66shc–/–) increased life span by 30%: control lifespan (129/sv), 750 d; shc +/–, 830 d; shc–/–, 970 d. Resistance to oxidative stress was increased in vivo: paraquat toxicity; lipid oxidation on high fat diet (Napoli et al, 2003b); and in vitro: DNA damage from UV-radiation, or H202 (Orsini et al, 2004; Migliaccio et al, 1999; Trinei et al, 2002). Sex differences include greater female resistance to paraquat; males have slight glucose elevation.
Two mouse mutants with altered fat cells have joined this longevous bestiary: the FIRKO (+12% life span) (Bluher et al, 2002; Bluher et al, 2003) and the C/EBPβ/β (+22% life span) (Chiu et al, 2004). FIRKO lacks the fat-specific insulin receptor, which decreases body fat by 50% and body weight by 20%. Despite higher food intake per gram body weight, blood glucose and glucose tolerance are normal. In FIRKO, unlike normal adults, body fat does not increase with age, nor does insulin resistance. Future comparisons could include knockouts of the insulin receptor in muscle (MIRKO) and liver (LIRKO); their insulin resistance may increase mortality (Rincon et al, 2004). Lastly, the C/EBPβ/β mutant carries a replacement of the C/EBPa gene, which alters adipocyte differentiation and blocks lipid accumulation (Chiu et al, 2004). Mitochondrial content and energy dissipation are increased. Mutants do not become obese on high fat diet. No details of aging or pathology were reported.
These exploratory studies are far from conclusive. Provisionally, I conclude that dwarfism is neither necessary nor sufficient for longevity, shown in Figure 5.8. Attenuated insulin-like signaling may be permissive of longevity in low stress environments. As discussed above, dwarf mice may be more vulnerable to wasting conditions and infections. Thus, attenuated insulin and IGF-1 signaling may be necessary, but not sufficient for increased life span in rodents, and possibly humans.
5.7.1.2 Human Hereditary Variations in Metabolic Genes
Would the mild to severe metabolic deficits in these rodent mutants also influence human longevity? At the least, inherited dwarfism and GH deficiency are compatible with normal life spans. In one of the first reports, isolated recessive GH deficiency was found in West Virginia kindred, with some dwarfs up to age 77 y, by self-report (Rimoin et al, 1966), which would have exceeded the life expectancy for that region and time.11 Mutations causing GH-receptor insensitivity are found in the Laron syndrome, first described in Israel (Laron, 2005). Several different GH-receptor mutations have GH signaling defects and elevated GH (Laron, 1999). Growth is impaired and adults are obese, glucose intolerant, and may be mentally retarded depending on the mutation (Shevah et al, 2005). In rural Ecuador, about 70 individuals share a point mutation in the GH receptor (E180 splice mutant in Exon 6) (Rosenbloom et al, 1999). These inbred and isolated communities include descendants of Jewish conversos from the Spanish Inquisition. The E180 mutation also occurs in Israel (Shevah et al, 2005). Findings are intriguing but incomplete. Adult dwarfs have elevated GH and very low IGF-1. Total cholesterol and LDL cholesterol may be elevated (Rosenbloom et al, 1999). Death from heart disease of a man at 55 and woman at 67 was considered “uncommon.” There may be 2-fold higher childhood mortality from infections (19% died before age 7 y vs. 9.7% deaths of unaffected sibs, due to diarrhea, meningitis, and pneumonia). Intelligence is considered normal (Kranzler et al, 1998), as in other E180 carriers (Shevah et al, 2005). Dwarf mice also have normal brain development, which does not depend on blood GH or IGF-1.
Two other groups with heritable GH deficiency were not treated with GH. A Swiss kindred with the entire GH-1 gene deletion lived about 25 y less than unaffected first and second degree relatives (Besson et al, 2003). The main causes of death in the dwarfs were “infectious diseases and heart problems,” not different from unaffected sibs. Other dwarfs from Krk in Croatia carry a prop1-like mutation causing deficits of GH and TSH, like the Ames mouse (Laron, 2005). Several dwarfs reported their ages as 70 y or older, and tombstones indicate others lived up to 91 y.
In sum, these preliminary findings do not allow firm conclusions about potential longevity effects of human mutant GH and IGF-1. More data are needed on the health history and cause of death in relation to the number of mutant gene copies. Glucose-insulin tolerance tests are needed. The Ecuadorian studies should continue to be very productive, but GH therapy will eliminate the dwarf phenotype sooner than later. However, for most elderly who do not have clinically recognized GH deficits, there is no recognized basis for GH supplements as a general ‘anti-aging’ intervention (Liu et al, 2007).
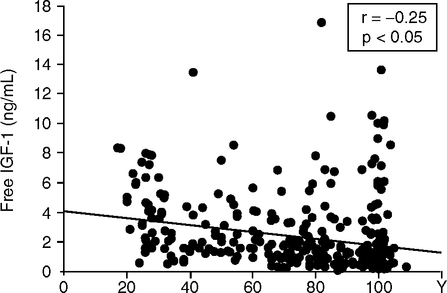
The race is on to find human gene variants that increase longevity through altered metabolism (Bartke, 2005; Katic and Kahn, 2005). Centenarians may have better glucose tolerance and a lower incidence of diabetes than those of average life span, as observed in small samples from southern Italy (Paolisso et al, 1996; Paolisso et al, 1997; Paolisso et al, 2001). Centenarians may also have body mass index in a lower range (Barbieri et al, 2004). In a larger Italian sample, blood IGF-1 levels tended to decrease age (Fig. 5.11) (Bonafe et al, 2003); nonetheless, some centenarians have higher plasma IGF-1 than young adults. Longitudinal sampling is needed to resolve this hetergeneity. The IGF-1R (receptor) homozygote A+/A+ was 30% more prevalent in the oldest (median age 99 y) than younger (median 60 y) and was associated with lower IGF-1. Nonetheless, about 30% of the oldest had elevated IGF-1, which cautions against sweeping conclusions.
