Disorders of Neuromuscular Transmission and of Motor Neurons
Myasthenia Gravis
Diana X. Bharucha-Goebel
Autoimmune Myasthenia Gravis
Myasthenia gravis (MG) is a chronic autoimmune disease of the postsynaptic endplate leading to abnormal neuromuscular transmission or blockade, characterized clinically by rapid fatigability of striated muscle, particularly extraocular and palpebral muscles and those of swallowing. It must be distinguished from congenital myasthenic syndrome, a genetic disorder of receptors on the presynaptic and postsynaptic membranes, as well as the synapse of the neuromuscular junction and toxin-induced disorders of neurotransmission, such as botulism (see below). In MG, the release of acetylcholine (ACh) into the synaptic cleft by the axonal terminal is normal, but the postsynaptic muscle membrane (i.e., sarcolemma ) or motor endplate is less responsive than normal. This is due to antibodies against the postsynaptic acetylcholine receptor (AChR), leading to an abnormal architecture/folding pattern of the postsynaptic membrane, as well as a decreased number of receptors to which acetylcholine can bind.
Infants born to myasthenic mothers can have a transient neonatal myasthenic syndrome secondary to placentally transferred anti-AChR antibodies, distinct from congenital myasthenic syndromes (Tables 630.1 to 630.3 ).
Table 630.1
Classification of the Congenital Myasthenia Syndromes*
| SYNDROME | PERCENTAGE |
|---|---|
| PRESYNAPTIC | |
| Choline acetyltransferase deficiency | 5 |
| SNAP25 deficiency | 0.3 |
| Synaptotagmin 2 deficiency† | 0 |
| SYNAPTIC BASAL LAMINA–ASSOCIATED | |
| Endplate AChE deficiency | 12.6 |
| Laminin β2 deficiency | 0.3 |
| POSTSYNAPTIC | |
| Primary AChR deficiency ± minor kinetic abnormality | 33 |
| Primary kinetic defect ± minor AChR deficiency | 17.5 |
| Na channel myasthenia | 0.3 |
| Plectin deficiency | 0.6 |
| DEFECTS IN EP DEVELOPMENT AND MAINTENANCE | |
| Agrin deficiency | 0.3 |
| LRP4 deficiency | 0.6 |
| MuSK deficiency | 0.3 |
| Dok-7 deficiency | 9.8 |
| Rapsyn deficiency | 14 |
| CONGENITAL DEFECT OF GLYCOSYLATION | |
| GFPT1 deficiency | 3 |
| DPAGT1 deficiency | 0.6 |
| ALG2† deficiency | 0 |
| ALG14† deficiency | 0 |
| OTHER MYASTHENIC SYNDROMES | |
| PREPL deletion syndrome | 0.3 |
| Defects in the mitochondrial citrate synthase carrier† | 0 |
| CMS associated with centronuclear myopathies | 0.3 |
| TOTAL | 100% |
* Classification based on cohort of 353 congenital myasthenic syndrome patients investigated at the Mayo Clinic between 1988 and 2014.
† Defects in ALG2 and ALG14, synaptotagmin 2, and the mitochondrial citrate synthesis carrier were identified at other medical centers.
Modified from Darras BT, Monani UR, De Vivo DC: Genetic disorders affecting the motor neuron: spinal muscular atrophy. In Swaiman KF, Ashwal S, Ferriero DM, et al (eds): Swaiman's pediatric neurology, 6/e, Elsevier, 2018, Philadelphia, Table 144-1.
Table 630.2
Distinctive Clinical and Electrodiagnostic Features of Congenital Myasthenic Syndrome
| PRESYNAPTIC | SYNAPTIC | POSTSYNAPTIC | |||||
|---|---|---|---|---|---|---|---|
| CHOLINE ACETYLTRANSFERASE DEFICIENCY | LMS-LIKE FORM | ACHE DEFICIENCY | PRIMARY ACHR DEFICIENCY | SLOW-CHANNEL CMS | FAST-CHANNEL CMS | DOK7 MUTATIONS | |
| Autosomal dominant inheritance | X (most mutations) | ||||||
| Episodic apnea triggered by stressors | X | ||||||
| Neonatal hypotonia and respiratory insufficiency | X | X | X (in severe cases) | X (in severe cases) | |||
| Skeletal deformities | X | X | X (in severe cases) | ||||
| Delayed pupillary light responses | X | ||||||
| Prominent neck, wrist, and finger extensor weakness | X | ||||||
| Repetitive CMAPs after single stimulus | X | X | |||||
| Progressive decrement with prolonged exercise or repetitive stimulation | X | X | |||||
| Marked increment (>200%) with high-frequency repetitive stimulation | X | ||||||
| Decrement repairs with AChE inhibitors | X | X | |||||
| Clinical improvement with AChE inhibitors | X | ||||||
| Clinical worsening with AChE inhibitors | X | X | X | ||||
AChE, acetylcholinesterase; AChR, acetylcholine receptor; CMAPs, compound muscle action potentials; CMS, congenital myasthenic syndrome; LEMS, Lambert-Eaton myasthenic syndrome.
From Muppidi S, Wolfe GI, Barhon RJ: Diseases of the neuromuscular junction. In Swaiman KF, Ashwal S, Ferriero DM, Schor NF, editors: Swaiman's pediatric neurology, ed 5, Philadelphia, 2012, Elsevier, Table 91-3.
Table 630.3
Differential Diagnosis of Congenital Myasthenic Syndromes
| NEONATAL PERIOD, INFANCY, CHILDHOOD |
| ADULT |
|
• Peripheral neuropathy* |
* This diagnosis was suspected in some cases of the slow-channel CMS.
From Darras BT, Monani UR, De Vivo DC: Genetic disorders affecting the motor neuron: spinal muscular atrophy. In Swaiman KF, Ashwal S, Ferriero DM, et al. (eds): Swaiman's pediatric neurology, 6/e, Elsevier, 2018, Philadelphia, 2018, Box 144-2.
Clinical Manifestations
The age of onset for immune-mediated MG ranges anywhere from 11 mo to 17 yr of age. In the prepubertal age-groups, the female:male ratio is about 1.5 : 1, and in the postpubertal age-groups, the female:male ratio is about 1 : 1. In juvenile autoimmune MG, unilateral or bilateral but usually asymmetric ptosis and some degree of extraocular muscle weakness are the earliest and most constant signs. Extraocular weakness is not confined to muscles innervated by just one or two of the three corresponding brainstem nuclei; it is progressive. Older children might complain of diplopia, and young children might hold open their eyes with their fingers or thumbs if the ptosis is severe enough to obstruct vision. Pupillary responses to light are preserved. Dysphagia and facial weakness also are common and, in early infancy, feeding difficulties are frequent as the cardinal sign of myasthenia; in severe cases, aspiration and airway obstruction may occur. Poor head control because of weakness of the neck flexors may be prominent. Involvement initially may appear to be limited to bulbar-innervated muscles, but the disease can be systemic and progressive weakness eventually involves limb–girdle muscles and distal muscles of the hands in many cases. Fasciculations of muscle, myalgias, and sensory symptoms do not occur. Tendon stretch reflexes may be diminished but rarely are lost. Ocular myasthenia gravis may prove to be transitory over time, but in some patients, weakness never progresses to involve the axial or appendicular muscles. This disorder accounts for approximately 25% of all juvenile MG patients and is most frequent in children of Chinese and southeastern Asian descent, suggesting an ethnic genetic predisposition. In addition, prepubertal patients are more likely to have ocular only myasthenia, whereas a majority of postpubertal patients with myasthenia will have generalized symptoms.
Rapid fatigue of muscles is a characteristic feature of MG that distinguishes it from most other neuromuscular diseases. Ptosis increases progressively as patients are asked to sustain an upward gaze for 30-90 sec. Holding the head up from the surface of the examining table while lying supine is very difficult (indicative of neck flexion weakness), and gravity cannot be overcome for more than a few seconds. Repetitive opening and closing of the fists produces rapid fatigue of hand muscles, and patients cannot elevate their arms for more than 1-2 min because of fatigue of the deltoids. Patients are more symptomatic late in the day or when tired. Dysphagia can interfere with eating, and the muscles of the jaw soon tire when an affected child chews. Reviewing activities of daily living helps determine the severity of symptoms (Table 630.4 ). Additional triggers for exacerbation of weakness may include heat and intercurrent illness.
Table 630.4
Myasthenia Gravis Activities of Daily Living Scale (MG-ADL)
| GRADE | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| Talking | Normal | Intermittent slurring or nasal speech | Constant slurring or nasal speech, but can be understood | Difficult to understand speech |
| Chewing | Normal | Fatigue with solid food | Fatigue with soft food | Gastric tube |
| Swallowing | Normal | Rare episode of choking | Frequent choking, modifications in diet | Gastric tube |
| Breathing | Normal | Shortness of breath with exertion | Shortness of breath at rest | Ventilator dependence |
| Impairment of ability to brush teeth or comb hair | None | Extra effort, but no rest periods needed | Rest periods needed | Cannot do 1 of these functions |
| Impairment of ability to arise from a chair | None | Mild, sometimes uses arms | Moderate, always uses arms | Severe, requires assistance |
| Double vision | None | Occurs, but not daily | Daily, but not constant | Constant |
| Eyelid droop | None | Occurs, but not daily | Daily, but not constant | Constant |
| TOTAL MG-ADL SCORE |
From Wolfe GI, Herbelin L, Nations SP, et al: Myasthenia gravis activities of daily living profile, Neurology 52:1487-1489, 1999.
Left untreated, MG is usually progressive and can become life-threatening because of respiratory muscle involvement and the risk of aspiration, particularly at times when the child is otherwise unwell, as during an upper respiratory tract infection. Familial myasthenia (congenital myasthenic syndrome) usually is not progressive, but may vary in severity from milder forms, limb–girdle forms, to more severe forms, including those with respiratory failure.
Myasthenic crisis is an acute or subacute severe increase in weakness in patients with MG, usually precipitated by an intercurrent infection, surgery, or even emotional stress. It may require intravenous cholinesterase inhibitors, immunoglobulin, plasma exchange, gavage feeding, and even transient ventilator support. It must be distinguished from cholinergic crisis secondary to overdosing with anticholinesterase medications. The muscarinic effects include abdominal cramps, diarrhea, profuse sweating, salivation, bradycardia, increased weakness, and miosis. Cholinergic crisis requires only supportive care and withholding of further doses of cholinergic drugs, and it passes within a few hours; the dose of medication to be restarted should be reconsidered, unless the patient had taken an overdose that was not prescribed.
