ENDOCRINE MYOPATHIES
ENDOCRINE MYOPATHIESMyopathies can occur in the setting of a variety of systemic diseases. Previous chapters have discussed inflammatory myopathies that can occur in the setting of connective tissue diseases (e.g., systemic lupus erythematosus, mixed connective tissue disease, Sjögren syndrome, and rheumatoid arthritis) and systemic infections (e.g., HIV). Myopathies occurring as complications of medications (toxic myopathies) are also dealt with elsewhere in the book. In this chapter, we will focus on myopathies related to endocrine disturbances, electrolyte imbalance, nutritional deficiency, and amyloidosis. We also discuss some other less well-defined syndromes such as fibromyalgia.
ENDOCRINE MYOPATHIESMyopathies can complicate various endocrinopathies.1,2 In this section, we review myopathies associated with thyroid, parathyroid, adrenal, pituitary, and pancreatic disorders.
Both hyperthyroidism and hypothyroidism can be associated with myopathy. In addition, polyneuropathy and neuromuscular junction disorders can occur with dysthyroid states and these need to be differentiated from one another.
The mean age of onset of thyrotoxicosis is in the fifth decade. The severity of the myopathy does not necessarily relate to the severity of the thyrotoxicosis. Muscle symptoms usually appear several months after the onset of other clinical symptoms associated with mild hyperthyroidism.3 Interestingly, thyrotoxicosis is more common in females; however, thyrotoxic myopathy occurs more commonly in men. Anywhere from 61% to 82% of patients with thyrotoxicosis have some degree of detectable weakness on examination, but only about 5% of patients with thyrotoxicosis present with muscle weakness as their chief complaint.1,3–5
Thyrotoxic myopathy is characterized by proximal muscle weakness and atrophy.2–4,6,7 Some individuals have severe shoulder-girdle atrophy and scapular winging.2 Distal extremity weakness can be the predominant feature in approximately 20% of patients.4 Myalgias and fatigue are common. Some patients develop dysphagia, dysphonia, and respiratory distress due to involvement of bulbar, esophageal–pharyngeal muscles, and ventilatory muscles.8,9 Weakness of extraocular muscles and proptosis occur in the setting of Graves’ disease but the sphincters are spared in hyperthyroidism. Rarely, rhabdomyolysis with myoglobinuria can develop in severe thyrotoxicosis.10
Muscle stretch tendon reflexes are often brisk. In addition, fasciculations and myokymia are occasionally seen which probably reflects thyrotoxicosis-induced irritability of anterior horn cells or peripheral nerves.11–13 Peripheral neuropathy in hyperthyroidism is quite rare, but a demyelinating polyneuropathy has been reported.11
Other manifestations of hyperthyroidism include nervousness, anxiety, psychosis, tremor, increased perspiration, heat intolerance, palpitations, insomnia, diarrhea, increased appetite, and weight loss. Common signs include goiter, tachycardia, atrial fibrillation, widened pulse pressure, as well as warm, thin, and moist skin.
Myasthenia gravis can develop in association with Graves’ disease. It can be a challenge distinguishing which neuromuscular symptoms are related to Graves’ disease or to myasthenia gravis. Muscle weakness associated with hyperthyroidism does not fluctuate or significantly improve with anticholinesterase medications.
Thyrotoxicosis is also associated with an unusual form of hypokalemic periodic paralysis. Thyrotoxic periodic paralysis (TPP) may occur sporadically, although a dominantly inherited mutation in a potassium channel has been recently identified in some patients. TPP has been commonly reported in Asians, but it is not restricted to this population.2,5 TPP is also more common in males. The attacks of weakness are similar in onset, frequency, duration, and pattern to familial hypokalemic periodic paralysis (Chapter 32). The one distinguishing feature is that familial hypokalemic periodic paralysis typically has its onset within the first three decades of life, while the onset of TPP usually develops later in adult life. Serum potassium levels tend to be low during the attacks of weakness, but levels can be normal. Muscle strength returns with treatment and normalization of thyroid function. β-adrenergic blocking agents also improve the myopathy.
Serum creatine kinase (CK) levels are usually normal in hyperthyroidism and can even be on the low side. Thyroid stimulating hormone (TSH) level is low in primary hyperthyroidism, while the thyroxine (T4) level and, occasionally, only the triiodothyronine (T3) level are elevated. In TTP serum potassium levels also are usually decreased. Routine motor and sensory nerve conduction studies (NCS) are normal.12,16 Electromyography (EMG) is usually normal, although fasciculation potentials and MUAP multiplets may be evident due to motor nerve hyperactivity.
Routine muscle biopsies are usually unremarkable, however, mild fatty infiltration, muscle fiber atrophy (types 1 and 2), variability in muscle fiber size, scattered isolated necrotic fibers, decreased glycogen, and increased internal nuclei can be noted.12,14–16 Nonspecific ultrastructural findings on electron microscopy (EM) may be seen including Z-band streaming, focal swelling of the T tubules, elongated mitochondria, decreased mitochondria, and subsarcolemmal glycogen deposition.17
In patients with TPP, muscle biopsies can reveal changes similar to that seen in familial hypokalemic periodic paralysis: Vacuoles may be appreciated, on routine light microscopy, while subsarcolemmal blebs filled with glycogen and dilated terminal cisternae of the sarcoplasmic reticulum might be apparent on EM.
The thyroid gland produces T4 that is converted to the more active T3 hormone in the periphery. These thyroid hormones are largely bound to plasma proteins. Free thyroid hormones bind to cytoplasmic receptors on target cells and are internalized into the nucleus, where they regulate the transcription of specific genes. Type 1 muscle fibers have a greater density of these thyroid receptors than do type 2 fibers.18
The pathogenic basis of thyrotoxic myopathy is unknown but is thought to be due to enhanced muscle catabolism. There is an increase in the basal metabolic rate with enhanced mitochondrial consumption of oxygen, pyruvate, and malate.18 Glucose uptake and glycolysis are stimulated in muscle independent of insulin.19 This can lead to an insulin-resistant state with fasting hyperglycemia and glucose intolerance and subsequent depletion of glycogen and reduced ATP production. Insulin resistance also may interfere with insulin’s anabolic effect on amino acid and protein metabolism.20 There is an inadequate level of protein synthesis to meet the demands of accelerated breakdown, which in turn, may be driven by increased lysosomal protease activity.21,22
A mutation in the KCNJ18 gene that encodes an inwardly rectifying potassium channel, Kir2.6 has been discovered in a cohort of TPP patients.23 Mutations were present in up to 33% of the unrelated TPP patients from the United States, Brazil, and France, but in only one of 83 patients from Hong Kong and 0 of 31 Thai patients. Thus, TPP is genetically heterogeneous. The demonstrated mutations appear to lead to muscle membrane inexcitability. In addition, thyroid hormones increase potassium efflux from muscle, which can lead to an increase in the number and activity of sodium-potassium ATPase pumps.24 This, in turn, results in partial depolarization of the muscle membrane, rendering it less excitable. Depolarization-induced sodium-channel inactivation6 and impaired propagation of the action potential across altered T tubules further renders the muscle membrane less excitable.25
Muscle strength improves gradually over several months with treatment of the hyperthyroidism.2 Propranolol can prevent and lessen the attacks of TPP. Unlike the familial form of hypokalemic periodic paralysis, acetazolamide is ineffective in preventing attacks of weakness associated with thyrotoxicosis.
Extraocular muscle weakness associated with Graves’ disease can persist for months or years after treatment. Artificial tears and ophthalmic ointments may be beneficial in preventing drying of the cornea and exposure keratitis that can result from severe lid retraction. Immunosuppression with corticosteroids and cyclosporine can be helpful in some patients but may be associated with significant side effects.26
Approximately one-third of individuals with hypothyroidism develop proximal arm and leg weakness along with myalgias, cramps, and generalized fatigue.2,7,27 Rare patients develop muscle hypertrophy; rhabdomyolysis may occur. Further, ventilatory muscles may be affected in severe cases.28
Delayed relaxation of the muscle stretch reflexes may be demonstrated, particularly at the ankle. This finding is best appreciated by having the patient kneel on a chair or bench while striking the Achilles tendon. Myoedema refers to painless and electrically silent mounding of muscle tissue when firmly percussed and is observed in approximately one-third of affected individuals.29 Myasthenia gravis can also occur in association with hypothyroidism.30
The serum CK levels are elevated as much as 10–100 times of normal. A TSH level should be checked in any patient with idiopathic CK-emia. In primary hypothyroidism, serum T4 and T3 levels are low, while TSH levels are elevated. The motor and sensory NCS are usually normal, unless they have a concomitant polyneuropathy. Needle EMG is also usually normal, although short-duration, low-amplitude polyphasic motor unit action potentials (MUAPs) may be appreciated in severely affected muscles.31–35
Muscle biopsy abnormalities are nonspecific and may include variability in muscle fiber size with atrophy of type 2 and occasionally type 1 fibers, hypertrophic muscle fibers, rare necrotic fibers, increased internalized nuclei, ring fibers, glycogen accumulation, vacuoles, and increased connective tissue.15,36,37 Mitochondrial swellings and inclusions, myofibrillar disarray with central core-like changes, autophagic vacuoles, glycogen accumulation, excess lipid, dilated sarcoplasmic reticulum, and T-tubule proliferation may be appreciated on EM.36
Hypothyroidism leads to reduced anaerobic and mitochondrial aerobic metabolism of carbohydrates and fatty acids decreasing ATP production. 38,39 Hypothyroidism also impairs adrenergic function and produces a concomitant insulin-resistant state. Protein synthesis and catabolism are reduced.