The Leiden 85+ Study examined six insulin pathway genes (1576 Ss, born 1883–1914) (van Heemst et al, 2005a). The strongest association was of short height with the GH-1 allele (intron SNP): female, height difference of –2 cm (p > 0.007); male, -1.9 cm (p > 0.078). The GH-1 allele also decreased mortality in females (RR, 0.80, range 0.67–0.96). Hormone levels were not reported. No allele associations were found for height or mortality with alleles of GHRHR (GH-releasing hormone receptor in the pituitary), IGF-1, insulin, and IRS1, or with two stress resistance genes, SOD2 and UPC2.
Genetic risk factors for type 2 diabetes should also be considered. The TCF7L2 gene, a putative transcription factor, has alleles that showed strong dose risk effects in a case control study in Iceland, with T/T homozygotes having 2.4-fold higher risk of diabetes (Grant et al, 2006). The T allele also predicted progression to diabetes during 3 years of observation with similar dose effects attributed to impaired pancreatic insulin secretion (Diabetes Prevention Program Study, DPP, 5847 Ss) (Florez et al, 2006). The diabetes-prone T/T allele may be the strongest genetic association with diabetes so far. The protective C allele should be examined for influences on longevity. The TCF7L2 gene is expressed in the developing pituitary and interacts with WNT signaling by binding to β-catenin downstream of PROP-1 (Douglas et al, 2001). Other candidates may be found in obesity. Kimura et al. (1997) noted that the daf-2 worm mutant was the same as an insulin receptor mutation associated with morbid obesity. Most recently, an obesity marker single nucleotide polymorphism was mapped near the insulininduced gene-2 (INSIG2) (Herbert et al, 2006).
The genetics of diabetes gives a useful calibration for longevity gene searchers. Despite huge efforts, few general genetic influences have been found. As holds for all adult onset diseases, dominant familial genes are rare, and polygenic contributions of modest risk effects may be the norm (Florez et al, 2003). There may be few consistently strong genetic effects across human populations because of the huge number of allele combinations among the 30 longevity gene candidates scattered across the chromosomes (Christensen et al, 2006). Some may be mutually canceling, while others may synergize.
5.7.1.3 Size and Longevity
The association of small size and longevity is controversial. On one hand, Rollo’s (2002) comprehensive meta-analysis of more than 1000 separate rodent groups, including diet restriction studies, showed statistically strong inverse correlations between maximum body size and maximal longevity (P > 0.00001). The regression coefficients account for 9–25% of the variance, depending on sex and species. As specific examples, in mice selected for different growth rates, the smallest also lived longer (Miller et al, 2000b; Miller, 2002b). It is interesting to compare the 10–15 g Ames dwarf with the 15––0 g wild-caught mice, which outlive the usual strains of inbred mice (Chapter 3, Fig. 3.3). (Here, we have a true genetic wildtype.) The Ames dwarf clearly wins, with 5 m longer average and maximum life spans than the wild mice.
However, not all of the longer-lived rodent mutants are small (Fig. 5.8). The IGF-1 receptor mutant is nearly normal in size, while the C/EBPβ/β and MCAT mutants are normal sized. Conversely, the mini and the Lewis dwarf rats have close to normal life spans, despite the reduction of body size in proportion to the Ames and Snell dwarfs. None of these mutants approach the Ames and Snell longevity.
Domestic dog breeds show strong inverse associations between plasma IGF-1 and body size versus longevity. Plasma IGF-1 ranges 10-fold across breeds, from the spaniel to the giant Newfoundland dog (Eigenmann et al, 1984; Eigenmann et al, 1988). Among these breeds, life spans vary inversely with adult size across a 3-fold range (Miller, 1999; Patronek et al, 1997). Similarly, some mouse strains at 15 m showed individual correlations of low IGF-1 with the remaining life span (Anisimov et al, 2004; Harper et al, 2003). The artificial selection of domestic dogs for size appears to parallel the rodent models, e.g., short-lived giant transgenic mice with high plasma GH (see above).
Nonetheless, robust and abundant evidence shows that human longevity has increased during the past 150 years at the same time as childhood growth increased (Section 4.4.1) (Crimmins and Finch, 2006a). Healthier children grow faster and are healthier and longer lived as adults. Small size is associated with smaller diameter coronary arteries that are at greater risk for occlusion, as is well known to vascular surgeons (Chapter 4.4.1). These observations are made in human populations exposed to challenges not experienced by lab models. These trends are also fully consistent with the allometric relationships of size and longevity in species comparisons of birds and mammals (Finch, 1990, Chapter 4) (Calder, 1984).
Nonetheless, in special cases larger size increases mortality risks. The statistics are convincing for inverse relationships of life expectancy to height in professional football and baseball players (Samaras and Elrick, 1999; Samaras et al, 2003a,b), and for premature mortality in sumo wrestlers (Section 3.4). A more general case is the association of breast cancer with higher body mass index and height (Barker et al, 1989a,b; Samaras et al, 2003a,b), e.g., the many-fold greater risk of breast cancer in Japanese women who grew up in Los Angeles versus Osaka (Henderson et al, 1984). As more humans live to greater post-reproductive, post-Darwinian ages, we should not be surprised to find many other exceptions.
5.7.1.4 The Insulin-Sensitivity Paradox
Nir Barzilai and colleagues describe the paradox that insulin insensitivity, e.g., metabolic syndrome increases human mortality, whereas mutant flies and worms with attenuated insulin-signaling live longer (Rincon et al, 2004). Adult humans with acquired insulin resistance (impaired insulin signaling) have increased chronic diseases (cancer, diabetes, hypertension, vascular disease) (Facchini et al, 2001). The adverse effects of insulin resistance in mammals versus the apparent benefit in fly and worm mutants may be explained by the different organization of insulin-signaling (Rincon et al, 2004). Recall from Section 5.4 that fly and worm insulin expression is largely restricted to neurons, whereas in mammals, insulin/IGF receptors are widely expressed in all tissues. Lacking information on the functions of most worm and fly insulin-like genes, it is not possible to make detailed comparisons with mammals. Even between flies and worms, different outcomes can be seen. Insulin receptor mutant worms with weak daf-2 mutants accumulate fat (Kimura et al, 1997). This association is consistent with a human insulin receptor mutation that caused insulin resistance and hyperinsulinemia (Kim et al, 1992); the insulin receptor mutant increased life span in the worm mutant, but should decrease it in human insulin resistance. Body fat is also accumulated in flies with lower neuronal insulin mRNA (dilp-2), which should lower haemolymph insulin, but in this case life span is longer (Hwangbo et al, 2004). As noted to me by Bartke, lower IGF-1 may benefit mice more than humans because of the higher incidence of cancer in inbred mouse strains. The insulininsensitivity paradox gives a useful platform for further discussion as additional species differences are recognized.
