Chapter 4
Primordial Soup
In 1951, Melvin Calvin (1911–1997), a professor at UC Berkeley, who, some years later, would win the Chemistry Nobel Prize for his studies of photosynthesis (more in chapter 7), published a paper in the journal Science that opened with the following: “The question of the conditions under which living matter originated on the surface of the earth is still a subject limited largely to speculation. The speculation has a greater chance of approaching the truth when it includes and is based upon the ever-wider variety of established scientific fact. One of the purposes of the observations reported herein is to add another fact that might have some bearing on this interesting question.”* The “another fact” his paper added? It was that upon bombarding aqueous solutions of carbon dioxide with highly ionizing radiation (helium nuclei accelerated to near the speed of light in the Berkeley cyclotron) in the presence of an iron catalyst, up to 22% of the carbon dioxide was converted into formic acid (HCO2H) by the reactive hydrogen species the radiation produced.
About the same time Calvin was conducting his work, Stanley Miller (1930–2007), then a graduate student at the University of Chicago, was in the audience when Harold Urey (1893–1981), who had won the 1934 Nobel Prize in Chemistry for his pioneering work on separating atomic isotopes, presented his thoughts regarding the chemical processes associated with the formation of the Solar System and how this chemistry might have driven and constrained the origins of life on Earth. Approaching Urey in 1952, Miller proposed experiments aimed at simulating the chemistry of the early Earth. At first Urey discouraged him, believing that a process that took geological eons might be a bit hard to re-create during the five or six years one typically spends earning a PhD. But Miller persisted and, in the end, triumphed.
Urey’s, Calvin’s, and Miller’s ideas were built on a hypothesis put forward in the early 1920s by the Soviet scientist Aleksandr Oparin (1894–1980) and, slightly later, independently proposed by the Scottish scientist J. B. S. Haldane (1892–1964).* Both had theorized that life arose through the slow “evolution” of chemical systems of increasing complexity. Haldane, specifically, postulated that, acting on an atmosphere of carbon dioxide, ammonia, and water vapor, UV light from the Sun could create key components of life, such as amino acids and sugars, which would have “accumulated till the primitive oceans reached the consistency of hot, dilute soup.”** In their own speculations on the process, Miller and Urey concluded that the early atmospheres of terrestrial planets—like the current atmospheres of Jupiter and Saturn—were made up of methane, ammonia, and hydrogen. Clearly, too, there would have been oceans with water still warm from the accretion of the planet, and thus the skies would have been filled with water vapor, clouds, and rain.
Miller built for his studies an apparatus consisting of two connected glass reservoirs (fig. 4.1). In the smaller, lower reservoir he placed water as a mimic of the primordial ocean. He filled the larger, upper sphere with methane, ammonia, and hydrogen—Urey’s hypothesized primordial atmosphere. Miller placed two electrodes in this “atmosphere,” through which he passed a high-voltage discharge mimicking lightning, in place of the UV light that Urey and Haldane had originally suggested was the likely energy source and the ionizing radiation that Calvin had employed. The connections between the two containers were such that, when heat was applied to the “ocean,” water vapor would rise into the lightning-filled atmosphere. A second tube connected the atmosphere and the ocean by way of a water-cooled condenser, with the condensed water mimicking rain and setting up a simple hydrological cycle.
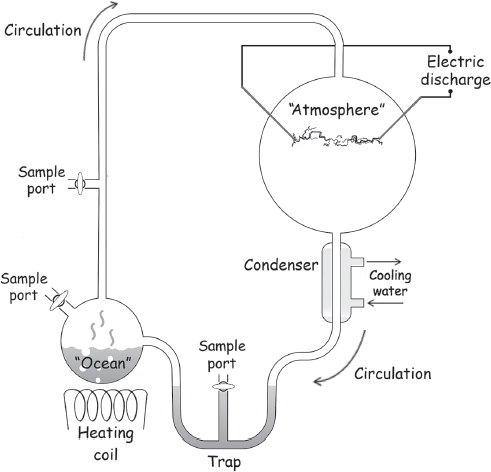
Figure 4.1 In 1953, Stanley Miller set up an experimental apparatus to simulate the conditions on the primordial Earth. After the experiment had been running just a few days, the “ocean” in the lower sphere became discolored and the “atmospheric” chamber (the upper sphere) became coated with a brown tar. Within a week, almost all the carbon originally introduced as methane had become fixed into larger, more complex molecules, including many of the amino acids on which present-day Earth life is built.
Legend has it that Miller sat by his experiment day and night for a week while the simulated lightning crackled and the faux ocean boiled, condensed, and fell back as “rain.” He didn’t have to wait long to see something happen: after just a day or two, the once-clear liquid became yellowish and then brown, and the discharge chamber became coated in an increasingly thick tar. After five days, Miller removed samples from the “ocean” for analysis. In a striking confirmation of his hypothesis, he found the water in his reaction vessel was now a rich solution of higher molecular weight carbon compounds accounting for 10% to 15% of the total carbon he had originally put in as methane. Moreover, this mixture contained several percent amino acids, including many that were known to be the building blocks of our proteins (fig. 4.2; see also sidebar 4.1). Under what seemed to be plausible prebiotic conditions, Miller had shown that even the simple single-carbon, single-nitrogen, and single-oxygen inputs of methane, ammonia, and water would spontaneously form many of the most fundamental chemical components of life on Earth.
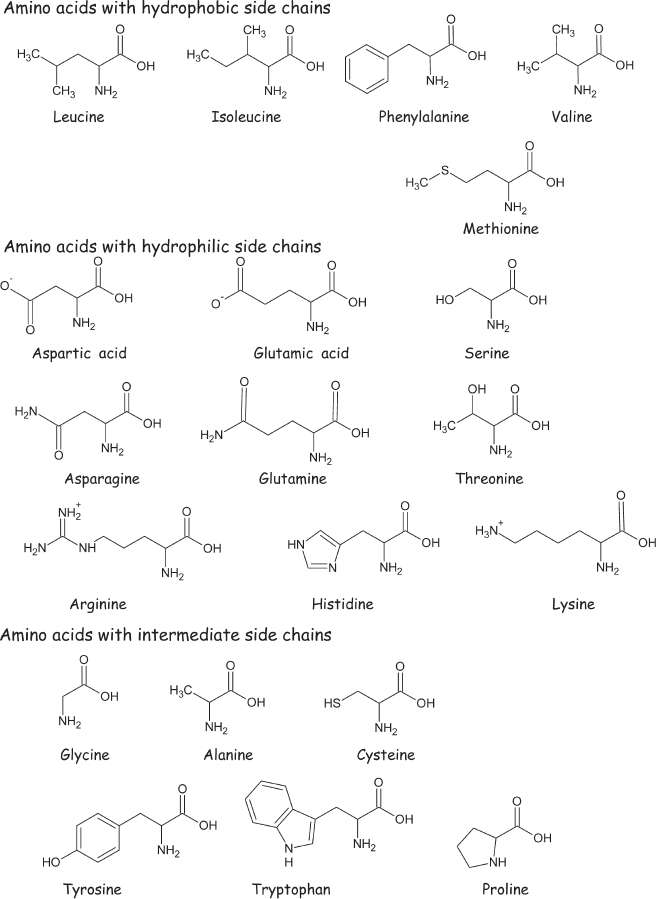
Figure 4.2 Amino acids are small organic molecules containing a carboxylic acid group (−CO2H) and an amino group (−NH2). The α-amino acids, shown here, are a subclass of amino acids in which the amino group is one carbon removed from the carboxylic acid group (i.e., placed on the carbon “alpha” to the carboxylate carbon). The α-amino acids play fundamental roles in Terrestrial biochemistry, both as important intermediates in metabolism and as the monomers out of which proteins are built. The 20 α-amino acids shown here, which are classified by their solubility in water (their “hydrophilicity”), are the amino acids from which our proteins are synthesized. (Note that the acidic amino acids, glutamic and aspartic acids, are often referred to as their negatively charged ions, glutamate and aspartate.) The unlabeled vertices in the structural formulas in this and later figures represent carbon atoms bound to enough hydrogen atoms to ensure that each participates in four bonds.