Approximately 70–80% of younger children and adolescents with immune-mediated MG will have elevated AChR antibodies. Approximately 30% of affected adolescents show elevations, but anti-AChR antibodies are only occasionally demonstrated in the plasma of prepubertal children. Some with negative titers of anticholinesterase exhibit anti–muscle-specific tyrosine kinase (MuSK) circulating antibodies. MuSK is localized at the neuromuscular junction and appears essential to fetal development of this junction. Additional autoantibodies related to immune MG include LRP4, titin, and ryanodine receptor (RyR) antibodies.
Infants born to myasthenic mothers can have respiratory insufficiency, inability to suck or swallow, and generalized hypotonia and weakness, a syndrome typically referred to as transient neonatal myasthenia. They might show little spontaneous motor activity for several days to weeks. The onset of symptoms typically occurs within the first 1-3 days of life. Some require ventilatory support and feeding by gavage during this period. Some patients may also require pyridostigmine (acetylcholinesterase inhibitor) management transiently. After the abnormal antibodies disappear from the blood and muscle tissue, these infants regain normal strength and are not at increased risk of developing MG in later childhood. Patients usually show full recovery by about 2 mo of age. A small minority develops fetal akinesia sequence with multiple joint contractures (arthrogryposis) that develop in utero from lack of fetal movement. AChR antibodies can usually be demonstrated in maternal blood, but at times maternal antibodies may not be detected. Rates of transient neonatal myasthenia are estimated to be as high as 10–20% of infants born to mothers with MG.
Congenital Myasthenic Syndromes
A heterogeneous group of genetic diseases of neuromuscular transmission is collectively called congenital myasthenic syndromes (CMSs). The etiology and pathogenesis of these syndromes are unrelated to either transitory neonatal myasthenia caused by placental transfer of maternal antibodies or to autoimmune MG, despite overlap of clinical symptoms. CMSs are nearly always permanent static disorders without spontaneous remission (see Tables 630.1 and 630.2 ). Several distinct genetic forms are recognized, nearly all with onset at birth or in early infancy with symptoms that may include hypotonia, external ophthalmoplegia, ptosis, dysphagia, weak cry, facial weakness, easy muscle fatigue generally, and sometimes respiratory insufficiency or failure, the last often precipitated by a minor respiratory infection. In the childhood-onset forms, findings such as fatigability, delayed motor milestones, and fluctuating ocular symptoms (ptosis and extraocular muscle weakness) are common. Cholinesterase inhibitors have a favorable effect in most, but in some forms the symptoms and signs are actually worsened. Children with most types of congenital MG do not experience myasthenic crises and rarely exhibit elevations of anti-ACh antibodies in plasma.
Mutations responsible for CMS have been identified in 24 different genes. The genetic mutations are known in less than half of children with CMS. The most common genes associated with CMS include CHAT, CHRNE, DOK7, COLQ, GFPT, and RAPSN. CMS can be caused by mutations affecting proteins involved in ACh synthesis, vesicle fusion into the synaptic cleft, ACh breakdown in the synaptic cleft, and reuptake of choline, within subunits of the postsynaptic acetylcholine receptor, as well as in postsynaptic glycosylation pathways. Basal lamina-associated proteins can lead to synaptic cleft abnormalities due to mutations in the COLQ, COL13A1, and LAMB2 genes. These pathways emphasize the role of the integrity of the extracellular matrix proteins in the formation and maintenance of the synapse. Anti-AChR and anti-MuSK antibodies are usually, but not always, absent in serum, unlike in autoimmune forms of MG affecting older children and adults.
There may be clinical clues that aid in the diagnosis (Table 630.5 ). In patients with apneic episodes, consider RAPSN, CHAT, and COLQ. Apneic episodes in patients with choline acyltransferase (CHAT) mutations can be episodic but can also be life-threatening. Although most CMS syndromes are inherited in a recessive fashion, there are several where an autosomal dominant pattern of inheritance or de novo dominant pattern of inheritance can be seen, including CHRNA1, CHRNB1, CHRND, CHRNE, and SYT2. Mutations in the RAPSN gene can lead to an early-onset hypotonia with respiratory failure and episodic apnea, but can also present in milder limb–girdle patterns of weakness with an onset in childhood or adolescence. Genes associated with a more limb–girdle myasthenic syndrome phenotype include GFPT1, DPAGT1, ALG2, ALG14, GMPPB, and PREPL. Genes affecting the postsynaptic AChR subunits may be associated with a slow-channel CMS in which patients may have variable weakness, typically with worsening with AChE inhibitors, as well as a fast-channel CMS syndrome; they can show improvement in symptoms in response to AChE inhibitors.
Rare Other Causes of Myasthenia
MG is occasionally associated with hypothyroidism, usually Hashimoto thyroiditis. Other collagen vascular diseases and also some centronuclear myopathies may be associated with defects in neuromuscular transmission. Thymomas, noted in some adults, rarely coexist with MG in children. Likewise, lung carcinomas that occur in adults associated with Lambert-Eaton myasthenic syndrome are not seen in children. Lambert-Eaton syndrome in children is rare but has been reported with lymphoproliferative disorders and with neuroblastoma. Postinfectious MG in children is transitory and usually follows a varicella-zoster infection by 2-5 wk as an immune response.
Laboratory Findings and Diagnosis
MG is one of the few neuromuscular diseases in which electromyography (EMG) is more specifically diagnostic than a muscle or nerve biopsy. A decremental response is seen to repetitive nerve stimulation; the muscle potentials diminish rapidly in amplitude until the muscle becomes refractory to further stimulation. Electrophysiologically, this response is due to endplate potentials decreasing with subsequent repetitive stimulations, such that stimuli are no longer resulting in endplate potentials that achieve a threshold to result in a propagating motor action potential. This results in a cumulative lowering of the compound muscle action potential (CMAP) amplitude with the repeated stimuli. A decline of greater than 10% between waves 1 : 4 on repetitive stimulation is diagnostic for a decremental response, and suggestive of a disorder of neuromuscular transmission. The motor nerve conduction velocity remains normal. This unique EMG pattern is the electrophysiologic correlate of the fatigable weakness observed clinically and is reversed after a cholinesterase inhibitor is administered. A myasthenic decrement may be absent or difficult to demonstrate in muscles that are not involved clinically. This feature may be confusing in early cases or in patients showing only weakness of extraocular muscles. Special electrophysiologic studies are required in the classification of CMS and involve estimating the number of AChRs per endplate and in vitro study of endplate function. These special studies and patch-clamp recordings of kinetic properties of channels are performed on special biopsy samples of intercostal muscle strips that include both the origin and insertion of the muscle but are only performed in specialized centers. If myasthenia is limited to the extraocular, levator palpebrae, and pharyngeal muscles, repetitive nerve stimulation of the distal and proximal muscles (e.g., abductor pollicis brevis muscle or trapezius muscle, respectively), although diagnostic in the generalized disease, is usually normal.
Anti-AChR antibodies should be assayed in the plasma but are inconsistently demonstrated. Antibodies against the MuSK receptor should be sought in children without circulating AChR antibodies, a diagnostic finding when elevated, which further delineates the etiology. Many cases of congenital MG result from failure to synthesize or release ACh at the presynaptic membrane. In some cases, the gene that mediates the enzyme choline acetyltransferase for the synthesis of ACh is mutated. In others, there is a defect in the quantal release of vesicles containing ACh. The treatment of such patients with cholinesterase inhibitors is futile. In some patients such as those with COLQ and DOK7 mutations as well as slow-channel myasthenia, acetylcholinesterase inhibitors (e.g., pyridostigmine) can lead to no response or even worsening of symptoms. Clinical genetic testing for congenital myasthenic syndrome can be done by panels that are commercially available and can test anywhere from 14-21 CMS-associated genes.
Other serologic tests of autoimmune disease, such as antinuclear antibodies and abnormal immune complexes, should also be sought. If these are positive, more extensive autoimmune disease involving vasculitis or tissues other than muscle is likely. A thyroid profile should always be examined. The serum creatine kinase level is normal in MG.
The heart is not involved, and electrocardiographic findings remain normal. Radiographs of the chest often reveal an enlarged thymus, but the hypertrophy is not a thymoma. It may be further defined by tomography or by CT or MRI of the anterior mediastinum if the radiographic findings are uncertain, but caution should be used when selecting the optimal imaging modalities because of radiation exposure for CT and anesthetic risk in a myasthenic patient if sedated MRI is needed for a younger myasthenic child.
The role of conventional muscle biopsy in MG is limited. It is not required in most cases, but approximately 17% of patients show inflammatory changes, sometimes called lymphorrhages, that are interpreted by some physicians as a mixed myasthenia–polymyositis immune disorder. Muscle biopsy tissue in MG shows a nonspecific type II muscle fiber atrophy, similar to that seen with disuse atrophy, steroid effects on muscle, polymyalgia rheumatica, and many other conditions. The ultrastructure of motor endplates shows simplification of the membrane folds; the AChRs are located in these postsynaptic folds, as shown by bungarotoxin (snake venom), which binds specifically to the AChRs.
A clinical test for MG is administration of a short-acting cholinesterase inhibitor, usually edrophonium chloride. Ptosis and ophthalmoplegia improve within a few seconds, and the fatigability of other muscles decreases.
Recommendations on the Use of Cholinesterase Inhibitors as a Diagnostic Test for MG in Infants and Children
Children Two Years of Age and Older
- ◆ The child should have a specific fatigable weakness that can be measured, such as ptosis of the eyelids, dysphagia, or inability of the cervical muscles to support the head. Nonspecific generalized weakness without cranial nerve motor deficits is not a criterion.
- ◆ An intravenous infusion should be started to enable the administration of medications in the event of an adverse reaction.
- ◆ Electrocardiographic monitoring is recommended during the test.
- ◆ A dose of atropine sulfate (0.01 mg/kg) should be available in a syringe, ready for intravenous administration at the bedside during the edrophonium test, to block acute muscarinic effects of the cholinesterase inhibitor, mainly abdominal cramps and/or sudden diarrhea from increased peristalsis, profuse bronchotracheal secretions that can obstruct the airway, or, rarely, cardiac arrhythmias. Some physicians pretreat all patients with atropine before administering edrophonium, but this is not recommended unless there is a history of a reaction to tests. Atropine can cause the pupils to be dilated for as long as 14 days after a single dose, and the pupillary effects of homatropine can last 4-7 days.