5.7.2 Inflammation
Inflammation gene variants are associated with mortality risks. Table 5.5 summarizes longevity gene candidates for inflammatory and lipoprotein genes, with chromosomal location. The histories of infectious exposure may have selected for certain combinations of alleles across different genes. For example, resistance to malaria may be mediated by hemoglobin variants and by the TNFa-promoter variant (-308) (Mombo et al, 2003). The genome projects may soon show the distribution of inflammatory gene haplotypes across human populations. IL-6 alleles at -174 may influence survival to advanced age. Further candidates may be sought in the genetic influences on plasma CRP, which is regulated by IL-6 and is a major risk indicator of vascular disease (Chapter 1, Fig. 1.16).
TABLE 5.5
Summary of Human Longevity Gene Candidates (see Text for References)
| Gene/Chromosome; Function | Alleles | Age-Association |
| apolipoprotein C3 apoC3/Ch11q23; lipid binding protein mediating transfer of cholesteryl esters from HDL | −641C/A | C/C associated with centenarians |
| apolipoprotein E apoE/Ch19q13; cholesterol transport protein | +112C, apoE3 +112R, apoE4 | E4, shorter life span; higher risk of cardiovascular and Alzheimer disease |
| −219G/T | T/T, myocardial infarct; G/G had | |
| −419A/T | higher plasma apoE and lower glucose | |
| cholesteryl ester transferase protein CETP/Ch16q21; | +405I/V | A/A, higher apoE +405V/V higher in centenarians |
| −629C/A | combination of −629A/A and apoE4 may be 3-fold higher in Alzheimer; apoE4, did not synergize with +405I/V | |
| interleukin-6 IL-6/Ch7p21; proinflam- matory regulator | −174G/C | G/G, lower plasma IL-6, better survival to old age, vascular health, and resistance to bacterial meningitis |
| interleukin-10 IL-10/CH1q32; anti- inflammatory cytokine | −1082G/A | G allele elevated IL-10; elevated IL-10 may increases risk of diabetes |
| peroxisome proliferator-activated receptor gamma PPARγ/Ch3p25; transcription factor influences insulin sensitivity | Pro12Ala | heterozygote may increase male survival to later ages |
| paraoxinase-1 PON1/Ch7q21.3; protects LDL from oxidative damage | 55L/M and 192R/Q | LR haplotype may increase survival to later ages |
The IL-6 promoter has two alleles (-174G/C) that may be risk factors in longevity and vascular disease. Among Danes, the prevalence of G/G was 30% in the elderly, suggesting survival advantage (G/G allele: 25% below age 50, with progressive increases to 32% at 100; 1710 Ss) (Christiansen et al, 2004). Finnish nonagenarians in a prospective study also had higher prevalence of the G/G allele of IL-6 (Hurme et al, 2005). Other studies indicate protective associations of 174G/G. Myocardial infarcts were 50% less frequent in older Italian G/G carriers (67 y mean) (Chiappelli et al, 2005). Older dialysis patients with G/G had lower diastolic blood pressure and left ventricular hypertrophy was 50% less frequent (Losito et al, 2003). G/G carriers were more likely to survive bacterial meningitis (Balding et al, 2003), which may be due to lower plasma IL-6 (Bonafe et al, 2001; Chiappelli et al, 2005). IL-6 elevations are risk indicators of vascular events (Chapter 1) and frailty (Ershler and Keller, 2000; Wilson et al, 2002; von Kanel et al, 2006).
IL-6 is also linked to plasma CRP by directly controlling CRP synthesis in the liver. Plasma CRP was lower in IL-6 -174G/G carriers (Ferrari, 2003). Moreover, plasma CRP levels showed strong heritability in association with IL-6 -174 G/G (Vickers et al, 2002). (The IL-1 and CRP genes are not chromosomally linked.) However, in younger ages, the IL-6 -174 G/C alleles have not been consistently associated with blood IL-6 levels (Qi et al, 2006). CRP levels are also influenced by alleles of IL-1 (Latkovskis et al, 2004) and of apoE (Austin et al, 2004; Rontu et al, 2006). CRP alleles at four different sites in the gene may influence plasma CRP levels (Kovacs et al, 2005; Szalai et al, 2002) but have not been associated with vascular events (Zee and Ridker, 2002). The apolipoprotein E4 genotype is proinflammatory relative to apoE3 (Section 1.3.2), which may be one mechanism in the associations of apoE4 as a risk factor in Alzheimer disease, vascular disease, and frailty (discussed below).
TGF-β1 variants are also candidates in longevity. The missense allele +915C in the signal peptide of TGF-β1, which does not influence the plasma level, had lower prevalence in Italian centenarians (Carrieri et al, 2004). Survival advantages are consistent with the slower development of coronary vascular pathology after cardiac transplantation in a younger group of +915C carriers (Densem et al, 2000; Densem et al, 2004).
TNFa alleles (-308G/A) influence TNFa synthesis, as discussed in Chapter 3 for links to adiposity, insulin resistance, and hypertension. These TNFa alleles also influence fetal growth (Section 4.9) but have not been consistently associated with longevity or coronary disease.
IL-10 alleles may influence aging through levels of plasma IL-10, which interacts with atherosclerosis, diabetes, and infections. IL-10 secretion by leukocytes in response to LPS is highly heritable (MZ twin, 0.75 heritability) (Eskdale et al, 1998; Westendorp et al, 1997a,b). Of three IL-10 promoter alleles, -1082G/A had the most effect on plasma IL-10 (Lio et al, 2004). Elevated plasma IL-10 represses IL-6 and TNFa synthesis, which are risk indicators in atherosclerosis. Elevated IL-10 predicted diabetes (Leiden 85-Plus Study) (van Exel et al, 2002). Meningococcal disease mortality was predicted by leukocyte cytokine production in first degree relatives: High IL-10 leukocyte production was associated with 20-fold more mortality; low TNFa production, 10-fold more mortality (Westendorp, 1997a,b). However, IL-10 alleles have not been consistently associated with heart disease or life span (Lio et al, 2004). We may anticipate a complex genetics, because the close proximity of IL-10 and CRP on Chromosome 1 (Section 1.3.2) generates multiple haplotypes of these common alleles.
