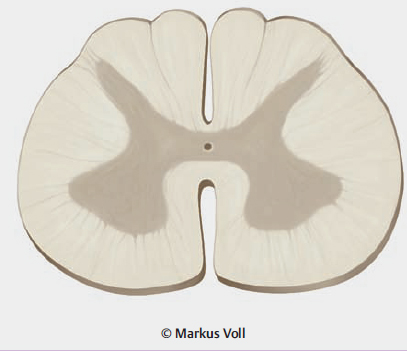
7.3 Slowly Progressive Spinal Cord Compression
7.4 Spinal Cord Ischemia and Hemorrhage
7.5 Infectious and Inflammatory Diseases of the Spinal Cord
7.6 Diseases Mainly Affecting the Long Tracts of the Spinal Cord
7.3 Slowly Progressive Spinal Cord Compression
7.4 Spinal Cord Ischemia and Hemorrhage
7.5 Infectious and Inflammatory Diseases of the Spinal Cord
7.6 Diseases Mainly Affecting the Long Tracts of the Spinal Cord
A Fall Brings the Answer
The patient, a 74-year-old retiree, had never been seriously ill before. Two years before admission, he began to have trouble walking. His legs felt stiff and he could not lift his feet fully off the ground, so that he shuffled while walking and sometimes fell. He thought this might be normal for his age and did not seek medical attention. He secretly feared he might have Parkinson disease, of which his father had died at age 75 years.
His gait rapidly worsened in the months before admission. He could no longer feel the soles of his feet when walking barefoot, and he had a strange sensation of walking on cotton. He fell more often. He also developed more frequent minor injuries on his feet, some of which he failed to notice. Finally, a fall led to a comminuted forearm fracture, and he was admitted to the hospital.
The orthopaedists repaired the fracture surgically and consulted a neurologist, who took a thorough history and performed a neurologic examination. The patient said that he had fallen more often in recent months and also (in response to a specific question) that he had sometimes failed to get to the toilet on time. His bladder control was poor at the moment, “but, after all, I have prostate trouble, too.” Further questioning brought out the family history of Parkinson disease.
On examination, there were no abnormalities of the head, cranial nerves, spine, or upper limbs, and there was no rigidity or tremor. Rapid alternating movements were dexterous. There was, however, a marked diminution of sensation to light touch and, to a lesser extent, of pain, from the level of the umbilicus downward. The lower limb extensors were spastically hypertonic. The knee- and ankle-jerk reflexes were brisk, with pathologic spreading. Babinski signs were present bilaterally.
The patient’s symptoms, age, and family history had made the orthopaedists think of Parkinson disease. The classic triad of hypokinesia/akinesia, rigidity, and tremor was missing, but repeated falls are indeed common in atypical parkinsonian syndromes. Yet the neurologic examination clearly showed that the cause had to lie elsewhere. The neurologist diagnosed an incomplete spinal cord transection syndrome at the T8 level. A sensory level (i.e., a band around the trunk below which multiple, or all, sensory modalities are impaired), spastic paraparesis with gait impairment, and bladder dysfunction are all typical findings of an incomplete spinal cord transection syndrome.
The slow progression of the deficits led the neurologist to suspect a slowly growing, and therefore probably benign, tumor compressing the spinal cord. The magnetic resonance imaging (MRI) scan that she ordered revealed the tumor, which was then completely resected by a neurosurgeon. Within a few months, the patient’s deficits resolved completely.
Key Point
Diseases of the spinal cord have manifestations in the limbs and trunk and sometimes affect micturition, defecation, and sexual function. Their symptoms and signs depend on the level of the lesion and the affected structures—conducting pathways, anterior horn cells, or the entire cross-sectional extent of the spinal cord.
The spinal cord is the component of the central nervous system (CNS) that connects the brain to the peripheral nerves; its anterior horn cells, though located in the CNS, are functionally a part of the peripheral nervous system. The spinal cord contains the following:
In the white matter, fiber pathways leading from the brain to the periphery and vice versa.
In the gray matter, an intrinsic neuronal system consisting of:
Interneurons, that is, relay neurons for the conducting pathways and reflex loops.
Motor neurons in the anterior horns.
Autonomic neurons in the lateral horns.
The somatosensory neurons are located outside the spinal cord, in the spinal ganglia.
The topographic relations of the spinal cord, vertebral column, and nerve roots are shown in ▶ Fig. 7.1, and the major ascending and descending pathways of the spinal cord are shown in ▶ Fig. 7.2. The blood supply of the spinal cord is described later.
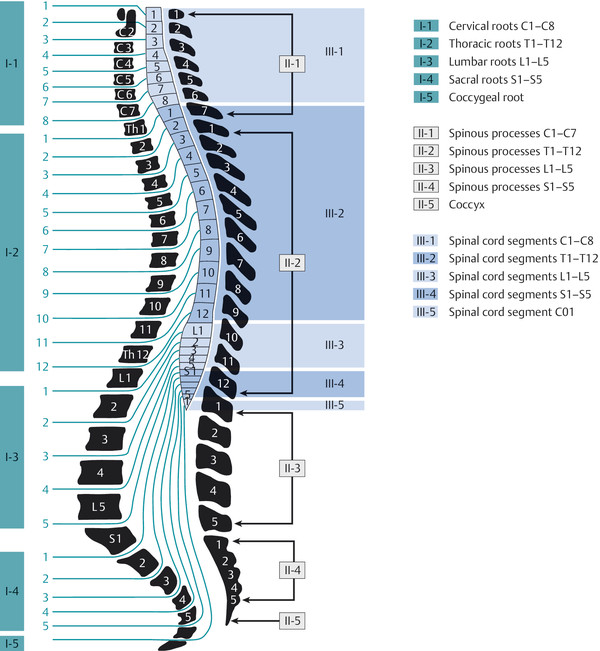
Fig. 7.1 Topographic relations of the vertebral column and nerve roots to the spinal cord. The growth of the spinal cord during embryonic development lags behind that of the spinal column; therefore, more caudally lying nerve roots traverse greater distances in the spinal subarachnoid space to reach their exit foramina. In the conventional numbering system, the cervical spinal nerves exit the spinal canal above the correspondingly numbered vertebra, while all spinal nerves from T1 downward exit below the correspondingly numbered vertebra. The conus medullaris generally ends at the upper L1 vertebral level but can also end more caudally, sometimes as far down as L3.
Key Point
The clinical manifestations of spinal cord lesions depend on their level and extent. Thus, the first step in diagnostic evaluation is topographic localization, that is, the deduction of the level and extent of the lesion from the patient’s constellation of neurologic deficits. The next step is the determination of the etiology, usually based on further criteria (accompanying nonneurologic manifestations, temporal course, findings of ancillary tests).
Note
Spinal cord transection syndrome is the pattern of neurologic deficits resulting from damage of the entire cross-section of the spinal cord at some level. In an incomplete spinal cord transection syndrome, only part of the cord is damaged.
Most acutely arising cases of spinal cord transection syndrome are of either traumatic or ischemic origin; rare cases are due to inflammation or infection (transverse myelitis) or nontraumatic compression (e.g., by a hematoma or tumor).
The clinical features of the spinal cord transection syndrome are:
Dysfunction of all of the ascending sensory pathways up to the level of the lesion, and of the posterior horns and posterior roots at the level of the lesion: there is a sensory level below which all sensory modalities of sensation are impaired or absent.
Bilateral pyramidal tract dysfunction: spastic paraparesis or paraplegia, or, with cervical lesions, spastic quadriparesis or quadriplegia (immediately after a trauma, in the phase of “spinal shock” [diaschisis], there is usually flaccid weakness, which subsequently becomes spastic).
Bladder dysfunction.
Dysfunction of the motor neurons of the anterior horn at the level of the lesion: possibly, flaccid paresis in the myotome(s) supplied by the cord at the level of the lesion, corresponding loss of reflexes and, later, muscle atrophy.
Practical Tip
Partial and incomplete transection syndromes are more common than complete transection syndrome.
To understand the pathogenesis of spinal cord syndromes, one needs to know the anatomic course of the pyramidal tract (cf. ▶ Fig. 5.1) and of the sensory afferent pathways (the posterior columns and lateral spinothalamic tract, cf. ▶ Fig. 5.2). Only the essential structures for clinical purposes are shown in ▶ Fig. 7.2.
