Pulmonary Hypertension
Primary Pulmonary Hypertension
Daniel Bernstein, Jeffrey A. Feinstein
Pathophysiology
Pulmonary hypertension (PH , elevated pressure in the pulmonary arteries) is characterized by pulmonary vascular obstructive disease and right-sided heart failure. The etiologies are varied, but all lead to similar symptoms (Tables 460.1 and 460.2 ). PH occurs at any age, although in pediatric patients the mean age at diagnosis is 7-10 yr. In patients with idiopathic or familial disease, females outnumber males 1.7 : 1; in other patients, both genders are represented equally. Mutations in the gene for bone morphogenetic protein receptor-2 (BMPR2 , a member of the transforming growth factor [TGF]-β receptor family) on chromosome 2q33 have been identified in 70% of patients with familial primary pulmonary hypertension (known as PPH1 ) and in 10–20% with idiopathic sporadic PH. Other potential disease causing genes include PPH2 , ALK1, ENG, SMAD9, CAV1, and KCNK3 , which cause a channelopathy in familial and sporadic cases of primary PH. Viral infection, such as with the vasculotropic human herpesvirus 8, has been suggested as a trigger factor in some patients.
PH is associated with precapillary obstruction of the pulmonary vascular bed as a result of hyperplasia of the muscular and elastic tissues and a thickened intima of the small pulmonary arteries and arterioles (Fig. 460.1 ). Secondary atherosclerotic changes may be found in the larger pulmonary arteries as well. In children, pulmonary venoocclusive disease may account for some cases of primary PH. Before a diagnosis of primary PH can be made, other causes of elevated pulmonary artery pressure must be eliminated; these include chronic pulmonary parenchymal disease, persistent obstruction of the upper airway, congenital cardiac malformations, recurrent pulmonary emboli, alveolar capillary dysplasia, liver disease, autoimmune disease, and moyamoya disease (Table 460.2 ). PH associated with congenital heart disease is currently the most common in pediatric patients (40–55%), followed by chronic respiratory disorders (20–35%) and idiopathic or familial disease (10–15%). PH associated with chronic lung disease (bronchopulmonary dysplasia) in premature infants is growing to encompass a larger portion of new cases.
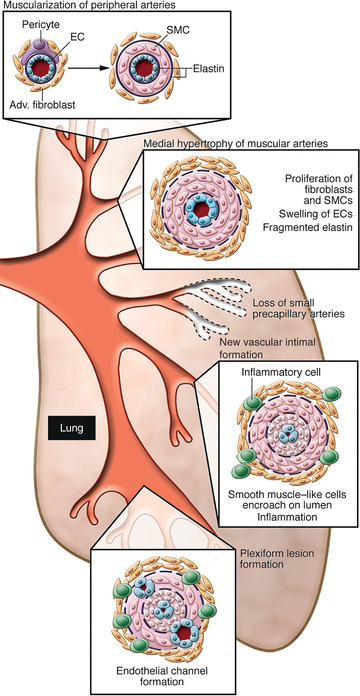
PH places an afterload burden on the right ventricle, which results in right ventricular hypertrophy (RVH). Dilation of the pulmonary artery is present, and pulmonary valve insufficiency may occur. In the later stages of the disease, the right ventricle dilates, tricuspid insufficiency develops, and cardiac output is decreased. Arrhythmias, syncope, and sudden death are known complications.
Clinical Manifestations
The predominant symptoms include exercise intolerance (dyspnea) and fatigability; occasionally, precordial chest pain, dizziness, or headaches are noted. Syncope may be noted in approximately 30% of pediatric patients. Patients often undergo an incorrect workup and are treated for asthma or seizures before a proper diagnosis is made. Peripheral cyanosis may be present, especially during exercise or in patients with a patent foramen ovale through which blood can shunt from right to left. In the late stages of disease, patients may have cold extremities and a gray appearance associated with low cardiac output. Arterial oxygen-hemoglobin saturation is usually normal unless there is an associated intracardiac shunt. If right-sided heart failure has supervened, jugular venous pressure is elevated, and hepatomegaly and edema are present. Jugular venous a waves are present, and in those with functional tricuspid insufficiency, a conspicuous jugular cv wave and systolic hepatic pulsations are manifested. The heart is moderately enlarged, and a right ventricular heave can be noted. The first heart sound is often followed by an ejection click emanating from the dilated pulmonary artery. The second heart sound (S2 ) is narrowly split, loud, and sometimes booming in quality; it is frequently palpable at the upper left sternal border. A presystolic gallop rhythm may be audible at the lower left sternal border. The systolic murmur is soft and short and is sometimes followed by a blowing decrescendo diastolic murmur caused by pulmonary insufficiency. In later stages, a holosystolic murmur of tricuspid insufficiency is appreciated at the lower left sternal border.