These scattered findings suggest potential benefits to longevity in some of the same gene inflammatory gene variants that influence vascular disease risk. The IL-6 and TGF-β1 alleles merit expanded screening.
5.7.3 Lipoproteins and Cholesterol Metabolism
Familial hypercholesterolemia was recognized 70 years ago as the cause of premature heart disease by Muller in Norway (Muller, 1938; Ose and Tolleshaug, 1989). The identification of LDL-receptor defects in a rare familial hypercholesterolemia by Brown and Goldstein (1974) has stimulated remarkable advances in vascular disease intervention and prevention, as well as the molecular mechanisms in normal vascular biology. However, lipoprotein system dominant mutants have minor contributions to vascular disease in most populations and account for <5% of variance in LDL cholesterol (Breslow, 2000). Nonetheless, elevated LDL cholesterol and low HDL cholesterol are strongly linked to vascular events and are becoming understood as complex environmental interactions of multiple genes (Knoblauch et al, 2004; Stengard et al, 2006). Further developments implicate several lipoprotein gene variants in human longevity and in human evolution (Chapter 6).
Apolipoprotein E (apoE) is the most common heritable variation in lipids with major associations of the apoE4 allele to heart disease, Alzheimer disease, and longevity (Mahley, 1988; Mahley and Rall, 2000). ApoE is a cholesterol transport protein that binds to the LDL receptor and is crucial to blood cholesterol levels, and for the transport of cholesterol for steroid synthesis and neuronal outgrowth. Coding changes that influence cholesterol binding in apoE2, -E3, and -E4 may account for 10% of the variance in total cholesterol and LDL-cholesterol (Breslow, 2000; Sing and Davignon, 1985). The apoE4 allele is associated with higher total blood cholesterol and LDL-cholesterol, e.g, LDL cholesterol is 10–20 mg/dl higher in E4/E3 than E3/E3, while E3/E2 are lower by the same amount. ApoE2 is associated with the uncommon familial type III hypercholesterolemia (E2/E2), but mysteriously, only 2% of E2/E2 develop this condition. When populations varying in apoE4 prevalence are considered, the level of E4 may account for 50% of the inter-population differences in average total cholesterol. Effects of E4 on triglycerides and HDL -cholesterol are more variable. Besides the structural differences in the apoE isoforms, apoE promoter variants influence blood apoE and other lipids, with differences by age and gender (Frikke-Schmidt et al, 2004; Stengard et al, 2006). Myocardial infarct risk may be higher in -219T carriers (Lambert et al, 2000).
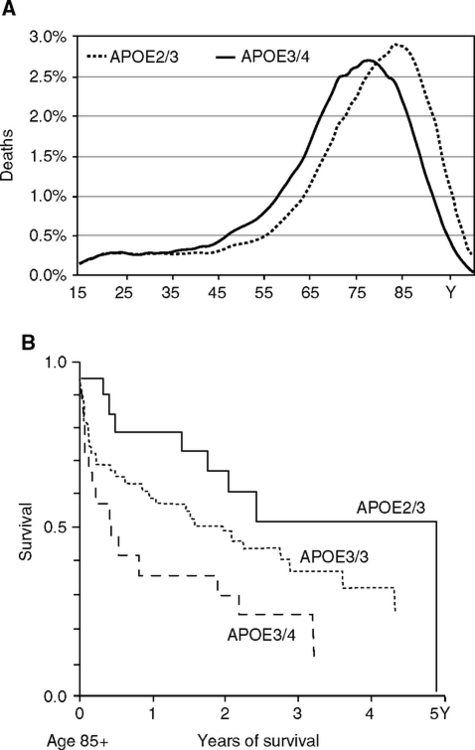
ApoE4 is associated with shorter life span and was the first human ‘aging’ gene identified by its near absence in centenarians (Schachter et al, 1994). The effects of E4 approximate the sex differences in life span; e.g., Danish life expectancy was 7 y shorter in E3/E4 than E3/E2 genotypes (Fig. 5.12A) (Ewbank, 2004). Of the 25% heritability of life span in Denmark, about 3.5% is attributable to apoE alleles, which is equivalent to a substantial 15% of total life span heritability. ApoE alleles also influence later age mortality. For example, at age 85, the remaining life span of those diagnosed with cardiovascular disease varied 2 years by apoE alleles (Fig. 5.12B). ApoE4 decreases in prevalence at advanced ages because of survival effects (Fig. 5.13) (Gerdes et al, 2000; Rontu et al, 2006). Relative to the E3/E3 and E4/E2 genotypes, mortality in E4 carriers is 10–14% higher, while E2 carriers have 4–12% lower mortality (Gerdes et al, 2000). For these reasons, apoE4 is considered a frailty gene (Corder et al, 2000; Gerdes et al, 2000).
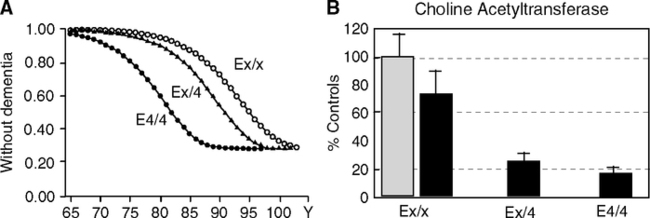
ApoE4 increases the risk of coronary heart disease by 42% (comprehensive meta-analysis of 48 studies representing 58,457 Ss) (Song et al, 2004). ApoE4 also increases the risk of Alzheimer disease (Fig. 5.14A) (Borenstein et al, 2005; Corder et al, 1993; Meyer and Breitner 1998; Poirier et al, 1993; Poirier, 2005; Roses, 2006) (Section 1.6). The E4 allele shows dose effects accelerating onset age (Khachaturian et al, 2004). ApoE4 carriers in end-stage Alzheimer disease have more severe cholinergic deficits, which are implicated in the early stages of memory impairments (Fig. 5.14B, C). Nonetheless, some centenarian E4/E4 homozygotes are not demented (Khachaturian et al, 2004; Sobel et al, 1995). Because some E4/E4 carriers reach advanced ages without dementia, apoE is not considered a genetic determinant, but rather a risk factor that influences the threshold for still mysterious early processes in neurodegeneration. The apoE4 allele show these adverse Alzheimer effects most consistently in temperate zonederived populations and shows weaker association in Latinos and African Americans (Section 1.6.3).