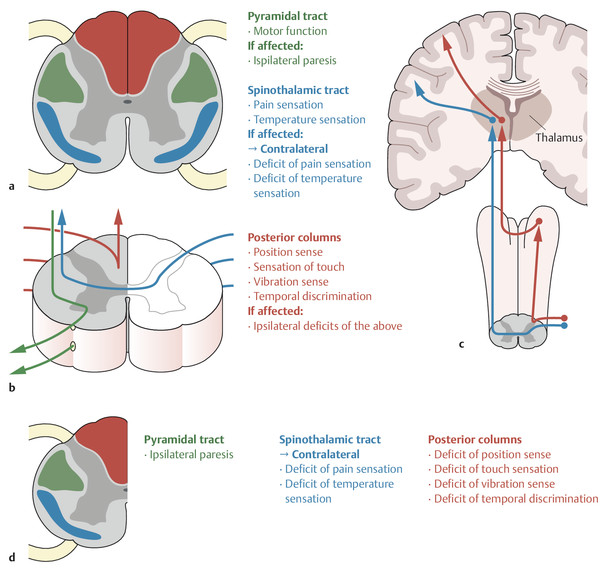
Fig. 7.2 Major fiber tracts of the spinal cord and the consequences of hemisection. a Clinically important structures are shown in cross-section, with color-coded descriptions of function. b The course of the nerve fibers and pathways, superimposed on a cross-sectional image. c The course of the somatosensory fibers and pathways in a coronal longitudinal section from the posterior root to the cortex. d Diagram of Brown-Séquard syndrome caused by hemisection of the spinal cord on the right side at one level. Strength, sensation to touch, position sense, and vibration sense are impaired ipsilaterally from the level of the lesion downward; pain and temperature sensation are impaired bilaterally at the level of the lesion and contralaterally below it.
Note
As the pyramidal tract descends, it crosses in the medulla and then travels down the anterolateral column of the spinal cord. A lesion of the pyramidal tract in the spinal cord causes ipsilateral paresis.
The somatosensory afferent fibers in the posterior columns ascend ipsilaterally and terminate in the nucleus gracilis and nucleus cuneatus of the medulla. A lesion of the posterior columns thus impairs fine sensation (touch, stereognosis), proprioception, and vibration sense ipsilaterally ( ▶ Fig. 7.2).
Nociceptive fibers from the periphery enter the spinal cord through the dorsal roots, cross to the other side on the same segmental level in the anterior commissure of the spinal cord, and then ascend in the lateral spinothalamic tract ( ▶ Fig. 7.2). A lesion impairs pain and temperature sensation contralaterally.
See also the overview of the anatomic basis of sensory and motor function in Chapter ▶ 5.
The symptoms and signs of spinal cord hemisection are described in ▶ Table 7.1 (see also ▶ Fig. 7.2b). An anatomic or functional disconnection of one half of the spinal cord exactly to the midline is a rare event. Incomplete unilateral lesions present with a subset of these manifestations; the most common type of unilateral lesion preferentially affects the anterolateral column.
|
Involved structure |
Deficits |
|
Ipsilateral deficits |
|
|
Pyramidal tract |
Paresis |
|
Vasomotor fibers of the lateral columns |
Initially, warmth and redness of the skin; sometimes absence of sweating |
|
Posterior columns |
Loss of proprioception and vibration sense, finely localized sensation of touch and pressure, two-point discrimination |
|
Anterior horns and anterior roots |
Segmental muscular atrophy and flaccid weakness |
|
Entering posterior roots |
Segmental anesthesia and analgesia |
|
Contralateral deficits |
|
|
Lateral spinothalamic tract |
Contralateral diminution or loss of pain and temperature sensation (dissociated sensory deficit) |
Note
Central cord syndrome reflects damage to the decussating nociceptive fibers in the anterior commissure of the spinal cord (second neuron of the lateral spinothalamic tract). A dissociated sensory deficit is found in the corresponding segment(s).
Central cord syndrome is the classic presentation of syringomyelia (see ▶ Fig. 7.11) but can also be due to an intramedullary hemorrhage or tumor. Its clinical features are:
Pyramidal tract dysfunction: spasticity of the limbs below the level of the lesion; cervical lesions tend to affect the upper limbs more than the lower limbs.
Interruption of the pain and temperature fibers of the anterior spinal commissure: bilateral impairment of pain and temperature sensation at the level of the lesion, with preservation of touch (segmentally restricted dissociated sensory deficit); in analogous fashion, concomitant involvement of the posterior horn(s), if present, causes segmental impairment of touch sensation, either uni- or bilaterally, depending on whether one or both posterior horns are affected.
Dysfunction of the lateral horn/intermediolateral tract: autonomic and trophic disturbances (disturbances of sweating, nail growth, and bone metabolism; hyperkeratosis and edema; all disturbances more pronounced in the upper limbs).
Possible concomitant involvement of the spinothalamic tracts: (bilateral) deficit of pain and temperature sensation below the level of the lesion, with intact touch sensation.
Possible concomitant involvement of the motor neurons of the anterior horns at the level of the lesion: segmental flaccid weakness, loss of reflexes, and muscle atrophy.
Sparing of the posterior horns and spinocerebellar tracts: touch, vibration sense, and proprioception usually remain intact.
Bladder dysfunction.
In this situation, the posterior columns are intact, and there is thus no impairment of sensation to touch, proprioception, or vibration sense.
The most common cause is ischemia or infarction in the territory of the anterior spinal artery (see section ▶ 7.4.2).
Pyramidal tract dysfunction: depending on the level of the lesion, spastic quadriparesis (quadriplegia) or paraparesis (paraplegia), with enhanced intrinsic muscle reflexes and pyramidal tract signs.
Involvement of the spinothalamic tracts and the pain and temperature fibers crossing in the anterior spinal commissure: dissociated sensory deficit in the entire region of the body at and below the level of the lesion; less commonly, the spinothalamic tracts are spared and there is a segmentally restricted dissociated sensory deficit.
Intact posterior columns: no impairment of touch or proprioception.
Bladder dysfunction.
Note
Sensations to touch, proprioception, and vibration sense are impaired, but the anterolateral columns are intact and there is thus no deficit of muscle strength or of pain and temperature sensation. The patient’s stance and gait are markedly ataxic.
A typical example is funicular myelosis due to vitamin B12 deficiency (see ▶ 7.6.3.1).
Below the lesion, impaired proprioception, vibration sense, fine touch and pressure sensation, and two-point discrimination.
Sensory ataxia that worsens in the dark or when the patient’s eyes are closed; positive Romberg test.
Sometimes, the is present (see section ▶ 8.2).
Note
In these situations, only some of the ascending or descending long tracts (or a single one) are affected.
The clinical syndromes vary correspondingly and include:
Pure spastic paraparesis (isolated lesion of the pyramidal tracts, e.g., in spastic spinal paralysis).
Impaired touch and position sense (posterior column lesion).
Ataxia (lesion of the spinocerebellar tracts and/or posterior columns).
Combinations of the above (e.g., pyramidal tract and posterior column dysfunction in funicular myelosis, dysfunction of these tracts and the spinocerebellar tracts in Friedreich ataxia).
Note
Isolated involvement of the motor anterior horn cells causes weakness and atrophy without any sensory deficit.
This group of diseases includes spinal muscular atrophy and acute poliomyelitis and is characterized by:
Flaccid weakness of various muscles and muscle atrophy (as well as fasciculations in chronic processes).
Diminution or loss or reflexes.
No sensory impairment or bladder dysfunction.
Note
For example, muscle atrophy may be combined with pyramidal tract dysfunction.
This type of problem is seen, for example, in amyotrophic lateral sclerosis (ALS; see section ▶ 7.7.3), with simultaneous involvement of the anterior horn ganglion cells and of the pyramidal and corticobulbar tracts due to upper motor neuron degeneration. The patient has weakness, muscle atrophy, and fasciculations, but also brisk reflexes.
Note
This syndrome affects the segment of the spinal cord above the conus medullaris, that is, levels T11 and/or T12.
Sacral and (sometimes) lumbar spinal functional deficits are seen, including:
Weakness of the muscles supplied by the affected lumbar and sacral segments.
Partial or total sensory loss in the affected lumbar and sacral dermatomes.
Usually, diminished lower limb reflexes (because the lesion destroys the sensory arm of the reflex loop as well as its motor arm); the Babinski sign is usually absent.
Bladder dysfunction.
Bowel dysfunction, sphincter weakness.
Sexual dysfunction.
Practical Tip
Epiconus syndrome is clinically indistinguishable from cauda equina syndrome ( ▶ Fig. 7.3).
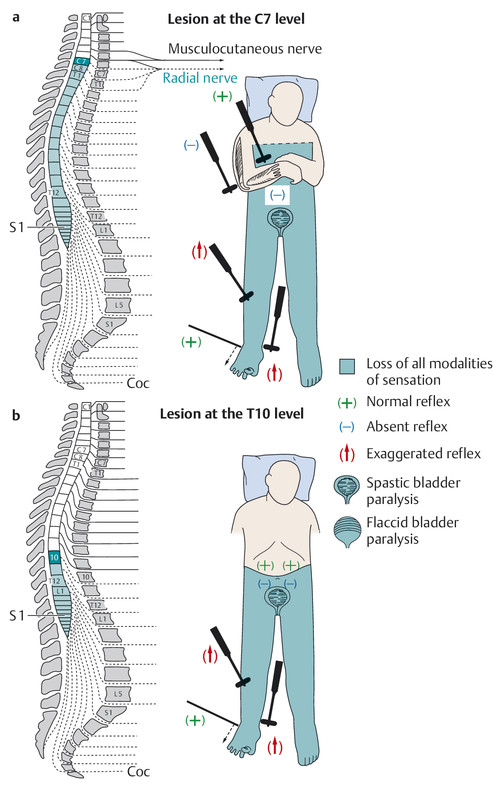
Fig. 7.3 Neurologic deficits resulting from spinal cord transection at various levels. Regarding the position of the conus medullaris, cf. legend to ▶ Fig. 7.1. a lesion at C7; b lesion at T10.
