Chapter 42
Skeletal Muscle Excitability
Chapter Outline
III. General Overview of Electrogenesis of the Action Potential
IV. Ion Channel Activation and Inactivation
V. Slow Delayed Rectifier K+ Current
VI. Mechanisms of Repolarization
VII. ATP-Dependent K+ Channels
VIII. Electrogenesis of Depolarizing Afterpotentials
IX. Ca2+-Dependent Slow Action Potentials
X. Developmental Changes in Membrane Properties
XI. Electrogenic Na+-K+ Pump Stimulation
XIII. Conduction of the Action Potential
XIV. Excitation Delivery to Fiber Interior by Conduction into the T-Tubular System
AI. More Information on Cl− Channels
AII. More Information on KATP Channels
AIII. Further Evidence that the T-Tubules Fire Na+-Dependent APS
AIV. Propagation Velocity in a Passive Cable
AV. Evidence for T-Tubule Communication with the SR across the Triadic Junction under Some Conditions
I Summary
The resting potential (RP) of skeletal muscle twitch fibers is about −80 mV (mammalian) or −90 mV (amphibian) and the action potential (AP) overshoots to about +40 mV. The maximal rate of rise of the AP is very fast, being about 500–700 V/s and is due to a large inward fast Na+ current (INa) which brings Em up close to ENa. The duration of the AP (at 50% repolarization or APD50) is brief, being about 1–3 ms and the falling phase of the AP is produced by several repolarizing factors: (1) increase in the delayed rectifier K+ conductance; (2) Na+ channel inactivation and deactivation (due to some repolarization); and (3) Cl− influx (outward ICl).
The skeletal muscle fibers are long, being formed by the fusion of myoblast cells and thereby producing a multinucleated long myotube or fiber. The fast-rising AP propagates at a velocity of about 5 m/s. Each skeletal muscle twitch fiber is normally closely controlled by the motor innervation, there being one or two motor end-plates (neuromuscular junctions) located near the midregion of each fiber. Excitation spreads in both directions from the neuromuscular junction.
The twitch fibers undergo developmental changes similar to those in cardiac muscle and neurons. In early development, there are few or no fast Na+ channels and the AP upstroke is slow and produced by an inward current through slow Ca2+ channels. The AP duration is also long because the delayed rectifier K+ conductance is not fully developed. During subsequent development, fast Na+ channels are gained, the RP increases (becomes more negative) and the AP shortens to a brief spike.
The skeletal muscle AP spike is immediately followed by a large and prominent early depolarizing afterpotential that slowly decays over 10–20 ms. This early afterpotential is caused in part by the persistence and slow decay of the delayed rectifier K+ conductance (that was turned on by the Na+ influx-caused depolarization), which has a Na+:K+ selectivity or PNa/PK ratio higher (e.g. 1/30) than that of the resting membrane (e.g. 1/100). This delayed rectifier K+ conductance holds Em for a time more depolarized than the normal RP.
After (and during) a tetanic burst (train) of AP spikes, a large prominent late depolarizing afterpotential is produced. This late afterpotential is caused by K+ accumulation in the T-tubules that acts to depolarize them due to the decrease in EK. Thereby, the surface sarcolemma is depolarized passively. In addition, a slow component of the delayed rectifier K+ conductance (less selective for K+ than the resting conductance) may persist during the train.
The AP invades into the T-tubules and propagates inward at a slower velocity of about 7–10 cm/s (Gonzalez-Serratos et al., 1978). At this velocity, it would take about 1.0–1.5 ms to propagate to the center of a myofiber having a radius of 30–40 μm. This serves to bring excitation deep into the fiber interior quickly. The depolarization of the T-tubules activates slow (L-type) Ca2+ channels located in them and this serves as a critical step in excitation–contraction (EC) coupling. The chapter on EC coupling provides a detailed discussion of a mechanism that involves the Ca2+ channels acting as voltage sensors that are coupled to and open the Ca2+ release channels in the TC-SR (surface facing the T-tubule).
Some skeletal muscles also contain a fraction of fibers that are non-twitch slow muscle fibers, which normally do not fire APs. They are multiply innervated by the motor neuron, with numerous motor end-plates spaced about 1 mm apart along the entire length of the fiber. Graded contraction of each fiber is produced by varying the frequency of axon APs that increase the amplitude of the end-plate potentials (EPPs) by temporal summation. The membrane potential change produced by the summed EPPs is carried passively into the T-tubules to bring about contraction.
II Introduction
The normal contraction of vertebrate twitch-type skeletal muscle fibers is always preceded by an AP. The AP depolarizes the sarcolemma beyond the membrane potential (Em) level at which contraction is triggered, i.e. the mechanical threshold (see the subsequent chapter on E-C coupling). This is the first step in the chain of events triggered by the initial excitatory process in the sarcolemma, linking it to the final mechanical response. This chain of events is known as excitation–contraction (E-C) coupling. In this chapter, we will study the sarcolemmal electrophysiological properties that are the basis of the AP generation. AP generation and excitability in neurons were covered in an earlier chapter. Most of the general electrophysiological principles discussed there also apply to skeletal muscle fibers and so are only briefly reviewed and summarized in this chapter. In most respects, the electrogenesis of the APs in nerve axons and skeletal muscle fibers is quite similar. Both are long fibers and have very brief and fast-rising APs whose inward current is carried by Na+ ions through fast Na+ channels. Skeletal muscle fibers, however, have the added complexity of an extensive internal transverse (T) tubular system formed by a periodic invagination of the surface cell membrane (Fig. 42.1), forming an orderly three-dimensional array of tubules that propagate excitation from the cell surface into the deep interior of the fiber for purposes of E-C coupling. In addition, electrogenesis in skeletal muscle fibers differs from nerve fibers in that (1) different types of afterpotentials are produced, (2) propagation is continuous (i.e. saltatory propagation does not occur) and (3) the membrane has a high Cl− conductance.
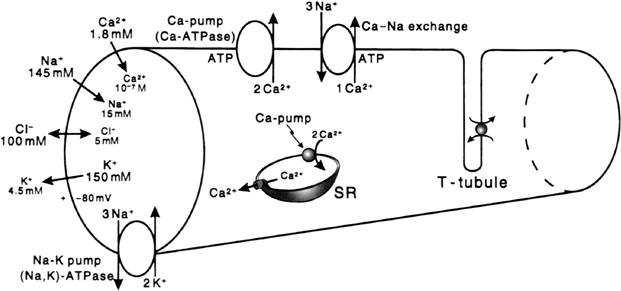
FIGURE 42.1 Intracellular and extracellular ion distributions in vertebrate skeletal muscle fibers. Also shown are the polarity and magnitude of the resting potential (RP). Arrows indicate direction of the net electrochemical gradient. The Na+-K+ pump and Ca2+-Na+ exchange carrier are located in the cell surface and in the T-tubule membranes. A calmodulin-dependent Ca2+-ATPase and Ca2+ pump, similar to that in the sarcoplasmic reticulum (SR), is located in the cell surface and T-tubule membranes.
The skeletal muscle AP is considerably different from the AP of cardiac muscle cells which have a very long duration with a pronounced plateau and a substantially lower rate of rise and propagation velocity. The myocardial cells are short and there is a slight delay in propagation at each cell-to-cell junction. Like skeletal muscle fibers, myocardial cells have a fast INa responsible for the rapid upstroke of the AP, but the delayed rectifier K+ conductance is turned on slowly and there is a substantial inward ICa during the entire plateau.
The APs of smooth muscle cells (SMCs) also are markedly different from those of skeletal muscle fibers, in that the RP (takeoff potential) is lower (more depolarized), the rate of rise of the AP is much slower, the AP overshoot is much less and the AP duration (APD) is considerably longer. Propagation velocity is much slower and the SMCs are short and small in diameter. The inward current for the APs in SMCs is primarily a slow Ca2+ current carried through L-type Ca2+ channels, but some cells do possess some functioning fast Na+ channels.
III General Overview of Electrogenesis of the Action Potential
The ion distributions and ion pumps and exchangers found in skeletal muscle fibers are similar to those of other types of cells, as described in the chapters on the RP and nerve APs (see Fig. 42.1). The APs in vertebrate skeletal muscle twitch fibers consist of a spike followed by a depolarizing (“negative”) afterpotential (Figs. 42.2 and 42.3). A large fast inward Na+ current, passing through voltage-dependent fast Na+ channels, is responsible for electrogenesis of the spike depolarization, which rises rapidly (500–700 V/s). Subsequently, a small slow inward Ca2+ current, passing through kinetically slow channels, may be involved in E-C coupling. Outward currents passing through K+ channels and Cl− channels are responsible for repolarization of the AP.
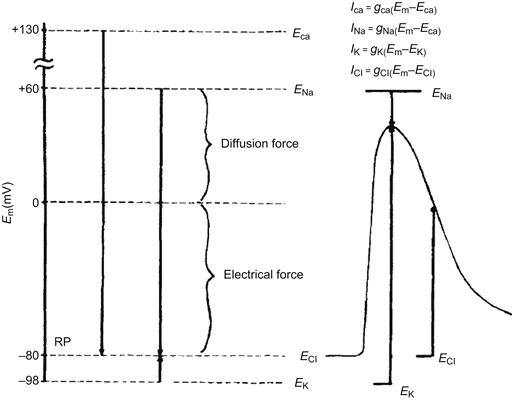
FIGURE 42.2 Representation of the electrochemical driving forces for Na+, Ca2+, K+ and Cl− at rest (left diagram) and during the AP in a skeletal muscle fiber (right diagram). Equilibrium potentials for each ion (e.g. ENa) are positioned vertically according to their magnitude and sign; they were calculated from the Nernst equation for a given set of extracellular and intracellular ion concentrations. Measured RP is assumed to be −80 mV. Electrochemical driving force for an ion is the difference between its equilibrium potential (Ei) and the membrane potential (Em), i.e. (Em−Ei). Thus, at rest, the driving force for Na+ is the difference between ENa and the resting Em; if ENa is +60 mV and resting Em is −80 mV, the driving force is 140 mV. The driving force is then the algebraic sum of the diffusion force and the electrical force and is represented by the length of the arrows in the diagram. Driving force for Ca2+ (about 210 mV) is even greater than that for Na+, whereas that for K+ is much less (about 18 mV). Direction of the arrows indicates the direction of the net electrochemical driving force, namely, the direction for K+ is outward, whereas that for Na+ and Ca2+ is inward. If Cl− is passively distributed, then for a cell sitting a long time at rest, ECl = Em and there is no net driving force. The driving forces change during the AP, as depicted. The equations for the different ionic currents are given in the upper right-hand portion of the figure. (Adapted from Sperelakis, 1979.)

FIGURE 42.3 (A) Action potential (AP) recorded with an intracellular microelectrode in a skeletal muscle fiber of frog semitendinosus muscle bathed in normal frog Ringer’s solution. Note the prominent depolarizing afterpotential. Shock artifact is at left of spike. (Modified fromSperelakis et al., 1973.) (B) Diagrammatic representation of the relative conductance changes for Na+ and K+ during an AP. The rising phase of the AP is caused by an increase in gNa, which brings the Em toward ENa. The falling phase of the AP is due to the rise in gK, the decrease in gNa, and to an outward Cl− current. The depolarizing afterpotential is explained in part by the fact that the delayed rectifier K+ channel is less selective for K+ (30:1 over Na+) than is the resting channel (100:1) and, in part, by the contribution of the AP traveling down the T-tubular network.
The skeletal muscle cell membrane has at least two types of voltage-dependent K+ channels (Fig. 42.4). One type allows K+ ions to pass more readily inward than outward, the so-called inward-going rectifier. This channel is responsible for anomalous rectification (i.e. decrease in gK with depolarization). There is a quick decrease in K+ conductance on depolarization and increase in K+ conductance with repolarization. The second type of K+ channel is similar to the usual K+ channel found in nerve membrane (e.g. squid giant axon), the so-called delayed rectifier. Its conductance turns on more slowly than gNa on depolarization. This channel allows K+ to pass readily outward down the electrochemical gradient for K+. The activation of this channel produces the large increase in total gK that helps to terminate the AP (see Fig. 42.3).
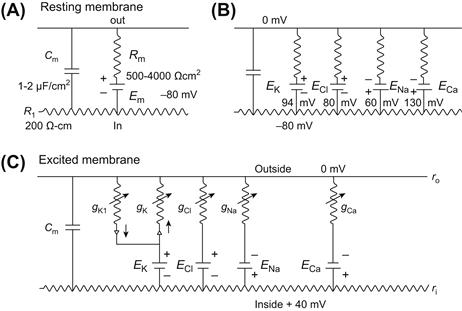
FIGURE 42.4 Electrical equivalent circuits for a skeletal muscle fiber cell membrane at rest (A and B) and during excitation (C). (A) Membrane as a parallel resistance-capacitance circuit, the membrane resistance (Rm) being in parallel with the membrane capacitance (Cm). RP (Em) is represented by an 80 mV battery in series with the membrane resistance, the negative pole facing inward. (B) Membrane resistance is divided into four component parts, one for each of the four major ions of importance: K+, Cl−, Na+ and Ca2+. Resistances for these ions (RK, RCl, RNa and RCa) are parallel to one another and represent totally separate and independent pathways for permeation of each ion through the resting membrane. These ion resistances are depicted as their reciprocals, namely, ion conductances (gK , gCl , gNa and gCa). Equilibrium potential for each ion (e.g. EK), determined solely by the ion distribution in the steady-state and calculated from the Nernst equation, is shown in series with the conductance path for that ion. RP of −80 mV is determined by the equilibrium potentials and by the relative conductances. (C) Equivalent circuit is further expanded to illustrate that, for the voltage-dependent conductances, there are at least two separate K+-conductance pathways (labeled here gK1 and gK). In series with the K+ conductances are rectifiers pointing in the direction of least resistance to current flow. There is one Na+ conductance pathway, the kinetically fast Na+ conductance (gNa). In addition, there is a kinetically slow pathway that allows Ca2+ to pass through. Arrows drawn through the resistors indicate that the conductances are variable, depending on membrane potential and time. (Adapted from Sperelakis, 1979.)
The AP amplitude is about 120 mV, from an RP of −80 mV in mammalian myofibers (−90 mV in amphibian) to a peak overshoot potential of about +40 mV (see Figs. 42.2 and 42.3). The duration of the AP (at 50% repolarization, or APD50) ranges between 3 and 6 ms, depending on the species and temperature. The threshold potential (Vth) for triggering of the fast Na+ channel conductance is about −65 to −55 mV; thus, a critical depolarization of about 25 mV is required to reach Vth. The turn-on of the fast gNa (fast INa) is very rapid (within 0.2 ms) and Em is brought rapidly toward ENa (see Figs. 42.2 and 42.3). There is an explosive (positive exponential initially) increase in gNa, caused by a positive feedback relationship between gNa and Em.
From the current versus voltage (I/V) curves, the maximum inward fast Na+ current occurs at an Em of about −20 mV. The current decreases at more depolarized Em levels because of the diminution in electrochemical driving force as the membrane is further depolarized, even though the conductance remains high. At the reversal potential (Erev) for the current, the current goes to zero; INa then reverses direction with greater depolarization.
As Em depolarizes, it crosses Vth for slow Ca2+ channels (also called L-type Ca2+ channels or dihydropyridine receptors), which is about −35 mV. These Ca2+ channels are primarily located in the transverse tubules. Turn-on of the Ca2+ conductance (gCa) and ICa is relatively slow and the peak ICa is considerably smaller than the peak INa. This Ca2+ influx is involved in E-C coupling.
The molecular rearrangements involved in activation of the slow Ca2+ channels are directly coupled to opening of Ca2+-release channels (or ryanodine receptors) in the sarcoplasmic reticulum (SR) membrane. However, the open probability of these channels is very low. Therefore, the resulting increase in Ca2+ conductance (gCa) and the peak ICa is relatively slow and considerably smaller than the peak INa. This Ca2+ influx is small during a single AP, but can contribute to E-C coupling during repetitive AP firing.
IV Ion Channel Activation and Inactivation
As discussed in the chapter on nerve excitability (see Chapter 19), the fast Na+ channels (and the slow Ca2+ channels) have a double gating mechanism; one gate is the activation gate (A-gate) and the second gate is the inactivation gate (I-gate). For a channel to be conducting, both the A-gate and I-gate must be open; if either one is closed, the channel is non-conducting. The A-gate is closed at the resting Em and opens rapidly on depolarization, whereas the I-gate is open at the resting Em and closes slowly on depolarization. In the Hodgkin–Huxley (1952) analysis, the opening of the A-gate requires simultaneous occupation of three negatively-charged sites by three positively-charged m+ particles. Therefore:
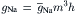 (42.1)
(42.1)
where m is the activation variable, h is the inactivation variable and 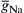 is the maximum conductance. A small gating current (Ig) has been measured that corresponds to the movement of the charged m particles (or rotation of an equivalent dipole). The outward Ig leads into the inward INa.
is the maximum conductance. A small gating current (Ig) has been measured that corresponds to the movement of the charged m particles (or rotation of an equivalent dipole). The outward Ig leads into the inward INa.
