Chapter 58 NURSING MANAGEMENT: chronic neurological problems
1. Compare and contrast tension-type, migraine and cluster headaches in terms of aetiology, clinical manifestations, multidisciplinary care and nursing management.
2. Describe the aetiology, clinical manifestations, diagnostic studies, multidisciplinary care and nursing management of seizure disorder, multiple sclerosis, Parkinson’s disease and myasthenia gravis.
3. Describe the clinical manifestations and multidisciplinary care of motor neuron disease and Huntington’s disease.
4. Explain the potential impact of chronic neurological disease on physical and psychological wellbeing.
5. Outline the major goals of treatment for the patient with a chronic, progressive neurological disease.
HEADACHE
Headache is probably the most common type of pain experienced by humans. The majority of people have functional headaches, such as migraine or tension-type headaches; the remainder have organic headaches that are caused by intracranial or extracranial disease. Headaches reduce social activities and work capacity in sufferers; the direct and indirect costs of migraine alone have been estimated to be in the order of $1 billion annually.1
Not all tissues of the cranium are sensitive to pain. The pain-sensitive structures in the head include the venous sinuses, dura, cranial blood vessels, three divisions of the trigeminal nerve (CN V), facial nerve (CN VII), glossopharyngeal nerve (CN IX), vagus nerve (CN X) and the first three cervical nerves. Thus, headache pain can arise from both intracranial and extracranial sources.
Headaches are classified using the International Headache Society (IHS) diagnostic criteria based on the characteristics of the headache and the facial pain. The primary classifications include tension-type, migraine and cluster headaches. Characteristics of these headaches are shown in Table 58-1. A patient may have more than one type of headache. History and neurological examination are diagnostic keys to determining the type of headache.

Tension-type headache
Tension-type headache, the most common type of headache, is characterised by a bilateral feeling of pressure around the head. It is estimated that up to 7 million people in Australia suffer with tension headaches.1 Tension-type headache has been called muscle-contraction, tension, psychogenic and rheumatic headache. Tension-type headaches are often subcategorised as acute or episodic and chronic.
AETIOLOGY AND PATHOPHYSIOLOGY
It was originally thought that tension-type headache was the result of sustained and painful contraction of the muscles of the scalp and neck. Recent evidence, however, does not support this mechanism in all patients with tension-type headaches. It is likely that neurovascular factors similar to those involved in migraine headaches play a role in the development of tension-type headaches.
CLINICAL MANIFESTATIONS
There is no prodromal stage (early manifestation of impending disease) in tension-type headache. The IHS classification system defines tension-type headache as involving at least two of the following characteristics: pressure or tightness sensation, mild-to-moderate severity, bilateral location or worsening with physical activity. The headache does not involve nausea or vomiting but may involve sensitivity to light (photophobia) or sound (phonophobia). The headache may occur intermittently for weeks, months or even years. Many patients can have a combination of migraine and tension-type headaches, with features of both headaches occurring simultaneously. Patients with migraine headaches may experience tension-type headaches between migraine attacks.
DIAGNOSTIC STUDIES
Careful history taking is probably the most important diagnostic tool for tension-type headache. Electromyography (EMG) may be performed. This test may reveal sustained contraction of the neck, scalp or facial muscles, but many patients may not show increased muscle tension with this test, even when the test is done during the actual headache. Conversely, patients with diagnosed migraine headaches may show increased muscle tension on EMG. If tension-type headache is present during physical examination, increased resistance to passive movement of the head and tenderness of the head and neck may be present.
Migraine headache
Migraine headache is a recurring headache characterised by unilateral or bilateral throbbing pain, a triggering event or factor, strong family history, and manifestations associated with neurological and autonomic nervous system dysfunction. The onset of migraine usually occurs in childhood or adolescence. A family history of migraine can be found in 40–60% of patients with migraine. Approximately 15% of the population in both New Zealand and Australia experience migraine headache.1,2 It is estimated that 23% of all households contain at least one migraine sufferer. The prevalence of migraine increases between the ages of 12 and 40 years and declines thereafter in both sexes.
AETIOLOGY AND PATHOPHYSIOLOGY
Although the exact cause of migraine headache is not known, evidence suggests that neurological, vascular and chemical factors are involved.3 The neurogenic model of migraine implies that a stimulus can trigger the trigeminovascular system (the trigeminal nerve and its connections to meningeal blood vessels), producing inflammation of the blood vessels and vasodilation. This vasodilation ultimately results in headache. The neurotransmitter serotonin produces cerebrovascular dilation and stimulates afferent pain fibre activation, both of which are important in promoting migraine progression.
In addition to the headache itself, migraines can be preceded by prodromal symptoms and aura. The prodromal symptoms may precede the headache phase by several hours or several days. The aura (sensation of light or warmth) of migraine is associated with ‘spreading depression’, a wave of oligaemia (diminished cerebral blood flow) beginning in the occipital lobe and spreading forwards in the brain at a rate of 2–3 mm per minute.
Migraine headaches, in many cases, have no known precipitating events. However, in some cases, the headache may be precipitated or triggered by stress, excitement, bright lights, menstruation, alcohol or certain foods (e.g. chocolate, cheese).
CLINICAL MANIFESTATIONS
Migraines are subdivided by the IHS into those with aura (formerly called classic migraine) and those without aura (formerly called common migraine). People who experience migraines with aura have been found to be at about twice the risk of having an ischaemic stroke when compared to the rest of the population (see Ch 57).
Migraine with aura is defined by the IHS as involving at least three of the following: (1) reversible aura involving brain dysfunction; (2) aura symptoms that develop gradually over more than 4 minutes, or two or more symptoms occurring in succession; (3) no aura lasts more than 60 minutes; and (4) headache follows aura within 60 minutes. Migraine with aura occurs in only 10% of migraine headache episodes. The sharply defined aura may last for 10–30 minutes before the start of the headache and may include sensory dysfunction (e.g. visual field defects, tingling or burning sensations, paraesthesias), motor dysfunction (e.g. weakness, paralysis), dizziness, confusion and even loss of consciousness. The classic aura symptom is perception of flashing lights in one quadrant of the visual field, often termed scintillating scotomata. Migraine with aura usually peaks in 1 hour and may last several hours.
The IHS classification defines migraine without aura as involving at least two of the following characteristics: (1) unilateral location; (2) pulsating quality; (3) moderate-to-severe intensity; (4) worsening with activity; and at least one of either: (a) nausea and vomiting; or (b) photophobia and phonophobia. Migraine without aura is the most common type of migraine headache. The headache itself may last several hours or days.
Clinical manifestations that might occur in migraine with and without aura are generalised oedema, irritability, pallor, nausea and vomiting, and sweating. In migraine with and without aura, the prodromal stage is not sharply defined. Prodromal symptoms can include psychic disturbances, gastrointestinal upset and changes in fluid balance.
During the headache phase, some patients with migraine may tend to ‘hibernate’; that is, they seek shelter from noise, light, odours, people and problems. The headache is described as a steady, throbbing pain that is synchronous with the pulse. However, the presentation of migraine is varied in its severity. Not all migraine headaches are disabling and many patients who have migraine headaches do not seek healthcare treatment for them. Although the headache is usually unilateral, it may switch to the opposite side in another episode.
DIAGNOSTIC STUDIES
There are no specific laboratory or radiological tests for migraine headache. The diagnosis of migraine headache is usually made from the history. Neurological and other diagnostic examinations are often normal. The IHS criteria are used as the clinical basis for migraine diagnosis. If atypical features are present, secondary headaches must be ruled out. Neuroimaging techniques (e.g. head computed tomography [CT], with or without contrast, and magnetic resonance imaging [MRI]) are not recommended for routine evaluation of headache unless abnormal findings are found on the neurological examination.
Cluster headache
Cluster headaches are characterised by repeated headaches that can occur for weeks to months at a time, followed by periods of remission. It is one of the most severe forms of head pain. Cluster headache occurs less frequently than migraine (the cluster headache to migraine frequency is 1:10) and is more frequent in men than in women by a ratio of 8:1. The onset is usually between 20 and 50 years of age.
AETIOLOGY AND PATHOPHYSIOLOGY
Neither the cause nor the pathophysiological mechanism of cluster headache is fully known. The vasodilation that occurs in the affected part of the face is extracranial. As with migraine headaches, the trigeminal nerve is implicated in the production of pain. Activation of this nerve causes release of substance P and other vasoactive substances that cause vasodilation, stimulation of afferent pain fibres and neurogenic inflammation with extravasation (movement of fluid out of the blood vessels). The periodicity (i.e. regularity in terms of timing) and autonomic symptoms of cluster headache indicate a dysfunction of the biological clock mechanisms of the hypothalamus.4 These headaches can also be triggered by alcohol ingestion.
CLINICAL MANIFESTATIONS
The IHS classification defines cluster headache as involving severe unilateral orbital, supraorbital or temporal pain and at least one of the following signs present on the pain side: conjunctival injection, lacrimation, nasal congestion, rhinorrhoea, forehead and facial swelling, miosis (constricted pupil), ptosis (eyelid dropping) or eyelid oedema. The headache has an abrupt onset, usually without prodromal symptoms. It peaks in 5–10 minutes and lasts 30–90 minutes. It is not uncommon for this type of headache to start at night, awakening the patient after a few hours of sleep. Headaches may recur several times a day over a period of several days, with each cluster lasting 2–3 months. It usually affects the upper face, the periorbital region and the forehead on one side of the face and the head. The headache may not recur for months or years.
The patient may also exhibit conjunctivitis, increased lacrimation (tearing) and nasal congestion on the side of the headache. Sweating may occur on the forehead of the affected side. A partial Horner’s syndrome (miosis and ptosis on the affected side) may be seen. The headache is described as deep, steady and penetrating but not throbbing.
Unlike the patient with migraine, who seeks isolation and quiet, the patient with a cluster headache paces the floor, cries out and resents being touched. The patient with a cluster headache does not experience the systemic manifestations that accompany a migraine headache, such as nausea or vomiting. As with migraine headaches, there are usually no complications with cluster headaches.
Other types of headaches
Although tension, migraine and cluster headaches are by far the most common types of headaches, other types of headache can also occur. These headaches may be the first symptom of a more serious illness. Headache can accompany subarachnoid haemorrhage; brain tumours; other intracranial masses; arteritis; vascular abnormalities; trigeminal neuralgia (tic douloureux); diseases of the eyes, nose and teeth; and systemic illness (e.g. bacteraemia, carbon monoxide poisoning, mountain sickness, polycythaemia vera). The symptoms vary greatly. Because of the variety of causes of headache, clinical evaluation must be thorough. It should include an evaluation of personality, life adjustment, environment and family situation, as well as a comprehensive evaluation of neurological and physical status.
MULTIDISCIPLINARY CARE FOR HEADACHES
If no systemic underlying disease is found, therapy is directed towards the functional type of headache. Box 58-1 outlines the diagnostic studies needed for a patient with headache to rule out any intracranial or extracranial disease. Table 58-2 summarises collaborative therapies for prophylaxis and symptomatic relief of common headaches. These therapies include drugs, meditation, yoga, biofeedback, cognitive–behavioural therapy and relaxation training.
DIAGNOSTIC STUDIES
History and physical examination
Neurological examination (often negative)
Special studies (e.g. CT scan, angiography, EMG, EEG, MRA, MRI)
CT, computed tomography; EEG, electroencephalography; EMG, electromyography; FBC, full blood count; MRA, magnetic resonance angiography; MRI, magnetic resonance imaging.

* Magnetic resonance imaging (MRI) should be considered in patients with non-acute headache, unexplained abnormal neurological examination, atypical headache, headache features or an additional risk factor, such as immune deficiency.
† only for patients suffering from one or more severe headaches per week.
Biofeedback involves the use of physiological monitoring equipment to give the patient information about muscle tension and peripheral blood flow (skin temperature of the fingers). The patient is trained to relax the muscles and raise the finger temperature and is given reinforcement (operant conditioning) in accomplishing these physiological alterations.
Cognitive–behavioural therapy and relaxation therapy used alone or in conjunction with drug therapy may be beneficial to some patients. Acupuncture, acupressure and hypnosis are also therapies that have worked well in some patients with headaches. Treatments for tension-type headache include physiotherapy (e.g. massage, hot packs, cervical collar), injection of local anaesthetic into spastic muscles and correction of faulty posture.
Drug therapy
Tension-type headache
Drug treatment for tension-type headache usually involves a non-opioid analgesic (e.g. aspirin, paracetamol) used alone or in combination with a sedative, muscle relaxant, tranquilliser or codeine. However, many of these drugs have serious side effects. The patient should be cautioned about the long-term use of aspirin and aspirin-containing drugs because they can cause gastric bleeding and coagulation abnormalities in susceptible patients. Drugs containing paracetamol can cause liver damage, especially when combined with alcohol, and kidney damage with chronic use.
Migraine headache
Drug treatment of the acute migraine attack is aimed at terminating or decreasing the symptoms of the attack. Many people with mild or moderate migraine can obtain relief with aspirin or paracetamol. Ergotamine is often used when simple analgesics do not relieve headache. Ergotamine inhibits the reuptake of noradrenaline into postganglionic nerve terminals of the sympathetic nervous system. This allows more noradrenaline to attach to α-adrenergic sites on smooth muscle in the artery wall, thereby causing prolonged vasoconstriction of cranial blood vessels. Ergotamine can be administered orally or rectally. The usual dosage is 1–2 mg (oral or rectal) at the onset of the headache, followed by 2 mg within 1 hour. No more than 6 mg is given for any single attack or 10 mg total in any week.