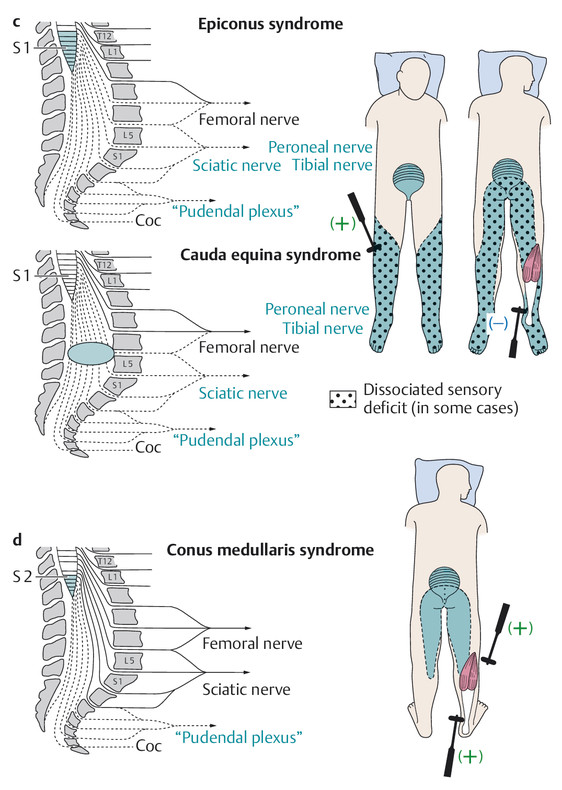
c epiconus syndrome and cauda equina syndrome, d conus medullaris syndrome.
Note
The conus medullaris is the lower end of the spinal cord and lies in the spinal canal at the L1 level ( ▶ Fig. 7.3).
An isolated conus lesion typically produces:
Bladder dysfunction.
Bowel dysfunction with sphincter weakness.
Sexual dysfunction.
Possibly a dissociated sensory deficit or complete loss of sensation in the cutaneous distribution of the sacral and coccygeal spinal cord segments (saddle anesthesia).
usually, intact motor function and absence of pyramidal tract signs.
Note
Cauda equina syndrome ( ▶ Fig. 7.3) results from compression of the nerve roots coursing through the spinal canal below the conus medullaris, that is, below the L1 level. Unlike conus medullaris syndrome, it involves a variably severe impairment of sensory and motor function in the lower limbs (see also Radicular Syndromes, section ▶ 13.1).
Its clinical manifestations are:
Flaccid weakness and areflexia of the lower limbs, without pyramidal tract signs.
Impairment of all sensory modalities in multiple lumbar and/or sacral dermatomes, usually most pronounced in the “saddle” area.
Impaired urination, defecation, and sexual function, with sphincter weakness.
An epiconus lesion can present with the same signs and symptoms as the cauda equina syndrome.
Practical Tip
A complete cauda equina syndrome can arise as the result of massive posteromedial intervertebral disk herniation ( ▶ Fig. 7.4). This condition demands immediate surgical treatment.
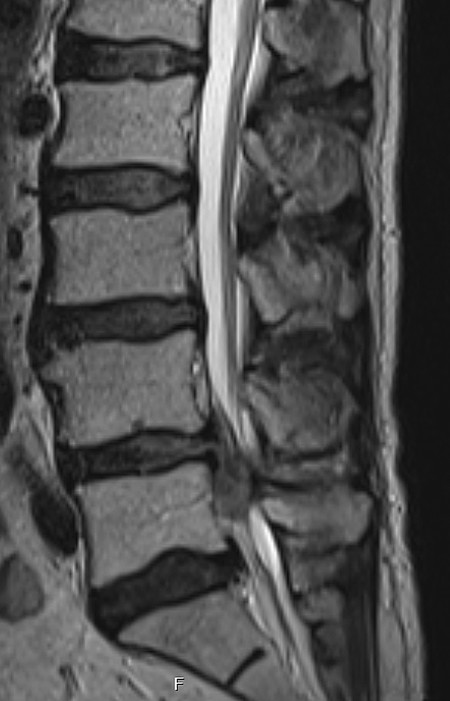
Fig. 7.4 Massive L4/L5 disk herniation causing acute cauda equina syndrome.
The further diagnostic evaluation of spinal cord lesions involves imaging studies (MRI, sometimes computed tomography [CT]) and laboratory testing of the cerebrospinal fluid (CSF) and blood. Electrophysiologic studies may also help determine the etiology. For details, see Chapter ▶ 4.
Key Point
Traumatic spinal cord lesions are usually due to fractures and dislocations of the spine causing displacement of fragments of bone and/or intervertebral disk. The spinal cord can also be compressed by a traumatic hemorrhage in the spinal canal or sustain a direct traumatic contusion in the absence of a fracture. Spinal cord trauma is often a component of polytrauma.
The signs and symptoms of spinal cord trauma depend on the level and severity of the lesion, as shown schematically in ▶ Fig. 7.3.
Like traumatic brain injury, spinal cord trauma can be classified by severity:
Spinal concussion Immediately after blunt trauma to the spine, a more or less complete spinal cord transection syndrome arises, usually at a cervical or thoracic level. The neurologic deficits regress completely within minutes.
Spinal contusion The traumatic event has caused extensive structural damage and contusion of the spinal cord, usually with hemorrhage. There is a partial or complete spinal cord transection syndrome (depending on the extent of the lesion), including bladder dysfunction (see section ▶ 7.1.2) and an initially flaccid paraparesis (paraplegia) or quadriparesis (quadriplegia) (phase of spinal shock, also called diaschisis). The transection syndrome usually improves no more than partially, if at all.
Sometimes the level of a high spinal cord lesion can be inferred from visual inspection of the patient alone ( ▶ Fig. 7.5).
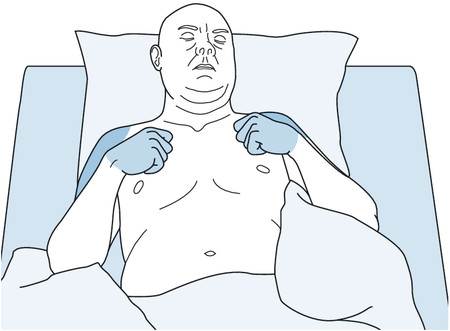
Fig. 7.5 A patient with a traumatic spinal cord lesion at the C7 level. The nerve supply to the elbow flexors, derived from the C6 root, is still intact; the triceps muscles, supplied by C7, are weak, as are the extensors of the wrist and finger joints.
Note
Spinal shock in the acute phase of severe spinal cord trauma: motor, sensory, and autonomic function are lost below the level of the lesion, and the muscle tone (including that of the bladder) is flaccid.
Function may (partially) return after a few days or weeks. The initially flaccid paresis becomes progressively spastic.
Spinal cord compression In traumatic spinal fractures and dislocations, the spinal cord can be compressed by fragments of bone or intervertebral disk, or by hemorrhage within the spinal canal. Compression can also be partly or even entirely due to enlargement of the cord from within by edema or hemorrhage in a severe contusion. As long as the acute spinal cord compression is not severe enough to choke off the cord’s blood supply and cause infarction, the neural tissue may be able to recover its function again once the external compressing elements have been surgically removed, the traumatic spinal cord edema has subsided, and any hemorrhage within the cord has been resorbed.
Practical steps In acute spinal cord trauma, these include the following:
Keep the airway free (to permit breathing and prevent aspiration), stabilize and monitor circulatory function, stop any ongoing hemorrhage.
Safely transport the patient to a trauma center (e.g., hard collar, back board, vacuum mattress).
Perform a gentle, nontraumatic neurologic examination to determine the level of the lesion.
Order targeted neuroimaging studies, usually CT possibly followed by MRI, to identify spinal fractures and dislocations of the vertebral column and assess damage of the intraspinal structures, including the spinal cord; objectively correlate the image findings with the clinically determined level, extent, and type of spinal cord injury.
Insert a bladder catheter.
Surgically decompress the spinal cord if indicated, in case of bony lesions, hematomas, etc.
Prevent decubitus ulcers from the beginning, with frequent repositioning of the patient.
High-dose steroids (given routinely in the past) are currently not recommended, as recent studies suggest that their complications may outweigh their modest benefit.
Patients should be transferred as soon as possible to a rehabilitation hospital specializing in spinal cord trauma where they can receive appropriate physiotherapy, ergotherapy, and psychological support.
Key Point
Spinal cord compression may develop acutely or by slow progression. Acute spinal cord compression is usually due to trauma (see earlier) or hemorrhage (e.g., epidural hematoma). Slowly progressive compression is usually due to a tumor.