The fast INa lasts only for 1–2 ms because of the spontaneous voltage inactivation of the fast Na+ channels, i.e. they inactivate quickly, even when the membrane remains depolarized. Inactivation is produced by the voltage-dependent closing of the I-gate. The voltage dependence of inactivation is given by the h∞ versus Em curve. The Na+ conductance (gNa) at any time is equal to the maximal value () times m3 h. Therefore, when h = 0, gNa = 0, and when h = 1.0, gNa = (if m = 1.0). At the normal RP, h∞ is nearly 1.0 and diminishes with depolarization, becoming nearly zero at about −30 mV. The maximal rate of rise of the AP (max dV/dt) is directly proportional to the net inward current or INa, which is directly proportional to gNa and can be expressed as:
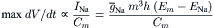 (42.2)
(42.2)
Therefore, a decrease in h∞ causes decrease in max dV/dt. Thus, depolarization by any means (e.g. elevated [K+]o or applied current pulses) decreases max dV/dt, and excitability disappears at about −50 mV.
The slow Ca2+ channels behave much the same way as the fast Na+ channels with respect to activation and inactivation, with one main difference being the voltage range over which the slow channels activate and inactivate. Slow channels inactivate between −45 mV and −10 mV, compared to −70 and −30 mV for the fast Na+ channels. Another major difference is that the slow Ca2+ conductance inactivates much more slowly than the fast Na+ conductance; i.e. they have a long inactivation time constant (τinact). (The h variable for the slow channel is sometimes referred to as the f variable and the m variable as the d variable.) Because slow Ca2+ channels are located in the T-tubular system, their function is affected by tubular Ca2+ depletion caused by the Ca2+ ions that flow into the myoplasm. The recovery process for the slow Ca2+ channels is slow compared to 1–2 ms for fast Na+ channels.
The K+ channel (delayed rectifier) may have only an activation gate, because it does not inactivate quickly. In the Hodgkin–Huxley analysis of squid giant axon, the A-gate opens when four positively-charged n+ particles simultaneously occupy four favorable positions (negatively-charged sites). If n is the probability that one site is occupied, then n4 is the probability that all four sites are occupied. Therefore,
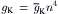 (42.3)
(42.3)
The fourth power to which n must be raised causes a delay (sigmoidal foot) in turn-on of the K+ conductance.
V Slow Delayed Rectifier K+ Current
Two types of K+ delayed rectifier currents occur in skeletal muscle. A slow IK was first described by Adrian and co-workers (1970a,b) in voltage-clamped frog sartorius fibers. The slow component of outward IK reached a maximum in about 3 s and declined with a time constant of about 0.5 s. In voltage-clamped frog toe muscle, Lynch (1978) observed that most fibers had both a slow component and a fast component of the outward IK (threshold of −55 mV). The voltage dependences of both K+ currents were shifted equally in the depolarizing direction by elevated [Ca2+]o or [H+]o, presumably due to altering the net negative outer surface charge of the membrane, thereby hyperpolarizing. Acidosis also increased the rate of turn-on of the slow delayed rectifier. The fast delayed current was relatively selectively blocked by TEA or by a sulfhydryl reagent, whereas the slow delayed current was selectively depressed by a histidine reagent. It was estimated that about 25% of the delayed rectifier channels are in the T-tubular membrane. The functional significance of the slow IK is unknown, although it may be partly responsible for the late depolarizing afterpotential (see Section VIII).
VI Mechanisms of Repolarization
The skeletal muscle AP is terminated by three processes: turn-on of gK, turn-off of gNa and influx of Cl− ions. The turn-on of the V-dependent K+ conductance (gK) (the delayed rectifier) (see Fig. 42.3) acts to bring Em towards EK (about −98 mV), since the membrane potential at any time is determined primarily by the ratio of gNa/gK. This type of gK channel is activated by depolarization and turned off by repolarization. Therefore, this gK channel is self-limiting, in that it turns itself off as the membrane is repolarized by its action.
In addition to the gK turn-on, turn-off of gNa occurs (see Fig. 42.3) (contributing to repolarization) for two reasons: (1) spontaneous inactivation of fast Na+ channels that had been activated, i.e. closing of their I-gate (inactivation τ of 1–3 ms) and (2) reversible shifting of activated channels directly back to the resting state (deactivation), because of the rapid repolarization that is occurring due to the gK increase (Fig. 42.5). Theoretically, it would be possible to have an AP that would repolarize (but more slowly) even if there were no gK mechanism, because the gNa channels would spontaneously inactivate and so the gNa /gK ratio and Em would be more slowly restored to their original resting values.
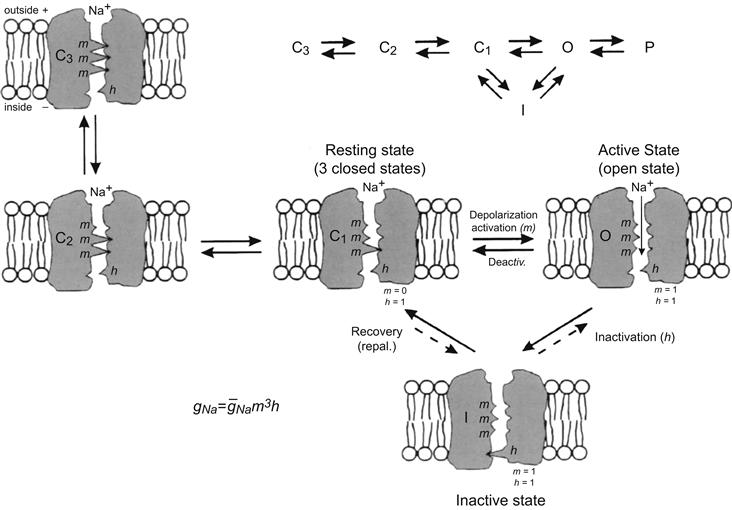
FIGURE 42.5 Illustration of the hypothetical states of the fast Na+ channel. The three states patterned after the Hodgkin–Huxley view were modified to reflect the fact that there is evidence for three closed states. As depicted, in the most closed state (C3), all three m gates (or particles) are in the closed configuration. In the mid-closed state (C2), two m gates are closed and one is open. In the least closed state (C1), one gate is closed and two are open. In the resting state, the activation gate (A) is closed and the inactivation gate (I) is open: m = 0, h = 1. Depolarization to the threshold activates the channel to the active state, the A-gate opening rapidly and the I-gate still being open: m = 1, h = 1. The activated channel spontaneously inactivates to the inactive state due to closure of the I-gate: m = I, h = 0. The recovery process on repolarization returns the channel from the inactive state back to the resting state, thus making the channel again available for reactivation. Na+ ion is depicted as being bound to the outer mouth of the channel and poised for entry down its electrochemical gradient when both gates are open. The reaction between the resting state and the active state is readily reversible and there is some reversibility of the other reactions. The fast Na+ channel is blocked by tetrotodoxin (TTX) binding to the outer mouth and plugging it.
In addition, there is an important third factor involved in repolarization of the AP in skeletal muscle: the Cl− current (see Fig. 42.2). The Cl− permeability (PCl) and conductance (gCl) are very high in skeletal muscle (and are not strongly V-dependent). In fact, PCl of the surface membrane is much higher than PK , the PCl/PK ratio being about 3–7. As discussed in the chapter on RP (see Chapter 9), the Cl− ion is passively distributed, or nearly so, and thus cannot determine the RP. However, net Cl− movements inwards (hyperpolarizing) or outward (depolarizing) do affect Em transiently until re-equilibration occurs and there is no further net movement. At the RP, there is no net Cl− current (ICl), since there is no electrochemical driving force for Cl− (since Em = ECl). However, during the AP depolarization, there is a larger and larger driving force for outward ICl (i.e. Cl− influx), since ICl = gCl (Em−ECl). In other words, the large electric field that was keeping Cl− out (i.e. [Cl−]i << [Cl−]o) diminishes during the AP and so Cl− ion enters the fiber. This Cl− entry is hyperpolarizing and so tends to repolarize the membrane more quickly than would otherwise occur. That is, AP repolarization is sharpened by the Cl− mechanism. (Note that influx of the negatively-charged Cl− ion is an outward Cl− current, which is repolarizing.)
To illustrate further some of the preceding points on the role of Cl−, when skeletal muscle fibers are placed into Cl−-free Ringer solution (e.g. methanesulfonate substitution), depolarization and spontaneous APs and twitches occur for a few minutes until most or all of the [Cl−]i is washed out. After equilibration, the resting Em returns to the original value ca. −90 mV for frog skeletal muscle and −80 mV for mammalian, clearly indicating that Cl− does not determine the RP and that net Cl− efflux produces depolarization. Re-addition of Cl− to the bath produces a rapid large hyperpolarization, e.g. to −120 mV, due to net Cl− influx; the Em then slowly returns to the original value (e.g. −90 mV) as Cl− re-equilibrates, i.e. redistributes itself passively. These same effects occur in cardiac muscle, smooth muscle and nerve, but to a lesser extent, because in these tissues PCl is much lower (e.g. PCl/PK ratio is only about 0.5 in vascular smooth muscle).
The importance of the Cl− current in repolarization in skeletal muscle fibers is illustrated by one type of myotonia in which an abnormally low PCl causes repetitive APs to occur. Because gCl is abnormally low, total membrane conductance Gm is also low. From the relationship between membrane current (Im) and Gm (Im /Gm = Em), it can be deduced that only a smaller outward depolarizing membrane current Im is necessary to reach threshold Eth for an AP. Since gCl is abnormally low, the membrane resistance Rm will be abnormally high, making the space constant λ larger than normal. Because gCl is abnormally low, the Cl− influx during AP repolarization is much less than normal and so the repolarization process is slowed, thus increasing the duration of the AP. As a consequence of the above, when depolarization occurs, the AP threshold is easily and quickly reached and the generated APs spread easily along the sarcolemma. These factors make the whole system unstable and oscillations trigger repetitive discharge of APs. That is, the muscle fibers lose their tight control by the motor neurons and so contraction becomes partly involuntary. For example, persons with myotonia find it difficult to release a handshake or to remove their hand from a drinking glass. There are several causes of myotonia, including genetic abnormalities in ion channels, as well as drug-induced conditions. Any agent that greatly lowers PCl or gCl will have the same effect. It has been shown that simply decreasing gCl causes repetitive firing in equivalent circuit models of skeletal muscle fibers. In addition, K+ ions tend to accumulate in the lumen of the T-tubules under normal conditions (see Section VIII). This accumulation is exaggerated with the prolonged APs and so tends partially to depolarize the fibers and increase their excitability. Some forms of myotonia are produced by abnormal fast Na+ channels; namely, a small fraction of these channels do not inactivate as quickly as usual (i.e. their I-gates do not close normally) and so causes a prolonged small depolarization after the AP and, consequently, a repetitive discharge.
In myotonia, AP repolarization is slowed and the duration of the AP is increased. As AP duration increases, more Na+ channels have time to return to the resting conformation by deactivation or recovery from inactivation (a process which has a time constant of 2–3 ms). This creates a window of instability during AP repolarization. The membrane potential remains depolarized above threshold, allowing some Na+ channels to reopen and trigger another AP. The skeletal muscles have a large safety factor with respect to the number of Na+ channels in the membrane and as few as 3–5% in the open state can trigger an AP. This instability in membrane repolarization is further enhanced by the high membrane resistance. Without the normal large gCl, only a smaller than normal outward depolarizing current is required to reach threshold. Because of the high membrane resistance Rm, the space constant λ is longer than normal. Consequently, the APs propagate at a faster velocity. These factors act synergistically to make the whole system unstable. Consequently, when depolarization occurs, the AP threshold is easily and quickly reached, the generated APs spread fast along the sarcolemma and the membrane is more excitable and susceptible to repetitive discharge of APs.
The high gCl in skeletal muscle fibers is due to a large number of voltage-dependent gated Cl− channels, which are outwardly rectifying. The major Cl− channel of skeletal muscle is the ClC-1 channel (Steinmeyer et al., 1991), a member of the ClC family of Cl− channels and Cl−/H+ antiporters (Zifarellli and Pusch, 2007). These Cl− channels are located both on the surface sarcolemma and T-rubule membrane in frogs and mammals. Denervation of mammalian fibers causes gCl to decrease almost to zero.
VII ATP-Dependent K+ Channels
KATP channels are among the most abundantly expressed K+ channels in the skeletal muscle sarcolemma, reaching densities of 10 channels per μm2 of surface membrane (Spruce et al., 1987), comparable to that of K+ delayed rectifier channels (Standen et al., 1985). KATP channel currents can be recorded from both surface (Spruce et al., 1985) and transverse tubular membranes (Heiny et al., 1983). However, their physiological role in skeletal muscle is not well understood (reviewed in Flagg et al., 2010). The functional KATP channel is an octomeric protein composed of four pore-forming Kir subunits (Kir 6.1 and Kir 6.2) and four regulatory SUR (sulfonylurea receptor) subunits assembled in a 4:4 stoichiometry. Functioning of the KATP channel requires the proper coupling between Kir and SUR subunits. More details on the subunits is given in the Appendix to this chapter.
ATP-dependent K+ (KATP) channels are characterized by the inhibition of channel openings by ATP (Yokoshiki et al., 1998). In addition to being ligand sensitive, these KATP channels are voltage-dependent; the open state probability increases with depolarization. Most studies of KATP channels have been performed using patch-clamp of isolated inside-out patches of skeletal muscle (Spruce et al., 1987). The unitary conductance of this channel varies with [K+]o, ranging from 15 pS in 2.5 mM [K+]o to 42 pS in 60 mM. KATP channels are closed when the ATP concentration in the intracellular myoplasm is in the range of 1.0 mM or higher, which is the normal physiological concentration. A decrease in ATP concentration or depolarization activates the channel. The half-maximum inhibition of this channel opening by ATP, measured at a constant [K+]o/[K+]i, is 0.135 mM at pH 7.2 (Fig. 42.6). Hydrolysis of ATP (into ADP + Pi) is not required for ATP to close KATP channels.
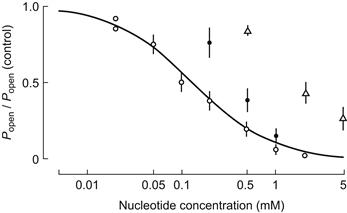
FIGURE 42.6 Effect of ATP (open circles), ADP (filled circles) and AMP (open triangles) on closing the ATP-regulated K+ channels. Ordinate: open-state probability (Po) of the channel relative to its value in nucleotide-free solution (Popen control). (Reproduced with permission from Spruce et al., 1987.)
Although ATP is the most specific ligand to close KATP channels, there are other ligands that modulate these channels. The metabolites produced during contraction, like ADP, Mg2+ and H+ also modulate the channels. The two adenine nucleotides, ADP and AMP (in the absence of ATP), also can block KATP channels in a dose-dependent manner. However, they are less effective than ATP (see Fig. 42.6). ATP analogs like GTP, ITP, XTP, CTP and UTP also have reduced effectiveness (about tenfold) in closing these channels. ADP shifts the ATP dose–response curve, raising the half-inhibition concentration, consistent with competition between ATP and ADP for the nucleotide binding site on the channel (Spruce et al., 1987; Vivaudou et al., 1991).
A decrease in pH at the cytoplasmic surface (pHi) reduces the degree of KATP channel inhibition caused by ATP. The ATP concentration for half-inhibition is 2.5 times and 15 times greater at pHi 6.8 and 6.3, respectively, as compared to that at pHi 7.2. Thus, during acidosis, a smaller decrease in ATP concentration will lead to a larger opening of KATP channels. It was proposed that proton binding to the channel prevents ATP binding (Davies et al., 1992). Mg2+ has a similar effect as protons. An increase of cytosolic Mg2+ reduces the ability of ATP to close KATP channels (Vivaudou et al., 1991). Mg2+ may bind to the channels and plug them (Woll et al., 1989). In addition, the inhibitory effect may be partly due to the ability of Mg2+ to bind to ATP.
KATP channels have been studied in excised patches of surface membrane blebs from muscles of frogs and mammals. These channels are, therefore, likely to be present in the sarcolemma. It is not known whether they are present in the T-tubular system. As stated earlier, the KATP channel density has been estimated to be as high as 10 channels per μm2 of surface membrane (Spruce et al., 1985), a density comparable to that for K+ delayed rectifier channels.