Sidebar 4.1
An Earth Biology Primer
An intellectual hurdle that the field of astrobiology faces is its extremely limited data set: we have only a single example of biology to study—as we discuss in chapter 6, every living thing on Earth arose from a single, fairly sophisticated, common ancestor. Given this, our views on the range of the possible must be at least somewhat suspect. Still, astrobiology being based on one example is a clear improvement over the earlier concept of exobiology, which had no examples at all: Earth life provides a tangible source of insights that we will constantly return to during our broader discussion of life in the Universe. Given this, we’re going to need everyone to be up to speed with the basics of our own chemistry.
Contemporary life on Earth is based on deoxyribonucleic acid (DNA), ribonucleic acid (RNA), and proteins. In cellular life, DNA serves as the genetic material and contains the information necessary to synthesize the RNA and proteins; however, some viruses, including the coronavirus that causes COVID-19, eschew DNA and use RNA to encode their genetic information. Proteins, and to a far lesser extent RNA, are the catalysts that perform the myriad of chemical and mechanical functions required for us to thrive and reproduce. They are, in effect, the tools that our genes have invented to ensure that they—the genes—reach the next generations. (Like it or not, every aspect of our biology is focused on ensuring that our genes are handed down to our descendants.)
DNA and RNA are long, information-containing molecules in which many nucleotide building blocks (monomers) are linked together by strong, covalent bonds to form linear chains called polymers. Each nucleotide in DNA or RNA consists of a sugar (ribose in RNA, and deoxyribose, a modified ribose missing one oxygen atom, in DNA) linked to the adjacent monomers in the chain via a phosphate group. Hanging like a pendant from the sugar is one of four nucleobases: adenine (A), guanine (G), cytosine (C), or thymine (T) in DNA and the same in RNA with the exception that thymine is exchanged for the closely similar uracil (U). It is the sequence of these nucleobases, lined up along the polymer chain, that encodes the information necessary for DNA to make RNA and for RNA to make proteins. The bases occur in complementary pairings: adenine binds weakly but specifically to thymine (or uracil), and guanine to cytosine. This allows two complementary polymer strands to come together to form a double helix. It also provides a means of copying one strand of DNA into another during replication or into a strand of RNA when it is needed to synthesize a protein.
Proteins are also polymers, but their building blocks are amino acids rather than nucleotides. An amino acid (more specifically, an α-amino acid) is an organic molecule containing an amino group (−NH2) on the carbon (called the α carbon) adjacent to a carboxylic acid group (−CO2H). Polymers of amino acids are generated by fusing the amino group of one amino acid to the carboxylic acid group of another to form a peptide bond with the following structure:
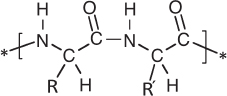
As the two functional groups involved in the polymerization are directly adjacent in the amino acid (both are on the α carbon), only the nitrogen and the two carbons closest to it form the backbone of the protein polymer. The rest of the amino acid molecule, called its side chain, is designated by the chemist’s shorthand “R,” which can represent any combination of atoms. Twenty different “proteinogenic” amino acids are commonly found in proteins on Earth (see fig. 4.2), with their 20 different side chains ranging in complexity from a single hydrogen atom to much larger chains and rings and spanning a wide variety of chemistries. Under the influence of selective pressures, evolution has come up with billions of different specific sequences of amino acids (around 25,000 different sequences are employed in us humans), each of which folds into a unique, three-dimensional shape that is defined by interactions between the side chains. Because proteins are, in effect, tools—they catalyze almost all the chemical and mechanical actions in a cell—these three-dimensional structures are critical to their activity. As with all tools, form defines function.
Miller’s “primordial soup” study was hailed on the front page of the New York Times as a breakthrough that would quickly lead to a deep understanding of our origins. His result, in some regards the second part of a one-two punch following elucidation of the structure of DNA published just a few short months earlier, produced tremendous optimism that the “origins” question would soon be answered. Alas, though, while the Miller-Urey experiment provides fascinating and tantalizing insights into possible prebiotic chemistry on the early Earth and elsewhere, it seems that some of this optimism was misplaced. A good fraction of a century later, many fundamental questions regarding prebiotic chemistry—and even more about our origins—remain unresolved.
The Volatile Inventory
Miller-Urey chemistry requires an atmosphere and an ocean, both of which are composed of materials far more volatile than the silicates and metals that make up the bulk of terrestrial planets. Thus, the origin of the “volatile inventory,” which we discussed in chapter 3, plays a fundamental role in defining the relevant prebiotic chemistry of a planet. And, as we’ll see, the chemical form of the volatiles is equally critical to our story.
Applying a cursory knowledge of chemistry to a table of the cosmological abundances of the elements (see fig. 1.2) suggests some likely candidate volatiles. Hydrogen is, of course, far and away the most common element in the Universe, but carbon, oxygen, and nitrogen are also relatively plentiful. Looking over likely (i.e., more chemically stable) “permutations” of these atoms, we can come up with a list of the chemically reasonable atmospheres of young terrestrial planets (table 4.1).
Look at table 4.1 closely and you’ll see that the likely volatile compounds of hydrogen, carbon, nitrogen, and oxygen are grouped into related columns. The relationship by which the compounds are organized is the degree to which they are oxidized—that is, the degree to which the core elements in these compounds have given up control over the electrons in a shared chemical bond, typically with an element such as oxygen (thus the term oxidized) that attracts the shared electrons more strongly. Conversely, if an atom forms a bond with hydrogen, it pulls in a larger share of the bonding electrons and is thus reduced, which is the opposite of oxidized.