Less common causes include abscess, granuloma, spinal deformities (kyphoscoliosis, ankylosing spondylitis), degenerative narrowing of the spinal canal (especially in the cervical region), and massive intervertebral disk herniation.
Clinical features Typical features of slowly progressive spinal cord compression include:
Increasing stiffness or fatigability of the lower limbs.
More or less rapidly progressive gait impairment.
Bladder dysfunction.
Impaired sensation in one or both lower limbs.
Band-like paresthesiae around the chest or abdomen.
Back pain.
Diagnostic evaluation Neuroimaging usually provides definitive evidence of spinal cord compression; MRI is generally superior to CT for this purpose.
General aspects of treatment The treatment is determined by the nature of the compressive lesion and is generally analogous to the treatment of corresponding lesions affecting the brain.
Practical Tip
A tumor must be included in the differential diagnosis of any patient with progressive spinal cord dysfunction.
Tumors in the spinal canal can arise from:
The spinal cord tissue itself (intrinsic spinal cord tumors).
The spinal meninges (meningioma).
The Schwann cells of the spinal nerve roots (neurinoma).
Tumors (particularly metastases) can also project into the spinal canal from the vertebral and paravertebral regions.
Intrinsic spinal cord tumors are intramedullary; leptomeningeal tumors are usually extramedullary, though still intradural (cf. ▶ Fig. 4.17). Tumors growing into the spinal canal from the surrounding structures are extramedullary and extradural. Some highly invasive tumors arising in an extramedullary location can infiltrate the substance of the spinal cord, thereby becoming partly intramedullary.
The more common varieties of spinal cord tumor are briefly described in the following paragraphs.
Metastases Metastases to the spine are usually derived from cancers of the lung and breast, followed by cancer of the prostate.
Metastases usually grow from the vertebral bodies into the spinal canal. Their initial symptom is usually pain, which may be restricted to the site of the tumor or else radiate in a radicular distribution. Paraparesis can arise quite rapidly thereafter, followed by bladder dysfunction. Clinical examination reveals the corresponding neurologic deficits (pyramidal tract signs, possible sensory level, radicular segmental deficits) and, often, focal tenderness of one or more spinous processes to percussion.
Neuroimaging studies are essential for the definitive diagnosis ( ▶ Fig. 7.6).
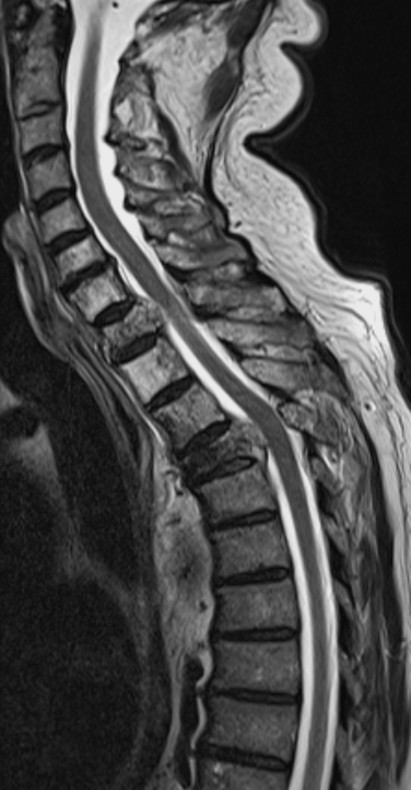
Fig. 7.6 Metastatic adenocarcinoma. Destruction of the T1 and T4 vertebral bodies by tumor, with tumor projecting into the spinal canal at these levels and compressing the spinal cord.
Meningiomas Meningiomas arise from the spinal arachnoid and account for one-third of all intraspinal masses. They are usually found in the thoracic and lumbar regions. They produce very slowly progressive gait impairment and spastic paraparesis, often over the course of several years.
MRI reveals meningiomas as well-demarcated, dural-based lesions with homogeneous contrast enhancement ( ▶ Fig. 7.7).
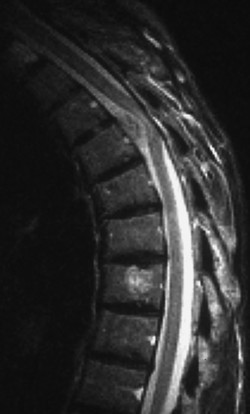
Fig. 7.7 Meningioma at T4, based on the ventral dura mater. Spinal cord compression is clearly visible on this T2-weighted MR image. The increased signal in the T7 vertebral body reflects an incidental, asymptomatic hemangioma.
Neurinomas Neurinomas (also called schwannomas) are nearly as common as meningiomas and, like them, are usually found in the thoracic and lumbar regions. They arise from the Schwann cells of the spinal nerve root sheaths. They nearly always present with radicular pain and radicular deficits. A neurinoma that arises from a nerve root and straddles an intervertebral foramen, so that it has both intra- and extraspinal portions, is called a dumbbell or hourglass tumor ( ▶ Fig. 7.8).
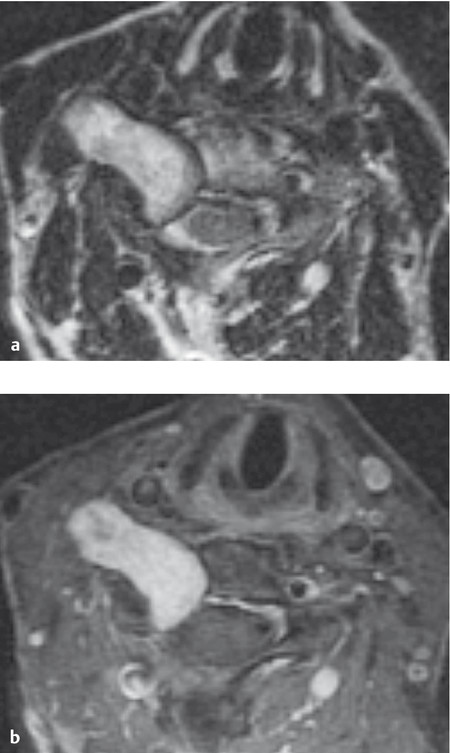
Fig. 7.8 Neurinoma of the right C5 root straddling the intervertebral foramen, with a small intraspinal portion and a large extraspinal portion. a The tumor has heterogeneous signal characteristics in this T2-weighted image. b Neurinomas are typically hyperintense in T1-weighted contrast-enhanced images.
Meningeal carcinomatosis and leukemic meningitis These can cause clinically evident spinal cord compression, in addition to pain (the most common symptom) and polyradicular neurologic deficits.
Intramedullary tumors are less common. Their symptoms and signs depend on their location. The two most common types are astrocytoma and ependymoma; imaging studies are essential for definitive diagnosis ( ▶ Fig. 7.9).
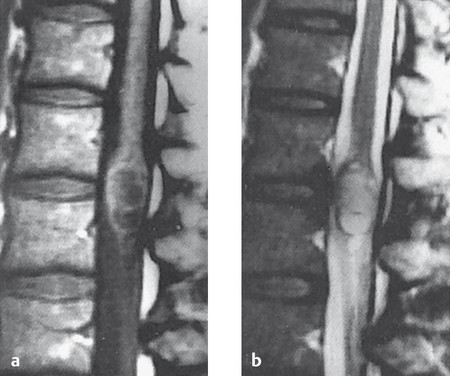
Fig. 7.9 Intramedullary ependymoma in the conus medullaris, as seen in T1-weighted (a) and T2-weighted (b) MR images.
Note
Cervical myelopathy is usually due to degenerative narrowing of the spinal canal and its lateral recesses, with resulting spinal cord compression. Patients are generally elderly and typically present with symptoms in the hands and legs that progress very slowly, over months to years.
Patients with inflammatory diseases of the spine, such as rheumatoid arthritis, are at elevated risk.
Clinical features The initial presentation is often with (poly)radicular deficits due to narrowing of the intervertebral foramina; as the central spinal canal becomes increasingly stenotic, clinically evident spinal cord compression develops. Patients typically complain at first of paresthesiae in the fingers and impairment of the sense of touch (examination reveals astereognosia). The intrinsic muscles of the hands may become atrophic. Ultimately—or, rarely, as the sole manifestation—involvement of the long white matter tracts produces spastic paraparesis, enhanced reflexes, and pyramidal tract signs.
Diagnostic evaluation Neuroimaging is essential for the establishment of the diagnosis; MRI is best ( ▶ Fig. 7.10), as it reveals not only spinal cord compression, but also intramedullary signal abnormalities.
