Nutrition and Infection in the Developmental Influences on Aging
Publisher Summary
This chapter considers developmental influences of infections, inflammation, and nutrition on aging and adult diseases. Birth size, overly small or excessively large, can adversely affect later health through complex pathways. Developmental influences attributed to maternal malnutrition in the Barker hypothesis are extended here to infections. Fogel’s emphasis on malnutrition as a factor in poor health can also be extended to include consequences of infection and inflammation. This chapter argues that infection and inflammation compromise fetal development by diverting maternal nutrients to host defense, with consequences to development that influence adult health and longevity. Low- and high-birthweight babies show higher vascular risk factors in the metabolic syndrome as adults. High birthweight (macrosomia) is common in obese and diabetic mothers. Malaria is prevalent in low birthweight, infecting the placenta, while diarrheas cause postnatal growth retardation, and the child suffers from chronic gastroenteritis, with endotoxin leaking into the circulation. The presence of endotoxin and IgG antibodies suggests useful markers for other populations in association with vascular disease. Nutritional supplements of malnourished populations improve pre- and postnatal growth. Breast cancer is increased (up to 250%) and the age of menopause decreased (up to 1.8 year) in those exposed postnatally to famine. Many factors at work during development can modify outcomes of chronic aging conditions.
4.1 INTRODUCTION
Developmental exposure to infections and poor nutrition can profoundly influence the outcomes of aging. The ‘Fetal Origins’ theory of Barker and colleagues, discussed below, has focused on links of adult metabolic and vascular disease to nutrition during development. In addition, the importance of infection and inflammation during development to later outcomes of aging is argued in this chapter. Inflammatory processes are prominent in arterial disease, Alzheimer disease, cancer, diabetes and obesity, and osteoporosis (Chapter 1). The Finch-Crimmins hypothesis proposes direct links of these chronic conditions to early inflammatory exposure (Finch and Crimmins, 2004, 2005; Crimmins and Finch, 2006a,b).
Aging begins during development in three concrete ways: (I) The adult capacity for somatic repair and regeneration is set during development. The extent of molecular and cell turnover (‘rejuvenation’) in each organ is controlled by the gene regulatory programs installed during differentiation (Chapter 1). Cell replacement varies widely. In some systems, cells are continually replaced (erythrocytes, hepatocytes, macrophages). However, there is little to no replacement of some cells (neurons, naive T cells, oocytes) and of some cell organizations (kidney nephrons, eye lens). Molecular replacement also is set during development through cell-specific patterns of gene expression. Arterial elastin and lens crystalins accumulate progressive damage during adult life because these molecules are made only during development and not replaced during adult life. Because of their molecular long life spans, these molecules progressively accumulate oxidative bystander damage, e.g., aortic elastin (Fig. 1.6D). (II) Fetal arteries have microscopic foci of macrophages and oxidized LDL that are early stages of atheromas (Fig. 1.6A, Fig. 1.15). Even before birth, synergies of oxidative stress and inflammation are at work. Maternal cholesterolemia can accelerate the postnatal accumulation of arterial lipids (Section 4.8). (III) Organ development is sensitive to environmental factors acting pre- and postnatally, including nutrition, infections, inflammation, and other stressors. Each organ system has critical periods during development when environmental factors have greatest impact on cell number and homeostatic set points. This chapter addresses the complex interactions of nutrition with infections during development that can alter outcomes of aging, and extends the themes of prior chapters.
Chapter 3 discussed evidence that diet restriction during adult life improves adult health, especially in sedentary lifestyles. The efficacy of drug interventions for vascular disease may involve anti-inflammatory effects, as well as the anti-coagulant and hypocholesterolemic activities (Chapter 2). The role of infections in stimulating arteriosclerosis through inflammatory processes is plausible, though not universally accepted. Inflammatory processes may be attenuated by diet restriction, particularly in obesity. As discussed in this chapter, maternal undernutrition is considered a cause of small birthweight; subsequent overnutrition that leads to catch-up growth with earlier onset obesity increases the risk of high blood pressure and diabetes. In turn, maternal obesity increases the risk of diabetes and obesity in the subsequent generation. The current theory of fetal origins of adult disease has focused on nutritional influences.
However, infections and inflammation may also have major roles. Infections at any age can cause energy deficits equivalent to malnutrition. Early infections can profoundly influence adult mortality from infections through immune pathways and accelerated immunosenescence (Fig. 1.2A). And, resistance to infections may be diminished by undernutrition, but also by ‘overnutrition’ that leads to hyperglycemia (Chapters 2 and 3). Atmospheric inflammogens also need consideration, e.g., maternal smoking also decreases birthweight and, independently of birthweight, increases the risk of childhood obesity (Section 4.5.3). These complex interactions are best understood from an ecological perspective.
This chapter begins with a historical synopsis of the Fetal Origins theory but does not attempt to give full details, which are hotly debated. The broad theory that adult diseases have fetal origins (developmental influences) is widely recognized through the leadership of David Barker, Clive Osmund, and many other colleagues in the past two decades. Review of those seminal papers shows a strong role of infections that, though well documented, is not widely known. Although no one denies developmental influences on chronic diseases of aging, the early influences are diverse and extend beyond low birthweight, the current focus of many studies.
The role of birthweight and adult height in adult health and longevity is then discussed, with a focus on nutrition and infections. Twins are an important example: Despite their low birthweight, adult twins have normal adult health and life expectancy. Other factors are at work in the associations of low singleton birthweights with later growth and mortality. Revisiting the classic Barker-Osmund studies shows influences of infection on later diseases. Many other studies clearly show the impact of maternal infections and early life infections on later adult health and mortality. Immune hyperstimulation during development, as well as in adult life, appears to deplete naive T cells used in instructive immunity and limit protective responses to new infections (Sections 1.2 and 2.8). Infections may also have an unrecognized role in the effects of pre- and postnatal exposure to malnutrition in Europe during World War II (‘epidemic shadows,’ discussed below). The efficacy of nutritional supplements pre- and postnatally may depend on the infectious load. These examples from unhygienic environments of the 20th century help to understand the remarkable recent increase of human life spans. The old world examples are relevant guides to future changes in the infectious and inflammatory environment, which may be progressively worsening (Chapter 6).
The mechanisms are multifarious in these hugely complex developmental variations. Infections during pregnancy can affect the placenta and fetal nutrient supply. Fetal arteries are also clearly influenced by maternal cholesterol, which can alter the rate of arteriosclerosis postnatally (Section 4.8). Additionally, fetal growth interacts (G x E) with alleles of TNFa and other inflammatory mediators that mediate resistance to infectious pathogens. Lastly, I consider maternal nutrition and infections in terms of fetal-maternal competition and imprinting, returning again to the critical role of the insulin/IGF system that regulates not only metabolism and aging, but also development.
4.2 SYNOPSIS OF THE FETAL ORIGINS THEORY
The concept that adult health is sensitive to environmental influences on development is not new. A good starting place is the remarkable 1934 study of Kermack, McKendrick, and McKinlay of 18th and 19th century cohorts of Britain and Sweden (Kermack et al., 1934). Survivors of early mortality in these birth cohorts retained characteristic mortality rates throughout the life span into old age. Mortality improved in successive generations, again across the life span. Another of their key insights is that these effects are transgenerational, i.e. the improved health of mothers preceded and enhanced the health and physique of their children. The reduction of virulent infections was indicated in the environmental improvements.
After the starvation in World War II, nutrition joined the discussion of environmental effects on long-term health. Many studies showed that growth can be irreversibly attenuated by caloric deficits during critical periods (Widdowson et al, 1964; Widdowson and McCance, 1975) and that puberty can be delayed, but not irreversibly, by disease and nutrition (Tanner, 1962, 1981). Infections can also attenuate postnatal growth of mice (Dubos et al, 1966). Subsequently, there seemed to be few lasting effects, with rapid rebound of health and fecundity after the Dutch Hunger Winter, 1944–1945 (Section 4.7) and in the Minnesota Starvation Experiment (Section 3.2.3).
A major effort was launched after World War II to improve maternal health and early growth by nutritional supplements in impoverished populations (Kramer, 1993; Pelletier, 1994; Ramakrishnan, 2004; Scrimshaw et al, 1968; Scrimshaw, 2003). Malnutrition and infection were recognized to synergize with effects that multiplied their individual contributions to mortality. Moreover, infections can cause malnutrition: by impairing ingestion and reallocating energy (Fig. 1.2B) for host defense, chronic infections or series of acute infections can slow growth. In many studies, nutritional supplements alone had limited benefit. The INCAP studies in Guatemalan villages, for example, provided either supplemental food or excellent medical care, 1959–1964. The nutritional supplement decreased respiratory and diarrheal infections by 70%, but the medical treatments had negligible benefit to preschool children (numerous reports summarized in (Scrimshaw and Guzman, 1995; Scrimshaw, 2003; Schroeder, 1995). At the end, these children’s height and weight did not differ between the villages, nor was the parasitic load changed. It is generally recognized that child health improvements require a full program that fundamentally alters the local environment, including education and hygiene, in addition to vaccination and drugs. Still, the explosive population growth during the Industrial Revolution is attributed to improved food and better distribution in McKeown’s The Modern Rise of Population (McKeown, 1976) and then Fogel and Costa’s Technophysio Revolution (Fogel and Costa, 1997), with less importance given to public health and hygiene.
In the early 1970s, Dorner proposed that metabolic hormonal regulation (ACTH, insulin, GH, TSH) and feeding behavior are epigenetically programmed at critical phases of development, with long-term impact on adult metabolism; papers in German are summarized in (Dorner, 1974; Dorner, 1976; Plagemann, 2005). Based on the recognized effects of maternal diabetes on fetal development, Dorner hypothesized critical phases of development in the hypothalamic metabolic and appetite centers, analogous to the time window of sensitivity to fetal sex steroids during hypothalamic sexual differentiation.
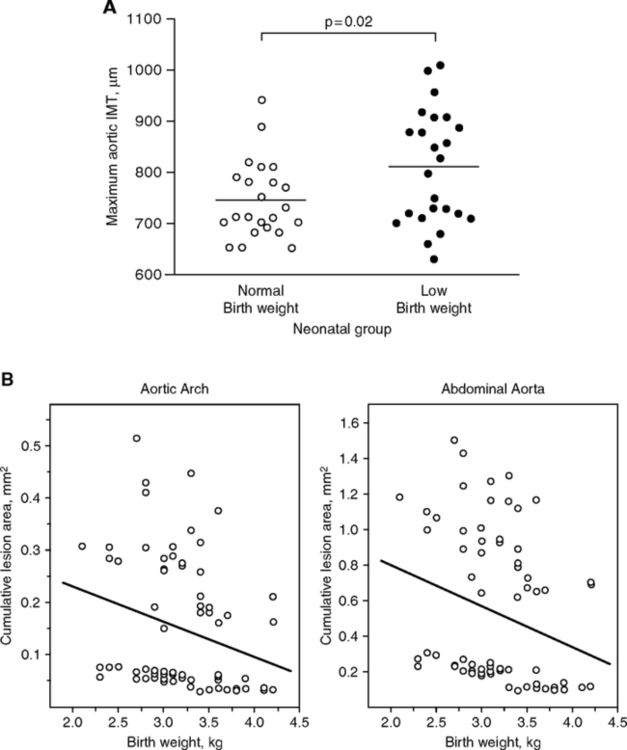
A decade later, Barker hypothesized that maternal malnutrition is the main influence on adult vascular health, through growth retardation leading to small birthweight (Barker and Osmond, 1986; Barker, 2004). This analysis of birth cohorts showed strong correlations between of infant mortality and later cardiovascular disease mortality for the survivors (Fig. 4.1A). Concurrently, Brenner noted links of low birthweight to adult hypertension, which he attributed to impaired kidney development (Brenner et al, 1988). Low birthweight babies have fewer nephron tubules, which places them at greater risk of cumulative damage to these irreplaceable structures. Deficits in functional nephrons can raise blood pressure, which is associated with increased risk of heart attack and stroke (Fig. 1.6C). Then, Hales with Barker further noted the association of diabetes at later ages with lower birthweight and early rapid postnatal growth (Hales et al., 1991; Hales and Barker, 2001). The diabetes was attributed to adult onset insulin insufficiency linked to pancreatic β-cell defects acquired during development.
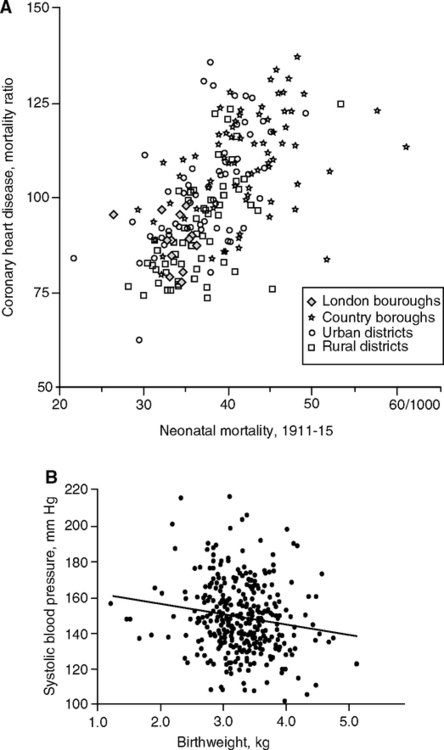
Further, the Barker group found associations of low birthweight with elevated adult systolic pressure (Fig. 4.1B). As observed a follow-up study of low birthweight babies from Sheffield, England, by age 50, systolic pressure was lower by 6 mm for each kg higher birthweight (Martyn et al., 1995). These findings, though controversial, were generally confirmed in a huge meta-analysis: systolic blood pressure varied inversely with birthweight in children, adolescents, and adults, with average effect of 2 mm higher pressure per kg lower birthweight (80 studies, 440,000 Ss worldwide up to age 80) (Huxley et al., 2000). Head size (circumference) at birth, however, was even more strongly associated with adult blood pressure than birthweight. The 2 mm pressure/kg birthweight effect seems modest, relative to the effects of aging on systolic blood pressure, which approximates 5 mm/decade during adult life (Chapter 1, Fig. 1.6B). Other long-lasting effects of the fetal environment may be adaptive responses to food availability that supports catch-up growth. Diabetes at later ages is also associated with lower birthweight and postnatal growth. According to the ‘thrifty gene hypothesis,’ type-2 diabetes is a ‘thrifty genotype rendered detrimental by progress’ (Neel, 1962; Neel, 1999). Rapid secretion of insulin could be adaptive during uncertain food supply, but maladaptive in affluent times when food is reliably available and physical demands are low. Hales and Barker (2001) further considered a ‘thrifty phenotype’ developed by the growth retarded fetus, which was hypothesized to program glucose-insulin metabolism during development in response to maternal malnutrition. Consequent insulin resistance would increase efficiency of fat storage, leading to obesity. Catch-up growth by those small-at-birth is a major co-variate in adult risk of metabolic disorders predisposing to vascular disease (Barker et al., 2005) (Fig. 4.1C). Genetic influences on fetal growth (Section 4.10) could be adaptive.
Another important human complexity came to light in these decades: socioeconomic status. Marmot and colleagues (1978) showed strong SES associations with heart disease, attributable to differences in smoking and sugar consumption (Marmot et al, 1978)1. SES is now well-known for lifelong effects on health (Berkman, 2005; Marmot, 2006).
In the current model of fetal origins of adult disease, development is adjusted to enable ‘predictive adaptive responses’ in anticipation of the postnatal environment (Barker, 2002, 2004; Bateson, 2007; Gluckman and Hanson, 2005; Plagemann, 2005; Vickers et al, 2005; Welles, 2007). Animal models show complex dose responses, in which maternal under- and overnutrition can both predispose to adult obesity and diabetes, depending on postnatal nutrients. Additionally, adult stress responses may also be developmentally modified. This plasticity, or programmability, is restricted to particular developmental stages and is often conditional depending on multiple factors (Vickers et al, 2005). Thus was ‘the Barker hypothesis’ extended to include multifarious influences of the maternal environment during critical or ‘plastic’ phases of development on outcomes of aging (Bateson, 2004; Gluckman, 2004; Plagemann, 2005). This synopsis represents the majority view that nutritional factors are paramount influences on chronic degenerative diseases of aging, principally vascular disease, cancer, and diabetes.
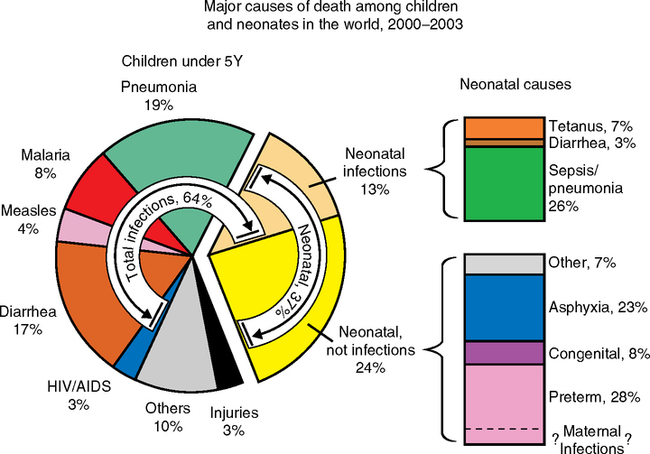
However, nutritional factors do not fully explain the early mortality, which is dominated by infections, as recognized by Kermack et al. (1934) and discussed below (Fig. 4.2). Crimmins and I have proposed a ‘cohort morbidity hypothesis’ to link early and later health through inflammatory processes (Finch and Crimmins, 2004; Crimmins et al 2006a,b). The level of chronic inflammation and infection from early in life influences later life health by interacting with the inflammatory processes in most chronic diseases. We consider that inflammation synergizes with malnutrition in the pathogenesis of adult chronic diseases (Fig. 1.2A). Acting at many levels within the gene regulatory matrix, the infectious load causes reallocation of nutrients, in turn attenuating organ development and growth. These insults may be superimposed on, and reciprocally modify, the developmental programming as studied in ‘clean’ animal models. Inflammatory mechanisms in later disease have not received as much emphasis as nutrition alone in the developmental theory of aging. Their consideration from the beginning could have developed a deeper research agenda that included the synergies of infection and nutrition during development, which were well recognized by the international development community.
4.3 THE BARKER STUDIES OF INFECTIONS AND VASCULAR DISEASE
Barker’s early papers are explicit in the importance of early infections to later age mortality. The initial Barker-Osmond report (Barker and Osmond, 1986), besides its main focus on ischemic heart disease, also analyzed mortality from respiratory infections by cohort and period. The cohort from England and Wales was born 1921–1925 and did not have access to antibiotics until adulthood. Adult deaths from rheumatic heart disease and bronchitis were as strongly associated with infant mortality2 as cardiovascular death (Table 4.1A, reformatted from this report). Infant mortality was attributed to two main causes of similar magnitude: infections (39%) and ‘congenital causes’ (41%). These figures for England-Wales of the mid-1920s approximate the current proportions of death causes worldwide before age 5 (Fig. 4.2), despite the much higher total mortality 80 years before. In the correlations of overall infant mortality with the adult cause of death in this population, adult bronchitis ranked highest, followed by adult stomach cancer, and then of equal weight, adult ischemic- and rheumatic heart disease (Table 4.1B). Most correlations are >0.5.
TABLE 4.1
Infant Mortality and Adult Death by Cause in 20th Century England and Wales
TABLE 4.1 A Causes of Infant Mortality (≤ 1 y) in England and Wales 1921–1925
| Cause of Death | Infant Deaths (Total 291, 082) |
| congenital | 41% |
| bronchitis & pneumonia | 21% |
| diarrhea | 11% |
| other infections | 7% |
| other causes | 20% |
| all infections 39% | |

Calculated from (Barker and Osmond, 1986).
TABLE 4.1B
Correlations of All Infant Mortality with Specified Adult Causes of Death, 35–74 Years
| Cause of Death | Correlation Coefficient, r |
| bronchitis | 0.82 |
| cancers: | |
| cervix | 0.60 |
| lung | 0.46 |
| stomach | 0.79 |
| ischemic heart disease | 0.73 |
| rheumatic heart disease | 0.72 |
| stroke | 0.54 |
From (Barker and Osmond, 1986), TABLE 1 and text.