On the left side of table 4.1 are the fully reduced compounds, such as ammonia (NH3), in which the central atom (nitrogen in this case) has electron density to spare and can therefore reduce other compounds, albeit at the price of becoming oxidized itself. In an environment dominated by reducing substances, nitrogen would be most likely to occur as ammonia, and carbon as methane (CH4). In the center column of the table are more neutral (meaning less reduced or, equivalently, more oxidized) materials, such as molecular nitrogen (N2), which are generally unwilling to give up or take on electrons and thus are rather chemically inert. Finally, at the far right we have the equilibrium mixture we would observe in the presence of free oxygen, which is very oxidized indeed: oxygen is the second most electronegative atom in the periodic table, which means that, in a bond with any other atom except fluorine (the only element that is more electronegative), oxygen will attract the shared electrons more strongly than does its binding partner. An atmosphere that is so strongly oxidized as to contain free oxygen is called an oxic atmosphere, a term that describes the present atmosphere of our planet. But the free oxygen in our atmosphere is due to photosynthesis, so the primordial atmosphere clearly lay to the left of the oxic column on the spectrum of possibilities shown in the table. The question we face in speculating on the origins of life is how far toward the left-hand, highly reduced end of this spectrum does the atmosphere of a newborn terrestrial planet typically lie?
Table 4.1
Volatile “sets” potentially available for young terrestrial planets
|
Reduced |
Oxidized |
Oxic |
Carbon (C) |
CH4 |
CO, CO2 |
CO2 |
Nitrogen (N) |
NH3 |
N2 |
N2 |
Oxygen (O) |
H2O |
H2O, CO, CO2 |
O2 |
Hydrogen (H) |
H2, CH4, NH3, H2O |
H2O |
H2O |
Miller-Urey Chemistry and the Early Earth
Although the Earth provides the best storehouse of information on the evolution of terrestrial planets, we have no direct record of the conditions prevalent during our planet’s earliest era; given Earth’s extraordinarily active geology, no rocks (and only a few small crystal grains) are known that unambiguously date from her first half billion years. Thus, we are left to speculate on the conditions present immediately after the accretion of a terrestrial planet.
Urey was one of the first to consider this. He reasoned that, while the crust and mantle of the terrestrial planets consist of fairly oxidized silicates, their bulk composition is reducing. That is, they are primarily composed of atoms and molecules that tend to give up electrons in chemical reactions, typically by forming bonds to very electronegative elements such as oxygen. The majority of the Earth’s mass, for example, is contained in its core, which is metallic iron and nickel. Free metals such as these are reduced. (Indeed, the word reduced originates from compounds that could reduce ore into free metal; we now know this occurs because the reducing compound provides electrons to the ores, which are typically oxides, and thus oxidized, to produce the electron-rich free metal.) If the process by which the Earth differentiated into a dense metallic core and rocky mantle and crust was slow, the metal core-forming elements would equilibrate with the rocky elements in the crust and mantle, leaving the crust partially reduced at the expense of oxidizing some of the iron in its core.
If, per Urey’s logic, the crust and mantle were reduced, then the ocean and atmosphere would be too. Even today, at age 4.5 billion years, the Earth is still geologically active enough to regularly cycle its volatile inventory into and out of the mantle. For example, the march of plate tectonics forces volatile-rich crustal plates down into the mantle at subduction zones, such as the Mariana Trench, where geothermal heat liberates the volatiles and sends them back up to belch out of volcanoes in a cycle that, were it not for the rapid production of oxygen by photosynthesis, would ensure that the Earth’s atmosphere remains more or less in chemical equilibrium with its crust.
In contrast to Urey’s speculations, however, the minerals present in the oldest rocks still to be found on Earth suggest that they formed in a relatively oxidized (though not oxic) environment. This, many geologists argue, implies that the dense metallic iron and nickel sank to form the core too quickly for chemical equilibration with the mantle to occur, and thus the Earth’s mantle and crust could have been, as they are today, composed of relatively oxidized silicates. Were this the case, volcanic outgassing would have led to a more oxidized atmosphere than would be expected from the bulk composition of the planet. Although without a good rock record of the first half billion years, we cannot tell which of these two scenarios came to pass, as we discussed with respect to Venus in chapter 3, photolysis does tend to oxidize terrestrial planets. Given this, while the rock record suggests that the Earth’s atmospheric composition was nearer the middle, neutral part of the spectrum from reduced to oxic by the time the planet was a billion years old, this is perhaps not proof that, a half billion years earlier, the primordial atmosphere was similarly oxidized.
Table 4.2
The more common Miller-Urey reaction products formed under reducing conditions
Compound |
Yield (% total fixed carbon) |
Formic acid |
4.0 |
Glycine |
2.1 |
Glycolic acid |
1.9 |
Alanine |
1.7 |
Lactic acid |
1.6 |
β-Alanine |
0.76 |
Propionic acid |
0.66 |
Acetic acid |
0.51 |
Iminodiacetic acid |
0.37 |
α-Hydroxybutyric acid |
0.34 |
α-Amino-n-butyric acid |
0.34 |
Succinic acid |
0.27 |
Sarcosine |
0.25 |
Iminoacetic propionic acid |
0.13 |
N-methylalanine |
0.07 |
Glutamic acid |
0.05 |
N-methylurea |
0.05 |
Urea |
0.03 |
Aspartic acid |
0.02 |
α-Aminoisobutyric acid |
0.01 |
Total |
15 |
Note: Proteinogenic amino acids shown in bold type.
In the decades since Miller’s initial work, his experiment has been repeated many times with many different atmospheric compositions and energy sources, including Miller’s electric discharge (simulating lightning), hot rocks (simulating volcanism), UV light (simulating sunlight), and ionizing radiation (simulating radioactive minerals and cosmic rays). As long as the experimental atmosphere is, as was true for the one Miller employed, fairly reducing, all these variations produce about the same results: a soup of amino acids and other small, simple organic compounds corresponding to about 10% to 15% of the total carbon input into the experiment, with the remaining carbon primarily ending up in a complex, high molecular weight tar lining the apparatus (typical reaction products are listed in table 4.2).
This is a pretty exciting observation, no? But it is not without its detractors. Historically, it was found that unless methane and ammonia or methane, nitrogen, and molecular hydrogen are present, Miller-type experiments produce relatively little in the way of biologically relevant small molecules. And, as noted above, geochemists have argued that the early Earth’s atmosphere was not as reduced as Urey had thought. Fortunately, however, Jeffrey Bada,* a geochemist at Scripps Institution of Oceanography near San Diego, California, has since demonstrated that, under some conditions, a relatively oxidized carbon dioxide and nitrogen atmosphere also supports Miller-Urey chemistry. Starting from this more oxidized atmosphere, Miller-Urey reactions create acids, which lower the pH of the “ocean” and reduce amino acid yields, and nitrites, which destroy what few amino acids are produced. These effects can be countered, however, by adding soluble reducing agents to remove the nitrites and carbonates to buffer the pH. For example, performing the Miller-Urey experiment using a carbon dioxide, nitrogen, and water vapor atmosphere in the presence of iron and calcium carbonate, Bada’s students obtained amino acids in modest yields.
