6.11 Cerebellar Diseases and Other Conditions Causing Ataxia
Cerebellar disturbances present clinically with disequilibrium, truncal
and/or appendicular ataxia, impaired coordination, and diminished muscle
tone (see also section ▶ 5.5.6).
6.11.1 Overview
Classification The different clinical types of ataxia can be classified in
several ways:
Clinical features Ataxia is characterized by impaired coordination of
movement and a dysfunctional interplay of agonist and antagonist
muscles. Thus, it typically manifests itself as poorly
controlled movements that tend to overshoot their target. The
additional manifestations of the individual diseases causing ataxia depend
on their etiology and the particular neural structure involved.
Ataxia can be caused not only by cerebellar disease, but
also by disease of the afferent and efferent pathways leading to and
from the cerebellum, or of any afferent somatosensory or special sensory
pathways. The underlying lesion may be in the cerebellum, but it may
also be in the brainstem, spinal cord, peripheral nerves, sensory
cortex, or thalamus.
An initial, clinically based classification of ataxia distinguishes symmetric from focal,
asymmetric types ( ▶ Table 6.36). The table can serve as an overview and
guide to the differential diagnosis of ataxia.
Table 6.36 The differential diagnosis of symmetric
and focal asymmetric ataxias
|
Symmetric ataxia
|
Focal asymmetric ataxia
|
-
Acute or chronic intoxication
-
Electrolyte disturbance
-
Acute viral cerebellitis
-
Miller Fisher syndrome
-
Postinfectious, ADEM
-
Alcoholic-nutritional cerebellar damage
-
Vitamin B1 deficiency
-
Vitamin B12 deficiency
-
Paraneoplastic cerebellar disease
-
Antigliadin antibody syndrome
-
Hypothyroidism
-
Creutzfeldt–Jakob disease
-
Hereditary cerebellar degeneration
-
Familial episodic ataxia
-
Multisystem atrophy
-
Tabes dorsalis
-
Psychogenic ataxia
|
-
Ischemic stroke
-
Cerebellar hemorrhage
-
Subdural hematoma
-
Abscess
-
Neoplasia (primary brain tumor, metastasis)
-
Demyelinating plaque, multiple sclerosis
-
Arteriovenous malformation, arteriovenous
fistula
-
Arnold–Chiari or Dandy–Walker malformation
-
Psychogenic ataxia
|
Diagnostic evaluation When ataxia is suspected, the age at
onset and the family history are
diagnostically relevant:
-
If ataxia arises in childhood or adolescence in a
patient with a positive family history, the diagnosis is probably an
autosomal recessive ataxia. The most
common of these is Friedreich ataxia.
-
If a parent is similarly affected, then the diagnosis
is probably an autosomal dominant ataxia,
for example, spinocerebellar ataxia.
-
Ataxia arising after age 40 years is less likely to be
hereditary and may be an acquired ataxia,
for example, of toxic origin (such as alcohol-induced cerebellar
degeneration).
Individual types of ataxia can be diagnosed by
molecular-genetic testing, imaging studies, or laboratory analysis.
Treatment The symptom complex called “ataxia” has no specific treatment,
although coordination may be improved by physiotherapy. Certain types of ataxia of known cause can be
correspondingly treated: examples include vitamin E administration in ataxia
due to vitamin E deficiency and in Aβ-lipoproteinemia, a diet that is low in
phytanic acid in Refsum disease, and a low-copper diet combined with
chelators or zinc in Wilson disease.
6.11.2 Selected Types of Ataxia
Disturbances of the cerebellum, like those of the cerebral
hemispheres, are usually due either to vascular processes (ischemia,
hemorrhage) or to tumors. Multiple sclerosis is a further common cause.
In this section, we will also discuss other diseases that may present
primarily with cerebellar dysfunction, including infectious,
parainfectious, (heredo-)degenerative, toxic, and paraneoplastic
conditions, as well as cerebellar involvement in general medical
diseases.
A thorough description of each ataxia syndrome would be beyond the
scope of this textbook, and we therefore discuss only the more important
ones briefly. Friedreich ataxia is described in greater detail in a
later chapter (section ▶ 7.6.2) among the other hereditary diseases
of the spinal pathways.
Cerebellar Heredoataxias
Cerebellar heredoataxias are of genetic
origin. The enzymatic defects and pathophysiologic mechanisms underlying
each have not yet been determined, except in a few cases. The main types
are listed in ▶ Table 6.37. Spinocerebellar
ataxias involve both the cerebellum and the spinal cord and
are associated with cognitive deficits as well (see
sections ▶ 7.6.1 and
▶ 7.6.2).