The infant mortality was also subdivided into deaths during the neonatal (0–27 d) and postneonatal periods (28–364 d) (not shown in Table 4.1). Adult death from ischemic heart disease was equally strongly correlated with neonatal and postneonatal deaths, with deaths from ‘bronchitis and pneumonia’ ranking highest over both periods (all r, 0.68–0.69). Stroke death, however, correlated better with neonatal (r, 0.66) than postneonatal (0.44) mortality. Adult deaths from bronchitis correlated more with postneonatal deaths (0.83) than neonatal deaths (0.58); deaths from ‘bronchitis and pneumonia’ again ranking highest (0.85), followed by diarrhea (0.74). Consistent with the major role of infections, adult death from rheumatic heart disease correlated more strongly with postneonatal deaths (0.72) than neonatal (0.55) deaths; the early causes of death were mainly from bronchitis and pneumonia (0.73).
While poor nutrition in early life has an undeniable role in mortality, these data on a pre-antibiotic cohort also show the importance of infection, to which even well-nourished infants are vulnerable. Among many examples, rheumatic heart disease is due to early streptococcal infection (Chapter 2). Another pathogenic pathway may also be considered (Fig. 1.2A) in which high infectious exposure early in life accelerates immunosenescence by depleting the pool of naive T cells (Section 2.8). The depletion of naive T cells is implicated in increased vulnerability to influenza among elderly (‘immune risk phenotype’) (Huppert et al, 2003; Wikby et al, 2005) (Section 1.2).
Further analysis of these 212 districts of England and Wales (Barker et al, 1989b) strengthened the role of neonatal and postneonatal influences. Adult ischemic heart deaths had independent trends with both neo- and postneonatal mortality. Bronchitis deaths trended mainly with postneonatal, whereas stroke trended mainly with neonatal mortality. Neonatal mortality was attributed to adverse intrauterine environment, which caused lower birth weights. A given example was a hospital with high maternal mortality, and neonatal mortality had average birth weights that were 289 g lower than in other hospitals with low neonatal and maternal mortality. These observations are consistent with infections causing deaths of lying-in mothers and babies, but also, I suggest, with prenatal effects of infection and nutrition on fetal retardation. The early-later age correlations in mortality with infections suggest that district differences in the infectious environment persisted for decades. As a possible mechanism, immune hyperstimulation is recognized to deplete naive T cells and is associated with oligoclonal memory T cells in unstable atheromas (Section 1.5.1) (De Palma et al, 2006).
Soon after, another paper described early body weight as a risk factor for ischemic heart disease (Barker et al, 1989b). In six districts of Hertfordshire among the above 212, the risk of ischemic heart disease in men up to age 70 was inversely correlated with weight at birth and at 365 days (Table 4.1C). Chronic obstructive lung disease, which accounts for 10% of adult deaths, had a similar relationship to birth weight. This report emphasized both pre- and postnatal growth retardation in the risk of later heart disease. Growth during the first year is very sensitive to infections (Fig. 1.2B) (Section 4.4), even with the breast-feeding that most of these men received. I suggest that postnatal growth retardation of these men reflects chronic infections that can accelerate immunosenescence with links to unstable atheromas, as noted above.
TABLE 4.1C
Standardized Mortality Ratios and Weight at Birth and at 1 Year
| Cause of Death (Number of Cases) | |||
| Age/Birth Weight Classes | Ischemic Heart Disease (434) | Chronic Obstructive Lung Disease (43) | All Causes (1186) |
| Birth Weight | |||
| ≤5.5 lb | 104 | 93 | 101 |
| 6 | 77 | 59 | 69 |
| 7 | 90 | 75 | 83 |
| 8 | 85 | 50 | 80 |
| 9 | 62 | 69 | 70 |
| ≥10 | 81 | 33 | 77 |
| 1-year weight | |||
| ≤18 lb | 111 | 129 | 89 |
| 19–20 | 81 | 86 | 89 |
| 21–22 | 98 | 41 | 85 |
| 23–24 | 71 | 61 | 68 |
| 25–26 | 68 | 52 | 73 |
| ≥27 | 42 | 29 | 58 |
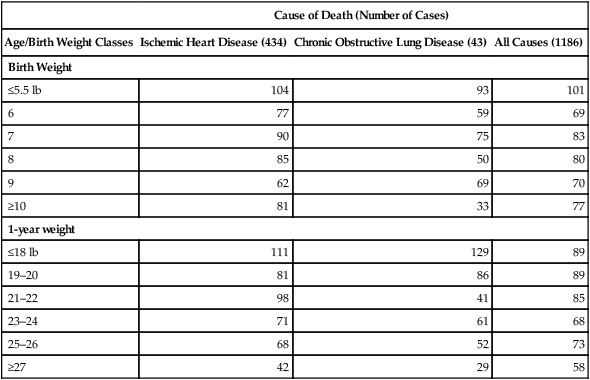
(Barker et al., 1989b), TABLE 1. The inverse trend of size and mortality from ‘all cause’ and ischemic heart disease was significant (P<0.001 and <0.002, respectively). Excluding lung and heart disease eliminated the ‘all cause’ significance. The lowest birth weight <5.5 lb corresponds to WHO standards <2500 g (Section 4.4.3) and comprised 4.4% of this sample.
The next report linked adult height with these same diseases (Barker, 1990). The counties with taller averages had lower mortality of infectious origins from chronic bronchitis and rheumatic heart disease (caused by infections) and from ischemic heart disease and stroke (not immediately linked to infections). The relative mortality risk of men for each cause of death varied by average height in counties, given as the ratio of shortest:tallest height class: bronchitis, 1.8-fold; rheumatic heart disease, 1.4; ischemic heart disease, 1.28; stroke, 1.33. Women showed similar trends. In both sexes, reproductive tract cancers had the opposite trend, with consistently higher incidence in counties with taller average height, an association that has been verified and extended, e.g., for breast cancer.
Infections were recognized for interactions with nutrition from the beginning: “… frequent intercurrent infections [can lead to] impaired nutrition” (Barker and Osmond, 1986, p. 1081). In many districts of Europe before WWII, average health was close to the current developing world in infectious disease. However, the subsequent focus sharpened to nutrition, and maternal infections were not discussed in a review on fetal nutrition and cardiovascular disease (Barker et al, 1993). The malnutrition-infection synergy has not been given much attention as the fetal origins field expanded, but is well recognized in health-poor developing countries where chronic infectious disease is more prevalent than in the health-rich developed countries.
Further findings are emerging from recent Barker-Osmond collaborations with the Helsinki Birth Cohort Study, which has detailed data on parental characteristics and childhood growth (Eriksson, 2005). In the cohort born 1934–1944, birth weights <2500 g predicted threefold higher adult risk for coronary heart disease (CHD) (O.R., 3.63). Slow growth up to 2 y also increased risk of CHD (Barker et al, 2005) (Fig. 4.1C) and stroke (Osmond et al, 2007). Catch-up growth (‘obesity rebound’) after age 2 increased adult risk of diabetes type 2 and CHD. Malnutrition is discussed as a likely factor in growth impairments, because these children and their mothers were exposed to food shortages before and during World War II: about 60% of the families were laborers, and some were known to be malnourished (Eriksson, 2005). Moreover, preceding generations were not well nourished, the average reaching 3000 kcal/d only after 1890 (Heikkinen, 1996). Adult height of Finns increased mostly in the 20th century (Silventoinen et al, 2000), approximating the trend for increased food consumption (Heikkinen, 1996). Nutritional deficits would also have been permissive for infections, a real possibility in this pre-antibiotic cohort which has not been considered in these Helsinki analyses.
These associations of early growth impairments and later vascular disease risk have been confirmed in many studies, but certainly not by all. Major issues beyond the methodological have been raised (Elford et al, 1991; Paneth and Susser, 1995; Paneth and Susser, 1996; Huxley et al, 2000). However, it seems rash to declare losers and winners, and wiser to focus on the emerging complex links between development and aging, and their gene-environment interactions.
Human health at any age is an outcome of multiple contingencies. For example, consider smoking, which all agree is harmful. Yet, most life-long smokers (85%) do not die of lung cancer (Fig. 2.11). Moreover, Jeanne Calment, who holds the record life span of 122, regularly smoked up to age 117 (Allard et al, 1998, p. 73). There may be constitutional factors (genetic and developmental variations, Section 5.2) that protect some smokers from lung cancer or premature mortality. Let’s now broaden the discussion from the focus on maternal nutrition as the main cause of fetal growth retardation to other environmental effects from infections and inflammation: gene-environmental interactions. The next sections consider interactions of infections, inflammation, nutrition, and growth to adult disease and mortality. This evidence further develops the cohort morbidity hypothesis (Finch and Crimmins, 2004; Crimmins and Finch, 2006a) and expands the domain of fetal origins into the real germy world.
4.4 SIZE, HEALTH, AND LONGEVITY
Larger-sized animal species generally live longer, whether feral or domestic. Among mammals, life span broadly scales with adult size according to allometric relationships of adult body mass and length (Calder, 1984; Finch, 1990, pp. 267–271). Yet, women are about 10% shorter than men, but live about 10% longer in most populations. Moreover, the allometry of life span, despite its statistical power, also includes major species deviations (Finch op. cit; Speakman, 2005), e.g., the naked mole rat, 30–80 g, that lives at least 28 y (Section 1.2.6), or even more extreme, the 6 g Brandt’s bat that lives at least 41 y (Podlutsky et al, 2005).3 We must be mindful that the allometry of life span is not derived from any known invariant property of cells or physiology.
Human height and weight vary widely across populations, modern and historical. Little is known about the genetic basis for these variations between populations. However, in all populations, pre- and postnatal development is very sensitive to environmental factors that influence adult height and weight: nutrition and infections. The following synthesis of diverse evidence shows that the levels of nutrition and infection during development are major determinants of adult size, but are also major determinants of adult mortality from infections and vascular disease. I will argue that small size at birth, which is very common in health-poor4 populations, is in response to energy restriction from the maternal load of infection and inflammation, in addition to nutritional deficits (Section 4.9). Responses to energy limitations include lower maternal metabolism during pregnancy and smaller food intake required by the small-sized offspring to reproduce within a shortened life expectancy. Identical twins in health-rich populations contrast starkly with pathological growth retardation: Despite the 900 g birthweight deficits, in healthy circumstances twins have ‘true’ catch-up growth and as adults have normal health and longevity.
4.4.1 Adult Height, Vascular Disease, and Longevity
In most modern populations, taller people live longer (Davey Smith et al, 2000; Davey Smith and Ebrahim, 2002). In the U.S. NHANES I survey (1971–1974), short height predicts mortality, with strongest effects in the range 50–75 y (Sunder, 2005). These relationships are represented in Waaler plots (Waaler, 1984; Fogel and Costa, 1997; Fogel, 2004) defined by contours (isoclines) of mortality rates as a function of height and body mass index (BMI) (Fig. 4.3A) (Sunder, 2005). The optimal BMI for minimum mortality is similar for both sexes. The asymmetry of the height-BMI contours in women indicates a subgroup with different pathophysiology.
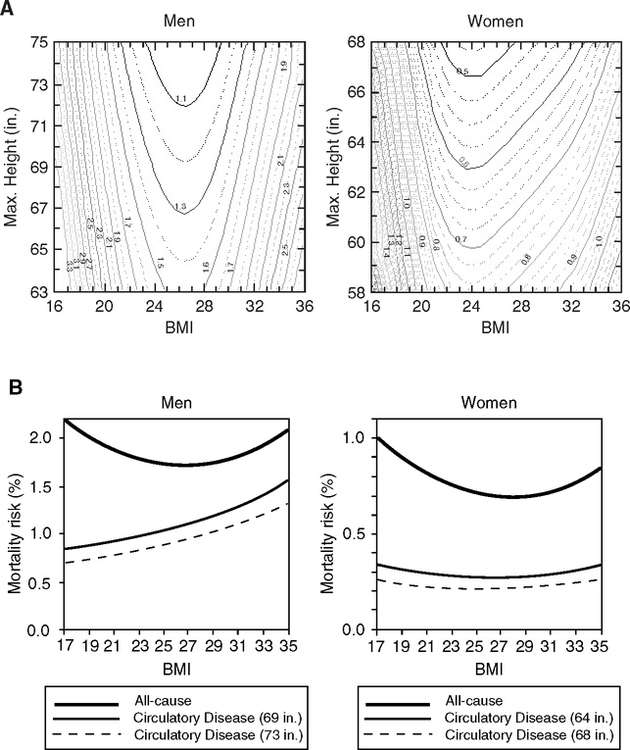
Cardiovascular mortality may be the most strongly correlated with height in men and women (Fig. 4.3B): In NHANES I, each inch of additional height reduced the risk of vascular death in men by 3.5% and in women by 5.25% (Sunder, 2005). The BMI has a small effect on mortality risk from vascular causes, consistent with overall causes of death in the NHANES for BMI of 20 to 30 (Fig. 3.4A, Chapter 3). Similarly, in the Physicians Healthy Study (United States), each inch of additional height reduced the risk of heart disease by 2–3% (Hebert et al, 1993). Most other studies concur (Langenberg et al, 2005; McCarron et al, 2002; Nwasokwa et al, 1997), including studies of twins (Silventoinen et al, 2003).
Dementia, both Alzheimer disease and vascular dementia, also varied inversely with height at midlife (Israeli Ischemic Study, 1,892 men) (Beeri et al, 2005). If replicated, these findings will further the broad overlap of risk factors and mechanisms in cardiovascular, cerebrovascular, and Alzheimer disease (Section 1.7.4).
The lower risks of being tall derive from many factors. First, relative tallness implies minimal growth retardation and better health during development. Other factors may follow from healthy development of a larger size. At birth, the diameter of coronary arteries scale in proportion to the height and weight: Larger arteries show less vulnerability to occlusions (Section 4.4.5). Additionally, glucose tolerance, a vascular risk factor in the metabolic syndrome (Chapter 1, Table 1.8), may be better in the taller (Brown et al, 1991; Leger et al, 1997). Glucose tolerance was impaired in men shorter than controls by 3.5 cm and women shorter by 3.0 cm (adjusted for BMI and age) (Brown et al, 1991). Other causes of death may have opposite relationships to height: Aortic aneurysms (McCarron et al, 2002) and cancer as noted by (Barker, 1990), among other conditions (Gunnell, 1998). Associations of height with different causes of death and life span may be expected to differ across populations because these conditions vary in prevalence.
Socioeconomic status (SES) deeply influences health throughout life, briefly mentioned in Section 4.2. In NHANES I and some other studies, low SES is linked to shortness and higher mortality, particularly from cardiovascular disease (Allebeck and Bergh, 1992; Floud et al, 1990; Marmot et al, 1978; Sunder, 2005). These interactions are broadly consistent with the Fetal Origins theory, because early age mortality and the inflammatory load are also worse in lower SES (Alley et al, 2005; Koster et al, 2006). Even in developed countries, low SES increases exposure to the same adverse influences of infection, inflammation, and poor nutrition, which once prevailed prior to the 20th century in the most developed countries of those times.
In historical cohorts from 18th and 19th century northern Europe, neonatal mortality was inversely correlated with adult height (Crimmins and Finch, 2006a) (Fig. 4.4), while both neonatal and childhood mortality correlated inversely with mortality rates at older ages (Chapter 2, Fig. 2.7B). These relationships extend the hypothesis of Kermack, McKendrick, and McKinlay (1934) (Section 4.2) that successive cohorts in England and Sweden had lower mortality throughout life because of improved health during childhood. They also noted a relationship to height (‘physique’) but provided no analysis. As discussed in Chapters 1 and 2, the reduction of early infections would also reduce the chronic inflammatory load, thereby attenuating the inflammatory processes in vascular disease along with other causes of morbidity during aging that share many inflammatory mechanisms.
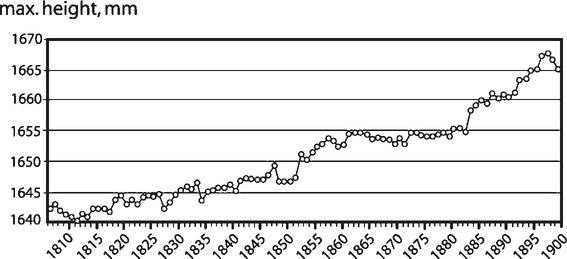
Adult height has increased progressively in most countries since the mid-18th century (Floud et al, 1990; Fogel, 2004; Tanner, 1981). Among many examples, during the past 250 years in Norway, male height increased by 21 cm from an average of 158 cm (62”) in 1761 to the current 179 cm (70.5”) (Waaler, 1984). The progressive increase of height and lowering of old age mortality in the 18th and 19th centuries was interpreted as the shared benefits of lower metabolic costs of infectious disease during the early years (Crimmins and Finch, 2006a). According to our hypothesis, the reduction of infections, together with improved nutrition, would enhance early growth, leading to taller adults. Infections have high energy costs from fever and other host defense responses that re-allocate nutrients at the expense of growth (Fig. 1.2B). Infections account for most mortality before age 2 when the rate of growth is also greatest (Fig. 4.5). The increase of adult height during improving conditions is included in the Fogel-Costa hypothesis of “technophysio evolution” (Fogel and Costa, 1997; Fogel, 2004). The crucial infection-nutrition relationships are discussed later for modern health-poor populations (Section 4.6.5).
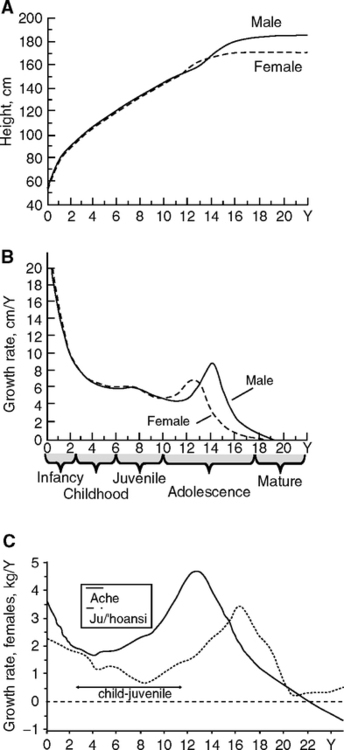
4.4.2 Size at Birth and Adult Height
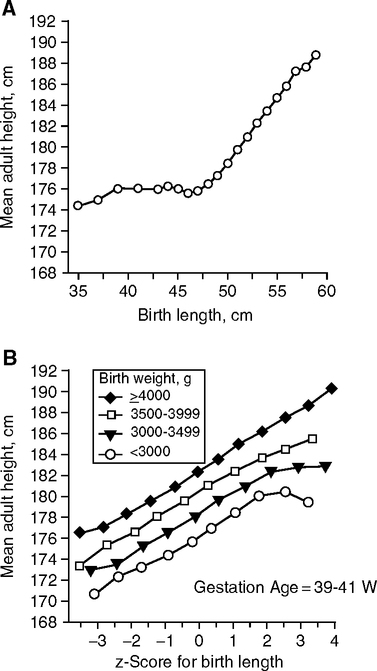
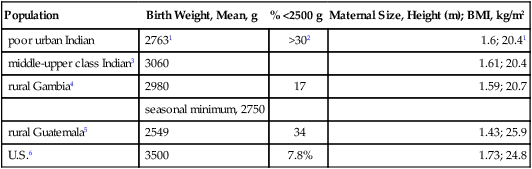
Adult height is strongly influenced by size differences that arise during development. Sex differences emerge after 32 weeks of gestation. By term, boys are 150g heavier than girls on average. This sex difference continues postnatally with 500g differences by 12 m (Thomson et al, 1968). In the largest study of birth size (length and weight) so far, (393,570 Norwegian males 1967––979) (Eide et al, 2005), the majority of births were >45 cm with normal gestation ages of 39–41 weeks; adult height closely followed birth length in each birth weight class (Fig. 4.6A, B). Among those short at birth (<45 cm, 2% of all births), the birth length did not predict adult height (Fig. 4.6A). Birth length accounted for up to 9% of adult height variation. Together, the birth weight and length accounted for 15% of adult height variation. Thus, most (85%) adult height variations were not accounted for by birth size.
Twins are particularly vulnerable to growth retardation from environmental hazards because of their small birth size. Finnish twin cohorts born before 1928 showed lower heritability of height, with progressive increases in heritability approximately in parallel with national economic improvements (Silventoinen et al, 2000). Other studies cited show lower heritability of height in twins from health-poor populations. Currently, Finnish twin adult height has a high heritability of about 90% (GenomeEUtwin cohorts) (Silventoinen et al, 2003a,b). These estimates may be also confounded by multigenerational effects of the environment, by which smaller mothers have smaller babies. The resolution of these gene-environment interactions is complicated by gene imprinting (Section 4.9).