Take a reduced (or even neutral), water-rich atmosphere, pass some lightning through it, sit back, and what do you get? Most of the amino acids that form our proteins (as shown in fig. 4.2). Pretty neat, eh? And, come to think of it, really all you need to know. But if you’re willing to hitch up your trousers and slog through a bit of chemistry, the story gets a little richer—and more complex—than we’ve let on so far.
Miller-Urey Mechanisms
Amino acids are less stable than a mixture of methane, ammonia, and water, and thus energy must be invested in order to synthesize them. In Miller’s experiment, the energy source was the lightning-like electric discharge, though, as we’ve noted, many other energy sources are likewise sufficient. In the case of the electric spark, high-energy electrons in the discharge tear other electrons out of the gas molecules, creating molecular species such as methyl and hydroxyl “radicals” that contain an odd number of electrons (and thus electrons not paired up with partners). As the main driving force of chemical reactions is pairing up electrons to fill up orbitals, radicals are usually unstable and extremely reactive. For example, the hydrogen radical (•H, where the dot represents the unpaired electron) can attack methane to produce hydrogen gas (H2) and the more stable methyl radical (•CH3). The methyl radical, in turn, reacts with water to produce another molecule of hydrogen and the still more stable methoxy radical (CH3O•). Of course, though, it doesn’t stop there: the methoxy radical can undergo a number of other reactions, including joining with a hydrogen radical to produce methanol (CH3OH) or reacting with a hydrogen radical to produce hydrogen gas and formaldehyde (H2C=O). Alternatively, the methyl radical can react with another carbon radical to produce larger, more complex carbon compounds, or it can bond with ammonia in the first step of a process that ultimately produces hydrogen cyanide (HC≡N). The theme here is that reaction of a radical with a stable “even-number-of-electrons” molecule, such as water, always produces another radical (with its odd number of electrons), and thus radical chemistry is characterized by chain reactions that build progressively more stable radicals until a second radical is encountered, ending the cascade. Moreover, as long as they remain in the gaseous atmosphere, the larger molecules produced in these cascades also fall prey to the electric discharge (or photolysis), forming still more complex radicals and still larger, more complex compounds. A smattering of some of the radical reactions thought to occur in the discharge is shown in fig. 4.3; similar “radical chain reactions” occur in flames and are a central feature of the chemistry of combustion (see fig. 1.1).
Hints as to the mechanism by which Miller-Urey chemistry forms amino acids from simpler precursors can be obtained by monitoring the concentration of various molecular species as the reaction proceeds (fig. 4.4). For example, the first 24 hours see a rise in the concentrations of both the simplest nitrogen-containing organic molecule, hydrogen cyanide, and the simple oxidized carbon compounds called aldehydes (containing an −HC=O grouping of atoms). After a few more days, however, the concentrations of these species fall to near zero. In contrast to this rise-from-zero-then-fall-to-zero behavior, the concentration of ammonia—which, as one of the starting materials, is initially at a very high concentration—falls steadily as the reaction proceeds, and the concentration of amino acids rises steadily for several days before reaching a plateau. These trends suggest that hydrogen cyanide and simple aldehydes are intermediates in a reaction that, in net, “fixes” ammonia into amino acids. A century before Miller’s experiment, the German organic chemist Adolph Strecker (1822–1871) had reported a synthetic route to the formation of amino acids that similarly employed hydrogen cyanide, ammonia, and aldehydes (fig. 4.5). Miller-Urey chemistry is now thought to be a variant of this “Strecker synthesis,” in which ammonia reacts with an aldehyde to produce an imine, a molecule containing a nitrogen-carbon double bond. The carbon in this bond is relatively susceptible to attacks from reactive species such as cyanide (−CN), with which it forms an α-aminocyanonitrile. The cyanonitrile group (−C≡N) is prone to hydrolysis, which is the breaking of a bond through its reaction with one or more water molecules. Specifically, the addition of two molecules of water (and the subsequent loss of a molecule of ammonia) causes conversion of cyanonitrile into carboxylic acid (−CO2H), forming, in this case, an amino acid in the process.
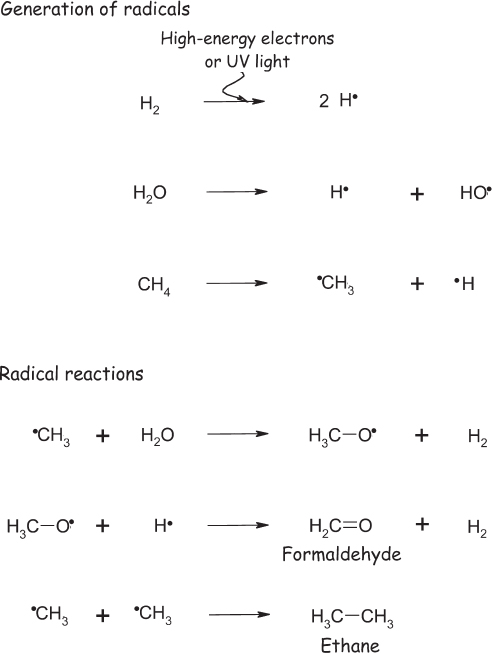
Figure 4.3 Miller-Urey chemistry is thought to proceed via reactions similar to those that take place in flames (see Figure 1.1). High-energy electrons in the spark strike atmospheric molecules, knocking an electron out of them to form radicals, energetic species containing an unpaired electron (shown as a dot). These highly reactive radicals quickly react with other atmospheric components, ultimately to produce larger, more complex molecules.
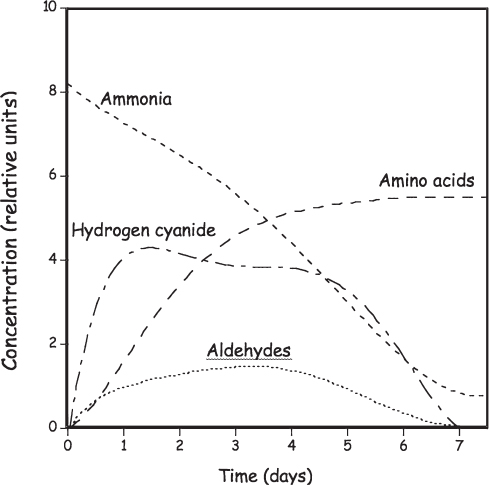
Figure 4.4 Insight into the mechanisms of Miller-Urey chemistry is provided by the time course of the consumption of ammonia (NH3) and formation of hydrogen cyanide (HC≡N), aldehydes (R−HC·O), and amino acids. The peak in the concentration of hydrogen cyanide and aldehydes at intermediate times suggests that these molecules are waypoints in a multistep reaction that converts small-molecule precursors into larger and more biologically relevant amino acids.