The myopathy improves with treatment of the hypothyroidism. However, some degree of weakness can persist even 1 year after return to a euthyroid state.
Myopathies are common in disorders of calcium and phosphate homeostasis. The regulation of calcium and phosphate levels requires a complex interaction of intestinal, renal, hepatic, endocrine, skin, and skeletal functions.2 Vitamin D regulates calcium absorption in the intestines. There are several forms of vitamin D: (1) vitamin D3 or cholecalciferol, which is derived from the skin; (2) vitamin D2 or ergocalciferol, which is dietary and absorbed through the intestines; and (3) 25-hydroxy-vitamin D, which is made in the liver and converted to the more potent metabolite 1,25-dihydroxy-vitamin D in the kidneys. Parathyroid hormone (PTH) assists in the regulation of serum calcium levels by promoting bone resorption, increasing renal calcium absorption and phosphate excretion, and enhancing 1,25-vitamin D conversion. Diet, intestinal absorption, and renal excretion contribute to serum phosphate levels. Increased PTH leads to increased levels of 1,25-dihydroxy-vitamin D, hypercalcemia, and hypophosphatemia. Persistently elevated PTH results in resorption of minerals within bone and replacement by fibrous tissue, a condition termed “osteitis fibrosa” or “osteitis fibrosa cystica” in severe forms.2
Muscle weakness is very common in osteomalacia, caused by vitamin D deficiency and secondary hyperparathyroidism in adults, occurring in as many as 72% of patients.40 Weakness develops however in only 2–10% of patients with isolated hyperparathyroidism.40,41 The earlier diagnosis and treatment of hyperparathyroidism and osteomalacia have led to fewer and less severe neuromuscular complications than appreciated in the past.40–45
The myopathy associated with primary hyperparathyroidism or osteomalacia is characterized by symmetric proximal weakness and atrophy, which are worse in the lower extremities. Concomitant bone pain is common due to associated microfractures. Involvement of the neck extensor muscles can lead to the so-called “dropped head syndrome.” There are rare reports of hoarseness, dysphagia, ventilatory involvement, and spasticity,41,46–49 although the majority of these cases were likely patients with amyotrophic lateral sclerosis and coincidental hyperparathyroidism.49
Muscle stretch reflexes are often brisk, but plantar responses are flexor. As many as 50% of patients complain of cramps and paresthesia. In addition, in 29–57% of patients there is stocking-glove loss of pain or vibratory sensation and decreased muscle stretch reflexes suggestive of an associated peripheral neuropathy.41 Finally, hypercalcemia can be associated with neurobehavioral abnormalities (memory loss, poor concentration, personality changes, inappropriate behavior including catatonia, anxiety, and hallucinations).
Secondary hyperparathyroidism and muscle weakness can develop in patients with chronic renal failure.50 Multifocal muscle infarcts and myoglobinuria due to calcification of the arteries (calciphylaxis) can develop in this setting.51–53 Calciphylaxis can also occur in patients with renal failure without overt hyperparathyroidism.54
Serum CK levels are usually normal in primary and secondary hyperparathyroidism and osteomalacia. CK may be slightly elevated in patients with muscle infarcts due to calciphylaxis.53 In primary hyperparathyroidism, serum calcium levels are usually elevated and serum phosphate levels are low, while urinary excretion of calcium is low and excretion of phosphate is high. In patients with concurrent hypoalbuminemia, serum calcium levels may be normal, so it is imperative to measure the ionized calcium levels which are typically elevated. Increased urinary excretion of cyclic adenosine monophosphate in the presence of hypercalcemia is also seen in hyperparathyroidism. Serum PTH levels and 1,25-dihydroxy-vitamin D levels are elevated in primary hyperparathyroidism. In contrast, 1,25-dihydroxy-vitamin D levels are low in secondary hyperparathyroidism due to renal failure. Noninvasive imaging techniques, such as ultrasound, thallium/technetium scintigraphy, computed tomography, and magnetic resonance imaging (MRI), may be useful in localizing abnormal parathyroid glands.55
Serum calcium level is low or normal, serum phosphate is variably low, and 25 OH vitamin D levels are also usually low in patients with osteomalacia. 1,25 OH vitamin D levels may be normal, however, as the body attempts to convert remaining vitamin D to this more potent form. Serum PTH levels are elevated in an attempt to normalize serum calcium levels that are reduced in response to vitamin D deficiency. Urinary excretion of calcium is low in an attempt to preserve serum calcium levels (except in cases secondary to renal tubular acidosis), while excretion of phosphate is high in response to secondary hyperthyroidism. In addition, serum alkaline phosphatase levels are elevated in 80–90% of cases of osteomalacia, again due to the body’s attempt to normalize serum calcium by increasing bone resorption.56 Skeletal survey reveals decreased bone density along with loss of trabeculae, blurring of trabecular margins, variably thinned cortices and microfractures that are most evident in the pelvis and proximal femur.42 EMG and NCS are normal unless the patients have a neuropathy related to their renal failure.
Muscle biopsies usually demonstrate nonspecific myopathic features with atrophy predominantly of type 2 fibers, but occasionally also of type 1 fibers. Muscle biopsies may reveal multifocal infarcts and calcium deposition primarily within vessel walls in patients with calciphylaxis.53
Primary hyperparathyroidism can be caused by parathyroid adenomas or hyperplasia as well as pituitary adenomas. Secondary hyperparathyroidism usually occurs in the setting of chronic renal failure which results in the reduction of 1,25-dihydroxy-vitamin D conversion or in malabsorption of vitamin D in disorders such as celiac disease. Vitamin D deficiency leads to diminished intestinal absorption of calcium and decreased renal phosphate clearance, which promotes secondary hyperparathyroidism and osteomalacia. In addition to acquired forms, there are hereditary forms of primary hyperparathyroidism57 and of vitamin D deficiency and osteomalacia.42
The mechanism(s) of weakness in hyperparathyroidism and osteomalacia are not known. PTH stimulates proteolysis in muscle58 and impairs energy production, transfer, and utilization.2,59 In addition, PTH may reduce the sensitivity of contractile myofibrillar proteins to calcium and activate a cytoplasmic protease, thus impairing the bioenergetics of muscle.1 Calcium and phosphate levels do not correlate well with the severity of muscle weakness.40,41,60 Vitamin D also has a direct effect on muscle by increasing muscle adenosine triphosphatase concentration, accelerating amino acid incorporation into muscle proteins,2,61 and enhancing the uptake of calcium by the sarcoplasmic reticulum and mitochondria.62,63
Hyperparathyroidism is diagnosed earlier than in the past because of routine screening of serum calcium levels. Thus, affected individuals are frequently asymptomatic or only mildly affected when they are diagnosed. Medical therapies and surgery are very effective for improvement of muscle weakness when detected within a few months.2,41,55,64
The treatment of choice of symptomatic patients with primary hyperparathyroidism is parathyroidectomy.55 If a patient has a parathyroid adenoma, the affected gland is removed, while additional glands may be biopsied. Individuals with hyperplasia of all four glands generally have subtotal (three and a half glands) parathyroidectomies. Those who are asymptomatic or have significant perioperative risk may be managed medically.64 Secondary hyperparathyroidism improves with vitamin D and calcium replacement or renal transplantation, if it is due to end-stage renal failure.65 Occasionally, subtotal parathyroidectomy may need to be performed in patients with secondary hyperparathyroidism. Likewise, the myopathy associated with osteomalacia responds well to vitamin D and calcium replacement and to treatment of the underlying responsible condition.40,42–44,56,66,67
Some authors have suggested that hyperparathyroidism can cause a neuromuscular syndrome that mimics amyotrophic lateral sclerosis and that patients may improve following resection of parathyroid adenomas.41,47 However, we suspect most of these patients who improved with parathyroidectomy did not have a motor neuron disorder, but rather, hyperparathyroid-related myopathy.49 In our experience, hyperparathyroidism in patients, who meet clinical and electrophysiologic criteria for amyotrophic lateral sclerosis, is rare and coincidental. These patients do not improve with parathyroidectomy.49
Hypoparathyroidism does not typically cause a myopathy, although a few patients do develop mild proximal weakness.68–70 In addition, painless myoglobinuria without objective weakness or tetany has been reported.71 On the other hand, paresthesia and tetany can develop in hypoparathyroidism secondary to hypocalcemia. The examiner may be able to demonstrate Chvostek sign (ipsilateral facial contraction upon tapping the facial nerve at the external auditory meatus) and Trousseau sign (thumb adduction, metacarpophalangeal joint flexion, and interphalangeal joint extension) in these hypocalcemic patients.