Drugs that affect selected serotonin receptors, the ‘triptans’, are aimed at treating the pathological process of migraine. These drugs reduce neurogenic inflammation of the cerebral blood vessels and produce vasoconstriction. They include sumatriptan, naratriptan and zolmitriptan. Because these drugs cause constriction of coronary arteries, they are avoided in patients with heart disease. Triptans should be taken at the first symptom of migraine headache. Other drugs that may relieve migraine headache include paracetamol, codeine phosphate, doxylamine succinate and opioids.
A variety of drugs are used to reduce the frequency and severity of tension-type and migraine attacks. They are taken on a daily basis and are usually used when headaches occur more than twice a month. Preventative drugs for migraine headaches include β-adrenergic blockers (e.g. propranolol, atenolol), tricyclic antidepressants (e.g. amitriptyline), selective serotonin reuptake inhibitors (e.g. fluoxetine), calcium channel blockers (e.g. verapamil), sodium valproate, clonidine and thiazides. Another drug that is rarely used, methysergide, competitively blocks serotonin receptors in the central and peripheral nervous systems. However, because of the risk of serious side effects, including retroperitoneal, pulmonary and cardiac fibrosis, the patient taking methysergide requires regular follow-up. It is recommended that a patient taking methysergide have a break (drug holiday) every 4–6 months.
Cluster headache
Cluster headaches occur suddenly, often at night, and are not long lasting, so drug therapy is not as useful as it is for the other types of headaches. Prophylactic drugs may include verapamil, lithium, ergotamine, sodium valproate or non-steroidal anti-inflammatory drugs (NSAIDs). Acute treatment of cluster headache is inhalation of 100% oxygen delivered at a rate of 7–9 L per minute for 15–20 minutes, which may relieve headache by causing vasoconstriction. It can be repeated after a 5-minute rest. However, a drawback to this treatment is that the patient must have continuous access to the oxygen supply. Sumatriptan is also effective in treating acute cluster headache. Methysergide may be used prophylactically when the cluster headache recurs at a known time.
Patients with frequent headaches may overuse analgesic drugs.5 Such overuse can lead to chronic daily headache, also called analgesic rebound headache or drug-induced headache. Drugs known to cause this problem are paracetamol, aspirin, NSAIDs (e.g. ibuprofen), sumatriptan and narcotics. Treatment involves abrupt withdrawal of the offending drug, except for opioids, which need to be tapered, and initiation of alternative drugs such as amitriptyline.
 NURSING MANAGEMENT: HEADACHES
NURSING MANAGEMENT: HEADACHES
Nursing assessment
Subjective and objective data that should be obtained from a patient with headache are presented in Table 58-3. Because the history provides the key to assessment of headache, it should include specific details of the headache itself, such as the location and type of pain, onset, frequency, duration, relation to events (emotional, psychological, physical) and time of day of the occurrence. Information about previous illnesses, surgery, trauma, allergies, family history and response to medication should also be obtained. The nurse can suggest that the patient keep a diary of headache episodes with specific details. This type of record can be of great help in determining the type of headache and the precipitating events. If the patient has a history of migraine, tension-type or cluster headaches, it is important to determine whether the character, intensity or location of the headache has changed. This may be an important clue as to the cause of the headache.

Nursing diagnoses
Nursing diagnoses for the patient with headache may include, but are not limited to, those presented in NCP 58-1.
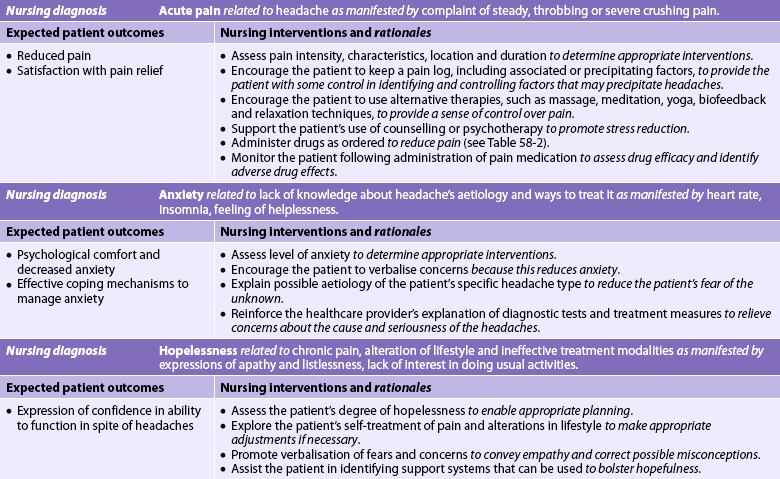
Planning
The overall goals are that the patient with a headache will: (1) have reduced or no pain; (2) experience increased comfort and decreased anxiety; (3) demonstrate understanding of triggering events and treatment strategies; (4) use positive coping strategies to deal with chronic pain; and (5) experience increased quality of life and decreased disability.
Nursing implementation
Patients with chronic headache present a great challenge to healthcare providers. Headaches may be related to an inability to cope with daily stresses. The most effective therapy may be to help patients examine their lifestyle, recognise stressful situations and learn to cope with them more appropriately. Precipitating factors can be identified, and ways of avoiding them can be developed. Daily exercise, relaxation periods and socialising can be encouraged because each may help decrease the recurrence of headache. The nurse can suggest alternative ways of handling the pain of headache through techniques such as relaxation, meditation, yoga and self-hypnosis. In addition to using analgesics and analgesic combination drugs for the symptomatic relief of headache, the patient should be encouraged to use relaxation techniques because they are effective in relieving tension-type and migraine headaches. The migraine sufferer often needs a quiet, dimly lit environment. Massage and moist hot packs to the neck and head can help a patient with tension-type headaches. The patient should learn about the drugs prescribed for prophylactic and symptomatic treatment of headache and should be able to describe the purpose, action, dosage and side effects of the drug. To prevent accidental overdose, the patient should make a written note of each dose of drug or headache remedy.
Does acupuncture help tension headaches?
EVIDENCE-BASED PRACTICE
Clinical question
For patients with tension-type headaches (P), does acupuncture (I) versus sham treatment or routine care (C) decrease headaches (O) over 3 months (T)?
Critical appraisal and synthesis of evidence
• 11 studies total (n = 2317). Six trials compared acupuncture to sham intervention with needles inserted at incorrect points or needles not penetrating skin. One half of patients receiving true acupuncture reported a decrease in headache days by at least 50% compared to 41% in sham acupuncture groups.
• Two trials compared acupuncture to acute headache treatment or routine care only. Approximately 47% of patients receiving acupuncture reported decrease in headache days compared with 16% of patients in control groups.
• Long-term effects of acupuncture on tension headache were not investigated.
P, patient population of interest; I, intervention or area of interest; C, comparison of interest or comparison group; O, outcome(s) of interest; T, timing.
For the patient whose headaches are triggered by food, dietary advice may be needed. The patient is encouraged to eliminate foods that may provoke headaches, such as vinegar, chocolate, onions, alcohol (particularly red wine), excessive caffeine, cheese, fermented or marinated foods, monosodium glutamate and aspartame. Active challenge and provocative testing with specific foods may be necessary to determine the specific causative agents. However, food triggers may change over time. Patients should avoid smoking and exposure to triggers such as strong perfumes, volatile solvents and petrol fumes. Cluster headache attacks may occur at high altitudes with low oxygen levels during air travel. Ergotamine, taken before the aeroplane takes off, may decrease the likelihood of these attacks. A teaching guide for the patient with a headache is presented in Box 58-2.
PATIENT & FAMILY TEACHING GUIDE
1. Keep a diary or calendar of headaches and possible precipitating events.
2. Avoid factors that can trigger a headache:
3. Describe the purpose, action, dosage and side effects of drugs taken.
4. Be able to self-administer sumatriptan subcutaneously if prescribed.
5. Use stress-reduction techniques, such as relaxation.
6. Participate in regular exercise.
7. Contact the healthcare provider if any of the following occurs:
Seizure disorders and epilepsy
Seizure is a paroxysmal, uncontrolled electrical discharge of neurons in the brain that interrupts normal function. Seizures are often symptoms of an underlying illness. They may accompany a variety of disorders or they may occur spontaneously without any apparent cause. Seizures resulting from systemic and metabolic disturbances are not considered epilepsy if the seizures cease when the underlying problem is corrected. In the adult, metabolic disturbances that cause seizures include acidosis, electrolyte imbalances, hypoglycaemia, hypoxia, alcohol and barbiturate withdrawal, dehydration and water intoxication. Extracranial disorders that can cause seizures are heart, lung, liver or kidney diseases; systemic lupus erythematosus; diabetes mellitus; hypertension; and septicaemia.
Epilepsy is a condition in which a person has spontaneously recurring seizures caused by a chronic underlying condition. The prevalence of epilepsy is approximately 70 per 100,000 persons per annum.6 It is higher in developing countries. The incidence rates are high during the first year of life, decline through childhood and adolescence, plateau in middle age and rise sharply again among the elderly.
AETIOLOGY AND PATHOPHYSIOLOGY
The most common causes of seizure disorder during the first 6 months of life are severe birth injury, congenital defects involving the central nervous system (CNS), infections and inborn errors of metabolism. In patients between 2 and 20 years of age, the primary causative factors are infection, trauma (including stroke), genetic factors and, less significantly, birth injury. In individuals between 20 and 30 years of age, seizure disorder usually occurs as the result of structural lesions, such as trauma, brain tumours or vascular disease. After 50 years of age, the primary causes of seizure disorders are cerebrovascular lesions and metastatic brain tumours. Many causes of seizure disorders have been identified; however, nearly three-quarters of all seizure disorder cases cannot be attributed to a specific cause and are considered idiopathic (primarily genetic) or cryptogenic (where undiscovered lesions are believed to be present).
The role of heredity in the aetiology of seizure disorders has been difficult to determine because of the problem of separating hereditary from environmental or acquired influences. In addition, some families carry a predisposition to seizure disorders in the form of an inherently low threshold to seizure-producing stimuli, such as trauma, disease and high fever. Nevertheless, at least 40 seizure disorder syndromes have been linked to specific genetic defects.7
In recurring seizures (epilepsy), a group of abnormal neurons (seizure focus) seems to undergo spontaneous firing. This firing spreads by physiological pathways to involve adjacent or distant areas of the brain. If this activity spreads to involve the whole brain, a generalised seizure occurs. The factor that causes this abnormal firing is not clear. Any stimulus that causes the cell membrane of the neuron to depolarise induces a tendency to spontaneous firing. Often the area of the brain from which the epileptic activity arises is found to have a proliferation of glial cells, referred to as scar tissue or gliosis. The scarring is thought to interfere with the normal chemical and structural environment of the brain neurons, making them more likely to fire abnormally.
Repetitive electrical discharges from an epileptic focus in experimental animals can produce long-lasting and possibly permanent changes in neuron excitability, both locally and in distant areas of the brain. This effect is called kindling and it presents an important implication for epilepsy in humans: seizures can beget more seizures. Clinical experience indicates that the longer a patient goes without good seizure control, the lower the likelihood that seizures will be controllable. Thus, a vigorous attempt must be made to control recurring seizures.
CLINICAL MANIFESTATIONS
The specific clinical manifestations of a seizure are determined by the site of the electrical disturbance. The preferred method of classifying recurring seizures is the International Classification System (see Box 58-3).8 This system is based on the clinical and electroencephalographic manifestations of seizures. In this system, seizures are divided into two major classes: generalised and partial (see Fig 58-1). Depending on the type, a seizure may progress through several phases, which include: (1) the prodromal phase with signs or activity, which precedes a seizure; (2) the aural phase with a sensory warning; (3) the ictal phase with full seizure; and (4) the postictal phase, which is the period of recovery after the seizure.
BOX 58-3 International Classification of Seizure Disorders
Source: Adapted from Task Force on Epilepsy Classification and Treatment. Seizure types. Brussels: International League Against Epilepsy; Revised 2010. Available at www.ilae-epilepsy.org/Visitors/Centre/ctf/ctfoverview.cfm, accessed December 2010.
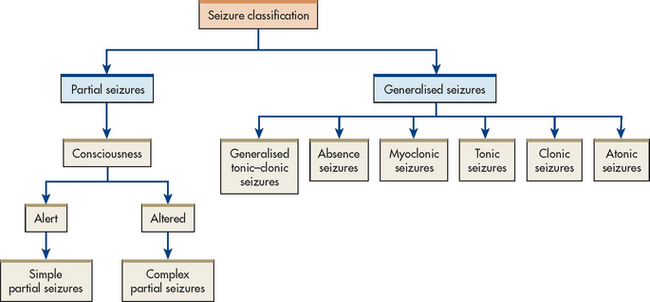
Generalised seizures
Generalised seizures are characterised by bilateral synchronous epileptic discharges in the brain from the onset of the seizure. Because the entire brain is affected at the onset of the seizures, there is no warning or aura. In most cases, the patient loses consciousness for a few seconds to several minutes.