Around 4% of the total carbon input (as methane) into a typical Miller-Urey experiment is converted into amino acids. Miller, using the analytical techniques of the time, identified five of these, three of which are among the 20 amino acids employed by life on Earth to make proteins. Follow-on experiments conducted decades later on Miller’s original samples identified more; we now know that 10 protein-forming (or “proteinogenic”) amino acids are produced under Miller’s conditions (listed in table 4.3) and that four more can be synthesized using slight “tweaks” on Miller’s original conditions. The 10 proteinogenic amino acids not found under Miller’s conditions include the aromatic amino acids phenylalanine, tyrosine, and tryptophan, which are characterized by their large, bulky, ring-containing side chains; the positively charged amino acids histidine, lysine, and arginine; the sulfur-containing amino acids cysteine and methionine; and the amide-containing amino acids asparagine and glutamine (amide group −CO−NH2). The latter four omissions, however, are technical rather than fundamental. For example, because Miller did not include any sulfur compounds in his reaction mixture, the lack of the two sulfur-containing amino acids is not surprising; when hydrogen sulfide (H2S) is included in the reaction mixture, both of these amino acids are produced. Likewise, the boiling “ocean” in Miller’s experiment destroyed the two amide-containing amino acids. The prebiotic synthesis of the other six proteinogenic amino acids, in contrast, seems a more fundamental problem. Those amino acids were almost certainly introduced by biochemistry after the origins of life. For the aromatic amino acids, the reason may be the difficulty in synthesizing the aromatic ring using gas-phase chemistry such as that seen in the Miller-Urey experiment.
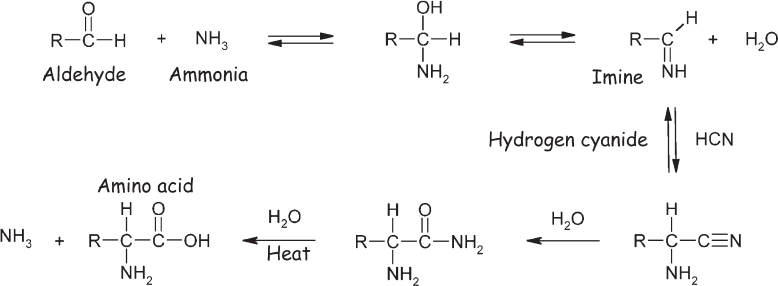
Figure 4.5 The Strecker synthesis is thought to parallel the mechanism by which amino acids are synthesized in the Miller-Urey experiment and perhaps to be the source of the amino acids delivered to Earth by the carbonaceous chondrite meteorites (discussed later in the chapter).
Table 4.3
Yields of the α-amino acids in a typical, hydrogen sulfide–free Miller-Urey experiment
Amino acid |
Yield (μM) |
Glycine |
440 |
Alanine |
790 |
α-Aminobutyric acid |
270 |
Norvaline |
61 |
Aspartate |
34 |
α-Aminoisobutyric acid |
30 |
Valine |
20 |
Leucine |
11 |
Glutamate |
8 |
Norleucine |
6 |
Isoleucine |
5 |
Serine |
5 |
Alloisoleucine |
5 |
Isovaline |
5 |
Proline |
2 |
Threonine |
1 |
Allothreonine |
1 |
Tert-Leucine |
0.02 |
Note: Proteinogenic amino acids shown in bold type.
The Prebiotic Synthesis of Sugars
Sugars are called carbohydrates because their elemental composition is that of a carbon atom combined with a water molecule: Cn(H2O)n, where n is in the range of three to seven (fig. 4.6). The pentoses, a family of sugars for which n = 5 (e.g., ribose), and the hexoses, for which n = 6 (e.g., glucose), are particularly important in biology on Earth. In defiance of the historical name, however, the hydrogen and oxygen atoms in carbohydrates do not take the form of water molecules (i.e., sugars are not “hydrates” of carbon). Instead, sugars are polyalcohols in which half of the hydrogens (plus one) are bound directly to a carbon and the other half (minus one) are bound to oxygen atoms to form alcohol groups (−OH). The “minus one” results from the fact that one carbon in every sugar molecule is bound to an oxygen by a double bond (C=O, where the oxygen has no binding capacity left to accommodate a hydrogen) to form an aldehyde or ketone functional group, which is located at the end of or within the chain of carbon atoms, respectively.
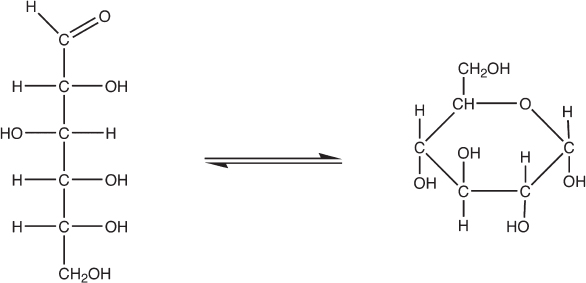
Figure 4.6 Sugars are polyalcohols of the general formula Cn(H2O)n. Shown here is glucose, an aldohexose (from aldo, designating the double-bonded oxygen at the end of the chain, and hexose, designating that n = 6). Sugars such as glucose typically exist in two or more interconverting forms. On the left is the linear form, and on the right is a cyclized conformation produced when one of the alcohol groups attacks the aldol carbon. For glucose, the cyclic form predominates in solution.
The trouble with sugars is that, even the simplest, those containing just three carbons and three oxygen atoms, are complex enough that Miller-Urey chemistry does not produce them. That is, although Miller-Urey chemistry forms simple alcohols, aldehydes, and ketones, even the smallest sugar is simply too large to be synthesized in the atmosphere; long before the simplest sugar could be produced, the precursor molecule would rain out of the sky.* Sugar formation, then, must have occurred in the oceans or on land through polymerization of the smaller precursors that Miller-Urey chemistry can produce. How might that have happened?
In 1861, the Russian chemist Alexander Butlerov (1828–1886) described the formose reaction, which would later be hailed as the most plausible prebiotic route to the sugars. This starts with formaldehyde dissolved at a concentration of 1% to 2% in water under alkaline (basic) conditions. (Although formaldehyde itself is not a sugar, with its formula H2C=O, it has the same “carbon-plus-water” composition as sugars.) After a short incubation, the reaction accelerates rapidly until, at its peak, up to 50% of the formaldehyde is converted into sugars. On further incubation, however, the sugar content decreases because the sugars that are formed are, ultimately, unstable in the presence of the strong base used to catalyze the reaction.