- ◆ Edrophonium chloride (Tensilon) is administered intravenously. The initial test dose is 0.01 mg/kg (no more than 1 mg [for children <30 kg], and no more than 2 mg initial dose [for children >30 kg]). After the initial dose, repeat doses may be given intravenously. For children <30 kg, repeat at a rate of 1 mg every 30-45 seconds to a maximum cumulative dose of 5 mg. For children >30 kg, repeat doses of 1 mg every 30-45 seconds to a maximum cumulative dose of 10 mg. In adults, the average edrophonium dose to show positive responses is approximately 3.3 mg for ptosis and approximately 2.6 mg for oculomotor symptoms. Side effects include nausea and emesis; light-headedness from bradycardia (atropine is the antidote) and bronchospasm are less common side effects. The edrophonium test may be done by intramuscular or subcutaneous injection but may require modification of dosing.
- ◆ Effects should be seen within 10 sec and disappear within 120 sec. Weakness is measured as, for example, the distance between the upper and lower eyelids before and after administration, degree of external ophthalmoplegia, or ability to swallow a sip of water.
- ◆ Long-acting cholinesterase inhibitors, such as pyridostigmine (Mestinon), are generally not as useful for the acute assessment of myasthenic weakness. The neostigmine (Prostigmin) test may be used (as outlined later) but might not be as definitively diagnostic as the edrophonium test.
Children Younger Than Two Years of Age
- ◆ Infants ideally should have a specific fatigable weakness that can be measured, such as ptosis of the eyelids, dysphagia, and inability of the cervical muscles to support the head. Nonspecific generalized weakness without cranial nerve motor deficits makes it less easy to assess results but may be a criterion at times.
- ◆ An intravenous line should be started as a rapid route for medications in the event of an adverse effect of the test medication.
- ◆ Electrocardiographic monitoring is recommended during the test.
- ◆ Pretreatment with atropine sulfate to block the muscarinic effects of the test medication is not recommended, but atropine sulfate should be available at the bedside in a prepared syringe. If needed, it should be administered intravenously in a dose of 0.01 mg/kg.
- ◆ Edrophonium is not recommended for use in infants; its effect is too brief for objective assessment, and an increased incidence of acute cardiac arrhythmias is reported in infants, especially neonates, with this drug.
- ◆ Prostigmin methyl sulfate (neostigmine) is administered intramuscularly at a dose of 0.04 mg/kg. If the result is negative or equivocal, another dose of 0.04 mg/kg may be administered 4 hr after the first dose (a typical dose is 0.5-1.5 mg). The peak effect is seen in 20-40 min. Intravenous Prostigmin is contraindicated because of the risk of cardiac arrhythmias, including fatal ventricular fibrillation, especially in young infants.
- ◆ Long-acting cholinesterase inhibitors administered orally, such as pyridostigmine (Mestinon), are generally not as useful for the acute assessment of myasthenic weakness because the onset and duration are less predictable.
The test should be performed in the emergency department, hospital ward, or intensive care unit; the important issue is preparation for potential complications such as cardiac arrhythmia or cholinergic crisis, as outlined.
Treatment
Some patients with mild MG require no treatment. Cholinesterase-inhibiting drugs are the primary therapeutic agents. Pyridostigmine bromide (Mestinon) may be given orally starting at 0.5-1 mg/kg per dose every 4-6 hr while the patient is awake, to a maximum of 60 mg per dose. The maximum daily recommended dose is 7 mg/kg/day, with most adults achieving effect with total daily doses of <960 mg per day, divided in 4-8 doses. Pyridostigmine is given in short-acting forms, and can also be used in a long-acting form at bedtime for patients with more weakness upon awakening in the morning. Overdoses of cholinesterase inhibitors produce cholinergic crises with symptoms such as increased secretions, diarrhea, and cramping; atropine blocks the muscarinic effects but does not block the nicotinic effects that produce additional skeletal muscle weakness. In the rare familial MG caused by the absence of endplate acetylcholinesterase, cholinesterase inhibitors are not helpful and often cause increased weakness; these patients can be treated with ephedrine or diaminopyridine, both of which increase ACh release from terminal axons.
Because of the autoimmune basis of the disease, long-term steroid treatment with prednisone may be effective. Thymectomy should be considered and might provide a cure. Thymectomy is most effective in patients who have high titers of anti-AChR antibodies in the plasma and who have been symptomatic for < 2 yr. Thymectomy is ineffective in congenital and familial forms of MG. Treatment of hypothyroidism usually abolishes an associated myasthenia without the use of cholinesterase inhibitors or steroids.
If the specific genetic mutation can be identified in a patient with one of the CMSs, specific therapeutic approaches are available for some that differ from the treatments listed above.
Plasmapheresis is effective treatment in some children, particularly those who do not respond to steroids, but plasma exchange therapy provides only temporary remission. Intravenous immunoglobulin is beneficial and should be tried before plasmapheresis because it is less invasive. Plasmapheresis and intravenous immunoglobulin appear to be most effective in patients with high circulating levels of anti-AChR antibodies. Refractory patients, as well as patients with MuSK-related MG, might respond more effectively to rituximab, a monoclonal antibody to the B-cell CD20 antigen.
Neonates with transient maternally transmitted MG require cholinesterase inhibitors for only a few days or occasionally for a few weeks, especially to allow feeding. No other treatment is usually necessary. In non–maternally transmitted congenital MG, identification of the specific molecular defect is important for treatment; Table 630.6 summarizes specific therapies for each type.
Table 630.6
Potential Therapies in Congenital Myasthenic Syndromes
Modified from Eyemard B, Hantaï D, Estounet B: Congenital myasthenic syndromes, Handb Clin Neurol 113:1469-1480, 2013.
Complications
Children with MG do not tolerate neuromuscular-blocking drugs, such as succinylcholine and pancuronium, and may be paralyzed for weeks after a single dose. An anesthesiologist should carefully review myasthenic patients who require a surgical anesthetic, and such anesthetics should be administered only by an experienced physician/anesthesiologist. Also, certain antibiotics can potentiate myasthenia and should be avoided; these include the aminoglycosides, beta blocking agents, procainamide, chloroquine, and fluoroquinolones.
Prognosis
Some patients with autoimmune MG experience spontaneous remission after a period of months or years; others have a permanent disease extending into adult life. Immunosuppression, thymectomy, and treatment of associated hypothyroidism might provide a cure. Genetically determined congenital myasthenic syndromes may show initial worsening in infancy but then remain static throughout childhood and into adult life.
Other Causes of Neuromuscular Blockade
Organophosphate chemicals, commonly used as insecticides, can cause a myasthenia-like syndrome in children exposed to these toxins (see Chapter 77 ).
Botulism results from ingestion of food containing the toxin of Clostridium botulinum, a Gram-positive, spore-bearing, anaerobic bacillus (see Chapter 237 ). The incubation period is short, only a few hours, and symptoms begin with nausea, vomiting, and diarrhea. Cranial nerve involvement soon follows, with diplopia, dysphagia, weak suck, facial weakness, and absent gag reflex. The mechanism is cleavage by the botulinum toxin of several of the structural glycoproteins of the wall (i.e., membrane) of synaptic vesicles within axonal terminals. These glycoproteins include synaptobrevin and synaptotagmin, but synaptophysin is resistant.
In infantile botulism, which classically presents between the ages of 4 and 7 mo, honey as well as spores from dirt (e.g., near construction sites) are common sources of contamination. The earliest signs are usually constipation, poor feeding, and then a weak cry. On evaluation, patients appear hypotonic, with facial weakness, dysphagia, and a poor gag. Generalized weakness with a risk of respiratory failure can occur. Generalized hypotonia and weakness then develop and can progress to respiratory failure. Neuromuscular blockade is documented by electromyography (EMG) with repetitive nerve stimulation. Slow repetitive nerve stimulation may show a decremental response, and baseline CMAP amplitudes may be low. With rapid repetitive nerve stimulation, there is an incremental response. EMG/repetitive nerve stimulation studies may help in confirming a diagnosis if the clinical presentation is not straightforward. However, when suspected, botulinum toxin studies should be sent preferentially from stool samples from the patient, and then treatment should be initiated as soon as possible with Botulinum Immune Globulin IV (Baby-BIG or BIG-IV). BIG-IV, which is human-derived antibotulism toxin antibodies, is approved by the United States Food and Drug Administration for the treatment of infant botulism types A and B. Early use of BIG-IV has shortened the overall length of hospitalization and improved the time to recovery. Respiratory and feeding/gavage support may be required for days or weeks until the toxin is cleared from the body.
Tick paralysis is a disorder of ACh release from axonal terminals due to a neurotoxin that blocks depolarization. It also affects large myelinated motor and sensory nerve fibers. This toxin is produced by the wood tick or dog tick, insects common in the Appalachian and Rocky Mountains of North America. The tick embeds its head into the skin, usually the scalp, and neurotoxin production is maximal about 5-6 days later. Motor symptoms include weakness, loss of coordination, and sometimes an ascending paralysis resembling Guillain-Barré syndrome. Tendon reflexes are lost. Sensory symptoms of tingling paresthesias can occur in the face and extremities. The diagnosis is confirmed by identification of the tick, and treatment involves the prompt removal of the entire tick. It is important to monitor patients closely, because some patients may show worsened respiratory symptoms for the first day after tick removal. Most patients will show rapid improvement within a few hours to a few days from the time of tick removal.
Bibliography
Abicht A, Muller JS, Lochmuller H. Congenital myasthenic syndromes . [In Gene Reviews [Internet], May 9] 2003 [updated July 14, 2016].
Ashfaq A, Bernes SM, Weidler EM, Notrica DM. Outcomes of thorascopic thymectomy in patients with juvenile myasthenia gravis. J Pediatr Surg . 2016;51:1078–1083.
Beeson D. Congenital myasthenic syndromes: recent advances. Curr Opin Neurol . 2016;29:565–571.
Castro D, Derisavifard S, Anderson M, et al. Juvenile myasthenia gravis: a twenty-year experience. J Clin Neuromuscul Dis. 2013;14:95–102.