The physiological role played by KATP channels in skeletal muscle is not clear. Under physiological conditions, the intracellular ATP concentration is about 5 mM at rest. Therefore, at the resting ATP level, almost all of the KATP channels should be inactive (Spruce et al., 1987; Davies et al., 1992). Even during repetitive contractions that lead to muscle fatigue, the ATP concentration is maintained at near normal levels by the action of creatine kinase and creatine phosphate (Carlson and Siger, 1960; Nassar-Gentina et al., 1978).
It has been proposed that KATP channels may be associated with the decrease in force development underlying muscle fatigue. A drug (SR44866) that opens KATP channels (in frog skeletal muscles) also reduces the AP duration, the early afterpotential and the peak twitch force (without affecting the RP) in intact muscles (Sauviat et al., 1991). The high K+ permeability found in muscle fibers that have undergone a permanent contracture (rigor) produced by repetitive stimulation when poisoned with cyanide and iodocetate (Fink and Lüttgau, 1976), may be caused by the decreased ATP concentration opening KATP channels. In metabolically-exhausted frog semitendinosus muscle fibers, the addition of tolbutamide and glyburide, two KATP channel antagonists, significantly reduces the K+ efflux rate (Castle and Haylett, 1987). Fatigued muscle fibers can contract when Ca2+ is released directly from the intracellular SR stores (Gonzalez-Serratos et al., 1978; Garcia et al., 1991). In intact animals, some of the skeletal muscle fatigue results from synaptic fatigue, including at the neuromuscular junction.
As stated previously, a decrease in pHi (e.g. from lactic acid production) reduces the inhibitory effect of ATP on KATP channels. During exercise, with fatigue development, pHi may decrease by about one unit (Renaud, 1989). A small decrease in ATP concentration is accompanied by an increase in ADP, H+ and Mg2+ concentrations and the overall combination of these chemical changes may then lead to the activation of KATP channels. As KATP channels open, they contribute to the increased K+ efflux found during repetitive muscle contraction. Because of the restricted diffusion out of the T-tubular system and the closeness of the intercellular fiber spacing, extracellular K+ concentration increases, especially inside the tubular system. This may cause a decrease in cell excitability, which may be reflected as decreased force development. This mechanism may protect skeletal muscle cells from large ATP depletions that would have deleterious effects.
VIII Electrogenesis of Depolarizing Afterpotentials
As mentioned previously, the AP spike in skeletal muscle fibers is followed by a prominent depolarizing afterpotential (also called a negative afterpotential based on the old terminology from external recording) (see Fig. 42.3). In addition to this early depolarizing afterpotential (i.e. emerging from the spike downstroke), there is a late depolarizing afterpotential that follows a tetanic train of spikes (e.g. 10 spikes). The electrogenesis of the early and late afterpotentials is different. The early afterpotential is due to a membrane conductance change, whereas the late afterpotential is due primarily to K+ accumulation in the T-tubules.
The early depolarizing afterpotential of frog skeletal fibers is about 25 mV in amplitude immediately after the spike component and gradually decays to the RP in 10–20 ms. This afterpotential results from the fact that the delayed rectifier K+ channel that opens during depolarization to terminate the spike is less selective for K+ (ca. 30:1, K+:Na+) than is the K+ channel in the resting membrane (ca. 100:1) (Adrian et al.,1970a). Therefore, the constant-field equation predicts that the membrane should be partly depolarized when EM is dominated by this K+ conductance that is turned on during the AP. Thus, the early depolarizing afterpotential is partly due to the persistence and slow decay of this less-selective K+ conductance. Adrian and Peachey (1973) were able to reconstruct the time course of the AP and the early depolarizating afterpotential by giving values to the access resistance of the T-tubular system, presence of Na+ and K+ tubular membrane currents and velocity of the tubular AP. The early depolarizing afterpotential reflects, in part, the tubular AP, as evidenced by the disappearance of the early depolarizing afterpotential in muscles in which the T-tubular system has been disrupted and disconnected from the surface membrane by the glycerol osmotic shock method1 (Eisenberg and Gage, 1969).
The late depolarizing afterpotential of frog skeletal fibers may result from accumulation of K+ ions in the T-tubules (Adrian and Freygang, 1962). During the AP depolarization and turn-on of gK (delayed rectifier), there is a large driving force for K+ efflux from the myoplasm coupled with a large K+ conductance, resulting in a large outward K+ current [IK = gK (Em−EK)] across all surfaces of the fiber, namely across the surface sarcolemma and T-tubule walls. The K+ efflux at the fiber surface membrane can rapidly diffuse away and mix with the relatively large interstitial fluid (ISF) volume, whereas the K+ efflux into the T-tubules (TT) is trapped in this restricted diffusion space. The resulting high [K+]TT decreases EK across the T-tubule membrane and thereby depolarizes this membrane. Because of cable properties, part of this depolarization is transmitted to the surface sarcolemma and is recorded by an intracellular microelectrode. The K+ accumulation in the T-tubules can only be dissipated relatively slowly by diffusion out of the mouth of the T-tubules and by active pumping back into the myoplasm (across the T-tubule wall) by the Na+-K+ pump sites located in the T-tubular membrane. Thus, the decay of the late depolarizing afterpotential will be a function of these two processes.
The amplitude and duration of the late depolarizing afterpotential is a function of the number of spikes in the train and their frequency. That is, the greater the spike activity, the greater its amplitude and duration. If the train consists of 20 spikes at a frequency of 50/s, a typical value for the amplitude of the late depolarizing afterpotential in frog fibers is about 20 mV. When the diameter of the T-tubules is increased by placing the fibers in hypertonic solutions2, the amplitude of the late afterpotential decreases as expected because of the greater dilution of the K+ ions accumulating in the T-tubule lumen. When the T-tubular system is disrupted and disconnected from the surface membrane by the glycerol osmotic shock method, the late afterpotentials disappear together with the early afterpotentials.
An alternative explanation for the late depolarizing afterpotential is that it may be due to the slow delayed rectifier gK change described above (Adrian et al., 1970b). The equilibrium potential for the slow IK is −83 mV and the sign (direction) of the late afterpotential reverses when the fiber is depolarized beyond −80 mV (e.g. to −70 mV). Hence, the late afterpotential could arise from the slow relaxation of a component of the K+ conductance increase, which is less selective for K+ than the K+ channels open in resting membrane. In this view, the electrogenesis of the late afterpotential would be similar to that for the early afterpotential.
All depolarizing afterpotentials, regardless of whether early or late, have physiological importance because they alter excitability and the propagation velocity of the fiber. A depolarizing afterpotential should enhance excitability (lower threshold) to a subsequent AP. This is because the critical depolarization required to reach the threshold potential would be decreased. A large late depolarizing afterpotential, such as that due to K+ accumulation in the T-tubules, can, under certain pathological conditions, trigger repetitive APs. The effect of depolarizing afterpotentials on velocity of propagation involves two opposing factors: (1) the decrease in critical depolarization required; and (2) the decrease in maximal rate of rise of the AP (max dV/dt), which is a function of the takeoff potential (h∞ versus Em curve). Therefore, what factor dominates will depend on the degree of depolarization and the shape of the h∞ curve. When frog skeletal fibers are depolarized slightly by elevating [K+]o, only a decrease in propagation velocity is observed (Sperelakis et al., 1970).
IX Ca2+-Dependent Slow Action Potentials
Slow APs are recorded under conditions in which the fast Na+ current is blocked by Na+-deficient solution, tetrodotoxin (TTX) or voltage inactivation of the fast Na+ channels in elevated [K+]o. Under these conditions, the only carrier of inward current available to produce an AP is Ca2+ ion. Spontaneously-occurring slow APs were first observed in frog sartorius fibers equilibrated in Cl−-free solution containing TTX (Sperelakis et al., 1967). Upon addition of Ba2+ ion (e.g. 0.5 mM), which is a potent blocker of K+ channels and PK, the fibers partially depolarize and spontaneously discharge slowly-rising (e.g. 1–10 V/s), overshooting APs of long duration (e.g. several seconds), having a prominent plateau component (resembling a cardiac AP in shape). An abrupt repolarization terminates the slow AP. Ba2+ depolarizes rapidly in Cl−-free solution, because the voltage-clamping effect of the Cl− distribution (ECl), due to the large PCl , is circumvented. Cl−-free solution raises the resistance of the cell membrane about sevenfold.
In a frog skeletal muscle fiber, using two intracellular microelectrodes, one for applying current intracellularly and the other for recording voltage a short distance away in the same fiber ([K+]o of 25 mM to depolarize the fiber to about −45 mV and thereby inactivate the fast Na+ channels and [Na+]o reduced to zero so that there could be no inward Na+ current), application of small hyperpolarizing current pulses during the slow AP indicated that membrane resistance increases progressively during the plateau (Kerr and Sperelakis, 1982). The rate of rise, overshoot and duration of the slow APs are a function of [Ca2+]o (Beaty and Stefani, 1976; Vogel et al., 1978; Kerr and Sperelakis, 1982). For example, the AP duration at 50% amplitude (APD50) was generally 2–8 s. The amplitude of the slow AP plotted against log [Ca2+]o gave a straight line with a slope of 28 mV/decade, which is close to the theoretical 29 mV/decade (at 21°C) from the Nernst relationship for a situation in which only Ca2+ ion carried the inward current. The slow APs were depressed and blocked by the Ca2+-antagonistic and slow-channel-blocking drugs, verapamil and bepridil, with an ED50 of about 5×10−8 M. The slow AP arises from the T-tubular system of the fiber (Vogel et al., 1978; Kerr and Sperelakis, 1982), based on their disappearance when the T-tubules were disrupted and disconnected from the surface membrane by the glycerol osmotic shock method. The normal fast APs are not affected by the glycerol treatment. These results indicate that the slow Ca2+ channels giving rise to the slow APs are located primarily in the tubular system.
Isotope flux measurements have shown that there is a net Ca2+ influx during contractions of phasic skeletal muscle fibers, suggesting that an influx from the extracellular space may initiate the contraction (Bianchi and Shanes 1959). Additionally, voltage-clamped muscle fibers have slow inward Ca2+ currents (ICa) (Stanfield, 1977; Sanchez and Stefani, 1978). Elevation of [Ca2+]o increased ICa, and ICa was depressed by the slow Ca2+ channel blockers D-600, nifedipine and Ni2+ (Stanfield, 1977; Sanchez and Stefani, 1978; Almers et al., 1981). Detubulation by the glycerol osmotic shock method abolishes ICa (Nicola-Siri et al., 1980; Potreau and Raymond, 1980). These results support the conclusion that ICa produces the slow APs. The various conformational states that the Ca2+ slow channels undergo during excitation are depicted in Fig. 42.7. These states are similar to those of the fast Na+ channels (see Fig. 42.5), except there are only two closed states.
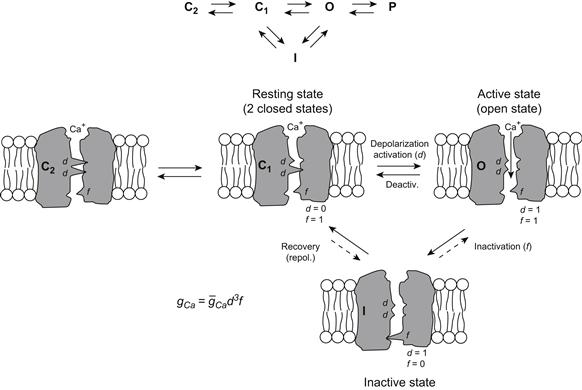
FIGURE 42.7 Illustration of the four hypothetical states of the slow Ca2+ channel. There is evidence for two closed states. As depicted, in the most closed state (C2), both d gates (or particles) are in the closed configuration. In the least closed state (C1), one gate is closed and one is open. In the resting membrane, the activation gate (A) is closed and the inactivation gate (I) is open: d = 0, f = 1. Depolarization to the threshold activates the channel to the active state, the A-gate opening rapidly and the I-gate still being open: d = 1, f = l. The activated channel spontaneously inactivates to the inactive state due to closure of the I-gate: d = 1, f = 0. The recovery process on repolarization returns the channel from the inactive state back to the resting state, thus making the channel again available for reactivation. Ca2+ ion is depicted as being bound to the outer mouth of the channel and poised for entry down its electrochemical gradient when both gates are open. The reaction between the resting state and the active state is readily reversible and there is some reversibility in the other reactions. The slow channels behave similarly to the fast channels, except that their gates appear to move more slowly on a population basis; i.e. the slow channels activate and recover more slowly. (Although the gates of any individual slow channel may move quickly, the stochastic behavior of the population of channels is such that their summed conductance changes slowly.) The slow channel gates operate over a different voltage range than the fast channels (i.e. less negative, more depolarized). TTX does not block the slow channels, but drugs such as nifedipine do block by binding to the channel.
Do slow inward calcium currents (ICaS), play a role in E-C coupling? A substantial contraction, of between 20 and 50% of the normal twitch tension, accompanies the slow APs (Vogel et al., 1978), suggesting that the Ca2+ channels in the tubular system may play a role during E-C coupling. However, skeletal muscle fibers contract for several minutes after [Ca2+]o is lowered to 10−8 M (Armstrong et al., 1972) and the Ca2+-channel blocker diltiazem did not depress twitch or tetanic force development (Gonzalez-Serratos et al., 1982). These results suggest that ICa may play no role in E-C coupling in normal amphibian muscles. Nevertheless, in dysgenic mice, in which contraction of skeletal muscles is weak, the Ca2+ channels in the T-tubules are few or absent.
Ca2+ influx during the slow AP could trigger the release of more Ca2+ from the nearby TC-SR via the Ca2+-trigger Ca2+-release mechanism (Fabiato, 1982). Because the time course of the slow AP is much longer than that of a twitch contraction, it was suggested that the inward Ca2+ current may play a role in K+ contracture, in tetanic contraction, or in long-term regulation of contraction, perhaps by increasing the Ca2+ concentration in the SR and thereby increasing the amount of internal Ca2+ available for release on subsequent activation (Nicola-Siri et al., 1980). [Ca2+]SR does increase following tetanic stimulation (Gonzalez-Serratos et al., 1982).
Slow APs were also recorded from mouse skeletal muscle fibers equilibrated in a solution that was Cl−-free, low Na+ (10 mM) and high K+ (20 mM) (Kerr and Sperelakis, 1982). As with frog muscle, the slow APs were abolished after detubulation and blocked by verapamil, bepridil, Mn2+ and La3+. Their rate of rise, amplitude and duration increased as a function of [Ca2+]o, with max dV/dt being about 0.5 V/s in 8 mM.
During the first 5 days in culture, embryonic skeletal muscle cells from Xenopus laevis need extracellular Ca2+ to contract when stimulated (in contrast to adult muscles). Thus, an inflow of Ca2+ from the extracellular space may be required in embryonic cells as the means to produce contraction. In whole-cell voltage-clamp studies, ICa currents have been observed in embryonic and neonatal skeletal muscle cells in culture (Moody-Corbett et al., 1989; Cognard et al., 1992; Gonzalez-Serratos et al., 1996; Cordoba-Rodriguez et al., 1997). The current density increased from 1.7 to 3.3 and to 7.9 pA/pF at 1, 5 and 15 days in culture, respectively. These results indicate that the T-tubules and SR are poorly developed or not functional in early stages of muscle development and that, in early development, ICa may be an important mechanism to trigger contraction.
X Developmental Changes in Membrane Properties
The cell membranes of most excitable cells apparently pass through similar stages of differentiation during development. For example, young (2- to 3-day-old) embryonic chick hearts (tubular) have few or no functional fast Na+ channels, but have a high density of slow channels (both Na+ and Ca2+) and fire slowly-rising TTX-insensitive APs. Fast Na+ channels then appear and progressively increase in number, reaching the maximal (adult) level at late embryonic development (e.g. day 20). The PNa/PK ratio is high in young hearts, due to a low PK , and accounts for the low RP and automaticity in nearly all the cells.
Skeletal muscle fibers and neurons also undergo developmental changes in membrane electrical properties (e.g. Spector and Prives, 1977; Spitzer, 1979) (see Chapter 25). In general, fast Na+ channels are absent in the young, less differentiated cells, but they do possess excitability because of a large number of slow channels. The AP is TTX-insensitive, slowly rising and of long duration (resembling a slow AP in cardiac muscle). Later during development, fast Na+ channels make their first appearance and the fast Na+ channels and slow channels coexist. During that period, TTX does not abolish the APs, but reduces max dV/dt (i.e. slow APs remain). At a later stage, the slow channels in the sarcolemma are lost (or greatly reduced in number) and the fast Na+ channels progressively increase in density. The APs become fast rising and of short duration and are completely abolished by TTX. As discussed previously, some functional slow Ca2+ channels remain in the T-tubular system.