Table 6.37 Types of cerebellar ataxia
|
Disease
|
Clinical features
|
Remarks
|
|
Autosomal
recessive hereditary ataxias
|
|
Friedreich ataxia
|
-
Lumbering, broad-based, unsteady gait,
progressive from the first or second decade
onward
-
Later, clumsiness of the hands, explosive
speech
-
Typical Friedreich foot (see ▶ Fig. 7.15)
-
Scoliosis, hypotonia
|
GAA triplet expansion on chromosome 9; impaired
synthesis of the protein frataxin
|
|
Refsum disease (heredopathia
atactica polyneuritiformis)
|
|
Lack of phytanic acid α-dehydrogenase
|
|
Aβ-lipoproteinemia
(Bassen–Kornzweig syndrome)
|
-
Progressive ataxia, nystagmus,
ophthalmoplegia, polyneuropathy
-
Acanthocytosis, low cholesterol and
triglyceride levels, vitamin E deficiency
|
Low serum lipoprotein, cholesterol, and triglyceride
levels
|
|
Ataxia telangiectasia
(Louis–Bar syndrome)
|
-
Onset in infancy with ataxia and
choreoathetosis
-
Frequent lung, ear, nose, and throat
infections
-
Slow eye movements
-
Telangiectases in conjunctiva and joint
creases
|
One of the chromosomal fragility syndromes
|
|
Spinocerebellar ataxia
(different varieties, e.g., deficiencies of
hexosaminidase, glutamate dehydrogenase, pyruvate
dehydrogenase, ornithine transcarbamylase, or vitamin
E)
|
-
Onset usually before age 10 y
-
Ataxia with variable accompanying deficits,
e.g., intellectual disability, visual or hearing
impairment, polyneuritis, myoclonus
-
Speech may be loud, deep, and harsh (“lion’s
voice”)
-
If hereditary, generally autosomal
recessive
|
|
|
Autosomal
dominant hereditary cerebellar ataxias
(ADCA)
|
|
Cerebellar cortical atrophy
(Holmes type = Harding type III)
|
|
Genetically heterogeneous, SCA 5 and SCA 6
|
|
Olivopontocerebellar atrophy
(Menzel type = Harding type I)
|
-
Onset at age 20 y or later
-
Cerebellar and noncerebellar signs including
optic nerve atrophy, basal ganglionic dysfunction,
pyramidal tract signs, muscle atrophy, sensory
deficits, and sometimes dementia
|
Genetically heterogeneous, SCA 1–SCA 4; SCA 3 =
Machado–Joseph disease
|
|
Cerebellar atrophy (Harding
type II)
|
|
Corresponds to SCA 7
|
|
Autosomal
dominant hereditary episodic
ataxias
|
|
Familial periodic paroxysmal
ataxia
|
|
|
|
Sporadic,
nonhereditary ataxias
|
|
“Atrophie cérébelleuse tardive à
prédominance corticale”
|
-
Onset in late adulthood with slowly
progressive gait and truncal ataxia, later arm
ataxia
-
Rarely, nystagmus, muscle hypotonia, and
pyramidal tract signs
|
Symmetric degeneration of Purkinje cells,
predominantly in the vermis; alcohol may be a
precipitating factor in persons with an underlying
genetic predisposition
|
The most common hereditary ataxia is Friedreich ataxia, an autosomal disorder with onset
usually in the first or second decade of life that generally leads
to death between the ages of 30 and 40 years. Its typical
manifestations are the following:
-
Unsteady gait, particularly with eyes closed; areflexia,
pyramidal tract signs.
-
Dysphagia, cerebellar dysarthria, oculomotor disturbances
(gaze-evoked nystagmus, square-wave jerks, abnormal
suppression of the vestibulo-ocular reflex,
▶ Fig. 12.6).
-
Optic nerve atrophy.
-
Early loss of proprioception and vibration sense.
-
Distal muscle atrophy, typical foot deformity (pes cavus),
kyphoscoliosis.
-
Cardiac manifestations (conduction abnormalities,
cardiomyopathy), diabetes mellitus.
-
Cognitive disturbances.
-
Dementia.
Symptomatic Types of Ataxia and Cerebellar
Degeneration
Aside from hereditary and idiopathic types of ataxia and cerebellar
degeneration, there are also types that reflect particular underlying
illnesses, including a wide variety of cerebellar and systemic diseases
( ▶ Table 6.38). The clinical manifestations vary
depending on the cause. Other systems and organs are often
involved.
