6.9 Parkinson Disease and Other Hypertonic–Hypokinetic Syndromes
The common feature of diseases of the basal ganglia is a
movement disorder in which there is either too much or too little movement,
that is, an excess or deficiency of movement impulse, movement automaticity,
and/or muscle tone (see section ▶ 5.5.2).
In general, such diseases are characterized by the following:
-
Disturbances of the process or movement (always).
-
Abnormally increased or diminished muscle tone (usually).
-
Involuntary movements (often).
-
Associated neuropsychological manifestations (sometimes).
Increased muscle tone is often associated with paucity of
movement, and, conversely, diminished muscle tone with excessive movement. Thus, there are two main classes of
extrapyramidal syndrome:
-
Hypertonic–hypokinetic extrapyramidal
syndromes (above all, parkinsonian syndromes and related
neurodegenerative disorders, which will be discussed in this
section).
-
Hypotonic-hyperkinetic extrapyramidal
syndromes (e.g., chorea, athetosis, ballism, and dystonia,
which will be discussed in the next section).
6.9.1 Overview
In hypertonic–hyperkinetic syndromes, elevated muscle tone is
typically manifest as rigidity. Paucity of
movement, depending on its severity, is termed either hypokinesia (= diminished movement) or akinesia (= complete lack of movement). A third so-called
“cardinal manifestation,” tremor, is also
commonly present. This clinical triad, called the parkinsonian syndrome (or parkinsonism), is typically found
in idiopathic Parkinson disease. Often, postural instability (= tendency to fall) occur as a fourth cardinal
manifestation.
Parkinson disease, however, is only
one possible cause of parkinsonism; there are many others besides.
Parkinsonism may be due to an underlying illness or condition other than
idiopathic Parkinson disease (symptomatic parkinsonian
syndromes). In addition, several systemic
neurodegenerative diseases cause parkinsonism. These rare
diseases are marked by a loss of neurons not only in the basal ganglia, but
also in other areas of the CNS, and thus are clinically characterized not
only by extrapyramidal manifestations, but also by neurologic deficits
localizable to other regions of the brain. The most important diseases in
this category, multisystem atrophy (MSA) and corticobasal degeneration
(CBD), will be discussed further in this chapter. Lewy body dementia belongs
in this category as well but will be discussed later in the subsection on
dementia (section ▶ 6.12).
6.9.2 Parkinson Disease (Idiopathic Parkinson Syndrome)
Definition Parkinson disease is defined by its clinical manifestations
(characteristic body posture and gait, with hypokinesia, rigidity, and,
usually, rest tremor) and their pathologic correlates in the brain: Lewy
bodies containing α-synuclein and degeneration of dopaminergic neurons in
the pars compacta of the substantia nigra (a pigmented nucleus in the
midbrain).
Epidemiology, Etiology, and Pathogenesis
Epidemiology and etiology Parkinson disease has an overall prevalence of 0.15%. Its age-specific
prevalence rises with increasing age, to 1% in persons older than 60
years and 3% in persons older than 80 years. Most cases are idiopathic, that is, without
any identifiable cause.
Familial clustering of Parkinson
disease is seen in 5 to 15% of cases (so-called hereditary Parkinson disease); patients who develop
Parkinson disease at an unusually young age are
particularly likely to have a problem of this type. To date, 18 genetic
loci (PARK1 through PARK18) and at least 7 genes have been identified
whose mutations can cause a hereditary parkinsonian syndrome. The mode
of inheritance can be autosomal dominant with variable penetrance or
autosomal recessive. A special type is the familial Parkinson–dementia
complex seen on the island of Guam. The combination of parkinsonism and
dementia also sometimes exhibits familial clustering.
Pathogenesis The neuropathologic hallmark of idiopathic Parkinson
disease is degeneration of the dopaminergic neurons of
the substantia nigra and the locus
ceruleus. Hyaline inclusion bodies, called Lewy bodies, are found within the degenerated neurons. The
loss of dopaminergic neurons leads to a degeneration
of the (inhibitory) nigrostriatal dopaminergic pathway and,
therefore, to dopamine deficiency in the
striatum. This, in turn, leads to enhanced activity of the
striatal glutamatergic neurons, which produces the clinical
manifestations of the disease.
In the Braak system of
Parkinson disease there are six neuropathologic
stages that trace the temporal progression of
intraneuronal Lewy body formation from lower to higher neural
centers in the brain. In stages 1 and 2, before any clinical
manifestations of the disease have arisen, Lewy bodies are present
only in certain areas of the brainstem and the olfactory system. The
first symptoms arise in stages 3 and 4 when Lewy bodies begin to
appear in the substantia nigra. Finally, in stages 5 and 6, Lewy
bodies are found in diffuse areas of the cerebral cortex.
Clinical Features
The clinical picture of idiopathic Parkinson disease and of hereditary
parkinsonian syndromes ( ▶ Fig. 6.55) is typically characterized by:
-
Hypokinesia, i.e., slowing of
movement.
-
Increased muscle tone
(rigidity).
-
Abnormal body posture (stooped head
and trunk, flexion at the knees).
-
Impaired postural reflexes, sometimes
leading to falls.
-
Often tremor.
-
Later, neuropsychological deficits
and certain vegetative/autonomic
disturbances such as oily (seborrheic) face and
bladder dysfunction.
The motor signs are often only unilateral, or more marked on one
side, when the disease first appears. They can be aggravated by
emotional stress.
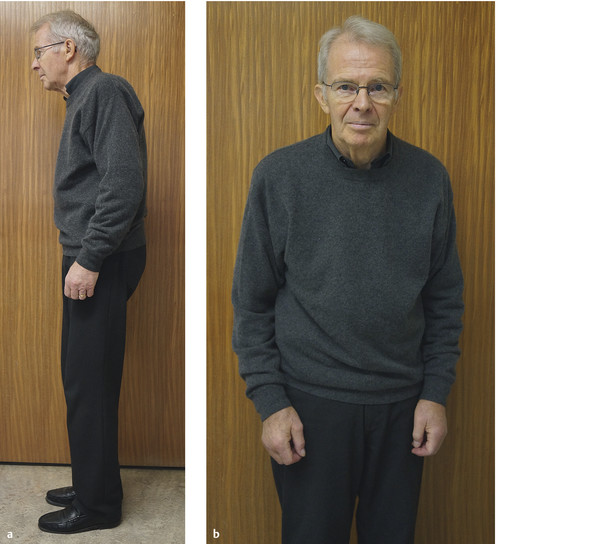
Fig. 6.55 Parkinson
disease. (a) Typical posture
with stooped head and upper body and lightly flexed elbows,
hips, and knees. (b) Hypomimia (paucity
of facial expression) and the asymmetry of manifestations that
is typical in idiopathic Parkinson disease (the right elbow is
somewhat more strongly flexed than the left).