4.4.3 Criteria for Growth Retardation
The focus on birth weight as a predictor of adult health and aging is motivated by two influences on mortality: (I) Small babies are more vulnerable to postnatal infections and higher mortality (Boulet et al, 2006; Karn and Penrose 1952); (II) smaller birth weight from <2500 g into the normative range is associated with adult systolic pressure and with other risk factors for cardiovascular disease included in the metabolic syndrome (Table 3.2). In a large meta-analysis, neonatal systolic pressure was positively associated with birth weight (80 studies, 440,000 Ss worldwide) (Huxley et al., 2000). The coronary risk of adults is increased by catch-up growth. Moreover, heavier babies (macrosomia) are also at risk for becoming obese as adults with metabolic disorders, particularly if their mothers were obese or had diabetes (Section 4.6). Much is still obscure about blood pressure regulation across the life span.
Most birth weights in the developed countries are >3000 g, with <10% below 2500 g—e.g., Norway (Fig. 4.7) (UNICEF, 2004). The criterion of low birthweight as <2500 g (5 lb, 8 oz) was proposed by Arvo Ylppö in 1919 and became widely used (World Health Organization, 2005a). The criterion weight for macrosomia is >4000 g. Not surprisingly, mortality risk curves vary between populations with different norms of birth weight (Rooth, 1980; Rooth and Ericson, 1980). In the developing world, particularly in rural areas and city slums, average birth weights are lower by up to 900 g (Table 4.2). India and Bangladesh are extremes with 30% of births <2500 g. The U.S. incidence of low birth weight in White non-Hispanics was 8.4%; and in Blacks, 4.8% (Hoyert et al, 2006). Birth length follows a similar distribution in Norway (Fig. 4.6B).
TABLE 4.2
Maternal Birth Weight and Size
| Population | Birth Weight, Mean, g | % <2500 g | Maternal Size, Height (m); BMI, kg/m2 |
| poor urban Indian | 27631 | >302 | 1.6; 20.41 |
| middle-upper class Indian3 | 3060 | 1.61; 20.4 | |
| rural Gambia4 | 2980 | 17 | 1.59; 20.7 |
| seasonal minimum, 2750 | |||
| rural Guatemala5 | 2549 | 34 | 1.43; 25.9 |
| U.S.6 | 3500 | 7.8% | 1.73; 24.8 |
1(Stein et al., 1996); Mysore, South India, born 1934–1954 measured at age 47 y.
2(Yajnik et al, 2003; Yajnik, 2004a,b).
3(Piers et al, 1995); Indian women, living in London; non-vegetarian.
4(Ceesay et al., 1997); West Kiang region, 1984–1990.
5(Mata, 1978); Santa Maria Cauque; 1964–1972, Tables 5.2, 6.3, 7.5.
6NHANES III.
Low birth weight arises from two main causes: (I) impaired fetal growth (intrauterine growth retardation) and/or (II) prematurity (preterm delivery, before 37 weeks). The relative importance of growth retardation and prematurity differs by the level of national development and the average income, which determines the quality of the diet, hygiene, housing, health, etc. In health-poor developing countries, retarded fetal growth accounts for most of the low birth weights, and prematurity has a minor contribution. In modern, health-rich countries of Europe and the Americas, low birth weight is <10% and is mainly due to prematurity (Villar and Belizan, 1982). Maternal age also strongly influences the incidence of low birthweight, with a U-shaped curve highest at early and later ages across ethnic groups (Ananth et al, 2004).
There is debate on the choice of threshold for low birthweight because of major differences between populations. Rooth (1980) suggested a new criterion normalized to the distribution as 2 standard deviations below the mean for countries with different mean birth weight. This is equivalent to a z-score of minus 2.
This relative criteria also adjusted for the neonatal mortality, which was less than expected in some countries for the absolute weight class. Implicit in this proposal is the controversial possibility that lower birth weight is an adaptation to local conditions (Section 4.8).
4.4.4 Maternal Metabolism and Fetal Growth
Direct evidence for adaptation of fetal growth to local conditions is the remarkable variation of maternal fat deposition and basal metabolic rates (BMR) during pregnancy (Prentice and Goldberg, 2000). Fat deposition during pregnancy ranged from large increases of maternal weight in health-rich Swedish and Dutch women (>10 kg) to the net losses in rural Gambian women in an extremely ‘health-poor’ population afflicted by malnutrition, malaria, and other infections (Fig. 4.8A). Birthweights ranged correspondingly, from Sweden (3300 g) to rural Gambia (2980 g) (Table 4.3).
TABLE 4.3
Birth Season Effects on Mortality in Gambia
TABLE 4.3 A Mortality
| Age Group | Hungry Season (Wet) | Harvest Season (Dry) | Odds Ratio of Premature Death |
| Birth Weight* | 2808 ± 41 g | 2944 ± 42 g | |
| Neonatal mortality+ 7% total births | 9–10% total births | ||
| 15–35 y# | 8.3% (38/460) | 2.4% (10/415) | 3.65 |
| 36–45 y# | 13.2% (9/68) | 1.4% (1/69) | 10.4 |

*(Prentice et al.,1987); +(Moore et al, 2004a); #(Moore et al, 1997).
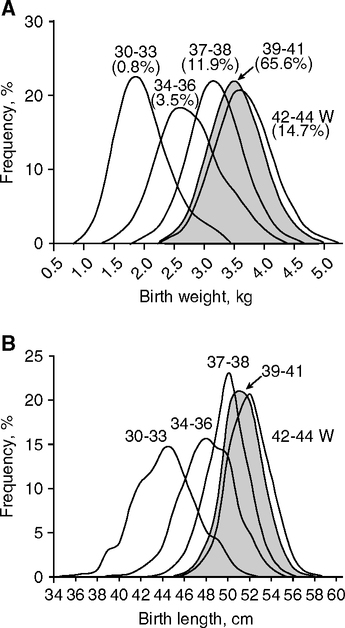
The BMR normatively increases during pregnancy but can decrease in the ‘health-poor’ (Fig. 4.8B). Swedish women have large increases in BMR early in gestation with further increases later when fetal mass accelerates. Their total body weight gain approaches 14 kg, of which 3.5 kg is fat (Lof et al, 2005; Lof and Forsum, 2006; Prentice and Goldberg, 2000). These large net gains in body tissue require net increase of energy (+523 MJ, or about 120,000 kcal) (Prentice and Goldberg, 2000). In great contrast, the rural Gambian women incur lower BMR early in pregnancy to an extent that offsets the later increase in BMR, so that there is a net energy reduction or ‘energy sparing’ during pregnancy (-30MJ, -7000 kcal). The 50% smaller weight gain (6.4 kg average) corresponds to the 300 g lower birthweight and smaller maternal fat gain of <0.5 kg (Lawrence et al, 1987; Poppitt et al, 1993; Poppitt et al, 1994). Food supplements partly corrected the decreased BMR (9 vs. 10). Other populations show intermediates in an evident adaptive continuum. In poor rural Philippine women, the BMR increased throughout gestation, but without increased food intake; nonetheless, 1.3 kg of fat was deposited; reduced physical activity may be the major strategy in this energy sparing. Birth weights averaged 2885 g (Tuazon et al, 1987). Moreover, ‘well-nourished and healthy’ middle- to upper-class Indian women have progressive increases in BMR during pregnancy that are close to the ‘health-rich’ European and English women, with increased BMR and fat deposits (3 kg) and food intake (Piers et al, 1995). Birth weights of 3060 g are 300 g above poor urban Indians (Table 4.2), reflecting better nutrition and probably lower infectious load. Because the placenta consumes 30% of maternal energy in healthy pregnancy (Tycko, 2006), the placental growth and metabolism may also be altered by maternal malnutrition and disease. These findings again point to the crucial role of energy allocation (Fig. 1.2B) in reproductive success. The hierarchy of restricted energy allocation during development is not known in detail at the cellular level in brain or other organs.
Humans appear to differ widely in the distribution of fat at birth. One typology is described in South India (Yajnik, 2004). Urban poor Mysore infants are unusual in two regards: (I) They are among the smallest worldwide, with mean birth weight of 2700 g, just above the criteria for low birth weight. More than 30% have birth weight <2500g. (II) They are born ‘thin but fat.’ Despite small bellies (abdominal circumference), they had normal skin fold thickness (trunk fat) and normal head circumference. Relative to English standards, the Mysore infants had relatively more fat and relatively less muscle. These potentially important differences in fat distribution are not evident from the gross birth weight, nor described by the ponderal index or abdominal circumference5 (Williams et al, 2000). In view of the importance of different anatomical fat depots to adult risk of diabetes and vascular disease (visceral vs. abdominal fat) (Chapter 2), a detailed anthropometric characterization of human fat diversity is urgently needed. Evolved specializations in fat may be found in traditional foraging societies with different patterns of postnatal growth and development (Section 4.9, Fig. 4.5C). The steatopygea of southern African Hottentots is well known to anthropologists, but scarcely studied with modern approaches. Time is running out to define the basic biology of these relict populations.
The total energy cost of pregnancy correlates strongly with adiposity before pregnancy (r, 0.80) and with weight gain during pregnancy (r,0.94) (Prentice and Goldberg, 2000). Around the world, thin women tend to gain less body fat during pregnancy. The extreme example may be the near absence of fat deposits in rural Gambian women. In the conventional view, low birth weight and maternal thinness, particularly in health-poor developing countries, is attributed to undernutrition (‘involuntary’ diet restriction). However, the prevalent infectious diseases also drain energy, as shown by the increase of birth weight after mothers were given anti-malarial drugs (Section 4.5).
Human populations also differ widely in postnatal growth and maturation rates that are postulated to be evolutionary adaptive responses to mortality rates (Walker et al., 2006). Analysis of 22 groups, including foragers, defined trajectories of fast growth relative to the adult size, with earlier menarche and first reproduction (Fig. 4.5C). Fast development was associated with higher post pubertal mortality in subadults, possibly due to poor nutrition and/or infectious load. Yet others had growth rates equaling U.S. standards, but still reaching smaller adult size. Some groups had delayed growth spurts, relative to developed nations, while others did not show a clear adolescent growth spurt. These remarkable variations could be due to local environmental influences of nutrition and infections, as well as genetic influences on growth and menarche, as observed in twins (Section 5.2).
4.4.5 Birth Size and Adult Vascular and Metabolic Disease
The coronary arterial diameter may directly determine the risk of ischemic occlusions. Smaller diameter coronary arteries in adults present more stenotic atherosclerotic lesions. The bottom tertile of coronary diameters had 50–300% more stenotic lesions than the top, varying by the particular coronary artery (sonography of 884 Ss, N.Y.C.) (Nwasokwa et al, 1996). Similarly, in cardiac stent patients, the risk of restenosis varied inversely with vessel diameter in several studies (e.g., Kastrati et al, 2006). These observations concur with a “maxim of interventional cardiology … bigger is better” (Nwasokwa et al, 1996). That is to say, larger coronary arteries have fewer critical stenotic occlusions after transplantation or introduction of stents. Restenosis and primary atherosclerosis share a key mechanism of endothelial smooth muscle cell proliferation.
Slower flow in smaller diameter arteries may be proatherogenic as an outcome of Poiseuille’s law that volume flow varies as the 4th power of the radius. Arterial blood flow velocity and turbulence have major effects on the proliferation of endothelial cells and the expression of inflammatory genes that influence the location of atheromas in different arterial beds (Section 1.5.3.2). This is directly observed in vein transplants in which slower flow causes more hyperplasia in clinical observations and canine models (Dobrin et al, 1989). According to this argument, the higher flow in a smaller diameter coronary at the same anatomic location will be more proatherogenic. Faster flow may also reduce the dwell-time of adherent LDL particles, monocytes, and platelets. Thus, minor developmental differences in arterial diameter could have exponentiated consequences later in life. However, this neat argument may be challenged by sex differences: Women have less clinical vascular disease than men at each adult age, yet their arterial size should be smaller. Data are needed.
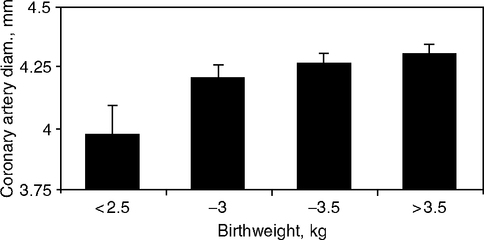
Arterial development may be altered in association with low birth weight. At age 9 years, coronary artery diameters varied in proportion to birth weight (Fig. 4.9) (Jiang et al, 2006). For each standard deviation (SD) unit increase in birth weight, the coronary artery diameter increased 0.1 mm. Information is also emerging on neonatal arteries. Neonatal aortic walls were 9% thicker in birth weights averaging 2713 g relative to controls averaging 3762 g; normalized for weight, the smaller babies had 45% thicker walls (Fig. 4.10) (Skilton et al, 2005). Moreover, fetal lipid aortic deposits (Chapter 1, Fig. 1.15A) vary inversely with birth weight (Fig. 4.10B), (Napoli et al, 1999b). Cord blood HDL cholesterol (vascular protective) varied directly with birth weight, while LDL cholesterol (proatherogenic) varied inversely (birth weight range 2625–4420 g; 480 infants) (Ophir et al, 2004), consistent with associations of low birth weight and prenatal atherosclerotic lesion size. Maternal lipids were not reported. These observations generally support the associations of low birth weight to adult coronary disease. However, low birth weight can arise from diverse causes that each could have different links to adult vascular disease.
Blood lipid and metabolic vascular risk factors are associated with birth weight and the ponderal index, but conclusions are less firm than for blood pressure. Placental cord and adult blood lipids may be more weakly linked to birth weight than adult systolic pressure. Each 1 kg lower birth weight is associated with 2 mg/dL higher total cholesterol. Not surprisingly, no consistent associations of birth weight were found with LDL or HDL cholesterol or triglycerides (meta-analysis of 79 studies) (Huxley et al., 2004). Moreover, identical twins, whose birth weight is typically <2500g at term, have normal lipid values as adults that do not differ from single births, despite the huge catch-up growth (Tuya et al, 2006).
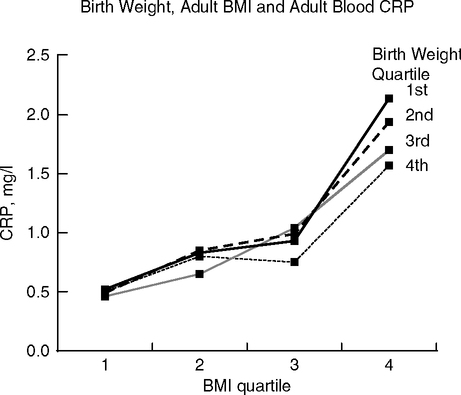
C-reactive protein (CRP), a major vascular risk indicator (Fig. 1.16A), may have stronger associations with birth size than the lipid risk factors. In the MIDSPAN Family Study (Scotland, 30–59 years), the most obese adults showed the strongest inverse correlation with CRP across all birth weights (Fig. 4.11) (Sattar et al, 2004). Each 1 kg increase in birth weight predicts 11% decrease in adult CRP in men and women. The association of low birth weight with elevated risk for vascular and metabolic disease in later life could involve other inflammatory pathways besides CRP.
Adult metabolic disorders have more consistent links. In a systematic review, low birth weight was associated with adult glucose-insulin dysregulation in about 75% of 233 reports (Newsome et al, 2003): fasting glucose (15/25 reports), fasting insulin (20/26), clearance of a glucose load (20/25), and prevalence of diabetes type-2 (13/16). In general, those born small who later grew obese as adults had the highest risk for adult vascular or metabolic disorders (metabolic syndrome). For example, a Swedish cohort born 1920–1924 showed weak inverse correlations between the ponderal index at birth and glucose tolerance at age 50 (r, -0.07); correlations were stronger in the tertile of BMI (r, -0.19) (Lithell et al, 1996). At age 60, the top fifth in ponderal index (fat babies) had 3-fold more diabetes than the others (12% vs. 4%). Adults of small birth weight show decreased insulin sensitivity and impaired insulin release (Levy-Marchal and Czernichow, 2006; Ong, 2006).
Low birth weight is not the only cause of adult metabolic dysfunctions. Larger birth weight is also associated with adult metabolic dysregulation (Kramer, 2004; Newsome et al, 2003; Yajnik, 2004). The varying strengths and directions of these effects across populations imply complex contingencies in the links of birth weight, high or low, to adult metabolism, with U-shaped and other multiphasic interactions.
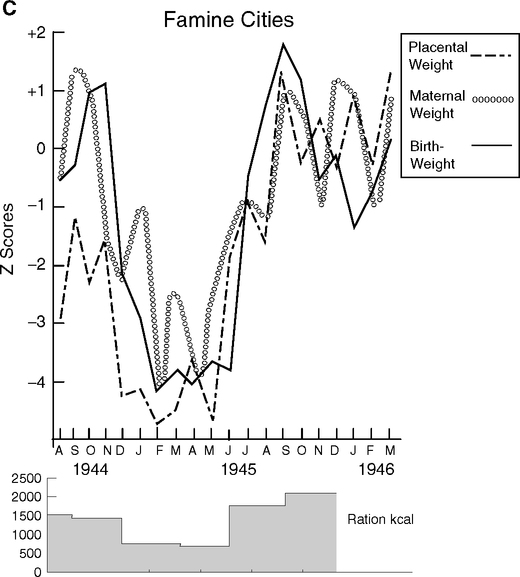
Since catch-up growth in those born small is also a risk factor of adult metabolic disorders, it may be more important as an indicator of future disease than birth weight per se (Barker et al, 2005; Hales and Ozanne, 2003; Levy-Marchal and Czernichow, 2006; Ong, 2006). These effects emerge early. As discussed in Section 4.3 for the Pre-WWII Helsinki cohort, slightly smaller birth size, thinness at age 2, and catch-up growth by age 11 increased the risk of coronary events as adults (Fig. 4.1C) (Barker et al, 2005). In a south Indian population, growth rates at ages 4–8 were strongly associated with insulin resistance at age 8, with the greatest effect seen in children who were small at birth but grew faster (Yajnik, 2004a,b). Experimental animal models support these conclusions (Gluckman, 2005; Hales and Ozanne, 2001; Hales and Ozanne, 2003). For example in rats, 30% diet restriction during pregnancy lowered birthweight by 35% and altered postnatal behavior: If given high-fat diets, the offspring of diet-restricted dams rapidly became obese and inactive (Vickers et al, 2005). Famine exposure gives further insights (Section 4.7).
4.4.6 Twins: Small Size at Birth and Catch-up Growth, but Normal Longevity
Twins are an important exception to the generally adverse effects of low birth weight and catch-up growth. Twin birth weights are typically <2500 g, due to growth retardation of 800–900 g and prematurity by 2–3 wks (Leon, 2001; Phillips et al, 2001; Rosello-Soberon et al, 2005). Monozygous twins have even lower birth weights by 100–200 g and higher perinatal mortality. The twin growth retardation is apparent in the third trimester by about 32 wks (when sex differences in fetal growth are detected), but may begin earlier (studies diverge). Postnatal catch-up growth is strong by 24 m and normal height is reached by age 9 y. Adult twins have normal height and weight distributions. Danish twins did not vary in adult cardiovascular mortality or all-cause mortality from the general population (Christensen et al, 2001). Nor did twins differ in atherogenic profiles of lipids (Tuya et al, 2006) or higher blood pressure (de Geus et al, 2001). Some, but not all, studies found higher diabetes and glucose intolerance in adult twins (reviewed in de Boo and Harding, 2006).
These findings suggest that the prenatal growth retardation experienced by twins has a different biology than in singletons (Leon, 2001; Phillips, 2001). As discussed below, exposure to inflammogens like maternal smoking and infections like malaria cause fetal growth retardation. Another key difference is that the next generation offspring of twins has normal size at birth, whereas women who were small at birth themselves tend to have smaller babies (Ounsted, 1986; Phillips et al, 2001) (Section 4.7). The type of chorion (outer fetal membrane) may be important. But the larger effect remains: The remarkable catch-up growth of twins from 800 g deficits at birth has fewer, if any, of the consequences to adult health than experienced by singleton births with birth weights much closer to the norm (Fig. 4.1C). One may ask how much fetal growth retardation in singleton births is due to subclinical infections or other perturbations of the maternal inflammatory environment.