Diagnosis
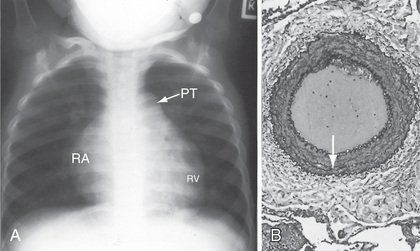
Chest radiographs reveal a prominent pulmonary artery and right ventricle (Fig. 460.2 ). The pulmonary vascularity in the hilar areas may be prominent, in contrast to the peripheral lung fields in which pulmonary markings are decreased. The electrocardiogram (ECG) shows RVH, often with spiked P waves. Echocardiography is used to screen for any congenital cardiac malformations. Doppler evaluation of the tricuspid valve, if insufficiency is present, will allow estimation of the right ventricular (and thus pulmonary arterial) systolic pressure.
At cardiac catheterization, the presence of left-sided obstructive lesions (pulmonary venous stenosis, mitral stenosis, restrictive cardiomyopathy) that result in pulmonary venous hypertension can be evaluated (see Chapters 454.9 , 458.7 , and 466.3 ). The presence of pulmonary arterial hypertension (PAH) with a normal pulmonary capillary wedge pressure is diagnostic of PAH. If the wedge pressure is elevated and left ventricular end-diastolic pressure (LVEDP) is normal, obstruction at the level of the pulmonary veins, left atrium, or mitral valve should be suspected. If LVEDP is also elevated, the diagnosis of restrictive cardiomyopathy should be entertained. The risks associated with cardiac catheterization are increased in severely ill patients with primary PH.
Prognosis and Treatment
Most forms of PH are progressive, and no cure is currently available. Fig. 460.3 provides a general treatment approach to PH. Some success has been reported with oral calcium channel blockers (CCBs ) such as nifedipine in children who demonstrate pulmonary vasoreactivity when these agents are administered during catheterization. Continuous intravenous infusion of the arachidonic acid metabolite prostacyclin (epoprostenol) provides relief as long as the infusion is continued. Despite the success of prostacyclin in reducing symptoms and improving quality of life, it slows but does not stop the progression of the disease. Treprostinil , a prostacyclin analog with a longer half-life, has also been shown to be effective. Nebulized forms of prostacyclin, oral pulmonary vasodilators such as endothelin receptor antagonists, and phosphodiesterase type 5 inhibitors have been used with success in adults and in a small number of clinical studies in children (Table 460.3 ).
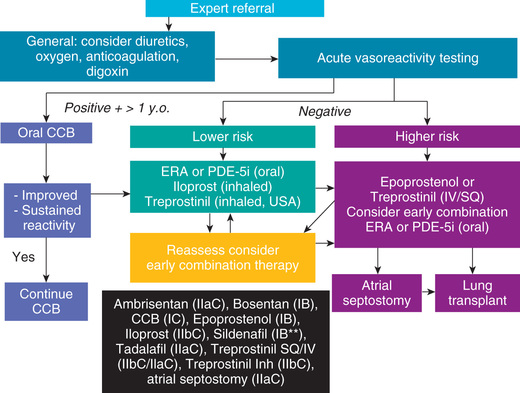
Table 460.3
| DRUG AND MECHANISM OF ACTION | DOSES USED IN PEDIATRIC STUDIES | COMMON SIDE EFFECTS |
|---|---|---|
| Epoprostenol (prostacyclin [PGI2 ], a potent vasodilator; also inhibits platelet aggregation) | 1 ng/kg/min initially. Increase based on clinical course and tolerance to 5-50 ng/kg/min. Some patients may require even higher doses. Must be given by continuous infusion that is not interrupted. | Flushing, headache, nausea, diarrhea, hypotension, chest pain, jaw pain |
| Iloprost (synthetic analog of PGI2 ) | 2.5-5.0 µg 6-9 times daily (not more frequently than every 2 hr) via inhalation | Flushing, headache, diarrhea, hypotension, jaw pain, exacerbation of pulmonary symptoms (cough, wheezing) |
| Treprostinil (synthetic analog of PGI2 ) | 1 ng/kg/min initially. Target dose ranges from 20-80 ng/kg/min. Given either IV or SC via continuous infusion. Longer half-life than epoprostenol. | Flushing, headache, diarrhea, hypotension, jaw pain. Pain at infusion site when given SC. |
| Ambrisentan (selective endothelin EtA receptor antagonist) | Target dose ranges from 1.25-10 mg. Use  dose for 1st mo.