Tonic–clonic seizures
The most common generalised seizure is the generalised tonic–clonic seizure. A tonic–clonic seizure is characterised by loss of consciousness and falling to the ground if the patient is upright, followed by stiffening of the body (tonic phase) for 10–20 seconds and subsequent jerking of the extremities (clonic phase) for another 30–40 seconds. Cyanosis, excessive salivation, tongue or cheek biting and incontinence may accompany the seizure. In the postictal phase, the patient usually has muscle soreness, is very tired and may sleep for several hours. Some patients may not feel normal for several hours or days after a seizure. The patient has no memory of the seizure.
Typical absence seizures
The absence seizure usually occurs only in children and rarely continues beyond adolescence. This type of seizure may cease altogether as the child matures or it may evolve into another type of seizure. The typical clinical manifestation is a brief staring spell that lasts only a few seconds, so it often occurs unnoticed. There may be an extremely brief loss of consciousness. When untreated, the seizures may occur up to 100 times a day. The electroencephalogram (EEG) demonstrates a 3 Hz (cycles per second) spike-and-wave pattern that is unique to this type of seizure. Absence seizures can often be precipitated by hyperventilation and flashing lights.
Atypical absence seizures
The atypical absence seizure is characterised by a staring spell that is accompanied by other signs and symptoms, including brief warnings, peculiar behaviour during the seizure or confusion after the seizure. The EEG demonstrates atypical spike-and-wave patterns, usually to greater or less than 3 Hz.
Other types of generalised seizures
Other types of generalised seizure include myoclonic and akinetic seizures. A myoclonic seizure is characterised by a sudden, excessive jerk of the body or extremities, which may be forceful enough to hurl the person to the ground. These seizures are very brief and may occur in clusters.
The terms akinetic (arrest of movement), atonic (loss of tone) and astatic (loss of balance) have been used interchangeably to describe drop attacks or falling spells. This type of seizure involves either a tonic episode or a paroxysmal loss of muscle tone and begins suddenly with the person falling to the ground. Consciousness usually returns by the time the person hits the ground and normal activity can be resumed immediately. Patients with this type of seizure are at a great risk of head injury and often have to wear a protective helmet. A less severe akinetic seizure involves brief loss of muscle tone without falling.
Partial seizures
Partial seizures are also referred to as partial focal seizures. They begin in a specific region of the cortex, as indicated by the EEG and usually by the clinical manifestations. For example, if the discharging focus is located in the medial aspect of the postcentral gyrus, the patient may experience paraesthesias and tingling or numbness in the leg on the side opposite the focus. If the discharging focus is located in the part of the brain that governs a particular function, sensory, motor, cognitive or emotional manifestations may occur.
Partial seizures may be confined to one side of the brain and remain partial or focal in nature, or they may spread to involve the entire brain, culminating in a generalised tonic–clonic seizure. Any tonic–clonic seizure that is preceded by an aura or warning is a partial seizure that generalises secondarily. Many tonic–clonic seizures that appear to be generalised from the outset may actually be secondary generalised seizures but the preceding partial component may be so brief that it is undetected by the person, by the observer or even on the EEG. Unlike the primary generalised tonic–clonic seizure, the secondary generalised seizure may result in a transient residual neurological deficit postictally. This is called Todd’s paralysis (focal weakness), which resolves after varying lengths of time.
Partial seizures are further divided into: (1) simple partial seizures (those with simple motor or sensory phenomena); and (2) complex partial seizures (those with complex symptoms). Simple partial seizures with elementary symptoms do not involve loss of consciousness and rarely last longer than 1 minute. They may involve motor, sensory or autonomic phenomena or a combination of these. The terms focal motor, focal sensory and jacksonian have been used to describe seizures of the simple partial type.
Complex partial seizures can involve a variety of behavioural, emotional, affective and cognitive functions. The location of the discharging focus is usually in the temporal lobe, hence the term temporal lobe seizure. These seizures usually last longer than 1 minute and are frequently followed by a period of postictal confusion. Complex partial seizures are distinct from simple partial (focal motor, focal sensory) seizures in that they involve some alteration in consciousness. The sole manifestation of complex partial seizures may be clouding of consciousness or a confused state without any motor or sensory components. This type of attack is sometimes termed temporal lobe absence. There is rarely the complete loss of consciousness that is typical of the generalised absence attack, nor does the patient snap back to the preseizure state as does the patient who has had a generalised absence attack.
The most common complex partial seizure involves lip smacking and automatisms (repetitive movements that may not be appropriate). These are often called psychomotor seizures. The patient may continue an activity that was initiated before the seizure, such as counting out change or picking items from a grocery shelf, but after the seizure does not remember the activity performed during the seizure. Other automatisms are less organised, such as picking at clothing, fumbling with objects (real or imaginary) or simply walking away.
A variety of psychosensory symptoms may occur during a complex partial seizure, including distortions of visual or auditory sensations and vertigo. There may be alterations in memory, such as a feeling of having experienced an event before (déjà vu) or alterations in thought processes. Alterations in sexual functioning can vary from hypo- to hypersexuality. Many patients with temporal lobe seizures have decreased sexual drive or erectile dysfunction. However, some may experience sexual sensations during their seizures. This is because the abnormal electrical activity arises from the brain centres responsible for these sensations. Some experience increased sexual drive just after a seizure. In addition, some antiepileptic drugs can cause a decrease in sexual drive because of sedation. Others can cause erectile dysfunction.
COMPLICATIONS
Physical
Status epilepticus is a state of continuous seizure activity or a condition in which seizures recur in rapid succession without return to consciousness between seizures. It is the most serious complication of epilepsy and is a neurological emergency. Status epilepticus can involve any type of seizure. During repeated seizures, the brain uses more energy than can be supplied. Neurons become exhausted and cease to function. Permanent brain damage may result. Tonic–clonic status epilepticus is the most dangerous because it can cause ventilatory insufficiency, hypoxaemia, cardiac arrhythmias, hyperthermia and systemic acidosis, all of which can be fatal.
Another complication of seizures is severe injury and even death from trauma suffered during a seizure. Patients who lose consciousness during a seizure are at greatest risk. Death can result from head injury incurred in a fall, from drowning in the bathtub or from severe burns.
Psychosocial
Perhaps the most common complication of seizure disorders is the effect it has on a patient’s lifestyle. Although attitudes have improved in recent years, epilepsy still carries a social stigma. It used to be associated with supernatural powers, possession by the devil and insanity. Today the stigma probably exists because the characteristics of seizures are in direct conflict with modern Western societal values of self-control, conformity and independence. The patient with epilepsy may experience discrimination in employment and educational opportunities. Transportation may be difficult because of legal sanctions against driving.
DIAGNOSTIC STUDIES
The most useful diagnostic tools are an accurate and comprehensive description of the seizures and the patient’s health history (see Box 58-4).
BOX 58-4 Seizure disorders and epilepsy
MULTIDISCIPLINARY CARE
Diagnostic studies
Collaborative therapy
Antiepileptic drugs (see Box 58-5)
Surgery (see Table 58-5)
CT, computed tomography; FBC, full blood count; MRA, magnetic resonance angiography; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; PET, positron emission tomography.
The EEG is a useful diagnostic adjuvant to the history but only if it shows abnormalities. Abnormal findings help determine the type of seizure and help pinpoint the seizure focus. Unfortunately, only a small percentage of patients with seizure disorders have abnormal findings on the EEG the first time the test is done. EEGs may need to be repeated often or continuous EEG monitoring may be needed to detect abnormalities. Abnormal discharges may not occur during the 30–40 minutes of sampling during EEG monitoring and the test may never indicate an abnormality. It is not a definitive test because some patients who do not have seizure disorders have abnormal patterns on their EEGs, whereas many patients with seizure disorders have normal EEGs between seizures. Magnetoencephalography may be done in conjunction with the EEG. This test has greater sensitivity in detecting small magnetic fields generated by neuronal activity.
A full blood examination, serum urea and electrolyte levels, studies of liver and kidney function, and urinalysis should be done to rule out metabolic disorders. A CT or MRI scan should be done in any new-onset seizure to rule out a structural lesion. Cerebral angiography, single photon emission CT (SPECT), magnetic resonance spectroscopy (MRS), magnetic resonance angiography (MRA) and positron emission tomography (PET) may be used in selected clinical situations.
MULTIDISCIPLINARY CARE
Most seizures do not require professional emergency medical care because they are self-limiting and rarely cause bodily injury. However, if status epilepticus occurs, if significant bodily harm occurs or if the event is a first-time seizure, medical care should be sought immediately. Table 58-4 summarises emergency care of the patient with a generalised tonic–clonic seizure, the seizure most likely to warrant professional emergency medical care. The multidisciplinary care of seizure disorders is summarised in Box 58-4.
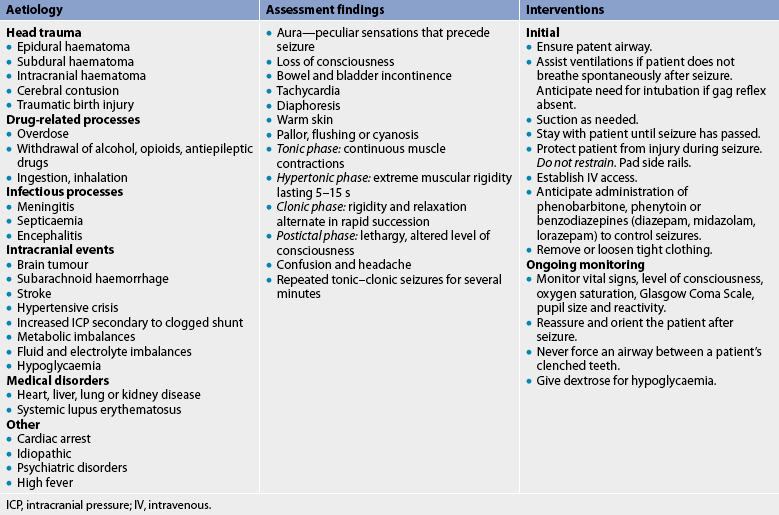
Drug therapy
Seizure disorders are treated primarily with antiepileptic drugs (see Box 58-5). Therapy is aimed at preventing seizures because cure is not possible.
Drugs generally act by stabilising nerve cell membranes and preventing spread of the epileptic discharge. In about 70% of patients, seizure disorders are controlled by medication. The primary goal of antiepileptic drug therapy is to obtain maximum seizure control with a minimum of toxic side effects. The principle of drug therapy is to begin with a single drug and increase the dosage until seizures are controlled or toxic side effects occur. Serum levels of the drug should be monitored if seizures continue to occur, if seizure frequency increases or if drug compliance is questioned. The therapeutic range for each drug indicates the serum level above which most patients experience toxic side effects and below which most continue to have seizures. Therapeutic ranges are only guides for therapy. If the patient’s seizures are well controlled with a subtherapeutic level, the drug dose need not be increased. Likewise, if a drug level is above the therapeutic range and the patient has good seizure control without toxic side effects, the drug dose need not be decreased. Many of the newer drugs do not require drug level monitoring because the therapeutic range is very large. If seizure control is not achieved with a single drug, the drug may be changed or a second drug may be added.
For many years the primary drugs for treatment of generalised tonic–clonic and partial seizures were phenytoin, carbamazepine, phenobarbitone and sodium valproate. For treatment of absence, akinetic and myoclonic seizures the drugs included ethosuximide, sodium valproate and clonazepam. Recently, many new drugs have become available, including gabapentin, lamotrigine, topiramate, tiagabine and levetiracetam. These drugs are effective for partial seizures and for some of the primary generalised seizure disorders as well.
Treatment of status epilepticus requires initiation of a rapid-acting antiepileptic drug that can be given intravenously. The drugs most commonly used are lorazepam and diazepam. Because these are short-acting drugs, they must be followed by administration of long-acting drugs such as phenytoin or phenobarbitone.
Drugs currently used in seizure management are shown in Box 58-5. Because many of these drugs (e.g. phenytoin, phenobarbitone, ethosuximide, lamotrigine, topiramate) have long half-lives, they can be given in once- or twice-daily doses. This increases the patient’s compliance with taking the drug by simplifying the drug regimen and avoiding the need to take medication at work or at school. Toxic side effects of antiepileptic drugs involve the CNS and include diplopia, drowsiness, ataxia and mental slowing. Neurological assessment for dose-related toxicity involves testing the eyes for nystagmus, hand and gait coordination, cognitive functioning and general alertness.
Idiosyncratic side effects involve organs outside the CNS, including the skin (rashes), gingiva (hyperplasia), bone marrow (blood dyscrasias), liver and kidneys. Nurses should be knowledgeable about these side effects so that patients can be informed and proper treatment can be instituted. A common side effect of phenytoin is gingival hyperplasia (excessive growth of gingival tissue), especially in children and young adults. This can be limited by good dental hygiene, including regular tooth brushing and flossing. If gingival hyperplasia is extensive, the hyperplastic tissue may have to be surgically removed (gingivectomy) and phenytoin may have to be replaced by another antiepileptic drug. Because phenytoin can also cause hirsutism in young people, other drugs are often used first.