Serum CK may be normal or mildly elevated in patients.72,73 Hypoparathyroidism is associated with low serum PTH and calcium levels and high serum phosphate levels. Motor and sensory NCS are normal. Needle EMG may reveal normal insertional activity. Fasciculation potentials result from motor nerve hyperexcitability induced by the hypocalcemia.74–76 Multiplets (clusters of MUAPs activated with voluntary effort with interdischarge intervals between 2 and 20 ms) are another manifestation of nerve hyperexcitability and is the most characteristic electrodiagnostic abnormality seen in hypoparathyroidism or tetany. Otherwise, MUAP morphology and recruitment are normal.
Muscle biopsies may be normal or demonstrate mild variability in fiber size and increased internalized nuclei that reflect previous muscle damage caused by episodes of tetany.2,15 Decreased glycogen phosphorylase activity of muscle biopsy specimens has also been described.1
Hypoparathyroidism is seen in a number of conditions including osteomalacia, complications of surgery, hypomagnesemia or hypermagnesemia, irradiation, drugs, sepsis, infiltrative diseases of the parathyroid, and autoimmune, hereditary, or developmental disorders of the parathyroid glands.77 Decreased PTH leads to reduced synthesis of 1,25-dihydroxyvitamin D, hypocalcemia, and hyperphosphatemia.
The pathogenic mechanism of muscle weakness associated with hypoparathyroidism is poorly understood. Decreased serum calcium concentration causes a shift in the resting membrane potential closer to threshold.71,78–80 Therefore, less current is required to elicit an action potential, which can lead to tetany. Elevated serum CK and mild histologic abnormalities on muscle biopsy are generally considered secondary to muscle damage from tetany.
Muscle weakness improves following correction of the hypocalcemia and hyperphosphatemia with vitamin D and calcium administration.70
The adrenal gland comprises three major regions: (1) zona fasciculata, (2) zona glomerulosa, and (3) zona reticularis.2 The zona fasciculata produces and secretes glucocorticoids, which when produced in excess by an adrenal tumor can cause a myopathy. Mineralocorticoids such as aldosterone are generated by the zona glomerulosa and when produced in excess can cause hypokalemia which in turn leads to muscle weakness. The zona reticularis generates androgens but excess or deficiency of these hormones does not result in a muscle weakness. In contrast, these so-called anabolic steroids may increase muscle strength and mass. In the following section, we discuss myopathies associated with glucocorticoid excess or deficiency.
Steroid myopathy is the most common endocrine-related myopathy. An excess of glucocorticoids may arise directly from adrenal tumors, indirectly from pituitary tumors or from iatrogenic sources (corticosteroid medications).
Approximately 50–80% of patients with Cushing disease develop some degree of proximal weakness prior to treatment.2,81 Distal extremity, oculobulbar, and facial muscles are spared. Patients classically have an increase in truncal adipose tissue, a rounded facial appearance, and thin, frequently ecchymotic and hyperpigmented skin (i.e., the so-called Cushingoid appearance).
The incidence of iatrogenic steroid myopathy is not at all clear. Women appear to be more at risk for developing a steroid myopathy than men, approximately 2:1 but the reasons are unclear. An increased risk of the myopathy is seen with prednisone doses of 30 mg/day or more (or equivalent doses of other corticosteroids).2 Fluorinated corticosteroids have a greater propensity for producing muscle weakness than the nonfluorinated compounds (e.g., risk for myopathy: Triamcinolone > betamethasone > dexamethasone).82 Alternate day therapy may reduce the risk of corticosteroid-induced weakness but this has never been proven in a clinical study. Weakness can develop within several weeks of starting high doses of corticosteroids but more typically develops after chronic administration. In addition, an acute onset of severe generalized weakness can occur in patients receiving high dosages of intravenous corticosteroids (e.g., 1 g of methylprednisolone/day for multiple consecutive days) with or without concomitant administration of neuromuscular blocking agents (see section on acute quadriplegic myopathy/critical illness myopathy in Chapter 35).
Serum CK is normal. Serum potassium can be low and sodium may be elevated. Motor and sensory NCS and EMG are normal.
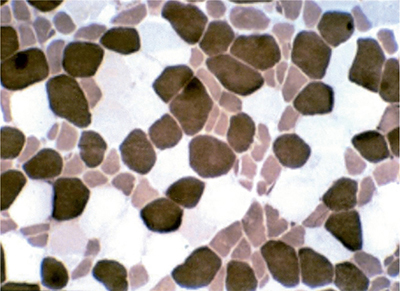
Muscle biopsy characteristically reveals preferential atrophy of type 2B fibers (Fig. 34-1).15,83 Milder degrees of atrophy and increased lipid deposition of type 1 muscle may be seen as well.
Figure 34-1. Steroid myopathy. Atrophy of type 2B fibers, which are intermediate staining, are appreciated on ATPase 4.5.
Corticosteroids bind to receptors on target cells and are subsequently internalized into the nuclei, where they regulate the transcription of specific genes. It is not known how corticosteroids lead to muscle dysfunction. Corticosteroids may result in diminished protein synthesis, increased protein degradation, altered carbohydrate metabolism, impaired mitochondrial function, or decreased sarcolemmal membrane excitability (i.e., in the setting of acute quadriplegic myopathy).1,2 In addition, hypokalemia associated with excess corticosteroid can also cause muscle weakness.
In cases of adrenal tumors, treatment is surgical when possible. In patients with iatrogenic steroid myopathy, treatment requires reduction in the corticosteroid dose, switching to an alternate day regimen, and encouraging exercise to prevent concomitant disuse atrophy.2 Experimental studies suggest that insulin-like growth factor-1 may have a prophylactic effect on preventing steroid myopathy.84 Increasing the dietary protein content of the diet is a suggested treatment of unproven benefit.
A common dilemma that physicians face is renewed or exacerbated weakness in a patient receiving corticosteroids for treatment of an immune-mediated neuromuscular disorder (e.g., inflammatory myopathy, inflammatory neuropathies, or myasthenia gravis).85,86 Following an initial improvement in their strength with corticosteroid treatment, some patients later experience a subsequent decline in muscle function. The question then arises: Is this a relapse/exacerbation of the disease or a steroid myopathy? Several clinical and laboratory features may be helpful in these situations. Patients with steroid myopathy usually have other manifestations of steroid excess, such as a Cushingoid appearance. If the weakness developed while the patient was tapering the corticosteroids, a relapse of the underlying disease process should be considered. In contrast, if weakness occurred while the patient was on a chronic high doses of steroids, a steroid myopathy is then perhaps more likely. In the case of an inflammatory myopathy, an increasing serum CK would point to an exacerbation of the myositis.87 An EMG can be useful in that it is usually normal in steroid myopathy, while abnormal insertion and spontaneous activity along with early recruitment of myopathic MUAPs would be expected in exacerbation of inflammatory myopathy. Abnormally increased signal on STIR images of skeletal muscle MRI scans would also favor active myositis. Likewise, in myasthenia gravis flare, one might expect to find fluctuation of clinical deficits on examination (ptosis and ophthalmoplegia are not seen in steroid myopathy) and an increase in decrement with repetitive stimulation tests, or increased jitter and blocking on single fiber EMG. Steroid myopathy typically affects the proximal muscles of the lower extremities first in a symmetric fashion, which can also help to distinguish it from aggravation of steroid responsive diseases. However, sometimes it is impossible to state with certainty whether the new weakness is related to a relapse of the underlying disease or secondary to the corticosteroid treatment. In such cases, the best approach is to taper the corticosteroid medication and closely observe the patient. If improvement in strength follows, one can assume the patient had a steroid myopathy. If the patient deteriorates, the worsening weakness is more likely related to the underlying autoimmune neuromuscular disorder, and they may require increased doses of corticosteroids or other forms of immunotherapy.
Adrenal insufficiency can result from adrenal or pituitary dysfunction and may be associated with subjective weakness (asthenia) and fatigue.2 Objective weakness is usually the result of electrolyte disturbances (e.g., hyperkalemia) or concurrent endocrinopathies.2 Serum CK levels, EMG, and muscle biopsies are usually normal. Symptoms improve with proper replacement of adrenal hormones.
PITUITARY DISORDERSPatients with acromegaly may develop slowly progressive proximal arm and leg weakness without muscle atrophy.2,88 If anything, muscle hypertrophy is appreciated. Acromegaly can cause bony overgrowth leading to nerve root or spinal cord compression. In addition, there is a predisposition for developing multiple entrapment neuropathies such as carpal and cubital tunnel syndromes. Degenerative joint changes can produce pain that limits activity which may result in disuse muscle atrophy as well.
Serum CK levels can be normal or mildly elevated. Motor and sensory NCS can be normal or demonstrate features of a mononeuropathy (i.e., carpal tunnel syndrome).89–91 Short-duration and low-amplitude MUAPs may be detected on EMG of proximal muscles of the arms and legs owing to the myopathy.88 In addition, the EMG can reveal neurogenic features of involved muscle groups, if a patient has a mononeuropathy or radiculopathy related to their acromegaly.