dose for 1st mo. |
Flushing, headache, hypotension, fluid retention/edema, nasopharyngitis, sinusitis, anemia, fluid retention, exacerbation of heart failure, anemia, palpitations |
| Bosentan (nonselective endothelin receptor EtA and EtB antagonist) | 2 mg/kg/dose bid. Use 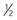 dose for 1st mo and check for LFT abnormalities before up-titrating.
dose for 1st mo and check for LFT abnormalities before up-titrating. |
Flushing, headache, nasopharyngitis, fluid retention, exacerbation of heart failure, anemia, elevated LFTs, palpitations |
| Macitentan (nonselective endothelin receptor EtA and EtB antagonist) | — | Flushing, headache, fluid retention, exacerbation of heart failure, anemia, nasopharyngitis, bronchitis, influenza, urinary tract infections |
| Sildenafil (inhibitor of cGMP-specific phosphodiesterase 5) | 1 mg/kg/dose given 3-4 times daily. Initial dosing should be 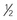 final target dose to evaluate for hypotension
final target dose to evaluate for hypotension |
Flushing, headache, diarrhea, myalgia, hypotension, priapism, visual disturbance (blue coloration), tinnitus |
| Tadalafil, a phosphodiesterase type 5 inhibitor | 1 mg/kg/dose given daily. Initial dosing should be 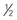 final target dose to evaluate for hypotension.
final target dose to evaluate for hypotension. |
Flushing, headache, diarrhea, myalgia, hypotension, priapism, visual disturbance (blue coloration), tinnitus |
| Calcium channel blockers (amlodipine, diltiazem, nifedipine) | Previously widely used. Now indicated only for patients who show a strong response to nitric oxide during cardiac catheterization. | Flushing, headache, edema, arrhythmia, headache, hypotension, rash, nausea, constipation, elevated LFTs |
* These medications should only be administered under the direction of a specialist in pulmonary hypertension.
cGMP, Cyclic guanosine monophosphate; IV, Intravenously; LFT, liver function test; SC, subcutaneously.
Anticoagulation may be of value in patients with previous pulmonary thromboemboli; some of these patients may respond to balloon angioplasty of narrowed pulmonary artery segments. Riociguat, a soluble guanylate cyclase stimulator, with vasorelaxation, antiproliferation, and antifibrotic properties, has proved effective in adults with chronic thromboembolic or idiopathic pulmonary hypertension. Despite many advances, definitive therapy is still heart-lung or lung transplantation (see Chapter 470.2 ). In patients with severe PH and low cardiac output, the terminal event is often sudden and related to a lethal arrhythmia. Patients with PH diagnosed in infancy, especially those in premature infants with chronic lung disease, or with pulmonary vein stenosis often have rapid progression and high mortality.
Pulmonary Vascular Disease (Eisenmenger Syndrome)
Daniel Bernstein, Jeffrey A. Feinstein
Pathophysiology
The term Eisenmenger syndrome refers to patients with an intracardiac defect or aortopulmonary connection through which blood is shunted partially or totally from right to left as a result of the development of pulmonary vascular disease. This physiologic abnormality can occur with ventricular or atrioventricular septal defects, patent ductus arteriosus, aortopulmonary window, or any other communication between the aorta and pulmonary artery, as well as in many forms of complex congenital heart disease with unrestricted pulmonary blood flow. Pulmonary vascular disease with an isolated atrial septal defect can occur, but this is less common and does not occur until late in adulthood.
In Eisenmenger syndrome, pulmonary vascular resistance (PVR) after birth either remains high or, after having decreased during early infancy, rises thereafter because of increased shear stress on pulmonary arterioles. Factors playing a role in the rapidity of development of pulmonary vascular disease include increased pulmonary artery pressure, increased pulmonary blood flow, and the presence of hypoxia or hypercapnia. Early in the course of disease, PH is the result of markedly increased pulmonary blood flow (hyperkinetic PH). This form of PH decreases with the administration of pulmonary vasodilators such as nitric oxide, or oxygen, or both. With the development of Eisenmenger syndrome, pulmonary hypertension is the result of pulmonary vascular disease (obstructive pathologic changes in the pulmonary vessels). This form of PH is usually only minimally responsive to pulmonary vasodilators or oxygen or unresponsive.
Pathology and Pathophysiology
The pathologic changes of Eisenmenger syndrome occur in the small pulmonary arterioles and muscular arteries (<300 µm) and are graded on the basis of histologic characteristics (Heath-Edwards classification):
- ◆ Grade I changes involve medial hypertrophy alone.
- ◆ Grade II consists of medial hypertrophy and intimal hyperplasia.
- ◆ Grade III involves near-obliteration of the vessel lumen.