Treatment Decompression of the spinal canal, often with spinal stabilization (fusion) at the same procedure, halts the progression of the neurologic deficits.
|
Symptoms/signs |
Localizing significance |
Remarks |
|
Spastic paraparesis/quadriparesis |
Pressure of syrinx cavity on the pyramidal tracts |
May be unilateral or asymmetric |
|
Muscle atrophy |
Loss of anterior horn ganglion cells |
Segmental, usually unilateral |
|
Sensory level |
Pressure of syrinx cavity on all ascending sensory pathways |
Differential diagnosis: extramedullary lesion compressing the spinal cord |
|
Uni- or bilateral dissociated sensory deficit below a given level |
Uni- or bilateral involvement of the ascending spinothalamic tract |
Highly characteristic |
|
Segmental loss of all modalities of sensation |
Syrinx involving posterior root entry zone |
Usually unilateral or asymmetric; predisposes to burns and other types of mutilating injury |
|
Pain |
Involvement of entering sensory fibers or ascending spinal cord pathways |
|
|
Segmental dissociated sensory deficit |
Syrinx involving the anterior spinal commissure, thereby interrupting the decussating fibers of the lateral spinothalamic tract |
Segmental, bilateral, less commonly unilateral |
|
Autonomic disturbances |
Involvement of the intermediolateral tract in the upper thoracic spinal cord or of the lateral horns |
Impaired sweating, edema, osteolytic bone lesions, arthropathy |
|
Trophic disturbances |
As above |
Severe spondylosis, mutilation of the fingers |
|
Kyphoscoliosis |
Secondary to weakness of the paravertebral muscles |
Usually a late finding, rarely congenital |
|
Associated anomalies |
Part of a disorder of prenatal development |
Basilar impression, Arnold–Chiari malformation, spina bifida, hydrocephalus |
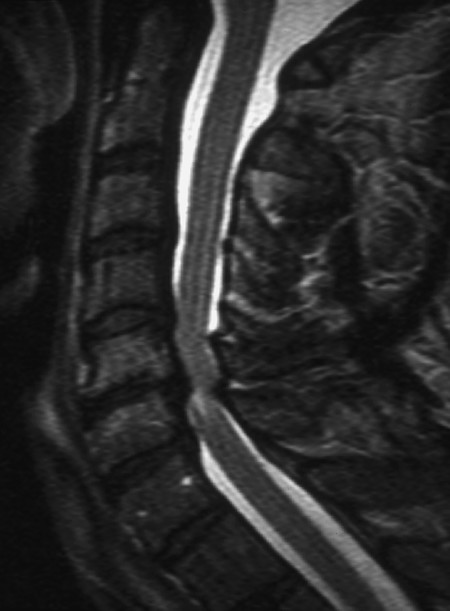
Fig. 7.10 Myelopathy in cervical spondylosis. The T2-weighted MR image reveals narrowing of the spinal canal at C4/C5 and C5/C6 both anteriorly and posteriorly because of degenerative spondylotic changes. A signal abnormality in the spinal cord below C5/C6 indicates a lesion induced by compression.
Key Point
Syringomyelia, a condition coming under the general heading of spinal dysraphism, is sometimes seen in combination with other congenital defects such as the Arnold–Chiari syndrome or spina bifida. It is characterized by the formation of a cavity inside the spinal cord.
Pathogenesis Syringomyelia is defined by the pathologic finding of a tubelike or cleft-like, fluid-filled cavity (syrinx) within the spinal cord, often lined by ependyma and usually extending over several spinal segments. The cavity may reach all the way up to the medulla or even the midbrain (syringobulbia, syringomesencephaly). Mere widening of the central canal of the spinal cord is called hydromyelia.
Clinical features The symptoms and signs of syringomyelia depend on the location of the syrinx within the spinal cord and on its vertical extent; they usually arise in the patient’s second or third decade. The typical clinical features are summarized in ▶ Table 7.2.
Diagnostic evaluation Syringomyelia can be diagnosed from its typical symptoms and physical findings, from which the presence of an intramedullary lesion can be inferred. The characteristic picture is of a dissociated sensory deficit combined with trophic disturbances. The diagnosis must be confirmed with neuroimaging, specifically MRI ( ▶ Fig. 7.11).
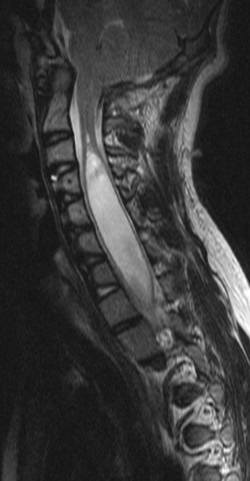
Fig. 7.11 Syringomyelia. The T2-weighted MR image reveals a cavity in the spinal cord from the C2/C3 level down to at least T1/T2. The spinal canal (and thus the syrinx) could not be included in the lower third of the image because of the patient’s scoliosis.
Clinical course and treatment Syringomyelia is usually slowly progressive. Neurosurgical treatment is occasionally successful. The options include the Puusepp operation (opening the posterior aspect of a large syrinx into the subarachnoid space), drainage of the syrinx with a shunt, or operation of an accompanying Arnold–Chiari malformation ( ▶ Fig. 6.3b) at the craniocervical junction.
Key Point
Vascular lesions of the spinal cord, as of the brain, are of two main types: hemorrhage and ischemia. The latter is due to blockage of either the arterial blood supply (e.g., because of thrombosis or embolism) or the venous outflow.
The spinal cord receives arterial blood from three vessels:
The unpaired anterior spinal artery, which runs down the anterior median fissure of the cord and supplies the anterior two-thirds of its cross-sectional area.
The paired posterolateral spinal arteries.
Each of these spinal arteries is made up of a series of individual segments that are linked with one another along the longitudinal axis cord and receive arterial blood from various sources. At cervical levels, the anterior spinal artery receives blood mainly from the vertebral artery and the costocervical and thyrocervical trunks; further down the spinal cord, it is supplied by segmental arteries arising from the aorta (spinal branches and radicular arteries, each of which has an anterior and a posterior branch). In the embryo, there is a radicular artery for each spinal segment; postnatally, only six to eight such arteries are still present. The largest of these, called the great radicular artery or the artery of Adamkiewicz, usually enters the spinal canal at some level from T10 to L2, more commonly on the left side.
The anatomy of the spinal vessels is shown in ▶ Fig. 7.12, and the intramedullary blood supply of a cross-section of the cord is shown in ▶ Fig. 7.13.
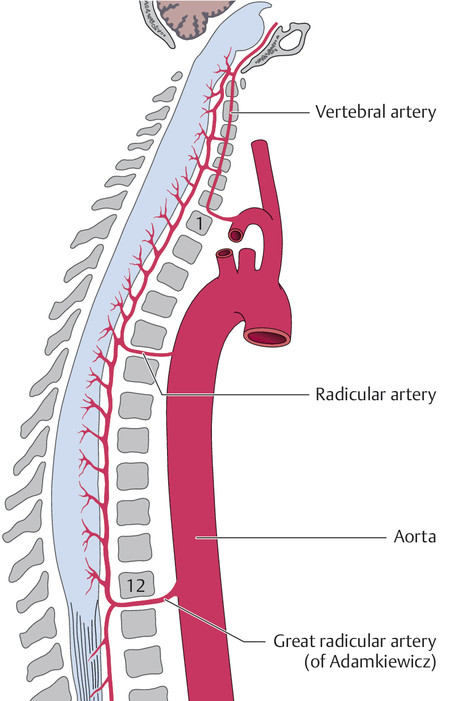
Fig. 7.12 Blood supply of the spinal cord (longitudinal view).
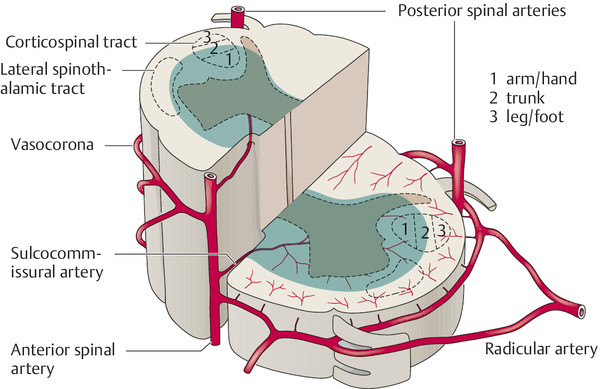
Fig. 7.13 Blood supply of the spinal cord (diagram, cross-sectional view). Occlusion of the anterior spinal a. produces infarction in the area shaded in blue.
The venous outflow of the spinal cord is through radicular veins and into the vena cava.
Note
The arterial blood supply of the spinal cord is derived from a small number of radicular arteries. An anastomotic vascular network links the territories of these vessels both vertically and ventrodorsally.
Note
Arterial hypoperfusion of the spinal cord can give rise to symptoms acutely (suddenly) or in a stuttering, progressive course over several hours or days. It can affect the entire cross-section of the spinal cord or only a part of it.
Infarction of the entire cross-section of the spinal cord at a particular level may be due to the occlusion of a local spinal artery or of a radicular artery, or due to extraspinal vascular pathology, such as an aortic aneurysm. The clinical presentation is usually an acute spinal cord transection syndrome (complete or partial, see section ▶ 7.1.2), although, in some patients, symptoms develop subacutely over the course of a few days or with stepwise progression. The affected patients usually remain paraplegic, particularly if the ischemic lesion is very extensive.