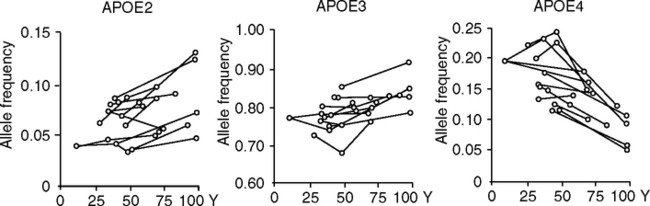
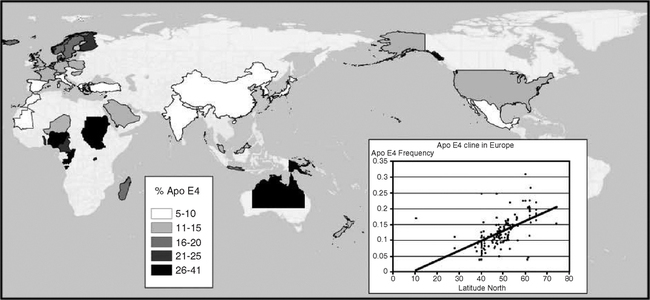
ApoE3 is the most prevalent allele in human populations, while apoE4 varies over a greater range (Fig. 5.15). Reported extremes of apoE4 range from 49% (Huli, New Guinea) to 0% (Ache, Paraguay) (Demarchi et al, 2005). ApoE2 is the least prevalent allele, and least understood. Because of its low prevalence (<5%), apoE2 is not emphasized here. ApoE4 is more prevalent in Northern Europe than in the Mediterranean, and some aboriginal populations tend to have high E4 (Corbo and Scacchi, 1999; Gerdes et al, 1992; Panza et al, 2003; Demarchi et al, 2005). The European longitudinal apoE4 gradients also correlate with heart attack risk (Stengard et al, 1998). Besides Nordic caucasians, other groups with apoE4 prevalence >15% include Greenland Inuit, Central African pygmies, and Australian and New Guinea aborigines (Demarchi et al, 2005; Sing et al, 2006). Thus, there is no global generalization of apoE allele frequency with longitude or climate. The regional gradients suggest migration and possibly natural selection from interactions with nutrition and pathogen resistance (Section 5.7.4; Chapter 6).
Other lipoprotein system genes have variants that may influence longevity. Barzilai and colleagues are searching for centenarian candidate genes of vascular and metabolic disease in Ashkenazi Jews. Their first gene hit was cholesteryl ester transferase protein (CETP): in centenarians, the +405V/V alleles were 2-fold more prevalent than controls, implying survival effect (Atzmon et al, 2006; Barzilai et al, 2004). This association was confirmed in samples of Northern Italians, average age 89 (Vergani et al, 2006), but not in Italian centenarians (Cellini et al, 2005). The concurring two studies observed lower plasma CETP and elevated HDL, which is considered anti-atherogenic in human and animal studies. CETP is a lipid binding protein that mediates the transfer of cholesteryl esters from HDL. The multiple CETP alleles may account for 10% of variation in HDL (Knoblauch et al, 2004). A CETP promoter allele (-629C/A) interacts with apoE4 in Alzheimer disease (Table 5.5): The combination of apoE4 and 629A/A may be 3-fold higher in Alzheimer’s; however, apoE4 did not synergize with +405I/V allele (Rodriguez et al, 2006).
The ApoC3 gene promoter allele -641 C/C homozygote was higher prevalence in centenarians (25%), than their F1 offspring (20%) or controls (10%) (Atzmon et al, 2006). The C/C genotype was associated with both longevity and lower plasma apoC, consistent with much evidence for elevated apoC as a risk of coronary disease. Plasma apoC3 is an exchangeable component of triglyceride-rich lipoproteins. By inhibiting lipoprotein lipase, blood apoC3 levels modulate the clearance of triglycerides after a meal, with low apoC3 associated with triglyceridemia. The centenarian apoC3 -C/C carriers also had favorable insulin sensitivity. There is a direct link of apoC3 to insulin regulation through an insulin response element seated in the gene cluster of apoA1, apoC3, and apoA4. Promoter alleles that influence transcription rate (Dallinga-Thie et al, 2001) are among haplotypes of apoC3 implicated in type 1 diabetes (Hokanson et al, 2006).
The transcription factor PPARγ regulates genes in adipocyte differentiation, insulin sensitivity, and inflammation. Two alleles at codon 12 (pro/ala) have associations with insulin sensitivity and obesity in younger groups (de Rooij et al, 2006a; Moon et al, 2005; Muller et al, 2003; Ostergard et al, 2005) and with birth weight (Eriksson et al, 2003). The ala allele reduces transcriptional activity of PPARγ and may protect against type 2 diabetes. In very old Italian men (average 97 y), the pro/ala heterozygote was 2-fold more prevalent than in younger men; women did not show this association (Barbieri et al, 2004). The paraoxinases (PON1, -2, -3) are longevity gene candidates because of associations with heart disease (Marchegiani et al, 2006). PON1 protects LDL from oxidative damage, a by-stander effect (Section 1.4.1) and has alleles at two loci: 55L/M and 192R/Q. The LR haplotype may be slightly overrepresented in later ages, suggesting survivor effects (Rea et al, 2004).
ApoC3 also has parallels to invertebrate longevity genes through insulin-signaling and lipoproteins (Atzmon et al, 2006). ApoC3 transcriptional responses to insulin (see above) involve the transcription factor FoxO1 (Dallinga-Thie et al, 2001), an orthologue of worm daf-16. Moreover, levels of the lipoprotein vitellogenin influence immunity and oxidative damage in the honeybee (Section 5.4) and can influence the worm life span (Section 5.5). CETP, as a lipid binder, is in a family of proteins that bind bacterial endotoxins (LPS) and might itself be directly involved in host defense. In mammals, lipoproteins protect against oxidative stress, through associated anti-oxidants, β-carotene, discussed below. Blood lipoproteins are also remodeled during acute phase responses (acute phase HDL, Section 1.5.5). The complex lipoprotein system genetics includes haplotypes that may also influence diabetes and life span. This brief synopsis neglects other lipoprotein system gene candidates for roles in longevity, metabolic disorders, and vascular disease.