Muscle biopsies reveal variation in muscle fiber size with hypertrophy and atrophy of all fiber types.92,93 Hypertrophy of satellite cells is often appreciated. In addition, rare necrotic fibers may be seen. Myofibrillar loss and abnormal glycogen accumulation may be found on EM.
The development and severity of muscle weakness correlate with the duration of acromegaly rather than the levels of serum growth hormone.88,94 Growth hormone increases protein synthesis within muscle fibers and may lead to muscle fiber hypertrophy.95,96 However, the pathogenic basis for the muscle weakness that develops despite increased muscle bulk is not known. Studies have demonstrated that the respiratory quotient of resting forearms muscles of patients with acromegaly is lower than normal (0.68 vs. 0.76).97 Administration of growth hormone increases fatty acid oxidation and decreases glucose utilization.98 It appears that growth hormone causes muscle to preferentially metabolize lipid as opposed to carbohydrates and this could alter dynamic muscle activity and fatigue. In addition, growth hormone may reduce myofibrillar ATPase activity.99 In addition, muscle membranes are slightly depolarized compared to normal resting activity which would make them less excitable.1
Surgical resection of the pituitary adenoma with subsequent reduction of the growth hormone levels leads to improved muscle strength.88
Pituitary failure in adults commonly leads to muscle weakness and fatigue, probably due to secondary deficiencies of thyroid and glucocorticoid hormones.100 The myopathy improves with replacement of these hormones. Prepubertal panhypopituitarism is associated with dwarfism and lack of sexual and muscular development. Deficiency of growth hormone may contribute to muscle weakness in this condition, as administration of only thyroid and adrenal hormones does not result in improved strength unless growth hormone is also replaced.101 However, it is less clear if growth hormone deficiency can contribute to muscle weakness in adults with panhypopituitarism.
Neuromuscular complications of diabetes are usually referable to peripheral neuropathies (see Chapter 21). The only myopathic disorder clearly associated with diabetes is muscle infarction.
Diabetic muscle infarction usually occurs in the setting of poorly controlled diabetes. Most patients have other evidence of end-organ damage (retinopathy, nephropathy, neuropathy).102–109 Patients most commonly present with acute pain and swelling in one thigh. Occasionally, the calf is affected and rarely an upper extremity. A tender mass may be palpated in the quadriceps (most often the vastus lateralis), biceps femoris, or thigh adductors, and occasionally in the gastrocnemius muscle. The focal swelling and MRI changes often lead to a misdiagnosis of a sarcoma or focal myositis. Muscle biopsy should be avoided, if possible, because of the risk of subsequent hemorrhage into the tissue.54
Serum CK levels may be normal or elevated. Hemoglobin A1C level and erythrocyte sedimentation rate (ESR) are elevated in the majority of patients. MRI or CT of the leg demonstrates signal abnormalities in areas of infarcted muscle. EMG demonstrates fibrillation potentials and positive sharp waves as well as small, polyphasic MUAPs with early recruitment in the involved muscles.103
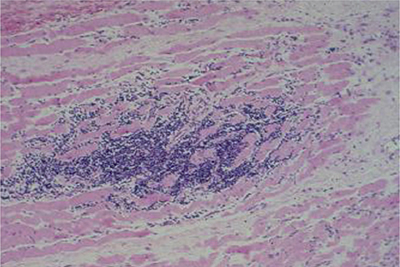
Muscle biopsies demonstrate large areas of necrosis, edema, hemorrhage, and inflammatory infiltrate consistent with muscle infarction (Fig. 34-2). This infarcted area is later replaced by connective and adipose tissue. Thickening of the basement membranes, hyperplasia of the media, and lumens occluded by fibrin, calcium, and lipid of small- and medium-sized blood vessels may be appreciated.54
Figure 34-2. Diabetic muscle infarct. Quadriceps muscle biopsy reveals widespread necrosis and endomysial inflammatory cell infiltrate. Paraffin section, stained with hematoxylin and eosin.
Ischemic damage and secondary hemorrhagic infarction result from long-standing, diabetic vasculopathy.
The muscle pain and swelling resolve after several weeks, although symptoms may recur in the contralateral leg. Treatment consists of immobilization and pain control. Sometimes we give a short course of prednisone to help with the pain by reducing edema and release of cytokines. However, one must closely monitor the serum glucose levels in such cases.
MYOPATHIES ASSOCIATED WITH ELECTROLYTE IMBALANCEHypokalemia is the most common electrolyte abnormality that causes muscle weakness.110 Clinical, laboratory, and electrophysiological features are similar to familial hypokalemic periodic paralysis (see Chapter 32). Patients must be evaluated for other etiologies of hypokalemia (Table 34-1) before assuming a diagnosis of familial hypokalemic periodic paralysis. Patients usually present with acute to subacute symmetric proximal or generalized weakness, although asymmetric muscle weakness can be seen. The presentation can be mistaken for Guillain–Barré syndrome. Weakness is often accompanied by complaints of myalgias and cramps. A severe complication of hypokalemia is rhabdomyolysis with myoglobinuria and secondary renal failure.
 TABLE 34-1. ETIOLOGIES OF SECONDARY HYPOKALEMIC AND HYPERKALEMIC PARALYSES
TABLE 34-1. ETIOLOGIES OF SECONDARY HYPOKALEMIC AND HYPERKALEMIC PARALYSES
Hypokalemic Paralysis
Thyrotoxic periodic paralysis
Renal tubular acidosis
Villous adenoma
Bartter syndrome
Hyperaldosteronism
Chronic or excessive use of diuretics, corticosteroids, licorice
Amphotericin B toxicity
Alcoholism
Toluene toxicity
Barium poisoning
Hyperkalemic Paralysis
Addison disease
Hypoaldosteronism (hyporeninemic)
Isolated aldosterone deficiency
Excessive potassium supplementation
Potassium-sparing diuretics (e.g., spironolactone, triamterene)
Chronic renal failure
Rhabdomyolysis
Usually the potassium levels are less than 2.5 mEq/L before any muscle breakdown and weakness occur. Serum CK levels are usually elevated in patients with hypokalemic myopathy. NCS are normal. EMG of weak muscles may demonstrate fibrillation potentials and positive sharp waves as well as early recruitment of small-duration, low-amplitude MUAPs. The EKG may demonstrate bradycardia, flattened T waves, prolonged PR and QT intervals, and notable U waves.
Biopsies of very weak muscles may demonstrate vacuoles and scattered necrotic fibers.
The mechanism of muscle fiber destruction and weakness is not fully known. Reduced extracellular potassium concentration may render the muscle membrane less excitable. Hypokalemia may also diminish blood flow and suppress the synthesis and storage of glycogen in muscles.
Muscle strength returns with correction of the hypokalemia. The patients need a medical workup to elucidate the underlying cause of the hypokalemia.
Hyperkalemia can also cause generalized muscle weakness. In addition, there is evidence of increased neuronal or muscle membrane excitability as manifested by the presence of Chvostek sign or myotonic lid lag. There are a number of causes of hyperkalemia that must be excluded before concluding a patient has familial hyperkalemic periodic paralysis (Table 34-1).
Most patients with severe generalized weakness have serum potassium levels greater than 7 mEq/L. Renal insufficiency and acidosis may accompany the hyperkalemia but serum CK levels are usually normal. EKG may demonstrate tall, peaked T waves.
Routine NCS are normal. EMG may demonstrate early recruitment of small “myopathic” MUAPs, but fibrillation potentials and positive sharp waves are atypical. Unlike, familial potassium–sensitive periodic paralysis, myotonic discharges are never seen in the acquired forms of hyperkalemic myopathy.
Muscle biopsies are typically normal.
Hyperkalemia causes a prolonged depolarization of the muscle membrane that in turn inactivates the sodium channel, inactivation reduces the excitability of the muscle membrane.
Muscle strength returns with correction of hyperkalemia. The underlying cause of the hyperkalemia must be elucidated and treated.
The muscle symptoms of hypercalcemia and hypocalcemia were discussed in the section regarding parathyroid myopathies. Hypercalcemia in the absence of parathormone excess usually manifests with primarily central nervous system rather than neuromuscular symptoms.
Hypophosphatemia can occur in diabetic ketoacidosis, acute alcohol intoxication, hyperalimentation with phosphate-poor preparations, severe diarrhea, and in patients taking phosphate-binding antacids. Serum phosphate levels less than 0.4 mM/L may lead to generalized muscle weakness potentially severe enough to produce ventilatory failure, rhabdomyolysis, and myoglobinuria.111 Some patients develop paresthesia and diminished muscle stretch reflexes. Severe hypophosphatemia is another potential Guillain–Barré syndrome mimic. In the authors, experience of a single case, the electrophysiologic signature was that of a sensorimotor axonopathy with muscle biopsy demonstrating type 2 fiber atrophy. Symptoms resolve with correction of the serum phosphate levels.