Thrombotic or embolic occlusion of the anterior spinal artery causes infarction in the anterolateral aspect of the spinal cord over one or more segments. The characteristic clinical features were described in section ▶ 7.1.2. An occlusion more distally along the course of the anterior spinal artery, for example, in a sulcocommissural artery (cf. ▶ Fig. 7.13), may cause a partial Brown-Séquard syndrome ( ▶ Table 7.1), with preservation of the sense of touch.
Infarction of the spinal cord, whether it involves the entire cross-section of the cord or only a part of it, is usually not restricted to a single cord segment in the vertical dimension, but rather tends to involve multiple segments, with loss of the motor neurons of the anterior horn. This causes flaccid paresis and areflexia at the level of the lesion. In a few weeks’ time, the flaccid muscles become atrophic. The clinical picture is, therefore, that of a “peripheral” paralysis at the level of the transection.
Intermittent spinal ischemia is very rare and causes a type of spinal intermittent claudication with fluctuating spastic paraparesis.
Chronically progressive vascular myelopathy can cause slowly progressive spastic paraparesis, as well as muscle atrophy owing to involvement of the anterior horns.
Note
Spinal cord ischemia due to impaired venous drainage is a rare cause of infarction. It is usually due to a spinal arteriovenous fistula or arteriovenous malformation.
Spinal arteriovenous malformations are usually found in the thoracolumbar region, while arteriovenous fistulae are usually found at lower lumbar levels (cf. ▶ Fig. 4.16). Both types of vascular anomaly are more common in men.
Patients generally become symptomatic between the ages of 10 and 40 years, often with (band-like) pain as the initial symptom. Neurologic deficits referable to the spinal cord are often only intermittent at first and are (partially) reversible at this stage; later, they take a chronic, progressive course and become permanent. A dural arteriovenous fistula, for example, can cause chronically progressive spastic paraparesis. These vascular anomalies also occasionally present with spinal subarachnoid hemorrhage.
MRI is the most important diagnostic study for the visualization of the vascular malformation and the congested veins distal to it. Spinal angiography can provide useful additional anatomic detail.
Note
Intramedullary, subdural, and (most commonly) epidural hemorrhage can arise spontaneously in an anticoagulated patient or can be caused by a ruptured vascular malformation or trauma.
The typical initial symptom of hemorrhage in or adjacent to the spinal cord is intense pain (possibly radiating in a radicular pattern), followed by a partial or complete spinal cord transection syndrome depending on the level and extent of the hemorrhage. Such hemorrhages always require immediate diagnostic evaluation with MRI or CT, sometimes followed by surgical decompression or interventional neuroradiologic treatment.
Key Point
The spinal cord and spinal nerve roots, like the brain, can be infected by bacteria, viruses, and other pathogens. Combined infection of the brain and intraspinal structures is common: simultaneous manifestations of encephalitis, meningitis, myelitis, and radiculitis (section ▶ 13.1) can be caused by spirochetes (borrelia, leptospira, treponemes; cf. section ▶ 6.7.5) and by many viruses. Acute anterior poliomyelitis, on the other hand, affects only the motor neurons of the anterior horns of the spinal cord.
Note
Any infectious or inflammatory disease of the spinal cord, whatever its etiology, is called myelitis. The causes of myelitis include direct infection, secondary autoimmune processes in the wake of an infectious disease, and chronic autoimmune inflammatory diseases of the CNS, such as multiple sclerosis and neuromyelitis optica.
Etiology The main causes of acute myelitis are viruses (measles, mumps, varicella zoster, herpes simplex, HIV, flaviviruses), rickettsiae, and leptospira. Postvaccinal and postinfectious myelitis have also been described, as has myelitis in the setting of granulomatous disease.
Clinical features and diagnostic evaluation The manifestations range from progressive spastic paraparesis to partial spinal cord transection syndrome. Myelitis can be visualized by MRI ( ▶ Fig. 7.14).
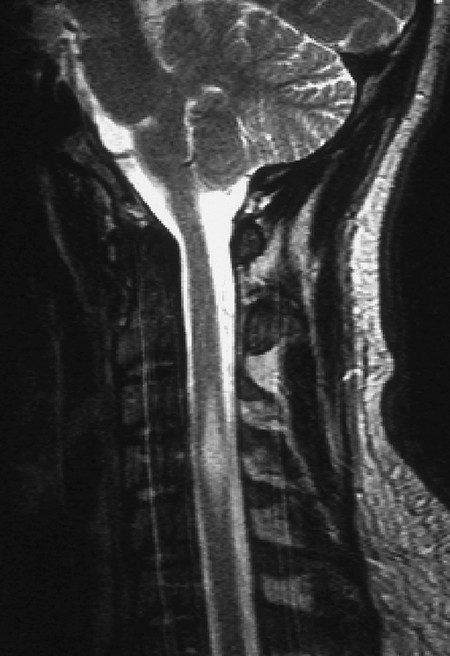
Fig. 7.14 Myelitis (T2-weighted MR image). A spindle-shaped signal anomaly extends from C3 to C5 and expands the spinal cord (which is wider here than the normal cervical enlargement).
Multiple sclerosis or neuromyelitis optica can manifest itself as acute myelitis, sometimes with marked changes in the spinal cord (cf. ▶ Fig. 8.6b).
Treatment and prognosis The treatment of myelitis is directed at its etiology (e.g., antibiotic or antiviral drugs). Patients who cannot walk because of myelitis need prophylactic treatment against deep venous thrombosis and thromboembolism, as well as physiotherapy.
Transverse myelitis is usually of parainfectious origin. It affects the entire cross-section of the spinal cord, producing a complete spinal cord transection syndrome.
Etiology and clinical features Transverse myelitis has a variety of causes; often, the cause cannot be determined (idiopathic transverse myelitis). In distinction to acute myelitis, transverse myelitis is thought to arise, in most cases, from an immune reaction (e.g., a parainfectious reaction after contact with a pathogen). The neurologic manifestations are often preceded by nonspecific flulike symptoms 1 to 3 weeks before onset. The spinal cord deficits usually arise acutely or subacutely and become maximally severe within a few days. Fever, back pain, and myalgia accompany the acute phase.
Diagnostic evaluation The CSF displays inflammatory changes (lymphocytic pleocytosis, elevated immunoglobulin G, and total protein concentrations), but contains CSF-specific oligoclonal proteins less commonly than in multiple sclerosis (cf. ▶ Fig. 8.7). A neuroimaging study (usually MRI) must be performed to rule out a mass or ischemic event.
Treatment and prognosis The pathogen is treated specifically if it can be identified; otherwise, only symptomatic treatment can be given (e.g., analgesic drugs, prednisone). The spinal cord transection syndrome resolves completely in only one-third of all patients. High transverse myelitis can also cause respiratory paralysis and death.
Etiology and epidemiology This disease, caused by a poliovirus, almost exclusively affects the motor neurons of the anterior horn of the spinal cord. Its incidence in countries with a well-developed public health system has been reduced nearly to zero by prophylactic vaccination. It is transmitted by the fecal–oral route under conditions of poor sanitation.
Clinical features After an incubation period ranging from 3 to 20 days, nonspecific prodromal manifestations arise, consisting of fever, flulike symptoms and, in some patients, meningeal signs. The prodrome may resolve without further consequence or be followed, within a few days, by a paralytic phase (likewise accompanied by fever): over the course of a few hours or days, flaccid paralysis arises in various muscles or muscle groups. The weakness is asymmetric, often mainly proximal, and of variable extent and severity. There is no sensory deficit, but the affected muscles may be painful and tender.
Diagnostic evaluation The diagnosis is based on the typical course and physical findings, combined with an inflammatory CSF pleocytosis: at first, there are several hundred cells per microliter, often mainly polymorphonuclear granulocytes. Later, there is a transition to a predominantly lymphocytic picture. The responsible pathogen (poliovirus) can be identified in the patient’s stool.
Treatment There is no specific etiologic treatment; the most important aspect of treatment is the management of respiratory failure (if present).
Prognosis Brain stem involvement and respiratory paralysis confer a poor prognosis; in the remainder of patients, paralysis may regress partially or completely in a few weeks or months. There is usually some degree of residual weakness.
Postpolio syndrome This term is used variously to refer to two very different syndromes.
Some authors use it for a symptom complex seen a few years after the acute illness in polio patients with residual weakness, characterized by fatigability, respiratory difficulties, pain, and abnormal temperature regulation (with negative polio titers).
Others use it for a syndrome with progressive worsening of residual weakness occurring decades after the acute illness. Before this problem can be ascribed to the earlier polio infection, other possible causes of weakness must be ruled out, for example, compression of the spinal cord or spinal nerve roots because of secondary degenerative disease of the spine.
Localization Spinal abscesses are most often epidural, less often subdural, and only rarely intramedullary.
Etiology The most common pathogen is Staphylococcus aureus, which reaches the spinal canal from a site of primary infection outside it by way of the bloodstream (hematogenous spread).
Clinical features Patients typically have general signs of infection (fever, elevated erythrocyte sedimentation rate, leukocytosis, sometimes shaking chills), pain, and neurologic deficits referable to the spinal nerve roots or spinal cord, depending on the specific anatomic situation.