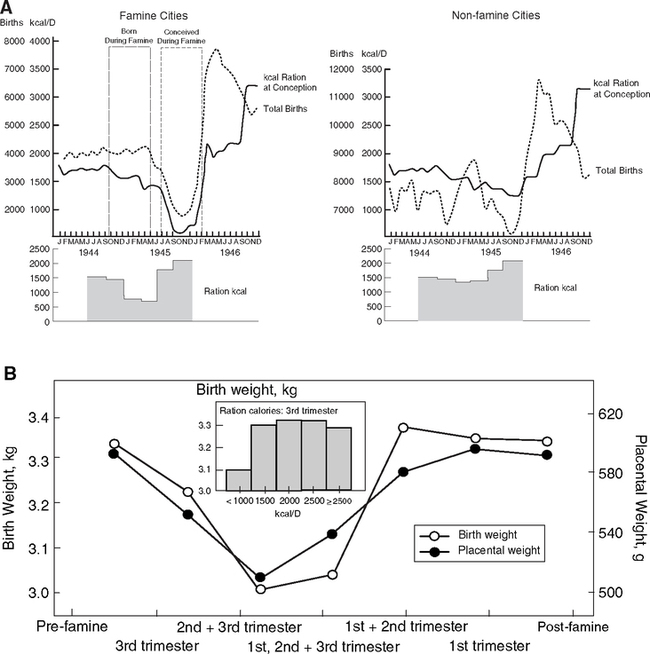
4.5 INFECTION AND UNDERNUTRITION ON BIRTH WEIGHT AND LATER DISEASE
Postnatal growth is highly sensitive to recurrent infections that were nearly unavoidable before the 20th century and are now much diminished in the health-rich developed countries. Infections can attenuate growth by draining energy through fever and anorexia during acute phase responses. Childhood infections are estimated to cost up to 30 g deficits in weight gain per day, if food is not sufficient (McDade, 2003).
4.5.1 The Tangle
The energy available for growth is determined by the balance of input from nutrition and the allocation for host defense and physical activity (Section 1.3, Fig. 1.2B), homeostatic adaptations that prevail throughout development. Maternal infections diminish the nutrients available for the ‘fetal supply line’ and may do so even if food is freely available. Maternal malnutrition, moreover, increases vulnerability to opportunistic infections. Both maternal malnutrition and maternal infections tend to retard fetal growth. At birth, a retarded or prematurely born fetus is more vulnerable to opportunistic infections and has more difficulty nursing. Infections of neonates and children retard growth, even if food is freely available, because of anorexia during acute phase responses. Even well-fed children are vulnerable to infections and growth retardation. Again, malnutrition decreases resistance to many infections. Thus, the analysis of malnutrition in limiting growth is inextricably entangled with the host defenses and synergies with infection and inflammation. Other causes of growth retardation include maternal exposure to smoke and stress.
Interactions of nutrition and infection are recognized as the key to interventions to maternal and children’s health in health-poor populations of the developing world. However, few researchers have recognized their potential importance in the healthier populations being studied for causes of adult disease. I suggest that subclinical infections may have a much larger role. The divergence between twin and singleton births in growth retardation discussed above could be due to subclinical infections or other inflammatory conditions.
4.5.2 Maternal Infections and Nutrition
Maternal infections are well known to cause fetal growth retardation. One of the best understood examples is in rural Gambia. These subsistence farmers are thin and obesity is rare, unlike urban Gambia (van der Sande et al, 2001): average BMI is 20.7, while 20% have BMI <18.5 and are at the margins of survival. There are remarkable seasonal cycles of low birth weight (8% range of variation) and higher mortality (Fig. 4.12A), which are driven by malnutrition and infections (Ceesay et al., 1997; Moore et al., 2001, 2006). A ‘hungry season’ during rains lasting 4–5 months, June-October, is followed by a longer dry season, December-April, with the harvest in April and May. The dry months of December to April have the highest birth weights and fewest premature births. The seasonal minimum of 2750 g birth weight approximates the annual mean in South India (Table 4.2).
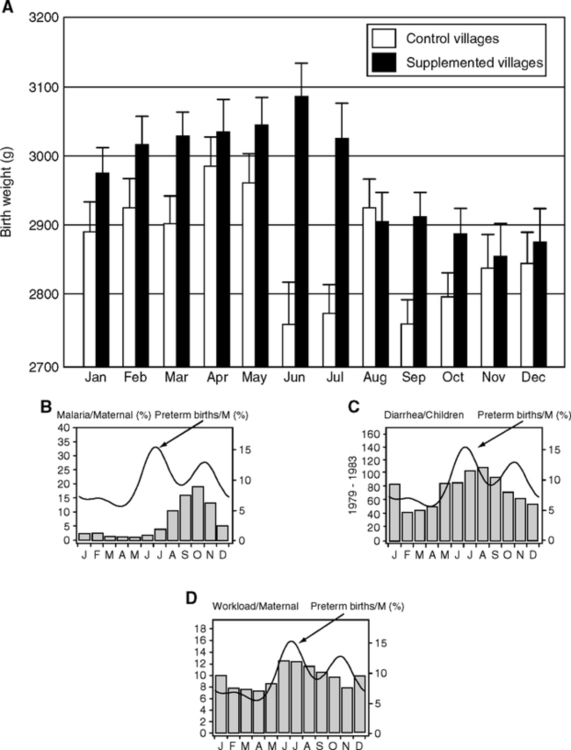
In Gambia during the hungry (wet) season of chronic negative energy balance, pregnant and lactating women lose up to 50% of body fat (-1.2 MJ/d). Women, even if pregnant, are expected to do physically demanding field work during the harvest. The fetal growth retardation develops later in gestation (after 37 wk, 1–3 wk before birth (Prentice et al, 1987). Thus, the retardation may be regarded as an ‘acute’ outcome of an impaired maternal nutrient supply, from maternal nutrient deficits and/or placental transfer toward the end of gestation. In contrast to these large effects on fetal growth and survival, the birth season did not alter body size of young children or adults (from longitudinal data on height, weight, waist-hip ratio, skinfold thickness). Thus, even in this highly stressful environment, catch-up growth eliminates the seasonal growth deficit seen in the birthweight. Nonetheless, childhood growth is slower than the norm. At age 8, children’s leptin levels are 2 ng/ml plasma, about 80% below an Italian comparison group, which is consistent with their thinness and poor nutritional state (Moore et al, 2002). Leptin is important as an immunomodulator (Section 1.3); e.g., leptin injections stimulated phagocytosis in starved mice infected with Streptococcus (Klebsiella pneumoniae) (Mancuso et al, 2006).
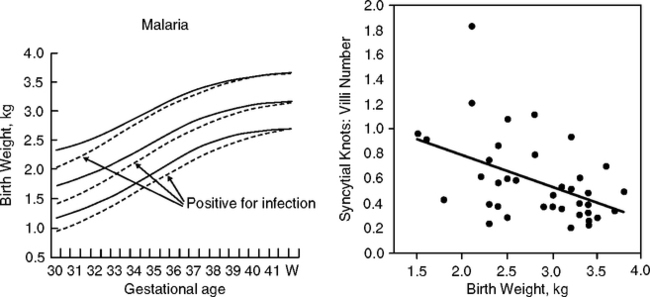
Infections are less rampant during the dry season, particularly diarrhea (Poskitt et al, 1999) and malarial parasitemia (Rayco-Solon et al., 2005). Two phases of premature births and low birth weight can be resolved. Phase I coincides with hard labor during the harvest (Fig. 4.12B, C, D). Phase II coincides with peaks of diarrhea and parasitemia. The relationship of these infections to growth retardation and prematurity is not resolved (the malaria data are from lactating women; diarrhea data, from children). However, some link of infections to fetal growth is likely because malaria directly infects the placenta (Crocker et al, 2004) and retards fetal growth in association with impaired placental blood flow (Fig. 4.13A, B). Helicobacter pylori (Section 2.9.1) is also associated with growth retardation in these rural Gambians (Thomas et al, 2004).
Nutrition and infections synergize in these seasonal effects. Diet supplements increased birth weights (Ceesay et al, 1997; Prentice et al, 1987) in one of the most striking successes of nutritional interventions. In a randomized control trial over 4 y, supplements of protein, fat, iron, and calcium were given midgestation for 82 d. The supplement had most benefits to birth weight and survival during the wet season, but little effect during the dry season when food is more abundant. Women supplemented during wet season pregnancies had larger birth weights by 230 g average (Fig. 4.12A), while the incidence of low birth weight <2500 g decreased 3-fold (23.7% to 7.5%). Mortality also decreased remarkably: stillbirths (-50%) and neonatal deaths (-35%). Moreover, the metabolic rate during pregnancy was increased. As discussed above (Fig. 4.8B), the basal metabolic rate (BMR) decreases early in pregnancy in these Gambian women, a pattern opposite to the increased BMR in well-nourished women. The nutrient supplements partly restored the BMR (Fig. 4.8C, right panel, 9 and 10) (Lawrence et al, 1987b) and increased birth weight about 230 g (Fig. 4.12A) (Ceesay et al, 1997). The birth weight distribution curves are shifted by nutrient supplement uniformly to the right in the wet season, but dry season supplements only improved the lowest weight <2500 g (Fig. 4.12A) (Prentice et al., 1987). The greatest birth weights in these populations are still 300 g or more below the median in healthy populations, e.g., Norway (Fig. 4.7A). This difference suggests a threshold of nutrient balance for fetal growth retardation, also observed in the Dutch Hunger Winter (Section 4.7; Fig. 4.15C, discussed below). Thus, three seasonal factors may synergize in growth retardation: the stress of hard physical work, active infections, and undernutrition. The effectiveness of the supplement did not differ by the level of maternal nutrition or weight-height interactions (Prentice et al., 1987), suggesting the greater role of active infections and physical stress.
4.5.3 Smoking and Aerosols
Smoke and aerosols are unregistered factors in these and most other studies of growth retardation. As described in Section 2.4, aerosols are important major causes of inflammation in adults. Low birth weight from intra-uterine growth retardation is associated with maternal smoking (Bakketeig et al, 1997; Lambers and Clark, 1996; Mitchell et al, 2002; Naeye, 1981), particularly with lower socioeconomic status (Dubois and Girard, 2006). Third trimester maternal smoking had the greatest effect, with 2-fold more low birth weight babies. Smoking dose effects on lowering birth weight are well known (Garn et al, 1978; Lieberman et al, 1994). In a smaller sample, children of maternal smokers were heavier at age 3 y; here, the critical period was the first trimester (Oken et al, 2005). Among many the mechanisms being considered in fetal growth retardation, nicotine crosses the placental barrier and may be 15% higher than in fetal than maternal blood (Lambers and Clark, 1996).
Air pollution may also be a factor in fetal growth retardation (Yang, 2003). In the developing world, urban and rural households are often very smoky, from heating fuel, which can cause ‘hut lung’ and other chronic respiratory conditions (Gold et al, 2000) (Section 2.4). Fecal aerosols from omnipresent domestic animals can induce systemic inflammatory responses, as observed in the elevated blood inflammatory proteins (CRP, complement C3) in sewage and garbage workers (Section 2.4).
4.6 INFECTION AND NUTRITION IN POSTNATAL DEVELOPMENT AND LATER DISEASE
The common childhood affliction of diarrhea is used to show mechanisms in growth retardation (next section) that are at work in the shorter life spans in health-poor populations.
4.6.1 Diarrheas in Growth Retardation
Diarrheas are major causes of childhood growth retardation and mortality in health-poor populations of the developing world (Lutter et al, 1989; Martorell et al, 1975; Poskitt et al, 1999; Rowland et al, 1977; Scrimshaw, 2003; Thapar and Sanderson, 2004). Energy deficits of diarrhea are 400––00 kcal/d (Hoyle et al, 1980; Lutter et al, 1989). Diarrheas cause ‘pathological caloric restriction’ by impairing food digestion and nutrient absorption and draining systemic energy in acute phase responses (Fig. 1.2B).
Infections are the main cause of diarrheas, particularly rotaviruses, and pathogenic forms of E. coli and epidemic cholera (Thapar and Sanderson, 2004). The intestinal flora also shifts in chronic diarrheas, with bacterial invasion of the jejunum (Mata et al, 1972), which is normally sterile (Section 2.4). Diarrhea decreases the intestinal transit time, as well as decreasing intestinal absorption of nutrients by damage to the microvilli and nutrient transporters. Bacterial endotoxins can enter the circulation and can stimulate antibody production (Campbell et al, 2003a,b; Voravuthikunchai et al, 2005), discussed below.
Chronic diarrhea also triggers acute phase responses including elevated CRP and IL-6 (Liu et al, 2005; Yeung et al, 2004) and inducing fever, which independently of diarrhea, can increases basal metabolism 25–100% (Section 1.3.3, Fig. 1.2B). Further compounding the energy drain, the anorexia of fever can reduce food intake by 20% (Butte et al, 1989; Lutter et al, 1989; Martorell et al, 1980). Adding further insult, diarrheas increase the risk of infection, reducing catch-up growth (Guerrant et al, 1992). Gene variants that influence inflammation and lipid metabolism may be important: Children with diarrhea who also carried apoE4 alleles showed better cognitive development (Oria et al, 2005) (Section 5.7.5).
The association of diarrhea with growth retardation is well known in health-poor populations, as shown in four examples. (I) Severe growth retardation (<3 SD below mean) was associated with 2-fold more total days of diarrhea and more frequent and longer episodes (Fontaleza, Brazil) (Guerrant et al, 1992). (II) Loss of growth was proportional to the number of diarrhea days (Bogota, Colombia) (Lutter, 1989; Martorell et al, 1975). (III) Nutritional supplements given to children with diarrhea eliminated growth retardation (Lutter et al, 1989). The total energy deficits from average 78 d of diarrhea before age 3 y (70,200 kcal total) approximate the nutritional supplements that eliminated the growth deficits (109,500 kcal total). However, in children without diarrhea, diet supplements did not improve growth. This finding suggests why diet supplements have variable benefits to growth. Lastly (IV), measles can precipitate diarrhea. Rural Guatemalan children in the 1960s had a high early prevalence of measles by age 2 y, accompanied by acute diarrhea, weight loss, and 5% mortality (Scrimshaw et al, 1966). Diarrhea was more frequent in undernourished children. Nutritional supplements nearly eliminated mortality and growth retardation during measles. Scrimshaw also documents that, in well-nourished populations, measles is rarely fatal and is rarely accompanied by diarrhea. In 19th century England, when malnutrition was common, diarrhea was ‘a usual accompaniment of measles’; this historical reference supports the association of early mortality and adult height in 19th century England (Crimmins and Finch, 2006a).
4.6.2 Seasonal Effects
We return to rural Gambia, where the birth season also has strong influences on adult mortality. As described above, those born in the hungry season had markedly higher seasonal death rates as neonates (first year). The hungry-season mortality bias persists to early adulthood (Table 4.3A) (Moore et al, 1997), with 10-fold higher risk of death after age 15, among which >40% are attributed to infections (Table 4.3B). Mortality from gastroenteritis and measles is more strongly associated with birth season than from malaria and acute respiratory infections (Moore et al, 1999; Moore et al, 2004). So far, the non-infectious causes of death have not differed by season of birth (Table 4.3). However, chronic infections and inflammation can cause cardiomyopathy, e.g., sudden death from clinically silent Chagas’ disease, caused by Trypanosoma cruzi (Baroldi et al, 1997). Thus, some mortality attributed to acute infections could also involve cardiomyopathy from chronic infections.
TABLE 4.3B
Causes of Death Ages After Age 15 Years
| Cause (Total Deaths) | Hungry Season (49) | Harvest Season (12) | All % |
| infections* | 21 | 7 | 46% (28) |
| other, not infections* | 21 | 4 | 43% (25) |
| unknown* | 7 | 1 | 20% (8) |
| chronic degenerative disease+ | 0 | 0 | 0 |
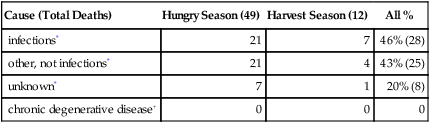
*From data in (Moore et al, 1997). Infections include 3 cases of preeclampsia, which is associated with malaria placenta (C.E. Finch assignment); 1 case of hepatoma, which is regionally locally associated with hepatitis B virus; 1 case of rheumatic heart disease; 1 case of constrictive carditis associated with tuberculosis. Other not infections includes; maternal (excluding preeclampsia), 6 cases; cancer, 2; kidney failure, 3; epilepsy, 3; accidents, 5; miscellaneous, 3. + Statement in (Moore et al, 2004)
The large seasonal mortality bias can be traced to the early months of life when infections are rampant in the hungry season, as discussed above. Most (75%) Gambian children suffer from gastroenteritis (enteropathy), which impairs intestinal transport of nutrients. The mucosa is damaged with villous atrophy and elevations of CD3+ T cells and cytokine-expressing monocytes (TNFa, IFNγ) by several-fold or more (Campbell et al, 2003a). Infant diarrhea is also common (7.3% of time, ages 3––5 m) (Campbell et al, 2003b). After normal postnatal growth up to 8 weeks, growth falls off sharply relative to norms. Moreover, unlike the normal decrease of intestinal permeability during the first year, these children have increased intestinal permeability with microbial leakage, as shown by serum elevations of bacterial endotoxin and IgG endotoxin antibodies. The growth impairments were correlated to varying degrees in each child by plasma endotoxin and IgG levels and gut permeability. Plasma C-reactive protein (CRP) and other acute phase proteins are also elevated in Gambian children, particularly before age 5 (Campbell and Kuo, 2003; Filteau et al, 1995). The increase of CRP during normal aging in health-rich populations might also be due to increased gut leakage of endotoxin during aging, suggested by evidence that aging rats have increased intestinal permeability (Section 2.3). The Gambian enteropathy is hypothesized by (Campbell et al, 2003a) to be acquired from unhygienically prepared weaning foods. Household hygiene is hard, if not impossible, to maintain during the rainy season for obvious reasons. The children failed to respond to nutritional interventions and had impaired digestion of lactose.
More generally, mucosal damage from poor hygiene is implicated as a major global cause of growth impairment and malnutrition. The link between early enteric infections and later vulnerability in infections could be included in Fig 1.2A. The high exposure to enteric antigens suggests a mechanism in the chronic antigenic stimulation of naive T cells, as has been associated with cytomegalovirus (CMV) infections that increase vulnerability to infections later in life in developed countries (Section 2.8). Ongoing studies may show whether diet supplements during pregnancy reduce the seasonal bias of deaths after age 15. So far, birth season in Gambia has not altered adult metabolic or/and vascular disease risk factors (Moore et al, 2001). The low incidence of cardiovascular risk factors in this rural population is consistent with the absence of obesity, the physically demanding lives, and the poor nutrition (van der Sande et al, 2001).
These seasonal effects in the later 20th century give insights into seasonal cycles of mortality that were once more extreme than in health-rich modern populations of Europe and North America. For those born in the early 20th century, spring season cohorts from Northern Europe lived 3–6 m longer than autumn births (Doblhammer and Vaupel, 2001). Australian mortality was also cyclic for the corresponding growth season, with a phase lag of 6 m. Besides seasonal variations in nutrition suggested by these authors, infections also vary seasonally. The higher prevalence of infections in the winter coincides with cyclic availability of bulk nutrients and vitamins to the general public.
Seasonal effects on immune function are proposed in an important expansion of the fetal origins hypothesis to include maternal effects on immune system development that influence adult vulnerability to infections (Collinson et al, 2003; Moore et al, 1999). As discussed above, the original study of Barker and Osmond also showed strong correlations of early and later mortality due to infections (Table 4.1). In Gambia, the neonatal thymus gland is about 10% smaller in the hungry-season. Correspondingly, a longitudinal study in the nearby country of Guinea Bissau showed associations of thymus size with infant mortality due to infectious disease (Aaby et al, 2002). A smaller thymus could cause developmental deficits in the initial endowment of naive T cells, and thus faster immunosenescence (Section 2.9). Undernutrition is considered the main seasonal factor in these African countries, because malarial parasitemia was not common in these women and because the timing of malarial transmission is not concurrent with the effects (Collinson et al, 2003). However, maternal parasitemia may be occult in peripheral blood because the placenta sequesters parasitized erythrocytes (Arbeille et al, 2002) (Section 4.4). Maternal cortisol elevations are probably prevalent during the hungry season, caused by negative energy balance from undernutrition (Section 3.3) and compounded by the nutrient drains from infection.