Eyemard B, Hantaï D, Estounet B. Congenital myasthenic syndromes. Handb Clin Neurol . 2013;113:1469–1480.
Felice KJ, DiMario F, Conway SR. Postinfectious myasthenia gravis: report of 2 cases. J Child Neurol . 2005;20:441–444.
Gadient P, Bolton J, Puri V. Juvenile myasthenia gravis: three case reports and a literature review. J Child Neurol . 2009;24:584–590.
Gilhus NE. Myasthenia gravis. N Engl J Med . 2016;375(26):2570–2581.
Harper CM. Congenital myasthenic syndromes. Semin Neurol . 2004;24:111–123.
Jayawant S, Parr J, Vincent A. Autoimmune myasthenia gravis. Handb Clin Neurol . 2013;113:1465–1468.
Hong Y, Skeie GO, Zisimopoulou P, et al. Juvenile-onset myasthenia gravis: autoantibody status, clinical characteristics and genetic polymorphisms. J Neurol . 2017;264:955–962.
Jovandaric MZ, Despotovic DJ, Jesic MM, Jesic MD. Neonatal outcome in pregnancies with autoimmune myasthenia gravis. Fetal Pediatr Pathol . 2016;35:167–172.
Liew WKN, Kang PB. Update on juvenile myasthenia gravis. Curr Opin Pediatr . 2013;25:694–700.
Maselli RA, Kong DZ, Bowe CM, et al. Presynaptic congenital myasthenic syndrome due to quantal release deficiency. Neurology . 2001;57:279–289.
Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol . 2009;8:475–490.
Milone M, Shen XM, Selcen D, et al. Myasthenic syndrome due to defects in rapsyn. Neurology . 2009;73:228–235.
Muller JS, Herczegfalvi A, Vilchez JJ, et al. Phenotypical spectrum of DOK7 mutations in congenital myasthenic syndromes. Brain . 2007;130:1497–1506.
Palace J, Lashley D, Newsom-Davis J, et al. Clinical features in a series of fast channel congenital myasthenia syndrome. Neuromuscul Disord . 2012;22:112–117.
Robb SA, Sewry CA, Dowling JJ, et al. Impaired neuromuscular transmission and response to acetylcholinesterase inhibitors in centronuclear myopathies. Neuromuscul Disord . 2011;21:379–386.
Rodriguez Cruz PM, Sewry C, Beeson D, et al. Congenital myopathies with secondary neuromuscular transmission defects: a case report and review of the literature. Neuromuscul Disord . 2014;24:1103–1110.
VanderPluym J, Vajsar J, Jacob FD, et al. Clinical characteristics of pediatric myasthenia: a surveillance study. Pediatrics . 2013;132:e939–e943.
Wargon I, Richard P, Kuntzer T, et al. Long-term follow-up of patients with congenital myasthenic syndrome caused by COLQ mutations. Neuromuscul Disord . 2012;22:318–324.
Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med . 2016;375(6):511–522.
Wylam ME, Anderson PM, Kuntz NL, et al. Successful treatment of refractory myasthenia gravis using rituximab: a pediatric case report. J Pediatr . 2003;143:674–677.
Zafeiriou DI, Pitt M, de Sousa C. Clinical and neurophysiological characteristics of congenital myasthenic syndromes presenting in early infancy. Brain Dev . 2004;26:47–52.
Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis. Neurology . 2007;68:837–841.
Spinal Muscular Atrophies
Goknur Haliloglu
Spinal muscular atrophy (SMA) is a degenerative disease of motor neurons that begins in fetal life and continues to be progressive in infancy and childhood. Among the autosomal recessive disorders in childhood, SMA is the most common cause of infant mortality, and is second in birth prevalence only to cystic fibrosis. The incidence of SMA is estimated to be 1 in 6,000-10,000 newborns, with a carrier frequency of approximately 1/40-1/60. It is a clinically heterogeneous, panethnic disorder. SMA is caused by a homozygous deletion in the survival motor neuron 1 (SMN1) gene on chromosome 5q13. Infrequent families with autosomal dominant inheritance are described, and a rare X-linked recessive form also occurs. There is also a separate group of clinically and genetically heterogeneous non-5q SMA forms (see Chapter 630.3 ).
The pathologic hallmark of SMA is the progressive denervation of muscle. This is compensated for in part by reinnervation from an adjacent motor unit, but giant motor units are thus created, with subsequent atrophy of muscle fibers when the reinnervating motor neuron eventually becomes involved. Motor neurons of cranial nerves III, IV, and VI to the extraocular muscles, as well as those of the sacral spinal cord innervating striated muscle of the urethral and anal sphincters, are selectively spared. The upper motor neurons (layer 5 pyramidal neurons in the cerebral cortex) also remain normal.
SMA is classified clinically into a severe infantile form, also known as Werdnig-Hoffmann disease or SMA type I; a late infantile and more slowly progressive form, SMA type II; a more chronic or juvenile form, Kugelberg-Welander disease, or SMA type III; and an adult-onset form (SMA type IV). A severe fetal form that is usually fatal in the perinatal period has been described as SMA type 0, with motor neuron degeneration demonstrated in the spinal cord as early as midgestation. These distinctions of types are based upon the age at onset, severity of weakness, maximum motor milestone achieved, and clinical course (Table 630.7 ). Some patients are transitional between types I and II or between types II and III in terms of clinical function. Of note, the SMN gene region comprises a centromeric copy containing the SMN2 gene. Although there is a correlation between the severity of disease, age at onset, and SMN2 copy number to an extent, it is believed that the phenotype of SMA spans a broad continuum without a clear delineation of subtypes.
Table 630.7
Clinical Classification of Spinal Muscular Atrophy
| SPINAL MUSCULAR ATROPHY TYPE | OTHER NAMES | AGE AT ONSET | LIFE EXPECTANCY WITH A NATURAL COURSE OF DISEASE | HIGHEST MOTOR MILESTONE ACHIEVED | OTHER FEATURES | SMN2 COPY NUMBER |
|---|---|---|---|---|---|---|
|
Type 0 (<1%) |
Very severe | Neonatal with prenatal signs | No survival beyond the first months after birth | Never sits | 1 | |
|
Type IA * (50–60%) |
Prenatal, congenital SMA, Werdnig-Hoffmann disease | Prenatal | <6 mo | Mostly unable to achieve motor milestones | 1-2 copies in 80% of the patients | |
|
Type IB * Type IC (50–60%) |
Werdnig-Hoffman disease, severe SMA (nonsitters) |
Type IB (0-3 mo) Type IC (3-6 mo) |
<2 yr without respiratory support | Never sits unsupported | 1-2 copies in 80% of the patients | |
|
Type II (30%) |
Intermediate SMA (sitters) | 6-18 mo | >2 yr ~ 70% alive at 25 yr of age | Sits independently, never stands or walks | 3 copies in >80% of patients | |
|
Type III (10%) |
Kugelberg-Welander disease, mild SMA (walkers) |
>18 mo Type IIIA (prior to 3 yr) Type IIIB (after 3 yr) |
Almost normal | Stands and walks | 3-4 copies in 96% of patients | |
|
Type IV (Adult SMA) (1%) |
Adult SMA | >21 yr | Normal | Normal | ≥4 copies |
* SMA types I, IA, IB, and IC all have a 60% proportion of total SMA.
Modified from Darras BT: Spinal muscular atrophies, Pediatr Clin North Am 62:743-766, 2015; Mercuri E, Bertini E, Iannaccone ST: Spinal muscular atrophy: controversies and challenges, Lancet Neurol 11:443-452, 2012.
Muscle biopsy does not distinguish types I and II, though type III shows a more adult than perinatal pattern of denervation and reinnervation. Type 0 can show biopsy features more similar to those of myotubular myopathy because of maturational arrest; scattered myotubes and other immature fetal fibers also are demonstrated in the muscle biopsies of patients with types I and II, but they do not predominate. Autonomic motor neurons of both the sympathetic and parasympathetic systems are not spared, but usually do not show clinical manifestations until late stages. Autonomic deficits may involve the detrusor muscle of the urinary bladder or the smooth muscle urethral and anal sphincters, in all three forms of SMA. In some patients with type I SMA and respiratory distress, there may be severe autonomic dysregulation with dysautonomia and cardiovascular collapse leading to death or to severe ischemic brain damage. The differential diagnosis is noted in Table 630.8 .
Table 630.8
ALS, amyotrophic lateral sclerosis; BMD, Becker muscular dystrophy; CIDP, chronic inflammatory polyneuropathy; DMD, Duchenne muscular dystrophy; LGMD, limb–girdle muscular dystrophy; SMARD1, spinal muscular atrophy with respiratory distress 1.
From Darras BT, Monani UR, De Vivo DC: Genetic disorders affecting the motor neuron: spinal muscular atrophy. In Swaiman KF, Ashwal S, Ferriero DM, et al (eds): Swaiman's pediatric neurology, 6/e, Elsevier, 2018, Philadelphia, Box 139-1.
Etiology
The cause of SMA is genetic as an autosomal recessive mendelian trait. It appears to be a pathologic continuation of a process of programmed cell death (apoptosis) that is normal in embryonic life. A surplus of motor neuroblasts and other neurons is generated from primitive neuroectoderm, but only about half survive and mature to become neurons; the excess cells have a limited life cycle and degenerate. If the process that arrests physiologic cell death fails to intervene by a certain stage, neuronal death can continue in late fetal life and postnatally. The survivor motor neuron gene (SMN) arrests apoptosis of motor neuroblasts. Unlike most genes that are highly conserved in evolution, SMN is a uniquely mammalian gene. An additional function of SMN, both centrally and peripherally, is to transport RNA-binding proteins to the axonal growth cone to ensure an adequate amount of protein-encoding transcripts essential for growth cone mobility, both during fetal development and in postnatal synaptic remodeling.
Clinical Manifestations and Course
The cardinal features of the classic, most common phenotype, SMA type I,
can be summarized as a presentation before the age of 6 mo with severe hypotonia (Fig. 630.1
); symmetric generalized muscle
weakness affecting the lower limbs more than the upper limbs, proximal more than distal; frog-leg posture; absence of deep tendon reflexes; tongue fasciculations; and selective involvement of the axial and intercostal muscles but sparing of diaphragm. SMA is in the differential diagnosis list of floppy infant syndrome (see Chapter 628
). Due to the involvement of the intercostal respiratory muscles, there is a typical paradoxical abdominal breathing pattern, bell-shaped chest, and weak cough (
Video 630.1
 ). Infants lie flaccid with little movement, unable to overcome gravity, and lack head control. These infants rarely achieve improvements of motor function and acquire motor developmental milestones (see Fig. 626.1
in Chapter 626
). In contrast to their severe weakness and floppiness, infants with SMA type I have an alert and bright expression with preserved cognitive functions. There is no involvement of the facial and extraocular muscles at presentation, although facial weakness does occur at later stages of the disease.