Table 6.38 Causes of symptomatic ataxia and
cerebellar degeneration
|
Cause or precipitating factor
|
Examples and remarks
|
|
Local disease: mass lesion or
ischemia
|
Malformation, cerebellar tumor, infarct, hemorrhage,
inflammation, trauma or other physical causes
|
|
Infection
|
Often in the aftermath of an infectious disease, e.g.,
mononucleosis
Acute cerebellar ataxia in
childhood arises a few days or weeks after a
chickenpox infection, less commonly after another viral
illness. The patient is usually a preschool-aged child.
Unsteady gait, ataxia, tremor, and nystagmus are the
characteristic signs; they usually resolve spontaneously
in a few weeks.
Acute cerebellitis is similar
to the foregoing and affects both children and adults.
In older patients, the manifestations can persist
|
|
Systemic diseases
|
For example, multiple
sclerosis, macroglobulinemias
|
|
Toxic conditions
|
Tardive cerebellar atrophy
in chronic alcoholism,
other toxic causes (diphenylhydantoin, lithium, organic
mercury, piperazine, methotrexate, 5-fluorouracil,
DDT)
|
|
Metabolic and hormone-associated
conditions
|
Familial AVED (an autosomal recessive condition that
clinically resembles Friedreich ataxia), vitamin B
deficiency
Hypothyroidism and myxedema
Malresorption syndrome, gluten intolerance
(gluten-induced ataxia with or without gastrointestinal
manifestations)
|
|
Paraneoplastic
conditions
|
Subacute cerebellar cortical atrophy
|
|
Further causes
|
-
Heatstroke
-
Miller Fisher syndrome (see
section ▶ 11.2.3)
-
Cranial polyradiculitis (see
section ▶ 11.2.3)
-
Creutzfeldt–Jakob disease (see
section ▶ 6.7.9): ataxia is
sometimes the first sign of this disease
|
Intermittent cerebellar manifestations
are found mainly in the following diseases:
The differential diagnosis of cerebellar ataxia must also include
disease processes that cause ataxia but do not involve the
cerebellum:
-
(Contralateral) frontal lobe
lesions.
-
Paresis of any cause.
-
Lesions of the afferent sensory
pathways (e.g., polyneuropathy or posterior column
lesion).
-
Lesions of the parietal sensory
cortex.
A prolonged beridden state (“bed ataxia”) and
psychogenic mechanisms are further possible
causes.
6.12 Dementia
Unlike the terms “intellectual disability” and “oligophrenia,”
which refer to congenital disturbances, the term “dementia” refers to an
acquired degeneration of intellectual and cognitive abilities that persists
for at least several months or takes a chronically worsening course, leading
to major impairment in the patient’s everyday life. The clinical picture is
dominated by personality changes as well as neuropsychological and
accompanying neurologic (particularly motor) deficits. Reactive changes,
including insomnia, agitation, and depression, are common.
In this chapter, we will first provide an overview of the general aspects of
the dementia syndrome and then describe the main types of degenerative brain
disease that cause dementia in greater detail.
6.12.1 Overview: The Dementia Syndrome
Etiology and classification
Unlike the various types of neuropsychological disturbance
caused by focal brain lesions, the dementia syndrome is caused by a
diffuse loss of functional brain tissue.
Neuroimaging usually discloses extensive brain
atrophy or multifocal brain
lesions.
Primary brain atrophy The loss of functional tissue is often due to primary
(degenerative) brain atrophy, which mainly affects the cerebral cortex,
progresses chronically, and causes irreversible cognitive impairment. In
such cases, dementia is the direct consequence and most obvious
expression of the causative pathologic process. Primary brain atrophy
characterizes dementing diseases in the narrow
sense of the term:
Symptomatic dementia In principle, however, any disease that damages the
structure or function of the brain can produce the dementia syndrome.
Dementia, in such cases, is often a possible but not
universal accompaniment of the main features of the disease
in question, and is generally not the only manifestation. It is
important to realize that nearly 10% of all cases of dementia are due to
diseases that can be reversed, or at least kept from progressing
further, by appropriate treatment. The timely diagnosis and treatment of
such patients is crucial for the prevention of further
deterioration.
Cortical and subcortical dementia In cortical types of dementia,
dementia is the main manifestation of the
disease (classic example: Alzheimer disease). In contrast, movement disorders are typically prominent in
patients with subcortical dementia.
Overview of causes ▶ Table 6.39
contains an overview of the causes of dementia, with an indication of
which among these conditions are irreversible and which are at least
partly treatable.