4.6.3 Serum Immune Response Markers of Chronic Infection in Health-Poor Children
Elevated CRP may be widespread in the developing world. The Tsimane of Amazonian (low-land) Bolivia are being studied because of their rare status as forager-agriculturalists with minimal access to modern medicine. The Tsimane may be as close representatives as still exist in the 21st century to pre-historic forager populations (Godoy et al, 2006; McDade et al, 2005; Gurven et al, in prep.). Their life expectancy is 42 y with infant mortality of 12% (Fig. 4.14A). These children have a high prevalence of elevated CRP—e.g., age 2–4 y, 23% had CRP >5 mg/L (McDade et al., 2005), a level which may be considered a clinical indicator of vascular disease in adults (‘high risk CRP’) (Chapter 1, Fig. 1.16). Tsimane may have elevations of CRP throughout their lives (Fig. 4.14 B). The proportion of 13% aged 2–15 y with high CRP >5m/L is far higher than health-rich populations in developed countries. In the U.S., about 7% of children aged 8–16 y had CRP elevations above a clinical threshold of 0.22 mg/L (NHANES III sample; different CRP cut-offs arise from different assays). Children with high CRP have growth retardation particularly if malnourished (preliminary analysis cited in McDade et al, 2005). Moreover, the prevalence of elevated CRP is several-fold higher than in the U.S. up to age 42 (Tsimane life expectancy) (Gurven et al, in prep.). In effect, Tsimane show 2-fold more years lived with high-risk CRP than in the U.S. up to this age (Fig. 4.14C). Repeat samplings are ongoing to evaluate whether CRP elevations are sustained over one or more years.
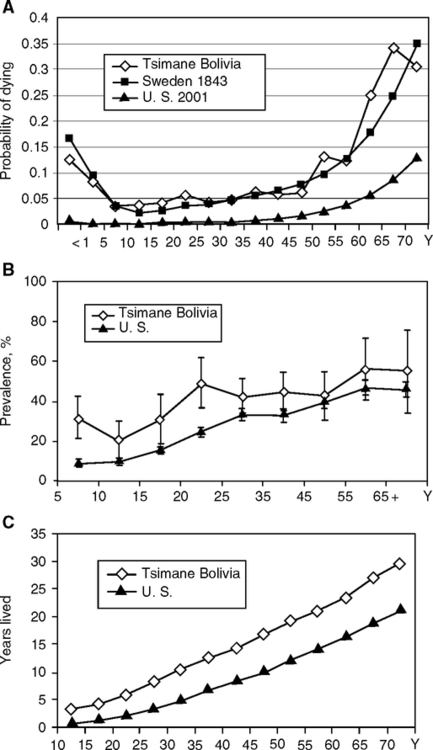
Adult immune responses are compromised by early exposure to unhealthy environments. In rural Filipino adolescents (Cebu Longitudinal Health and Nutrition Survey, CLHNS) (McDade et al, 2001a,b; McDade et al, 2004), successful response to typhoid vaccine was 44% in a group with the healthiest history (normal birth weight, long breast-feeding, rapid growth, higher BMI). In contrast, in those with poorer health by these criteria, only one-third as many (14%) responded to the vaccine. In all of these trait combinations, lower birth weight (<10th percentile for gestational age) was associated with poor later response to immunization. Immunoglobulin E (IgE) levels in adolescents were inverse to the number of childhood infectious episodes, mostly gut and lung (McDade et al, 2004). These findings support the hygiene hypothesis of allergies, which associates the elevation of IgE in atopic immune diseases (asthma, hay fever, eczema) with reduced antigen exposure during childhood (Strachan, 1989). Strachan’s original hypothesis emphasized household density, cross-infections, and hygiene and was expanded to include other infections and inflammogens, pre- and postnatally (Tantisira and Weiss, 2001). Bacterial endotoxin may be protective for atopic diseases, even during adult exposure (Douwes et al, 2004).
4.6.4 Infections During Development
In malarial pregnancies, low maternal weight before pregnancy was the largest factor associated with low birth weights (Kramer, 1987). Maternal malaria, as noted above, retards fetal growth (Fig. 4.13) (Kalanda et al., 2005) and may account for 5% of low birth weights in exposed populations (meta-analysis of 895 studies) (Kramer op. cit.). Maternal malaria also increased the risk of spontaneous abortions, stillbirths and preeclampsia, a systemic inflammatory condition associated with placental pathology (Menendez, 1995). The real prevalence of malarial impairments of fetal growth may be higher than estimated because maternal blood parasitemia can be low or undetectable, as noted previously.
Although maternal transmission of malaria to the fetus is rare, there are many direct effects through maternal support (Beeson and Duffy, 2005; Brabin et al, 2004; Crocker et al, 2004; Duffy and Fried, 2005; Menendez, 1995) shown in four aspects: (I) Maternal anemia from lysis of parasitized maternal erythrocytes causing degrees of hypoxia; (II) maternal hypoglycemia; (III) direct damage to the placenta by adherent parasitized maternal erythrocytes (‘placenta malaria’). The parasitized maternal erythrocytes accumulate in pockets on the placenta and damage the villi that transfer maternal nutrients in association with inflammatory reactions and increased IL-8 and IFNγ, deposits of fibrous proteins, and necrosis (‘syncytiotrophoblast degradation’ and ‘fibrinoid villar necrosis’). Birth weight varied inversely with the number of ‘syncytial knots’ on the placenta (Fig. 4.13B), suggesting graded impairments of nutrient transfer (Arbeille et al, 2002). (IV) The hypoglycemia of pregnancy is worsened by malaria, consistent with the high metabolic demand of fever and host defense responses to parasitemia (Menendez, 1995). Anti-malarial treatment (Maloprim) begun at midgestation increased birth weight by 153 g, confirming a prior study (Menendez et al, 1994). Nutritional supplements alone also remarkably improved birth weights during the malarial season in Gambia (Fig. 4.12A), consistent with the compensating for the nutrient drain of malaria.
TNFa gene variants are associated with susceptibility to malaria in Gambia and other populations, and are also associated with fetal growth retardation and insulin resistance (Casano-Sancho et al, 2006). For example, TNFa promoter alleles, -308G/A, alter susceptibility to malaria (Knight, 2004; Ubalee et al, 2005), hepatitis virus B (Niro, 2005), and septicemia (Nakada et al, 2005; Sipahi et al, 2006). These alleles also influence TNFa synthesis and possibly adiposity, insulin resistance, and hypertension (metabolic syndrome) (Table 3.2). In non-infected pregnancies, these alleles also influence fetal growth (Section 4.9). The genetics is complex, because the TNFa gene resides among other immunoregulatory genes of the highly polymorphic MHC complex (Section 1.3.2).
Although the above low birth weight group (2300 g mean) in rural Gambia was chosen to exclude ‘infectious etiology” (Casano-Sancho et al, 2006), subclinical infections may be elusive. For example, in developed countries periodontal disease is associated with small birth weight, premature delivery, and other adverse outcomes of pregnancy: a meta-analysis showed higher risk of adverse outcomes in 18/25 studies, while 3 clinical trials showed that periodontal treatments lowered the risk of low birth weight by 57% (Xiong et al, 2006). In rodent models, the oral pathogen Campylobacter rectus (gram-negative) induced placental inflammation (Offenbacher et al, 2005). Similarly, systemic LPS injections (Gram-negative endotoxin) induced placental inflammatory responses including elevated amniotic fluid IL-6 and IL-10 (Beloosesky et al, 2006). This study also showed the close link of oxidative stress to inflammation: The anti-oxidant N-acetyl cysteine (NAC) attenuated the IL-6 elevations in the amniotic fluid and maternal serum, whether given before LPS or up to 2 h after.
4.6.5 The Cost of Infections to Postnatal Growth: Evidence from Migration and Antibiotics
Examples just discussed show that infections stunt the growth of children in health-poor populations in direct relation to the intensity and duration of infections. When children from these backgrounds grow up in a better environment, the improvement can be large within one generation. The children of rural Guatemalan immigrants to the U.S. in the 1990s are 8–10 cm taller at ages of 5–12 y, relative to a Guatemalan reference population in 1998 (Bogin, 1999a; Bogin, 2002). About 70% of the total increase is due to leg length, which may be particularly sensitive to infection and malnutrition. Similarly, Ache children (Fig. 4.5C) that were adopted and lived in the U.S. had faster motor development, walking 9 m earlier, and grew 10 cm taller (Hill and Hurtado, 1996, p. 220).
Likely causes of improved development in the U.S. include access to better medical care during pregnancy and early childhood (the W.I.C. Program includes vaccinations and antibiotics; better sanitation and clean water; and nutrition, especially free school breakfast and lunch). Concurrently in Guatemala, the rural villagers have not shown such large increases between generations, although conditions are improving (Mata, 1978; Rivera and Ruel, 1997). In a well-studied group of Guatemalan villages (INCAP Study), birth length was 2 cm less than the U.S. reference and growth up to 3 y was markedly retarded, with infants <12 months showing the greatest impairments (Martorell et al, 1975; Rivera et al, 1997). The gap of 10 cm at age 3 y was almost as large as the adult difference of 13 cm gap below the U.S. mean. Diarrhea consistently impaired growth up to 3 y with a dose response such that each 10% more diarrhea days impaired growth by 0.2–0.7 cm. Respiratory conditions independently reduced growth, but less than diarrhea (Martorell et al, 1975). Children aged 6–7 y experienced 115 d of illness/year. The birth weights and incidence of infectious disease should be analyzed in the U.S. immigrants.
Intrauterine infections have an unknown role in growth retardation. In rural Guatemala and probably many other health-poor settings, intrauterine infections are indicated by elevations of immunoglobulins IgG and IgA, and complement C3 (acute phase protein) in both umbilical cord blood and maternal blood (Lechtig and Mata, 1971a,b, 1972; Mata, 1972). In the absence of chronic maternal infections (tuberculosis, rubella), fetal IgA is very low (Allansmith et al, 1968; Berg, 1969). Elevations of fetal cord blood IgA, IgG, and C3 have been attributed to fetal immune responses to infections; by midpregnancy, the fetus can make C3 and IgM (Berg and Nilsson, 1969; Lechtig and Mata, 1972; Mata, 1972) (Section 4.5).
Even without overt infections, postnatal growth can be retarded by chronic inflammatory conditions. Juvenile diabetes, juvenile rheumatoid arthritis, and uremia share the feature of retarded growth and elevated plasma IL-6 and lower IGF-1 in the absence of GH deficiency. In mice engineered for high systemic IL-6 expression, IL-6 elevations in the absence of infections cause skeletal muscle atrophy soon after maturation (Tsujinaka et al, 1995). One hypothesis is that IL-6 elevations suppress GH signal transduction via the SOCS family proteins (suppressors of cytokine signaling) that modulate GH receptor activation (Lieskovska et al, 2003). Protein synthesis may be impaired by elevated glucocorticoids in maternal stress (Jackman and Kandarian, 2004). Malnutrition, like diet restriction (Fig 3.2A), elevates blood cortisol as an adaptive homeostatic response to increase glucose availability through gluconeogenesis (Fig. 3.10). Diarrhea also increases blood cortisol (Zin Thet et al., 1992), which may be either or both a stress or gluconeogenic response. In a rat model of sterile diarrhea, cardiac muscle protein synthesis was inhibited (Hunter et al, 2001), possibly linking infant diarrhea with later cardiovascular disease (Buck and Simpson, 1982).
Other evidence pointing to the cost of infections comes from antibiotics. The major increase of adult height after the mid 20th century in Japan and other developed countries coincided with mass production of antibiotics (Ternak, 2004). The 10 cm added to height of Japanese men by the 1980s is hard to explain by improved nutrition alone. Liberal use of antibiotics may also be a factor in the obesity epidemic. While there cannot be controlled experiments in humans, antibiotics are widely used for growth promotion in the fowl and swine, with up to 30% greater growth during maturation (Lochmiller and Deerenberg, 2000).
Another model is germ-free animals (rodents, fowl, swine), which usually grow faster (Lochmiller and Deerenberg, 2000). Although germ-free status can eliminate bacteria and parasites, it does not eliminate viruses (Section 2.2.2). Mouse colonies can also differ in postnatal growth rates because of different enteric organisms. Transfer of microbiota at weaning also conferred the donor colony characteristic growth rate (Dubos et al, 1966). The intestinal microbiome is extremely diverse and interacts throughout life with systemic functions (Eckburg et al, 2005; Ley et al, 2006). Commensal bacteria are essential for immune system development, yet cost the host by draining resources that attenuate growth below its maximum. The energetic cost to the organism in the commensal bacterial load can be regarded as a set of trade-offs, which are part of the evolutionarily ‘design’ that allows the immune system to be programmed by ecological information (McDade, 2003).
4.6.6 Unknowns
Much remains unexplained about low birth weight that is not simply accounted for by poor maternal nutrition and infections. Some women have a series of low birth weight babies, despite apparent health. In a huge sample from Norway, women whose first child was <2500 g had 5-fold higher risks of second low birth weight (Bakketeig et al, 1979, 1997). These associations were not explained by maternal smoking or medical complications of pregnancy. Fetal growth was attenuated throughout, possibly more at the end of gestation in those with repeatedly retarded fetal growth than in those with sporadic retardation. Maternal weight and diet were not analyzed.
These examples show the complexity of the nutrition-infection interaction on developmental links to later health and aging. The seasonal variations in nutrition and infections with long-term consequence to adult health and mortality may seem exotic and even irrelevant to researchers on aging processes in much healthier populations in the developed countries. I suggest that they are highly relevant because subclinical infections occur in even the healthiest populations (the prevalence of subclinical infections in pregnancy is not well characterized). The role of maternal infections seems particularly important to further define as a cause of fetal growth retardation. Another major question is the epigenetics and genetics of birth size. Could small size be an evolutionary response to energy limitations or other diet deficiencies that allows an adaptive phenotype? This controversial concept is discussed together with epigenetic imprinting of maternal and paternal genes that influence fetal growth in Sections 4.8 and 4.9.
4.7 FAMINE
Famine exposure in 19th century Finland and Sweden and during World War II is being studied intensively for effects of malnutrition during development on later aging. In addition, there is evidence for epidemics of infectious disease that have not been much considered in these analyses. Overall, the impact of famine and epidemic disease during development on lifespan and chronic diseases of aging seems relatively modest, as summarized in Table 4.4. Up to age 57, prenatal exposure to the Dutch famine has not altered adult mortality (Painter et al., 2005a). Nor has birth weight been found to have a major influence on adult health in the ‘Dutch Hunger Winter’ cohort.
TABLE 4.4
Summary of Main Famine Effects on Adult Disease and Mortality and Link to Birth Weight
| Event (see text for references) | Birth Weight Link |
| Swedish famine, 1809–1849 | no birth weight data |
| 1.2-fold higher death from stroke; no overall change in mortality | |
| Finnish famine, 1866–1868 | no birth weight data |
| no effect on mortality 17–80 y | |
| Dutch Hunger Winter, 1944–1945 | |
| 1. no effect on mortality, age 17–57 | |
| 2. obesity increased (+90%) from exposure in first and second trimesters | |
| 3. coronary disease by age 55 y: 3-fold higher in first trimester exposure. | no |
| 4. blood vascular risk factors LDL:HDL-C in first trimester exposure | no |
| 5. systolic pressure up to age 50 varied inversely with birth weight: 2.6 mm decrease in bp per kg birth weight. | yes |
| 6. adult 50–100% more respiratory disease, any trimester exposure | no |
| 7. mild glucose intolerance at 50 and 58 y; inverse birth weight association. | yes |
| 8. postnatal famine exposure: breast cancer increased 2-fold | |
| Leningrad Siege, 1941–1942 | no birth weight data |
| 1. exposure age 9–15 y: 1.5-fold more hypertension, 1.4-fold more ischemic heart disease, and 1.65-fold more stroke ages 67–74 y | |
| 2. no increased glucose intolerance or dyslipidemia at 52–3 y | |
| 3. no increase of obesity |
4.7.1 World War II (WWII)
The ‘Dutch Hunger Winter’ of November 1944-May 1945 occurred during German reprisals, which imposed severe food deprivation upon the civilian population, including about 40,000 pregnant women with surviving children (Burger et al, 1948; Stein et al, 1975) (Table 4.5A, note 1). By the beginning of WWII, the Dutch were considered well nourished (Bogin, 2005; Stein et al, 1975).
TABLE 4.5A
Caloric Intake during the WWII Dutch Hunger Winter
| Period1 | Rations | |
| May 1940–December 1943 | 1800 kcal/d | |
| German Occupation | ||
| December 1943–October 1944 | gradual decrease from 1800 to 1400 kcal/d | |
| German Reprisals | Famine Zone (west)5 | Control Zone (north) |
| 26 November 1944 | June–August 1944, 1512 kcal/d | 1512 kcal |
| September–November 1944, 1414 | 1450 | |
| December–February 1945, 740 | 1345 | |
| March–May 1945, 670 Amsterdam (400–800) | 1392 | |
| 12 May 1945 (Liberation) | Restoration and Rebound | |
| June–August 1945, 1757 kcal/d | 1755 | |
| September–November, 2083 | 2083 | |
| December–February 1946, 2270 | 2273 | |
| March–April 1946, 3200 | 3200 | |
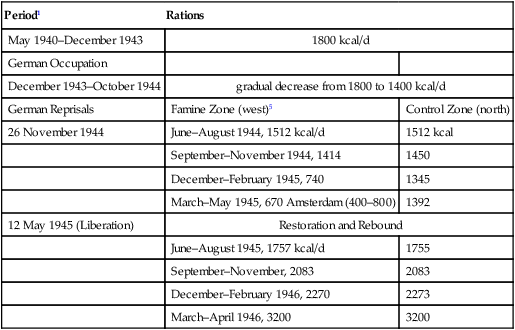
2 Emergency supplements were sometimes officially available to those who had lost >25% of normal body weight (Burger et al., 1948). As the food shortage worsened, the criterion was raised up to 40% weight loss (Burger op. cit., p.21). Special needs were recognized (footnote 5). Additional ‘extra-legal’ food was sought from charitable organizations, foraging trips to the agricultural areas, and on the black market (Burger et al, 1948; Roseboom et al, 2001b; Stein et al, 1975). These supplements may have nearly doubled the official rations, according to a detailed survey made just after liberation (Burger op. cit., pp. 76–77). This ‘extra-legal’ food clearly delayed and blunted the full effects of starvation that would have otherwise been predicted throughout the country by mid-1944, when official rations had decreased from 1800 to 1400 kcal/d. For comparison, most in the Minnesota Starvation Experiment developed visible edema on 1800 kcal/d within 4–6 m (Keys et al, 1950, p. 956; Keys, 1994). However, edema of starvation was not reported until January 1945, 13 m after the official ration fell below 1800 kcal/d (Burger op. cit., p. 20; Stein et al, 1975, p. 45). Not long after, hunger edema was widespread. Thus, the real food intake for most of 1944 must have exceeded the strict 1800 kcal/d in the Minnesota Starvation ration by several hundred calories. Another burden not experienced in Minnesota was the cold interior of homes and workplaces.
3 There were major SES differences during the war and even after liberation; e.g., in Amsterdam in June 1945, the poor class had 55% less milk than the upper class (Burger op. cit., Tables 13 and 14 of Appendix in Vol. II). The poor also had up to 5-fold more diarrhea (Burger, op. cit., Table 6).
4 Average, 3 mo (Roseboom et al, 2001a,b; Stein et al, 1975); Amsterdam, December 1944-May 1945 (Painter et al 2005a).
1The Dutch population was considered well nourished at outbreak of war. During the German occupation after May 1940, official rations were about 1800 kcal/d (7650 kJ), which gradually decreased after December 1943. In 1944, Netherlands was still under German control, except for a liberated zone south of the Rhine. Then in November, the German occupation imposed an embargo on transported goods including food in reprisal for a general strike by railroad workers (‘German Reprisals’). The Western urban area (Amsterdam, Rotterdam) suffered the worst food shortages. In contrast, rations were larger and more supplements were available in the northern and southern agricultural areas and their cities (Enschede, Gronigen), designated for study as the ‘Northern and Southern famine control areas’.
5When possible, children up to 1 y were ‘protected’ by official rations of ≥1000 kcal/d, with composition “always above the Oxford Nutrient Survey” (Painter et al, 2005a; Burger op. cit., p. 22). Pregnant and lactating women and men required to do heavy physical labor were also eligible for official supplements. However, the allotted rations were difficult for most to obtain, and supplements to women were not available during the worst of the famine (Painter et al, 2005a) (Stein et al, 1975, p. 50).