). Infants lie flaccid with little movement, unable to overcome gravity, and lack head control. These infants rarely achieve improvements of motor function and acquire motor developmental milestones (see Fig. 626.1
in Chapter 626
). In contrast to their severe weakness and floppiness, infants with SMA type I have an alert and bright expression with preserved cognitive functions. There is no involvement of the facial and extraocular muscles at presentation, although facial weakness does occur at later stages of the disease.
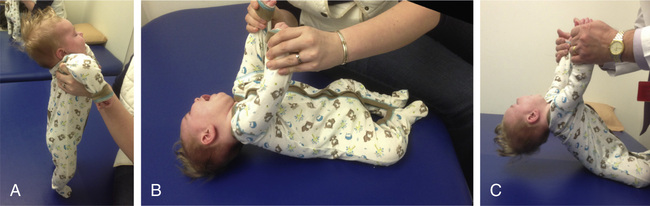
SMA type I is not homogeneous within itself. At least three clinical subgroups can be defined as (1) severe weakness from birth or the neonatal period; head control is never achieved; (2) presentation after the neonatal period, within the first 2 mo; head control is never achieved; and (3) onset after the neonatal period but head control is achieved, and some of the infants may gain the ability to sit with support. There may be a range of clinical presentations and courses of respiratory involvement and swallowing and sucking difficulties in this fragile group of SMA type I patients.
Infants with SMA type I develop respiratory failure within the first 2 yr of life, and without respiratory and nutritional support, they usually do not survive beyond their second birthday. A multidisciplinary approach (respiratory, gastrointestinal, and orthopedic interventions) combined with noninvasive ventilatory support (NIV) and enteral feeding have changed the natural course of the disease over the years. To date, the median time to either death or full-time noninvasive ventilation (NIV > 16 hr/day) is 13.5 mo with improved supportive respiratory and nutritional care. Infants who are symptomatic prenatally or at birth are classified as having a rare phenotype, SMA type 0 (<1%); they can present with severe muscle weakness, respiratory distress, feeding problems, and cranial nerve involvement. Congenital contractures, ranging from simple clubfoot to generalized arthrogryposis, occur in approximately 10% of severely involved neonates (see Chapter 626.10 ). There is a perception of decreased intrauterine movements by the mother, and these infants usually die within the first months of life. Although motor neurons are primarily affected tissue in SMA, other tissues, including those of the brain, cardiac system, vascular system, and even sensory nerves, may also contribute to the overall phenotype, especially in the most severe forms of the disease. Early-stage developmental congenital heart defects described in severe SMA patients, generally carrying one copy of SMN2, include atrial septal defects, a dilated right ventricle, ventricular septal defects, and hypoplastic left heart syndrome. These patients are also prone to possible involvement of the autonomic nervous system, which may result in arrhythmia and sudden death. Vasculopathy can be another rare presentation, and ulceration and necrosis of the fingers and toes have also been described in two severe type I SMA patients.
In type II SMA, affected infants are usually able to suck and swallow, and respiration is adequate in early infancy. Developmental delay in gross motor milestones or stagnation of motor development between the ages of 6 and 18 mo is rather typical for this form. Proximal muscle weakness is again more prominent in the lower extremities compared with the upper extremities. Patients can sit without support but are unable to walk independently. These children show progressive weakness, but many survive into the school years or beyond, although they are confined to an electric wheelchair and severely handicapped. Nasal speech and problems with deglutition develop later. Respiratory complications are less severe and develop later during the course of the disease. Scoliosis becomes a major complication in many patients with long survival times. Gastroesophageal reflux may lead to malnutrition or to aspiration with acute airway obstruction or pneumonia.
Kugelberg-Welander disease
is the mildest SMA (type III),
and patients can appear normal in infancy. The progressive weakness is proximal in distribution, particularly involving the shoulder girdle muscles. Patients are ambulatory and develop a variable course of proximal muscle weakness after the age of 18 mo. There may be a transition to SMA type II, and loss of ambulation can occur at some point during the course of the disease. Symptoms of bulbar muscle weakness are rare. Patients with this form of SMA may have muscular hypertrophy rather than atrophy, and it may easily be confused with a muscular dystrophy (
Video 630.2
). Longevity can extend well into adult life. Fasciculations are a specific clinical sign of the denervation of muscle. In thin children, they may be seen in the deltoid and biceps brachii muscles and occasionally the quadriceps femoris muscles, but the continuous, involuntary, worm-like movements may be masked by a thick pad of subcutaneous fat. Fasciculations are best observed in the tongue, where almost no subcutaneous connective tissue separates the muscular layer from the epithelium. If the intrinsic lingual muscles are contracted, as in crying or when the tongue protrudes, fasciculations are more difficult to see than when the tongue is relaxed. Cramps and myalgias of appendicular and axial muscles are common, especially in later stages, and problems of micturition may be present, though adolescent patients may be too embarrassed to state them unless the physician directly inquires.
The outstretched fingers of children with SMA often show a characteristic tremor (polyminimyoclonus)
owing to fasciculations and weakness (
Video 630.3
). It should not be confused with a cerebellar tremor.
The adult phenotype of the disease is SMA type IV, which is characterized by a mild muscle weakness with an onset usually in the second or third decade of life.
There may be an intrafamilial variability in the clinical expression of the disease.
The intelligence is normal, and children often appear brighter than their normal peers because the effort they cannot put into physical activities is redirected to intellectual development, and they are often exposed to adult speech more than to juvenile language because of the social repercussions of the disease. Progressive deterioration of ambulation and the high risk of falling and fracturing long bones or the pelvis eventually require use of a wheelchair; an electric wheelchair often is needed because weakness of the upper extremities does not allow the patient to manually push the wheels. Progressive scoliosis is another serious complication and may have a further adverse effect on respiration.
Laboratory Findings
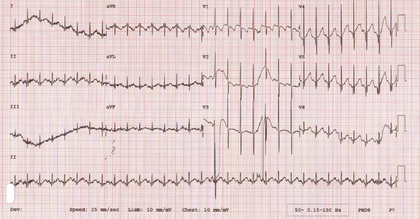
The serum creatine kinase (CK) level may be normal, but more commonly is mildly elevated (up to 2- to 4-fold), but usually not more than 10 times the normal upper limit. The chest x-ray in early-onset disease may demonstrate thin ribs. Electrocardiography (EKG) may serve as a simple and practical tool in patients with SMA to demonstrate a baseline tremor as an artefact representing muscle fibrillations more prominent on lead II (Fig. 630.2 ). Although seen in mainly lower motor neuron diseases, including poliomyelitis, recognition of this EKG pattern may prevent further electrophysiologic tests (electromyography [EMG] and nerve conduction studies [NCSs]) in SMA patients. Electrophysiologic studies (EMG-NCS) should be reserved for selected atypical patients. The results of motor nerve conduction studies are normal, except for mild slowing in terminal stages of the disease, an important feature distinguishing SMA from peripheral neuropathy. EMG shows fibrillation potentials and other signs of the denervation of muscle. There is no need for a muscle biopsy, which demonstrates a neurogenic pattern with group atrophy in all forms of SMA.
Diagnosis
The simplest, most definitive first-step diagnostic test in a patient with a clinical suspicion of SMA and normal and/or mildly elevated serum CK levels, is a molecular genetic marker in the blood for the homozygous deletion in SMN1 (Table 630.9 ). The current gold standard is SMN1 deletion/mutation and SMN2 copy number testing, with a minimal standard of SMN1 deletion testing. The absence of SMN1 exon 7 (with or without deletion of exon 8) confirms the diagnosis of SMA. The genetic test for SMA has a 95% sensitivity and nearly 100% specificity (see Table 630.9 ). Real-time polymerase chain reaction (PCR) or multiplex ligation-dependent probe amplification (MLPA) tests give quick and reliable SMN1 gene copy numbers. Semiquantitative assays improve the diagnostic sensitivity up to 98%. According to different scenarios, for example, if the patient has a single SMN1 copy, the coding region of the second undeleted allele should be sequenced to identify the second causative mutation, including point mutations, insertions, and deletions. Of note, in ~ 30% of patients with a clinical picture, mutations are not detected in the SMN1/SMN2 coding region, which is more common for type III SMA patients. Direct sequencing of the gene is also recommended in patients with a clinical diagnosis, two SMN1 copies, and a consanguineous background.
Table 630.9
| TYPE OF MUTATION | TEST APPLIED | MUTATION DETECTION RATE |
|---|---|---|
| Homozygous deletion of exon 7 * |
SMN1 Targeted mutation analysis Polymerase chain reaction/restriction enzyme analysis or multiplex ligation probe amplification methodologies |
Approximately 95–98% |
|
Compound heterozygosity (deletion of SMN1 exon 7 [allele 1] and an intragenic mutation of SMN1 † [allele 2]) |
Targeted mutation analysis combined with SMN1 sequence analysis ‡ | 2–5% |
| SMN2 copy number ‖ | Quantitative polymerase chain reaction analysis and other methodologies ¶ | Not available |
* Testing for exon 8 deletion is not necessary.
† Small intragenic deletions/insertions and nonsense, missense, and splice site mutations.
‡ Whole-gene deletions/duplications are not detected.
‖ SMN2 copy number ranges from 0 to 5.
¶ MLPA, long-range PCR, CMA that includes the SMN1 , SMN2 chromosomal segment.
From Darras BT: Spinal muscular atrophies, Pediatr Clin North Am 62: 743-766, 2015; Adapted from Markowitz JA, Singh P, Darras BT: Spinal muscular atrophy: a clinical and research update, Pediatr Neurol 46:1-12, 2012.