Table 6.39 Causes of dementia
|
Type
|
Treatability
|
Diseases
|
|
Degenerative diseases of the nervous
system
|
Partly treatablea
|
|
|
Irreversible
|
-
Frontotemporal dementia (Pick disease)
-
Frontal lobe degeneration
-
Pantothenate kinase–associated
neurodegeneration
-
Heredoataxias
-
Lewy body disease
-
Corticobasal degeneration
-
Fragile-X syndrome
|
|
Cerebrovascular
diseases
|
Partly treatablea
|
|
|
Irreversible
|
-
Amyloid angiopathy
-
CADASIL
|
|
Infectious dementias
|
Treatableb
|
|
|
Partly treatablea
|
|
|
Irreversible
|
-
Prion diseases:
-
Kuru
-
Creutzfeldt–Jakob disease
-
Gerstmann–Sträussler–Scheinker
syndrome
-
Familial fatal insomnia
-
Familial progressive subcortical
gliosis
|
|
Metabolic disorders affecting the
brain
|
Treatableb
|
|
|
Partly treatablea
|
|
|
Irreversible
|
|
|
Neoplasia
|
Partly treatable
|
|
|
Irreversible
|
|
|
Autoimmune
encephalopathies
|
Treatableb
|
|
|
Partly treatablea
|
|
|
Epilepsy
|
Treatableb
|
|
|
Partly treatablea
|
|
|
Mental illnesses
|
Treatableb
|
-
Depression
-
Schizophrenia
-
Hysteria
|
|
Hydrocephalus
|
Treatableb
|
|
|
Trauma
|
Partly treatablea
|
|
|
Irreversible
|
|
|
Demyelination
|
Partly treatablea
|
|
All patients with dementia deserve a thorough diagnostic
evaluation, because a treatable cause may be discovered.
Epidemiology
One percent of persons aged 60 to 64, and more than 30% of
persons older than 85 years, suffer from dementia. The most common cause is
Alzheimer disease, which accounts for 40 to 50%
of all patients. The second most common cause, and the most common cause of
symptomatic dementia, is vascular (i.e., multi-infarct)
dementia (15%); the third most common cause is alcoholism.
General clinical features
The dementia syndrome is characterized by
neuropsychological deficits, personality changes, and behavioral
abnormalities. In particular, the following are seen:
-
Impairment of short-term and long-term memory:
-
Impairment of thinking,
particularly with respect to:
-
Judgment.
-
Problem-solving.
-
Symbol comprehension.
-
Impairment of visuospatial and spatial-constructive
functions.
-
Aphasia and apraxia.
-
Impairment of attention and concentration.
-
Reduced drive, initiative, and motivation; easy
fatigability.
-
Affect lability and impaired affect control.
-
Impairment of emotionality and social behavior.
-
In some patients, confusion and impairment of
consciousness.
General diagnostic evaluation
The diagnosis of the dementia syndrome is based on thorough
history-taking from the patient and family
members, a comprehensive general medical and
neurologic examination, and neuropsychological testing.
Screening tests The following can be used as screening tests for dementia,
although they are not very informative or specific:
-
Mini-Mental State Examination
( ▶ Table 3.1).
-
Clock test (the patient is asked to draw a clock face
indicating a particular time).
-
Montreal Cognitive Assessment (MoCA) test: this test
simultaneously assesses various cognitive skills with a sequence
of simple tasks that can be scored on a point system. It takes
approximately 10 minutes to administer. See
www.mocatest.org.
Imaging studies and electrophysiology Neuroimaging (usually MRI) should
be performed in every case as part of the search for the underlying
etiology. PET, SPECT,
and EEG can be performed as well.
Laboratory tests Depending on the specific clinical situation and the
suspected causes, targeted laboratory testing
should be performed, including, for example, complete blood count,
electrolytes, hepatic and renal function tests, thyroid hormones,
vitamin B12, folic acid, TPHA test, HIV serology, and CSF
examination.
Differential Diagnosis at the Syndrome Level
The dementia syndrome may be hard to differentiate from certain other
psychopathologic states, especially the following:
-
Nonpathologic cognitive decline
in old age (can be diagnosed on the basis of history
obtained from family members and clinical testing).
-
Depression with severely reduced drive, so-called
depressive pseudodementia
(neuropsychological and psychiatric examination).
-
An isolated neuropsychological
disturbance, especially aphasia, apraxia, and/or
agnosia (clinical testing, neuropsychological
examination).
-
Congenital intellectual
disability, that is, oligophrenia (history).
-
Cognitive impairment by medications or drugs of abuse (history, general
physical examination, laboratory testing).
-
Status epilepticus with
partial complex seizures or absences (EEG).