Early in the German occupation before the Hunger Winter, the official ration was 1700–1800 kcal/d, with allowances for pregnancy and nursing (Table 4.5A, note 2) (Fig. 4.15). This ration was about the same or slightly less than in the Minnesota Starvation Experiment, which was sustainable for barely 6 m under much better conditions of temperature (Section 3.2.3; Table 4.5B). The official rations varied regionally, reaching recognized starvation levels (<1500 cal/d) in some urban areas for about 6 months, and diminishing to as low as 400 kcal/d in Amsterdam. However, this average food intake was unevenly available, and total daily caloric intake was estimated at nearly twice the official rations through undocumented ‘extra-legal’ food (Table 4.5A, note 2). Nonetheless, several hundred thousand became emaciated, and malnutrition-related death rates soared, particularly among the elderly. About 10,000 died because of hunger and associated causes (Stein et al, 1975, p. 167). Fuel for heating was scarce and the difficulties of staying warm increased the stress of the food shortages. Hygiene declined for lack of soap and warm water. Infectious disease rose sharply throughout the Netherlands. Skin abscesses and other infections were common. Although mortality of infants and children increased (Table 4.5B), it was attributed to the cold and infections (dysentery, diptheria); with few exceptions, infants were given sufficient food to avoid emaciation (Stein et al, 1975, p. 52, pp. 93–94). Despite the severity of diet restriction, vitamin deficiencies were rare (op cit, p. 77). Thus, the extent of starvation varied widely, particularly affecting the poor who had limited goods to trade for food and less access (Table 4.5A, note 3).
TABLE 4.5B
| Famine District | Control District | |
| stillbirths (cohort) | 20/1000 | 35/1000 |
| <6 d | 15 | 15 |
| 7–29 d | before famine (B): 5–10 | B: 5 |
| during famine (F): 20 | F: 10 | |
| 30–89 d | B: 10 | B: 15 |
| F: 30 | F: 30 | |
| 90–364 d | B: 10 | B: 30 |
| F: 40 | F: 60 |
The higher mortality in the control district may be associated with epidemics that swept southward in 1944–1945 (Stein et al, 1975).
TABLE 4.5C
Comparisons with Other Studies of Starvation and Diet Restriction
| Food Intake, kcal/d | |
| typical Western diet | 2000–3000 |
| South Asian* | 2110 average; 1810, lowest range |
| Minnesota, 1944** | 1800 |
| Biosphere, 1991–1993*** | 1750–2200 |
| Caloric Restriction Society 2004+ | participants, 1112–1958 controls 1976–3537 |
*FAO norms for South Asia (Chandrasekhar and Ghosh, 2003).
**Minnesota Starvation Experiment: (Keys et al, 1950; Keys, 1994).
***Biosphere 2: (Walford et al, 2002); approximate mean values.
+Caloric Restriction Society (Fontana et al 2004; Meyer et al, 2006). See Section 3.2.3.
Zena Stein, Mervyn Susser, and colleagues looked for evidence of impaired mental development from famine exposure by the trimester of pregnancy, which developed criteria for path analysis and cohort data formats that are still widely used. Everyone should read their classic monograph, Famine and Human Development (Stein et al, 1975). The Dutch famine cohort includes 2254 live births, November 1943 through February 1947, with data on birth and placental weight; maternal weight postpartum; stillbirths and mortality up to age 18; and head circumference and body length at birth (Stein et al, 1975; Stein et al, 2004a,b) (this Stein, Aryeh, is Zena’s nephew). Contrary to expectations, there was no increase of mental retardation, or of cognitive impairments in young men exposed to famine before birth or shortly after. However, famine exposure clearly increased developmental defects and psychiatric disorders. These unique data reveal critical periods for energy deficits during human development that influence adult health and reproduction, some effects extending to the next generation.
Few physical effects of prenatal famine exposure during early life have been described in those who survived to adulthood. Height and BMI were normal (Lopuhaa et al, 2000), as was female fecundity (Lumey et al, 1995). However, 90% more obesity was found in young men exposed to famine during trimesters 1 and 2, relative to ‘famine control regions’ (Ravelli et al, 1976). Birth weight does predict systolic pressure up to age 50 (inverse correlation, 2.6 mm decrease/kg) (Roseboom et al, 1999) approximating the global meta-analysis value of 2 mm/kg noted above (Section 4.2) (Huxley et al, 2000). Vascular blood lipid and clotting risk factors increased with first trimester exposure (Painter et al, 2005b; Roseboom et al, 2000). The ratio of LDL:HDL cholesterol was slightly elevated, but not associated with birth weight (Fig. 4.16B). Adult blood pressure, while not related to caloric intake during pregnancy, was sensitive to the protein carbohydrate ratio in the third trimester (Roseboom et al, 2001d). Maternal diet composition had a stronger influence on systolic pressure than birth weight. In the third trimester, each 1% increase in protein: carbohydrate ratio lowered adult systolic pressure 0.5 mm Hg.
Then 30 y later by age 55, first trimester exposure was associated with 3-fold more coronary artery disease (Roseboom et al, 2000) (Fig. 4.16A). As expected, the subgroup of adults with more body fat, higher systolic pressure, and impaired glucose tolerance had more coronary disease. Even so, birth weight, maternal weight, and SES have not shown links to coronary disease in this population. Nor has prenatal famine exposure influenced adult mortality up to age 57, of which 20% deaths are attributed to coronary disease and 40% to cancer (Roseboom et al, 2001; Painter et al, 2005a). However, the success of modern clinical interventions for vascular risk factors must be considered in the apparent lack of famine impact on mortality. The above evidence for increased coronary disease and obesity suffice for a ‘cohort morbidity’ effect of prenatal famine.
Urinary albumin elevations (‘microalbuminuria,’ an index of kidney dysfunction) at age 50 were 1.7-fold more prevalent for midgestation famine exposure, relative to the other trimesters or the non-exposed (12% vs. 7%) (Painter et al., 2005b). Microalbuminuriacs had more electrocardiographic abnormalities (25% vs. 14%), higher systolic blood pressure (135 vs. 125 mm Hg), poorer glucose clearance (40% vs. 16%), and higher glycated hemoglobin (HbA1c, an index of elevated glucose and systemic oxidative stress; Section 1.4.2). At ages 50 and 58 y (repeated testing), none of those exposed in any trimester had abnormal glucose regulation (de Rooij et al, 2006). In the standard oral glucose tolerance test, 120-min glucose levels were below the clinical threshold for pre-diabetes. Nor was there any difference in diagnosed diabetes in relation to famine exposure during pregnancy. Respiratory symptoms (wheeze, cough) and obstructive airways disease were 50–100% more frequent in those from second trimester exposure (Lopuhaa et al, 2000). Here again, birth weight was not a factor.
Birth weight declined definitively in the famine area with the onset of famine, with smaller decreases in the control areas (Fig. 4.15C) (Stein et al, 1975). Famine exposure in the third trimester accounted for 64% of the variance in birth weight. At the most affected times, mean birth weights of 3011 g had fallen by 9% below the postfamine norm. Note that this decrease is still 10% above the 2700 g baseline of South Indian current birth weights discussed above. The diet threshold for smaller birth weight may be 1500–1750 kcal/d (Susser, 1991); again, this may underestimate the real consumption (Table 4.5A, note 2). Later pregnancy was the most susceptible to effects on the birth weight: third trimester (-9%); second trimester (-4%); first trimester, no effect (A. Stein et al, 2004). The protection of fetal weight during maternal malnutrition is remarkable.
Birth weight was also linked to maternal weight changes during pregnancy and decreased when maternal gain was ≤ 0.5 kg/wk (Stein et al, 1995). Statistical resolution of the causal pathways (caloric intake to maternal weight and/or to birth weight) is not easy. Non-famine controls did not show strong links of caloric intake and birth weight. Only below some caloric threshold was the link of caloric intake to maternal weight gain to birth weight significant (Susser, 1991). Smaller birth weight was not due to shortened gestation, nor was gestation length altered. Those in the famine ‘control regions’ exposed to milder diet restriction (Table 4.5A) maintained normal birth weights of about 3400 g. Maternal weight postpartum was most impacted by caloric deficits in the third trimester: Each 100 kcal/d intake predicts maternal weight change of 0.37 kg postpartum, explaining 77% of the variance. Famine exposure in first and second trimesters had little effect on maternal weight.
Head size (circumference), like birth weight, was smaller in third trimester famine exposure (Stein, 1975, p. 104; Stein et al, 2004). Small head size may be a marker for risk of later neurodegeneration. Alzheimer patients showed an excess of small heads above age-matched elderly controls in two studies (Borenstein et al, 2005; Mortimer et al, 2003). The Dutch hunger populations did not show adverse effects of small head size on mental status up to age 18 (Stein et al, 1975).
To summarize effects of birth weight on later health, low birth weight had small effects on glucose tolerance (de Rooij et al, 2006). Although no associations with cardiovascular disease have been detected (Roseboom et al, 2000), later effects may be anticipated because of effects on systolic pressure, which varied inversely with birth weight up to age 50; a 2.6 mm decrease/kg increased birth weight (Roseboom et al, 1999).
Postnatal exposure to famine has shown immediate and delayed effects on reproduction. Reproduction decreased sharply at all ages during the famine (Stein et al, 1975). Menstrual irregularities occurred in 50% of women (Burger et al, 1948, p. 77) and were more common in the poor. At the worst time, births decreased by 65%. Stillbirths and perinatal mortality increased with third trimester exposure. The decline of births conceived during the famine was delayed by 2 m after the reduction of official rations below 1500 kcal/d, both in the famine cities, where the drop was greater, and in the control cities (Fig. 4.15A). This asynchrony could represent the depletion of endogenous body reserves required for fertility, or the depletion of food hoards, or both. A strong SES gradient of impaired fertility (Stein et al, 1975, p. 79) corresponds to the worse health and nutrition in the urban poor. With improved food, fertility immediately recovered, with a transient surge of births in 1946 that doubled the prefamine incidence (Fig. 4.18).
Early exposure to famine impaired adult reproduction in unexpected ways. First, no critical age of exposure was evident for later effects on reproduction. Second, women exposed to severe famine before puberty were 14% less likely to have a first or second child and had more reproductive problems requiring surgically induced menopause (Elias et al, 2005). Third, menopause was slightly earlier, showing ‘dose effects’ in proportion to the level of famine exposure during childhood (Elias et al, 2003). Menopause was 1.83 y earlier in those exposed to famine at ages 2–6 y. Later exposures had progressively less effect on menopause. These effects are opposite in direction to effect of diet restriction (DR) in rodents, which slows ovarian oocyte loss (Chapter 3, Fig. 3.18). However, the mouse exposure to DR was much longer in proportion to the life span (50%). Lastly, and the most curious, is a transgenerational effect: In the next generation from women exposed to famine in the first trimester, the grandchildren’s birth weights are markedly lower in the second and later births (Lumey, 1997), despite the lack of effects of first trimester exposure on birth weight. These and other transgenerational effects are discussed in Section 4.8.
Postnatal famine exposure increased cancer up to 250% in proportion to the severity of famine, with breast cancer accounting for most of the effect (Fig. 4.17A) (Elias et al, 2005; van Noord, 2004). The average age of famine exposure was post pubertal (15–20 y). The increase of breast cancer would not be predicted by the earlier mean age of menopause. Delayed menarche, which was observed throughout the war, particularly in those aged 14, might also be protective (van Noord and Kaaks, 1991). However, other risk factors, IGF-1 and IGF binding protein-3 (IGFBP-3), showed modest elevations in proportion to the severity of famine exposure at ages 2–20 (Elias et al, 2004) (Fig. 4.17B). Elevated IGF-1 and IGFBP-3 may enhance the risk of breast cancer (Chapter 1), particularly after age 50 (Rinaldi et al, 2006). Because diet restriction decreases IGF-1 and IGFBP-3 (Chapter 3), it was hypothesized that the elevations represent a neuroendocrine disturbance. In contrast to breast cancer, colon and prostate cancer showed weak reductions (Dirx et al, 2001; Dirx et al, 2003). van Noord (2004) is correct in viewing the famine exposure and cancer relationship as very different from diet restriction in the laboratory because of the rebound eating and weight gain when food became available.
These cohorts are being followed closely. Adults exposed to famine in the first trimester had 2-fold higher self-reported poor health (Roseboom et al, 2003). In longitudinal studies, self-rated health strongly predicts future longevity (Mossey and Shapiro, 1982) and adds further statistical power to blood risk indicators such as blood albumin and leukocyte levels (Jylha et al, 2006). Vulnerable subgroups may be defined, especially among those with self-rated ‘poor health.’
Besides undernutrition, I suggest infections may also have an impact on future outcomes of aging in the Dutch cohorts. Acute gastrointestinal and respiratory infections are recognized as major causes of civilian deaths during and immediately after the war (Stein et al, 1975, p. 181). Mortality of infants and children from infections increased strikingly during 1944––946 throughout the country (Stein et al, 2004a,b). Stillbirth rates and first-week deaths rose higher than ‘control’ than in the ‘famine areas’ (Stein et al, 1975, p. 152). A rubella epidemic spread country-wide in 1943 and the first half of 1944, and was implicated in a surge of congenital heart disease (op. cit., p. 225). Although this epidemic preceded the full famine, the already diminished food rations may have increased vulnerability to infections. Severe typhus epidemics arose locally during the end of the war, with 3-fold increase in cases in 1944 versus 1941–1943 (Burger et al, 1948; Hemmes, 1945).6 Bacillary dysentery also increased several-fold during the war. Many households kept increased numbers of household rabbits and goats for food, which increased bacillus-carrying flies (op. cit., p.112). Diarrhea was common, 25% across ages and social classes (Burger et al, 1948, p. 77). Diptheria may have caused the most casualties, with 20-fold increase of morbidity 1943–1944 (op. cit., p. 120). Puerperal fever increased 2-fold 1940–1944 (op. cit., p. 127). Tuberculosis death increased up to 3-fold by the winter of 1945 (op. cit., p. 403). After liberation, there was an increase of stillbirths spreading from North to South, “… compatible with the spread of a maternal infection that had greater impact in nutritionally deprived areas (Stein et al., 1975, p. 154; also p. 163). “… Taken together with reports of rampant infections, these [mortality] patterns suggest that debility produced by prenatal stressors predisposed infants to … postnatal infections current at the time of epidemic mortality” (op. cit., pp. 185, 224). The surge of infections during increased food availability recalls the Murrays’ observations in refeeding of starving Africans (Section 3.2.4).
Given the greater privations of the lower social classes, we might expect distinct morbidity subgroups during future aging. The possible impact of these infections on adult health has not been included in subsequent analyses, as far as I can find. One approach might be screening for antibodies to endotoxin, such as found in Gambians with chronic gastroenteritis (Campbell et al, 2003a,b) (Section 4.6.2). Acceterated immunosenescence may be predicted (Section 2.8).
Leningrad (St. Petersburg) experienced an even more terrible famine, 1941–1942 from the German blockade (Siege of Leningrad). Conditions were so dire that almost half of the population died, far more than in the Netherlands (Antonov, 1947; Pavlov, 1965; Stanner et al, 1997). In addition to dire malnutrition, the population was subjected to the stress of ceaseless bombardment and shelling (Antonov, 1947; Bell, 2004), not experienced by the Dutch. During the worst phase November 1941-February 1942, rations averaged 300 kcal/d. This diet was mostly carbohydrate and much less in amount and composition than in the Dutch Hunger Winter (Table 4.5A). Nutrition gradually improved after the siege.
During the first half of 1942, birth weights dropped by 500 g, and half of the live-born weighed <2500 g (Antonov, 1947). Birth weight information by personal recall indicates a 700 g deficit: intrauterine exposure, 2700g, not exposed 3200 g (Stanner and Yudkin, 2001). Nonetheless, 4% of births were >3500 g above the median normal weights, implying that some had privileged access to food during the siege.
Vascular risks are higher in men who were exposed to famine during ages 9–15 (‘around puberty’) (Sparen et al, 2004). Relative to a wartime reference group outside the siege zone, the famine-exposed had 1.56-fold more hypertension (>160 mm Hg), 1.39-fold more ischemic heart disease, and 1.65-fold more stroke in 1999 (ages 67–74; my calculation). However, exposure at earlier or later ages had less or no effect. Effect of famine exposure during trimester of pregnancy has not been reported. Exposure to famine during pregnancy versus infancy did not alter coronary artery disease indicators, glucose intolerance, or dyslipidemia at age 52–53 (Stanner and Yudkin, 2001). Nor did early famine exposure influence height and weight at age 52, relative to those outside Leningrad. Overall, cardiovascular mortality was slightly higher than in controls, not besieged, who experienced milder undernutrition.
Effects of the Leningrad siege and Dutch Hunger Winter are similar in the decrease of birth weight and increase of vascular disease at middle age. However, there are important differences: Leningrad survivors have not shown intrauterine effects on vascular disease, but this may represent the success of medical interventions. Obesity and overweight in adults were not increased from the Leningrad siege. An important gap is the lack of reliable data from Leningrad on birth weight and postnatal growth. In other populations, as discussed above, catch-up growth is linked to adult obesity and vascular disease. Table 4.4 summarizes similarities and differences.
4.7.2 19th Century Famines
Famine was horribly recurrent and widespread in many countries that are now highly developed and well nourished. In 18th century Finland, for example, crop failures occurred about twice a decade, slightly improving by the mid-19th century (Pitkanen, 1993). The ‘Great Finnish Famine of 1866–1868’ was made even worse by a series of intermittently poor crops, beginning in 1857 (the ‘decade of misery’) (op. cit. p. 51), which caused about 150,000 excess deaths. The lower SES landless class suffered the most from starvation and disease (Ikonen, 1990; Pitkanen, 1993).7 About 100,000 left their homes in treks of endless begging (Hakkinen, 1992). Moreover, infections raged throughout Finland, particularly typhus and dysentery (Table 4.6): typhus rose 30-fold above prior levels and was still 5-fold higher the year after (Pitkanen, 1993). In 1865–1866 infant mortality transiently doubled to 400/1000 (Vuorinen, 2006). Nonetheless, these severe health challenges show remarkably little impact on adult mortality. Those born during the famine who survived to age 17 had mean lifespans of about 43 more years (Kannisto et al, 1997). Their mortality rates did not differ up to age 80 from those born 1866–1868, relative to those born 4 years before or after the famine. Kannisto and colleagues (1997) noted that the postfamine conditions in the 19th century were worse than in the 20th century. Nonetheless, there could have been cognitive and physical impairments, as in the 1918–1919 Influenza Pandemic. My comparison of fertility shows the great difference in these postfamine environments. The postfamine fertility (Fig. 4.18) showed much smaller rebound in the Finns (<1.5-fold increase), barely regaining 1861 fertility levels; whereas the Dutch fertility boomed immediately by 4-fold. This huge difference could be caused by poor diet and persistent infections in the rural Finns. The average Finnish diet in the 1860s was 2300 kcal/d, reaching 3000 kcal/d only after 1890 (Heikkinen, 1996).
TABLE 4.6
Deaths from Infectious Disease During the Decade of the Great Finnish Famine (Famine Years in Bold)
| Total Deaths | Typhus | Dysentery | Pulmonary Tuberculosis | Smallpox | Measles (Rubella) | |
| 1862 | 48,639 | 1,437 | 878 | 4,660 | 302 | 2,439 |
| 1863 | 51,556 | 1,574 | 771 | 4,594 | 307 | 4,179 |
| 1864 | 39,914 | 2,038 | 728 | 4,460 | 321 | 852 |
| 1865 | 45,743 | 3,747 | 835 | 4,788 | 3,033 | 1,215 |
| 1866 | 61,894 | 14,151 | 1,229 | 5,253 | 4,264 | 1,327 |
| 1867 | 69,774 | 21,026 | 1,038 | 5,895 | 3,103 | 808 |
| 1868 | 137,720 | 59,582 | 7,855 | 8,048 | 4,159 | 2,322 |
| 1869 | 42,474 | 7,471 | 848 | 4,674 | 712 | 727 |
| 1870 | 31,071 | 2,819 | 615 | 6,758 | 205 | 172 |
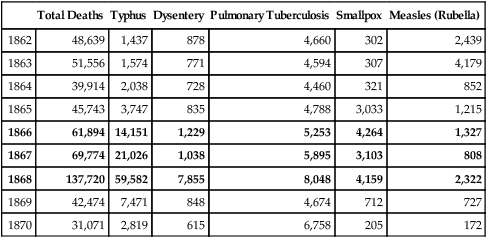
(From Pitkanen, 1993, TABLE 5.1). ‘Typhus’ deaths may be underestimated because some medical officers of the time separately classified deaths from dysentery and other secondary causes of typhus. The old name ‘typhus” represents three diseases, each caused by a different bacterium with common symptoms of fever and headache: louse-borne typhus (‘epidemic typhus’) due to Rickettsia prowazakii was probably spread by desperate and parasite-ridden refugees from the famine zones. However, the endemic typhus (R. felis) is transmitted by mice and fleas. Typhoid fever (typhus abdominalis, enteric fever) is caused by Salmonella bacteria and may be distinguished by abdominal pain and higher fevers.