Surgical therapy
A significant number of patients whose epilepsy cannot be controlled with drug therapy are candidates for surgical intervention to remove the epileptic focus or prevent spread of epileptic activity in the brain (see Table 58-5). The major types of surgery are removal of one lobe (usually the temporal lobe), removal of cortex or separation of the two hemispheres (corpus callosotomy).9
TABLE 58-5 Surgical procedures for seizure disorders and epilepsy
| Type of seizure | Surgical procedure | Results |
|---|---|---|
| Complex partial seizure of temporal lobe origin | Resectioning of epileptogenic tissue | Absence of seizures 5 years postoperatively in 55–70% of patients |
| Partial seizures of frontal lobe origin | Resectioning of epileptogenic tissue (if in resectable area) | Absence of seizures 5 years postoperatively in 30–50% of patients |
| Generalised seizures (Lennox-Gastaut syndrome or drop attacks) | Sectioning of corpus callosum | Persistence of seizures, less violent, less frequent, less disabling events |
| Intractable unilateral multifocal epilepsy associated with infantile hemiplegia | Hemispherectomy or callosotomy | Reduction in seizure frequency and type, improvement in behaviour |
The benefits of surgery include a cessation or reduction in the frequency of the seizures, but not all types of epilepsy benefit from surgery. An extensive preoperative evaluation is important, including continuous EEG monitoring and other specific tests to ensure the precise localisation of the focal point. Before surgery is performed, three requirements must be met: (1) the diagnosis of epilepsy must be confirmed; (2) there must have been an adequate trial with drug therapy without satisfactory results; and (3) the electro-clinical syndrome (type of seizure disorder) must be defined.
Other therapies
Another treatment for seizure disorders is vagal nerve stimulation. An electrode is surgically placed around the left vagus nerve in the neck. It is connected to a battery placed beneath the skin in the upper chest. The device is programmed to deliver intermittent electrical stimulation to the brain to reduce the frequency and intensity of seizures. The exact mechanism of action is unknown, although the stimulation may interrupt synchronisation of epileptic brain-wave activity. This method is currently used in only a small number of patients.
Biofeedback to control seizures is aimed at teaching the patient to maintain a certain brain-wave frequency that is refractory to seizure activity. This method is still in the experimental stage.
NURSING MANAGEMENT: SEIZURE DISORDERS AND EPILEPSY
Nursing assessment
Subjective and objective data that should be obtained from a patient with a seizure disorder are presented in Table 58-6. Data related to a specific seizure episode can be obtained from a witness.
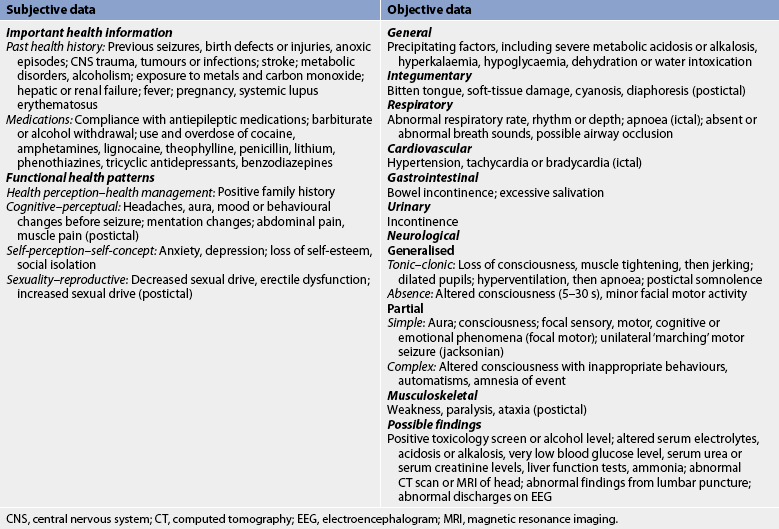
Nursing diagnoses
Nursing diagnoses for the patient with seizure disorders and epilepsy may include, but are not limited to, those presented in NCP 58-2.
Planning
The overall goals are that the patient with seizures will: (1) be free from injury during a seizure; (2) have optimal mental and physical functioning while taking antiepileptic drugs; and (3) have satisfactory psychosocial functioning.
Nursing implementation
Health promotion
Many cases of seizure disorders can be prevented by promotion of general safety measures, such as the wearing of helmets in situations involving risk of head injury. In the Western world improved perinatal, labour and delivery care have reduced fetal trauma and hypoxia and thereby have reduced brain damage leading to seizure disorders.
The patient with a seizure disorder should practise good general health habits (e.g. maintaining a proper diet, getting adequate rest, exercising). The patient should be helped to identify events or situations that precipitate the seizures and they should be given suggestions for avoiding them or handling them better. Excessive alcohol intake, fatigue and loss of sleep should be avoided, and the patient should be helped to handle stress constructively.
Acute intervention
The nursing care for a hospitalised patient with a seizure disorder or a patient who has had seizures as a result of metabolic factors involves several responsibilities, including observation and treatment of the seizure, education and psychosocial intervention.
When a seizure occurs, the nurse should carefully observe and record details of the event because the diagnosis and subsequent treatment often rest solely on the seizure description. All aspects of the seizure should be noted. What events preceded the seizure? When did the seizure occur? How long did each phase (aura [if any], ictal, postictal) last? What occurred during each phase?
Both subjective data (usually the only type of data in the aural phase) and objective data are important. Objective data should include the exact onset of the seizure (which body part was affected first and how); the course and nature of the seizure activity (loss of consciousness, tongue biting, automatisms, stiffening, jerking, total lack of muscle tone); the body parts involved and their sequence of involvement; and the presence of autonomic signs, such as dilated pupils, excessive salivation, altered breathing, cyanosis, flushing, diaphoresis or incontinence. Assessment of the postictal period should include a detailed description of the level of consciousness, vital signs, memory loss, muscle soreness, speech disorders (aphasia, dysarthria), weakness or paralysis, sleep period and the duration of each sign or symptom.
During the seizure, it is important to maintain a patent airway. This may involve supporting and protecting the head, turning the patient to the side, loosening constrictive clothing or easing the patient to the floor, if seated. The patient should not be restrained and no objects should be placed in the mouth. After the seizure, the patient may require suctioning and oxygen may be needed.
A seizure can be a frightening experience for the patient and for others who may witness it. The nurse should assess the level of their understanding and provide information about how and why the event occurred. This is an excellent opportunity for the nurse to dispel many common misconceptions about seizures.
Ambulatory and home care
Prevention of recurring seizures is the major goal in the treatment of epilepsy. Because seizure disorders cannot be cured, drugs must be taken regularly and continuously, often for a lifetime. The nurse should ensure that the patient knows this, as well as the specifics of the drug regimen and what to do if a dose is missed. Usually the dose should be made up if the omission is remembered within 24 hours. The patient should be cautioned not to adjust drug doses without professional guidance because this can increase seizure frequency and even cause status epilepticus. The patient should be encouraged to report any medication side effects and to keep regular appointments with the doctor.
Nurses play an important role in teaching the patient and the family. Guidelines for teaching are shown in Box 58-6. Nurses should teach family members and significant others the emergency management of tonic–clonic seizures (see Table 58-4). They should be reminded that it is not necessary to call an ambulance or send a person to the hospital after a single seizure unless the seizure is prolonged, another seizure immediately follows or extensive injury has occurred.
BOX 58-6 Seizure disorders and epilepsy
PATIENT & FAMILY TEACHING GUIDE
The patient should be taught the following:
1. Take drugs as prescribed. Report any and all side effects to the healthcare provider. When necessary, blood is taken to ensure that therapeutic levels are maintained.
2. Use non-drug techniques, such as relaxation therapy and biofeedback training, to potentially reduce the number of seizures.
3. Be aware of the availability of resources in the community.
4. Wear a medical alert bracelet, necklace and identification card.
5. Avoid excessive alcohol intake, fatigue and loss of sleep.
6. Eat regular meals with snacks in between if feeling shaky, faint or hungry.
Family members should be taught the following:
1. For first aid treatment of tonic–clonic seizure, it is not necessary to call an ambulance or send the patient to the hospital after a single seizure unless the seizure is prolonged, another seizure immediately follows or extensive injury has occurred.
2. During an acute seizure, it is important to stay with the patient and protect them from injury. This may involve supporting and protecting the head, turning the patient to the side, loosening constrictive clothing and easing the patient to the floor, if seated.
Family members also need to be shown how to place the person into a recovery position post-seizure. They need to stay with the patient until they have recovered from the seizure or until other help arrives.
Patients with a seizure disorder also experience concerns or fears related to recurrent seizures, incontinence or loss of self-control. The nurse can provide support for the patient through education and by helping to identify coping mechanisms. Perhaps the greatest challenge that a seizure disorder presents to the patient is adjusting to the personal limitations imposed by the illness. Discrimination in employment may be one of the most serious problems facing the person with a seizure disorder. For issues relating to job discrimination, patients can be referred to the Australian Human Rights and Equal Opportunity Commission, state anti-discrimination and equal opportunities bodies or the New Zealand Human Rights Commission (Te Kahui Tika Tangata).
A variety of other resources can be offered to the patient with a seizure disorder who has a specific problem. If the nurse believes that associating with others who have a seizure disorder would be beneficial, the patient can be referred to the local group of the Epilepsy Action Australia or Epilepsy New Zealand, which are voluntary agencies that offer a variety of services to people with epilepsy. Sometimes people with chronic epilepsy are unable to find relief from their condition even with medications or surgery; this means that they may be unable to work or to participate in the normal activities of daily living.
New Zealand is one of three countries in the world that has developed a program to help people who are in this position. It has done this through a program that provides dogs to live with people who have chronic, poorly controlled epilepsy, in the same way that dogs assist blind people. The New Zealand Epilepsy Assist Dogs Trust (see the Resources on p 1677) trains dogs to assist their owners after they have had a seizure by fetching a phone or a towel and staying with them. Dogs are matched to owners and training can take up to 2 years. Epilepsy Assist dogs help their owners to become more independent and many people with the dogs have successfully returned to work. Some dogs seem to detect seizures before they happen. Stella (see Fig 58-2) recently saved the life of her owner by alerting his family when he fell onto a barbeque.

Figure 58-2 Epilepsy Assist dogs are specially trained to assist people with severe epilepsy to lead safer and more independent lives.
Photo used with permission from the New Zealand Epilepsy Assist Dogs Trust.
Patients should be informed that medical alert bracelets, necklaces and identification cards are available through the Epilepsy Associations, local pharmacies or companies specialising in medical alert identification devices. However, the use of these medical identification tags is optional. Some patients have found them beneficial but others have found them to be more of a burden than helpful because they prefer not to be identified as having a seizure disorder.
Social workers and welfare agencies, such as Centrelink Australia and Work and Income New Zealand (WINZ), can link patients and their families to disability employment assistance, advise on medical eligibility for disability and carer-related payments, and refer to other disability and carer organisations that can help with non-work related issues.
The patient should be encouraged to learn more about epilepsy through self-education materials. The Epilepsy Associations provide several information pamphlets and may facilitate support groups. Many agencies that offer services to epileptic patients, as well as local groups of Epilepsy Associations, have these available as teaching aids.
Multiple sclerosis
Multiple sclerosis (MS) is a chronic, progressive, degenerative disorder of the CNS characterised by disseminated demyelination of nerve fibres of the brain and spinal cord. It is not known exactly how many people have MS. High prevalence rates (over 30 per 100,000) occur in northern Europe, the northern United States, southern Canada, and southern Australia and New Zealand. Low prevalence rates (less than 5 per 100,000) occur in southern Europe, Japan, China and South America. This difference may be related to climate or genetic differences, or both. MS is five times more prevalent in temperate climates (between 45 and 65° of latitude), such as those found in the northern United States, Canada and Europe, than in tropical regions.10 Reflecting the worldwide trend of higher incidence being associated with latitude, the prevalence of MS in the South Island of New Zealand is over twice that in Waikato in the North Island. MS is considered a disease of young to middle-aged adults, with the onset usually being between 15 and 50 years of age. Women are affected more often than are men.
AETIOLOGY AND PATHOPHYSIOLOGY
The cause of MS is unknown, although research findings suggest that MS is related to infectious (viral), immunological and genetic factors and is perpetuated as a result of intrinsic factors (e.g. faulty immunoregulation). The susceptibility to MS appears to be inherited. First-, second- and third-degree relatives of patients with MS are at a slightly increased risk. Multiple genes confer susceptibility to MS.
The role of precipitating factors, such as exposure to pathogenetic agents, in the aetiology of MS is controversial. It is possible that their association with MS is random and that there is no cause-and-effect relationship. Possible precipitating factors include infection, physical injury, emotional stress, excessive fatigue, pregnancy and a poorer state of health.
MS is characterised by chronic inflammation, demyelination and gliosis (scarring) in the CNS. The primary neuropathological condition is an autoimmune disease orchestrated by autoreactive T cells (lymphocytes). This process may be triggered initially by a virus in genetically susceptible individuals. The activated T cells in the systemic circulation migrate to the CNS, causing blood–brain barrier disruption. This is the likely initial event in the development of MS. Subsequent antigen–antibody reaction within the CNS results in activation of the inflammatory response and through multiple effector mechanisms leads to demyelination of axons. The disease process consists of loss of myelin, disappearance of oligodendrocytes and proliferation of astrocytes. These changes result in characteristic plaque formation, or sclerosis, with plaques scattered throughout multiple regions of the CNS.