-
Cognitive impairment due to endogenous psychosis (neuropsychological and
psychiatric examination).
General Aspects of Treatment
Cause-directed treatment If the dementia syndrome is found to be due to a treatable condition,
cause-directed treatment can be given,
leading to a cure or, at least, the prevention of further progression.
In all other cases, however, dementia responds poorly to treatment, if
at all. The current medications for Alzheimer disease provide only
modest clinical benefit, often at the cost of side effects.
Accompanying manifestations Various manifestations that accompany dementia can be treated symptomatically, with major benefit to the
patient and his or her family, including depression, delusions,
insomnia, and agitation. Training of (temporarily)
preserved cognitive abilities is also advisable to keep the
patient functionally independent for as long as possible.
Nursing care In advanced stages of disease, patients often require home nursing visits, or care in a suitable day clinic; removal from the home to a permanent
care facility should be deferred for as long as this is practically
achievable. The patient’s family is an important component of treatment.
Family members should receive early and thorough information about the
patient’s disease and, where appropriate, counseling in the ways they
can help care for the patient.
6.12.2 Alzheimer Disease (Senile Dementia of Alzheimer Type)
Alzheimer disease is the paradigmatic example of cortical
(as opposed to subcortical) dementia, with dementia itself as the main
clinical manifestation. It is the most common cause of dementia in the
elderly.
Synonyms
Senile dementia of Alzheimer type; Alzheimer dementia.
Epidemiology Women are more commonly affected than men, and the prevalence rises
with age. The mean age at the onset of Alzheimer disease is 78 years.
Persons under age 65 with the disease are said to have “presenile
dementia.”
Etiology and genetics
Genetic factors are clearly
contributory, as first-degree relatives of Alzheimer patients are much
more likely to develop the disease than persons with no family history
of it. Yet, as most affected persons have no such history, environmental factors must also play a role. It
seems that the likelihood of developing the disease is higher in persons
of low educational attainment and those whose intellectual abilities are
not regularly put to use; hypercholesterolemia, diabetes mellitus, and
other brain diseases also increase the risk.
Genetics Hereditary cases of Alzheimer
disease (which are often early-onset cases) are associated with defects
in:
-
Chromosome 21q, which contains the amyloid precursor protein
(APP) gene.
-
Chromosome 14: mutation of the presenilin-1 gene.
-
Chromosome 1: mutation of the presenilin-2 gene.
Familial clustering of Alzheimer
disease is associated with defects in:
Persons with trisomy 21 (Down
syndrome) are generally demented after age 30 years.
Pathogenesis and Pathology
The neuropathologic lesion in Alzheimer disease
consists of neuronal loss in the cerebral
cortex, particularly in the basal
temporal lobe (hippocampus) and the temporoparietal region. Histologic examination reveals
cell necrosis and an accumulation of
neuritic (“senile”) plaques and Alzheimer neurofibrillary tangles. Amyloid angiopathy is often present as
well.
Amyloid and tau protein deposition In Alzheimer disease, degradation of the membrane protein
APP (amyloid precursor protein) leads to the increased production of a
neurotoxic protein called β-amyloid (more
precisely, Aβ1–42, which is longer than the physiologic degradation
product). This pathologic protein forms perivascular
deposits and cortical amyloid
plaques. In addition, hyperphosphorylated tau protein also accumulates within neurons in Alzheimer fibrils and tangles. Amyloid and tau
protein deposition has multiple consequences:
-
Neuron degeneration, mainly
temporobasal (hippocampal) and temporoparietal.
-
Axon degeneration and loss of synapses.
-
A cholinergic deficit in the cerebral
cortex.
-
In the late stage of the disease, immune
reactions of glial cells to plaques, resulting in
further cell loss.
Cholinergic deficit Neuron degeneration is regularly seen in the nucleus basalis of Meynert, which sends a diffuse
cholinergic projection to the frontal cortex. This and the diminished amount of acetylcholine found in the
brain of persons with Alzheimer disease suggest that the cholinergic system plays a role in the
pathogenesis of the disease. These observations provide the motivation
for treatment with cholinergic drugs (as described later).
Clinical Features, Course, and Diagnostic Evaluation
Clinical features and course The nonspecific early manifestations
can include depression, insomnia, agitation, anxiety, and excitability.
Early signs of dementia include impairments of memory, word-finding ability,
and spatial orientation.
Persons who have a demonstrable memory deficit that impairs their
performance of complex everyday tasks (but not of simpler ones) are said
to have “mild cognitive impairment.” This can be an early manifestation
of Alzheimer disease.