XI Electrogenic Na+-K+ Pump Stimulation
The Na+, K+-ATPase pump is electrogenic in skeletal muscle fibers (both mammalian and amphibian). The pump produces a net outward current, because three Na+ ions are pumped out to every two K+ ions pumped in. The electrogenic pump potential contribution to the RP (see Chapter 11 to 9) is very large, about 12–16 mV, in rat skeletal muscle fibers (Sellin and Sperelakis, 1978). The net pump current can be stimulated by increasing the number of pump sites per unit area of cell membrane or by increasing the turnover rate of each pump site. β-Adrenergic agonists (e.g. isoproterenol) rapidly hyperpolarize skeletal muscle fibers by 7–9 mV within 5 min. Insulin has a similar effect, but hyperpolarizes more slowly (e.g. peak reached by 10 min) and to a smaller degree (e.g. 5–7 mV) (Iannaccone et al., 1989). Since cAMP also hyperpolarizes, the action of β-agonists is believed to be mediated by elevation of cAMP and phosphorylation of the Na+-K+ pump (or an associated regulatory protein) by protein kinase A (PKA). The action of insulin is thought to be mediated by the incorporation of spare membrane from an internal pool, which contains Na+-K+ pumps, into the cell membrane.
The pump current (Ip) can be directly measured in single fibers (cultured skeletal myotubes rounded by use of colchicine, a microtubule disrupter) by doing whole-cell voltage clamp under conditions in which all the ionic conductances are blocked. When this is done, the pump current can be measured at different voltages and normalized for unit membrane capacitance (hence membrane area). Values of about 1 pA/pF or 1.0 μA/cm2 were obtained, with a reversal potential (or zero current) of about −140 mV (Li and Sperelakis, 1994).
When [K+]o is lowered below the normal physiological level, e.g. from 4.5 mM to about 0.1 mM, a large depolarization occurs in mammalian skeletal muscle fibers (Fig. 42.8). This depolarization is caused, in part, by inhibition of the Na+-K+ pump current. The Km value for [K+]o for the Na+, K+-ATPase is about 2 mM and the relationship between Na+, K+-ATPase activity and [K+]o is very steep. Therefore, inhibition of the Na+-K+ pump occurs.
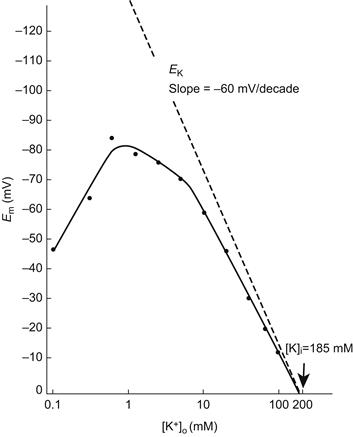
FIGURE 42.8 The mean resting membrane potential (Em) of normal mouse skeletal muscle plotted as a function of the extracellular K+ concentration ([K+]o) on a logarithmic scale. The straight line drawn through the data points for 20 mM [K+]o and above has a slope of 50 mV/decade. Extrapolation of this line to zero potential gives the intracellular K+ concentration ([K+]i) of 185 mM. The dashed line gives the calculated EK values (slope of 61 mV/decade). Note the “fold-over” of the Em curve at [K+]o, levels below 1 mM, presumably due to inhibition of the electrogenic pump potential (Vp) and to a decrease in PK and gK at low [K+]o levels. (Reproduced from Sellin and Sperelakis, 1978.)
XII Slow Fibers
One type of skeletal muscle fibers, known as slow fibers, subserves tonic functions, including posture. Slow fibers should not be confused with “slow twitch fibers”. The true slow fibers do not fire APs, whereas all types of twitch fibers do. The slow fibers are usually smaller in diameter than twitch fibers and they exhibit a less distinct myofibrillar arrangement (so-called “felden” structure). Slow fibers have been found in a number of vertebrate muscles, e.g. in the frog rectus abdominus muscle, frog ileofibularis muscle and mammalian extraocular muscles. It is probable that careful searching will reveal some slow fibers in other mammalian muscles as well.
The slow fibers have multiple innervation by a series of motor end-plates (spaced about 1 mm apart), all from a single motor neuron. As with twitch fibers, acetylcholine (ACh) is the synaptic transmitter. The force of contraction of the slow fibers is controlled by graded end-plate potentials (EPPs). That is, an increase in frequency of impulses in the motoneuron produces a larger EPP (by temporal summation) and this, in turn, produces a greater contraction in the vicinity of the end-plate. Since the end-plates are spaced closely together – at a distance of about one length constant – the entire fiber becomes nearly uniformly depolarized, even though there are no propagated APs. Therefore, the entire length of the slow fiber contracts almost uniformly.
The slow fibers do possess T-tubules which abut at the triadic junctions with the terminal cisternae of the SR (TC-SR). Therefore, the T-tubules may act as passive conduits in the slow fibers to bring the depolarization (produced in the surface membrane by the EPP) deep into the fiber interior. Thus, depolarization of the T-tubule occurs by their cable properties. This depolarization, in turn, could bring about the influx of Ca2+ by activation of voltage-dependent slow Ca2+ channels located in the T-tubules.
APs normally cannot be induced to occur in vertebrate slow fibers under a variety of experimental conditions. However, denervation of frog slow fibers does allow an AP-generating mechanism to appear (Miledi et al., 1971). APs can be induced in slow fibers of invertebrates (e.g. crustacean skeletal muscles) (Fatt and Ginsborg, 1958). Similarly, in the neurogenic horseshoe crab (Limulus) heart, which normally is activated by summating excitatory postsynaptic potentials, propagating (ca. 5 cm/s) and overshooting spontaneous APs can be rapidly induced by Ba2+ (0.1–10 mM) (Rulon et al., 1971). These slowly-rising (ca. 1.0 V/s) APs are resistant to TTX and these voltage-dependent slow channels can pass Ba2+, Sr2+ and Ca2+.
XIII Conduction of the Action Potential
When the EPP, generated at the neuromuscular junction, reaches threshold for eliciting an AP in the vertebrate twitch skeletal muscle fiber, an AP is propagated down the muscle fiber in both directions from the end-plate. (In some muscle fibers, there is a second end-plate innervated by a motoneuron exiting the spinal cord at another level.) The AP is overshooting (to about +40 mV) and propagates at a constant velocity of about 5 m/s over the surface sarcolemma. Propagation occurs by means of the local-circuit currents that accompany the impulse, as discussed in Chapter 19. The reader is referred to that chapter for details on the radial (transmembrane) currents and the longitudinal (axial) currents. The external longitudinal currents can use the entire ISF space (since current takes the path of least resistance), allowing the electromyogram (EMG) to be recorded from the skin overlying an activated skeletal muscle. The amplitude of the EMG potentials becomes larger when more fibers within the muscle are activated (fiber summation), because of summation of the IR voltage drops produced by each fiber activated simultaneously. The frequency of the EMG potentials reflects the frequency and asynchrony of activation of the muscle.
The skeletal muscle fibers are formed by myoblast cells that have fused end to end to become long multinucleated myotubes and then cylindrical fibers later in development. They behave as semi-infinite cables. That is, an AP can propagate from one end of the fiber to the other, uniformly and unimpeded. The space constant or length constant (λ) of the fiber cable is about 1.5 mm for frog sartorius fibers (Sperelakis et al., 1967) and about 0.76 mm for the rat EDL muscle (Sellin and Sperelakis, 1978). The length constant is the distance over which a voltage applied at one region would decay to 1/e (1/2.717 = 0.368) or 36.8% of the initial value. That is, in a passive cable, voltage decays exponentially with a certain length constant as given by:
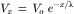 (42.4)
(42.4)
where Vx is the voltage at the distance x and Vo is the voltage at the origin (x = 0). λ is given by:
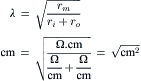 (42.5)
(42.5)
Assuming that ro (the outside longitudinal resistance) is negligibly small compared to ri (this would be true for a superficial fiber in a bundle immersed in a large bath):
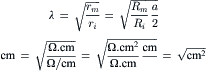 (42.6)
(42.6)
where rm (Ω·cm) and ri (Ω/cm) are the membrane resistance and the internal longitudinal resistance normalized for unit length of fiber, Rm (Ω·cm2) is the membrane resistance normalized for both fiber radius and length, Ri (Ω·cm) is the resistivity of the myoplasm (normalized for length and cross-sectional area), and a (cm) is the fiber radius. Rm is often loosely called membrane resistivity, but this is not accurate because for true membrane resistivity (ρm) there must be correction for membrane thickness δ:
 (42.7)
(42.7)
The factors that determine active velocity of propagation (θα) include the intensity of the local-circuit current, threshold potential and the passive cable properties, λ and τm. As discussed previously, the greater the rate of rise of the AP, the greater the intensity of the local-circuit current, hence the greater the θα. In addition to its dependence on the density of the fast Na+ channels (determinant of the maximum Na+ conductance, ), the kinetic properties of the channel gating and the threshold potential (Vth), max dV/dt is determined also by the RP (or takeoff potential) (related to the h∞ versus Em curve), as discussed previously (Fig. 42.9). In addition, because cooling decreases Na channel activation (Q10 3), max dV/dt and θα are slowed accordingly. Rm is increased by cooling, the Q10 for RK in frog sartorius fibers being about 2.8 (ion diffusion in free solution has a Q10 of about 1.2) (Sperelakis, 1969). For a description of passive conduction, see Appendix III to this chapter.
3), max dV/dt and θα are slowed accordingly. Rm is increased by cooling, the Q10 for RK in frog sartorius fibers being about 2.8 (ion diffusion in free solution has a Q10 of about 1.2) (Sperelakis, 1969). For a description of passive conduction, see Appendix III to this chapter.
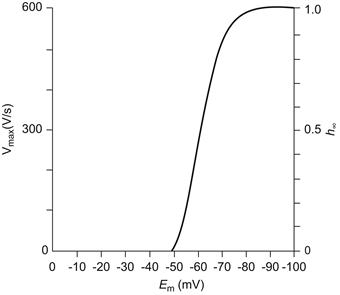
FIGURE 42.9 Graphic representation of the maximal rate of rise of the APl (max dV/dt) as a function of resting Em or takeoff potential. Max dv/dt is a measure of the inward current intensity (membrane capacitance being constant), which is dependent on the number of channels available for activation; h is the inactivation factor of Hodgkin–Huxley as gNa = 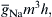 where gNa is the Na+ conductance, is the maximal conductance and m and h are variables; h∞ represents h at t = ∞ or steady state (practically, after 20 ms). The fast Na+ channels begin to inactivate at about −75 mV and nearly complete inactivation occurs at about −30 mV (h∞ low). Therefore, max dV/dt decreases because h∞ decreases.
where gNa is the Na+ conductance, is the maximal conductance and m and h are variables; h∞ represents h at t = ∞ or steady state (practically, after 20 ms). The fast Na+ channels begin to inactivate at about −75 mV and nearly complete inactivation occurs at about −30 mV (h∞ low). Therefore, max dV/dt decreases because h∞ decreases.
XIV Excitation Delivery to Fiber Interior by Conduction into the T-Tubular System
The experiments of Huxley and Taylor (1958) were the first to provide evidence that there was some structure, located at the level of the Z-lines in frog skeletal muscle fibers, which is involved in E-C coupling. This structure allows relatively fast conduction of the excitatory process (AP) from the surface membrane to the center of the muscle cells. These investigators applied current pulses at different points along the length of the sarcomeres in isolated fibers and found that when the microelectrode tip was opposite the Z-line, graded contractions of the two half-sarcomeres occurred. The greater the current, the greater was the inward spread of the contraction. In addition, they discovered that there were sensitive spots located around the perimeter of the fiber at the Z-line level; i.e. the membrane was not uniformly sensitive. At about the same time, it was discovered by electron microscopy that transverse (T-) tubules were located at the level of the Z-lines in amphibian skeletal muscle (and at the level of the A-I junctions of the sarcomere in mammalian skeletal muscle). Thus, the T-tubules probably represent the morphological conduit for the findings of Huxley and Taylor. The morphological arrangements of the sarcotubular system of skeletal muscle fibers are illustrated in Fig. 42.10.
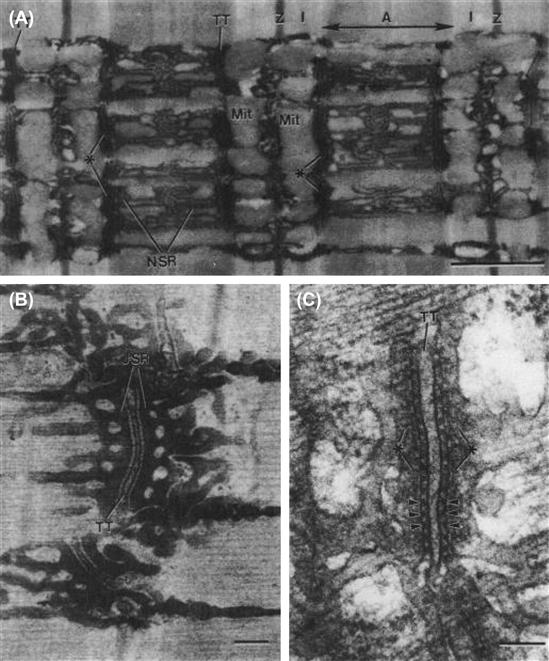
FIGURE 42.10 Sarcotubular system of skeletal muscle fibers from tibialis anterior muscle of mouse (A,C) and iliotibialis muscle of lizard (B). (A) Longitudinal section showing the sarcomere structure of several myofibrils: A-band, I-band, Z-line. The network sarcoplasmic reticulum (N-SR), also known as the longitudinal SR, appears as a torn sleeve surrounding the surface of each myofibril. The N-SR is continuous with the junctional SR (J-SR) that abuts close to the transverse tubules (TT). The TT membranes are invaginations of the cell surface membrane at the level of the A-I junctions in mammalians (or at the level of the Z-line in lizards and amphibians). The J-SR and TT form the complex coupling known as a triad (∗). The N-SR is continuous across the I-band, but this is obscured in this section by the presence of paired mitochondria (Mit) over the I-bands. The TT and SR are both selectively filled with osmium tetroxide precipitate, causing their profiles to be more electron opaque than the other structures. Scale bar at lower right represents 1.0 μm. (B) Higher magnification of a triad to show more detail. As shown, the triad consists of a single T-tubule sandwiched between two cisternae of the J-SR. Scale bar = 0.1 μm. (C) High magnification of a triadic junction to illustrate the array of regularly-spaced junctional processes or SR foot processes (several indicated by arrowheads) that project between the TT membrane and the J-SR membrane. There are dense granules within the lumen of the J-SR cisternae (∗). Scale bar = 0.1 μm. (Electron micrographs provided courtesy of Dr Mike Forbes, University of Virginia.)
Diffusion of some substance from the surface membrane into the skeletal muscle fiber interior is much too slow to account for the relatively short latent period of about 1–3 ms between the beginning of the AP and the beginning of contraction. That is, the diameter of the fibers (mean value of about 70 μm in frog sartorius fibers) is much too large for a diffusion mechanism from the fiber surface to be involved. Diffusion time (for 95% equilibration) increases by the square of the distance and would require about 2.5 s for a small molecule freely diffusing across a cell radius of 50 μm; estimates for Ca2+ diffusion time are considerably longer than this (Podolsky and Costantin, 1964). Therefore, the T-tubular system serves as an electrical conduit to bring excitation deep into the fiber interior rapidly and thereby reduces the required diffusion distance to an average value of about 0.7 μm (Sperelakis and Rubio, 1971). It was reported that disruption of the T-tubule system (by a glycerol osmotic-shock method) uncouples contraction from excitation (Eisenberg and Gage, 1969).
Estimates of the length constant of the T-tubules (λTT), assuming the T-tubule membrane has about the same resistivity (Rm) as the surface membrane 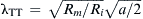 give values of about 50 μm. Because the resistivity of the T-tubule membrane of frog muscle is probably higher than that of the surface sarcolemma because of lower gCl (Hodgkin and Horowicz, 1959; Adrian and Freygang, 1962; Sperelakis and Schneider, 1968), this would give a longer value. Therefore, it is theoretically possible for the T-tubules to serve as passive conduits to bring the depolarization from the surface membrane (during its AP) into the fiber interior. (This mechanism does operate when small depolarizing voltage changes [below the AP threshold] occur on the surface sarcolemma, and conversely any voltage change originating in the T-tubules can be conducted passively to the surface sarcolemma.)
give values of about 50 μm. Because the resistivity of the T-tubule membrane of frog muscle is probably higher than that of the surface sarcolemma because of lower gCl (Hodgkin and Horowicz, 1959; Adrian and Freygang, 1962; Sperelakis and Schneider, 1968), this would give a longer value. Therefore, it is theoretically possible for the T-tubules to serve as passive conduits to bring the depolarization from the surface membrane (during its AP) into the fiber interior. (This mechanism does operate when small depolarizing voltage changes [below the AP threshold] occur on the surface sarcolemma, and conversely any voltage change originating in the T-tubules can be conducted passively to the surface sarcolemma.)