It is cogent that most Finns were rural farm workers, with endless heavy daily physical work loads. The diet available to most in 19th century Finland, even in good crop years, would be considered uneven in micronutrients and energy and was almost certainly below Dutch norms after WWII (Marja Jylhä, pers. comm.). The rebound physical growth of famine-exposed Dutch infants after the major improvement of food is considered a major factor in the subsequent increase of metabolic disorders, as discussed above. Considering the smaller fertility rebound after the Finnish famine as an index of more modest improvements in a basically stringent diet, it is likely that there was also less rebound growth. The combination of poor diet and the infections might both be synergizing factors in the small fertility rebound.
Another example is from Överkalix parish in Sweden, which suffered intermittent crop failure from 1809–1849 (Bygren et al, 2000). Although exposure to famine during pregnancy did not influence overall mortality up to age 70, sudden death from stroke was 2-fold higher in those exposed to variable food supply during pregnancy (these deaths were 11% of the total). Thus far, stroke incidence has not been reported in the Dutch famine population. There is also evidence for transgenerational effects from alternating famine and feast years (Kaati, 2002): Surfeit of food during the paternal grandfather’s growth period increased his grandchildren’s mortality from cardiovascular and diabetes by 2-fold. Male germ line gene imprinting of IGF-2 is a possible mechanism (Section 4.10).
In sum, episodes of famine (with or without epidemic infections) experienced during development in several northern European populations have not shown major effects on subsequent mortality or life expectancy. Further analysis may show how episodic malnutrition and infection have different impact on arterial disease and immunosenescence than chronic life-long exposures, such as persist in health-poor populations of Africa and Asia (Section 4.6). Survivors of the 1919 Great Influenza Pandemic might also yield valuable information.8 But these 20th century populations can not be compared with the earlier Finnish and Swedish populations discussed above and those in (Finch and Crimmins, 2004; Crimmins and Finch, 2006a,b) because of advances in public health and medicine in the 20th century, and because of the confounds of smoking (Finch and Crimmins, 2005). One general feature seems clear: Early growth impairments from whatever cause, when followed by catch-up growth, increase the risk of adult vascular disease and diabetes. We are just beginning to appreciate the multiple pathways by which development can influence outcomes of aging.
4.8 MATERNAL PHYSIOLOGY, FETAL GROWTH, AND LATER CHRONIC DISEASE
The adverse conditions that retard growth in health-poor populations with shorter life expectancy also give some clues to growth retardation in developed countries. The mechanisms linking the maternal environment to chronic diseases are poorly known and certainly extend far beyond immediate effects on birth weight. The main current focus is on how maternal nutrient physiology alters metabolic setpoints that favor greater postnatal obesity, leading to metabolic syndrome risk factors for vascular disease. Maternal diet restriction and diabetes-obesity have opposite effects on prenatal growth, but appear to converge postnatally to favor catch-up growth and overgrowth, and increasing the risk of the metabolic syndrome (Chapter 3, Table 3.2). Several other aspects of maternal physiology have received less attention: maternal cholesterol and maternal stress; and how any or all of these factors impose growth limitation on fetal structures, including kidney and the arterial bed architecture and elastin. In addition, I argue that maternal infections warrant greater attention because of direct pathogen impact on feto-placental function and because the pathogens themselves and maternal host defense responses compete with the fetus for energy and nutrients. Moreover, tobacco smoke and other environmental inflammogens alter fetal growth through unknown pathways that may interact with others above.
Maternal and fetal blood do not directly mix; gases and nutrients are exchanged across the feto-placental-maternal barrier. Maternal glucose enters the placental circulation, but maternal insulin is excluded. Fetal oxygen is much lower than the maternal, particularly early in gestation when the embryo is highly sensitive to oxidant damage. Many other factors may intervene between maternal and fetal blood nutrients that can alter growth and that can alter fetal development without altering the birth weight (Cetin et al, 2005; Regnault et al, 2005). Fetal growth is controlled by insulin-like growth factors (IGFs) that enhance the uptake of AA and glucose. Fetal IGF production does not depend on pituitary secretion of growth hormone (GH), unlike the postnatal regulation. Fetal blood amino acid (AA) levels exceed those in maternal arteries. Most maternal plasma AA levels decrease after the first trimester, when fetal growth is accelerating, whereas fetal plasma AA levels are normally higher than the maternal. The placenta is highly sensitive to the maternal condition of nutrients, stress, and infections, and is directly attacked by malaria, among other pathogens associated with retarded fetal growth (Section 4.5.2).
Fetal blood levels of amino acids (AAs) may be lower in growth-retarded fetuses. A large sample of growth-retarded fetuses 26–40 weeks (<10th percentile for gestational age; Milan, Italy) had umbilical vein blood AA levels about 30% below maternal arterial levels for most AA (Cetin et al, 1996). Yet, the maternal plasma AA levels in these cases were about 50% higher than in normal pregnancies, confirming (Economides et al, 1989) and others cited by (Cetin et al, 1996). Thus, fetal growth can be retarded despite the evident lack of maternal deficits in AA, which implicates placental dysfunction. Fetal growth retardation has been largely attributed to maternal malnutrition by the Barker group, but pathological conditions may also be suspected, particularly preeclampsia, toxemia, or infections. Preeclampsia is a toxic condition of pregnancy associated with placental inflammation (Jauniaux et al, 2006; Matthiesen et al, 2005), in which some maternal AAs are increased (Lopez-Quesada et al, 2003). Again, some of these AAs are elevated in models of endotoxemia, e.g., acute response of rats to LPS (Hallemeesch et al, 2000). In view of the diverse conditions that can afflict pregnancies and placental functions, particularly in unhealthy environments, it seems rash to attribute growth retardation simply to malnutrition.
Birth size has a strong tendency to be repeated in the next generation for both small and large size extremes (Klebanoff et al, 1997; Ounsted, 1986). For example, 20th century Swedish mothers were more likely to give birth to small babies if the mothers were born small for gestational age (2.68-fold higher) or if the mothers had low BMI (1.24-fold); influences of adult height were not reported (Selling et al, 2006). The maternal line shows a ‘prepotency’ of influences on fetal growth of greater impact than the paternal (Ounsted, 1986). The effects may extend for three generations: In 20th century UK, each 10 cm of the grandmother’s height added 53 gm to her grandchildren’s birth weight (Emanuel et al, 1992). These effects are coming to have major importance because of maternal obesity, which may impose a generalized set of burdens on the fetus with later consequences.
Birth weight increases with maternal weight, with few exceptions (Dietl, 2005; King, 2006; Oken and Gillman, 2003; Sebire et al, 2001). Birth weights over 4000g (macrosomia) increase the risk of elevated BMI of adolescents and, in some studies, increase the risk of type-2 diabetes (King, 2006; Oken and Gillman, 2003). Maternal obesity and diabetes before pregnancy are independent risk factors in macrosomia (Sebire et al, 2001). Fetal macrosomia also increases fetal death and other birth complications (Jolly et al, 2003). Moreover, children of normal birth weight who were exposed to gestational diabetes had an increased risk of obesity and glucose intolerance (Malcolm et al, 2006). On the other hand, growth retardation and low birth weight are lessened in maternal obesity (Sebire et al, 2001).
Obesity increases the risk of gestational diabetes with hyperglycemia to 6––0%, many-fold above the risk in all pregnancies of 1–3%. The normal increase of insulin resistance during the third trimester is greater in obese women (Table 4.7), which may promote a cascade of greater postprandial elevations of glucose and other nutrients that enhance fetal growth and adiposity (King, 2006; Ramsay et al, 2002). Maternal hyperglycemia later in pregnancy increases fetal growth, whereas first trimester (embryonic period) hyperglycemia is associated with slower growth. Gestational diabetes has differential effects on fetal growth, with increased fetal adipocyte formation, inferred from greater abdominal circumference, and impaired bone growth, as indicated by shorter legs (Lampl and Jeanty, 2004) . Gestational diabetes and obesity may increase the risk of diabetes and obesity, which are major risk factors in vascular disease (Boney et al, 2005; Malcolm et al, 2006); e.g., maternal obesity during pregnancy increased the risk of childhood obesity up to 4-fold (Berkowitz et al, 2005).
TABLE 4.7
Lipid and Inflammatory Factors in Obese Pregnancy, Third Trimester; Median Values (Interquartile Range); * Significant Difference
| Lean | Obese | Obese:lean (%) | |
| Body mass index (BMI) | 22.1 (20–24) | 31 (29.1–34) | +140%* |
| Cholesterol, total (mmol/L) | 6.35 (5.87–6.95) | 6.25 (5.6–7.0) | 0 |
| Cholesterol, VLDL (mmol/L) | 0.52 (0.31–0.64) | 0.75 (0.6–1.0) | 30%* |
| Triglycerides (mmol/L) | 2.17 (1.9–2.6) | 2.7 (2.3–3.2) | 120%* |
| Insulin (mU/liter) | 6.15 (4.47–9.5) | 14.2 (11.3–27) | 230%* |
| Leptin (ng/ml) | 23.4 (12.4–30.9) | 56.8 (46.2–65.2) | 240%* |
| CRP (mg/ml) | 2.13 (0.89–3.29) | 4.45 (3.09–6.78) | 210% |
| IL-6 (pg/ml) | 2.1 (1.7–2.8) | 3.15 (2.36–3.59) | 150%* |
(From Ramsay et al, 2002). Healthy, normotensive. Ages: lean, 20–24 y, median 22 y; obese, 25—34 y, median 31 y. BMI, first trimester. Leptin correlated more strongly with fasting insulin (r = 0.74), than with CRP (r = 0.53).
Maternal obesity increases metabolic fuels used by the fetus, particularly glucose. In contrast, maternal diabetes increased certain fetal blood AAs (Cetin et al, 2005) . The increased obesity may be an intensification of the normal increase of circulating nutrients needed for the increased fetal growth later in gestation. Hyperlipidemia also develops (Table 4.7), with elevated VLDL cholesterol, CRP and IL-6, and triglycerides. Gestational diabetes accelerates atherosclerosis (carotid artery thickening) in the mother herself (Tarim et al, 2006). In these cases, VLDL was elevated, while total cholesterol and LDL were not. Some impact on fetal arteries might be expected in view of the greater fetal atherogenesis in maternal hypercholesterolemia (Fig. 4.19, discussed below).
Materno-fetal hyperglycemia and hyperinsulinism are hypothesized to alter the postnatal setpoints for appetite and metabolism in the hypothalamus of the developing brain (Dorner, 1976; Horvath and Brunins, 2006; Plagemann et al, 2006; Ong, 2006). Altered hypothalamic leptin regulation is also implicated in obesity, which is associated with leptin insensitivity (resistance) (Chapter 3). In a rat model of maternal diabetes, fetal glucose was elevated 5-fold (Singh et al, 1997). The elevated maternal insulin did not alter fetal glucose, which is expected because maternal glucose, but not insulin, crosses the placental barrier. Hypothalamic neuropeptide Y (NPY) was lowered in the fetus exposed to maternal hyperglycemia, whereas isolated maternal hyperinsulinemia had no effect. NPY is strongly implicated in neuroendocrine setpoints (Section 3.3.1) and could be a mechanism in fetal programming by maternal glucose.
Inflammation is also at work in obese pregnancies. As King (2006) emphasized, obese women bring higher basal levels of many inflammatory factors to their pregnancy. Table 4.7 compares healthy normotensive obese and normal weight women. In the third trimester, obese women had >2-fold elevation of plasma CRP and leptin, and 50% higher IL-6 (Ramsey et al, 2002). In another study, obese women with elevated IL-6 during pregnancy had fatter babies (Radaelli et al, 2006). Moreover, their neonatal lean body mass varied inversely with maternal insulin-like growth factor binding protein-I (IGFBP-I). Rat models support a direct link of maternal systemic IL-6 to fetal adipogenesis. IL-6 is transported across the placenta into the fetal circulation, whereas IL-1 and TNFa are not (Zaretsky et al, 2004). Thus, the effect of exogenous IL-6 and TNFa to increase fetal adipose depots by ≥ 30% (Dahlgren et al, 2001) could be mediated by different pathways, in which maternal elevations of IL-6 act directly on fetal adipogenesis, whereas elevations of TNFa would be indirect.
Maternal diabetes also markedly increases the risk of infections, as shown in many studies. Group B streptococcus colonization of the vagina was 3.5-fold higher for diabetic women (Ramos et al, 1997). Group B streptococcus is found in 10–30% of women and is a significant cause of neonatal morbidity. Vaginal fungal infections were 4-fold more prevalent in diabetic pregnancies (Nowakowska et al, 2004). These findings are consistent with the higher incidence of infections in diabetics, which may be a direct consequence of elevated glucose (Section 3.2.4).
Preeclampsia, another inflammatory condition associated with obesity, is characterized by hypertension and toxemia (Jauniaux et al, 2006; King, 2006). The placenta becomes oxidatively damaged and releases debris into the maternal circulation, causing systemic inflammatory responses, with elevations of cytokines IL-6 (4-fold) and IL8 (>10-fold) (Jonsson et al, 2006). Lipid changes include increased oxidized LDL and decreases of paroxinase (PON1) (Jauniaux, 2006; Uzun et al, 2005), which are also observed in lipid remodeling during infections (Section 1.5.4). Consistent with this proatherogenic lipid profile, preeclampsia increases subsequent maternal cardiovascular death by about 3-fold (Roberts and Gammill, 2005). Preeclampsia is associated with many inflammatory and infectious conditions, including elevated blood (Qiu et al, 2004); malaria, which infects the placenta (Section 4.4.4, above); cytomegalovirus infections (CMV); and periodontal disease (Jauniaux, 2006).
As a further example, maternal elevations of cholesterol may be directly linked to the earliest stages of atherogenesis in fetal arteries (Section 1.3, Fig. 1.15). In normal pregnancies, maternal plasma cholesterol is modestly elevated, up toward the current clinical criterion of 200 mg/dL (Table 4.8) (Herrera et al, 2006; Napoli et al, 1997; Saarelainen et al, 2006; Woollett, 2005). However, some pregnancies develop moderate to extreme hypercholesterolemia >300mg/dl (the prevalence is not well defined). A unique set of fetal and postnatal arteries from normal and hypercholesterolemic pregnancies in Naples, Italy, was assembled by Napoli, Palinski, and colleagues (Napoli et al, 1997; Napoli et al, 1999a,b; Napoli and Palinski, 2001). Mean birth weights in normal hypercholesterolemic and normal groups were identical, and there was no evidence of malnutrition. Maternal hypercholesterolemia increased the accumulation of fetal and postnatal arterial lipids (Fig. 4.19A, B). Fetal arteries from hypercholesterolemic mothers had bigger fatty streaks and more intimal layer lipid accumulations with oxidized LDL and macrophage/foam cells (Fig. 4.19A). The nascent atheromas apparently grow larger in an environment of maternal hypercholesterolemia and oxidized lipids (see Table 4.8 note for interactions of fetal and maternal cholesterol). Moreover, the children of hypercholesterolemic pregnancies had faster accumulations of arterial lipid (Fig. 4.19B), despite their own normal blood cholesterol. As discussed in Section 4.3, atherosclerotic lesion size was inversely correlated with birth weight in children of normal cholesterol mothers only (Fig. 4.10) (Napoli et al, 1999a). Animal models of maternal hypercholesterolemia also increased fetal atherogenesis (Napoli et al, 2002; Palinski and Napoli, 2002; Yamashita et al, 2006) and increased expression of genes associated with atherogenesis, e.g., SOD3 and FGF binding protein (Napoli et al, 2002). Although it is difficult to accumulate such cases of maternal hypercholesterolemia and fetal postmortem material, high-resolution ultrasound imaging may further develop these findings.
TABLE 4.8
| Hypercholesterolemia | Hypercholesterolemia | ||
| Normal (27% subjects) | Pregnancy Induced (33%) | Pre-existing (40%) | |
| before pregnancy | 155 ± 28, mg/dl | 178 ± 30 | 292 ± 41 |
| pregnancy | 175 ± 20 | 325 ± 44 | 385 ± 50 |
From (Napoli et al, 1997) and (Wulf Palinski, pers. comm.). Women from Naples, Italy (N=82) of similar age (mean 28 y) and dietary and smoking habits. Maternal and fetal cholesterol were significantly correlated (r = 0.37, p<0.02), but correlations were stronger when analyzed by fetal age up to 6 months (r = 0.88, P <0.01; accounting for 77% of the variance vs. 14%). These statistics support the clinical understanding that maternal and fetal cholesterol at birth are not well correlated. In the two hypercholesterolemic groups, plasma lipoxides and cholesterol were strongly correlated, as expected. Also see Fig. 4.19. Maternal blood lipids (but not lipoproteins) are transported across the placental barrier (Herrera et al, 2006). Cholesterol and non-esterified fatty acids are directly transferred to the fetus, while transport of long-chain polyunsaturated fatty acids (LC-PUFA) is mediated by placental receptors. In humans, maternal cholesterol may contribute more to fetal cholesterol during early development.
The impact of maternal cholesterol on fetal arteries may be one of the strongest examples of ‘fetal origins’ hypothesis, but a different version than originally proposed. Maternal obesity may become of great general importance in the developed nations as a transgenerational factor predisposing to obesity and early chronic diseases. The obesity-linked condition of preeclampsia also favors premature maternal cardiovascular disease.
4.9 GROWTH IN ADAPTIVE RESPONSES TO THE ENVIRONMENT
As discussed above, fetal development is highly sensitive to the maternal systemic and uterine environments. Diverse phenotypic variations in arterial, endocrine, and neural systems have multifarious links to adult health. Evidently, many attenuated developmental phenotypes are compatible with subsequent reproductive success. Otherwise, we would not see the large populations of small people whose birth weights can be reasonably inferred to have also been small for innumerable generations. The remarkable variations of maternal metabolism during gestation (Section 4.4.4) also make this clear. On one extreme are Swedish women, with progressive increases of metabolism throughout pregnancy and 14 kg of body weight gain (Fig. 4.8B). Rural Gambian women are at the other extreme in this study, with decreased maternal basal metabolism during most of gestation and smaller body weight gain of 6 kg, also shown in Fig. 4.8. Their ability to maintain pregnancy is remarkable, given the burden of malnutrition, infectious disease, and physically demanding work. Humans may have evolved special homeostatic adaptive mechanisms not known in the great apes to maintain successful pregnancies at this level of duress. Despite the smaller birth size and higher incidence of stillbirths than in developed countries and despite the poor diet and high load of infections, this traditional society shows no sign of reproductive collapse. Similarly, the 2700 g average birth weights in Southern India suggest similar adaptations (metabolic data not reported).
The ergonomics of growth approximates caloric availability in these differing environments and shows the tight margin of this adaptation. The current average daily intake in Southern India is about 2100 kcal/d and the “lowest range of food” is 1810 kcal/d (FAO norms for South Asia). By various estimates, 30–60% of Indians are below the FAO norm (Chandrasekhar and Ghosh, 2003). The 1810––100 kcal/d intakes are close to the margin of energy needed for sustained physical labor. The trade-value (equivalent purchase cost) of these calories as 3 kg wheat matches the smallest wage that Indian day laborers are willing to accept (Seckler et al, 1984; Clarke and Haswell, 1970). The 3 kg wheat at 3150 kcal/kg wheat yields 9450 kcal, which the day laborer would share with his household (average 5.3 dependants), equivalent to 1783 kcal/d per individual. Recall from the Minnesota starvation experiment (Section 3.2.3 and above) that this caloric input is at the level of 1800 kcal/d (Table 4.3B), which caused drastic weight loss and imminent death by starvation within 6 m. Although Indian laborers weigh about 25% less than the Minnesota volunteers at the start of the starvation experiment (52 vs. 69 kg), their caloric input may be just above starvation levels. Nonetheless, this small margin of caloric safety has still enabled sufficient life expectancy and reproduction for rural population growth in the 19th and 20th centuries. There may be other metabolic adaptations, besides those during pregnancy in malnourished women (Section 4.4.4, Fig. 4.8).