Within a year, patients manifest gradually
worsening forgetfulness,
fatigability, poor concentration, and
lack of initiative. Nonetheless, motor
functioning, including erect body posture, remains normal for a long time,
so that the superficial appearance of good health is preserved.
There are often also focal neuropsychological
deficits such as aphasia, apraxia, impaired temporal and spatial
orientation, and the applause sign (described in
section ▶ 5.5.1). The patient loses the ability to
think abstractly or grasp complex situations, and confusion, lack of
interest, and the progressive loss of language ultimately lead to the loss of functional independence and the need for nursing care.
Diagnostic evaluation Early recognition and interpretation of the psychopathologic
deficits described earlier is crucial for the diagnosis of the
disease.
Often, the diagnosis is further supported by typical neuroimaging findings (MRI: cortical brain atrophy, especially in the temporal lobe, and
ventriculomegaly; see ▶ Fig. 6.61).
Neuroimaging is also mandatory because it can rule out some of the other
causes of the dementia syndrome (cf. ▶ Table 6.39).
Further studies (hematologic biochemical, and serologic
blood tests, CSF examination, EEG) may be indicated, depending on
the specific differential diagnostic considerations in the individual
patient.
A differential diagnostic overview of the main clinical and radiologic
features of Alzheimer disease and other conditions resembling it is provided
in ▶ Table 6.40.
Table 6.40 Distinguishing features of different
types of dementia
|
Alzheimer dementia
|
Lewy body dementia
|
SAE (vascular dementia)
|
Frontotemporal dementia (Pick disease)
|
|
Type of dementia
|
Cortical
|
Both cortical and subcortical
|
Subcortical
|
Frontal
|
|
Main cognitive
manifestations
|
Disturbance of recent memory, visuospatial orientation,
word-finding
typical: normal “façade”
|
Combined manifestations of cortical and subcortical
dementia
|
Slowing, forgetfulness, impaired concentration and
drive
|
Personality change, loss of drive, sometimes
aphasia
|
|
Accompanying neurologic or other bodily
manifestations
|
Sometimes hyposmia, otherwise no particular bodily
manifestations at first
physical impairment, only in the late stage
|
Fluctuating attention and wakefulness, visual
hallucinations, parkinsonism
|
Increased muscle tone, hypokinesia, small-stepped and
broad-based gait (lower-body parkinsonism), dizziness,
falls, sometimes TIAs, urinary incontinence
|
Early urinary incontinence, sometimes parkinsonism
|
|
Mental disturbances
|
Depression, later restlessness, anxious agitation,
paranoia, insomnia
|
Insomnia, visual hallucinations, delusions
|
Sometimes depression, irritability, “lazy” speech
|
Variable; personality change, abnormal behavior,
progressively “lazy” speech
|
|
Neuroimaging
|
CT, MRI: cortical brain atrophy, ventriculomegaly
|
SPECT, PET: low dopaminergic activity in occipital
lobes
|
CT, MRI: marked SAE lesions and multiple lacunar
infarcts
|
CT, SPECT, PET: frontal hypoperfusion and
hypometabolism
|
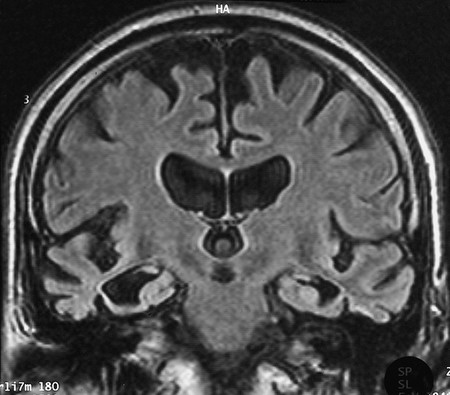
Fig. 6.61 Brain atrophy in
dementia. High-grade, symmetric, mainly frontal atrophy
of the cerebral hemispheres in a 64-year-old man. Note the marked
atrophy of the temporal lobes as well. The lateral ventricles,
including the temporal horns, are markedly dilated, as is the third
ventricle. Both external hydrocephalus and internal hydrocephalus
are present (“ex vacuo,” i.e., as a result of brain atrophy).
6.12.3 Treatment and Prognosis
Treatment Cholinomimetic drugs (acetylcholinesterase
inhibitors: donepezil, galantamine, rivastigmine) and antiglutamatergic NMDA-receptor antagonists (memantine) improve
neuropsychological deficits symptomatically but do not halt the progression
of dementia.
A possible beneficial effect of acetylsalicylic acid is currently being
studied, as patients with Alzheimer disease often have concomitant
small-vessel disease as a contributory cause of dementia.