However, direct microscopic observations of the degree of myofibril activation across the width of the fiber caused by raised [K+]o depolarization suggested otherwise (Gonzales-Serratos, 1975). Also, during the foot of the AP, only a small outer ring of the tubular network depolarizes beyond the mechanical threshold (Hodgkin and Nakajima, 1972). These results imply that in order for all the myofibrils in the cross-section of a fiber to be activated, which had been demonstrated previously (Gonzalez-Serratos, 1971), there must be a T-tubular AP (TT-AP).
There is evidence that the T-tubules actually do fire APs, i.e. they actively propagate impulses inward and so bring large depolarization deep into the fiber interior. The evidence for this includes the observation of a threshold for sudden initiation of localized contraction (Costantin and Podolsky, 1967; Costantin and Taylor, 1973). The TT-AP is sensitive to TTX and is Na+ dependent and, therefore, is apparently similar in nature to the surface membrane AP. By use of high-speed cinemicrography to measure sequential activation of the myofibrils in a radial direction, Gonzalez-Serratos (1971) estimated the propagation velocity of the TT-AP (θTT) to be about 10 cm/s, with a Q10 of 2.2 (which is similar to the Q10 of the surface membrane for AP conduction). This velocity is sufficient to account for the short latent period before contraction begins.
Early evidence for the existence of active propagation in the T-tubules came from a number of indirect measurements (reviewed in Caputo, 1978). The speed and Q10 (2.2) of the spread of mechanical activation were greater than expected for passive conduction (Gonzalez-serratos 1971). Moreover, twitch tension was reduced by TTX or a Na+-deficient medium (Costantin, 1970). Depolarization by elevated [K+]o, which inactivates Na+ channels, fails to activate contraction in deeper myofibrils (Gonzales-Serratos, 1975). These results suggested that in order for all the myofibrils in the cross-section of a fiber to be activated, there must be fast Na+ channels in the T-tubules and a T-tubular AP (TT-AP). Subsequently, tubular Na+ currents have been measured directly (Hille and Campbell, 1976) and direct experimental confirmation of propagating APs in the T-tubules have been demonstrated in amphibian (Nakajima and Gilai, 1980) and mammalian muscle fibers (DiFranco et al., 2005). These direct recordings of the propagating AP in the T-tubules were achieved using potential-sensitive dyes, since the T-tubule membranes are not accessible to conventional microelectrode methods.
In muscles placed in low [Na+]o and stimulated briefly at high frequency, the normal tetanic tension rapidly falls, simultaneous with the central myofibrils becoming inactive. These results are due to Na+ depletion in the T-tubule network, particularly in the deeper parts far from the orifice at the fiber surface (Bezanilla et al., 1972). It is thought that the Na+ influx (the inward fast Na+ current) with each AP in the T-tubule produces a progressive decline in [Na+]TT, which slows propagation velocity down the T-tubules and eventually leads to loss of excitability when [Na]TT drops below some critical level (e.g. 30 mM). Na+ depletion should occur more rapidly deep in the T-tubule network because there would be less diffusion of Na+ in from the mouth of the T-tubule to replenish the Na+ loss. Active Na+-K+ pumping in the T-tubules may not occur fast enough to keep up with the Na+ loss into the fiber myoplasm.
There are also voltage-dependent slow Ca2+ channels in the T-tubule membrane and slow APs that arise from the T-tubule can be recorded under appropriate conditions (Sperelakis et al., 1967; Vogel et al., 1978). The evidence for the existence of this type of channel and some of its properties was discussed above. The Ca2+ influx into the myoplasm through these Ca2+ channels could play a role in E-C coupling. For a discussion of the relationship between the T-tubules and the terminal cisternae of the SR, see Appendix IV.
Appendix
AI More Information on Cl− Channels
In frog, there are several subtypes of Cl− channels that have single-channel conductances ranging between 40 and 70 pS and each channel may exhibit several subconductance states. But there is usually a main gate that opens or closes the entire channel. In fetal mammalian fibers, Cl− channels with conductances of about 40, 60 and 300 pS have been observed. Myoballs cultured from muscle biopsies of patients having one form of myotonia had a reduced (ca. 50%) single-channel conductance for the Cl− channel, which would contribute to the myotonia (Fahlke et al., 1993). In primary cultures of rat skeletal muscle, the fast Cl− channel showed a behavior consistent with six closed states and two open states (Weiss and Magleby, 1992). The Cl− channel in myoblasts and myotubes of the L6 cell line derived from rat skeletal muscle had a high conductance of about 330 pS (Hurnak and Zachar, 1992). Voltage-gated Cl− channels have also been found in the SR membrane of skeletal muscle.
Some Cl− channels described for other tissues include: (1) Ca2+-dependent Cl− channels; (2) stretch-activated Cl− channels; and (3) cyclic AMP-stimulated Cl− channels. The receptor-operated Cl− channels apparently have a G-protein (e.g. Gs or Gi) as intermediate for coupling.
The voltage-dependent Cl− channels can be blocked relatively selectively by several methods, including acidosis and use of compounds such as the stilbene derivatives (DIDS and SITS) and 9-anthracene carboxylic (9-AC) acid. The Cl− channels in frog skeletal muscle are relatively insensitive to 9-AC acid, whereas those in adult mammalian muscle are highly sensitive. The anion selectivity sequence for some voltage-dependent Cl− channels is I− > Br− > Cl− > F−.
AII More Information on KATP Channels
It has been proposed that KATP channels may be associated with the decrease in force development underlying muscle fatigue. A drug (SR44866) that opens KATP channels (in sarcolemmal membrane patches from frog skeletal muscles) also reduces the AP duration, the early afterpotential and the peak twitch force (without affecting the RP) in intact frog muscles (Sauviat et al., 1991). The high K+ permeability found in muscle fibers that have undergone a permanent contracture (rigor) produced by repetitive stimulation while metabolically-poisoned with cyanide and iodocetate (Fink and Lüttgau, 1976), may be caused by the decreased ATP concentration opening KATP channels. In metabolically-exhausted frog semitendinosus muscle fibers, the addition of tolbutamide and glyburide, two KATP channel antagonists, significantly reduce the K+ efflux rate (Castle and Haylett, 1987). As said previously, even under fatigue induced by prolonged repetitive stimulation, the decrease in intracellular ATP is small (Nassar-Gentina et al., 1978) and fatigued muscle fibers can contract when Ca2+ is released directly from the intracellular SR stores (Gonzalez-Serratos et al., 1978; Garcia et al., 1991). In intact animals, some of the skeletal muscle fatigue results from synaptic fatigue, including fatigue at the neuromuscular junction.
The Kir6.x channel is a typical inward-rectifier type K+ channel protein. It has two transmembrane helices that form the pore and K+ selectivity filter. The cytoplasmic NH2 and COOH termini interact to form an ATP binding site. A key property of Kir channels is inhibition of channel opening by ATP (Spruce et al., 1987). Each Kir subunit can bind one molecule of ATP. Inhibition by ATP is not a consequence of phosphorylation or ATP hydrolysis, but of direct binding to intracellular domains on the Kir channel (Kakei et al., 1985). That is, ATP is not consumed in this action.
SUR is a regulatory protein that is linked to the C-terminus of Kir. SUR is an ATP-binding cassette (ABC) protein, which itself (unlike other ABC proteins) has no intrinsic transport function, but associates with Kir6.x K+ channels to form the functional KATP channel. SUR serves as a regulatory subunit which fine-tunes the activity of Kir6.x in response to changes in cell metabolism. Each SUR subunit contains two nucleotide-binding folds which contain binding sites for Mg2+-adenosine nucleotides. Cytosolic free Mg2+ is high in resting skeletal muscles (ca 1 mM). In the absence of Mg2+, nucleotides (such as ADP) act on SUR to inhibit KATP activity. Therefore, nucleotides can regulate KATP channels through interactions with both Kir and SUR.
As with other members of the Kir family, the KATP channel exhibits inward rectification (ir). That is, the open channels pass inward current with increasing hyperpolarization, but outward K+ current does not flow when the membrane is depolarized by more than about 10 mV above the EK potential. This property of inward rectification is caused by cytoplasmic ions such as Mg2+ and polyamines plugging the pore pathway upon depolarization and thereby obstructing the outward flow of K+ (Woll et al., 1989).
The half-maximum inhibition of opening of KATP channels by ATP in skeletal muscle (measured at a constant [K+]o/[K+]i) is 0.135 mM at pH 7.2 (see Fig. 42.6). Intracellular [ATP] is about 5 mM in resting muscle (Dawson et al., 1978, 1980) and it is maintained at near basal levels even during repetitive contractions that lead to muscle fatigue (Carlson and Siger, 1960; Nassar-Gentina et al., 1978). Therefore, the KATP channels are largely inactive (or masked) under normal physiological conditions.
In searching for a possible role of the KATP channel under other conditions, a modest role was demonstrated in maintaining membrane polarization under sustained muscle use and fatigue. Although ATP is the controller of KATP opening, KATP activity can be modulated by a number of metabolites through interactions with Kir, SUR or both. These include: K+, H+ and phospholipids (such as PIP2), all of which are known to increase during muscle contraction.
Extracellular K+ concentration ([K]o) rises dramatically in the muscle extracellular space during repetitive AP activity and can reach 15–50 mM in the transverse tubules (Almers, 1980; Clausen, 2008). The unitary conductance of the KATP channel increases with [K+]o, from 15 pS in 2.5 mM to 42 pS in 60 mM. Therefore, at higher [Ko], KATP channels are more conductive at the same ATP concentration.
Kir channels are also regulated by pH at the cytoplasmic surface (pHi) (reviewed in Jiang et al., 2002; Qu et al., 2000). Intracellular pH may drop by up to one unit during intense muscle use (Renaud, 1989). A decrease in pHi reduces the inhibitory effect of ATP on KATP channels, leading to an increase in the probability of KATP channel opening (Davies et al., 1992). The ATP concentration required for half-inhibition increases by 2.5 times and 15 times, respectively, at pHi 6.8 and 6.3, compared to that at pHi 7.2. That is, a decrease in pHi increases the ATP concentration required for half-inhibition of channel activity. During acidosis, a small decrease in ATP concentration leads to a greater opening of KATP channels.
The decrease in pHi during contraction is matched by an increase in cytosolic free Mg2+. A rise in cytosolic Mg2+ reduces the ability of ATP to close KATP channels (Vivaudou et al., 1991). This is most likely related to the ability of Mg2+ to bind to ATP and to the stimulatation of KATP channels via the SUR regulatory subunit. Although the increases in [K]o, pHi and [Mg2+]i may act collectively and synergistically to activate KATP channels during repetitive muscle contraction, this effect is modest at normal muscle [ATP]i.
On the other hand, KATP channels may become activated when the muscle is stressed or metabolically compromised (Hussain et al., 1994). Adenosine, which is released from metabolically-compromised muscle, stimulates KATP (Bartlett-Jolley and Davies, 1997) and, as said previously, intracellular acidosis is a potent activator of KATP channels. Although the skeletal muscles of genetically-altered mice which lack Kir (Kir6.2 knockout mice) develop fatigue more rapidly than control muscles (Cifelli et al., 2007), the more rapid onset of fatigue is not associated with KATP channel activation (Boudreault et al., 2010). These findings indicate that KATP channels are not essential for maintaining force during fatigue.
The activation of KATP channels after fatigue has developed helps preserve a polarized membrane potential, keeping it near EK. This function prevents voltage-dependent Ca2+ entry that can occur following fatigue and cause fiber injury. Consistent with this idea, both Kir6.2 knockout mice and SUR knockout mice show extensive muscle fiber damage when subjected to exercise training (Thabet et al., 2005; Stoller et al., 2007). Therefore, KATP channels in muscle serve a myoprotective role to prevent fiber damage following intense fatigue or during metabolic exhaustion. In this respect, the role of KATP channels in skeletal muscles is similar to that in other cell types, i.e. KATP channels serve as molecular sensors of cellular metabolism, linking metabolism to membrane excitability.
AIII Further Evidence that the T-Tubules Fire Na+-Dependent APS
Further evidence of a Na+-dependent TT-AP came from observations in which muscles placed in low [Na+]o and stimulated briefly at high frequency, initially develop normal tetanic tension that rapidly falls and steadily decreases to a lower sustained tension; simultaneously the central myofibrils becoming inactive. These results are due to Na+ depletion in the T-tubule network, particularly in the deeper parts far from the orifice at the fiber surface. Na+ depletion occurs because gNa increases during the TT-AP generation, causing an inward Na+ flow into the myoplasm (Bezanilla et al., 1972). The fatigue occurs more rapidly in fibers pre-equilibrated in low [Na+]o (e.g. 60 mM). There is a progressive decline in [Na+]TT, which slows propagation velocity down the T-tubules and eventually leads to loss of excitability when [Na]TT drops below some critical level (e.g. 30 mM). Na+ depletion should occur more rapidly deep in the T-tubule network because there would be less diffusion of Na+ in from the mouth of the T-tubule to replenish the Na+ loss. Active Na+-K+ pumping in the T-tubules may not occur fast enough to keep up with the Na+ loss into the fiber myoplasm.
There is evidence that the T-tubules actually do fire APs, i.e. they actively propagate impulses inward and so bring large depolarization deep into the fiber interior. The evidence for this includes the observation of a threshold for sudden initiation of localized contraction (Costantin and Podolsky, 1967; Costantin and Taylor, 1973). The TT-AP is sensitive to TTX and is Na+ dependent and, therefore, is apparently similar in nature to the surface membrane AP. By use of high-speed cinemicrography to measure sequential activation of the myofibrils in a radial direction, Gonzalez-Serratos (1971) estimated the propagation velocity of the TT-AP (θTT) to be about 10 cm/s. The Q10 of 2.2 is similar to that of the surface membrane AP. Although this velocity is about 16 times slower than propagation down the fiber longitudinally (about 1.6 m/s), it is sufficient to account for the short latent period before contraction begins. Additional data supporting the presence of APs in the T-tubules has been obtained more recently using V-sensitive dyes.
AIV Propagation Velocity in a Passive Cable
In a passive cable, such as in a resting skeletal muscle fiber, the passive propagation velocity (θρ) is directly proportional to the length constant and inversely proportional to the time constant:
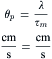 (42A.1)
(42A.1)
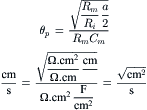 (42A.2a)
(42A.2a)
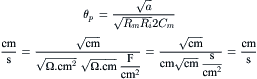 (42A.2b)
(42A.2b)
Therefore, propagation velocity is directly proportional to the square root of the fiber radius (a) and inversely proportional to membrane capacitance (Cm) and to the square root of Ri and the square root of Rm. For example, propagation velocity is greater in large-diameter muscle fibers. The length constant for sinusoidally-varying applied currents (λac) is shorter than λdc, depending on the ac frequency.
The relationship between propagation velocity and membrane current density (Im) is given by:
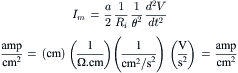 (42A.3)
(42A.3)
where d2V/dt2 is the second time derivative of the AP. As indicated, membrane current is proportional to d2V/dt2, whereas the longitudinal current (Il) or the capacitative current (Ic) is proportional to dV/dt:
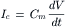 (42A.4)
(42A.4)
AV Evidence for T-Tubule Communication with the SR across the Triadic Junction under Some Conditions
This Appendix section is included to let the student know that there are data that do not fit with currently accepted hypotheses.
Ca2+ for contraction in skeletal muscle is primarily released from the TC-SR (Winegrad, 1968) and there is an internal cycling of Ca2+ ion. Changes in [Ca2+]o of the bathing solution take a relatively long time (e.g. 30 min) before exerting a large effect on the force of contraction. In contrast, in cardiac muscle, the effect of lowered [Ca2+]o is significant within a few seconds, indicating that the primary determinant of the force of contraction is the Ca2+ influx across the sarcolemma through the slow Ca2+ channels. Therefore, in skeletal muscle, excitation propagates actively down the T-tubules and Ca2+ is released from the TC-SR, but it is controversial as to how the signal is transferred from the T-tubule to the TC-SR across the triadic junction.
Electron-opaque tracer molecules, like horseradish peroxidase (HRP) (ca. 60 Å diameter), enter into the T-tubules and from there can enter into some of the TC-SR of frog skeletal muscle (Rubio and Sperelakis, 1972; Kulczycky and Mainwood, 1972) (Fig. 42A.1A,B). Exposure of the fibers to hypertonic solutions facilitates the entry of HRP into the TC-SR, so that nearly 100% of the TC-SR become filled (Fig. 42.A.1C,D). Thus, there may be a functional connection between the SR and the extracellular space (Sperelakis et al., 1973). If so, there may be lumen-to-lumen continuity between the T-tubules and TC-SR during excitation, allowing the AP in the T-tubules to invade directly into the TC-SR to depolarize and bring about the release of Ca2+. The depolarization of the TC-SR could activate voltage-dependent Ca2+ channels, allowing Ca2+ influx into the myoplasm down an electrochemical gradient.