The formose reaction is thought to proceed via the two-carbon intermediate glycolaldehyde, which is formed from two molecules of formaldehyde (fig. 4.7). Glycolaldehyde is also a catalyst in the formose reaction, and this autocatalysis accounts for the short delay observed before the reaction takes off; once a few molecules of glycolaldehyde have formed, they speed up the formation of yet more molecules of glycolaldehyde. Under alkaline conditions, the glycolaldehyde in turn reacts with a third molecule of formaldehyde to form the three-carbon sugar glyceraldehyde. Glyceraldehyde, in turn, can undergo a base-catalyzed isomerization reaction (a reaction that moves atoms around in a molecule but does not change its overall formula) that converts it into the other three-carbon sugar dihydroxyacetone. These two sugars, with their C=O functionality on an end carbon and middle carbon, respectively, are the only possible three-carbon sugars.
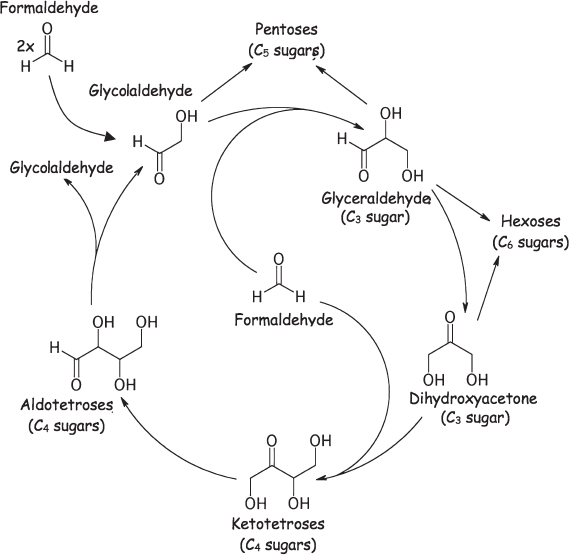
Figure 4.7 The base-catalyzed formose reaction converts one-carbon formaldehyde into higher molecular weight sugars. After the slow formation of the first molecule of glycolaldehyde from two molecules of formaldehyde (upper left), a reaction cycle is started that synthesizes both sugars and, with each turn of the cycle, more glycolaldehyde, which serves to accelerate the reaction in the next round. The reaction is catalyzed by reasonably plausible prebiotic catalysts, such as calcium hydroxide, but it is fairly nonspecific, producing a complex mixture of high molecular weight sugars.
From glyceraldehyde and dihydroxyacetone, each only three carbons long, the formose reaction can produce many other larger and more complex sugars. For example, two of these three-carbon sugars can combine by an aldol condensation reaction (so named because it results in an aldehyde-alcohol) to form most of the six-carbon hexoses. Alternatively, dihydroxyacetone can condense with another molecule of formaldehyde to form the four-carbon ketotetrose (four carbons, with C=O within the chain) erythrulose. Erythrulose, in turn, isomerizes to form the aldotetroses (also four carbons, but with the C=O at the end of the chain), which can undergo a reverse version of the aldol condensation, splitting the molecule into two glycolaldehydes and starting the cycle anew. Of note, the isomerizations and aldol condensations thought to occur in the formose reaction mimic the chemistry by which present-day Earth life produces sugars from smaller precursors. Whether this coincidence represents a historical artifact of prebiotic chemistry that was later hijacked by life or simply represents the most convenient chemistry by which sugars can be made (and thus the most likely chemistry for evolution to independently discover) remains an open question.
Although the formose reaction has some of the traits we are looking for—it is spontaneous and can produce high yields—it is not without its problems as an explanation for the prebiotic synthesis of sugars. The first problem is the formaldehyde. Not that it can’t be made prebiotically; formaldehyde can be synthesized by the action of UV light on carbon dioxide and water in the atmosphere. Light with a wavelength of less than 240 nm (relatively far into the UV spectrum) splits water into the hydroxyl radical (•OH) and a free hydrogen atom. Similarly, short wavelength (and thus high-energy) UV light splits carbon dioxide into carbon monoxide and atomic oxygen, itself a potent radical generator. In the presence of carbon monoxide and water, the radicals formed in these processes generate formaldehyde, although ever so slowly. Current estimates suggest that, by this mechanism, it would take around 10 million years for formaldehyde to accumulate to the levels required for the formose reaction to proceed. Formaldehyde, though, is a fairly reactive species, and thus it seems unlikely that such a slow process could, at the end of the day, generate a high enough formaldehyde concentration to drive the reaction forward. Moreover, even if sufficient formaldehyde were available, the problem remains that the formose reaction is nonspecific and produces only small quantities of each of the many different types of sugars. In particular, the formose reaction produces very little ribose, which in many regards is the most fundamental sugar for life on Earth and, as we will see in the next chapter, is intimately coupled to the most promising theories of life’s origins. Worse still, what little ribose is produced by the formose reaction is relatively unstable. Even at modest temperatures, the half-life of ribose in water is just decades. Given these issues, it is hard to imagine that much ribose could have built up simply from the formose-induced polymerization of formaldehyde in a primordial ocean.
Fortunately, a potential solution to at least this last “ribose” problem has been demonstrated in the laboratory. As a catalyst for the formose reaction, researchers have traditionally used calcium hydroxide, which is a fairly powerful base. Very little ribose accumulates under strongly basic conditions because the sugars produced further polymerize into a complex, brown goo. Steven Benner at the University of Florida has shown, however, that calcium borate, which occurs in nature as the minerals borax and colemanite, stabilizes sugars, such as ribose, in which two neighboring hydroxyls point in the same direction. It does so by forming a cyclic boron “ester”—a ring-shaped molecule with boron bridging the oxygens of the former alcohol groups. As this renders the sugar unreactive, formation of the ester terminates the formose reaction. Of course, while this pushes up the yield of ribose and a few other, structurally similar sugars, it does nothing to stabilize the ribose once the free sugar is released from the boron (the reaction is easy to reverse—just add water). And the chemical half-life of ribose remains quite short relative to the required lengths of time we’d expect for prebiotic chemistry to build up usable quantities of life’s precursors.
The Prebiotic Synthesis of Nucleic Acids
Miller-Urey chemistry also fails when it comes to producing significant amounts of purine (two-ring) and pyrimidine (one-ring) nucleobases (fig. 4.8), important ingredients in life on Earth that serve as key components of DNA and RNA. In 1961, however, John “Juan” Oro (1923–2004) found that amino acids and small amounts of the purine base adenine form spontaneously from mixtures of hydrogen cyanide and ammonia in water. Following this, other researchers have explored Oro’s chemistry in more detail and found that small amounts of several different nucleobases are detectable when solutions of ammonium cyanide, the compound formed when ammonia and hydrogen cyanide combine, are allowed to react and then boiled with acid.