No clear benefit has been shown for high-dose vitamin E, Ginkgo biloba preparations, calcium antagonists, or
nootropic drugs (= drugs that putatively improve brain performance), such as
piracetam.
The most important aspect of treatment in all patients is the management
of the accompanying manifestations:
-
Depression (preferably with selective
serotonin reuptake inhibitors).
-
Psychosis (preferably with clozapine or
olanzapine).
-
Insomnia, agitation, and aggressiveness
(preferably with low-potency neuroleptic agents such as pipamperone,
melperone, and clomethiazole).
Patients with advanced Alzheimer disease, and their families, can benefit
from referral to special outpatient and day care
facilities.
Prognosis Alzheimer disease always progresses.
The average life expectancy from the time of diagnosis is 8 to 9
years.
6.12.4 Dementia with Lewy Bodies
Epidemiology Vascular dementia is the second most common type of dementia after
Alzheimer disease, but the second most common neurodegenerative dementing
disease is Lewy body dementia.
Lewy body disease overlaps with Parkinson
disease (section ▶ 6.9) in
its neuropathologic and clinical features but differs in having dementia
as a prominent early manifestation, appearing at the same time as the
motor disturbances or even before them.
Pathology The neuropathologic hallmark of this disease is the presence of Lewy inclusion bodies (deposits of α-synuclein) in the neurons of the cerebral cortex and
brainstem.
Clinical features In these patients, progressive dementia (of a
combined cortical and subcortical type; cf. ▶ Table 6.38) is
accompanied by certain other characteristic findings: there are fluctuating deficits of attention and concentration,
as well as frequent, objective visual
hallucinations and motor parkinsonism
(particularly in patients with early disease onset). Patients often suffer
from repeated falls, syncope, brief episodes of unconsciousness, and
hallucinatory experiences.
Diagnosis and treatment
-
SPECT and PET reveal diminished perfusion and metabolism in the
occipital lobes.
-
An L-DOPA challenge test may alleviate motor parkinsonism in
patients with this disease (although it often does not), while
tending to intensify the hallucinations. Neuroleptic drugs make all
manifestations worse.
-
Acetylcholinesterase inhibitors can improve both dementia and
hallucinations.
6.12.5 Frontotemporal Dementia (Pick Disease)
Classification Frontotemporal dementia (Pick disease) is the
commonest type of focal cortical atrophy. The
dementing diseases belonging to this category, all of them much rarer than
Alzheimer disease, are characterized by localized atrophy
of circumscribed areas of the cerebral cortex. They are
classified among the system atrophies. Histopathologic examination reveals
gliosis and spongiform
changes.
Aside from frontotemporal dementia, focal cortical atrophy
can also be of the following kinds:
-
In primary progressive aphasia, the
language disturbance may precede the development of generalized
dementia by several years.
-
In posterior cortical atrophy,
dementia may be accompanied by the specific neuropsychological
deficits of Gerstmann syndrome (see
section ▶ 5.5.1,
▶ 5.5.1.4).
Frontotemporal dementia is also classified by some authors as a type of
frontal lobe degeneration.
Etiology and epidemiology Some cases are hereditary, with an autosomal dominant inheritance pattern.
Most patients are less than 65 years old; in this age group, frontotemporal
dementia is, in fact, nearly as common as Alzheimer disease. Overall,
however, the focal cortical atrophies account for only approximately 5% of
all cases of dementia.
Clinical features See also ▶ Table 6.40. Patients with frontotemporal dementia often
manifest frontal personality changes and abnormal
social conduct (see section ▶ 5.5.1).
They may have reduced drive, aphasia, and
difficulty initiating speech, as well as an affect disturbance. The type of
cognitive deficit depends on the particular area of cerebral cortex that is
affected. Patients often develop urinary
incontinence early in the course of this disease.
6.12.6 Vascular Dementia: SAE-Associated Dementia and Multi-Infarct
Dementia
SAE stands for subcortical arteriosclerotic encephalopathy. On this topic,
see also the discussion of lacunar infarction in
section ▶ 6.5.6.
Etiology and epidemiology Vascular dementia, the second most common etiologic category of dementia,
is caused either by multiple subcortical lacunar infarcts due to cerebral
microangiopathy (SAE) or, less commonly, by multiple cortical and
subcortical infarcts due to macroangiopathy or
recurrent embolic stroke (multi-infarct
dementia). These two mechanisms often operate in combination. The site and
extent of the infarcts determine the severity and rate of progression of the
dementia syndrome.
Clinical features See ▶ Table 6.38.