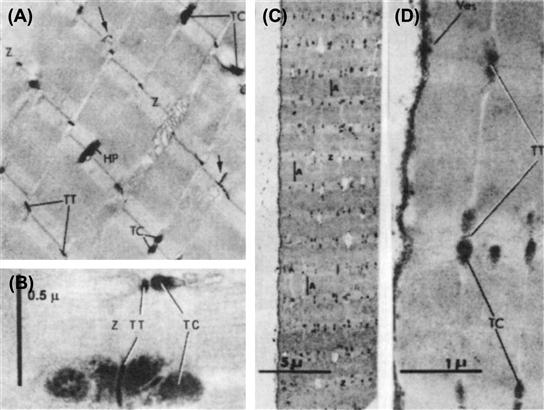
FIGURE 42A.1 Evidence that large molecules of horseradish peroxidase (HP) can enter into the terminal cisternae (TC) of the SR via the transverse tubules (TT) of frog sartorius fibers. Electron micrographs of longitudinal sections. (A,B) Fiber was exposed to HP under isosmotic conditions. (A) Section through several myofibrils showing presence of HP activity (as a dense electron-opaque material) in the TT and in some of the TC at triadic junctions. In amphibian muscle, the TT occur at the level of the Z-lines (Z) of the sarcomeres. Arrows point to two branches of the TT running longitudinally. (B) Higher magnification of two triads, one with both cisternae filled with HP and the other with only one cisterna filled. (C,D) Fiber was exposed to HP under hypertonic condition (3 X isotonic, using NaCl), showing that almost all cisternae were filled with peroxidase. (C) Section at low magnification. (D) Portion of same section as in Part C shown at higher magnification. The surface vesicles (Ves) also became filled with HP. (Modified from Figs. 2 and 4 of Rubio, R. and Sperelakis, N. (1972). Z. Zellforsch. 124, 57–72.)
If the longitudinal SR (L-SR) were electrically isolated from the TC-SR by a substantial resistance (e.g. zippering between the two SR compartments, described later), this would account for the fiber capacitance measured being relatively low (Mathias et al., 1980). The effect of this would be to remove the very large membrane surface area of the L-SR and hence greatly reduce the capacitance that would be measured.
It has been suggested that the SR is depolarized during the release of Ca2+ in E-C coupling. For example, optical signals (e.g. birefringence and fluorescence changes) can be recorded from the SR membranes during contraction (e.g. Baylor and Oetliker, 1975; Bezanilla and Horowicz, 1975). In addition, Natori (1965) demonstrated that propagation of contraction (1–3 cm/s) triggered by electrical stimulation can occur in muscle fiber regions that had been denuded (skinned) of their sarcolemma, the propagation of excitation presumably occurring by means of the SR membranes.
It was demonstrated that E-C uncoupling could be produced by exposing frog skeletal muscle fibers to Mn2+ (1 mM) or La3+ (1 mM) while in hypertonic solution (to facilitate entry of the blockers into the TC-SR) (Sperelakis et al., 1973). After the fibers were returned to normal Ringer solution, normal fast APs could be elicited, but there were no contractions accompanying them; i.e. a “permanent” E-C uncoupling was produced. These results were interpreted as suggesting that Mn2+ and La3+ entered into the lumen of the TC-SR and blocked the Ca2+ channels. A similar exposure of frog sartorius fibers to Mn2+, La3+ or to Ca2+-free solution blocked the caffeine-induced contracture as well (Rubio and Sperelakis, 1972). Thus, from these physiological and ultrastructural studies, it was suggested that the lumen of the SR is continuous with that of the T-tubule under conditions of hypertonicity and that substances can enter into the TC-SR to exert an effect on Ca2+ release into the myoplasm.
Compartmental analysis of skeletal muscle has also suggested that the SR is open to the ISF. (In contrast, in cardiac muscle, there is no evidence that the SR is open to the ISF [Rubio and Sperelakis, 1972].) For example, Conway (1957), Harris (1963) and Keynes and Steinhardt (1968) concluded that Na+ inside frog skeletal muscle fibers is distributed in two separate compartments. Harris (1963) suggested that the Na+, K+ and Cl− concentrations in one compartment (presumably the SR) were about equal to those of the ISF. Rogus and Zierler (1973) concluded that the Na+ concentration in the SR of rat skeletal muscle approximates that of the ISF. The volume of the SR compartment was 14.3% of fiber volume and, in hypertonic solution, the SR volume increased and the washout of the SR compartment was faster. Tasker et al. (1959) also had reported a large sucrose space of 26.5% for frog sartorius fibers.
Other researchers (Birks and Davey, 1969) have demonstrated that the volume changes of the SR of skeletal muscle in hypertonic (sucrose) and hypotonic solutions were always opposite of those occurring within the myoplasmic compartment. They concluded that sucrose must enter into the SR, pulling in water osmotically from the myoplasm, to produce the marked swelling of the SR that occurred in hypertonic solutions. Vinogradova (1968) concluded from the distribution of non-penetrating sugars in frog sartorius muscle that the SR compartment is continuous with the ISF; the inulin space was 19.0% and increased in hypertonic solution and decreased in hypotonic solution and in glycerol-treated fibers (for disruption of the T-tubules).
The total [3H]-sucrose space of frog sartorius muscles was found to be 18.0% in isotonic solution and 22.6% in twofold hypertonic solution (Sperelakis et al., 1978). The relative SR volume (including the small T-tubule volume) was 12.4% and 17.0% of fiber volume, respectively. This value for SR volume of frog skeletal muscle is close to that measured by ultrastructural techniques (Peachey, 1965; Mobley and Eisenberg, 1975). Evidence that the TC-SR and L-SR may not be freely connected to one another under resting conditions comes from the observations that: (1) the L-SR did not fill with HRP, whereas the TC-SR did (Rubio and Sperelakis, 1972); and (2) there is a zippering of the membranes connecting these two components of the SR in mouse and frog skeletal muscle (Howell, 1974; Wallace and Sommer, 1975; Forbes and Sperelakis, 1979).
In 45Ca washout experiments on frog muscles, Kirby et al. (1975) found three compartments, similar to the three sucrose compartments described previously, except the half-times were about two- to threefold shorter. They suggested that the first compartment was the ISF space, the second was the T-tubule plus the TC-SR and the third was the L-SR. Bianchi and Bolton (1974) also found a transient increase in 45Ca efflux and a marked loss of muscle Ca2+ from frog sartorius muscles exposed to hypertonic solutions (twice isotonicity) and suggested that hypertonicity produces transient communication between the TC-SR and the T-tubules, thus allowing their Ca2+ to be lost to the ISF. In addition, it has been reported in a human muscle disease, polymyositis, that the T-tubules are spatially continuous with the SR, as visualized with lanthanum tracer, and that enzymes leak from the TC-SR into the T-tubules and ISF (Chou et al., 1980).
Frog skeletal muscle fibers have an osmotically inactive volume of about 32% when placed into Ringer solution made hypertonic with sucrose or other non-penetrating solutes; i.e. fiber diameter does not shrink to the theoretical value expected if it were a perfect osmometer (Sperelakis and Schneider, 1968; Sperelakis et al., 1970). For example, in twofold hypertonic solution, there should be a decrease in fiber volume to one-half and fiber radius to 0.707  of the original value. The observed change is to only 0.81 of the original diameter. Because the SR volume increases in hypertonic solution (Huxley et al., 1963; Sperelakis and Schneider, 1968; Birks and Davey, 1969), it is likely that the osmotic inactive volume is due to the SR. The swollen SR would prevent the fiber volume from decreasing to one-half in twofold hypertonic solution, even if the volume of the myoplasm proper were to decrease to one-half.
of the original value. The observed change is to only 0.81 of the original diameter. Because the SR volume increases in hypertonic solution (Huxley et al., 1963; Sperelakis and Schneider, 1968; Birks and Davey, 1969), it is likely that the osmotic inactive volume is due to the SR. The swollen SR would prevent the fiber volume from decreasing to one-half in twofold hypertonic solution, even if the volume of the myoplasm proper were to decrease to one-half.
In cardiac muscle, an osmotically-inactive volume is not present (Sperelakis and Rubio, 1971), electron-opaque tracers do not enter the SR (Sperelakis et al., 1974) and the SR volume does not increase with hypertonicity (Sperelakis and Rubio, 1971).
AVI Invertebrate Striated Muscle Fibers
This Appendix was prepared by Hugo Gonzalez-Serratos and Hector Rasgado-Flores and was edited by Nicholas Sperelakis.
Introduction
In 1902, Overton observed that frog skeletal muscle cells lost their excitability when the extracellular Na+ concentration ([Na+]o) was decreased below 10% of normal. This was the first indication that excitability of vertebrate skeletal muscle fibers was Na+-dependent. Using intracellular microelectrodes, Nastuck and Hodgkin demonstrated, in 1950, that skeletal muscle fibers of the frog have a clear relationship between the amplitude of the AP and [Na+]o. They concluded that vertebrate striated skeletal muscle APs are generated by mechanisms similar to those in squid giant nerve axons. In an attempt to test this Na+ hypothesis in striated muscle fibers of the crustacean, Fatt and Katz (1953) bathed crayfish muscles with different external Na+ concentrations. However, against their prediction, they found that there was no effect on the amplitude of the APs. That is, striated invertebrate crayfish muscles were excitable and capable of developing full APs in the absence of external Na+ ions. Thus, they concluded that invertebrate striated muscle fibers did not have an Na-dependent excitability. Instead, the APs were generated by currents using other ions. The APs of invertebrate striated muscle fibers, compared to vertebrates, are longer in duration and some of them even have a plateau (similar to cardiac muscle) (Fig. 42A.2).
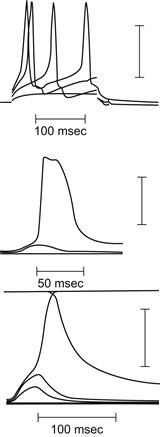
FIGURE 42A.2 Different action potentials in invertebrate striated muscle fibers of different crab species. (A) Portunus (fromAtwood, 1965 ). (B) Carcinus maenas (fromFatt and Katz, 1953 ). (C) Portunus deporatus (fromFatt and Katz, 1953 ). Voltage calibration bars:40 mV.
Calcium Hypothesis
What are the ionic currents that generate the APs in invertebrate striated muscle fibers? With zero extracellular Na+, the invertebrate muscles were capable of generating APs as long as there were Ca2+ ions in the bathing solution. It was concluded that these fibers produced Ca2+-dependent APs (Fatt and Ginsborg, 1958). These APs persisted when Sr2+ or Ba2+ (which have some physicochemical properties similar to Ca2+) replaced the Ca2+. This led to the calcium hypothesis. Hagiwara and Naka (1964) induced APs in the giant barnacle striated muscles fibers (Balanus nubilus) in which they changed the intracellular Ca2+ concentration by injecting the fibers with Ca2+chelating substances. As the internal free Ca2+ concentration was decreased progressively, the AP amplitude increased progressively. At an internal Ca2+ concentration of 8×10−8 M, the AP became all-or-none (Hagiwara and Nakajima, 1966). Thus, they confirmed the generation of Ca2+ APs in invertebrate striated muscle fibers.
Three types of experiments have proven the Ca2+ hypothesis for invertebrate striated muscle cells: (1) the Ca2+-dependence of the AP amplitude; (2) the lack of APs in zero Ca2+ bathing solution; and (3) the entry of Ca2+ ions during the APs.
Ca2+ dependence of the APs. Fatt and Ginsborg in 1958 and Pilgrim and Wiersma in 1963 demonstrated that the striated myofibers of the crayfish (Orconectes virilis) and lobster (Homarus americanus) were capable of generating propagated all-or-none APs. These APs did not disappear when the muscles were immersed in bathing solutions without Na+, K+ or Mg2+. However, the amplitude of the APs depended on the extracellular Ca2+ concentration [Ca2+]o. The AP amplitude increased with a slope of 25 mV per decade of [Ca 2+]o. These results strongly supported the proposal that the APs of invertebrate striated skeletal muscle fibers are generated by inward Ca2+ currents. In the crayfish (Astacus fluvilis), the potentiating effect of [Ca2+]o was strongly enhanced in the presence of quaternary ammonium ions like tetraethylammonium (TEA, an inhibitor of outward IK) (Fig. 42A.3). The maximum rate of depolarization of the APs increased from 8 V/s to 15 V/s when [Ca2+]o was changed from 4 to 16 mM. In the giant muscle fibers from the barnacle (Balanus nubilus), Hoyle and Smith (1963) and Hagiwara and Naka (1964) found that the usual graded responses were converted to all-or-none responses when the gradient between extracellular and intracellular [Ca2+] increased by injecting the cells with Ca2+-chelating substances like EDTA and K2SO4. The amplitudes of the APs were a function of the extracellular and intracellular [Ca2+] ratios.
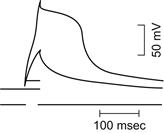
FIGURE 42A.3 Potentiation of invertebrate striated muscle of the crayfish (Astacus fluvialitis) with TEA. Upper trace: action potential. Lower trace: applied current. (From Fatt and Ginsborg, 1958.)
Calcium influx. Hagiwara and Naka (1964) found that, during prolonged stimulations that produced repetitive APs, there was a substantial amount of radioactive Ca2+ influx into barnacle giant muscle. They estimated that the intracellular Ca2+ concentration increased by an average of 75 pmole/cm2/impulse and calculated that, during each AP, there was an influx of 6 pmoles of Ca2+/cm2. This is a very high Ca2+ influx compared with the values reported for vertebrate skeletal muscle fibers (Bianchi and Schanes, 1959).
Prolonged depolarization (elicited with high concentrations of extracellular K+) above the mechanical threshold leads to sustained mechanical activation (contractures). When K+-contractures were elicited in a bathing solution without Ca2+ ions, the K+-contractures disappeared after a short time, even though the membrane remained depolarized. This indicates that, during prolonged depolarization of invertebrate striated muscles, there is a substantial inflow of Ca2+ that brings about the mechanical activation.
Potentiation of APs.Fatt and Ginsborg (1958) observed that when all the extracellular Ca2+ was removed and substituted with equimolar concentrations of SrCl2 or BaCl2, the crayfish striated muscles not only produced all-or-none APs, but they were of higher amplitude and longer duration (Fig. 42A.4). The RPs were unaffected. These results were confirmed by Werman and Grundfest (1961) in lobster muscle fibers and by Werman et al. (1961) in the muscle of the grasshopper (Romalea microptera). Thus, inward Sr2+and Ba2+ currents are also capable, like Ca2+ ions, of generating APs. The increased AP durations were the result of increased durations of the plateau. The control muscle fibers had an average duration of only 3–6 ms. The duration of the Ba2+ APs was 15–100 ms, compared with 400–12 000 ms for the Sr2+ APs (Fig. 42A.4). The AP overshoot was about 20 mV larger with Ba2+ compared with Sr2+ in striated muscles from crayfish (Astacus fluvialis) and lobster (Homarus americanus) (Fatt and Ginsburg, 1958; Werman and Grundfest, 1961). The RP of −90 mV did not change with Sr or Ba ions.
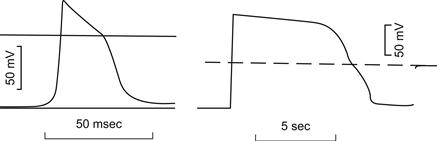
FIGURE 42A.4 Crayfish striated muscles fibers. Action potentials of fibers bathed with strontium (left panel) and barium (right panel). (From Fatt and Ginsborg, 1958.)
Ionic currents. Hagiwara and Naka (1964), by using a voltage-clamp method, showed that the currents developed during the AP were composed of early inward Ca2+ currents, since they varied in amplitude with extracellular Ca2+ concentration and were not affected by TTX. It was concluded that, in invertebrate striated muscle fibers, the propagated AP was the consequence of inward Ca2+ current. Hen ek et al. (1969) measured Sr2+ current in the crayfish (Astacus fluviaitilis) using a sucrose-gap voltage-clamp. They found that the currents consisted of early inward current followed by outward current. The Ca2+ currents were smaller than the Ba2+ currents (Fig. 42A.5). The late outward current was decreased by the K+-current blocker TEA (Fig. 42A.6).
ek et al. (1969) measured Sr2+ current in the crayfish (Astacus fluviaitilis) using a sucrose-gap voltage-clamp. They found that the currents consisted of early inward current followed by outward current. The Ca2+ currents were smaller than the Ba2+ currents (Fig. 42A.5). The late outward current was decreased by the K+-current blocker TEA (Fig. 42A.6).