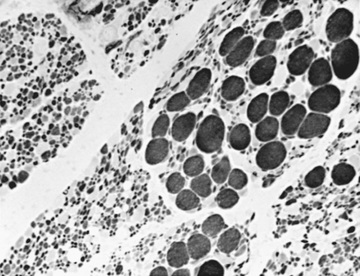
Muscle biopsy used to be the diagnostic test before the genetic marker from blood samples became available, and muscle biopsy now is used more selectively in patients showing equivocal or negative genetic findings. The muscle biopsy in infancy reveals a characteristic pattern of perinatal denervation that is unlike that of mature muscle. Groups of giant type I fibers are mixed with fascicles of severely atrophic fibers of both histochemical types (Fig. 630.3 ). Scattered immature myofibers resembling myotubes also are demonstrated. In juvenile SMA, the pattern may be more similar to adult muscle that has undergone many cycles of denervation and reinnervation. Neurogenic changes in muscle also may be demonstrated by EMG, but the results are less definitive than by muscle biopsy in infancy. Sural nerve biopsy is now performed only occasionally, but shows mild sensory neuropathic changes, and the sensory nerve conduction velocity may be slowed; hypertrophy of unmyelinated axons also is seen. At autopsy, mild degenerative changes are seen in sensory neurons of dorsal root ganglia and in somatosensory nuclei of the thalamus, but these alterations are not perceived clinically as a sensory loss or paresthesias. The most pronounced neuropathologic lesions are the extensive neuronal degeneration and gliosis in the ventral horns of the spinal cord and brainstem motor nuclei, especially the hypoglossal nucleus. On rare instances, the clinical features of an SMA-like presentation may be a feature of mitochondrial diseases (SCO2, DGUOK, and TK2 mutations). SCO2 encodes one of the COX assembly proteins, and the latter two gene mutations are associated with mitochondrial DNA depletion syndromes. Unexpectedly elevated serum CK levels at some point in the clinical course of these patients can be a clue to considering a mitochondrial disease in the differential diagnosis. Depending on the stage and progression of the disease, a muscle biopsy demonstrating ragged red fibers and COX-deficient fibers, may help in the differential diagnosis.
Genetics
Molecular genetic diagnosis by DNA probes in blood samples or in muscle biopsy or chorionic villi tissues is available for the diagnosis of suspected cases and for prenatal diagnosis. Most cases are inherited as an autosomal recessive trait.
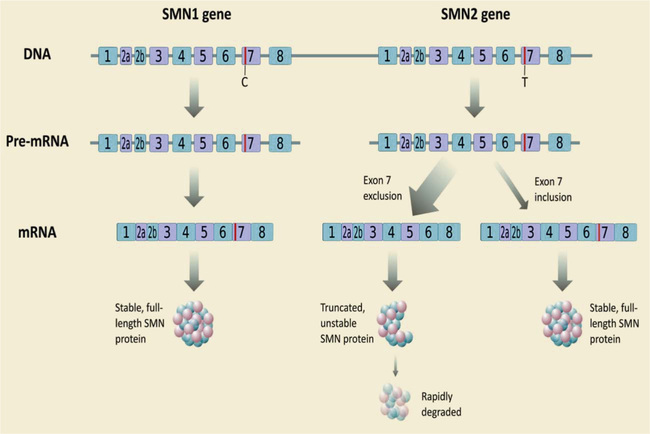
The genetic locus for all three of the common forms of SMA is on chromosome 5, a deletion at the 5q11-q13 locus, indicating that they are variants of the same disease rather than different diseases. The affected SMN1 gene has a molecular weight of 38 kDa and contains 8 exons that span 20 kb and telomeric and centromeric exons that differ only by 5 bp and produce a transcript encoding 294 amino acids. SMN1 is duplicated in a highly homologous gene called SMN2, and both genes are transcribed. SMN2 remains present in all patients with SMA, but cannot fully compensate the SMN1 defect. However, a molecular basis for correlation between the SMN2 copy number and clinical severity of the SMA is the capability of SMN2 to encode a small amount of an identical SMN protein. The critical difference between SMN1 and SMN2 is a cytosine (C) to thymine (T) transition in exon 7 of SMN2 (Fig. 630.4 ).
The SMN complex has a role in the formation of small nuclear ribonucleoproteins (snRNPs), through assembly of Sm-proteins (a distinctive family of RNA-associated small proteins) onto small nuclear RNAs (SnRNAs). SMN deficiency and reduced snRNP assembly capacity are hypothesized to cause aberrant splicing or transport of RNPs to motor neurons. Dysregulation of genes involved in synaptogenesis and the maintenance of neuromuscular junctions in animal studies possibly explains the special vulnerability of motor neurons. A second view is that, independent from snRNPs assembly, SMN may have a motor neuron–specific role such as mRNA transport along the axon. Considering the length of axons, integrity of neuromuscular junctions, and interactions with skeletal muscle, SMN protein deficiency may be detrimental for motor neurons. SMN is localized in bright-dot–like structures, called gems (gemini of Cajal bodies) in the nucleus. It is also present in other cellular structures such as Golgi bodies, cell membranes, and especially the axon and growth cone compartments of motor neurons. Owing to its localization in ribonucleoprotein granules in neurites and growth cones in neurons, SMN modulates axonal growth and localization of β-actin messenger ribonucleic acid (mRNA) in growth cones of motor neurons. Early functional impairment of sensory-motor connectivity in animal models showed that motor neuron loss follows afferent synapse loss with the same temporal and topographic pattern, with changes occurring first in motor neurons innervating the proximal muscles and axial muscles, and then the distal muscles. The third view connects SMN function, in a direct or indirect manner, to actin dynamics and actin-dependent processes. There is an expansion in the spectrum of SMN function, including actin dynamics, vesicular transport, protein translation and trafficking, mRNA transport, apoptosis, and many others, which are reflected to widespread pathophysiologic findings described in humans and animal models (Fig. 630.5 ).
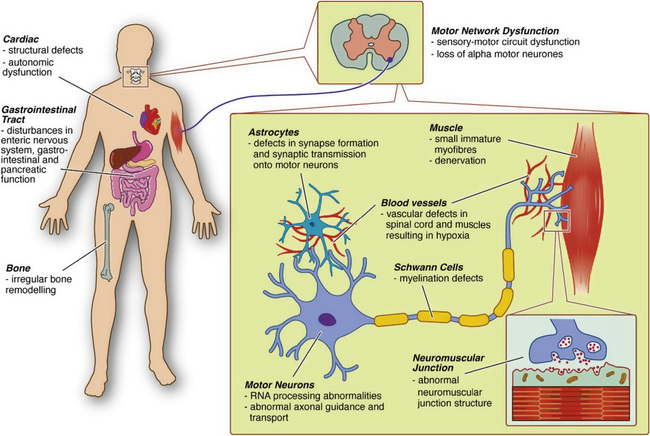
The severity of the disease is inversely correlated with the amount of functional SMN protein. In that sense, other than the SMN2 copy number, which is the major protective modifier, the severity of the phenotype can be also influenced by other genetic modifiers, including plastin 3 and neurocalcin. Nutritional deficiency, oxidative stress, and hypoxia may contribute to widespread splicing alterations, including SMN2, and affect the disease progression.
Carrier testing by dose analysis is available and is based on semiquantitative real-time PCR or MLPA. In this context, limitations of the molecular testing, difficulties in predicting the offspring's phenotype based solely on the SMN2 copy number, and the effect on reproductive planning should be considered.
Newborn screening is aimed at identifying presymptomatic SMA patients. Deoxyribonucleic acid (DNA) extraction from newborn blood spots, followed by either liquid microbead array or real-time PCR techniques, have been developed, which helps to identify SMN1 homozygous deletions. Challenges in newborn screening include the inability to detect carriers of heterozygous deletions of SMN1 , and SMN2 copy numbers.
Management
A multidisciplinary and supportive approach is the key in the management of a patient with SMA. Follow-up coordination should be managed by an expert in neuromuscular disorders, and the team ideally should include a pediatric and an adult neurologist, respiratory physicians, geneticists, gastroenterologists, palliative care physicians, rehabilitation specialists, orthopedic surgeons, and allied healthcare professionals. The consensus statement for the standard of care in SMA includes ethical and palliative care sections. Despite increased standards and technologic advances, there is a high variability in terms of ventilatory support, nutritional support, and scoliosis surgery. In terms of advances in disease-modifying treatments that will change the natural course of the disease, care and treatment options should be discussed clearly with the family and/or patients to define expectations, the quality of life, and palliative care issues. Because SMA is a dynamic disease by nature, a proactive plan should be introduced in almost every care subtopic (Table 630.10 ). Overall, supportive therapy should aim to help the patient to be as functionally independent as possible.
Table 630.10
Management of Spinal Muscular Atrophy
| PROBLEMS | ASSESSMENTS | INTERVENTIONS | |
|---|---|---|---|
| Respiratory |
Referral to respiratory specialist Airway clearance techniques and cough assistance (chest physiotherapy, postural drainage, mechanical or manual cough assistance) Respiratory devices–noninvasive ventilation (nocturnal and/or daytime if indicated)* Antibiotics intensified airway clearance Increased ventilation support* |
||
| Nutritional | |||
| Orthopedic physiotherapy |
Equipment to assist with mobility, self-care, and function Physiotherapy, standing frames, orthoses Spinal surgery † Stretching, adequate positioning Exercises with low resistance or high repetition Evaluation with functional motor scales developed for SMA patients |
||
| Other organ involvement | |||
| Psychological | Assess for depression/anxiety |
* The appropriate level of interventional support to prolong life, particularly in SMA type I, is controversial, and the consensus statement recognizes the importance of discussions with the family to explore and define the potential quality of life and palliative care issues. The philosophy and introduction of proactive respiratory support in patients with SMA type I varies considerably and practice varies internationally.
† There is no consensus on management of scoliosis or hip subluxation/dislocation in nonambulant patients. If there is no fast progression of scoliosis, surgery should be delayed until at least 10-12 yr of age to allow for optimum growth. Otherwise, growing rods and vertical expandable prosthetic titanium ribs should be considered. Possibility of intrathecal administration of drugs should be taken into account.
The management of SMA incorporates a multidisciplinary and supportive approach, including neurologists (adult and pediatric), respiratory physicians, geneticists, gastroenterologists, palliative care physicians, rehabilitation specialists, orthopedic surgeons, and allied health care professionals.
Modified from Farrar MA, Park SB, Vucic S, et al: Emerging therapies and challenges in spinal muscular atrophy, Ann Neurol, 81:355-368, 2017; and from Pechman A, Kirschner J: Diagnosis and new treatment avenues in spinal muscular atrophy, Neuropediatrics 48(4):273-281, 2017.