Hypermagnesemia most often occurs secondary to over usage of magnesium-containing laxatives, particularly if the patient has renal insufficiency.112 It can also develop during treatment of eclampsia with magnesium sulfate. Severe generalized and ventilatory muscle weakness may ensue but resolves with correction of the serum magnesium levels.
Muscle and nerve hyperexcitability, as characterized by Chvostek and Trousseau signs as well as tetany, may be seen in hypomagnesemia. However, hypocalcemia and other electrolyte disturbances typically accompany hypomagnesemia, and therefore, it is difficult to attribute the neuromuscular abnormality solely to the low serum magnesium levels.
MYOPATHIES ASSOCIATED WITH MALIGNANCYPatients with malignancies frequently develop generalized weakness, although most do not represent a true paraneoplastic syndrome. Muscle weakness in patients with cancer are much more likely related to impaired nutrition, increased catabolic state induced by the tumor, disuse atrophy, and perhaps toxic effects of chemotherapeutic agents. There are a few well-defined paraneoplastic syndromes, including sensory neuronopathies or sensorimotor neuropathies (e.g., anti-Hu syndrome as discussed in Chapter 19) and Lambert–Eaton syndrome (see Chapter 26), resulting in generalized weakness. Inflammatory and necrotizing myopathies can occur in the setting of cancer (as discussed in more detail in Chapter 33). Rarely, patients with malignancy can have spread of the tumor into a region of muscle.113,114 Any muscle group can be invaded by resulting in pain, swelling, and weakness in the local region. EMG of the affected muscles may reveal membrane instability and MUAPs with short duration and low amplitudes. Muscle biopsy can demonstrate evidence of tumor emboli.
OTHER MYOPATHIES SECONDARY TO SYSTEMIC DISEASEAmyloid myopathy usually occurs in the setting of primary amyloidosis (light-chain amyloidosis, AL).115–122 and is less frequent with familial amyloidosis.123,124 Amyloid myopathy does not typically occur in secondary amyloidosis (AA), however we have seen it in rare cases of senile amyloidosis.
With primary and familial amyloidosis, cardiac muscles, peripheral nerves, skin, kidneys, and other organs can also be affected in addition to skeletal muscle. In fact, most patients present with non–muscle-related symptoms. Approximately 20% of patients have a coexistent generalized peripheral neuropathy; mononeuropathies such as carpal tunnel syndrome and ulnar neuropathy also occur.112 Amyloid myopathy usually manifests with an insidious onset of progressive proximal weakness, although distal muscles can also be affected.120,122 The distal muscle weakness may be in part related to a superimposed amyloid neuropathy. Hypertrophy of involved muscle groups can be appreciated; the tongue is often involved with notable macroglossia. However, other patients develop atrophic muscles; again this could be related to the associated neuropathy. Rare patients have been reported presenting with neck extensor weakness (dropped head syndrome).125 Ventilatory failure can occur due to involvement of the diaphragm muscle and phrenic nerves. Muscle induration, stiffness, and pain are also variably present.
Serum CK is usually elevated two- to fivefold but has been as high as 70-fold in a patient with familial amyloidosis due to gelsolin mutation FA.120 AL is associated with monoclonal light chain immunoglobulins (λ greater than κ) in the serum or urine. Renal insufficiency and proteinuria result from amyloid deposition in the kidneys.
NCS are abnormal in patients with coexistent peripheral neuropathy. They often reveal reduced motor and sensory amplitudes and mild slowing of conduction velocities.117,119–121 Superimposed carpal tunnel syndrome is a common finding. EMG reveals muscle membrane irritability with frequent fibrillation potentials and positive sharp waves, particularly in the paraspinal and proximal extremity muscles.115,117–122,126 Complex repetitive discharges and myotonic discharges may also be appreciated. Early recruitment of short-duration, low-amplitude, polyphasic MUAPs is present in weak proximal muscles. In addition, patients with superimposed amyloid neuropathy often have decreased recruitment of long-duration, large-amplitude potential MUAPs in distal muscles.
MRI scans may reveal hypointense reticulum in the subcutaneous fat with or without increased T2 and STIR signal in affected extremities.127–130 Furthermore, MRI may show decreased T1 signaling in the bone marrow, suggesting hematologic malignancies.127
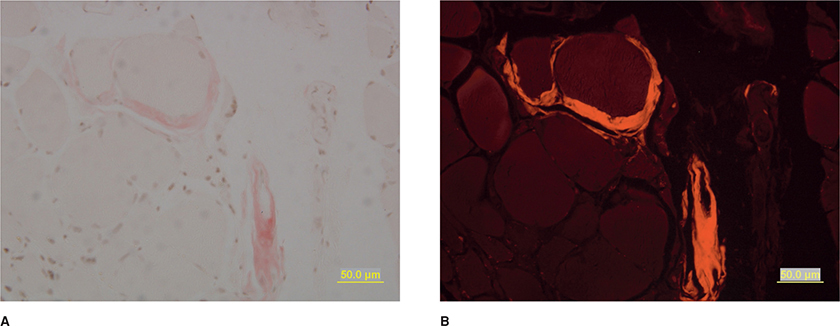
Muscle biopsies demonstrate variability in fiber size with an admixture of hypertrophic and atrophic fibers.120 Scattered necrotic and regenerating fibers and increased internalized nuclei may be seen. Group atrophy related to denervation may be appreciated. Amyloid deposition is best visualized using rhodamine optics on Congo-red stained section (Fig. 34-3).120 After employing this technique in the routine evaluation of all muscle specimens, the Mayo Clinic demonstrated a 10-fold increase in the diagnosis of amyloid myopathy, suggesting it is probably an underdiagnosed entity.120
Figure 34-3. Amyloid myopathy. Amyloid deposition is appreciated surrounding blood vessels and occasionally encasing individual muscle fibers on Congo red staining. The deposits are pink on routine light microscopy (A) and bright red using rhodamine optics (B).
The amyloid deposits are best appreciated surrounding small arterioles and venules. In addition, muscle fibers are also partially or completely encased by amyloid deposits. In primary amyloidosis, immunohistochemical studies reveal that the amyloid deposits are composed of λ or κ light chains.120 Immunohistochemistry employing antibodies directed against gelsolin and transthyretin are useful in excluding or diagnosing familial amyloidosis. Mass spectroscopy can also be utilized to identify the subtype of the amyloid deposits. Membrane attack complex may colocalize with amyloid deposition. ApoE was deposited in all patients regardless of the type to systemic amyloidosis in one large series of patients.120 EM confirms the deposition of short, nonbranching 10-nm amyloid filaments around small blood vessel and muscle fibers.
The exact mechanism by which amyloid deposition causes muscle fiber damage is not known. Ischemic damage may arise due to deposition of amyloid in blood vessel walls. Encasement of muscle fibers by the amyloid may interfere with the transport of oxygen, nutrients, and wastes into and out of muscle fibers. There may also be mechanical interference of muscle contraction secondary to amyloid infiltration. Alternatively, the amyloid may interfere with electrical conduction along the sarcolemma.
Although autologous stem bone marrow transplant has been attempted, there is no proven effective medical therapy for the myopathy secondary to systemic amyloidosis.
This entity is usually associated with high dose steroids with or without nondepolarizing neuromuscular agents and is discussed in detail in Chapter 35 regarding Toxic Myopathies.
ILL-DEFINED DISORDERSPolymyalgia rheumatica usually occurs in patients over the age of 50 years (peak incidence of 70–79 years).7,131,132 The prevalence is approximately of 1 case for every 133 people older than 50 years.133 There is a female predilection for the development of polymyalgia rheumatica. Patients usually complain of an insidious or acute onset of diffuse nonarticular pain and stiffness, particularly in the morning, beginning about the neck and shoulder region. Other body regions may be affected. A low-grade fever, anorexia, and malaise may accompany the myalgias. Affected individuals may complain of feeling weak or fatigued but on manual muscle testing their strength should be normal. Approximately 16% of patients develop temporal arteritis which can be complicated by acute vision loss.131 Temporal artery biopsy should be performed on all such patients with headaches or visual disturbances to look for evidence in giant-cell arteritis.
The ESR is usually abnormal and elevated; typically over of 40 mm/h. Serum CK should be normal. Likewise, EMG and NCS should be normal.
The muscle biopsies should be normal, but mild nonspecific findings (e.g., type 2 fiber atrophy, fiber size variation, and moth-eaten fibers) have been reported.132 Also note there is no evidence of significant inflammation in the muscle or overlying fascia.
The exact pathogenic basis for polymyalgia rheumatica is unclear. The elevated ESR and excellent response to corticosteroids suggest an immunologic mechanism. Some cases are clearly associated with arteritis/vasculitis but it is not usually related to a true myositis or fasciitis.
The administration of corticosteroids, usually at low dose, results in considerable symptom relief within a few days.