Further, Beaton (1989) calculated the energy involved in the greater growth in developed countries. Depositing 1 kg of tissues during 1 y requires 5000 kcal of energy at 5 kcal/g tissue for normal growth (13.5 kcal/d; FAO/WHO). Maintaining this 1 kg of tissue also costs about 5000 kcal/y. Then compare Swedish women, weighing 55 kg, with poor Indian women, weighing 40 kg. The 15 kg body weight difference would require 75,000 additional kcal/y, which is about 10% of the 766,500 kcal annual average intake (average daily intake 2100 kcal/d; FAO norm for South Asia). Thus, a major increase in energy availability would be needed to maintain the Swedish body weight, which could be achieved by reducing the load of infections, as well as increased food.
These variations in growth and metabolism (Gambia vs. Sweden) may be considered alternate pathways within the evolutionary reaction norm of phenotypic variations (Stearns and Koella, 1986; Walker et al, 2006). Others have considered small birth weight as an adaptation to the low nutritional energy available over the life history (Beaton, 1989; Ounsted, 1986; Walker, 2006). In considering small birth size and small adult size as adaptations to the energy available, I do not suggest that these lives should be considered optimal or benign from humanistic or ethical perspectives. These people suffer hunger, infections, and many other stigma and insults of low SES. While the concept of ‘small but healthy’ (Seckler, 1984) is challenged by the reality of malnutrition and many infectious diseases, long-term evolutionary processes may have enabled successful postnatal maturation despite fetal growth retardation. In our pre-history, adult size decreased by 15% after 50,000 years ago (Chapter 6, Fig. 6.7).
We do not know how many generations in improved environments are required to eliminate prior environmental effects. While data are fragmentary, they show fast responses to improvements. As discussed in Section 4.6.5, in only one generation, the children of Guatemalans who immigrated to the United States in the 1990s grew 8–10 cm taller. The birth weight trends for these children are not known, but would be expected to improve. In the second generation of Indian immigrants to the UK, birth weights were 280 g (8%) heavier than the first in 1989 (Dhawan, 1995). Comparisons of two generations of Illinois residents 1950–1990 showed modest increases of birth weight in the second generation (Chike-Obi et al, 1996).
Transgenerational effects of the environment are well recognized in animals. Zoos around the world record the increased size and fecundity of primates in successively healthier generations from feral origins. Rhesus monkeys have been observed for 5 generations after import from India to the Wisconsin Primate Laboratory. Birth weights of females increased by 8% after the third generation (Price and Coe, 1999; Price et al, 2000a). Surprisingly, male birth weights were unchanged in these same lineages, which might be consistent with gene imprinting, as discussed below. Maternal weights were also unchanged. The matrilineal associations of birth weight extend to half-sibs for both small and large birth weight. The age at first birth decreased progressively by 6 m per generation, associated with earlier attaining of adult weight. These changes might be further informed by individual records of health and infectious load.
Effects of food restriction, micronutrient deficiencies, and maternal obesity-diabetes with three or more transgenerational effects are well described in animal models (Aerts and Van Assche, 2006; Campbell and Perkins, 1988; Reusens and Remacle, 2006). In an exemplary study, rats from malnourished pregnancies were cross-fostered on normally fed nurses (Zamenhof et al, 1971a,b). As adults, their brain cell density (DNA content) was lower. The offspring of mating with normally fed controls still had lower brain cell density. Importantly, these effects were transmitted maternally and not found in males from the same litters. Such maternal transmission suggests gene imprinting, as discussed in the next section.
These models test the many hypotheses of fetal origins of adult disease on different organ systems, aptly described as the ‘fetal matrix’ (Gluckman, 2005). The fetal oocyte is another element in this matrix with potential transgenerational environmental effects. Oocytes are fully formed before birth in the fetal ovary, as recently confirmed (Eggan et al, 2006). Thus, the egg from which we stemmed was present in our mother’s ovaries before her birth, exposed to our grandmaternal environment (Finch and Loehlin, 1998).
These reversible variations in fetal growth are matched by the varying schedule of growth and maturation in traditional foragers, e.g., in the timing of the adolescent growth spurt and age at first birth (Walker et al, 2006) (Fig. 4.5C, legend). The progressively earlier age at menarche and greater adult size in the past 5–10 generations in Europe and America are comparable to the differences seen in these foragers (Floud et al, 1990; Tanner, 1962; Tanner, 1981). In the larger national populations, the historical changes are clearly environmentally driven. New mutations influencing growth are unlikely to have major roles in this number of generations. However, there could be different allele frequency distributions of genes that alter growth during these historical changes or between the forager populations with different developmental schedules.
More generally, the plasticity of schedules for growth and reproduction for rapid responses to the environment is seated in the ability of these systems to respond soon after their differentiation. The male and female secondary sex characteristics that emerge at puberty, e.g., pubic hair, can be induced in neonates by sex steroids (Leung et al, 2005). Thus, life history scheduling depends on two main parameters (Finch and Rose, 1995): (I) the developmental acquisition of receptors and other cell machinery that enables response to hormonal and neural signals at the next life history stage; (II) the arrival of the appropriate stimuli. The gonads are able to produce sex steroids far before puberty. The hypothalamic-pituitary axis, in turn, can produce sufficient gonadotrophins to support precocious puberty; or depending on nutrition, stress, and health, puberty may delayed for years. The hypothalamic-pituitary-gonadal axis is also sensitive to metabolic hormones and cytokines, including leptin and insulin and hypothalamic TGF-α and TNFa (Gamba and Pralong, 2006). The system level of regulation allows rapid evolution of the reproductive schedule. If each component had its own genetically controlled clock, change would be very slow (Finch and Rose, 1995). This ‘physiological architecture’ enables the manifested plasticity in the timing and extent of adaptive responses to energy resources.
4.10 GENOMICS OF FETAL GROWTH REGULATION
The variations of growth, pre- and postnatal, are mediated by the genome in two fundamental mechanisms: genetic variations encoded in DNA base sequences and epigenetic variations, which are reversible, including gene imprinting, which alters the expression of the parental genes.
4.10.1 Inherited Genetic Variations
Inherited genes clearly contribute to the tendency of small mothers to have small babies (Knight et al, 2005) but have less influence than environmental factors. In 20th century Sweden, genetics contributed 33–46% of the incidence of small-for-gestational-age births (Svensson et al, 2006) [familial and twin studies, based on the Swedish Multigeneration Register of 8.5 million children born since 1932 with links to the biological parents and at least one set of grandparents]. This range of heritability approximates that of life expectancy in these and other populations from health-rich populations (Section 5.2). In other populations with high burden of disease and malnutrition, genetic influences on birth weight would be even more masked by the adverse environment.
Two gene candidates have come to light that have plausible links to inflammation and nutrition in fetal growth. The TNFa promoter alleles, discussed in Section 4.6.4 for influences on malaria resistance, also influence fetal growth. The -308 G/G homozygotes had 2.3-fold higher risk of being small-for-gestational age, with lower birth weight across the full birth weight range (Casano-Sancho et al, 2006). In very low birth weight infants, the -308 alleles were associated with different durations of required breathing support (mechanical ventilation) (Bokodi et al, 2005). The lower TNFa expression in -308 G/G carriers may influence fetal growth, because TNFα is mitogenic for myocytes and possible adipocytes (Casano-Sancho et al, 2006). Responsiveness to endotoxin by TNFα alleles could directly link maternal infections to birth weight. In mice, LPS increased TNFα in maternal blood and amniotic fluid, increased lipid oxidation (TBARS) in maternal and fetal liver 2-fold, and caused fetal growth retardation and death (Xu et al, 2006).
The apoE receptor-2 was implicated in small birth weight by an SNP screen of candidate genes for preeclampsia in Black women (SNP, single nucleotide polymorphisms in 129 candidate genes chosen for possible associations with fetal growth retardation and preeclampsia) (Wang et al, 2006). The apoE2 allele, which is protective for adult vascular disease, was underrepresented in small birth weight in a multi-ethnic study (Infante-Rivard et al, 2003). It is thus possible that apoE2 may protect against low birth weight, which is a risk factor in adult heart and metabolic disease. Preeclampsia has not been associated with any apoE allele.
These two gene candidates in regulating fetal growth return us to the possibility that environmental conditions could alter allele frequency by differential survival, within a few generations. The 10-fold decrease in early mortality during the 19th and 20th centuries could have allowed survival of those with allele sets that would have been eliminated in the not-too-distant generations that were conceived and born under harsher conditions.
4.10.2 Gene Imprinting: Inherited but Epigenetic Influences on Development
Fetal growth is highly dependent on IGF-1, IGF-2, and insulin, and their receptors. IGF-2 at its receptor IGF-2R in particular is influenced by epigenetic gene imprinting, in which one parental allele of a gene is silenced or not transcribed in somatic cells. Of the tens of thousands of mammalian genes, fewer than 100 are imprinted. Other genes that regulate growth are over-represented in this small group Akt-1/2 (Fig. 1.3A,B), metabolite transporter (Slc22a18), and cyclin-ckd inhibitor (Cdkn1c) (Reik et al, 2003; Smith et al, 2006; Tycko and Efstratiadis, 2002; Tycko, 2006). The longevity pathway links of IGF-1 and Akt-1/2 thus may extend to fetal growth regulation through imprinting. Nematodes and insects also employ imprinting by parent-of-origin, but genes that influence longevity (Fig. 1.3A) have not shown imprinting.
Imprinted genes are clustered in human chromosome 11p15.5 and 15q11–13 and the corresponding mouse chromosome. In addition, mice have imprinting of X-chromosome genes not known in humans. The silent imprinted gene, while not expressed in somatic cells, continues to be transmitted through the germ line. Imprinted genes are silenced by enzymatic methylation of cytosines, which can alter accessibility of the gene to transcription regulators by altering chromatin packing. Acetylation and phosphorylation also mediate imprinting and its converse, loss of imprinting (Kaneda and Feinberg, 2005; Mancini-Dinardo et al, 2006). The complexity of imprinting mechanisms continues to emerge.
Imprinting may be a fundamental epigenetic mechanism in the regulation of fetal growth by the parental genomes (Devriendt, 2000; Solter, 1988). Embryos derived from only one parent fail, despite having complete diploid genomes: Parthenogenetic embryos derived from maternal genes fail to develop the trophoblast and other extra-embryonic components, becoming teratomas; conversely, androgenetic embryos develop only the extra-embryonic tissues, becoming hydatidiform moles. In the Beckwith-Wiedemann syndrome, the fetus grows abnormally large and differentially; neonates are hypoglycemic and, if they survive, have a high incidence of Wilms tumor and other childhood cancers. The Beckwith-Wiedemann syndrome is caused by segmental trisomy and other defects of chromosome 11p15, where IGF-2 is located (DeChiara et al, 1991). IGF-2, the first imprinted gene identified, regulates the growth of both placenta and fetus (Smith et al, 2006; Tycko and Efstratiadis, 2002). Gene knockout experiments for IGF-2 and some other imprinted genes also show the importance of maternally expressed genes to placental growth and fetal development (Tycko and Efstratiadis, 2002). IGF-2 with a placental specific promoter activates placental trophoblast cells through AKT phosphorylation and is a regulator of amino acid transport.
The parental contributions to imprinting differ among the imprinted genes: IGF-2, which promotes growth, is mainly expressed from the paternal allele; its receptor, IGF-2R, which inhibits growth, is expressed from the maternal allele. Some maternally expressed genes encode placental transporters of amino acids and other solutes. While all known imprinted genes are active in the placenta, not all imprinted genes control growth. These parental asymmetries in the imprinting of key metabolic genes suggest counterbalancing evolutionary strategies and are interpreted as an outcome of fetal-maternal competition for resources (Haig, 1993; Haig, 1996). From the ‘selfish gene’ perspective, success of the paternal genome has depended on evolution of mechanisms that favor its genes in competition with maternal genes for nutritional and other resources during development. The maternal genome, according to this model, counterattacks by expressing genes that inhibit growth. The exclusive paternal expression of IGF-2 is countered by the exclusive maternal expression of IGF-2 receptor, which suppresses growth by trafficking IGF-2 for lysosomal degradation.
Imprinted genes have major influences on mammalian development by controlling placental growth, and thence the nutrient supply to the fetus, but also fetal growth itself (Angiolini et al, 2006). Placental nutrient flow is influenced by multiple imprinted genes, including IGF-2 (Constancia et al, 2005) and PHLDA2 (Salas et al, 2004), as well as other genes that have not shown imprinting, e.g., TOR (Fig. 3.10) (Jansson, 2006). Moreover, imprinted genes influence maternal postpartum behaviors, including nursing (Davies et al, 2005; Isles and Holland, 2005). Gene imprinting is implicated in low birth weight, preeclampsia, and other pathological conditions (Haig, 1993; Tycko, 2006). For example, PHLDA2 (maternally silenced) had increased expression in the placentas of growth-retarded human fetuses (McMinn et al, 2006). Loss of imprinting of PHDLA, which increases its expression, also caused fetal growth retardation in mice (Salas et al, 2004). The importance of loss of imprinting extends to cancer, as observed for IGF2, which shows increased cellular expression in colorectal and other cancers (Kaneda and Feinberg, 2005). There is no direct evidence for maternal infections and placental or fetal gene imprinting. However, preeclampsia, which is associated with malaria and other infections (see above), can involve maternally expressed imprinted genes expressed in the placenta (Oudejans et al, 2004).
Neonatal behavioral experiences can also modify neuroendocrine gene imprinting functions with potentially important later effects as shown by Meaney and colleagues. Experimental handling of rat pups altered adult levels of plasma corticosterone and glucocorticoid receptor (GR) in the hippocampus, a critical locus of feedback control of the adrenal cortex (Meaney et al, 1988). During aging, the handled rats had lower corticosterone and less hippocampal neuron loss. As noted in Section 2.5.2, hippocampal neuron loss during aging was more prominent in earlier rodent colonies. Further studies show that maternal grooming also alters hippocampal GR gene methylation at a binding site for the NGFA1 binding site (Weaver et al, 2007). These findings suggest that infections that alter neonatal-maternal behavioral interactions could have consequences to adult brain functions in the vulnerable hippocampal neurons.
Gene imprinting may be sensitive to nutrition. Mice fed a diet deficient in methyl donors (low in folic acid, methionine, and vitamin B12) showed loss of imprinting of the maternal IGF-2 allele, with decreased cytosine DNA methylation and increased gene expression. The effect persisted after restoration of a balanced diet for 100 days (Waterland et al, 2006). Thus, childhood dietary deficiencies can have persisting effects on gene imprinting, with potential importance to fetal growth. The scope of dietary imprinting is unknown but could be of major importance. Maternal protein-deficient diets that cause growth retardation also alter placental amino acid transport by the transport ‘system A’ (Malandro et al, 1996), which includes imprinted genes. Imprinting effects from early nutritional conditions may have caused the transgenerational effects of famine on grandchildren noted above on birth weight (Section 4.7.1) (Lumey, 1997) and on mortality from cardiovascular disease/diabetes (Section 4.5.2 (Kaati et al, 2002). As discussed in Section 4.9, in monkeys during adaptation to captivity, female but not male birth weights increased (Price, 1999; Price and Coe, 2000), suggestive of imprinting. Moreover, the trends of small mothers to have small babies for several generations could also involve imprinting (Section 4.6). Future studies may consider infections that directly affect the placenta and cause fetal growth retardation, such as malaria (Fig. 4.13) and HIV (McDonagh et al, 2004).
4.11 SUMMARY
Review of Barker’s initial papers, 1986–1991, shows the important evidence that infections in birth cohorts were correlated with adult mortality to nearly the same extent as birth weight. Immunosenescence, accelerated by chronic antigenic stimulation, may contribute as much to adult mortality as the vascular risk factors. Adult height, which is determined by fetal and postnatal growth, was identified by Barker as protective for vascular events. It is robustly confirmed that taller people live longer, as seen in the historical increase of height and longevity. Crimmins and I hypothesize that both the increases of height and longevity resulted from the overall decrease of infection and inflammation (Crimmins and Finch, 2006a).
Birth weight is an important indicator of maternal health and nutrition, and predicts health of the adult in a U-shaped distribution. Low and high birthweight babies show higher vascular risk factors in the metabolic syndrome as adults. High birth weight (macrosomia) is more common in obese and diabetic mothers. The future of maternal effects will be dominated by maternal obesity and diabetes. Such pregnancies increase not only the risk of the same conditions in their offspring, but also the risk of infections.
I have questioned the focus on maternal malnutrition as the main cause of low birth weight. Twins typically have birth weight 800 g below single births in well nourished mothers, yet usually catch up by age 9 y. Because adult twins show many fewer levels of the vascular risk factors than low birth weight singletons, I suggest a different pathobiology is at work than in twins. Could maternal infections cause a substantial portion of low birth weights, even in health-rich countries? Subclinical maternal infections may be more prevalent than recognized. Survey of maternal blood for endotoxin antibodies might yield clues (Campbell et al, 2003a,b).
Infections and poor nutrition clearly synergize to cause low birth weight in health-poor tropical populations. Maternal metabolism is remarkably different in these poverty-struck populations. Unlike well-fed mothers, health-poor mothers fail to increase basal metabolism during pregnancy and gain 50% less weight. This may be a metabolic adaptation to the energy challenge. In rural Gambians, who experience a hungry season with high infections, food supplements during pregnancy increased birth weight by 230 g, but only partly restored the maternal metabolic upregulation. Moreover, birth weights are still 300 g or more below the median in healthy populations. Prevalent malaria is a factor in low birth weight by directly infecting the placenta, while diarrheas cause postnatal growth retardation. These children also suffer chronic gastroenteritis, with endotoxin leaking into the circulation. Those born in the hungry season of high infections had higher adult mortality from infections than those born 4–6 m earlier. The presence of endotoxin and IgG antibodies in adults (Campbell et al, 2003a,b) suggests useful markers for other populations in association with vascular disease. Even in health-rich populations, maternal infections may have unrecognized roles in fetal retardation.
Moreover, widening international trade can spread infections. As will be developed in Section 6.4, we live in a globally confluent microbial system. Moreover, modern health-rich populations are only 2–3 generations removed from highly infectious environments. With few exceptions, nutritional supplements of malnourished populations improve pre- and postnatal growth. However, broad generalizations are unlikely to be sustained because of the heterogeneity of populations in infectious disease load and nutritional history. Individual variations in intestinal flora may reflect host genome-environment interactions in the life history of nutrition and pathogen exposure.
Infection-nutrition interactions in health-poor tropical populations may be pertinent to ongoing studies of famine and malnutrition in the 19th century and World War II. Many studies show relationships of adult health to caloric deficits at different times in pre- and postnatal development. Moreover, infections were also raging in the Netherlands at that time, as well as in the Finnish Famine of the 19th century. Survivors of the WWII famines have shown modest trends for glucose dysregulation, but inconsistent effects on cardiovascular disease (present in the Dutch Hunger Winter, exposed first trimester; absent in Siege of Leningrad). However, breast cancer is notably increased (up to 250%) and the age of menopause decreased (up to 1.8 y) in those exposed postnatally to famine (each change shows its own critical time). Overall, the effects of maternal starvation on development seem relatively mild in populations that had been relatively healthy and well nourished. However, exposure to infections predicts accelerated immunosenescence (Section 2.8).
Clearly, many factors at work during development can modify outcomes of chronic aging conditions. Besides infections, inflammation, and nutritional factors from the environment, genetic factors are at work. For example, TNFa promoter alleles, which influence susceptibility to malaria, may also influence fetal growth, adult adiposity, and other metabolic syndrome risk factors. Other genetic variations in inflammatory responses may be viewed as balancing selection in the context of nutrition, infection, and delayed health consequences.
Another genetic effect on development is through imprinted alleles of the insulin/IGF pathways that may be modified by diet. But there is another major source of variation, through chance events during development, illustrated by the wide variation in ovarian oocyte numbers that, in turn, determine the onset of reproductive senescence (Finch and Kirkwood, 2000). The next chapter considers these chance variations as a factor in the low overall heritability of the life span.
The evidence assembled shows major roles of infection and inflammation on human development. Similar associations were described in Barker’s original papers (1986–1991). I hope to have challenged the focus on maternal nutrition in the Fetal Origins theory to include the omnipresent burden of infections and inflammation, which contribute on the same level of magnitude to adult chronic diseases with inflammatory processes.