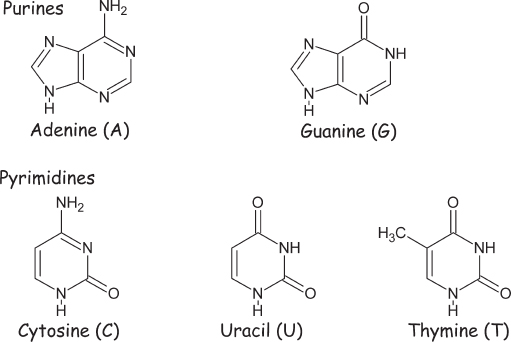
Figure 4.8 Five different nucleobases (often simply referred to as bases) are employed throughout Terrestrial biochemistry. Three of these, adenine, guanine, and cytosine, appear in both RNA and DNA. Rounding out the set we have uracil as the fourth nucleobase in RNA and the closely similar thymine as the fourth nucleobase in DNA.
Key to the chemistry of these reactions is the fact that hydrogen cyanide is a fairly reactive molecule and thus contains the energy necessary to drive a multistep prebiotic chemical pathway. Nevertheless, because four molecules of the compound are condensed in the first steps of the proposed adenine synthetic reaction, relatively high concentrations of hydrogen cyanide are required. Indeed, the concentrations typically employed in the laboratory experiments were hundreds of thousands of times higher than the hydrogen cyanide concentration calculated to exist on the surface of the early Earth. Worse still, because hydrogen cyanide is more volatile than water, it cannot be concentrated by, for example, the evaporation of seawater in a tidal pool. A year after Oro’s experiment, however, Leslie Orgel (1927–2007) suggested a possible solution to this problem when he proposed that the requisite high concentrations can be achieved by freezing a solution of hydrogen cyanide, which concentrates it in the voids between ice crystals. To test this, in 1975 Miller placed a fairly dilute sample of ammonium cyanide in cold storage at −78°C. Some 27 years later (tenure provides the breathing room required for professors to take the long view), Miller thawed his samples for analysis and was able to detect small quantities of several nucleobases, including adenine. This suggests that their prebiotic synthesis may have happened only in the Earth’s polar regions or in icy planetesimals in the outer Solar System.
The abiological synthesis of the purine nucleobases has been explored in some detail. The process is thought to involve the multistep condensation of four molecules of hydrogen cyanide to form diaminomaleonitrile (fig. 4.9). Under the influence of UV light (sunlight is sufficient), this compound rearranges and reacts with yet another molecule of hydrogen cyanide to produce the nucleobase adenine in about 7% overall yield. That is, 7% of the starting cyanide is converted into adenine, and the other 93% either does not react or reacts to produce other molecules. Alternatively, four molecules of hydrogen cyanide can react with the salt ammonium formate to produce adenine with a yield of better than 90%. To achieve this yield, however, the reaction mixture must be heated to dryness: the reaction occurs with the liberation of two water molecules, and thus driving off the water with heat favors formation of the nucleobase.
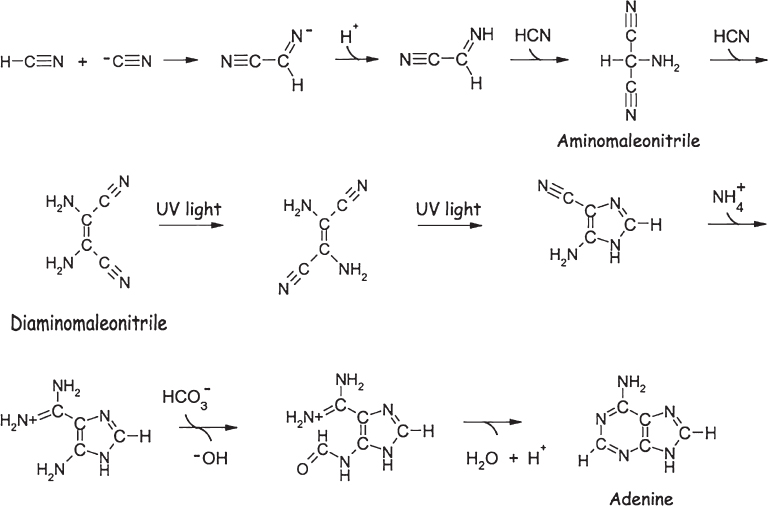
Figure 4.9 One of several pathways proposed for the prebiotic synthesis of the nucleobase adenine (a purine) from bicarbonate (HCO3−), ammonium (NH4+), and hydrogen cyanide (HCN), the latter of which is produced in Miller-Urey reactions.
The other purine nucleobase guanine can also be synthesized by variations on the above synthesis of adenine. Guanine can be synthesized, for example, by the polymerization of ammonium cyanide, which, again, is formed by dissolving both ammonia and hydrogen cyanide in water (fig. 4.10). The synthesis works effectively over a broad range of temperatures, but even at ammonium cyanide concentrations far higher than those plausible on the prebiotic Earth, the overall yield of this nucleobase is less than 1%. Likewise, when ammonium cyanide polymerizes in ice, guanine is produced in parallel to adenine, albeit at yields 10 times lower. In contrast, an interesting gas-phase route has been described that may achieve more of the desired nucleobase. Specifically, Svatopluk Civis of the Czech Academy of Sciences used an ultra-high-power laser to simulate the effects of a meteorite impact on a primordial Earth with the Miller-Urey product formamide. From this, he recovered guanine at levels of a few milligrams per liter.
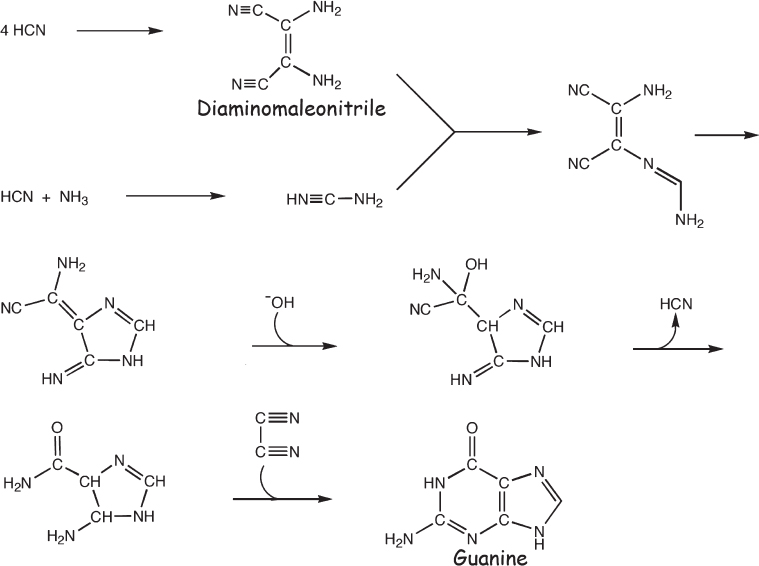
Figure 4.10 A proposed prebiotic synthetic route to the nucleobase guanine from ammonia (NH3) and hydrogen cyanide (HCN). Even under optimized laboratory conditions, however, the yield of this reaction is low.