Hypokinesia Hypokinesia manifests itself as paucity
of facial expression (mask-like facies), reduced frequency of blinking, and speech
disturbances (dysphonia, i.e., slow, monotonous, unmodulated
speech, and repetitions). There is little spontaneous movement, and the
normal accessory movements (e.g., arm swing during walking) are
diminished or absent. The patient’s handwriting becomes progressively
smaller (micrographia). Repeated or alternating
movements (e.g., finger-tapping) are performed slowly and with smaller
excursions (dysdiadochokinesia; cf. ▶ Fig. 3.19). Axial
movements, such as turning around while standing or turning over in bed,
are difficult to perform. Very severe hypokinesia is sometimes called
akinesia.
Gait A parkinsonian gait is characterized by a mildly stooped posture, with the head jutting forward,
and a small-stepped, often
shuffling gait, without accessory arm
movements ( ▶ Fig. 6.56). To turn around while standing, the
patient makes many small turning steps.
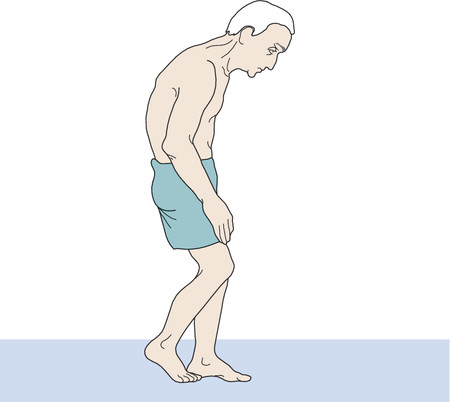
Fig. 6.56 Typical
parkinsonian posture while walking: inclined head,
slightly stooped upper body, flexed elbows, and lightly flexed
hips and knees.
Increased muscle tone This is primarily evident as rigidity ( ▶ Fig. 3.24), felt by the
examiner during large-amplitude, passive flexion and extension of the
joints. Rigidity is sometimes easier to detect when the patient
voluntarily contracts the muscles on the opposite side of the body.
Often, during passive movement, the examiner may feel a small, brief,
periodically recurring diminution of muscle tone, known as the cogwheel phenomenon, which is usually most
evident at the wrist ( ▶ Fig. 3.25). The patient’s
postural tone, too, is elevated; if, for
example, the head is lifted off the bed and let go, it may remain
suspended in midair for some time (the Wartenberg sign; the classic
literature spoke of a “coussin psychique,”
i.e., a virtual pillow).
Another test for the objectification of rigidity is the
so-called swinging test: the examiner grasps and shakes the
patient’s forearm back and forth. Rigidity markedly diminishes the
swinging (pendular) motion of the wrist. The test can also be
performed at the elbow or knee joint.
Tremor Three-quarters of patients with Parkinson disease have
tremor sooner or later in the course of their disease, typically a distal rest tremor at a frequency of 5 Hz. A
pronation–supination (“pill-rolling”)
tremor is highly characteristic. The tremor is present at
rest and generally disappears on voluntary movement; it is sometimes
increased by mental exertion, deep concentration, or walking. Some
patients have postural and intention tremor in addition to rest tremor (see
▶ Fig. 3.22).
The risk of falling An impairment of postural
reflexes, combined with hypokinesia, has the consequence that
changes of body posture and orientation in space can no longer be
compensated for by reflexive, rapid corrective movements. The most
obvious manifestations of this problem are pro- and
retropulsion. If the patient is pushed while standing still,
or stumbles over an obstacle, the movements made to regain balance are
too small and too slow, and a fall may
result.
The patient’s postural reflexes and possible tendency
to fall can be tested with the pull test and the push-and-release
test. In the former, the examiner stands behind the patient and
pulls back on both shoulders; in the latter, the patient is propped
up in a standing position from behind by the examiner’s hands, which
are then suddenly released. (Obviously, when performing these tests,
the examiner must make sure that the patient can be caught in case
of a fall.)
Impaired olfaction An impaired sense of smell is almost universal in patients
with idiopathic Parkinson disease but rare in patients with symptomatic
parkinsonism.
neuropsychological deficits When the first symptoms of Parkinson disease arise, the
patient’s cognitive functions are generally normal or only mildly
impaired. As the disease progresses, however, neuropsychological
deficits almost always arise. Memory is impaired, cognitive processes
are slowed (bradyphrenia), and there is a tendency toward perseveration.
Rapid changes in thought content are difficult to achieve, and the
planning and execution of actions and behaviors is impaired (so-called
dysexecutive syndrome).
Psychiatric manifestations Depression affects one-third to one-half of all patients
over the course of the disease and is treatable. Isolated apathy
(without depression) can also arise. Impulse-control disorders, such as
compulsive shopping, gambling, or hypersexuality, are usually side
effects of dopaminergic drugs. The patient’s perceptions and thought
processes can become abnormal over the course of the disease, because of
either the disease itself or its dopaminergic treatment; hallucinations
and overt psychoses can result.
Disturbances of autonomic and vegetative function Such disturbances arise partly as a by-product of
hypokinesia and partly because of direct involvement of the autonomic
nervous system. These include seborrhea (an
oily face, caused by excessive fat production in the skin),
hypersalivation, cold intolerance, a tendency toward orthostatic hypotension and constipation, urinary urgency
(possibly causing incontinence), and sexual dysfunction (altered libido,
erectile dysfunction). Insomnia and
behavioral disturbances during REM sleep (see
section ▶ 10.4) are often seen
early on in the course of disease; the patient’s sleep can also be
disturbed by restless legs syndrome (see
section ▶ 10.2.2) or spontaneous
pain in the limbs.
The nonmotor manifestations of Parkinson disease are summarized in
▶ Table 6.30.
Table 6.30 Nonmotor manifestations of Parkinson
disease
|
Autonomic/vegetative
|
Cognitive
|
Psychiatric
|
-
Hyperhidrosis
-
Hypersalivation
-
Seborrhea
-
Obstipation
-
Cold intolerance
-
Circulatory dysregulation, orthostatic
hypotension
-
Sexual dysfunction (loss of libido, abnormally
increased libido, erectile dysfunction)
-
REM-sleep behavioral disorder
-
Insomnia
-
Daytime somnolence, sleep attacks
-
Pain, paresthesiae
-
Hyposmia, anosmia
|
-
Slowed thinking (bradyphrenia)
-
Perseveration
-
Impaired planning, strategy, and execution
(dysexecutive syndrome)
-
Cognitive impairment, ranging from mild to
severe frontal dementia in advanced disease
|
-
Depression
-
Apathy
-
Hallucinations, illusions
-
Psychosis (mainly as a drug effect)
-
Impulse-control disorder (compulsive shopping,
gambling, or sexual behavior; mainly as a drug
effect)
|
Classification and grading of manifestations The manifestations described are present to variable
extents in different patients with Parkinson disease. Generally
speaking, the disease has three main clinical variants:
-
The akinetic-rigid type (without
tremor).