Treatment Spinal abscesses usually require prompt surgical treatment, followed by high-dose antibiotics for several weeks.
Key Point
The diseases described in this section are those that remain confined to the white matter and primarily involve one or more of its long tracts. Many of these diseases are of genetic origin (e.g., the spinocerebellar ataxias); some are of metabolic (e.g., vitamin B12 deficiency), endocrine, paraneoplastic, or infectious origin.
Hereditary diseases
Note
Hereditary diseases can affect ascending or descending pathways, or both, and can thus manifest themselves with spasticity, sensory dysfunction, and/or incoordination.
The genetic basis of most of the hereditary cerebellar and spinocerebellar atrophies (cf. ▶ Table 6.37) has been thoroughly studied. For example, the disease previously known as olivopontocerebellar atrophy, now called SCA1 and SCA2, has been traced to an extended CAG trinucleotide repeat at 6p22–p23. Friedreich ataxia, the most common type of hereditary ataxia, will be described in greater detail later as a prominent member of the class of diseases with degeneration of the spinocerebellar tracts.
Aside from the spinocerebellar ataxias, there are other diseases that cause degeneration of specific fiber tracts in the spinal cord. The pathogenesis of these diseases is only partly understood. For example, the pyramidal tract degenerates in familial spastic spinal paralysis, which will also be described in greater detail later.
Adrenoleukodystrophy (see ▶ Table 6.23) and certain other congenital metabolic disorders can affect individual fiber tracts of the spinal cord.
Nonhereditary diseases The tracts of the spinal cord can also become dysfunctional more or less acutely because of vitamin B12 deficiency or toxic, autoimmune (e.g., paraneoplastic), or metabolic processes. The clinical features resemble those of the hereditary disorders. An important member of this class, described in greater detail later, is funicular myelosis due to vitamin B12 deficiency.
Note
This autosomal recessive disease, the most common hereditary form of ataxia, is due to a defect on chromosome 9 (an extended repeat of the triplet GAA).
Pathogenesis The main pathologic findings include cell loss in the dentate nucleus and combined degeneration of the spinocerebellar tracts, pyramidal tracts, and posterior columns.
Clinical features and diagnostic evaluation The disease usually manifests itself in the second decade, initially with signs of posterior column degeneration and then with spasticity and cerebellar signs.
Typical findings early in the course of disease include:
Progressive (spinal) ataxia with disequilibrium, particularly when walking with the eyes closed.
Diminution or loss of intrinsic muscle reflexes.
Impaired proprioception.
In advanced stages of the disease, cerebellar dysarthria is found as well.
The diagnosis is based on the typical symptoms and signs, including:
A typical deformity of the foot due to the pathologic increased muscle tone ( ▶ Fig. 7.15).
Intracardiac conduction abnormalities.
Often kyphoscoliosis.
Sometimes optic nerve atrophy and nystagmus.
Pyramidal tract signs, distal muscle atrophy.
Cognitive deficits tending toward dementia.
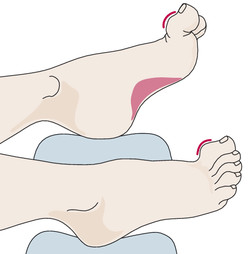
Fig. 7.15 The typical foot deformity in Friedreich ataxia (“Friedreich foot”).
Course Friedreich ataxia is chronically progressive and causes invalidism within a few years of onset. It sometimes takes a protracted course.
Treatment No effective treatment is known.
Synonyms Hereditary spastic paraplegia, spastic paraplegia.
Etiology This genetically heterogeneous syndrome can be inherited in X-linked, autosomal dominant, or (most commonly) autosomal recessive fashion.
Pathogenesis Its pathophysiologic hallmark is degeneration of the pyramidal tracts, more severe at caudal levels, due to diffuse loss of neurons in the primary motor cortex. This condition is thus caused by isolated disease of the first (upper) motor neuron, as opposed to the spinal muscular atrophies, which involve isolated disease of the second (lower) motor neuron (as will be described further in section ▶ 7.7.2).
Clinical features and course Spastic spinal paralysis is characterized by spastic paraparesis, usually beginning in childhood and then progressing slowly over many years, with brisk reflexes, pyramidal tract signs, and increasing gait impairment (“scissors gait” due to adductor spasticity).
Treatment Only symptomatic treatment is now available, consisting mainly of physiotherapy.
Synonym Funicular spinal disease.
Etiology and pathogenesis Funicular myelosis is caused by vitamin B12 deficiency. The latter, in turn, may be due either to inadequate dietary intake or to impaired resorption because of insufficient gastric intrinsic factor (e.g., in atrophic gastritis, or after gastrectomy). The pathologic findings include demyelination of the posterior columns, posterior roots, and pyramidal tracts; in later stages of the disease, other tracts of the spinal cord, and the cerebral white matter, can be affected as well.
Clinical features There is often, but by no means always, an accompanying hyperchromic, megaloblastic anemia with macrocytosis, and the patient’s skin is pale yellow. Neurologic examination reveals an ataxic gait, impaired proprioception, and, rarely, other sensory deficits. These abnormalities may arise subacutely (over a few weeks) or acutely (over a few days). The intrinsic muscle reflexes are diminished because of posterior root involvement, pyramidal tract signs are common, and cognitive and emotional disturbances may be seen, ranging to dementia.
Diagnostic evaluation The diagnosis rests on the demonstration of vitamin B12 deficiency in patients with typical clinical features.
Treatment and course Vitamin B12 must be given as rapidly as possible, by the intramuscular route. The neurologic deficits will then resolve at least in part, but not always completely.
Key Point
The anterior horns of the spinal cord contain the motor neurons whose axons exit the spinal cord in the ventral roots and travel via the peripheral nerves to innervate the skeletal musculature. The pyramidal and extrapyramidal motor pathways terminate in the anterior horns, which also mediate the proprioceptive muscle reflexes (deep tendon reflexes). Diseases of the anterior horns thus manifest themselves in flaccid paresis, attenuation or loss of reflexes, progressive muscle atrophy, and fasciculations.
Although the best-known acute anterior horn disease is acute anterior poliomyelitis (see section ▶ 7.5.1), most diseases in this class are of genetic origin and take a chronic progressive course. The typical clinical features of diseases involving chronic loss of anterior horn cells are summarized in ▶ Table 7.3. Some of these diseases are described in further detail in this section.
|
Disease |
Affected structures |
Symptoms and signs |
Remarks |
Etiology |
|
Infantile SMA (Werdnig–Hoffmann) |
Anterior horn ganglion cells of the spinal cord (and bulbar motor neurons) |
Muscle atrophy and weakness, hypotonia, fasciculations of the tongue |
Affects infants and small children; rapidly fatal |
Autosomal recessive inheritance; mutation of the SMN-1 gene on chromosome 5q13.2 |
|
Pseudomyopathic SMA (Kugelberg–Welander) |
Anterior horn ganglion cells of the spinal cord |
Muscle atrophy and fasciculations, progressive gait disturbance, no bulbar involvement |
Children and adolescents, proximal, usually begins in the lower limbs, slowly progressive |
Autosomal dominant inheritance due to a heterozygous mutation in the DYNC 1H1 gene on chromosome 14q32.31 |
|
Adult SMA (Aran–Duchenne) |
Anterior horn ganglion cells of the spinal cord |
Muscle atrophy, weakness, and fasciculations |
Young adults; begins distally (hands) |
Usually sporadic, of unknown etiology; occasionally due to syphilis |
|
Proximal SMA of the shoulder
girdle
|
Anterior horn ganglion cells of the spinal cord |
Muscle atrophy, weakness, and fasciculations in the shoulder girdle |
Adults; slowly progressive |
Unknown; occasionally due to syphilis |
|
ALS (sometimes including true bulbar palsy) |
Anterior horn ganglion cells of the spinal cord, perhaps also motor cranial nerve nuclei, pyramidal tracts, and corticobulbar tracts |
Muscle atrophy and weakness, fasciculations, bulbar palsy with dysarthria and dysphagia, spasticity, pyramidal tract signs |
Adults, rapidly progressive and lethal; juvenile (familial) cases are less common and have a relatively benign course |
Usually sporadic, rarely familial |
|
Abbreviations: ALS, amyotrophic lateral sclerosis; SMA, spinal muscular atrophy. Note: Several rarer neurologic diseases affect the anterior horn ganglion cells as one component of a wider disease process: these include, but are not limited to, Creutzfeldt–Jakob disease, orthostatic hypotension, diabetic amyotrophy, recurrent hypoglycemia due to an insulin-secreting tumor, metacarcinomatous myelopathy, and organic mercury poisoning. |
||||
The best-known degenerative disease affecting the anterior horns is amyotrophic lateral sclerosis, in which there is simultaneous degeneration of the anterior horn cells and of the first motor neurons that give rise to the corticospinal and corticobulbar tracts. It thus has spastic manifestations as well: brisk reflexes and pyramidal tract signs.
Note
The first motor neuron has its cell body in the motor cortex and sends its axon down the pyramidal tract to terminate in the anterior horn of the spinal cord.