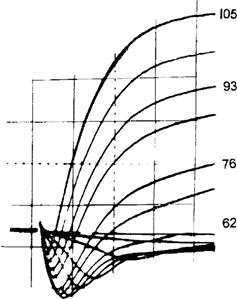
FIGURE 42A.5 Membrane currents under voltage clamp from crayfish striated muscle (Astacus Fluviatilis) bathed with extracellular strontium chloride isotonic solution. The clamped membrane potentials are shown in mV. Records below the zero line are inward currents, above are outward currents.
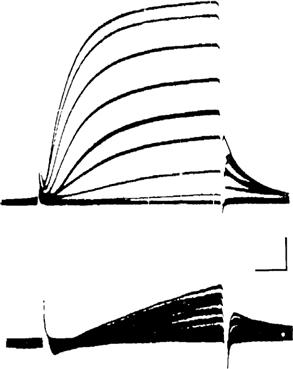
FIGURE 42A.6 Effect of TEA on membrane currents from the crayfish striated muscle (Astacus Fluviatilis) under voltage clamp. Top records, bathed with normal extracellular solution. Bottom records, after addition of 50 mM of TEA to the bathing solution. Calibrations: 0.15 mA/cm2 and 5 ms. (From Henek et al., 1969.)
The AP in invertebrate striated muscles, like in vertebrate striated muscles, fulfills two main roles. One role is to propagate a depolarization wave all along the length of the entire muscle fiber. Another role is to increase the myoplasmic Ca2+ to levels that induce the binding of myosin with actin to bring about activation of the contractile proteins (Fig. 42A.7). The mechanical activation develops either shortening, force, or both (producing muscle work).

FIGURE 42A.7 Myoplasmic calcium transients recorded with the luminescent ptotein aequorin during stimulations of barnacle (Balanus nubilis) striated muscle. Traces: 1: membrane potentials; 2: light output, myoplasma calcium transients; 3: isometric forces; 4: stimulating electrical pulses. On each set of recording traces, the recordings from top to bottom correspond to each other. Vertical calibrations: 20 mV, 3.8×10−9 lumens, 5 g. Horizontal calibration: 100 ms. (From Ashley and Ridgway, 1968.)
Regulation of Intracellular Ca2+
Vertebrate skeletal muscle fibers bathed in Ringer’s solution containing 1 mM [Ca2+] had an average Ca2+ influx at rest of 0.26 pmoles/cm2/s and, during stimulation, the Ca2+ influx increased to 0.73 pmoles/cm2/impulse (Curtis,1966). Blocking the inward Ca2+ current has no effect on the contraction or E-C coupling mechanism (Gonzalez-Serratos et al., 1982). Thus, the main role of inward Ca2+ current in vertebrate skeletal muscles fibers is to maintain long-term adequate amounts of intracellular Ca2+ (mainly in the SR2+).
In contrast, invertebrate striated muscle fibers have an average Ca2+ influx of 75 pmoles/cm2/impulse (Hagiwara and Naka, 1964). Since the SR is sparse, there must be another mechanism to decrease the free myoplasmic Ca2+ after activation. Otherwise, these muscles would develop permanent and powerful contractures and suffer deleterious effects. The mechanism by which invertebrate striated muscles decrease and regulate the free intracellular Ca2+ concentration is via the sarcolemmal Ca2+/H+ exchanger mediated by Ca2+-ATPase (DeSantiago et al., 2007) and the Na+/Ca2+ exchanger (Rasgado-Flores et al., 1991). The Na+/Ca2+ exchanger mediates the coupling of the movement of external Na+ ions down their electrochemical gradient, with the movement of internal Ca2+ ions (in the opposite direction) across the sarcolemma. In giant striated muscle of the barnacle (Balanus nubile) (like most excitable cells), the Na+/Ca2+ exchanger has a stoichiometry of three Na+ ions exchanged per each Ca2+ ion (Rasgado-Flores et al., 1989).
Innervation of Invertebrate Muscle Fibers
One important characteristic of the innervation of non-vertebrate striated muscle fibers is that the nerve terminals on the muscle fibers form terminal buttons spread all along the muscle fiber. This is in contrast with vertebrate muscle where a motor nerve ends in one button localized near the middle of the fiber. The branching of invertebrate motor nerves leads to multiple nerve terminals that are distributed along the length and around the perimeter of the muscle fibers. There are some differences as a consequence of the differences in the topological distribution of the nerve terminals. The excitatory responses to motor nerve APs can trigger in the muscle fiber either local APs or full-size APs all along the muscle fiber. The transmission of the excitatory process leads to multiple postsynaptic potentials. The regulation of force development during contractions of vertebrate muscles is under central nervous system control. In contrast, in invertebrate muscles, the regulation of the degree of contractions is done peripherally and depends on the number of either excitatory or inhibitory nerve terminals that are activated.
Another feature of invertebrate neuromuscular junctions is that they are structurally simpler than vertebrate motor end-plates. The motor nerves of the crustaceans have short nerve terminals that have periodical swellings in series. The swellings make contact with the surface membrane of the muscle fiber. The nerve terminals are very close together. The response of the invertebrate neuromuscular junction to a nerve AP is called the junctional potential (JP). Evoked JPs are due to a large and synchronized release of neurotransmitters triggered by the nerve terminal AP. The JPs of invertebrates are either excitatory (EJPs) or inhibitory (IJPs). The excitatory neurotransmitter is glutamate. The inhibitory neurotransmitter is GABA (γ-aminobutyric acid). The release of these transmitters will lead either to excitation and mechanical activation of the muscle fiber or inhibition of the excitation and thus producing relaxation. The RP can be hyperpolarized due to the IJPs. Therefore, the amount of current necessary to reach the mechanical threshold voltage (for E-C coupling) can also increase as a consequence of the IJPs hyperpolarizing the membrane.
The electrical responses (postsynaptic potentials) at the neuromuscular junctions are, in most cases, small and graded. But when the small responses are summated, the muscle membrane reaches threshold that generates a full AP that propagates along the muscle fiber. Another important feature of invertebrate muscle innervations is that a single myofiber can be innervated by more than one motor neuron, i.e. polyneural innervation. The polyneural innervation leads to complex electrical and mechanical responses. The topological distribution of nerve junctions and the graded activation permit the existence of fast and slow contractions, as well as excitation and inhibition (often within a single myofiber).
When an EJP reaches the mechanical threshold, a local contraction is produced. When several EJPs are elicited in synchrony, they generate a graded contraction that can show summation. This summation causes a large contraction. IJPs cause hyperpolarization, moving the membrane potential away from the mechanical threshold, thereby causing relaxation. The multiple innervations characteristics of invertebrate muscles cause one muscle fiber to produce EJPs while its neighbor may produce IJPs. The same muscle fiber can show an excitatory response that develops force and an inhibitory response that reduces the force developed (Dudel and Kuffler, 1961). From one to nine excitatory nerves may innervate a single muscle fiber, while the number of inhibitory nerves can be up to three (Kennedy et al., 1966). The size of EJPs (initially of low magnitude) increases progressively during repetitive stimulation and this is called facilitation (Hoyle and Wiersma, 1958). In contrast, the large EJPs decrease in size during the repetitive activation due to transmitter depletion or fatigue, a process known as fatigue or anti-facilitation (Hoyle and Burrows, 1973). Another feature of invertebrate muscle fibers is that the faster fibers display higher E-C thresholds. Therefore, they require EJPs of larger magnitude to reach the E-C threshold. They also develop stronger twitches.
There is relevant information about crayfish muscle stretch receptors given in a previous chapter.
BIBLIOGRAPHY
1. Adrian RH, Freygang WH. The potassium and chloride conductance of frog muscle membrane. J Physiol (London). 1962;163:61–103.
2. Adrian RH, Peachey LD. Reconstruction of the action potential of frog sartorius muscle. J Physiol (London). 1973;235:103–131.
3. Adrian RH, Chandler WK, Hodgkin AL. Voltage clamp experiments in striated muscle fibers. J Physiol (London). 1970a;208:607–644.
4. Adrian RH, Chandler WK, Hodgkin AL. Slow changes in potassium permeability in skeletal muscle. J Physiol (London). 1970b;208:645–668.
5. Almers W, Fink R, Palade PT. Calcium depletion in frog muscle tubules: the decline of calcium depletion in frog muscle tubules: the decline of calcium current under maintained depolarization. J Physiol (London). 1981;312:177–207.
6. Armstrong CM, Bezanilla FM, Horowicz P. Twitches in the presence of ethylene glycol bis(β-aminoethyl ether)-N, N′tetraacetic acid. Biochem Biophys Acta. 1972;267:605–608.
7. Ashley CC, Ridgway EB. Simultaneous recording of membrane potential, calcium transient and tension in single muscle fibers. Nature. 1968;219:1168–1169.
8. Atwood HL. Characteristics of fibres in the extensor muscle of a crab. Comp Biochem Physiol. 1965;14:205–207.
9. Baylor SM, Oetliker H. Birefingence experiments on isolated skeletal muscle fibres suggest a possible signal from the sarcoplasmic reticulum. Nature. 1975;253:97–101.
10. Beaty GN, Stefani I. Calcium dependent electrical activity in twitch muscle fibers of the frog. Proc R Soc London (Biol). 1976;194:141–150.
11. Bezanilla F, Horowicz P. Fluorescence intensity changes associated with contractile activation in frog muscle stained with Nile Blue A. J Physiol (London). 1975;246:709–735.
12. Bezanilla F, Caputo C, Gonzalez-Serratos H, Venosa RA. Sodium dependence of the inward spread of activation in isolated twitch muscle fibres of the frog. J Physiol (London). 1972;223:507–523.
13. Bianchi CP, Bolton TC. Effect of hypertonic solutions and glycerol treatment on calcium and magnesium movements of frog skeletal muscle. J Pharmacol Exp Ther. 1974;188 536–522.
14. Bianchi CP, Shanes AM. Calcium influx in skeletal muscle at rest, during activity, and during potassium contracture. J Gen Physiol. 1959;42:803–815.
15. Birks RI, Davey DF. Osmotic responses demonstrating the extracellular character of sarcoplasmic reticulum. J Physiol (London). 1969;21:171–188.
16. Boudreault L, Cifelli C, Bourassa F, Scott K, Renaud JM. Fatigue preconditioning increases fatigue resistance in mouse flexor digitorum brevis muscle with non-functioning K(ATP) channels. J Physiol. 2010;588:4549–4562.
17. Caputo C. Excitation and contraction processes in muscle. Annu Rev Biophys Bioeng. 1978;7:63–83.
18. Carlson ED, Siger A. The mechanochemistry of muscular contraction I The isometric twitch. J Gen Physiol. 1960;44:33–60.
19. Castle NA, Haylett DG. Effect of channel blockers on potassium efflux from metabolically exhausted frog skeletal muscle. J Physiol. 1987;383:31–43.
20. Chou SM, Nonaka I, Voice GF. Anastomoses of transverse tubules with terminal cisternae in polymyositis. Arch Neurol. 1980;37:257–266.
21. Cifelli C, Bourassa F, Gariepy L, Banas K, Benkhalti M, Renaud JM. KAPT channel deficiency in mouse flexor digitorum brevis causes fiber damage and impairs Ca2+ release and force development during fatigue in vitro. J Physiol. 2007;582:843–857.
22. Clausen T. Clearance of extracellular K+ during muscle contraction–roles of membrane transport and diffusion. J Gen Physiol. 2008;131:473–481.
23. Cognard C, Rivet-Bastide M, Constantin B, Raymond G. Progressive predominance of ‘skeletal’ versus ‘cardiac’ types of excitation-contraction coupling during in vitro skeletal myogenesis. Pflügers Arch. 1992;422:207–209.
24. Conway EJ. Nature and significance of concentration relations of potassium and sodium ions in skeletal muscle. Physiol Rev. 1957;37:84–132.
25. Cordoba-Rodriguez R, Gonzalez-Serratos H, Matteson DR, Rozycka M. Ica is important in E-C coupling in developing cultured embryonic amphibian skeletal muscle cells. Biophys J. 1997;72:A119 H5.
26. Cordoba-Rodriguez R, Matteson DR, Gonzalez-Serratos H. Embryonic E-C coupling in cultured skeletal muscle cells. Biophys J. 1996;70:A390.
27. Costantin LL. The role of sodium current in the radial spread of contraction in frog muscle fibers. J Gen Physiol. 1970;55:703–715.
28. Costantin LL, Podolsky RJ. Depolarization of the internal membrane system in the activation of frog skeletal muscle. J Gen Physiol. 1967;50:1101–1124.
29. Costantin LL, Taylor SR. Graded activation in frog muscle fibers. J Gen Physiol. 1973;61:424–443.
30. Curtis RA. Ca fluxes in single twitch muscle fibers. J Gen Physiol. 1966;50:225–267.
31. Davies NW, Standen NB, Stanfield PR. The effect of intracellular pH on ATP-dependent potassium channels of frog skeletal muscle. J Physiol. 1992;445:549–568.
32. Dawson MJ, Gadian DG, Wilkie DR. Muscular fatigue investigated by phosphorus nuclear magnetic resonance. Nature. 1978;274:861–866.
33. Dawson MJ, Gadian DG, Wilkie DR. Mechanical relaxation rate and metabolism studied in fatiguing muscle by phosphorus nuclear magnetic resonance. J Physiol. 1980;299:465–484.
34. DeSantiago J, Batlle D, Khilnani M, et al. Ca2+/H+ exchange via the plasma membrane Ca2+ ATPase in skeletal muscle. Front Biosci. 2007;12:4641–4660.
35. DiFranco M, Capote J, Vergara JL. Optical imaging and functional characterization of the transverse tubular system of mammalian muscle fibers using the potentiometric indicator di-8-ANEPPS. J Memb Biol. 2005;208:141–153.
36. Dudel J, Kuffler SW. Presynaptic inhibition at the crayfish neuromuscular junction. J Physiol. 1961;155:543–562.
37. Eisenberg RS, Gage PW. Ionic conductances of the surface and transverse tubular membranes of frog sartorius fibers. J Gen Physiol. 1969;53:279–297.
38. Fabiato A. Mechanism of calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cardiac cells studied with potential-sensitive dyes. In: Ohnishi ST, Endo M, eds. The Mechanism of Gated Calcium Transport Across Biological Membranes. New York: Academic Press; 1982;:237–255.
39. Fahlke C, Zachar E, Rudel R. Chloride channels with reduced single-channel conductance in recessive myotonia congenita. Neuron. 1993;10:225–232.
40. Fatt P, Ginsborg BL. The ionic requirements for the production of action potentials in crustacean muscle fibers. J Physiol (London). 1958;142:156–543.
41. Fatt P, Katz B. The electrical properties of crustacean muscle. J Physiol. 1953;120:171–204.
42. Fink R, Lüttgau HC. An evaluation of the membrance constants and the potassium conductance in metabolically exhausted muscle fibers. J Physiol. 1976;263:215–238.
43. Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. Muscle KATP channels: Recent insights to energy sensing and myoprotection. Physiol Rev. 2010;90:799–829.
44. Flucher BE, Takekura H, Franzini-Armstrong C. Development of the excitation-contraction coupling apparatus in skeletal muscle: association of sarcoplasmic reticulum and transverse tubules with myofibrils. Dev Biol. 1993;160:135–147.
45. Forbes MS, Sperelakis N. Ruthenium red staining of skeletal and cardiac muscles. Z Zellforsch Cell Tissue Res. 1979;200:367–382.
46. Garcia Mdel C, Gonzalez-Serratos H, Morgan JP, Perreault CL, Rozycka M. Differential activation of myofibrils during fatigue in phasic skeletal muscle cells. J Musc Res Cell Motil. 1991;12:412–424.
47. Gonzalez-Serratos H. Inward spread of activation in vertebrate muscle fibers. J Physiol (London). 1971;212:777–799.
48. Gonzalez-Serratos H. Graded activation of myofibrils and the effect of diameter on tension development during contractures in isolated skeletal muscle fibers. J Physiol (London). 1975;253:321–339.
49. Gonzalez-Serratos H, Cordoba-Rodriguez R, Matteson DR, Rozycka M. Role of calcium currents in excitation-contraction coupling in developing cultured embryonic amphibian skeletal muscle cells. J Physiol, 1996;:494P.
50. Gonzalez-Serratos H, Somlyo AV, McClellan G, Shuman H, Borrero LM, Somlyo AP. Composition of vacuoles and sarcoplasmic reticulum in fatigued muscle: electron probe analysis. Proc Natl Acad Sci USA. 1978;75:1329–1333.
51. Gonzalez-Serratos H, Valle-Aguilera R, Lathrop DA, Garcia Mdel. Slow inward calcium currents have no obvious role in muscle excitation-contraction coupling. Nature. 1982;298:292–294.