Initially the myelin sheaths of the neurons in the brain and spinal cord are attacked (see Fig 58-3, A and B). Early in the disease the myelin sheath is damaged but the nerve fibre is not affected and nerve impulses are still transmitted (see Fig 58-3, C). At this point the patient may complain of a noticeable impairment of function (e.g. weakness). However, the myelin can regenerate and the symptoms can disappear, resulting in a remission.
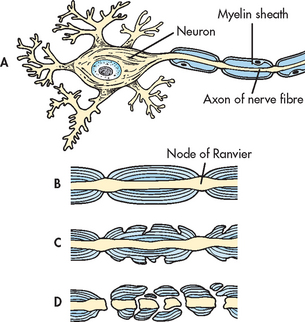
Figure 58-3 Pathogenesis of multiple sclerosis. A, Normal nerve cell with myelin sheath. B, Normal axon. C, Myelin breakdown. D, Myelin totally disrupted; axon not functioning.
In addition to myelin disruption, the axon also becomes involved (see Fig 58-3, D). Myelin is replaced by glial scar tissue, which forms hard sclerotic plaques in multiple regions of the CNS (see Fig 58-4). Without myelin, nerve impulses slow down, and with destruction of nerve axons, impulses are totally blocked, resulting in permanent loss of function. In many chronic lesions, demyelination continues with progressive loss of nerve function.
CLINICAL MANIFESTATIONS
Because the onset is often insidious and gradual, with vague symptoms that occur intermittently over months or years, the disease may not be diagnosed until long after the onset of the first symptom. The disease process has a spotty distribution in the CNS, so the signs and symptoms vary over time. The disease is characterised by chronic, progressive deterioration in some persons and by remissions and exacerbations in others. With repeated exacerbations, however, progressive scarring of the myelin sheath occurs and the overall trend is progressive deterioration in neurological function.
The clinical manifestations vary according to the areas of the CNS involved. Some patients have severe, long-lasting symptoms early in the course of the disease. Others may experience only occasional and mild symptoms for several years after onset. A classification scheme that identifies the various courses of MS has been developed (see Table 58-7).11
TABLE 58-7 Clinical courses of multiple sclerosis
| Category | Characteristics |
|---|---|
| Relapsing– remitting | Clearly defined relapses with full recovery or sequelae and residual deficit on recovery |
| Primary-progressive | Disease progression from onset with occasional plateaus and temporary minor improvements |
| Secondary-progressive | A relapsing–remitting initial course, followed by progression with or without occasional relapses, minor remissions and plateaus |
| Progressive–relapsing | Progressive disease from onset, with clear acute relapses, with or without full recovery; periods between relapses are characterised by continuing progression |
Common signs and symptoms of MS include motor, sensory, cerebellar and emotional problems. Motor symptoms include weakness or paralysis of the limbs, trunk or head; diplopia; scanning speech; and spasticity of the muscles that are chronically affected. Patients with MS experience a variety of sensory abnormalities, including paraesthesias, patchy blindness (scotomas), blurred vision, vertigo, tinnitus, decreased hearing and chronic neuropathic pain. Radicular (nerve root) pains may be present, particularly in the low thoracic and abdominal regions. Lhermitte’s phenomenon is a transient sensory symptom described as an electric shock radiating down the spine or into the limbs with flexion of the neck. Cerebellar signs include nystagmus, ataxia, dysarthria and dysphagia.
Bowel and bladder function can be affected if the sclerotic plaque is located in areas of the CNS that control elimination. Problems with defecation usually involve constipation rather than faecal incontinence. Urinary problems are variable. A common problem in MS patients is a spastic (uninhibited) bladder. This indicates a lesion above the second sacral nerve, which cuts off suprasegmental inhibiting influences on bladder contractility. As a result, the bladder has a small capacity for urine and its contractions are unchecked. This is accompanied by urinary urgency and frequency and results in dribbling or incontinence. On the other hand, a flaccid (hypotonic) bladder indicates a lesion in the reflex arc governing bladder function. The bladder has a large capacity for urine because there is no sensation or desire to void, no pressure and no pain. Generally, there is urinary retention, but urgency and frequency may also occur with this type of lesion. Another urinary problem is a combination of the previous two problems. Urinary problems cannot be adequately diagnosed and treated unless urodynamic studies are done.
Sexual dysfunction occurs in many persons with MS. Physiological erectile dysfunction may result from spinal cord involvement in men. Women may experience decreased libido, difficulty with orgasmic response, painful intercourse and decreased vaginal lubrication. Diminished sensation can prevent a normal sexual response in both sexes. The emotional effects of chronic illness and the loss of self-esteem also contribute to loss of sexual response.
MS has no apparent effect on the course of pregnancy, labour, delivery or lactation. Some women with MS who become pregnant experience remission or an improvement in their symptoms during the gestation period. The hormonal changes associated with pregnancy appear to affect the immune system. However, during the postpartum period, women are at greater risk of exacerbation of the disease.12
Although intellectual functioning generally remains intact, emotional stability may be affected. Cognitive sequelae can produce significant disability for some patients with MS. They may experience anger, depression or euphoria. Signs and symptoms of MS are aggravated or triggered by physical and emotional trauma, fatigue and infection.
The average life expectancy after the onset of symptoms is more than 25 years. Death usually occurs because of infective complications of immobility (e.g. pneumonia) or because of an unrelated disease.
DIAGNOSTIC STUDIES
Because there is no definitive diagnostic test for MS, diagnosis is based primarily on history, clinical manifestations and the presence of multiple lesions over time as measured by MRI (see Box 58-7). Certain laboratory tests are currently used as adjuncts to the clinical examination. In some patients, cerebrospinal fluid (CSF) analysis may show an increase in oligoclonal immunoglobulin G. The CSF also contains a high number of lymphocytes and monocytes. Evoked responses are often delayed in people with MS because of decreased nerve conduction from the eye and the ear to the brain. MRI scans may be helpful because they can detect sclerotic plaques as small as 3–4 mm in diameter. Characteristic white-matter lesions, scattered through the brain or spinal cord, are also evident on such scans. MRS may be used to evaluate patients with MS.
MULTIDISCIPLINARY CARE
Drug therapy
Because there is no cure for MS, multidisciplinary care is aimed at treating the disease process and providing symptomatic relief (see Box 58-7). The disease process is treated with drugs (see Table 58-8) and the symptoms are controlled with a variety of drugs and other forms of therapy.13

ACTH, adrenocorticotrophic hormone; CNS, central nervous system; FBC, full blood count; MAO, monoamine oxidase. SOB, shortness of breath.
* see Chapter 49 for effects of long-term corticosteroid therapy.
† Urodynamic studies must be done before initiation of therapy because patients with MS have multiple lesions, and type of bladder dysfunction cannot be diagnosed from symptoms alone.
Adrenocorticotrophic hormone (ACTH), methylprednisolone and prednisone are helpful in treating acute exacerbations of the disease, probably by reducing oedema and acute inflammation at the site of demyelination. Although the dose and route of administration may vary, these drugs are used in patients with all types of MS. However, these drugs do not affect the ultimate outcome or degree of residual neurological impairment from the exacerbation.
Immunosuppressive drugs, such as azathioprine, methotrexate and cyclophosphamide, have been shown to produce some beneficial effects in patients with progressive–relapsing, secondary-progressive and primary-progressive MS. However, the potential benefits of these drugs must be counterbalanced against the potentially serious side effects.
Immunomodulator drugs modify the disease process. Interferon β-1b is used for ambulatory patients with relapsing–remitting MS. Interferon β-1a is similar to interferon β-1b in efficacy and is used in similar patient groups with MS. Interferon β-1a is given intramuscularly once a week or subcutaneously three times a week. Interferon β-1b is administered subcutaneously on alternate days. Glatiramer acetate, formerly known as copolymer-1, is unrelated to interferon. It is given subcutaneously every day in patients with relapsing–remitting MS. In clinical trials, all these drugs reduced the frequency and severity of exacerbations. MRI scans have demonstrated a reduction in active lesions.
Many other drugs are used to treat the symptoms of MS. Antispasmodics are used for spasticity. Amantadine and CNS stimulants (e.g. methylphenidate, modafinil) are used for fatigue. Anticholinergics are used to treat bladder symptoms. Tricyclic antidepressants and antiepileptic drugs are used for chronic pain syndromes.
Surgical therapy
Spasticity is primarily treated with antispasmodic drugs. However, surgery (e.g. neurectomy, rhizotomy, cordotomy), dorsal-column electrical stimulation or an intrathecal baclofen pump may be required. Tremors that become unmanageable with drugs are sometimes treated by thalamotomy or deep brain stimulation.
Other therapies
Neurological function sometimes improves with physiotherapy and speech therapy. Physiotherapy is important in keeping the patient as functionally active as possible. The purpose of therapy is to relieve spasticity, increase coordination and train the patient to substitute unaffected muscles for impaired ones. An especially beneficial type of therapy is water exercise (see Fig 58-5). Water gives buoyancy to the body and allows the patient to perform activities that would normally be impossible. In water, the patient experiences more control over the body.

Nutritional therapy
Various nutritional measures have been used in the management of MS, including megavitamin therapy (vitamin B12, vitamin C) and diets consisting of low-fat and gluten-free food and raw vegetables. These particular dietary measures have not come into widespread use because of lack of proof of their effectiveness.
A nutritious, well-balanced diet is essential. Although there is no standard prescribed diet, a high-protein diet with supplementary vitamins is often advocated. A diet high in roughage may help relieve the problem of constipation. Vitamins are merely supplemental and not curative.
What is the effect of exercise on quality of life in patients with multiple sclerosis?
EVIDENCE-BASED PRACTICE
Clinical question
In adults with multiple sclerosis (P), what is the effect of exercise (I) as compared to no exercise therapy (C) on activity limitations and quality of life (O)?
Conclusions
• Exercise strongly affects isometric strength, physical fitness and mobility-related activities when compared to no exercise therapy.
• Exercise therapy also improves a person’s mood (anxiety and depression).
• One type of exercise was not superior to another in improving activity or participation.
Implications for nursing practice
• Inform patients that daily functioning can be improved with exercise.
• Collaborate with patients to determine the best ‘dose’ and type of exercise to reach optimum beneficial effects.
P, patient population of interest; I, intervention or area of interest; C, comparison of interest or comparison group; O, outcome(s) of interest.
NURSING MANAGEMENT: MULTIPLE SCLEROSIS
Nursing assessment
Subjective and objective data that should be obtained from the patient with MS are presented in Table 58-9.

Nursing diagnoses
Nursing diagnoses for the patient with MS may include, but are not limited to, those presented in NCP 58-3.


Planning
The overall goals are that the patient with MS will: (1) maximise neuromuscular function; (2) maintain independence in activities of daily living for as long as possible; (3) optimise psychosocial wellbeing; (4) adjust to the illness; and (5) reduce factors that precipitate exacerbations.
Nursing implementation
The patient with MS should be aware of triggers that may cause exacerbations or worsening of the disease. Exacerbations of MS are triggered by infection (especially of the upper respiratory tract and urinary tract), trauma, immunisation, giving birth, stress and change in climate. Of these, the best documented are upper respiratory tract infections, the postpartum period and head trauma.11 Each person responds differently to these triggers. The nurse should help the patient identify particular triggers and develop ways to avoid them or minimise their effects.
The most common reasons for hospitalisation of the patient with MS are for a diagnostic examination and treatment of an acute exacerbation. During the diagnostic phase the patient needs reassurance that even though there is a tentative diagnosis of MS, certain diagnostic studies must be done to rule out other neurological disorders. The nurse should assist the patient in dealing with the anxiety caused by a diagnosis of a disabling illness. The patient with recently diagnosed MS may need assistance with the grieving process.
During an acute exacerbation, the patient may be immobile and confined to bed. The focus of nursing intervention at this phase is to prevent major complications of immobility, such as respiratory and urinary tract infections and pressure ulcers.
Patient teaching should focus on building general resistance to illness, including avoiding fatigue, extremes of heat and cold, and exposure to infection. The last measure involves avoiding exposure to cold climates and to people who are sick, as well as vigorous and early treatment of infection when it does occur. It is important to teach the patient to: (1) achieve a good balance of exercise and rest; (2) eat nutritious and well-balanced meals; and (3) avoid the hazards of immobility (e.g. contractures, pressure ulcers). The patient should understand their treatment regimen, the side effects of drugs and how to watch for the effects, as well as drug interactions with over-the-counter medications. The patient should consult a doctor or pharmacist before taking non-prescription drugs.
Bladder control is a major problem for many patients with MS. Although anticholinergics may be beneficial for some patients to decrease spasticity, other patients may need to be taught self-catheterisation (see Ch 45). Bowel problems, particularly constipation, occur frequently in patients with MS. Increasing the dietary fibre may help some patients achieve regularity in bowel habits.
The patient and family must make many emotional adjustments because of the unpredictability of the disease, the need to change lifestyles and the challenge of avoiding or decreasing precipitating factors. The Multiple Sclerosis Societies of New Zealand and Australia can offer a variety of services to meet the needs of patients with MS (see the Resources on p 1677).