-
The tremor-dominant type (with little
hypokinesia and rigidity).
-
The mixed or “equivalence” type (with
roughly equal severity of all three cardinal
manifestations—rigidity, hypokinesia, and tremor).
Individual clinical manifestations can be graded on
pseudoquantitative scales, if this is desired for long-term follow-up or
for research purposes, for example, with the Webster
Rating Scale ( ▶ Table 6.31) or the very detailed Unified Parkinson's Disease Rating Scale (UPDRS), which is
not reproduced here. Cognitive function can be assessed with the MOCA
test or the Mini-Mental State Examination (MMSE; see
▶ Table 3.11).
Table 6.31 Simplified scale for evaluating the
severity of individual signs of Parkinson disease
|
1. Bradykinesia of hands, including handwriting
|
0 = normal
1 = mild slowing
2 = moderate slowing, handwriting severely
impaired
3 = severe slowing
|
|
2. Rigidity
|
0 = none
1 = mild
2 = moderate
3 = severe, present despite medication
|
|
3. Posture
|
0 = normal
1 = mildly stooped
2 = arm flexion
3 = severely stooped; arm, hand, and knee
flexion
|
|
4. Arm swing
|
0 = good bilaterally
1 = unilaterally impaired
2 = unilaterally absent
3 = bilaterally absent
|
|
5. Gait
|
0 = normal, turns without difficulty
1 = short steps, slow turn
2 = markedly shortened steps, both heels slap on
floor
3 = shuffling steps, occasional freezing, very slow
turn
|
|
6. Tremor
|
0 = none
1 = amplitude < 2.5 cm
2 = amplitude > 10 cm
3 = amplitude > 10 cm, constant, eating and writing
impossible
|
|
7. Facial expression
|
0 = normal
1 = mild hypomimia
2 = marked hypomimia, lips open, marked
drooling
3 = mask-like facies, mouth open, marked
drooling
|
|
8. Seborrhea
|
0 = none
1 = increased sweating
2 = oily skin
3 = marked deposition on face
|
|
9. Speech
|
0 = normal
1 = reduced modulation, good volume
2 = monotonous, not modulated, incipient dysarthria,
difficulty being understood
3 = marked difficulty being understood
|
|
10. Independence
|
0 = not impaired
1 = mildly impaired (dressing)
2 = needs help in critical situations, all activities
markedly slowed
3 = cannot dress him- or herself, eat or walk
unaided
|
The Neurologic Examination and Other Diagnostic
Tests
The diagnosis of Parkinson disease is based on the typical
clinical manifestations and characteristic
findings on neurologic examination.
History Important points to be addressed in clinical history-taking include
the following:
-
Has the patient had difficulty with fine motor activities such
as writing, getting dressed, or eating?
-
Is the patient’s gait less steady than before, perhaps with
stumbling or falls?
-
Has the patient noticed any difference between the right and
left sides of the body?
-
Does the patient suffer from pain or disturbed sleep?
-
Has there been any impairment of the sense of smell or any
difficulty swallowing?
Neurologic examination In addition to hypokinesia, rigidity, tremor, and propulsion/retropulsion, the examination generally reveals
the following:
-
Weak convergence (movements to focus
the eyes are slowed).
-
A persistent glabellar reflex (i.e.,
lack of habituation of the reflex after repeated glabellar
tapping).
-
Saccadic ocular pursuit
movements.
-
Impaired olfaction.
The intrinsic muscle reflexes are normal, however, as are all
somatosensory modalities.
For numerical grading, see the preceding paragraphs and
▶ Table 6.31.
Imaging studies CT and MRI of the head reveal no abnormalities and are generally
performed only to rule out competing diagnoses, for example, symptomatic
parkinsonian syndromes. The loss of dopaminergic
afferent input to the striatum can be demonstrated with positron emission tomography (PET) or single-photon emission computed tomography
(SPECT) after the administration of 18fluorodopa
( ▶ Fig. 6.57). Cerebral
ultrasonography can reveal early changes in the substantia nigra.

Fig. 6.57 An
18F-DOPA-PET scan in a normal person (left,
top and bottom) and in a patient with incipient Parkinson
disease, worse on the left side of the body (right, top and
bottom). The basal ganglia are seen in axial and
coronal section (upper and lower rows of images, respectively).
The patient with Parkinson disease has a more than 20% reduction
in the activity of dopamine decarboxylase in the right putamen
(particularly in its dorsal aspect), with relatively normal
activity in the caudate nucleus. (Image provided courtesy of Dr.
F. Jüngling, PET/CT-Zentrum NW-Schweiz, St. Claraspital, Basel,
Switzerland.)
Idiopathic Parkinson disease is always a diagnosis of exclusion,
that is, all varieties of symptomatic parkinsonism must be ruled out before this diagnosis can be
made.
Testing of olfaction Impairment of the sense of smell early on in the course of
disease is supporting evidence for idiopathic or genetically triggered
Parkinson disease. Smell is tested with small samples of various
substances (coffee, etc.).
Genetic testing In young patients with a positive family history, genetic
testing can help secure the diagnosis and enable a more accurate
prognosis.
Treatment, complications, and prognosis Effective treatment alleviates the manifestations of the
disease, moving the symptomatic progression curve to the right by some 3
to 5 years, but does not affect the disease process as such. The
putative early neuroprotective effect of
certain antiparkinsonian drugs has not yet been confirmed.
Pharmacotherapy The goal of drug therapy is to replace
the missing dopamine in the striatum.
The most important antiparkinsonian drug is still levodopa (L-DOPA), which is metabolized to
dopamine in the CSF. At the beginning of treatment, only small doses
are needed to alleviate the clinical manifestations; later on,
however, higher doses are needed, and side effects such as
dyskinesia and on–off motor fluctuations commonly arise. Therefore,
efforts are often made to delay the administration of L-DOPA to
younger patients by giving alternative drugs first.
-
MAO-B inhibitors are
increasingly used as drugs of first choice because of their
putative neuroprotective effect. These include selegiline and
rasagiline (a long-acting selegiline derivative). They inhibit
the degradation of dopamine to homovanillic acid and thereby
ameliorate parkinsonian manifestations.