Therapeutic Advances
SMN-antisense oligonucleotide (ASO), nusinersen, administered intrathecally is approved by the U.S. FDA and by the European Medicines Agency for all types of SMA patients. It modifies the splicing of SMN2 by inducing an increase in exon 7 retention in SMN2 pre-mRNA, which finally allows a protein product similar to SMN1 . Phase 1 to phase 3 studies in SMA type I (0-6 mo) and SMA type II/III patients (2-14 yr), showed favorable safety, tolerability, and encouraging clinical efficacy. The primary endpoint was met in each study at interim analysis with statistically significant improvement in motor milestones. In an ongoing, open-label clinical trial, the effect has also been tested in presymptomatic patients with SMA, with favorable results so far. Long-term follow-up is necessary to evaluate the effect of this treatment at different stages of the disease. Scoliosis, surgical interventions, and severe respiratory disease may complicate the lumbar puncture procedure.
Orally administered small molecules (RG7916 and LMI070) are also able to promote exon 7 inclusion and are currently under investigation.
Another therapeutic approach is gene therapy (AVXS-101), to replace SMN1 and thus increase the production of the full-length SMN protein. Adeno-associated viral vector (AAV-9) is able to transport a functional copy of SMN1 crossing the blood–brain barrier. An interim analysis of a phase I clinical trial of intravenously administered AVXS-101 in SMA type I patients revealed a safety profile and efficacy with achievement of motor milestones.
In terms of neuroprotective strategies, phase 2 studies with oral olesoxime (TRO19622) in the SMA type II or nonambulant type III patient population showed stabilization or improvement compared with placebo. Although the primary endpoint was not met, olesoxime was safe and might be used in combination with other drugs targeting other mechanisms of the disease. The role of exercise as a neuroprotective measure is also under investigation. Current clinical trials also include fast skeletal troponin activator (CK2127107), pyridostigmine, and 4-aminopyridine to enhance nerve or muscle function.
Genetic counseling, depending on carrier screening tests, or in the presence of a previously affected child with SMA, may help with reproductive planning (prenatal diagnosis or preimplantation diagnosis). Prenatal diagnosis should be offered to families with an index patient in the family (recurrence risk is 25%), and antenatal screening by chorionic villus sampling between the 10th and 12th gestational week of pregnancy may serve for SMN1 deletion/mutation analysis.
Bibliography
Andersson HC. Newborn screening for spinal muscular atrophy and lysosomal storage disorders takes advantage of novel therapies. J Pediatr . 2017;190:9–10.
Bertini E, Dessaud E, Mercuri E, et al. Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type III spinal muscular atrophy: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol . 2017;16(7):513–522.
Castro D, Iannaccone ST. Spinal muscular atrophy: therapeutic strategies. Curr Treat Options Neurol . 2014;16(11):316.
Chanprasert S, Wang J, Weng SW, et al. Molecular and clinical characterization of the myopathic form of mitochondrial DNA depletion syndrome caused by mutations in the thymidine kinase (TK2 ) gene. Mol Genet Metab . 2013;110:153–161.
Chien YH, Chiang SC, Weng WC, et al. Presymptomatic diagnosis of spinal muscular atrophy through newborn screening. J Pediatr . 2017;190:124–129.
Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a phase 1 study of nusinersen (ISIS-SMNRX ) in children with spinal muscular atrophy. Neurology . 2016;86:890–897.
D'Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis . 2011;6:71.
Darras BT. Spinal muscular atrophies. Pediatr Clin North Am . 2015;62:743–766.
De Sanctis R, Coratti G, Pasternak A, et al. Developmental milestones in type I spinal muscular atrophy. Neuromuscul Disord . 2016;26:754–759.
Farrar MA, Kiernan MC. The genetics of spinal muscular atrophy: progress and challenges. Neurother . 2015;12:290–302.
Farrar MA, Park SB, Vucic S, et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol . 2017;81:355–368.
Finkel RS, Bertini E, Muntoni F, et al. Outcome measures and clinical trial readiness in spinal muscular atrophy 7-9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord . 2015;25:593–602.
Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open label, dose escalating study. Lancet . 2016;388:3017–3026.
Finkel RS, McDermott MP, Kaufmann P, et al. Observational study of spinal muscular atrophy type I and implementations for clinical trials. Neurology . 2014;83:810–817.
Finkel RS, Mercuri E, Darras AM, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med . 2017;377(18):1723–1732.
Finkel RS, Sejersen T, Mercuri E, et al. Revisiting the consensus on standards of care in SMA February 19-21 2016, Naarden, The Netherlands. Neuromuscul Disord . 2017;27(6):596–605.
Food and Drug Administration. FDA approves first drug for spinal muscular atrophy . https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm534611.htm .
Grohmann K, Varon R, Stolz P, et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann Neurol . 2003;54:719–724.
Hache M, Swoboda KJ, Sethna N, et al. Intrathecal injections in children with spinal muscular atrophy: nusinersen clinical trial experience. J Child Neurol . 2016;31:899–906.
Haliloglu G, Gungor M, Anlar B. The role of electrocardiography in the diagnosis of spinal muscular atrophy type III. Pediatrics . 2015;166:1092.
Harding BN, Kariya S, Manani UR, et al. Spectrum of neuropathophysiology in spinal muscular atrophy type I. J Neuropathol Exp Neurol . 2014;74:15–24.
Kaifer KA, Villalon E, Osman EY, et al. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight . 2017;2(5):e89970.
Kolb SJ, Coffey CS, Yankey JW, et al. Baseline results of the NeuroNEXT spinal muscular atrophy infant biomarker study. Ann Clin Transl Neurol . 2016;21:132–145.
Markowitz JA, Singh P, Darras BT. Spinal muscular atrophy: a clinical and research update. Pediatr Neurol . 2012;46:1–12.
Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med . 2017;377(18):1713–1722.
Mercuri E, Bertini E, Iannaccone SI. Spinal muscular atrophy: controversies and challenges. Lancet Neurol . 2012;11:443–452.
Messina MF, Meddina S, Gaeta M, et al. Infantile spinal muscular atrophy with respiratory distress type I (SMARD 1): an atypical phenotype and review of the literature. Eur J Paediatr Neurol . 2012;16:90–94.
Nurputra DK, San Lai P, Harahap NIF, et al. Spinal muscular atrophy: from gene discovery to clinical trials. Ann Hum Genet . 2013;77:435–463.
Pechmann A, Kirschner J. Diagnosis and new treatment avenues in spinal muscular atrophy. Neuropediatrics . 2017;48(4):273–291.
Phan HC, Taylor JL, Hannon H, Howell R. Newborn screening for spinal muscular atrophy: anticipating an imminent need. Semin Perinatol . 2015;39:217–229.
Roper H, Quinlivan R. On behalf of workshop participants: implementation of “the consensus statement for the standard of care in spinal muscular atrophy” when applied to infants with severe type 1 SMA in the UK. Arch Dis Child . 2009;95:845–849.
Sarnat HB, Trevenen CL. Motor neuron degeneration in a 20-week male fetus: spinal muscular atrophy type 0. Can J Neurol Sci . 2007;34:215–220.
Scoto M, Finkel RS, Mercuri E, Muntoni F. Therapeutic approaches for spinal muscular atrophy (SMA). Gene Ther . 2017;24(9):514–519.
Shababi M, Habibi J, Yang HT, et al. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Molec Genet . 2010;19:4059–4071.
Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol . 2005;57:704–712.
Teoh HL, Solyom A, Schuchman EH, et al. Polyarticular arthritis and spinal muscular atrophy in acid ceramidase deficiency. Pediatrics . 2016;138(4):e20161068.
The Medical Letter. Nusinersen (spinraza) for spinal muscular atrophy. Med Lett . 2017;59(1517):50–52.
Toshihiro N, Takenouchi T, Fukushima H, et al. Catastrophic autonomic crisis with cardiovascular collapse in spinal muscular atrophy with respiratory distress type 1. J Child Neurol . 2013;28:949–951.
Viollet L, Melki J. Spinal muscular atrophies. Handb Clin Neurol . 2013;113:1395–1411.
Wang CH, Finkel RS, Bertini E, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol . 2007;22:1027–1049.
Other Motor Neuron Diseases
Goknur Haliloglu
Motor neuron diseases (MNDs) are a heterogeneous group of progressive neurodegenerative disorders characterized by upper and lower neuron dysfunction, with an onset from birth to adulthood. A variety of causes, including hereditary, immune-mediated, infectious, paraneoplastic, and sporadic diseases, should be considered.
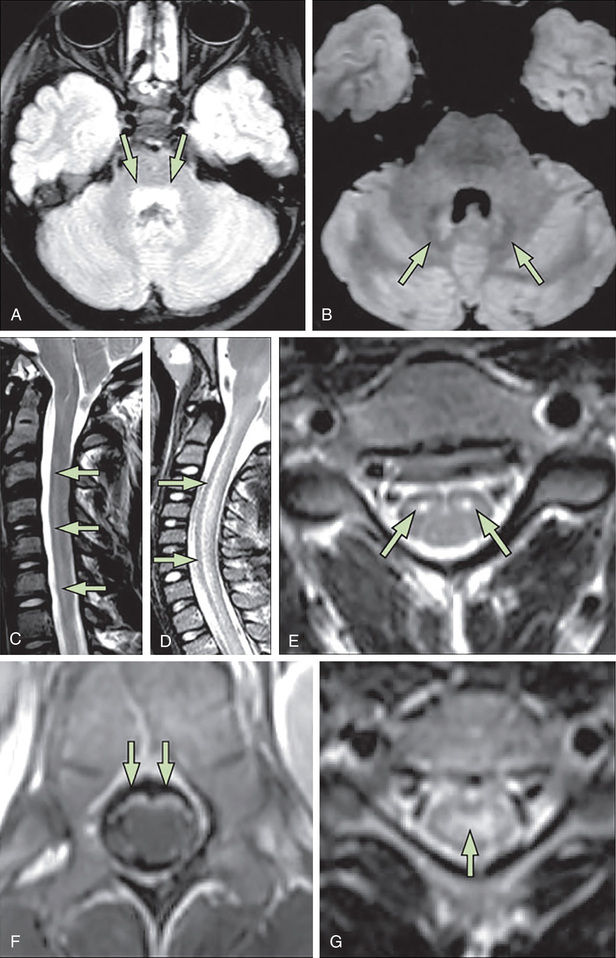
Acute flaccid paralysis is the most common presentation of MND in children; it may occur in outbreaks. Poliomyelitis used to be a major cause of chronic disability, but with the routine use of polio vaccine, this viral infection is rare (see Chapter 276 ). Other enteroviruses, such as coxsackievirus and echovirus, or the live polio vaccine virus can also cause an acute infection of motor neurons, with symptoms and signs similar to poliomyelitis, although usually milder. Specific PCR tests and viral cultures of cerebrospinal fluid are diagnostic. A clustering of cases of acute flaccid paralysis has been reported during outbreaks of enterovirus D68 in multiple states in children (mean age 7-11 yr). Limb weakness is often asymmetric and includes bulbar weakness, as well as cranial nerve VI and VII involvement. MRI may demonstrate longitudinal spinal cord lesions with dominant anterior horn cell involvement (Fig. 630.6 ). Cerebrospinal fluid pleocytosis and elevated protein are common. Treatments have included steroids and intravenous immunoglobulin; persistent paresis is a common sequelae. Motor neuron infection with the West Nile virus also occurs.