5.7.4 ApoE4 Interactions with Diet, Cognition, and Vascular Aging
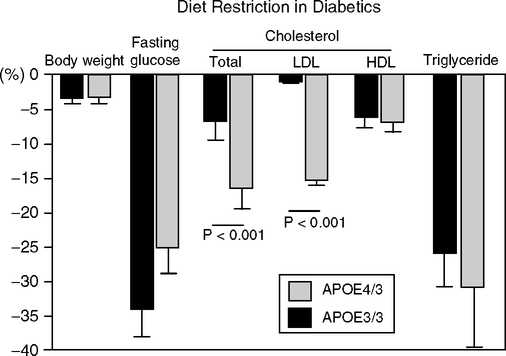
Diet was not explicitly included in any of the above studies of genetic effects on longevity. Few lipoprotein gene variants have consistent effects on blood cholesterol responses to dietary changes (meta-analysis of >2500 studies) (Masson et al, 2003). ApoE4 carriers had the largest decreases of LDL cholesterol in response to reductions of dietary fat and cholesterol (nine studies); HDL cholesterol responded most in apoE4 carriers (3 of 4 studies) (Lopez-Miranda et al, 1994). The apoE promoter allele -219G/T in combination with apoE3/E3 did not influence the plasma LDL or HDL cholesterol responses to a diet shift from saturated to unsaturated fats (Moreno et al, 2005). However, glucose regulation in –219T/T genotype was relatively less sensitive to fat intake. The –219G/G carriers had lower glucose on all diets, confirming (Viitanen et al, 2001). Two other gene longevity candidates, apoC3 and CETP, also show allelic differences in LDL cholesterol responses to diet (Masson et al, 2003), but the active alleles are at different sites in these genes than those associated with longevity (apoC3, exon4 SsI; CETP, intron Taq1). Diet restriction of type 2 diabetics who were apoE4 carriers showed the most improvement in blood LDL-cholesterol (Fig. 5.16) (Saito et al, 2004). This observation recalls that mouse strains and mutants differ in responses to diet restriction; e.g., DBA/J (Fernandes et al, 1976) and GHRKO (Bonkowski et al, 2006b) show little, if any, increased life span.
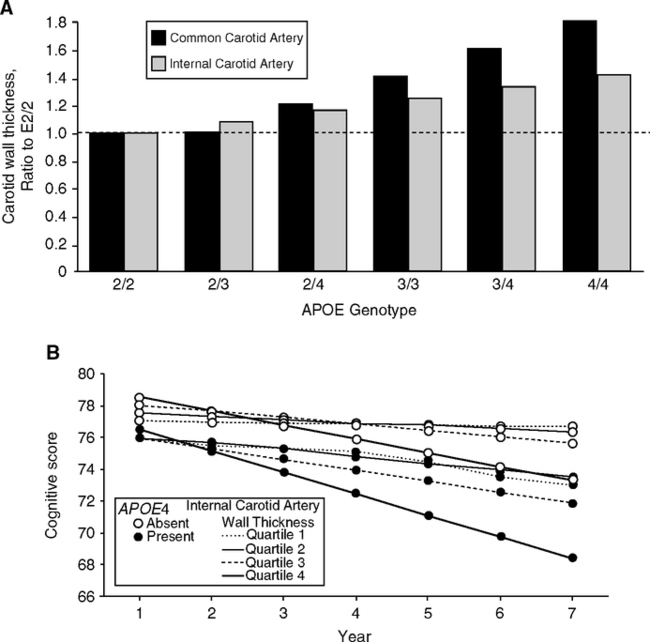
ApoE4 carriers have faster carotid artery thickening after age 65 (Fig. 5.17A) (Haan et al, 1999), which is a cardiovascular risk indicator (Section 1.5, Fig 1.14). Carotid thickening may contribute to cerebral deterioration directly and indirectly. Cognitive decline after age 65 is accelerated in apoE4 carriers (Cardiovascular Healthy Study) (Fig. 5.17B) (Haan et al, 1999). ApoE4 carriers with diabetes, carotid atherosclerosis, or peripheral vascular disease showed the most cognitive loss. Similarly, E4 carriers in the healthy and high-performing elderly of the MacArthur Study of Successful Aging showed higher risk of cognitive decline and greater declines over 7 years (Hu et al, 2006b). The majority of elderly did not have measurable cognitive decline during 7 years in both studies. ApoE4 carriers in middle age already have reduced glucose metabolism in cerebral cortex regions (Reiman et al, 2005; Small et al, 2000) and use mnemonics extensively in compensation (Ercoli et al, 2006). Synapse density in the hippocampus granule neurons of cognitively normal elderly is also lower in apoE4 than E3 carriers (Ji, 2003). It is possible that these later deficits were present earlier.
Older apoE4 carriers with cognitive decline most likely included early Alzheimer disease and the syndrome of ‘mild cognitive impairment’ (MCI) (Section 1.6.3). ApoE4 is more prevalent in MCI than elderly controls and associated with greater hippocampal atrophy in proportion to the E4 allele dose; women may be more vulnerable to a single apoE4 allele than men (Farlow et al, 2004; Flatt and Kawecki, 2004; Fleisher et al, 2005). In the Cache County Study, elderly with MCI and other mild disorders were 2–5-fold more likely to develop Alzheimer disease if they were E4 carriers (Tschanz et al, 2006). These later life changes are departures from the slower and more general brain aging changes beginning during middle-age, which include synaptic atrophy and reduced blood flow (Chapter 1, Fig. 1.7). The transitions of the general brain aging changes to Alzheimer disease are not well resolved.
While apoE4 is the most common genetic risk factor for Alzheimer disease, it also synergizes with other rarer dominant genotypes to accelerate the age of onset, including the familial Alzheimer mutations in the amyloid precursor protein (APP), the presenilin mutations (PS-1 and PS-2), and in trisomy 21 (Downs syndrome). Moreover, a meta-analysis of the new AlzGene database indicates more than 10 potential other risk factor candidates, at least some of which interact with apoE alleles, e.g., apoC1, which is closely linked to apoE on chromosome 19 (Bertram et al, 2007). Combinations of risk factors on different chromosomes (Section 1.3.2) could reclassify some of the apparently sporadic cases of Alzheimer disease.