Fibromyalgia and myofascial pain syndrome, and the fatigue that often accompany them, are commonly diagnosed disorders that are controversial in regard to their nature because of the lack of objective evidence of organic disease.134,135 Fibromyalgia is often dominated by subjective complaints of generalized muscle pain in addition to other somatic complains including headaches, fatigue, and abdominal cramps. In this regard, it shares many features with the somatoform disorders and the dubious “chronic fatigue syndrome.”136,137
There is no “gold standard” for diagnosing fibromyalgia. Fibromyalgia may be diagnosed if a sufficient although arbitrary number of “tender points” at specific locations on the body are found.138 Unfortunately, the study by which these criteria were based was scientifically flawed.134,135 The investigators predetermined that tender points were necessary for the diagnosis and they each received training in how to identify such tender points. Patients diagnosed with fibromyalgia based on the presence of tender points were then evaluated by other investigators to confirm their presence. That these tender points were reproducible (good intraobserver reliability), served as a validation of the diagnosis for the investigators. Skeptics criticized the study for confirming the established bias of the investigators.134,135,139 Detecting tender points is dependent on the patient’s subjective input and is in no way a truly objective marker. The neurological examination including muscle strength testing and laboratory evaluation is otherwise normal in fibromyalgia. Likewise, serum CK, NCS, and electromyography (even of in the areas of tender points) are normal. Finally, there is no difference in the frequency of abnormal histologic findings compared to control populations.
Myofacial pain syndrome (MPS) is similar to fibromyalgia, but the pain is described as being more focal as opposed to generalized.134,135 Rather than tender points, advocates of the disorder suggest patients have “trigger points.” These trigger points have been associated with “taut bands,” “nodules,” and “local twitch responses.” However, blinded, controlled studies have demonstrated a low sensitivity and specificity of this so-called diagnostic marker of MPS.140 One study described abnormal “spontaneous EMG” activity in the area of the trigger points.141 However, review of the published figures suggest this was just normal end plate spike activity. The majority of electromyographers, including ourselves, have not been able to verify the presence of any abnormalities in MPS.134,135 As with fibromyalgia, the clinical examination, serum CK, EMG, and NCS, and muscle biopsies are normal.
Regardless of the organicity of the pain related to fibromyalgia or MPS, the patients’ symptoms are often quite distressing and disabling to them. We often recommend treatment with tricyclic antidepressant medications, pregabalin, or gabapentin, as we do with other chronic pain syndromes, in addition to maintenance of as normal a lifestyle as possible. Patients may also benefit from physical therapy program to increase their endurance and tolerance.
SUMMARYMany systemic disorders can be associated with a myopathy. The myopathy may be a direct effect of the systemic process or may be toxic related to drugs used to treat the underlying condition. The regenerative capability of muscle allows for improvement in strength with effective treatment of the underlying cause in many cases. This underlines the importance of a detailed evaluation in patients referred for possible myopathy.
1. Kaminski HJ, Ruff RL. Endocrine myopathies (hyper- and hypofunction of adrenal, thyroid, pituitary, and parathyroid glands and iatrogenic corticosteroid myopathy). In: Engel AG, Franzini-Armstrong C, eds. Myology, 2nd ed. New York, NY: McGraw-Hill; 1994:1726–1753.
2. Kissel JT, Mendell JR. The endocrine myopathies. In Rowland LP, DiMauro S, eds. Handbook of Clinical Neurology, Vol 18(62). Amsterdam: Elsevier Science Publishers BV; 1992:527–551.
3. Ramsay I. Thyroid Disease and Muscle Dysfunction. Chicago, IL: William Heinemann Medical Books; 1974.
4. Puvanendran K, Cheah JS, Naganathan N, Wong PK. Thyrotoxic myopathy: a clinical and quantitative analytic electromyographic study. J Neurol Sci. 1979;42:441–451.
5. Sataysohi E, Murakami K, Kowa H, Kinishita M, Nishiyama Y. Periodic paralysis in hyperthyroidism. Neurology. 1963;13: 746–752.
6. Ruff RL, Weissmann J. Endocrine myopathies. Neurol Clin. 1988;6:575–592.
7. Ludin HP, Spiess H, Koenig MP. Neuromuscular dysfunction associated with thyrotoxicosis. Eur Neurol. 1969;2:269–278.
8. McElvaney GN, Wilcox PG, Fairborn MS, et al. Respiratory muscle weakness and dyspnea in thyrotoxic patients. Am Rev Respir Dis. 1990;141(5 pt 1):1221–1227.
9. Mier A, Brophy C, Wass JA, Besser GM, Green M. Reversible muscle weakness in hyperthyroidism. Am Rev Respir Dis. 1989;139:529–533.
10. Bennet WR, Huston DP. Rhabdomyolysis in thyroid storm. Am J Med. 1984;77:733–735.
11. Feibel JH, Campa JF. Thyrotoxic neuropathy (Basedow’s paraplegia). J Neurol Neurosurg Psychiatry. 1976;39:491–497.
12. Havard CW, Campbell ED, Ross HB, Spence AW. Electromyographic and histologic findings in the muscles of patients with thyrotoxicosis. Q J Med. 1963;32:145–163.
13. McCommas AJ, Sica RE, McNabb AR, Goldberg WM, Upton AR. Evidence for reversible motoneurone dysfunction in thyrotoxicosis. J Neurol Neurosurg Psychiatry. 1974;37:548–558.
14. Engel AG. Neuromuscular manifestations of Grave’s disease. Mayo Clin Proc. 1972;47:919–925.
15. Hudgson P, Kendall-Taylor P. Endocrine myopathies. In: Mastaglia FL, Walton JN, eds. Skeletal Muscle Pathology. Edinburgh: Churchill Livingstone; 1992:493–509.
16. Waldstein SS, Bronsky D, Shrifter HB, Oester YT.et al. The electromyogram in myxedema. AMA Arch Intern Med. 1958;101:97–102.
17. Engel AG. Electron microscopic observations in thyrotoxic and corticosteroid-induced myopathies. Mayo Clin Proc. 1966;41:785–796.
18. Janssen JW, Delange-Berkout IW, Van Hardeveld C, Kassenaar AA. The disappearance of l-thyroxine and triiodothyronine from plasma, red and white skeletal muscle after administration of one subcutaneous dose of l-thyroxin to hyperthyroid and euthyroid rats. Acta Endocrinol (Copenh). 1981;97:226–230.
19. Celsing F, Blomstrand E, Melichna J, et al. Effect of hyperthyroidism in fibre-type composition, fibre area, glycogen content, and enzyme activity in human muscle protein activity in human skeletal muscle. Clin Physiol. 1986;6:171–181.
20. Dubaniewicz A, Kaciuba-Uscilko H, Nazar K, Budohoski L. Sensitivity of the soleus to insulin in resting and exercising with experimental hypo- and hyperthyroidism. Biochem J. 1989;263:243–247.
21. Brown JG, Millward DJ. The influence of thyroid status on skeletal muscle protein metabolism. Biochem Soc Trans. 1980;8:366–367.
22. Morrison WL, Gibson JN, Jung RT, Rennie MJ. Skeletal muscle and whole body protein turnover in thyroid disease. Eur J Clin Invest. 1988;18:62–68.
23. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88–98.
24. Everts ME, Dørup I, Flyvberg A, Marshall SM, Jørgensen KD. Na(+)-K(+) pump in rat muscle: Effects of hypophysectomy, growth hormone, and thyroid hormone. Am J Physiol. 1990;259(2 pt 1):E278–E283.
25. Dulhunty AF, Gage PW, Lamb GD. Differential effects of thyroid hormone on T-tubules and terminal cisternae in rat muscles: an electrophysiological and morphometric analysis. J Muscle Res Cell Motil. 1986;7:225–236.
26. Prummel MF, Mourits MP, Berout A, et al. Prednisone and cyclosporine in the treatment of severe Graves’ disease. N Engl J Med. 1989;321:1353–1359.
27. Salick AI, Colachis SC Jr, Pearson CM. Myxedema myopathy: Clinical, electrodiagnostic, and pathologic findings in advanced case. Arch Phys Med Rehabil. 1968;49:230–237.
28. Martinez FJ, Bermudez-Gomez M, Celli BR. Hypothyroidism. A reversible cause of diaphragmatic dysfunction. Chest. 1989;96:1059–1063.
29. Salick AI, Pearson CM. Electrical silence of myoedema. Neurology. 1967;17(9):899–901.
30. Takamori M, Gutman L, Crosby TW, Martin JD. Myasthenic syndromes in hypothyroidism. Electrophysiological study of neuromuscular transmission and muscle contraction in two patients. Arch Neurol. 1972;26:326–335.
31. Afifi AK, Najjar SS, Mire-Salam J, Bergman RA. The myopathology of the Kocher-Debre’-Se’me’laigne syndrome: Electromyography, light- and electron-microscopic study. J Neurol Sci. 1974;22:445–470.
32. Astrom KE, Kugelberg E, Muller R. Hypothyroid myopathy. Arch Neurol. 1961;5:472–482.
33. Emser W, Schimrigk K. Myxedema myopathy: A case report. Eur Neurol. 1977;16:286–291.
34. Klein I, Parker M, Shebert R, Ayyar DR, Levey GS. Hypothyroidism presenting as muscle stiffness and pseudohypertrophy: Hoffman’s syndrome. Am J Med. 1981;70:891–894.