Vascular dementia often strikes patients with preexisting
arterial hypertension and/or other vascular
risk factors. There may be a history of transient neurologic deficits in the past, due to TIAs or minor
strokes. Dementia can arise suddenly or progress in
spurts. There may be accompanying neuropsychological deficits, such as
aphasia, as well as marked incontinence of affect: involuntary laughing and
crying are common. The neurologic findings include
enhanced perioral reflexes, signs of pseudobulbar palsy (e.g., dysarthria
and dysphagia), a tripping, small-stepped gait (old person’s gait, “marche à
petits pas”), and, sometimes, pyramidal and extrapyramidal signs. The psychopathology of subcortical vascular dementia
includes apathy, depression, and slowness. Patients can recall old
information more easily than they can store new information.
Diagnostic evaluation Neuroimaging reveals brain atrophy and evidence of multiple focal lesions, often old lacunar infarcts; these are
usually found in the subcortical white matter
( ▶ Fig. 6.62).
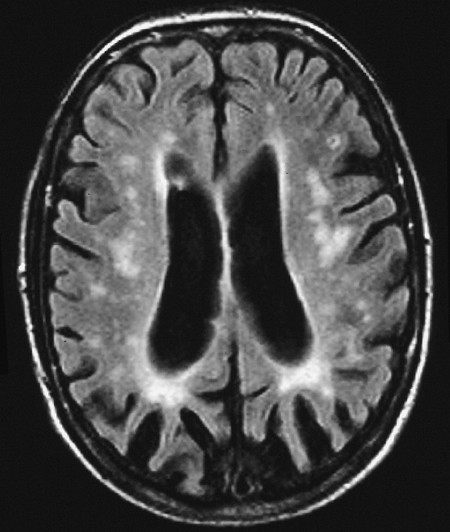
Fig. 6.62 Vascular
encephalopathy as seen by MRI. There are multiple focal
signal abnormalities in the deep white matter, the subcortical
region, and the cerebral cortex. The ventricles and subarachnoid
space are dilated (“hydrocephalus ex vacuo”).
Treatment The intermediate goal of treatment is vascular risk
reduction (treatment of arterial hypertension, cardiac
arrhythmias, diabetes mellitus, and hypercholesterolemia, if present;
inhibition of platelet aggregation with aspirin and/or other to lower the
risk of thrombosis). Generally speaking, the treatment is the same as that
discussed earlier for the prevention of ischemic stroke
(section ▶ 6.5.9). The symptomatic and supportive
measures are the same as for Alzheimer disease: cholinomimetic drugs (donepezil, galantamine, rivastigmine) and
the antiglutamatergic drug memantine alleviate the
cognitive disturbances.
Course and prognosis Vascular dementia is a progressive illness, typically with stepwise
progression. The rate of progression is variable, however, as it depends on
the type and extent of the underlying arteriopathy.
6.12.7 Dementia due to Malresorptive Hydrocephalus
Synonym This disorder is often incorrectly called (idiopathic or secondary) normal pressure hydrocephalus.
Epidemiology The peak incidence is between the ages of 50 and 70 years.
Etiology This disorder can arise in the aftermath of meningitis, aneurysmal or
traumatic subarachnoid hemorrhage, or venous sinus thrombosis; as a
consequence of elevated CSF protein concentration; or spontaneously, that
is, without any known risk factor. The common pathophysiologic mechanism is
impaired CSF outflow through the arachnoid
granulations and nerve root pouches, leading to a buildup of CSF
and ventricular dilatation (see ▶ Fig. 6.2).
Clinical features The classic clinical triad of malresorptive hydrocephalus includes a progressively severe gait disturbance, urinary incontinence, and psycho-organic syndrome. Neurologic examination reveals
paraparesis, ataxia, impaired memory, and, in advanced stages, dementia. Some patients also have headache.
Diagnostic evaluation CT or MRI characteristically reveals enlarged lateral ventricles, while the subarachnoid
space appears tight ( ▶ Fig. 6.2c). The CSF pressure
measured via LP is normal or, at most, mildly
elevated (hence the alternative name of this condition, “normal pressure
hydrocephalus”).
The removal of CSF via LP can bring about prompt, but transient,
improvement of the patient’s gait and memory. The “sticky” and
small-stepped gait can suddenly become much more fluid. A good response
to CSF removal (the “CSF tap test”) supports the diagnosis of
malresorptive hydrocephalus and implies a likely benefit from treatment
with a shunt (see later).
Treatment If the patient responds well to the CSF tap test, a ventriculoperitoneal shunt is neurosurgically implanted. This
generally leads to an improvement of the symptomatic triad.