-
Amantadine, an
NMDA-receptor antagonist, is sometimes used very early in the
course of disease; it is thought to enhance dopamine release
from nerve terminals.
-
If these agents are no longer sufficiently
effective, nonergot dopamine agonists
(e.g., ropinirole, pramipexole, or rotigotine) can be used in
younger patients; their effectiveness, however, matches that of
L-DOPA only in the early stages of the disease.
-
In older patients, L-DOPA
is used from the outset. This agent, unlike dopamine itself,
crosses the blood–brain barrier; it is converted to dopamine in
the CSF.
-
L-DOPA is always given in combination with a decarboxylase inhibitor (e.g., benserazide [HCl] or carbidopa) to prevent its premature degradation in
the periphery.
-
The combination of L-DOPA with a COMT inhibitor, such as entacapone or tolcapone,
can further increase dopamine bioavailability. Tolcapone,
however, is occasionally hepatotoxic and is therefore reserved
for otherwise intractable cases. Entacapone is sold as a
combination drug together with L-DOPA and a decarboxylase
inhibitor.
-
Anticholinergic drugs,
such as biperiden, are mainly effective
against tremor. Note: their
anticholinergic effect can also induce or worsen confusion
and/or dementia.
The following should be borne in mind as rules of thumb
for the dosing of antiparkinsonian drugs, particularly
L-DOPA:
Drug side effects and complications Prolonged treatment with L-DOPA and other antiparkinsonian
drugs can cause several problems:
-
Fluctuations in drug effect (on–off
fluctuations, end-of-dose akinesia) can often be
improved by the use of sustained-release L-DOPA preparations,
division of the daily dose into smaller individual doses at more
frequent intervals (perhaps with the use of water-soluble L-DOPA
preparations), and/or the addition of dopamine agonists or COMT
inhibitors. Water-soluble L-DOPA takes effect very rapidly and
can shorten off-phases or prevent imminent off-phases. Deep
brain stimulation is another therapeutic possibility (see
later).
-
Drug-induced dyskinesias,
for example, peak-dose dyskinesia or
hyperkinesia (often involving
choreiform involuntary movements; see
section ▶ 6.10), are
seen in 40% of patients after 6 months of L-DOPA treatment, in
60% after 2 years, and in nearly all after 6 years. They are
usually more disturbing to patients’ families than to the
patients themselves, but they can be disabling if severe and may
be untreatable except by deep brain stimulation.
-
Painful foot dystonia can
be managed with the use of sustained-release preparations in the
evening and perhaps with apomorphine injections.
-
“Freezing,” that is,
sudden arrest of movement, is not directly related to the serum
concentration of L-DOPA. Various mental techniques can help
(carrying a briefcase, stepping over real or imagined obstacles,
etc.).
-
Psychosis and other
psychiatric disturbances (e.g., hypersexuality, illusions,
hallucinations, delusions, compulsive gambling, food cravings)
usually arise as side effects of drugs, especially dopamine agonists (ropinirole,
pramipexole). They may respond to a reduction of the dose or to
the addition of an atypical neuroleptic drug (clozapine,
risperidone).
-
An akinetic crisis is a
prolonged phase of extreme rigidity causing complete immobility and accompanied by hyperthermia,
hyperhidrosis, other autonomic disturbances, and dysphagia. Such
crises can be precipitated, for example, by abrupt
discontinuation of antiparkinsonian drugs, errors in drug-taking
or drug-prescribing, the use of neuroleptic drugs, surgical
procedures, or infection. They can be treated with water-soluble
L-DOPA (given by nasogastric tube), amantadine (by intravenous
infusion), or apomorphine (by subcutaneous injection).
-
Further side effects that can be induced by any
dopaminergic drug include nausea
(especially at the beginning of treatment; it can be
counteracted with an antiemetic dopamine antagonist that has a
purely peripheral effect, such as domperidone), fatigue, and
orthostatic hypotension.
Movement disorders that are caused by L-DOPA:
On-dyskinesia (also called peak-dose or
plateau hyperkinesia); choreatiform movements that arise as the
serum L-DOPA concentration increases (see
section ▶ 6.10).
Off-dystonia (also called early-morning
dystonia): painful dystonia, for example, in one or both feet, that
arises as the serum L-DOPA concentration falls.
Deep brain stimulation Neurosurgical treatment, consisting of the stereotactic implantation of stimulating
electrodes into the thalamus (nucleus ventrointermedius),
globus pallidus, or subthalamic nucleus for chronic electrical
stimulation, can markedly alleviate the manifestations of the disease;
its indication depends on their severity and intractability in the
individual patient. Each stimulating electrode has multiple metallic
contacts, at which the intensity, pulse width, and frequency of the
applied current can be independently controlled to optimize the clinical
effect. This method has now largely replaced earlier destructive methods
involving the creation of permanent lesions. It was once considered a
treatment of last resort but is now increasingly used for patients in
intermediate stages of the disease. An overview of the expected effects
of deep brain stimulation in each of the three currently used target
structures is provided in ▶ Table 6.32.
Table 6.32 Current target structures for deep
brain stimulation in the treatment of Parkinson disease, and the
expected effects of stimulation at each structure
|
Target structure
|
Best effect
|
Remarks
|
|
Subthalamic nucleus
|
Reduction of hypokinesia, less frequent and less
intense off-phases
|
The L-DOPA dose can be markedly reduced. Dyskinesia
and tremor improve as well
|
|
Globus pallidus internus
|
Reduction of dyskinesia during on-phases
|
Hypokinesia and rigidity improve as well, but less
than with stimulation in the subthalamic nucleus
|
|
Nucleus ventrointermedius of the thalamus
|
Reduction of tremor
|
Little effect on hypokinesia or dyskinesia
|
Further types of treatment Aside from drugs and surgery, physical
therapy and regular exercise (sports, walking, hiking) can
help improve and maintain mobility. Speech
therapy may be helpful as well.
Moreover, adequate psychological support for
patients and their families is important. Self-help
groups can be valuable in this regard.
Course and prognosis L-DOPA treatment can shift the
symptomatic progression curve to the right by 6 to 7 years. It is hard
to predict which patients will eventually become dependent on the help
of others or on around-the-clock nursing care. This tends to occur after
approximately 20 years of illness.
The tremor-dominant type has a
relatively favorable prognosis. The prognosis is worse for older
patients, men as opposed to women, and patients with severe disease (in
terms of both motor and nonmotor manifestations). Parkinson disease can
shorten the patient’s life span.
Parkinson disease is a progressive disease whose manifestations
can be alleviated by drugs, deep brain stimulation, and
physiotherapy.