Cerebral metabolism may be slightly impaired as early as by age 30 in apoE4 carriers who have not shown clinical abnormalities (Reiman et al, 2004). Smaller head circumference increased the risk of dementia in E4 carriers 2-fold above non-E4 carriers (Borenstein et al, 2005). Because head size is driven by brain growth, we must consider the portentous question of apoE alleles on brain development. In a transgenic mouse with targeted substitution of apoE, the synaptic density was lower in E4 carriers than E3 (Wang et al, 2005a). Thus, it is possible that the lower synapse density in older E4 carriers (see above) and the trend to lower cerebral metabolism by age 30 represent developmental impairments.
ApoE alleles influence the effects of elevated plasma CRP and cholesterol, which are major vascular risk indicators. In the Finnish Vitality 90+ project (Rontu et al, 2006), plasma CRP and cholesterol varied with the apoE4 dose in opposite directions. We may anticipate additional complexities in the relationships of these risk factors to life span. In general, elevated plasma CRP and LDL-cholesterol are risk factors for vascular disease (Fig. 1.16), as is apoE4. In Finnish nonagenarians, however, some were observed to have elevated LDL-cholesterol, but low CRP. The apoE alleles partly explained the CRP-LDL status. Plasma CRP varied inversely with the apoE4 allele dose, while LDL cholesterol increased with E4 dose. The inverse relation of the apoE4 allele dose to CRP in this age group differs from the proinflammatory associations of apoE4 in experimental and clinical contexts involving young ages (Section 1.3.2), suggesting a survivor effect that differs from the norm at advanced ages. CRP also co-varied with IL-6 over a 3-fold range, as expected from the direct control of CRP synthesis by IL-6 (Lehtimaki et al, 2005). Both reports excluded those with acute infections.
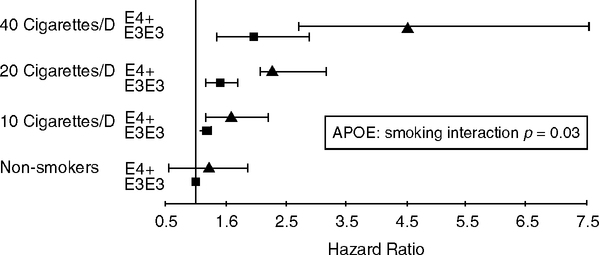
ApoE4 smokers may have higher risks of cardiovascular events than E3 smokers (Fig. 5.18) (Talmud et al, 2005). This finding if verified suggests that smoking, a powerful inflammatory stimulation, synergizes with apoE4 to accelerate atherosclerosis more than the apoE alleles (Fig. 5.17A). The incidence of atherosclerotic carotid plaques showed further synergies: apoE4, non-smokers, 1.7-fold higher risk than non-E4; E4-smokers, 3.7-fold higher risk (Djousse et al, 2002). In a different group (Caucasian diabetics), oxidized LDL cholesterol was 28% higher in E4 smokers than other genotypes or non-smokers (op. cit.). Furthermore, apoE4-smokers had lower serum β-carotene, an anti-oxidant (MacArthur Study of Successful Aging) (Hu et al, 2006b). One may speculate about the apoE alleles of Jeanne Calment, a regular smoker who achieved the record life span of 122 (Allard et al, 1998). The greater vulnerability of E4 carriers to oxidant damage from smoke should be examined for other proinflammatory aerosols (Chapter 2).
Alcohol consumption may also have different vascular risks by apoE allele. Modest regular alcohol consumption is recognized to lower risk of vascular events according to a J-shaped dose response. The coronary disease risk is about 30% lower with 1–2 drinks/d (Gronbaek, 2006), which is equivalent to 10––0 gm pure ethanol/d. Heavy drinking (>100 g/d) may increase the risk beyond heavy smoking. The mechanisms in alcohol benefit may derive from the consistently observed elevations of HDL cholesterol. However, there may be interactions of apoE4 and alcohol, because HDL elevations were smaller in apoE4 carriers of the NHLBI Family Heart Study (Djousse et al, 2004). In the Framingham Study population, apoE4 males had higher LDL cholesterol in proportion to alcohol consumption (controlled for energy intake, age, BMI, and smoking) (Corella et al, 2001; Ordovas, 2002). These effects of alcohol on LDL cholesterol could contribute to the greater ‘frailty’ of apoE4 carriers. Evidence is inconclusive about possible benefits from different types of alcoholic beverage and the content of resveratrol, an antioxidant and antibiotic (Section 5.4) (Baur and Sinclair, 2006; Gronbaek, 2006).
Could the benefits of exercise to vascular health and mortality (Section 3.4) be influenced by apoE alleles? Sedentary E4 carriers may have more atherogenic lipid profiles. However, findings on vascular correlates of exercise and the alleles vary between studies (Bernstein et al, 2002; Hagberg et al, 2000).
5.7.5 ApoE Alleles, Infection, and Reproduction
If apoE4 has so many adverse effects, what maintains this allele in all human populations? Collaborations with Robert Sapolsky and with Craig Stanford considered the role of apoE alleles in the evolution of the human reproductive schedule (Finch and Sapolsky, 1999; Finch and Stanford, 2004). The cognitive and vascular changes that are more severe in apoE4 carriers during middle age would impair the uniquely human multi-generational care giving. From other perspectives, Brian Charlesworth (1996) hypothesized that apoE4 is selected by some early life benefit, while George Martin (1999) proposed in a discussion of the Finch-Sapolsky article that apoE4 is a resistance factor for lipophilic pathogens. The ‘Charlesworth-Martin’ hypothesis thus considers that apoE4 may persist in human populations because of balancing selection with antagonistic pleiotropy. Balancing selection is a major factor in life-history gene evolution (Finch and Rose, 1995) (Section 1.2.8).