The second motor neuron has its cell body in the anterior horn and sends its axons down the spinal nerve roots and peripheral nerves to innervate the skeletal musculature.
Only first neuron (pyramidal tract) affected: familial spastic spinal paralysis.
Only second neuron affected: spinal muscular atrophy (SMA).
First and second motor neurons affected: amyotrophic lateral sclerosis (ALS).
These diseases are due to a genetic defect on chromosome 5 that causes isolated degeneration of the second (lower) motor neurons, that is, the motor neurons of the anterior horn cells and the cranial nerve nuclei. The result is the typical clinical syndrome of anterior horn degeneration:
Flaccid weakness.
Diminution or loss of reflexes.
Progressive muscle atrophy.
Fasciculations.
All types of SMA are genetically linked to chromosome 5q11.2–13.3.
The main clinical types of SMA are classified according to their age of onset and the pattern of motor deficits that they cause:
Werdnig–Hoffmann type (early childhood type) Neonates and infants with this disease suffer from rapidly progressive muscle weakness, beginning in the muscles of the pelvic girdle. They can survive for no more than a few years.
Kugelberg–Welander type (pseudomyopathic spinal muscular atrophy) The clinical onset is between the 2nd and 10th years of life. The pelvic girdle is most severely affected at first, as in the early childhood type, but the weakness and atrophy progress more slowly, and the overall prognosis is much more favorable. The first signs of disease are progressive quadriceps weakness, loss of the patellar tendon reflex, and, sometimes, pseudohypertrophy of the calves.
Generalized types Types that become symptomatic from the third decade onward tend to be generalized, though the initial presentation tends to be either mainly distal (Aran–Duchenne) or mainly proximal (Vulpian–Bernhardt). The Aran–Duchenne type often presents with atrophy of the intrinsic muscles of the hand, and the Vulpian–Bernhardt type with scapulohumeral atrophy. The latter is now considered a subtype of familial ALS (see later); it affects not only the muscles of the limbs, but also those of the trunk and respiratory apparatus ( ▶ Fig. 7.16).
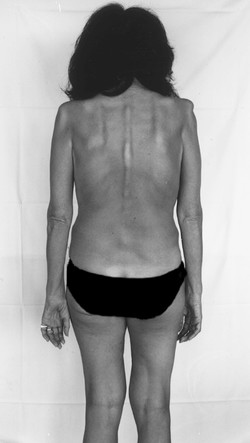
Fig. 7.16 Spinal muscular atrophy in a 46-year-old woman. There is marked atrophy of the muscles of the shoulder girdles, arms, and hands, as well as of the paravertebral musculature.
Note
ALS is characterized by combined degeneration of the anterior horn cells and the corticospinal (pyramidal) and corticobulbar tracts. Loss of the anterior horn cells causes muscle atrophy, weakness, and fasciculations, while loss of the upper-motor-neuron tracts causes brisk reflexes and pyramidal tract signs.
Synonyms This disease is also known as myatrophic lateral sclerosis, as motor neuron disease, and (in North America) as Lou Gehrig disease, after a prominent sufferer.
Epidemiology and Etiology Three-quarters of patients are men, most of them between the ages of 40 and 65. More than 90% of cases are sporadic; the rare familial cases are thought to be due to a defect of the Cu/Zn superoxide dismutase gene.
Pathogenesis The neuropathologic hallmark of this disease is loss of anterior horn cells, combined with degeneration of the pyramidal and corticobulbar tracts and of the Betz’s pyramidal cells in the precentral gyri.
Clinical features The following are typical:
Weakness and atrophy of the muscle groups of the limbs and trunk (including the respiratory apparatus) and/or the bulbar muscles (tongue, throat), progressing slowly over months.
Fasciculations.
Brisk reflexes.
(In some patients) pyramidal tract signs.
Intact sensation.
Often muscle cramps and pain.
Course At first, there is circumscribed, asymmetric, mainly distal muscle atrophy, which is usually most apparent in the intrinsic muscles of the hands. There may be accompanying pain or fasciculations, which are often evident only on prolonged observation. As the disease progresses, muscle atrophy spreads proximally ( ▶ Fig. 7.17). Spasticity gradually appears as well; it is usually only mild at first and may indeed remain so over the ensuing course of the disease. The intrinsic muscle reflexes are usually brisk, more so than one would expect in view of the concomitant atrophy and weakness, but pyramidal tract signs are not necessarily demonstrable. The bulbar muscles are also involved in approximately 20% of patients, as manifested by atrophy, weakness, and fasciculations of the tongue ( ▶ Fig. 7.18), dysarthria, and dysphagia (true bulbar palsy). Involvement of the corticobulbar tracts leads to brisk nasopalpebral, perioral and masseteric reflexes, and, frequently, involuntary laughing and crying. Further progression often leads to frontal dementia.
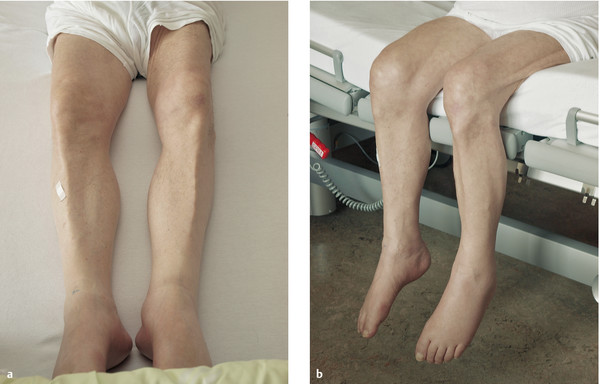
Fig. 7.17 Muscle atrophy and dorsiflexor weakness in ALS. a A 72-year-old man with bilateral atrophy of the thigh and calf muscles, more marked on the left. b When the patient sits, his feet hang limply because of weakness of the foot and toe dorsiflexors.
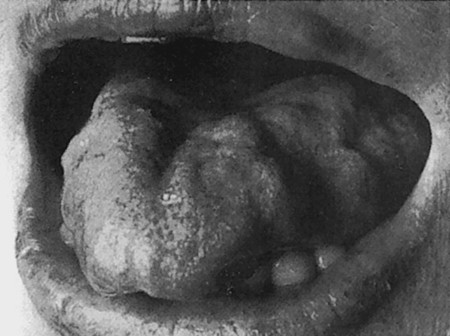
Fig. 7.18 Atrophy of the tongue due to true bulbar palsy in a 65-year-old woman with ALS.
Rarely, the course of ALS deviates from the typical one just described. For example, spasticity may dominate the clinical picture for several months at first, or a patient with a slowly progressive SMA may not develop signs of a corticospinal lesion until years after the onset of disease.
Diagnostic evaluation ALS can generally be recognized by its typical clinical features. Ancillary tests are important mainly for the exclusion of competing differential diagnoses.
EMG: Electromyography reveals pathologic spontaneous activity, both in the paretic muscles and in those that still seem normal.
Motor evoked potentials (MEP, see section ▶ 4.3.3) are used to demonstrate dysfunction of the upper motor neuron.
Pulmonary function tests typically reveal alveolar hypoventilation with low vital capacity.
Laboratory tests: Creatine kinase (CK) activity is mildly elevated. Further laboratory values of interest include vitamins B12 and D, immune electrophoresis, thyroid hormones, and parathyroid hormone.
Lumbar puncture for the exclusion of other diseases. The CSF is normal in ALS.
Imaging studies: For example, MRI of the head and spine can be used to rule out cervical spinal cord compression as the cause of the patient’s problem.
The El Escorial criteria for ALS are a selection of important clinical, neurophysiologic, and neuropathologic criteria that can be used to characterize the diagnosis of ALS as definite, probable, possible, or presumptive.
Treatment and prognosis ALS takes a chronically progressive course. Death usually ensues within one or two years, although a minority of patients survives longer. The treatment of ALS consists of numerous symptomatic measures, which vary as the disease progresses.
Riluzole is a sodium-channel blocker with an antiglutamatergic effect. It marginally slows the progression of ALS.
Adequate nutrition is important to prevent weight loss and dehydration. Dysphagic patients will need to be fed via gastrostomy. This measure should be discussed and implemented early in the course of disease.
When hypoventilation causes hypercapnia and daytime somnolence, mask ventilation may improve the patient’s quality of life. When bulbar palsy is present, adequate ventilation requires a tracheostomy, which can prolong the patient’s life. This should be discussed with the patient and his or her family before respiratory failure occurs. If a tracheostomy is performed in this situation, the patient may survive until progressive weakness finally results in a locked-in state. The patient and his or her family should be aware of this possibility, and the patient’s instructions should be recorded in an advance care directive.
Patients with severe dysarthria can be helped to communicate with electronic communication aids.
psychological care of the patient and family is important at all times, optimally by the same person for the entire duration of the illness.
Additional Information
Fasciculations are a typical feature of ALS but should only be interpreted as a sign of disease when accompanied by other abnormalities (e.g., weakness, atrophy). Fasciculations can be seen in patients with other neurologic diseases and even in healthy medical students and hospital staff.