Differential Diagnosis
Table 6.33 The differential diagnosis of
idiopathic Parkinson disease
|
Cause of parkinsonism
|
Examples
|
|
Arteriosclerotic
parkinsonism
|
|
|
Drug-induced
parkinsonism
|
-
Neuroleptic agents (most common cause)
-
Antiemetic drugs (mainly dopamine agonists
such as metoclopramide)
-
Lithium
-
Valproic acid
-
Reserpine
-
Calcium antagonists such as flunarizine
|
|
Parkinsonism of infectious
origin
|
|
|
Normal pressure hydrocephalus
(also called malresorptive hydrocephalus)
|
|
|
Toxic parkinsonism
|
-
Carbon monoxide poisoning
-
Manganese poisoning
-
MPTP
(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a
by-product of the synthesis of the designer drug
MPPP)
-
Cyanide
-
Methanol
|
|
Trauma
|
|
|
Metabolic diseases
|
-
Hypo- and pseudohypoparathyroidism with basal
ganglionic calcification
-
Idiopathic basal ganglionic calcification,
Fahr disease
|
|
Neurodegenerative diseases
(see also ▶ Table 1.1)
|
-
Progressive supranuclear palsy
-
Multisystem atrophy
-
Corticobasal degeneration
-
Lewy body disease
-
Frontotemporal dementia
-
Alzheimer disease with parkinsonian
manifestations
|
|
Hereditary diseases that can have
prominent parkinsonian manifestations
|
-
Wilson disease
-
Pantothenate kinase–associated
neurodegeneration
-
SCA3 spinocerebellar ataxia
-
Fragile-X-tremor-ataxia syndrome
(FXTAS)
-
Westphal variant of Huntingtonchorea
-
Prion diseases
|
|
Further causes
|
|
Tremor, hypokinesia, and rigidity can also be expressions of diseases
other than Parkinson disease, including symptomatic parkinsonian
syndromes.
The main differential diagnoses of idiopathic Parkinson
disease are: neuroleptic side effects and
other neurodegenerative systemic
diseases causing abnormal movements: Lewy body dementia,
MSA, progressive supranuclear palsy (PSP), malresorptive hydrocephalus, and subcortical arteriosclerotic encephalopathy.
Neuroimaging now enables the recognition of some of these
entities by their typical MRI findings:
-
Ventricular enlargement in malresorptive hydrocephalus (also
called “normal pressure hydrocephalus”).
-
“Hot-cross-bun” sign in MSA.
-
Midbrain atrophy with the “Mickey Mouse” and/or “hummingbird”
signs, along with frontal atrophy, in PSP.
An overview of these and other differential diagnoses is provided in
▶ Table 6.33.
6.9.3 Symptomatic Parkinsonian Syndromes
There are several clinical conditions that resemble idiopathic
Parkinson disease but have a different underlying cause or pathophysiologic
mechanism. The clue to such a condition may be a history of a precipitating event (e.g.,
intoxication, drug use, trauma, or infection) or a structural abnormality of the basal ganglia or other
brain areas (e.g., multiple arteriosclerotic
changes, hydrocephalus) revealed by CT
or MRI. A further characteristic of symptomatic parkinsonism is its relative resistance to treatment with L-DOPA, in
contrast to idiopathic Parkinson disease, which usually responds very well
to L-DOPA, at least at first. Moreover, some forms of symptomatic
parkinsonism present with symmetric manifestations, while idiopathic
Parkinson disease generally presents asymmetrically.
6.9.4 Progressive Supranuclear Palsy
This disease is also known as Steele–Richardson–Olszewski syndrome.
Etiology and pathology A polymorphism in the tau protein gene (chromosome 17) causes deposition
of an abnormally phosphorylated tau protein in the cells of the basal
ganglia. This “tauopathy,” in turn, leads to cellular degeneration in the
substantia nigra, globus pallidus, subthalamic nucleus, periaqueductal area
of the midbrain, and other brain nuclei.
Clinical features The clinical features of PSP include the following:
-
Paucity of movement.
-
Gait disturbance early in the course of
the disease.
-
Predominantly axial rigidity, often with
a permanently extended cervical spine (head
turned upward).
-
Frequent falls with a tendency to fall
backward.
-
Progressive dementia.
-
Impaired vertical gaze movements (particularly downward), with
nystagmus.
Diagnostic evaluation PSP is diagnosed from its typical clinical features. MRI reveals midbrain atrophy.
Treatment and course This disease presents between ages 50 and 70 years, mainly in men. Its
manifestations tend to respond only weakly to L-DOPA, or not at all. PSP
progresses rapidly and causes death within a few years.
6.9.5 Multisystem Atrophy
This term subsumes a collection of rare diseases
that were previously described as separate entities:
-
Olivopontocerebellar atrophy (OPCA), now
also called MSA-C (cerebellar type).
-
Striatonigral degeneration (SND), now
also called MSA-P (parkinsonian
type).
-
Shy–Drager syndrome (SDS).
-
Mixed forms of these.
Pathology The neuropathologic lesion consists of intracellular inclusion bodies
(with α-synuclein; synucleinopathy) in glial cells as well as cellular
degeneration and gliosis in the substantia nigra, striatum, pons, inferior
olive, and cerebellum.
Clinical features The main clinical features of MSA are present to varying extents in its
different forms of MSA, each of which has its own characteristic initial
presentation:
-
Parkinsonism:
bradykinesia, akinesia, rigidity, and rest
tremor (seen early in the course of MSA-C and
MSA-P).
-
Autonomic dysfunction, including
orthostatic hypotension, incontinence, and erectile dysfunction in
men (seen early in the course of SDS).
-
Ataxia and other cerebellar signs (prominent in OPCA, i.e.,
MSA-C).
-
Pyramidal tract signs such as pathologic
reflexes and spasticity.
-
In some cases, dementia and/or frontal signs.
Diagnostic evaluation These conditions are diagnosed from their clinical
features. The diagnosis of MSA may be supported by an MRI finding of focal brain atrophy, for example,
cerebellar atrophy or a loss of fiber connections in the pons (the
“hot-cross-bun” sign with cross-shaped hypointensity in the pons).
Treatment MSA generally responds poorly to treatment; dopamine agonists tend to be
more effective than L-DOPA. The disease usually leads to severe disability
and death within a few years of onset.
6.9.6 Corticobasal Degeneration
Pathology The neuropathologic lesion in this disease consists of cellular
degeneration and gliosis in the substantia nigra and in the pre- and
postcentral gyri. The cerebral peduncles are correspondingly diminished in
size. Like PSP, CBD is a type of tauopathy.
Clinical features The manifestations of CBD, which are asymmetrically distributed,
include:
-
Impaired fine motor control of an arm
(early in the course of the disease).
-
Progressive rigidity and akinesia.