Parkinson’s disease
Parkinson’s disease is a disease of the basal ganglia characterised by a slowing down in the initiation and execution of movement (bradykinesia), increased muscle tone (rigidity), tremor at rest and impaired postural reflexes. It is the most common form of parkinsonism (a syndrome characterised by similar symptoms). Parkinson’s disease is named after James Parkinson, who, in 1817, wrote a classic essay on ‘shaking palsy’, a disease whose cause is still unknown.
AETIOLOGY AND PATHOPHYSIOLOGY
The prevalence of Parkinson’s disease is about 160 per 100,000 and the incidence is about 20 per 100,000. The diagnosis of Parkinson’s disease increases with age, with the peak onset in the sixth decade of life. Onset of Parkinson’s disease before age 50 is more likely to be related to a genetic defect.14 Parkinson’s disease is more common in men by a ratio of 5:4.
There are many forms of parkinsonism other than Parkinson’s disease. Encephalitis lethargica, or type A encephalitis, has been clearly associated with the onset of parkinsonism. However, the incidence of postencephalitic parkinsonism has now dwindled. Parkinsonism-like symptoms have occurred after intoxication with a variety of chemicals, including carbon monoxide and manganese (among copper miners) and the product of meperidine-analogue synthesis, MPTP. Drug-induced parkinsonism can follow reserpine, methyldopa, lithium, haloperidol and phenothiazine therapy. Parkinsonism can also be seen following the use of illicit drugs, including amphetamine and methamphetamine. Other causes of parkinsonism include hydrocephalus, hypoxia, infections, stroke, tumour and trauma.14
The pathological process of Parkinson’s disease involves degeneration of the dopamine-producing neurons in the substantia nigra of the midbrain (see Figs 58-6 to 58-8), which in turn disrupts the normal balance between dopamine (DA) and acetylcholine (ACh) in the basal ganglia. DA is a neurotransmitter essential for normal functioning of the extrapyramidal motor system, including control of posture, support and voluntary motion. Symptoms of Parkinson’s disease do not occur until 80% of neurons in the substantia nigra are lost.

Figure 58-6 Nigrostriatal disorders produce parkinsonism. Left-sided view of the human brain showing the substantia nigra and the corpus striatum (shaded area) lying deep within the cerebral hemisphere. Nerve fibres extend upwards from the substantia nigra, divide into many branches and carry dopamine to all regions of the corpus striatum.

Figure 58-7 Dopaminergic synaptic activity is mediated by dopamine. Cholinergic synaptic activity is mediated by acetylcholine. A balance between the two kinds of activity produces normal motor function. A relative excess of cholinergic activity produces involuntary movements. Neurons in the caudate nucleus contain gamma-aminobutyric acid (GABA) and possibly control dopaminergic neurons in the substantia nigra through a feedback pathway.

CLINICAL MANIFESTATIONS
The onset of Parkinson’s disease is gradual and insidious, with a gradual progression and a prolonged course. It may involve only one side of the body initially. The classic manifestations of Parkinson’s disease often include the triad of tremor, rigidity and bradykinesia. In the early stages, only a mild tremor, slight limp or decreased arm swing may be evident. Later the patient may develop a shuffling, propulsive gait with arms flexed and loss of postural reflexes. Some patients may have a slight change in speech patterns. None of these alone is sufficient evidence for a diagnosis of the disease.
Tremor
Tremor, often the first sign, may be minimal initially, so the patient is the only one who notices it. This tremor can affect handwriting, causing it to trail off, particularly towards the ends of words. Parkinsonian tremor is more prominent at rest and is aggravated by emotional stress or increased concentration. The hand tremor is described as ‘pill rolling’ because the thumb and forefinger appear to move in a rotary fashion as if rolling a pill, coin or other small object. Tremor can involve the diaphragm, tongue, lips and jaw but rarely causes shaking of the head. Unfortunately, in many people a benign essential tremor has mistakenly been diagnosed as Parkinson’s disease. Essential tremor occurs during voluntary movement, has a more rapid frequency than parkinsonian tremor and is often familial.
Rigidity
Rigidity is the increased resistance to passive motion when the limbs are moved through their range of motion. Parkinsonian rigidity is typified by a jerky quality, as if there were intermittent catches in the movement of a cogwheel, when the joint is moved. This is termed cogwheel rigidity. The rigidity is caused by sustained muscle contraction and this consequently elicits a complaint of muscle soreness; feeling tired and achy; or pain in the head, upper body, spine or legs. Another consequence of rigidity is slowness of movement because it inhibits the alternating of contraction and relaxation in opposing muscle groups (e.g. biceps and triceps).
Bradykinesia
Bradykinesia is particularly evident in the loss of automatic movements, which is secondary to the physical and chemical alteration of the basal ganglia and related structures in the extrapyramidal portion of the CNS. In the unaffected patient, automatic movements are involuntary and occur subconsciously. They include blinking of the eyelids, swinging of the arms while walking, swallowing of saliva, self-expression with facial and hand movements, and minor movement of postural adjustment. The patient with Parkinson’s disease does not execute these movements and there is a lack of spontaneous activity. This accounts for the stooped posture, masked facies (deadpan expression), drooling of saliva and shuffling gait (festination) characteristic of a person with this disease. There is also difficulty in initiating movement and impaired fine movement, especially of the fingers.
COMPLICATIONS
Many of the complications of Parkinson’s disease are caused by the progressive deterioration and loss of spontaneity of movement. Swallowing may become very difficult (dysphagia) in severe cases, leading to malnutrition or aspiration. General debilitation may lead to pneumonia, urinary tract infections and skin breakdown. Mobility is greatly decreased. The gait slows and turning is especially difficult. The gait usually consists of rapid, short, shuffling ministeps. The posture is that of the ‘old man’ image, with the head and trunk bent forwards and the legs constantly flexed (see Fig 58-9). The lack of mobility may lead to constipation, ankle oedema and, more seriously, contractures. Dementia is up to six times more common in the elderly person with Parkinson’s disease.
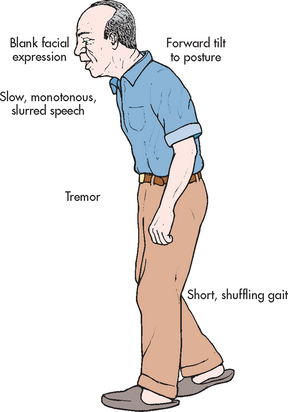
Orthostatic hypotension may occur in some patients and, along with loss of postural reflexes, may result in falls or other injury. Troublesome complications include seborrhoea (increased oily secretion of the sebaceous glands of the skin), dandruff, excessive sweating, conjunctivitis, difficulty reading, insomnia, incontinence and depression.
Many of the apparent complications of Parkinson’s disease are the result of the side effects of drugs, particularly levodopa. These include dyskinesias (e.g. fidgeting movements of limbs), hallucinations, orthostatic hypotension, weakness and akinesia (total immobility). These complications become apparent after prolonged levodopa therapy.
DIAGNOSTIC STUDIES
Because there is no specific diagnostic test for Parkinson’s disease, the diagnosis is based solely on the history and the clinical features. A firm diagnosis can be made only when at least two of the three characteristic signs of the classic triad are present: tremor, rigidity and bradykinesia (slow or retarded movement). Dementia occurs in up to 40% of patients with Parkinson’s disease.15 The ultimate confirmation of Parkinson’s disease is a positive response to antiparkinsonian drugs.
MULTIDISCIPLINARY CARE
Because there is no cure for Parkinson’s disease, collaborative management (see Box 58-8) is aimed at relieving the symptoms.
MULTIDISCIPLINARY CARE
Diagnostic studies
History and physical examination
Positive response to antiparkinson drugs (see Table 58-10)
Rule out side effects of phenothiazines, benzodiazepines, haloperidol
Collaborative therapy
Antiparkinson drugs (see Table 58-10)
Surgical destruction or deep brain stimulation of ventrolateral nucleus of the thalamus or posteroventral globus pallidus
Drug therapy
Drug therapy for Parkinson’s disease is aimed at correcting the imbalance of neurotransmitters within the CNS. Anti-parkinsonian drugs either enhance the release or supply of DA (dopaminergic) or antagonise or block the effects of the overactive cholinergic neurons in the striatum (anticholinergic). Levodopa with carbidopa is often the first drug to be used. Levodopa is a precursor of DA and can cross the blood–brain barrier. It is converted to DA in the basal ganglia. Levodopa can be combined with carbidopa or benserazide, which are agents that inhibit the enzyme dopa-decarboxylase in the peripheral tissues. This enzyme breaks down levodopa before it reaches the brain. The net result of the combination of levodopa and carbidopa is that more levodopa reaches the brain and therefore lower doses are required.
Many patients are given levodopa early in the disease course. However, some healthcare providers believe that after a few years of therapy, the effectiveness of levodopa wears off, so they prefer to initiate therapy with a DA receptor agonist instead. These drugs include bromocriptine and cabergoline pergolide. These drugs directly stimulate DA receptors. When more moderate-to-severe symptoms are present, levodopa with carbidopa is added to the drug regimen.
Anticholinergic drugs are also used to manage Parkinson’s disease. These drugs act by decreasing the activity of ACh, thus providing a balance between cholinergic and dopaminergic actions. Antihistamines with anticholinergic properties or a β-adrenergic blocker (e.g. propranolol) are used to manage tremors. The antiviral agent amantadine is also an effective antiparkinsonian drug. Although its exact mechanism of action is not known, amantadine promotes the release of DA from neurons.
Selegiline is a monoamine oxidase (MAO) inhibitor that is sometimes used in combination with carbidopa-levodopa. By inhibiting MAO, the degradative enzyme for DA, the levels of DA are increased. Entacapone and tolcapone block the enzyme catechol-O-methyl transferase (COMT), which breaks down levodopa in the peripheral circulation, thus prolonging the effect of carbidopa-levodopa. This helps to manage symptoms caused by ‘wearing off’ of carbidopa-levodopa before the next dose is due.
Table 58-10 summarises the drugs commonly used in Parkinson’s disease, the symptoms they relieve and their common side effects. The use of only one drug is preferred because there are fewer side effects and the drug dosage is easier to adjust than when several drugs are used. However, as the disease progresses, combination therapy is often required. Excessive amounts of dopaminergic drugs can lead to paradoxic intoxication (aggravation rather than relief of symptoms).

Surgical therapy
Surgical procedures are aimed at relieving the symptoms of Parkinson’s disease and are usually used in patients who are unresponsive to drug therapy or who have developed severe motor complications. Surgical procedures fall into three categories: ablation (destruction), deep brain stimulation (DBS) and transplantation. Ablation surgery involves stereotactic ablation of areas in the thalamus (thalamotomy), globus pallidus (pallidotomy) and subthalamic nucleus (subthalamic nucleotomy). Ablative procedures have been used in patients with Parkinson’s disease for more than 50 years but recently they have been replaced by DBS. DBS involves placing an electrode in one of the thalamus, globus pallidus or subthalamic nucleus and connecting it to a generator placed in the upper chest (like a pacemaker). The device is programmed to deliver a specific current to the targeted brain location. Unlike ablation procedures, DBS can be adjusted to control symptoms better and is reversible—that is, the device can be removed. These ablative and DBS procedures work by reducing the increased neuronal activity that is produced by DA depletion.16 Transplantation of fetal neural tissue into the basal ganglia is designed to provide DA-producing cells in the brains of patients with Parkinson’s disease. This form of therapy is still in the experimental stages.
Nutritional therapy
Diet is of major importance to the patient with Parkinson’s disease because malnutrition and constipation can be serious consequences of inadequate nutrition. Patients who have dysphagia and bradykinesia need appetising foods that are easily chewed and swallowed. The diet should contain adequate roughage and fruit to avoid constipation. Food should be cut into bite-sized pieces before it is served and it should be served on a warmed plate to preserve its appeal. Eating six small meals a day may be less exhausting than eating three large meals a day. Ample time should be planned for eating to avoid frustration and encourage independence. In addition, absorption of levodopa can be impaired by protein ingestion. Some patients are advised to limit their protein intake to the evening meal to avoid this problem.
NURSING MANAGEMENT: PARKINSON’S DISEASE
Nursing assessment
Subjective and objective data that should be obtained from the patient with Parkinson’s disease are presented in Table 58-11.
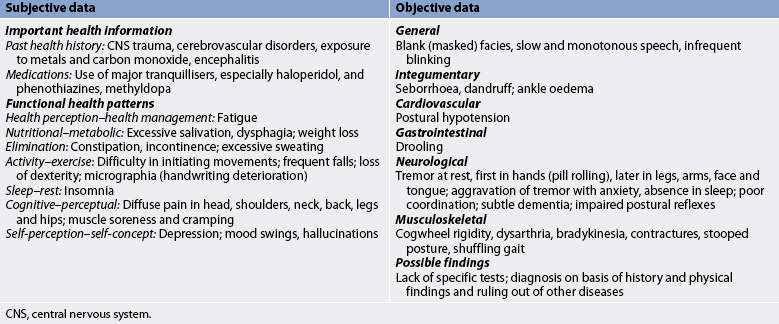
Nursing diagnoses
Nursing diagnoses for the patient with Parkinson’s disease may include, but are not limited to, those presented in NCP 58-4.

Planning
The overall goals are that the patient with Parkinson’s disease will: (1) maximise neurological function; (2) maintain independence in activities of daily living for as long as possible; and (3) optimise psychosocial wellbeing.