A strong case is emerging for the protective effect of apoE4 in infections by hepatitis C virus (HCV). ApoE4 carriers had milder liver disease in two independent studies (Fabris et al, 2005; Wozniak et al, 2002). The progression to hepatic fibrosis was worsened in proportion to E4 dose. Moreover, female E4 carriers progressed faster to fibrosis than males. Hepatitis virions associate with plasma LDL and HDL, and cell uptake is mediated by LDL receptor. We do not know if apoE isoforms alter virion transport or uptake.
Other examples indicate apoE allele interactions with the brain during childhood and later in life. In a Brazilian favela with poor hygiene, preadolescent apoE4 carriers had better cognitive scores (Oria et al, 2005). The apoE4 carriers had 15% fewer episodes of diarrhea and less Giardia. The cognitive benefits may be due to higher plasma cholesterol of E4 carriers, which might support brain neuronal outgrowth and myelination (Oria et al, 2005), particularly in lower birth weights (Garces et al, 2002). Although head circumference did not differ by apoE genotype in this study, childhood growth can be stunted by heavy diarrhea (Section 4.6). Later in life, Alzheimer disease shows a higher prevalence of apoE4 carriers with HSV-1 infections (Itzhaki, 2004). However, HSV-1 is neither necessary nor sufficient for Alzheimer disease. The HSV-1 load in Alzheimer disease may be a bystander consequence of HSV-1, which is a ubiquitous and opportunistic infection. ApoE4 carriers with Alzheimer disease also had higher brain load of Chlamydia pneumoniae (Gerard et al, 2005), which may represent opportunistic infections during brain degeneration, as indicated for associations of Chlamydia with arterial disease (Section 2.2.2).
The hypothesis that ApoE4 is maintained by balancing selection requires some evidence of associations with fecundity. The apoE gene is deeply linked to reproduction because the apoE protein is the major transporter of the cholesterol used by gonadal cells for sex steroid synthesis. However, apoE alleles have not shown consistent associations with sex steroid levels or age at menarche (about 10 papers, not cited). Adult fertility may differ slightly by allele according to mostly small samples from different populations: Danish men (Gerdes et al, 1996), women from Southern Italy (Corbo, 2004), African-Ecuadorian women (Corbo et al, 2004b). These European E3/3 carriers had slightly greater fertility than E3/4. In possible contrast, African-Ecuadorian women with E3/4 tended to have more children (significance only in a subgroup with 9–17 children). While this might suggest a later age at menopause in apoE3/4 carriers, others found little association of menopause and the apoE alleles: Iranian E4 carriers had earlier menopause by about 1 year (Koochmeshgi et al, 2004), whereas no E3/E4 difference in age at menopause was found in Down’s syndrome (Schupf et al, 2003) or in large sample of German women (Tempfer et al, 2005). Postmenopausal plasma sex steroids do not differ consistently by apoE alleles, e.g., in a large U.S. study (Barrett-Connor and van Muhlen, 2003).
In sum, apoE4 allele benefits to hepatitis C virus infections support the Charlesworth-Martin hypothesis that apoE4 is maintained by balancing selection. The neurocognitive protection of apoE4 during childhood diarrhea could account for the higher prevalence of E4 in some equatorial populations (Fig. 5.15). The major effect of apoE alleles is on vascular and cognitive health during middle age and later. It is within this uniquely human ‘aging’ domain that the human apoE allele system evolved (Finch and Sapolsky, 1999; Finch and Stanford, 2004), discussed next in Chapter 6. Other allele systems with disease-longevity effects (Table 5.5) could also be interpreted by balancing selection.
5.8 SUMMARY
Genetic influences on aging in general populations are clear in the lower mortality risk of women throughout life. We do not know which genes on the sex chromosomes determine disease and mortality risks. The best characterized human ‘aging’ gene is apoE on chromosome 19 with two alleles, apoE3 and E4. The less common apoE4 allele consistently increases the risk of arterial disease and Alzheimer disease. Many other adverse effects characterize apoE4 as a frailty gene with an impact during mid-life and later. The persistence of the apoE4 allele may be due to balancing selection for pathogen resistance, as shown for hepatitis virus C and possibly other infections. Two lipoprotein system gene variants are associated with survival to advanced ages, apoC3 and CETP. All three genes influence blood cholesterol responses to diet. IL-6 alleles influence vascular disease and possibly survival to advanced age. The main gene candidates for longevity thus are lipoprotein and inflammatory system genes, which are strongly associated with vascular disease. Although the heritability may be minor in the individual variations, careful analysis of gene x environment interactions will be fruitful in identifying potential interventions, as precedented by responses to diet in the lipoprotein gene variants.
Animal model studies, however, have focused on a different set of genes, those connected with growth and metabolism. Mutants causing decreased insulin-IGF-1 signaling in the worm, fly, and mouse are long-lived and often accumulate fat. In humans, impaired insulin signaling (insulin insensitivity) is characteristic of diabetes, which increases mortality in association with vascular disease. Small size is not consistently associated with longevity and may increase vascular mortality. Historical improvements in adult life span are strongly associated with faster growth during childhood and larger adult height (Chapter 2). Future studies may develop further links of the insulin-IGF-1 model mutants to vascular disease.
Genetic influences on life span are now clearly seen in a physiological context that involves nutrition, hormones, and especially, interactions with pathogens in my view. A remarkable convergence and overlap of gene systems is emerging. Interactions of p53 and sirtuins with insulin signaling in responses to pathogens may be anticipated. Neuroendocrine seats of regulation of these complex interactions are also recognized by the clinical and experimental communities, in the importance of hypothalamic regulation of eating behavior and spontaneous activity, and in the neuronal expression of mutant genes that influence longevity of flies and worms. These recent findings support the concept that neuroendocrine systems can be pacemakers of aging (Dilman, 1971; Finch, 1973a; Finch and Rose, 1995; Finch and Ruvkun, 2001).
The longevity of the insulin system mutants may depend on a highly protected environment; e.g., hypopituitary humans and mice may have higher mortality from infections. Insulin resistance, as modeled in worms and fly mutants, also decreases resistance to stress and infectious disease in humans. Long-lived worm mutants with impaired insulin-like signaling were out-competed by non-mutants in two studies. We must consider the possibility that the long-lived mutants are a ‘hot-house’ phenomenon with genotypes that may not enhance longevity in dirtier and more complex ‘real’ environments. The next, and last, chapter considers the dirty and invasive environments within which humans evolved and which may be worsening through global warming and urbanization.