-
Weakness.
-
Central sensory disturbances.
-
(Sometimes) apraxia.
-
(Sometimes) dystonia.
Treatment and course L-DOPA is generally not very effective and patients usually become
severely disabled within a few years of the onset of the disease.
6.9.7 Lewy Body Dementia
This disease is described later in the section on dementia
(section ▶ 6.12.3).
6.10 Chorea, Athetosis, Ballism, Dystonia: Hyperkinetic Syndromes
These disturbances, unlike Parkinson disease, are characterized
by “too much” movement, often in combination with diminished muscle tone.
The different clinical types of hyperkinesia include chorea, athetosis,
ballism, and dystonia, and mixed forms. Each of these movement disturbances
may be due to a variety of underlying diseases. Thus, the hyperkinetic
extrapyramidal syndroms are a diverse group both in their clinical features
and in their causes.
An overview of the hyperkinetic extrapyramidal syndroms is provided in ▶ Table 6.34. The more
important ones are described in greater detail in the text that follows.
Table 6.34 The diagnostic evaluation of hyperkinetic
extrapyramidal syndromes
|
Syndrome
|
Etiology
|
Remarks
|
|
Chorea: sudden, usually
rapid, distal, brief, irregular involuntary movements;
hypotonia
|
|
Chorea minor
|
Autoimmune; streptococcal infection
|
Often a sequela of streptococcal pharyngitis, most commonly in
girls aged 6 to 13 y
|
|
Chorea mollis
|
Autoimmune; streptococcal infection
|
Hypotonia is prominent
|
|
Chorea gravidarum
|
Third to fifth month of pregnancy
|
Usually during first pregnancy, often with prior history of
chorea minor
|
|
Chorea due to antiovulatory drugs
|
Antiovulatory drugs
|
Rare, reversible with discontinuation of the drug
|
|
Huntington disease
|
Autosomal dominant
|
Onset usually at age 30 to 50 y; associated with progressive
dementia
|
|
Benign familial chorea
|
Autosomal dominant
|
Onset in childhood, no further progression, no dementia
|
|
Choreoacanthocytosis
|
Autosomal recessive
|
Mainly orofacial, tongue-biting, elevated CK, hyporeflexia,
acanthocytosis
|
|
Postapoplectic chorea
|
Vascular (prior stroke)
|
Sudden hemichorea and hemiparesis, often combined with
hemiballism
|
|
Senile chorea
|
Vascular and degenerative
|
Occasional presenile onset, often more severe on one side,
occasionally with dementia
|
|
Athetosis: slow,
exaggerated movements against resistance of antagonist
muscles, mainly distal; seem uncomfortable and
cramped
|
|
Status marmoratus
|
Perinatal asphyxia
|
Soon after birth, increasingly severe athetotic hyperkinesia,
often cognitive impairment, sometimes also spasticity
|
|
Status dysmyelinisatus
|
Kernicterus of the newborn
|
Onset immediately after birth, often with other signs of
perinatal brain damage; further progression
|
|
Pantothenate kinase–associated neurodegeneration
|
autosomal recessive disorder of pigment metabolism
|
Choreoathetotic movements beginning at age 5 to 15 y,
rigidity, dementia, and retinitis pigmentosa in one-third of
cases; progressive, with characteristic joint
hyperflexion/hyperextension; death by age 30 y
|
|
Hemiathetosis
|
Focal lesion of pallidum and striatum
|
Unilateral, may come about some time after the causative
lesion
|
|
Ballism/hemiballism: unilateral,
lightning-like, high-amplitude flinging movements of multiple
limb segments
|
Ischemic or neoplastic lesion of the subthalamic
nucleus
|
Sudden onset, usually with hemiparesis as well
|
|
Dystonic syndromes
|
|
Torsion dystonia: slow, tonic contractions of muscles or
muscle groups, of shorter or longer duration, usually against
the resistance of antagonist muscles
|
Familial types
|
Often in Ashkenazi Jewish families, onset before age 20 y with
focal dystonia; later, rotatory movements of the head and trunk,
as well as limb movements and athetotic finger movements
|
|
Symptomatic types
|
For example, in Wilson disease, Huntington disease,
pantothenate kinase–associated neurodegeneration
|
|
L-DOPA-sensitive dystonia: usually arises in childhood with
dystonia of variably localization and fluctuation over the
course of the day; progresses over the years
|
Sometimes due to familial tyrosine hydroxylase
deficiency
|
Responds well to L-DOPA; can be difficult to distinguish from
juvenile Parkinson disease with dystonia
|
|
Spasmodic torticollis: slow contraction of cervical and nuchal
musculature against antagonist resistance, with rotatory
movements of the head
|
Idiopathic, occasionally after cervical spine trauma and
various other causes
|
One-third spontaneous recovery, one-third no change, one-third
progression to torsion dystonia
|
|
Localized dystonia (see section
▶ 6.10.5)
|
|
For example, writer's cramp, faciobuccolingual dystonia,
oromandibular dystonia
|
6.10.1 HuntingtonChorea
Etiology Huntington disease (chorea major) is a genetic disorder of autosomal
dominant inheritance caused by an unstable CAG trinucleotide repeat
expansion on the short arm of chromosome 4.
Pathology The neuropathologic correlate of the disease is loss of small ganglion
cells, mainly in the putamen and the caudate nucleus.
Clinical features The disease generally becomes symptomatic between the ages of 30 and 50
years. As a rule, choreiform movements appear
first, followed by progressive dementia. Patients
who inherited the defective gene from their father
tend to develop overt disease at an earlier age, sometimes with rigidity and
pyramidal tract signs as the initial manifestations.
Chorea consists of irregular,
sudden, involuntary movements that are usually more
pronounced at the distal end of the limbs.
-
In some patients, these movements are of low amplitude and
look almost normal, resembling nonpathologic “fidgetiness;” in
others, they are massive and highly disturbing.
-
Chorea can be unilateral (“hemichorea,” ▶ Fig. 6.58) or bilateral.
-
Muscle tone is normal or diminished.
-
There is no weakness or sensory deficit, and pyramidal tract
signs are absent.
-
The intrinsic muscle reflexes are normal, except that they may
have a second extension phase (Gordon
phenomenon) if elicited at the same time as an
incipient choreiform movement.
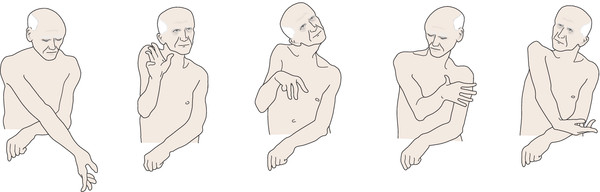
Fig. 6.58 Senile
hemichorea. Drawings from video stills.