Nursing implementation
Promotion of physical exercise and a well-balanced diet are major concerns for nursing care. Exercise can limit the consequences of decreased mobility, such as muscle atrophy, contractures and constipation. Parkinson’s Australia (see the Resources on p 1677) publishes a series of fact sheets and videotapes that are helpful in terms of exercises that can be used by family members and healthcare professionals. A physiotherapist may be consulted to design a personal exercise program aimed at strengthening and stretching specific muscles. Overall muscle tone, as well as specific exercises to strengthen the muscles involved with speaking and swallowing, should be included. Although exercise will not halt the progress of the disease, it will enhance the patient’s functional ability.
Because Parkinson’s disease is a chronic degenerative disorder with no acute exacerbations, nurses should note that teaching and nursing care are directed towards maintenance of good health, encouragement of independence and avoidance of complications such as contractures.
Problems secondary to bradykinesia can be alleviated by relatively simple measures. The following are helpful hints for patients who tend to ‘freeze’ while walking: consciously think about stepping over imaginary or real lines on the floor, drop rice kernels and step over them, rock from side to side, lift the toes when stepping, take one step backwards and two steps forwards. The patient should be assessed for the possibility of levodopa overdose because it is a common cause of akinesia (‘freezing’). A brief period of dyskinesia, usually athetosis (slow, writhing, continuous and involuntary movement) of the neck, should alert the nurse to this possibility.
Getting out of a chair can be facilitated by using an upright chair with arms and placing the back legs on small (6 cm) blocks. Other aspects of the environment can be altered. Rugs and excess furniture can be removed to avoid stumbling. An ottoman can be used to elevate the legs and avoid dependent ankle oedema. Clothing can be simplified by the use of slip-on shoes and Velcro hook-and-loop fasteners or zippers on clothing, instead of buttons and hooks. An elevated toilet seat can facilitate getting on and off the toilet. The nurse should work closely with the patient’s family in exploring creative adaptations that allow maximum independence and self-care.
Myasthenia gravis
Myasthenia gravis (MG) is an autoimmune disease of the neuromuscular junction characterised by the fluctuating weakness of certain skeletal muscle groups. The prevalence rate of 400 per million has been estimated to grow to 500 per million with improving survival rates.17 MG can occur at any age but most commonly occurs between the ages of 10 and 65. The peak age at onset is 20–30 years in women and 60+ years in males.18 Overall, both sexes are equally affected.
AETIOLOGY AND PATHOPHYSIOLOGY
MG is caused by an autoimmune process in which antibodies attack ACh receptors, resulting in a decreased number of ACh receptor sites at the neuromuscular junction. This prevents ACh molecules from attaching and stimulating muscle contraction. Anti-ACh receptor antibodies are detectable in the serum of 85–90% of patients with generalised MG and in 50–60% of patients with ocular myasthenia.18 Thymic tumours are found in about 15% of patients and abnormal thymus tissue is found in most others.
CLINICAL MANIFESTATIONS AND COMPLICATIONS
The primary feature of MG is fluctuating weakness of skeletal muscle. Strength is usually restored after a period of rest. The muscles most often involved are those used for moving the eyes and eyelids, chewing, swallowing, speaking and breathing. The muscles are generally the strongest in the morning and become exhausted with continued activity. Consequently, by the end of the day, muscle weakness is prominent.
In 90% of cases, the eyelid or extraocular muscles are involved. Facial mobility and expression can be impaired. There may be difficulty in chewing and swallowing food. Speech is affected and the voice often fades after a long conversation. The muscles of the trunk and limbs are less often affected. Of these, the proximal muscles of the neck, shoulder and hip are more often affected than the distal muscles. No other signs of neural disorder accompany MG; there is no sensory loss, reflexes are normal and muscle atrophy is rare.
The course of this disease is highly variable. Some patients may have short-term remissions, others may stabilise and others may have severe, progressive involvement. Restricted ocular myasthenia, usually seen only in men, has a good prognosis. Exacerbations of MG can be precipitated by emotional stress, pregnancy, menses, secondary illness, trauma, temperature extremes and hypokalaemia. Ingestion of drugs, including aminoglycoside antibiotics, β-adrenergic blockers, procainamide, quinidine and phenytoin, can aggravate MG. Psychotropic drugs (e.g. lithium carbonate, phenothiazines, benzodiazepines, tricyclic antidepressants) have also been associated with worsening of MG, as have neuromuscular blocking agents (pancuronium, suxamethonium chloride). In some cases, the onset of MG occurs after one of these events.
Myasthenic crisis is an acute exacerbation of muscle weakness triggered by infection, surgery, emotional distress or overdose of or inadequate drugs. The major complications of MG result from muscle weakness in areas that affect swallowing and breathing, resulting in aspiration, respiratory insufficiency and respiratory tract infection.
DIAGNOSTIC STUDIES
The diagnosis of MG is based on a history and physical examination. However, other tests may be used if the diagnosis is still in doubt. Blood tests show that antibodies to ACh receptors are found in 85–90% of patients with generalised MG. EMG may show a decreasing response to repeated stimulation of the hand muscles, indicative of muscle fatigue. Use of drugs may also aid in the diagnosis. The Tensilon test in a patient with MG reveals improved muscle contractility after intravenous injection of the anticholinesterase agent edrophonium chloride, which blocks the enzyme acetylcholinesterase. This test also aids in the diagnosis of cholinergic crisis (secondary to overdose of anticholinesterase drug). In this condition, Tensilon does not improve muscle weakness but may actually increase it. Atropine, a cholinergic antagonist, should be readily available to counteract Tensilon effects when it is used diagnostically.
MULTIDISCIPLINARY CARE
Drug therapy
Drug therapy for MG includes anticholinesterase drugs, alternate-day corticosteroids and immunosuppressants (see Box 58-9).
Anticholinesterase drugs are aimed at enhancing function of the neuromuscular junction. Acetylcholinesterase is the enzyme that breaks down ACh in the synaptic cleft. Thus, inhibition of this enzyme by an anticholinesterase inhibitor will prolong the action of ACh and facilitate transmission of impulses at the neuromuscular junction. Neostigmine and pyridostigmine are the most successful drugs of this group in treating MG. Tailoring the dose to avoid a myasthenic or cholinergic crisis often presents a clinical challenge. Because of the autoimmune nature of the disorder, corticosteroids (specifically prednisone) are used to suppress the immune response. Drugs such as azathioprine and cyclophosphamide may also be used for immunosuppression.
Many drugs are contraindicated or must be used with caution in patients with MG. Classes of drug that should be evaluated cautiously before use include anaesthetics, antiarrhythmics, antibiotics, quinine, antipsychotics, barbiturates and sedative–hypnotics, cathartics, diuretics, opioids, muscle relaxants, thyroid preparations and tranquillisers.
Surgical therapy
The presence of the thymus gland in the patient with MG appears to enhance the production of ACh receptor antibodies, so removal of the thymus gland results in improvement in a majority of patients. Thymectomy is indicated for almost all patients with thymoma, for patients with generalised MG between the ages of puberty and about 65 years, and for patients with purely ocular MG.19
Other therapies
Plasmapheresis can yield a short-term improvement in symptoms and it is indicated for patients in crisis or in preparation for surgery when corticosteroids must be avoided. (Plasmapheresis is discussed in Ch 13.) Intravenous immunoglobulin G has been used with some success and is recommended as a second-line treatment for MG.20
NURSING MANAGEMENT: MYASTHENIA GRAVIS
Nursing assessment
The nurse can assess the severity of MG by asking the patient about fatiguability, which parts of the body are affected and how severely they are affected. The patient’s coping abilities and understanding of the disorder should also be assessed. Some patients become so fatigued that they are no longer able to work or even move around.
Objective data should include respiratory rate and depth, oxygen saturation, arterial blood gas analyses, pulmonary function tests and evidence of respiratory distress in patients with acute myasthenic crisis. Muscle strength of all face and limb muscles should be assessed, as should swallowing, speech (volume and clarity), and cough and gag reflexes.
Nursing diagnoses
Nursing diagnoses for the patient with MG may include, but are not limited to, the following:
• ineffective breathing pattern related to intercostal muscle weakness
• ineffective airway clearance related to intercostal muscle weakness and impaired cough and gag reflex
• impaired verbal communication related to weakness of the larynx, lips, mouth, pharynx and jaw
• imbalanced nutrition: less than body requirements related to impaired swallowing
• disturbed sensory perception: visual related to ptosis, decreased eye movements and disconjugate gaze
• activity intolerance related to muscle weakness and fatiguability
• disturbed body image related to inability to maintain usual lifestyle and role responsibilities.
Planning
The overall goals are that the patient with MG will: (1) have a return of normal muscle endurance; (2) avoid complications; and (3) maintain a quality of life appropriate to the disease course.
Nursing implementation
The patient with MG who is admitted to the hospital usually has a respiratory tract infection or is in an acute myasthenic crisis. Nursing care is aimed at maintaining adequate ventilation, continuing drug therapy and watching for side effects of therapy. The nurse must be able to distinguish cholinergic from myasthenic crisis (see Table 58-12) because the causes and treatment of the two conditions differ greatly.
TABLE 58-12 Comparison of myasthenic crisis and cholinergic crisis
| Myasthenic crisis | Cholinergic crisis | |
|---|---|---|
| Causes | Exacerbation of myasthenia following precipitating factors or failure to take drug as prescribed or drug dose too low | Overdose of anticholinesterase drugs resulting in increased ACh at the receptor sites, remission (spontaneous or after thymectomy) |
| Differential diagnosis | Improved strength after IV administration of anticholinesterase drugs; increased weakness of skeletal muscles manifesting as ptosis, bulbar signs (e.g. difficulty in swallowing, difficulty in articulating words) or dyspnoea | Weakness within 1 h after ingestion of anticholinesterase; increased weakness of skeletal muscles manifesting as ptosis, bulbar signs, dyspnoea; effects on smooth muscle include pupillary miosis, salivation, diarrhoea, nausea or vomiting, abdominal cramps, increased bronchial secretions, sweating or lacrimation |
ACh, acetylcholine; IV, intravenous.
As with other chronic illnesses, care focuses on the neurological deficits and their impact on daily living. A balanced diet with food that can be chewed and swallowed easily should be prescribed. Semisolid foods may be easier to eat than solids or liquids. Scheduling doses of drugs so that peak action is reached at mealtimes may make eating less difficult. Diversional activities that require little physical effort and match the interests of the patient should be arranged. Teaching should focus on the importance of following the medical regimen, potential adverse reactions to specific drugs, planning activities of daily living to avoid fatigue, the availability of community resources, the complications of the disease and therapy (crisis conditions), and what to do about them. Contact with a support group may be helpful and should be explored.
Restless legs syndrome
AETIOLOGY AND PATHOPHYSIOLOGY
Restless legs syndrome (RLS) is characterised by unpleasant sensory (paraesthesias) and motor abnormalities of one or both legs. Prevalence rates vary from 1% to 15%, although the numbers may be higher because the condition is underdiagnosed.21 Although the exact cause of RLS is not known, probably more than half of all cases are transmitted in an autosomal dominant pattern.22 RLS can be seen in metabolic abnormalities associated with iron deficiency, renal failure, polyneuropathy associated with diabetes mellitus, thyroid disorders, rheumatic disorders (e.g. rheumatoid arthritis) and pregnancy. However, the majority of cases are idiopathic. Idiopathic RLS may be related to nervous system dysfunction. Although the exact aetiology remains to be determined, theories include: (1) an alteration in dopaminergic transmission in the basal ganglia; (2) axonal neuropathy; or (3) a brainstem disinhibition phenomenon resulting in motor and sensory disturbances.
CLINICAL MANIFESTATIONS
The severity of RLS sensory symptoms ranges from infrequent minor discomfort (paraesthesias including numbness, tingling, ‘pins and needles’ sensation) to severe pain. Sensory symptoms often appear first and they are manifested as an annoying and uncomfortable (but usually not painful) sensation in the legs that may be compared with the sensation of bugs creeping or crawling on the legs. The leg pain is localised within the calf muscles. Patients can also experience pain in the upper extremities and trunk. The discomfort occurs when the patient is sedentary and usually occurs in the evening or at night.
The pain at night can produce sleep disruptions and is often relieved by physical activity, such as walking, stretching, rocking or kicking. In the most severe cases, patients sleep only a few hours at night, resulting in daytime fatigue and disruption of the daily routine. The motor abnormalities associated with RLS consist of voluntary restlessness and stereotyped, periodic, involuntary movements. The involuntary movements usually occur during sleep. Symptoms are aggravated by fatigue. Over time, RLS advances to more frequent and more severe episodes.
DIAGNOSTIC STUDIES
RLS is a clinical diagnosis and is based in large part on the patient’s history or the report of the bed partner related to night-time activities. The International Restless Legs Study Group has proposed four minimum diagnostic criteria: (1) desire to move the limbs; (2) motor restlessness; (3) symptoms that are worse or exclusively present at rest with at least partial and temporary relief by activity; and (4) symptoms that are worse in the evening or at night.21 Polysomnography studies during sleep may be performed for the patient with RLS to distinguish the problem from other clinical conditions (e.g. sleep apnoea) that can disturb sleep. However, periodic leg movements in sleep are a common feature in RLS patients. The patient’s history of diabetes mellitus and its management may provide information to determine whether paraesthesias are caused by peripheral neuropathy or RLS.