In children, an insidious onset, slow progression, and family history can be clues for a genetic basis. Although the most common MND in children is 5q13-associated SMA, with a typical or predominant lower motor neuron phenotype, there is a clinically and genetically heterogeneous group of MNDs that overlap with hereditary spastic paraplegias (HSPs), hereditary sensory-motor neuropathies (HSMN), and juvenile forms of amyotrophic lateral sclerosis (ALS).
A less common group of MNDs, not associated with SMN1, are called non-5q13-associated SMAs; this heterogeneous group can be associated with X-linked, autosomal dominant or autosomal recessive SMAs, distal SMAs, segmental SMAs, or distal hereditary motor neuropathies or neuronopathies. Additional features, such as deafness; epilepsy; encephalopathy; spasticity; visual impairment; or brainstem, cerebellar, gastrointestinal, or rheumatologic disorders may be indicative of a widespread involvement. These atypical SMA phenotypes can also be called SMA-plus syndromes, and they show extensive phenotypic overlap and molecular genetic heterogeneity (Table 630.11 ). Primary involvement of the upper motor neuron, with a progressive upper and lower motor neuron loss, characterize juvenile amyotrophic lateral sclerosis, which is rare and ultimately fatal.
Table 630.11
Main Forms of SMA Not Linked to SMN1 (non-5q SMA, SMA-plus Syndromes, Atypical SMAs)
AD, autosomal dominant; AR, autosomal recessive; RNA, ribonucleic acid.
Modified from Teoh HL, Carey K, Sampaio H, et al: inherited pediatric motor neuron disorders: beyond spinal muscular atrophy, Neural Plast 2017:6509493, 2017.
Parallel to advances in next-generation techniques, there has been an increase in the molecular diagnostic yield and expansion of clinical phenotypes. This further helps not only to understand the natural course and common pathophysiologic mechanisms involved, but also indicates the appropriate genetic counseling and prenatal diagnosis.
A pattern of weakness, amyotrophy, and progression (proximal or distal, bulbar or respiratory involvement), the presence of spasticity, deep tendon reflexes, and the family history should be evaluated. In contrast to typical SMA, electrophysiologic studies and electromyography (EMG) may serve as important tools to demonstrate a neurogenic basis. Multisystem assessment, including vision, hearing, and cognitive development, is required. Clinical evaluation and recognition of distinctive features will help to classify MND and consider treatable MND forms in the differential diagnosis.
SMA with respiratory distress (SMARD) is a rare autosomal recessive disease due to mutations in the gene encoding IGHMBP2 on chromosome 11q13. In contrast to classical SMA type I, predominant distal weakness with diaphragmatic palsy results in severe respiratory failure. There is usually an early presentation between 6 wk and 6 mo of age, with intrauterine growth retardation, a weak cry and suck, and congenital foot deformities. Routine chest X-ray may reveal diaphragmatic eventration, which causes early respiratory failure. Atypical patients with peripheral neuropathy and no respiratory involvement have also been described. Beyond the core symptoms, sensory and autonomic dysfunction (excessive sweating, urinary retention, constipation, and cardiac arrhythmia), seizures, and progressive cranial nerve involvement can be additional features.
Brown-Vialetto-Van Laere (BVVL) syndrome is a rare heterogeneous neurodegenerative disorder characterized by involvement of cranial nerves VII to XII, progressive facial weakness, sensorineural deafness, dysphagia, tongue amyotrophy, fasciculations, bulbar palsy, and respiratory insufficiency. It may present at all ages. Weakness of the arms and hands, optic atrophy, and ataxia may be additional presentations. The clinical presentation of Fazio-Londe syndrome is the same, and characterized by progressive bulbar palsy resulting from motor neuron degeneration more in the brainstem than the spinal cord, without sensorineural deafness.
Identification of mutations in the riboflavin transporter genes (Table 630.11 ) provided a targeted therapeutic strategy with oral or intravenous high-dose riboflavin supplementation with the customary dose being 10 mg/kg/day. The clinical response in this group may be variable, ranging from a rapid response to gradual improvement over 12 mo, clinical stabilization, or rarely no response. Recognition of abnormal acylcarnitine profiles mimicking multiple acyl-CoA dehydrogenase deficiency in BVVL should also be taken into account. A biochemical response to treatment is also evident. The common core phenotype of this treatable MND includes progressive axonal sensorimotor neuropathy (manifesting with sensory ataxia and severe weakness of the upper limbs and axial muscles with distinctly preserved muscle power of the lower limbs), hearing loss, optic atrophy, and respiratory insufficiency.
The classic form of pontocerebellar hypoplasia with SMA (PCH1) is characterized by severe hypotonia, areflexia, muscle weakness, central visual impairment, dysphagia, respiratory insufficiency, and acquired microcephaly, with a presentation in the first months of life and death in infancy. There is a wide clinical spectrum, with a severe antenatal onset representing the severe end of the spectrum with arthrogryposis and polyhydramnios.
SMA with progressive myoclonic epilepsy (SMAPME) is characterized by treatment-resistant progressive myoclonic epilepsy combined with proximal muscle weakness, areflexia, atrophy, progressive weakness, and dysphagia, followed by normal developmental milestones. Mild facial weakness, tongue fasciculations, sensorineural hearing loss, and tremor may be additional features. Rare variants include the recently described polyarticular arthritis with SMA, mild SMA without seizures, eyelid myoclonic status epilepticus, and absence and atonic seizures in adolescence.
Lethal arthrogryposis with anterior horn cell disease (LAAHD) and SMA with congenital arthrogryposis and fractures are atypical SMA forms within the fetal akinesia/hypokinesia spectrum (see Chapter 626.10 ).
A variety of mitochondrial diseases may present with an SMA-like clinical phenotype. In addition to hypotonia, weakness, and respiratory failure, there is a more extensive spectrum of multisystem involvement, such as infantile hypertrophic cardiomyopathy, hepatic failure, spasticity, Leigh-like syndrome, encephalopathy, seizures, brainstem dysfunction, global developmental delay, ptosis, and ophthalmoplegia. Lactic acidosis and increased serum CK levels may further help to include genes involved in COX assembly proteins and mitochondrial depletion syndromes in the molecular genetic workup.
SMA with lower extremity predominance (SMALED) is characterized by congenital or early-onset, proximal lower limb–predominant muscle weakness and atrophy. There is again a wide range of clinical presentations from the antenatal to adulthood periods. Spasticity and cognitive impairment can be a part of the clinical picture in some patients.
Scapuloperoneal SMA is an autosomal dominant condition defined by its selective muscle involvement, progressive distal weakness, and atrophy. Laryngeal palsy, sensorineural deafness, short stature, scoliosis, and mild limb and skeletal dysplasia may accompany the picture.
Motor neurons become involved in several metabolic diseases of the nervous system, such as gangliosidosis (Tay-Sachs disease), ceroid lipofuscinosis (Batten disease), and glycogenosis II (Pompe disease), but the signs of denervation may be minor or obscured by the more prominent involvement of other parts of the central nervous system or of muscle. Amyotrophy related to lower motor neuron degeneration is a prominent future of some multisystem disorders, such as infantile neuroaxonal dystrophy (INAD), achalasia–addisonianism–alacrima (AAA, triple A, or Allgrove syndrome), and Chédiak-Higashi syndrome (CHS).
Bibliography
Bonwitt J, Poel A, DeBolt C, et al. Acute flaccid myelitis among children—Washington, September-November 2016. MMWR . 2017;66(31):826–828.
Bosch AM, Stroek K, Abeling NG, et al. The Brown-vialetto-van laere and fazio londe syndrome revisited: natural history, genetics, treatment and future perspectives. Orphanet J Rare Dis . 2012;7:83.
Centers for Disease Control. Acute flaccid myelitis among persons aged ≤ 21 years—United States, August 1-November 13, 2014. MMWR . 2015;63(53):1243–1244.
Centers for Disease Control. Cluster of acute flaccid myelitis in five pediatric patients—maricopa county, Arizona, 2016. MMWR . 2017;66(28):758–760.
D'Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis . 2011;6:71.
Darras BT. Spinal muscular atrophies. Pediatr Clin North Am . 2015;62:743–766.
Foley AR, Menezes MP, Pandraud A, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain . 2014;137:44–56.
Greninger AL, Naccache SN, Messacar K, et al. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012-2014): a retrospective cohort study. Lancet . 2015;15(6):671–682.
Messacar K, Schreiner TL, Maloney JA, et al. A cluster of acute flaccid paralysis and cranial nerve dysfunction temporally associated with an outbreak of enterovirus D68 in children in Colorado, USA. Lancet . 2015;385:1662–1671.
Nelson GR, Bonkowsky JL, Doll E, et al. Recognition and management of acute flaccid myelitis in children. Pediatr Neurol . 2016;55:17–21.
Peeters K, Chamova T, Jordanova A. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrofies. Brain . 2014;137:2879–2896.
Teoh HL, Carey K, Sampaio H, et al. Inherited paediatric motor neuron disorders: beyond spinal muscular atrophy. Neural Plast . 2017;2017:6509493.
Van Haren K, Ayscue P, Waubant E, et al. Acute flaccid myelitis of unknown etiology in California, 2012-2015. JAMA . 2015;314(24):2663–2671.