Chorea, like other hyperkinesias (see later), is typically enhanced by
goal-directed movement, mental stress, or concentration, and subsides in
sleep and under general anesthesia.
Treatment, course, and prognosis Huntingtonchorea progresses chronically,
generally ending in death 10 to 15 years after the
onset of symptoms. There is no treatment other than palliative, symptomatic
management. The abnormal movements can be alleviated to some extent with
perphenazine,
tetrabenazine, tiapride, and other
neuroleptic drugs. Depression can be treated with an SSRI or sulpiride; anxiety, agitation,
and insomnia with benzodiazepines; and psychosis
with neuroleptic drugs, preferably atypical ones
such as olanzapine.
6.10.2 Chorea Minor (Sydenham Chorea)
Epidemiology Chorea minor is the most common disease
associated with choreiform movements. It mainly strikes school-aged girls.
Etiology and pathogenesis This disease arises after an infection with β-hemolytic
group A streptococci and is caused by an autoimmune reaction in
which antibodies are generated that cross-react with neurons.
Clinical features Within a few days or weeks after an attack of “strep throat,” or within a
few weeks or months of an attack of rheumatic fever, the patient develops
choreiform motor unrest (mainly in the face,
pharynx, and hands), combined with irritability and
other mental abnormalities.
Treatment, course, and prognosis The treatment is with high-dose penicillin for at
least 10 days. The manifestations resolve spontaneously in a few weeks or
months.
6.10.3 Athetosis
Pathology The neuropathologic basis of athetosis is loss of neurons in the striatum,
the globus pallidus, and, less commonly, the thalamus.
Etiology and types of athetosis Congenital and perinatally acquired lesions of the basal
ganglia (status marmoratus, status dysmyelinisatus, severe
neonatal jaundice = kernicterus) cause bilateral athetosis (athétose double), sometimes in conjunction with other
signs of brain damage. Choreoathetosis and dystonia are prominent
manifestations of iron deposition in the basal ganglia in pantothenate kinase–associated neurodegeneration. Focal brain lesions, too, for example, an infarct,
can produce hemiathetosis.
Clinical features Athetosis generally consists of slow, irregular
movements mainly affecting the distal ends of the limbs, causing
extreme flexion and extension at the joints and correspondingly bizarre
postures, particularly of the hands ( ▶ Fig. 6.59). The interphalangeal joints
may be hyperextended to the point of subluxation (“bayonet
finger”). Athetosis is often found in combination with chorea (“choreoathetosis”).
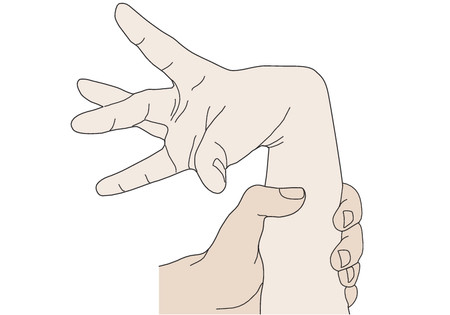
Fig. 6.59 Hand posture in
athetosis.
Athetosis can be hard to distinguish from dystonia and is often
designated as such. Athetosis can be considered a form of dystonia that
is most prominent at the distal end of the limbs.
6.10.4 Ballism
Pathology The neuropathologic substrate of ballism is a lesion of the contralateral
subthalamic nucleus (corpus Luysii) and/or its fiber connections to the
thalamus.
Etiology Ballism is usually due to focal ischemia, and
less commonly due to a space-occupying lesion. It
may also be the result of severe neonatal jaundice or of a hereditary
degenerative disease; it is typically bilateral in such patients.
Clinical features Rapid, propulsive, large-amplitude, unbraked flinging
movements of the limbs are seen on one side of the body
(hemiballism) or both. Unlike chorea, these movements occur mainly in the
proximal joints. The limbs may be hurled
against walls, etc., causing injury.
Treatment Haloperidol and chlorpromazine can alleviate ballism. Stereotactic neurosurgical
procedures are sometimes necessary.
6.10.5 Dystonic Syndromes
Pathology and etiology There are no characteristic neuropathologic abnormalities in dystonia. To
date, only a few of the disorders that cause dystonia have a known
pathophysiologic basis (e.g., L-DOPA-sensitive dystonia). Precipitating
factors of symptomatic dystonia are likewise only rarely identifiable.
Often, the etiology of dystonia remains unclear.
Clinical features Dystonia consists of slow, long-lasting contractions of
individual muscles or muscle groups. The trunk, head, and limbs
assume uncomfortable or even painful positions and maintain them for long
periods of time. The various clinical types of dystonia are classified as
either focal, that is, affecting isolated,
individual (small) muscle groups, or generalized.
Treatment In the treatment of generalized dystonia, baclofen, carbamazepine, and
trihexyphenidyl are used, either as monotherapy or in combination, often
with only limited success. For some types of dystonia, a trial of L-DOPA
treatment may be worthwhile. Focal dystonias can be successfully treated
with botulinum A toxin injections. In some cases of
dystonia, deep brain stimulation can be highly effective; the preferred
target structure is the globus pallidus internus.
Types of Generalized Dystonia
Torsion dystonia This category of dystonia is characterized by slow, forceful, mainly rotatory movements of the trunk
and head, usually accompanied by athetotic finger movements.
Muscle tone is diminished at the onset of the disease. In some cases,
hyperkinesia gradually ceases and gives way to hypertonia with a rigidly
maintained dystonic posture (myostatic type).
The various types of primary torsion dystonia are mostly of autosomal dominant inheritance, with low
penetrance, and have been localized to genes on various chromosomes. The
early-onset form is particularly common among Jews of Ashkenazi (Eastern
European) ancestry and is due to a genetic defect at the 9p34
locus.
L-DOPA-sensitive dystonia (Segawa disease) This is an autosomal recessive
disorder due to a genetic defect on chromosome 14q. It usually presents
in young girls as a gait
disturbance characterized by dystonic postures or movements
of the legs that fluctuate widely in severity over the course of the
day. It is liable to misdiagnosis as a psychogenic disorder. It
typically responds to low doses of L-DOPA (250 mg, or a little more, per
day). A therapeutic test of L-DOPA is worthwhile in any young patient
with dystonia, even if no other family members are affected.
Focal Dystonia
Focal dystonia is much more common than generalized dystonia. The
abnormal movements are restricted to individual parts of the body or
muscle groups. The main types of focal dystonia are the
following:
Spasmodic torticollis In this disorder, slow contraction of individual muscles of
the neck and shoulder girdle produce tonic rotation of
the head to one side or the other ( ▶ Fig. 6.60). It
is usually the contralateral sternocleidomastoid muscle that is most
strongly contracted. Only one-third of all patients with “wry neck” due
to spasmodic torticollis undergo spontaneous remission; a further third
go on to develop other dystonic manifestations. The etiology usually
remains unclear; there are probably multiple causes.