NURSING AND COLLABORATIVE MANAGEMENT: RESTLESS LEGS SYNDROME
The goal of collaborative management is to reduce patient discomfort and distress and to improve sleep quality. When RLS is secondary to uraemia or iron deficiency, correction of these conditions will decrease symptoms. Non-pharmacological approaches to RLS management include establishing regular sleep habits, encouraging exercise, avoiding activities that cause symptoms and eliminating aggravating factors, such as alcohol, caffeine and certain drugs (neuroleptics, lithium, antihistamines and antidepressants).
If non-pharmacological measures fail to provide symptom relief, drug therapy may be started. The main drugs used in RLS are dopaminergic agents, opioids and benzodiazepines. Dopaminergic agents such as carbidopa-levodopa and DA agonists (e.g. pergolide, bromocriptine) are the drugs of choice as they are effective in managing sensory and motor symptoms. Dopaminergic agents have a number of side effects, including hypotension and gastric irritation.
Other agents that may be used include antiepileptic drugs, such as gabapentin, sodium valproate, lamotrigine and carbamazepine. Clonidine and propranolol are also effective in some patients. Opioids (e.g. oxycodone) are usually reserved for those patients with severe symptoms who fail to respond to other drug therapies. When used, opioids given in low doses have been found to be effective in reducing symptoms. The main side effect of opioids is constipation, so the patient may need to take a stool softener or laxative.
Motor neuron disease
Motor neuron disease (MND) is a rare, progressive neurological disorder characterised by loss of motor neurons and usually leads to death within 2–6 years after diagnosis. (MND is also known as amyotrophic lateral sclerosis in the US.) The onset is usually between 40 and 70 years of age. MND is more common in men than women by a ratio of 2:1.
For unknown reasons, motor neurons in the brainstem and spinal cord gradually degenerate in MND (see Fig 58-10). Dead motor neurons cannot produce or transport vital signals to muscles. Consequently, electrical and chemical messages originating in the brain do not reach the muscles to activate them.

Figure 58-10 Pathogenesis of motor neuron disease. This disease is characterised by degeneration of the pyramidal tract and the motor cells in the anterior grey horns. In cases with corticobulbar involvement, the motor nuclei of cranial nerves V, VII, IX, X, XI and XII also undergo degeneration.
The typical symptoms are weakness of the upper extremities, dysarthria and dysphagia. However, weakness may begin in the legs. Muscle wasting, fasciculations and exaggerated reflexes result from the denervation of the muscles and lack of stimulation and use. Death usually results from respiratory tract infection secondary to compromised respiratory function. Unfortunately, there is no cure for MND. Riluzole slows the progression of MND.23,24 This drug works to decrease the amount of glutamate (an excitatory neurotransmitter) in the brain. In clinical trials, riluzole has been shown to delay the need for tracheostomy and death by a few months.24
The illness trajectory for MND is devastating because the patient remains cognitively intact while wasting away. The challenge of nursing care is to support the patient’s cognitive and emotional functions. This is achieved by facilitating communication, reducing the risk of aspiration, decreasing pain secondary to muscle weakness, decreasing risk of injury related to falls, providing diversional activities such as reading and human companionship, and helping the person and family with advance care planning and anticipatory grieving related to loss of motor function and ultimately death.
Huntington’s disease
Huntington’s disease (HD) is a genetically transmitted, autosomal dominant disorder that affects both men and women of all races. The offspring of a person with this disease have a 50% risk of inheriting it (see the Health disparities box). The onset of HD is usually between 30 and 45 years of age.25 Often, the diagnosis is made after the affected individual has had children.
In Australia, the incidence of HD is 6–7 in 100,000.25 Most analysts agree that migration out of Europe brought HD to Australia, New Zealand, North and South America and other regions with European contact.26 The prevalence of HD in these regions is similar to that in Europe (40–100 cases per million people). Among the Caucasian populations of Australia and New Zealand there is a uniform, widespread distribution of HD. Tasmania represents an exception to this distribution: there is an unusually high frequency of HD in one large clan of English origin. These people are descendants of a single ancestor with HD from Somerset in the UK. There have been no documented cases of HD in the Indigenous populations of Australia or New Zealand.26
Diagnosis in the past was based on family history and clinical symptoms. However, since the gene for HD has been discovered, people now can be tested for the presence of the gene. Those who are asymptomatic but who have a positive family history of HD face the dilemma of whether or not to be tested. If the test is positive, the person will develop HD, but when and to what extent the disease develops cannot be determined.
Like Parkinson’s disease, the pathological process of HD involves the basal ganglia and the extrapyramidal motor system. However, instead of a deficiency of dopamine, HD involves a deficiency of the neurotransmitters ACh and gamma-aminobutyric acid (GABA). The net effect is an excess of DA, which leads to symptoms that are the opposite of those of parkinsonism. The clinical manifestations are characterised by abnormal and excessive involuntary movements (chorea). These are writhing, twisting movements of the face, limbs and body. The movements get worse as the disease progresses. The facial movements that are involved with speech, chewing and swallowing are affected and they may cause aspiration and malnutrition. The gait deteriorates and ambulation eventually becomes impossible. Perhaps the most devastating deterioration is in mental function, which includes intellectual decline, emotional lability and psychotic behaviour. Death usually occurs 10–20 years after the onset of symptoms.
Because there is no cure, multidisciplinary care is palliative. Antipsychotic drugs (e.g. haloperidol) and antidepressants (fluoxetine, clonazepam) are prescribed and have some benefit. However, they do not alter the course of the disease. Transplantation of fetal striatal neural tissues into the striatum (caudate and putamen) of the brain is a treatment that has been found to be effective in some circumstances.27
HD presents a great challenge to healthcare professionals. The goal of nursing management is to provide the most comfortable environment possible for the patient and family by maintaining physical safety, treating the physical symptoms, and providing emotional and psychological support. Because of the choreic movements, energy requirements are high. Patients may require as many as 16,000–20,000 kJ per day to maintain body weight. As the disease progresses, meeting energy needs becomes a greater challenge as the patient has difficulty swallowing and holding the head still. Depression and mental deterioration can also compromise nutritional intake.
The patient with epilepsy with headache
CASE STUDY
Patient profile
Julie Perry is a 24-year-old woman who was diagnosed with epilepsy at age 11. At that time, she had a tonic–clonic seizure and was given a prescription for phenobarbitone. Her family moved to another state and follow-up was forgotten until she had a second witnessed seizure at age 13. She had not taken medication for seizures until phenytoin was begun at age 13. She now complains of headaches and says she is afraid that her seizures are going to return. She is single, lives alone and describes her job as stressful.
CRITICAL THINKING QUESTIONS
2. What is the pathophysiology of epilepsy?
3. What is the significance of the laboratory and diagnostic findings?
4. Is the headache related to seizure activity?
5. What is a priority nursing intervention for this patient?
6. What should be included in the teaching plan regarding the course of the disease that should be developed for this patient?
7. Based on the assessment data, what are the priority nursing diagnoses?
1. The nurse plans care for the patient with a migraine headache based on the knowledge that during a migraine the patient is most likely to:
2. The triad of symptoms the nurse would expect to find during assessment of the patient with Parkinson’s disease is:
3. During assessment of the patient with motor neuron disease, the nurse would expect to find:
4. The emotional response of the patient with a chronic neurological disease is often:
5. A major goal of treatment for the patient with a chronic, progressive neurological disease is:
1 Headache Australia. Prevalence and cost of headache. Available at http://headacheaustralia.org.au/what-is-headache/11-prevalence-and-cost-of-headache. accessed 21 November 2010.
2 Neurological Foundation of New Zealand. Migraine and headache. Available at www.neurological.org.nz/News/General-Articles/Article/Migraine+and+headaches/. accessed 21 November 2010.
3 Lipton RB, Bigal ME. Migraine: epidemiology, impact, and risk factors for progression. Headache. 2005;45(suppl 1):S3–S13.
4 May A. Cluster headache: pathogenesis, diagnosis, and management. Lancet. 2005;366(9488):843–855.
5 Hagan K, Albertsen C, Vilming VT, Salvesen R, Grønning M, Helde G. Management of medication overuse headache: 1-year randomised multicentre open-label trial. Cephalgia. 2009;29(2):221–232.
6 Epilepsy Review Committee. WA Epilepsy Services Model of Care: neurosciences and the senses health network. Available at www.wagpnetwork.com.au/client_images/164442.pdf. accessed 21 November 2010.
7 Gurnett CA, Hedera P. New ideas in epilepsy genetics: novel epilepsy, copy number alterations, and gene regulations. Arch Neurol. 2007;64:324–328.
8 Engel J. Report of the ILAE Classification Core Group, 2006. Epilepsia. 2006;47(9):1558–1568.
9 Smellie-Decker M, Berlin J, Leonard T, Salazar C, Wall Strother K. Surgical management of epilepsy. Available at www.springerlink.com/content/lj4v0l4t49hm3781/fulltext.pdf, 2007. accessed 21 November 2010.
10 World Health Organization/Multiple Sclerosis International Foundation. Atlas: multiple sclerosis resources in the world. Available at www.who.int/mental_health/neurology/Atlas_MS_WEB.pdf, 2008. accessed 21 November 2010.
11 Murray TJ. Diagnosis and treatment of multiple sclerosis. Br Med J. 2006;332(7530):25–27.
12 Salemi G, Callari G, Gammino M, et al. The relapse rate of multiple sclerosis changes during pregnancy: a cohort study. Acta Neurol Scand. 2004;110(1):23–26.
13 Ryan M. Drug therapies for the treatment of multiple sclerosis. J Infusion Nursing. 2009;32(3):137–144.
14 Fahn S, Przedborski S. Parkinsonism. In Rowland LP, ed.: Merritt’s neurology, 12th edn., Philadelphia: Lippincott Williams & Wilkins, 2009.
15 Glosser G. Neurobehavioral aspects of movement disorders. Neurol Clin. 2001;19(3):535–551. v.
16 Benabid AL, Chabardes S, Mitrofamis J, Pollak P. Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson’s disease. Lancet Neurol. 2009;8(1):67–81.
17 Angelini C. Diagnosis and management of autoimmune myasthenia gravis. Clin Drug Investig. 2010;31(1):1–14.
18 Penn AS, Rowland LP. Myasthenia gravis. In Rowland LP, ed.: Merritt’s neurology, 12th edn., Philadelphia: Lippincott Williams & Wilkins, 2009.
19 Spillane J, Kullmann DM, Taylor C, Hirsch NP, Havard R. Thymectomy: its role in the management of myasthenia gravis. J Neurol Neurosurg Psychiatry. 2010;81(11):e62–e63.
20 Kondo K, Monden Y. Myasthenia gravis appearing after thymectomy for thymoma. Eur J Cardiothorac Surg. 2005;28:22–25.
21 Salas RE, Garnaldo CE, Allen R. Update in restless leg syndrome. Curr Opin Neurol. 2010;23(4):401–406.
22 National Institute of Neurological Disorders and Stroke. NINDS restless legs syndrome information page. Available at www.ninds.nih.gov/disorders/restless_legs/restless_legs.htm. accessed 23 November 2010.
23 Bento-Abreu A, Van Damme P, Van Den Bosch L, Robberecht W. The neurobiology of amyotrophic lateral sclerosis. Eur J Neurosci. 2010;31(12):2247–2265.
24 Miller RG, Mitchell JD, Lyon M, et al. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. (1):2007. CD001447.
25 Huntingtons Australia. Available at http://huntingtonsaustralia.asn.au/ accessed 23 November 2010.
26 Stanford University Population Huntington’s Outreach Project. Population genetics and Huntington’s disease. Available at http://hopes.stanford.edu/n3421/hd-genetics/population-genetics-and-huntingtons-disease-text-and-audio#What_is_the_frequency_of_HD_around_the_world, Updated 30 August 2010. accessed 2 December 2010.
27 Kelly CM, Dunnett SB, Rosses AE. Medium spiny neurons for transplantation in Huntington’s disease. Biochemic Soc Trans. 2009;37(Pt.1):323–328.
Australasian Neuroscience Nurses’ Association. www.anna.asn.au
Brain Foundation. www.brainaustralia.org.au
Epilepsy Action Australia. www.epilepsy.org.au
Epilepsy Foundation of Victoria. www.epinet.org.au
Epilepsy New Zealand. www.epilepsy.org.nz
Headache Australia. www.headacheaustralia.org.au
Huntingtons Australia. www.huntingtonsaustralia.asn.au
Huntington’s Disease Associations of New Zealand. www.huntingtons.org.nz
Migraine Support New Zealand. www.migraine.co.nz
MND Australia. www.mndaust.asn.au
Multiple Sclerosis Society of Australia. www.msaustralia.org.au
Multiple Sclerosis Society of New Zealand. www.msnz.org.nz/
Neurological Foundation of New Zealand. www.neurological.org.nz
New Zealand Epilepsy Assist Dogs Trust. http://epilepsyfoundation.org.nz/index.asp?pageID=2145841675
Parkinson’s Australia. www.parkinsons.org.au
Parkinson’s New Zealand. www.parkinsons.org.nz
Restless Legs Syndrome. www.rls.org.au