35. Norris FH Jr, Panner BJ. Hypothyroid myopathy. Arch Neurol. 1966;14:574–589.
36. Evans RM, Watanabe I, Singer PA. Central changes in hypothyroid myopathy: A case report. Muscle Nerve. 1990;13:952–956.
37. Laycock MA, Pascuzzi RM. The neuromuscular effects of hypothyroidism. Semin Neurol. 1991;11:288–294.
38. Ho KL. Basophilic bodies of skeletal muscle in hypothyroidism: enzyme histochemical and ultrastructural studies. Hum Pathol. 1989;20:1119–1124.
39. Schwartz HL, Oppenheimer JH. Physiologic and biochemical actions of thyroid hormone. Pharmacol Ther B. 1978;3:349–376.
40. Smith R, Stern G. Muscular weakness in osteomalacia and hyperparathyroidism. J Neurol Sci. 1969;8:511–520.
41. Patten BM, Bilezikian JP, Mallette LE, Prince A, Engel WK, Aurbach GD. Neuromuscular disease in primary hyperparathyroidism. Ann Intern Med. 1974;80:182–193.
42. Goldring SR, Krane SM, Avioli LV. Disorders of calcification: Osteomalacia and rickets. In: De Groot, ed. Endocrinology. 3rd ed. Philadelphia, PA: WB Saunders; 1994:1204–1227.
43. Mallette LE, Patten BM, Engel WK. Neuromuscular disease in secondary hyperparathyroidism. Ann Intern Med. 1975;82:474–483.
44. Smith R, Stern G. Myopathy, osteomalacia and hyperparathyroidism. Brain. 1967;90:593–602.
45. Vicale CT. The diagnostic features of a muscular syndrome resulting from hyperparathyroidism, osteomalacia, owing to renal tubular acidosis, and perhaps to related disorders of calcium metabolism. Trans Am Neurol Assoc. 1949;74:143–147.
46. Berenbaum F, Rajzbaum G, Bonnchon P, Amor B. Une hyperparathyroide revelee une chute de la tete. Rev Rhum Mal Osteoartic. 1993;60:467–469.
47. Gelinas DF, Miller RG, McVey AL. Reversible neuromuscular dysfunction associated with hyperparathyroidism [abstract]. Neurology. 1994;44(Suppl 2):A348.
48. Gentric A, Pennec YL. Fatal primary hyperparathyroidism with myopathy involving respiratory muscles. J Am Geriatr Soc. 1994;42:1306.
49. Jackson CE, Amato AA, Bryan WW, Wolfe GI, Sakhee K, Barohn RJ. Primary hyperparathyroidism and ALS. Is there a relation? Neurology. 1998;50:1795–1799.
50. Floyd M, Ayyar DR, Barwick DD, Hudson P, Weightman D. Myopathy in chronic renal failure. Q J Med. 1974;53:509–524.
51. Richardson JA, Herron G, Reitz R, Layzer R. Ischemic ulcerations of the skin and necrosis of muscle in azotemic hyperparathyroidism. Ann Intern Med. 1969;71:129–138.
52. Randall DP, Fisher MA, Thomas C. Rhabdomyolysis as the presenting manifestation of calciphylaxis. Muscle Nerve. 2000;23:289–293.
53. De Luca GC, Eggers SD. A rare complication of azotemic hyperparathyroidism: Ischemic calcific myopathy. Neurology. 2010;75:1942.
54. Banker BQ, Chester CS. Infarction of thigh muscle in the diabetic patient. Neurology. 1973;23:667–677.
55. Norton JA, Sugg SL. Surgical management of hyperparathyroidism. In: De Groot, ed. Endocrinology. 3rd ed. Philadelphia, PA: WB Saunders; 1994:1106–1122.
56. Russell JA. Osteomalacic myopathy. Muscle Nerve. 1994;17(6):578–580.
57. Habener J, Arnold A, Potts JT. Hyperparathyroidism. In: De Groot, ed. Endocrinology. 3rd edn. Philadelphia, PA: WB Saunders; 1994:1044–1060.
58. Garber AJ. Effects of parathyroid hormone on skeletal muscle protein and amino acid metabolism in the rat. J Clin Invest. 1983;71:1806–1821.
59. Baczynski R, Massry SG, Magott M, El-Belbessi S, Kohan R, Brautbar N. Effect of parathyroid hormone on energy metabolism of skeletal muscle. Kidney Int. 1985;28:722–727.
60. Frame B, Heinze EG Jr, Block MA, Manson GA. Myopathy in primary hyperparathyroidism. Observations in three patients. Ann Intern Med. 1968;68:1022–1027.
61. Birge SG, Haddad JG. 25-hydroxycholecalciferol stimulation of muscle metabolism. J Clin Invest. 1975;56:1100–1107.
62. Curry OB, Basten JF, Francis MJ, Smith R. Calcium up-take by the sarcoplasmic reticulum of muscle from vitamin D deficiency in rabbits. Nature. 1974;249:83–84.
63. Pointon JJ, Francis MJO, Smith R. Effect of vitamin D deficiency on sarcoplasmic reticulum and troponin C concentration of rabbit skeletal muscle. Clin Sci (Lond). 1979;57: 257–263.
64. Nussbaum SR, Neer RM, Potts JT Jr. Medical management of hyperparathyroidism and hypercalcemia. In: De Groot, ed. Endocrinology. 3rd ed. Philadelphia, PA: WB Saunders; 1994:1094–1105.
65. Probhala A, Garg R, Dandona P. Severe myopathy associated with vitamin D deficiency in western New York. Arch Intern Med. 2000;160(8):1119–1203.
66. Irani PF. Electromyography in nutritional osteomalacic myopathy. J Neurol Neurosurg Psychiatry. 1976;39:686–693.
67. Schott GD, Wills MR. Myopathy in hypophosphataemic osteomalacia presenting in adult life. J Neurol Neurosurg Psychiatry. 1975;38:297–304.
68. Kruse K, Scheunemann W, Baier W, Schaub J. Hypocalcemic myopathy in idiopathic hypoparathyroidism. Eur J Pediatr. 1982;138:280–282.
69. Snowdon JA, Macfie AC, Pearce JB. Hypocalcemic myopathy and parathyroid psychosis. J Neurol Neurosurg Psychiatry. 1976;38:48–52.
70. Yamaguchi H, Okamoto K, Shooji M, Morimatsu M, Hirai S. Muscle histology of hypocalcemic myopathy in hypoparathyroidism. J Neurol Neurosurg Psychiatry. 1987;50:817–818.
71. Akmal M. Rhabdomyolysis in a patient with hypocalcemia due to hypoparathyroidism. Am J Nephrol. 1993;13:61–63.
72. Hower J, Struck H, Tackman W, Stolecke H. CPK activity in hypoparathyroidism. N Engl J Med. 1972;287:1098.
73. Shane E, McClane KA, Olarte MR, Bilezikian JP. Hypoparathyroidism and elevated serum enzymes. Neurology. 1980;30:192–195.
74. Kugelberg E. Neurologic mechanism for certain phenomena in tetany. Arch Neurol Psychiatry. 1946;56:507–521.
75. Kugelberg E. Activation of human nerves by ischemia; Trousseau’s phenomenon in tetany. Arch Neurol Psychiatry. 1948;60:140–164.
76. Kugelberg E. Activation of human nerves by hyperventilation and hypocalcemia. Arch Neurol Psychiatry. 1948;60:153–164.
77. Fitzpatrick LA, Arnold A. Hypoparathyroidism. In: De Groot, ed. Endocrinology. 3rd ed. Philadelphia, PA: WB Saunders; 1994:1123–1135.
78. Brink F. The role of calcium ion in neural processes. Pharmacol Rev. 1954;6:243–298.
79. Frankenhaeuser B. The effect of calcium on the myelinated nerve fiber. J Physiol. 1957;137:245–260.
80. Frankenhaeuser B, Hodgkin AL. The action of calcium on the electric properties of squid axons. J Physiol. 1957;137:218–244.
81. Muller R, Kugelberg E. Myopathy in Cushing’s syndrome. J Neurol Neurosurg Psychiatry. 1959;22:314–319.
82. Faludi G, Gotlieb J, Meyers J. Factors influencing the development of steroid-induced myopathy. Ann N Y Acad Sci. 1966;138:62–72.
83. Pleasure DE, Walsh GO, Engel WK. Atrophy of skeletal muscle in patients with Cushing’s syndrome. Arch Neurol. 1970;22: 118–125.
84. Kanda F, Takatani K, Okuda S, Matsushi T, Chihara K. Preventive effects of insulin-like growth factor-1 on steroid-induced muscle atrophy. Muscle Nerve. 1999;22:213–217.