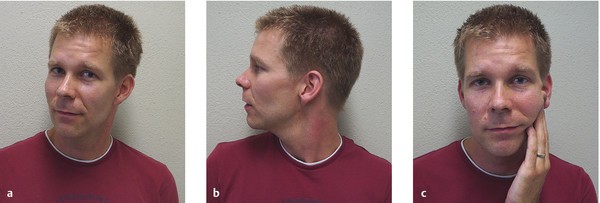
Fig. 6.60 Spasmodic
torticollis. (a) A
32-year-old man whose head is spontaneously lightly turned to
the right. (b) Torticollis with tonic,
involuntary head rotation to the right. Note the hypertrophic
left sternocleidomastoid muscle. (c)
The patient can bring his head back to the neutral position by
pressing gently on the lower jaw with a fingertip.
Blepharospasm This consists of bilateral tonic contraction of the
orbicularis oculi muscles, often with very
long-lasting involuntary eye closure, during which the
patient cannot voluntarily open his or her eyes. Blepharospasm tends to
affect older patients, mainly women. Eye closure may be forceful, with
visible contraction of the orbicularis oculi muscle, or weak, with a
relatively normal external appearance. Cases of the latter type are
alternatively designated lid-opening apraxia.
Misdiagnosis as a psychogenic disturbance is, unfortunately,
common.
Dystonia affecting multiple muscles of the head The various types of focal dystonia that come under this
heading are not rare when taken together; they include facio-bucco-lingual dystonia,
oromandibular dystonia, and Brueghel
or Meige syndrome. There may also be a
relatively isolated dystonia of the mouth, pharynx,
and tongue, particularly in patients who have been treated
with neuroleptics. An acute form can appear as a complication of
antiemetic drugs such as metoclopramide.
Isolated dystonia Isolated dystonias have been described for practically
every muscle group in the body. Dystonia of this type may be idiopathic
or may arise in connection with occupational
overuse of the muscle group in question. Well-known examples
include writer’s cramp, hand
dystonia in musicians, and foot
dystonia in certain other occupations. Spastic dysphonia is a focal dystonia of the laryngeal
musculature.
6.10.6 Essential Tremor and Other Types of Tremor
Types of tremor The main phenomenological distinction is between
the following:
-
Rest tremor.
-
Action tremor.
Action tremor, in turn, is subdivided into the following:
-
Postural tremor.
-
Isometric tremor (appearing when a muscle is contracted against
constant resistance).
-
Kinetic tremor (appearing only during movement). Intention tremor
is a kinetic tremor that worsens as the limb nears its
target.
Tremor can also be classified etiologically as
shown in ▶ Table 6.35.
Table 6.35 Types of tremor
|
Designation
|
Characteristics
|
|
Physiologic tremor
|
Seen in normal individuals; intensified by pain, anxiety,
cold, caffeine ingestion, etc.
|
|
Thyrogenic tremor
|
Hyperthyroidism also intensifies physiologic tremor
through the mediation of catecholamines. The tremor appears
as a fine trembling of the hands
|
|
Essential tremor
|
See text. This hereditary condition is sometimes
misdiagnosed as Parkinson disease
|
|
Orthostatic tremor
|
Similar to essential tremor bur arises only when the
patient is standing, manifesting itself as shaking of the
legs; mainly seen in the elderly. Other muscle groups can be
similarly affected when under tonic stress
|
|
Parkinsonian tremor
|
See the discussion of Parkinson disease (see
section ▶ 6.9.2).
Usually consists of rest tremor, e.g., of the hand or
fingers when resting on a solid surface, or of the hand when
the arm is dependent
|
|
Psychogenic tremor
|
Sudden attacks of tremor, e.g., in acute stress disorder;
often highly variable in intensity; coarse; of demonstrative
quality
|
|
Holmes tremor
|
Unilateral, low-frequency rest, postural, and intention
tremor (becomes stronger as the movement nears its
target)
|
|
Cerebellar tremor
|
Intention tremor arising after cerebellar injury (becomes
stronger as the movement nears its target)
|
|
Dystonic tremor
|
Tremor and movement disturbance due to dystonia
(section ▶ 6.5.10)
|
|
Asterixis (“flapping tremor”)
|
Sudden loss of postural tone, mainly seen in hepatic
encephalopathy but also in other liver diseases, e.g.,
Wilson disease
|
|
Alcohol-related tremor
|
Fine rest and intention tremor, worse during alcohol
withdrawal, improves after consumption of alcohol (but note:
essential tremor often improves with alcohol as well)
|
|
Tremor induced by drugs, hormones, or toxic
substances
|
Resembling physiologic tremor, but more intense
|
Essential tremor This is the most common type of tremor
and often runs in families. Genetic defects causing
familial essential tremor have been found on chromosomes 2p22– p25 and 3q.
It usually arises between the ages of 35 and 45 years. It is a predominantly
postural and sometimes also kinetic tremor of the hands; a pure intention
tremor is seen in 15% of patients (see
▶ Fig. 3.22). It may also affect the head in isolation (nodding or shaking tremor of the
“yes” or “no” type), sometimes including the chin and/or vocal cords.
Essential tremor typically improves after the consumption of a small
amount of alcohol and worsens with nervousness or stress.
“Essential tremor plus” is a
combination of this entity with another neurologic disorder (e.g., Parkinson
disease, dystonia, myoclonus, polyneuropathy, restless legs
syndrome).
Diagnostic evaluation Thorough history-taking and a precise neurologic examination often yield
important clues to the etiology of tremor; further studies (e.g., imaging
studies) are only rarely necessary. In some cases, electrophysiologic testing is needed to pinpoint the correct
diagnosis (e.g., in suspected orthostatic tremor). Laboratory testing may
also be needed to exclude underlying disease.
Treatment If the tremor is severe enough to interfere with the patient’s
everyday activities, a β-blocker such as
propranolol can be tried; this drug is particularly effective against
essential tremor. Primidone,
benzodiazepines, and clozapine are
further alternatives. A tremor accompanied by dystonia may respond well to
botulinum toxin injections. Deep brain stimulation
through an electrode that has been stereotactically implanted in the nucleus
ventrointermedius of the thalamus is highly effective but is reserved for
severe, medically intractable cases.
Differential diagnosis Involuntary movements arising from diseases of the basal ganglia must be
differentiated from a variety of other movement disorders, which are listed
in ▶ Table 5.3.