- ◆ Grade IV includes arterial dilation.
- ◆ Grades V and VI include plexiform lesions, angiomatoid formation, and fibrinoid necrosis.
Grades IV-VI indicate irreversible pulmonary vascular obstructive disease. Eisenmenger physiology is usually defined by an absolute elevation in pulmonary arterial resistance to >12 Wood units (resistance units indexed to body surface area) or by a ratio of pulmonary-to-systemic vascular resistance of ≥1.0.
Pulmonary vascular disease occurs more rapidly in patients with trisomy 21 who have left-to-right shunts. It also complicates the natural history of patients with elevated pulmonary venous pressure secondary to mitral stenosis or left ventricular dysfunction, especially in those with restrictive cardiomyopathy (see Chapter 466.3 ). Pulmonary vascular disease can also occur in any patient with transmission of systemic pressure to the pulmonary circulation via a shunt at the interventricular or great vessel level, as well as in patients chronically exposed to low partial pressure of oxygen (because of high altitude). Patients with cyanotic congenital heart lesions associated with unrestricted pulmonary blood flow are at particularly high risk.
Clinical Manifestations
Symptoms do not usually develop until the 2nd or 3rd decade of life, although a more fulminant course may occur. Intracardiac or extracardiac communications that would normally shunt from left to right are converted to right-to-left shunting as PVR exceeds systemic vascular resistance. Cyanosis becomes apparent, and dyspnea, fatigue, and a tendency toward dysrhythmias begin to occur. In the late stages of the disease, heart failure, chest pain, headaches, syncope, and hemoptysis may be seen. Physical examination reveals a right ventricular heave and a narrowly split S2 with a loud pulmonic component. Palpable pulmonary artery pulsation may be present at the left upper sternal border. A holosystolic murmur of tricuspid regurgitation may be audible along the left sternal border. An early decrescendo diastolic murmur of pulmonary insufficiency may also be heard along the left sternal border. The degree of cyanosis depends on the stage of the disease.
Diagnosis
On chest radiograph, the heart varies in size from normal to greatly enlarged; the latter usually occurs late in the course of the disease. The main pulmonary artery is generally prominent, similar to other causes of PAH (see Fig. 460.2A ). The pulmonary vessels are enlarged in the hilar areas and taper rapidly in caliber in the peripheral branches. The right ventricle and atrium are prominent. The ECG shows marked RVH. The P wave may be tall and spiked. Cyanotic patients have various degrees of polycythemia that depend on the severity and duration of hypoxemia.
The echocardiogram shows a thick-walled right ventricle and demonstrates the underlying congenital heart lesion. 2D echocardiography assists in eliminating from consideration lesions such as obstructed pulmonary veins, supramitral membrane, mitral stenosis, and restrictive cardiomyopathy. Doppler studies demonstrate the direction of the intracardiac shunt and the presence of a typical hypertension waveform in the main pulmonary artery. Tricuspid and pulmonary regurgitation can be used in the Doppler examination to estimate pulmonary artery systolic and diastolic pressures.
Cardiac catheterization usually shows a bidirectional shunt at the site of the defect. Systolic pressure is generally equal in the systemic and pulmonary circulations. Pulmonary capillary wedge pressure is normal unless a left-sided heart obstructive lesion or left ventricular failure is the cause of the PAH. Arterial oxygen-hemoglobin saturation is decreased depending on the magnitude of the right-to-left shunt. The response to vasodilator therapy (oxygen, prostacyclin, nitric oxide) may identify patients with less severe disease. Selective pulmonary artery injections may be necessary if pulmonary venous obstruction is suspected because of high wedge pressure and low LVEDP.
Treatment
The best management for patients who are at risk for the development of late pulmonary vascular disease is prevention by early surgical elimination of large intracardiac or great vessel communications during infancy. Some patients may be missed because they have not shown early clinical manifestations. Rarely, PVR never decreases at birth in these infants, and therefore they never acquire enough left-to-right shunting to become clinically apparent. Such delayed recognition is a particular risk in patients with congenital heart disease who live at high altitude. It is also a risk in infants with trisomy 21, who have a propensity for earlier development of pulmonary vascular disease. Because of the high incidence of congenital heart disease associated with trisomy 21, routine echocardiography is recommended at the time of initial diagnosis, even in the absence of other clinical findings.
Medical treatment of Eisenmenger syndrome is primarily symptomatic. Many patients benefit substantially from either oral (CCB, endothelin antagonist, phosphodiesterase inhibitors) or chronic intravenous (prostacyclin) therapy. Combined heart-lung or bilateral lung transplantation is the only surgical option for many of these patients (see Chapter 470.2 ).