85. Afifi AK, Bergman RA, Harvey JC. Steroid myopathy. Johns Hopkins Med J. 1968;123:158–173.
86. MacLean K, Schurr PH. Reversible amyotrophy complicating treatment with fludrocortisone. Lancet. 1959;1:701–702.
87. Askari A, Vignos PJ Jr, Moskowitz RW. Steroid myopathy in connective tissue disease. Am J Med. 1976;61:485–492.
88. Pickett JB, Layzer RB, Levin SR, Scheider V, Campbell MJ, Sumner AJ. Neuromuscular complications of acromegaly. Neurology. 1975;25:638–645.
89. Low PA, McLeod JG, Turtle JR, Donnelly P, Wright RG. Peripheral neuropathy in acromegaly. Brain. 1974;97:139–152.
90. Lundberg PO, Osterman PO, Stalberg E. Neuromuscular signs and symptoms in acromegaly. In: Walton JN, Canal N, Scarlato G, eds. Muscle Diseases: Proceedings of an International Congress Milan, 19–21, May, 1969. Amsterdam: Excerpta Medica; 1970:531–534.
91. Stewart BM. The hypertrophic neuropathy of acromegaly. Arch Neurol. 1966;14:107–110.
92. Mastaglia FL, Barwick DD, Hall R. Myopathy in acromegaly. Lancet. 1970;2:907–909.
93. Stern LZ, Payne CM, Hannapel LK. Acromegaly: Histochemical and electron microscopic changes in deltoid and intercostal muscle. Neurology. 1974;24:589–593.
94. Naglesparen M, Trickey R, Davies MJ, Jenkins JS. Muscle changes in acromegaly. Br Med J. 1976;2:914–915.
95. Bigland B, Jehring B. Muscle performance in rats, normal and treated with growth hormone. J Physiol. 1952;116:129–136.
96. Prysor-Jones RA, Jenkins JS. Effect of excessive secretion of growth hormone ion tissues of the rat, with particular reference to the heart and skeletal muscle. J Endocrinol. 1980;85:75–82.
97. Rabinowitz D, Zierler KL. Differentiation of active from inactive acromegaly by studies of forearm metabolism and response to intra-arterial insulin. Bull Johns Hopkins Hosp. 1963;113:211–224.
98. Winckler B, Steele R, Altszuller N, De Bodo RC. Effect of growth hormone on free fatty acid metabolism. Am J Physiol. 1964;206:174–178.
99. Florini JR, Ewton DZ. Skeletal muscle fiber types and myosin ATPase activity do not change with age or growth hormone administration. J Gerontol. 1989;44:B110–B117.
100. Brasel JA, Wright JC, Wilkins L, Blizzard RM. An evaluation of seventy-five patients with hypopituitarism. Am J Med. 1965;38:484–498.
101. Raben MS. Growth hormone: 2. Clinical use of growth hormone. N Engl J Med. 1962;266:82–86.
102. Anglada M, Vidaller A, Bolao F, Ferrer I, Olive M. Diabetic muscular infarction. Muscle Nerve. 2000;23:825–826.
103. Barohn RJ, Kissel JT. Painful thigh mass in a young woman: diabetic thigh infarction. Muscle Nerve. 1992;15:850–855.
104. Bjornskowve EK, Carry MR, Katz FH, Lefkowitz J, Ringel SP. Diabetic muscle infarction: a new perspective on pathogenesis and management. Neuromuscul Disord. 1995;5(1):39–45.
105. Bodner RA, Younger DS, Rosoklija G. Diabetic muscle infarction. Muscle Nerve. 1994;17:949–950.
106. Chester CS, Banker BQ. Focal infarctions of muscle in diabetics. Diabetic Care. 1986;9:623–630.
107. Umpierrez GE, Stiles RG, Kleinbart J, Krendel DA, Watts NB. Diabetic muscle infarction. Am J Med. 1996;101:245–250.
108. Huang BK, Monu JU, Doumanian J. Diabetic myopathy: MRI patterns and current trends. AJR Am J Roentgenol. 2010;195:198–204.
109. Joshi R, Reen B, Sheehan H. Upper extremity diabetic muscle infarction in three patients with end-stage renal disease: a case series and review. J Clin Rheumatol. 2009;15:81–84.
110. Comi G, Testa D, Cornelio F, Comola M, Canal M. Potassium depletion myopathy: a clinical and morphological study of six cases. Muscle Nerve. 1985;8:17–21.
111. Knochel JP. The clinical status of hypophosphatemia: an update. N Engl J Med. 1985;313:447–449.
112. Mordes JP, Wacker WE. Excess magnesium. Pharmacol Rev. 1977;29:273–300.
113. Doshi R, Fowler T. Proximal myopathy due to discrete carcinomatous metastases in muscle. J Neurol Neurosurg Psychiatry. 1983;46:358–360.
114. Heffner RR Jr. Myopathy of embolic origin in patients with carcinoma. Neurology. 1971;21:840–846.
115. Jennekens FG, Wokke JH. Proximal weakness of the extremities as a main feature of amyloid myopathy. J Neurol Neurosurg Psychiatry. 1987;50:1353–1358.
116. Nardkarni N, Freimer M. Mendell JR. Amyloidosis causing a progressive myopathy. Muscle Nerve. 1995;18:1016–1018.
117. Ringel SP, Claman HN. Amyloid-associated muscle pseudohypertrophy. Arch Neurol. 1982;39:413–417.
118. Roke ME, Brown WF, Boughner D, Ang LC, Rice GP. Myopathy in primary systemic amyloidosis. Can J Neurol Sci. 1988;15:314–316.
119. Rubin DI, Hermann RC. Electrophysiologic findings in amyloid myopathy. Muscle Nerve. 1999;22:355–359.
120. Spuler S, Emslie-Smith A, Engel AG. Amyloid myopathy: an underdiagnosed entity. Ann Neurol. 1998;43:719–728.
121. Whitaker JN, Hashimoto K, Quinones M. Skeletal muscle pseudohypertrophy in primary amyloidosis. Neurology. 1977; 27:47–54.
122. Smetstad C, Monstad P, Lindboe CF, Mygalns A. Amyloid myopathy present with distal atrophic weakness. Muscle Nerve. 2004;29:605–609.
123. Bruni J, Bilbao JM, Prtzker PH. Myopathy associated with amyloid angiopathy. Can J Neurol Sci. 1977;4:77–80.
124. Yamada M, Tsukagoshi H, Hatakeyama S. Skeletal muscle amyloid deposition in AL- (primary or myeloma-associated), AA- (secondary), and prealbumin-type amyloidosis. J Neurol Sci. 1988;85:223–232.
125. Chuquilin M, Al-Lozi M. Primary amyloidosis presenting as “dropped head syndrome”. Muscle Nerve. 2011;43:905–909.
126. Chapin JE, Kornfeld M, Harris A. Amyloid myopathy: characteristic features of a still underdiagnosed disease. Muscle Nerve. 2005;31(2):266–272.
127. Hull KM, Griffith L, Kuncl RW, Wigley FM. A deceptive case of amyloid myopathy. Clinical and magnetic resonance imaging features. Arthritis Rheum. 2001;8:1954–1958.
128. Mandl LA, Folkerth RD, Pick MA, Weinblatt ME, Gravallese EM. Amyloid myopathy masquerading as polymyositis. J Rheumatol. 2000;27:949–952.
129. Metzler JP, Fleckenstein JL, White CL 3rd, Haller RG, Frenkel EP, Greenlee RG Jr. MRI evaluation of amyloid myopathy. Skeletal Radiol. 1992;21:463–465.
130. Tuomaala H, Kärppä M, Tuominen H, Remes AM. Amyloid myopathy: A diagnostic challenge. Neurol Int. 2009;1:e7.
131. Chuang TY, Hunder GG, Ilstrup DM, Kurland LT. Polymyalgia rheumatica. A 10-year epidemiologic and clinical study. Ann Intern Med. 1982;97:672–680.
132. Coomes EN. The rate of recovery of reversible myopathies and the effects of anabolic agents in steroid myopathy. Neurology. 1965;18:523–530.
133. Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med. 2002;347(4): 261–271.
134. Bohr T. Problems with myofascial pain syndrome and fibromyalgia syndrome. Neurology. 1996;46:593–597.
135. Bohr TW. Fibromyalgia syndrome and myofascial pain syndrome: Do they exist? Neurol Clin. 1995;13:365–384.
136. Goldenberg DL. Fibromyalgia, chronic fatigue syndrome, and myofascial pain syndrome. Curr Opin Rheumatol. 1993;5:199–208.
137. Komaroff AL, Goldenberg D. The chronic fatigue syndrome: Definition, current studies, and lessons for fibromyalgia research. J Rheumatol. 1989;16:23–27.
138. Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology. 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990;33(2):160–172.
139. Cohen ML, Quinter JL. Fibromyalgia syndrome, a problem of tautology. Lancet. 1993;342:906–909.
140. Wolfe F, Simons DG, Fricton J, et al. The fibromyalgia and myofascial pain syndromes: A preliminary study of tender points and trigger points in persons with fibromyalgia, myofascial pain syndrome and no disease. J Rheumatol. 1992;19(6):944–951.
141. Hubbard D, Berkoff G. Myofascial trigger points show spontaneous needle EMG activity. Spine (Phila Pa 1976). 1993;18:1803–1807.