52. Hagiwara S, Naka K. The initiation of spike potential in barnacle-muscle fibers under low intracellular Ca++. J Gen Physiol. 1964;48:141–162.
53. Hagiwara S, Nakajima S. Difference in Na and Ca spikes as examined by application of tetrodotoxin, procaine, and manganese ions. J Gen Physiol. 1966;49:793–806.
54. Hagiwara S, Chichibu S, Naka K. The effect of various ions on resting and spike potentials of barnacle muscle fibers. J Gen Physiol. 1964;43:163–179.
55. Harris EJ. Distribution and movement of muscle chloride. J Physiol (London). 1963;166:87–109.
56. Heiny JA, Ashcroft FM, Vergara J. T-system optical signals associated with inward rectification in skeletal muscle. Nature. 1983;301:164–166.
57. Hen ek M, Nonner W, Stämpfli R. Voltage clamp of a small muscle membrane area by means of a circular sucrose gap arrangement. Pflügers Arch. 1969;313:71–79.
ek M, Nonner W, Stämpfli R. Voltage clamp of a small muscle membrane area by means of a circular sucrose gap arrangement. Pflügers Arch. 1969;313:71–79.
58. Hille B, Campbell DT. An improved vaseline gap voltage clamp for skeletal muscle fibers. J Gen Physiol. 1976;67:265–293.
59. Hodgkin AL, Horowicz P. The influence of potassium and chloride ions on the membrane potential of single muscle fibers. J Physiol (London). 1959;148:127–160.
60. Hodgkin AL, Huxley AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol (London). 1952;116:449–472.
61. Hodgkin AL, Nakajima S. Analysis of the membrance capacity in frog muscle. J Physiol (London). 1972;221:121–136.
62. Howell JN. Intracellular binding of ruthenium red in frog skeletal muscle. J Cell Biol. 1974;62:242–247.
63. Hoyle G, Burrows M. Correlated physiological and ultrastructural studies on specialized muscles 3 neuromuscular physiology of the power-stroke muscle of the swimming leg of portunus sanguinolentus. J Exp Zool. 1973;185:83–95.
64. Hoyle G, Smith T. Neuromuscular physiology of giant muscle fibres of a barnacle, Balanus nubilus Darwin. Comp Biochem Physiol. 1963;10:219–314.
65. Hoyle G, Wiersma CAG. Excitation at neuromuscular junctions in Crustacea. J Physiol. 1958;143 493–425.
66. Hurnak O, Zachar J. Maxi chloride channels in L6 myoblasts. Gen Physiol Biophys. 1992;11:389–400.
67. Hussain M, Wareham AC, Head SI. Mechanism of action of a K+ channel activator BRL 38227 on ATP-sensitive K+ channels in mouse skeletal muscle fibres. J Physiol., 478 Pt. 1994;3:523–532.
68. Huxley AF, Taylor RE. Local activation of striated muscle fibres. J Physiol (London). 1958;144:426–441.
69. Huxley HE, Page S, Wilkie DR. Appendix An electron microscopic study of muscle in hypertonic solutions. J Physiol (London). 1963;169:312–329.
70. Iannaccone ST, Li K-X, Sperelakis N, Lathrop DA. Insulin-induced hyperpolarization in mammalian skeletal muscle. Am J Physiol. 1989;256:C368–C374.
71. Jiang C, Qu Z, Xu H. Gating of inward rectifier K(+) channels by proton-mediated interactions of intracellular protein domains. Trends Cardiovasc Med. 2002;12:5–13.
72. Kakei M, Noma A, Shibasaki T. Properties of adenosine-triphosphate-regulated potassium channels in guinea-pig ventricular cells. J Physiol. 1985;363:441–462.
73. Kennedy D, Evoy WH, Fields. HL. The unit bases of some crustacean reflexes. Symp Soc Exp Biol. 1966;20:75–109.
74. Kerr LM, Sperelakis N. Effects of the calcium antagonists verapamil and bepridil (CERM-1978) on Ca2+-dependent slow action potentials in frog skeletal muscle. J Pharmacol Exp Ther. 1982;222:80–86.
75. Keynes RD, Steinhardt RA. The components of the sodium efflux in frog muscle. J Physiol (London). 1968;198:581–599.
76. Khan AR. Influence of ethanol and acetaldehyde on electromechanical coupling of skeletal muscle fibres. Acta Physiol Scand. 1981;111:425–430.
77. Kirby AC, Lindley BD, Picken JR. Calcium content and exchange in frog skeletal muscle. J Physiol (London). 1975;253:37–52.
78. Kulczycky S, Mainwood GW. Evidence for a functional connection between the sarcoplasmic reticulum and the extracellular space in frog sartorius muscle. Can J Physiol Pharmacol. 1972;50:87–98.
79. Li K-X, Sperelakis N. Electrogenic Na-K pump current in rat skeletal myoballs. J Cell Physiol. 1994;159:181–186.
80. Lueck JD, Rossi AE, Thornton CA, Campbell KP, Dirksen RT. Sarcolemmal-restricted localization of functional ClC-1 channels in mouse skeletal muscle. J Gen Physiol. 2010;136:597–613.
81. Lynch III C. Kinetic and biochemical separation of potassium currents in frog striated muscle PhD thesis. New York: University of Rochester; 1978.
82. Mathias RT, Levis RA, Eisenberg RS. Electrical models of excitation-contraction coupling and charge movement in skeletal muscle. J Gen Physiol. 1980;76:1–31.
83. Miledi R, Parker RI, Schalow G. Measurement of calcium transients in frog muscle by the use of arseno III. Proc R Soc London (Biol). 1977;198:201–210.
84. Miledi R, Stefani E, Steinbach AB. Induction of the action potential mechanism in slow muscle fibres of the frog. J Physiol (London). 1971;217:737–754.
85. Mobley BA, Eisenberg BR. Sizes of components in frog skeletal muscle measured by methods of stereology. J Gen Physiol. 1975;66:31–45.
86. Moody-Corbett F, Gilbert R, Akbarali H, Hall J. Calcium current in embryonic Xenopus muscle cells in culture. Can J Physiol Pharmacol. 1989;67:1259–1264.
87. Nakajima S, Gilai A. Action potentials of isolated single muscle fibers recorded by potential-sensitive dyes. J Gen Physiol. 1980;76:729–750.
88. Nassar-Gentina V, Passonneau JV, Vergara JL, Rapoport SI. Metabolic correlates of fatigue and of recovery from fatigue in single frog muscle fibers. Gen Physiol. 1978;72:593–606.
89. Nastuk WL, Hodgkin AL. The electrical activity of single muscle fibres. J Cell Comp Physiol. 1950;35:39–74.
90. Natori R. Propagated contractions in isolated sarcolemma-free bundle of myofibrils. Jikeidai Med J. 1965;12:214–221.
91. Nicola-Siri L, Sanchez JA, Stefani E. Effect of glycerol treatment on calcium current of frog skeletal muscle. J Physiol (London). 1980;305:87–96.
92. Ortega A, Gonzalez-Serratos H, Lepock J. Effect of organic calcium channel blocker D-600 on sarcoplasmic reticulum calcium uptake in skeletal muscle. Am J Physiol. 1997;272:C310–C317.
93. Overton E. Beitrage zur allgemeinen muskel- und nervenphysiologie I Abh I Ueber die osmotischen figenschatten der muskeln. Pflugers Arch. 1902;92:115–280.
94. Peachey LD. The sarcoplasmic reticulum and transverse tubules of the frog’s sartorius. J Cell Biol. 1965;25:209–231.
95. Peachey LD, Huxley AF. Transverse tubules in crab muscles. J Cell Biol. 1964;23:70A.
96. Pilgrim RIC, Wiersma CAG. Observations on the skeletal and somatic musculature of the abdomen and thorax of Procambaru clarkii (Girard) with notes on the thorax of Panulirus interruptus and Astacus. J Morphol. 1963;113:453–487.
97. Podolsky RJ, Costantin LL. Regulation by calcium of the contraction and relaxation of muscle fibers. Fed Proc. 1964;23:933–939.
98. Potreau D, Raymond G. Calcium-dependent electrical activity and contraction of voltage-clamped frog single muscle fibers. J Physiol (London). 1980;307:9–22.
99. Qu Z, Yang Z, Cui N, et al. Gating of inward rectifier K+ channels by proton-mediated interactions of N- and C-terminal domains. J Biol Chem. 2000;275:31573–31580.
100. Rasgado-Flores H, DeSantiago J, Espinosa-Tanguma R. Stoichiometry and regulation of the Na-Ca exchanger in barnacle muscle cells. Ann NY Acad Sci. 1991;639:22–33.
101. Rasgado-Flores H, Santiago EM, Blaustein MP. Kinetics and stoichiometry of coupled Na efflux and Ca influx (Na/Ca exchange) in barnacle muscle cells. J Gen Physiol. 1989;93:1219–1241.
102. Renaud JM. The effect of lactate on intracellular pH and force recovery of fatigued sartorius muscles of frog, Rana pipiens. J Physiol. 1989;416:31–47.
103. Rogus E, Zierler KL. Sodium and water contents of sarcoplasm and sarcoplasmic reticulum in rat skeletal muscle: Effects of anisotonic media, ouabain, and external sodium. J Physiol (London). 1973;233:227–270.
104. Rubio R, Sperelakis N. Penetration of horseradish peroxidase into the terminal cisternae of frog skeletal muscle fibers and blockade of caffeine contracture by Ca++ depletion. Z Zellforsch. 1972;124:57–71.
105. Rulon R, Hermsmeyer K, Sperelakis N. Regenerative action potentials induced in the neurogenic heart of Limulus polyphemus. Comp Biochem Physiol. 1971;39A:333–335.
106. Sanchez JA, Stefani E. Inward calcium current in twitch muscle fibers of the frog. J Physiol (London). 1978;283:197–209.
107. Sauviat M-P, Ecault E, Faivre J-F, Finlay I. Activation of ATP-sensitive K channels by a K channel opener (SR 44866) and the effect upon electrical and mechanical activity of frog skeletal muscle. Pflügers Arch. 1991;418:261–265.
108. Sellin LC, Sperelakis N. Decreased potassium permeability in dystrophic mouse skeletal muscle. Exp Neurol. 1978;62:609–617.
109. Spector I, Prives JM. Development of electrophysiological and biochemical membrane properties during differentiation of embryonic skeletal muscle in culture. Proc Natl Acad Sci USA. 1977;74:5166–5170.
110. Sperelakis N. Changes in conductance of frog sartorius fibers produced by CO2, ReO4, and temperature. Am J Physiol. 1969;217:1069–1075.
111. Sperelakis N. Origin of the cardiac resting potential. In: Berne R, Sperelakis N, eds. Handbook of Physiology, the Cardiovascular System, Vol 1: the Heart. Bethesda: American Physiological Society; 1979;:187–267.
112. Sperelakis N, Gonzalez-Serratos H. Skeletal muscle action potentials. In: Sperelakis N, ed. Cell Physiology Sourcebook. San Diego: Academic Press; 2001;:865–886.
113. Sperelakis N, Mayer G, Macdonald R. Velocity of propagation in vertebrate cardiac muscles as functions of tonicity and [K+]. Am J Physiol. 1970;219:952–963.
114. Sperelakis N, Rubio R. Ultrastructural changes produced by hypertonicity in cat cardiac muscle. J Mol Cell Cardiol. 1971;3:139–156.
115. Sperelakis N, Schneider MF. Membrane ion conductances of frog sartorius fibers as a function of tonicity. Am J Physiol. 1968;215:723–729.
116. Sperelakis N, Forbes MS, Rubio R. The tubular systems of myocardial cells: ultrastructure and possible function. In: Dhalla NS, Rona G, eds. Recent Advances in Studies on Cardiac Structure and Metabolism, Myocardial Biology, Vol 4. Baltimore: University Park Press; 1974;:163–194.
117. Sperelakis N, Schneider MF, Harris EJ. Decreased K+ conductance produced by Ba++ in frog sartorius fibers. J Gen Physiol. 1967;50:1565–1583.
118. Sperelakis N, Shigenobu K, Rubio R. 3H-Sucrose compartments in frog skeletal muscle relative to sarcoplasmic reticulum. Am J Physiol. 1978;234:C181–C190.
119. Sperelakis N, Valle R, Orozco C, Martinez-Palomo A, Rubio R. Electromechanical uncoupling of frog skeletal muscle by possible change in sarcoplasmic reticular content. Am J Physiol. 1973;225:793–800.
120. Spitzer NC. Ion channels in development. Annu Rev Neurosci. 1979;2:363–397.
121. Spruce AE, Standen NB, Stanfield PR. Voltage-dependent ATP-sensitive potassium channels of skeletal muscle membrane. Nature. 1985;316:736–738.
122. Spruce AE, Standen NB, Standfield PR. Studies of the unitary properties of adenosine-5′-triphospate-regulated potassium channels of frog skeletal muscle. J Physiol. 1987;382:213–236.
123. Standen NB, Stanfield PR, Ward TA. Properties of single potassium channels in vesicles formed from the sarcolemma of frog skeletal muscle. J Physiol. 1985;364:339–358.
124. Stanfield PR. A calcium-dependent inward current in frog skeletal muscle fibers. Pflügers Arch. 1977;368:267–270.
125. Stephenson EW. Activation of fast skeletal muscle: contributions of studies on skinned fibers. Am J Physiol. 1981;240:C1–19.
126. Steinmeyer K, Ortland C, Jentsch TJ. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature. 1991;354:301–304.
127. Stoller D, Kakkar R, Smelley M, et al. Mice lacking sulfonylurea receptor 2 (SUR2) ATP-sensitive potassium channels are resistant to acute cardiovascular stress. J Mol Cell Cardiol. 2007;43:445–454.
128. Tasker P, Simon SE, Johnstons BM, Shankly KH, Shaw FH. The dimensions of the extracellular space in sartorius muscle. J Gen Physiol. 1959;43:39–53.
129. Thabet M, Miki T, Seino S, Renaud JM. Treadmill running causes significant fiber damage in skeletal muscle of KATP channel-deficient mice. Physiol Genomics. 2005;22:204–212.
130. Vinogradova NA. Distribution of nonpenetrating sugars in the frog’s sartorius muscle under hypo- and hypertonic conditions. Tsitologiya. 1968;10:831–838.
131. Vivaudou MB, Arnoult C, Villaz M. Skeletal muscle ATP-sensitive K+ channels recorded from sarcolemmal blebs of split fibers: ATP inhibition is reduced by magnesium and ADP. J Membr Biol. 1991;122:165–175.
132. Vogel S, Harder D, Sperelakis N. Ca++ dependent electrical and mechanical activities in skeletal muscle. Fed Proc. 1978;37:517.
133. Wallace N, Sommer JR. Fusion of sarcoplasmic reticulum with ruthenium red. Proc Electron Microsc Soc A. 1975;33:500–501.
134. Weiss DS, Magleby KL. Voltage-dependent gating mechanism for single fast chloride channels from rat skeletal muscle. J Physiol (London). 1992;453:279–306.
135. Werman R, Grundfest H. Graded and all-or-none electrogenesis in arthropod muscle II The effects of alkali-earth and strontium ions on lobster muscle fibers. J Gen Physiol. 1961;44:997–1027.
136. Werman R, McCann FV, Grundfest H. Graded and all-or-none electrogenesis in arthropod muscle I The effects of alkali-earth cations on the neuromuscular system of Romalea microptera. J Gen Physiol. 1961;44:979–995.
137. Winegrad S. Intracellular calcium movements of frog skeletal muscle during recovery from tetanus. J Gen Physiol. 1968;51:65–83.
138. Woll KH, Lonnendonker U, Neumcke B. ATP-sensitive potassium channels in adult mouse skeletal muscle: different modes of blockage by internal cations, ATP, and tolbutamide. Pflügers Arch. 1989;414:622–628.
139. Yokoshiki H, Sunagawa M, Seki T, Sperelakis N. ATP-sensitive K+ channels in pancreatic, cardiac, and vascular smooth muscle cells Invited Review. Am J Physiol. 1998;274:C25–C37.
140. Zifarelli G, Pusch M. CLC chloride channels and transporters: A biophysical and physiological perspective. Rev Physiol Biochem Pharmacol. 2007;158:23–76.
1 To produce glycerol osmotic shock, about 300 mOsm glycerol is added to Ringer’s solution (Eisenberg and Gage, 1969). The glycerol rapidly permeates into the fiber interior (so the fiber shrinks transiently) and equilibrates. But when the glycerol is washed out, there is a great hypotonic shock produced that disrupts the T-tubules.
2 In hypertonic solutions, skeletal muscle fibers shrink (fiber diameter decreases) like a perfect osmometer (but with an osmotically-inactive volume of about 32%), but their T-tubules swell.