The prebiotic synthesis of the pyrimidine nucleobases has also been explored, with many of the proposed routes starting with the precursor cyanoacetylene, a minor product of the reactions induced in methane-nitrogen mixtures under the influence of a spark discharge (fig. 4.11). This precursor reacts with water to form cyanoacetaldehyde, and this in turn can react with the small, nitrogen-containing organic compound urea, which is also formed via Miller-Urey chemistry. The intermediate so formed spontaneously rearranges to form the more stable cytosine, one of the three pyrimidine bases used by biology on Earth. As nucleobases go, however, cytosine is relatively unstable; its half-life in water is estimated to be around a century at room temperature. The good news about this reactivity is that the hydrolysis product of cytosine is uracil, another of the three pyrimidines found in our biochemistry, thus accounting for the prebiotic synthesis of this critical component. The bad news, however, is that the short half-life of cytosine means that it would have been difficult for significant quantities of this nucleobase to build up over a geological time frame.
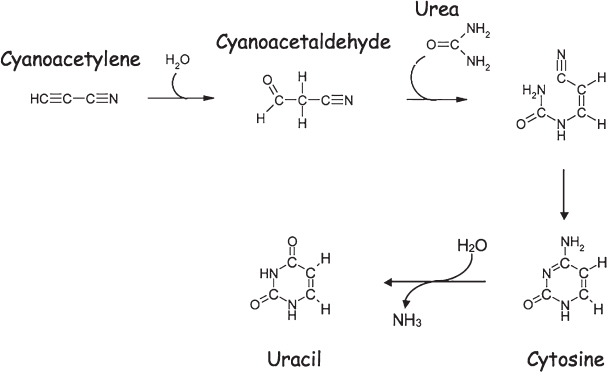
Figure 4.11 The proposed prebiotic synthesis of the nucleobase cytosine (a pyrimidine) from cyanoacetylene and urea, both of which are produced in Miller-Urey reactions. Cytosine, in turn, reacts with water and loses ammonia to form uracil.
The above is all fine and dandy but, by and large, Terrestrial biology doesn’t employ free nucleobases so much as three-part structures consisting of a nucleobase covalently attached to a ribose (or deoxyribose) sugar that, in turn, is linked to one or more phosphate groups (fig. 4.12). Specifically, nucleobase-sugar combinations come in two general forms. The nucleosides consist of a nucleobase covalently linked to a sugar by a glycosidic bond, and nucleotides consist of nucleosides plus phosphates. Nucleosides and nucleotides are synthesized from free sugars, nucleobases, and phosphates by dehydration reactions, which are the reverse of the hydrolysis reactions described above. That is, this linkage chemistry proceeds with the removal of a water from the molecule. Because of this, these reactions are generally unfavorable when they take place in liquid water; you’ll remember from chapter 1 that there are a whopping 55.5 mol of water in each liter of water, which is an amazingly high density of molecules. With all this water around in aqueous environments, chemical reactions tend to want to go in the direction that consumes water molecules rather than the direction that liberates them. This, in part, is why the formation of nucleosides and nucleotides, both of which occur by dehydration reactions, is energetically unfavorable in water and must be “driven” with some energy input in order to move forward efficiently. Today, our bodies use the energy obtained from oxidizing carbohydrates to drive these reactions. But this, of course, was not available before the advent of life. How, then, might the prebiotic synthesis of nucleotides have been achieved?

Figure 4.12 Nucleosides are a covalent combination of a sugar (either ribose, as shown here, or, in DNA, deoxyribose, which we discuss in the next chapter) and a nucleobase. A nucleotide is a nucleoside plus one or more phosphate groups. Shown here is the ribonucleotide adenosine diphosphate (abbreviated ADP).
Leslie Orgel, who spent several decades at the Salk Institute, demonstrated a relatively straightforward route to the synthesis of nucleosides of the nucleobase hypoxanthine. If a mixture of hypoxanthine, ribose, and magnesium ions is heated to dryness, the nucleoside inosine is formed (fig. 4.13). Even better, the nucleoside produced is in the so-called β configuration, in which the nucleobase is on the opposite side of the sugar ring from the two remaining hydroxyls, which is the structure observed biologically on Earth. How does this reaction work? The magnesium ion binds to two of the hydroxyls on the ribose, thus “activating” the carbon to which the nucleobase will link. A reactive nitrogen atom in the hypoxanthine attacks this activated carbon to form a nucleoside. Heating to dryness pushes the linkage reaction forward by driving off the water that is liberated as the reaction progresses.
Unfortunately—isn’t it starting to appear as though there’s always an “unfortunately” in this business?—this reaction is neither general enough nor specific enough to plausibly account for the prebiotic synthesis of nucleosides. It is insufficiently general because hypoxanthine is the only nucleobase that is reactive enough to form nucleosides by this mechanism, and of the four nucleobases for which we have plausible prebiotic syntheses, it’s the one that is least relevant, at least for contemporary Earth life. It is insufficiently specific because many of the possible five-carbon sugars react equally well, which would waste precious nucleobases without producing the desired ribose-containing nucleoside.
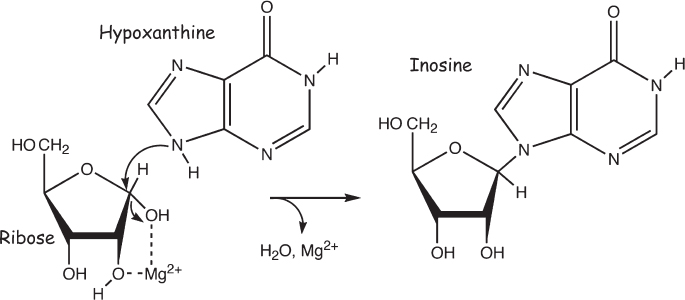
Figure 4.13 If a magnesium ion is present to act as a catalyst, the nucleoside inosine spontaneously forms from ribose and the nucleobase hypoxanthine. No other nucleobase, however, has been shown to participate in this coupling reaction.
And that’s not all. Orgel’s reaction is an example of what synthetic organic chemists pejoratively call a “relay synthesis.” That is, Orgel’s group never succeeded in going from the basic precursors, cyanide and ribose, to a fully formed nucleoside. Instead, they started with pure hypoxanthine and ribose (which they purchased, commercially, taking the “baton” only at this late stage in the game), rationalizing that previous work had demonstrated that these molecules can be synthesized under prebiotic conditions. Moving forward, they showed that hypoxanthine can be linked to ribose under other plausible prebiotic conditions. The problem here, though, is that the conditions that support the formation of the ribose and of the nucleobase are very different from one another and from the conditions that support their linkage to form the nucleoside. The experimenters thus had to hypothesize that one part of the prebiotic Earth provided the right conditions to synthesize the sugar, a second part provided the base, and, after they were transported there, a third place would foster their linkage to form the nuleoside. And while this is plausible (the Earth’s a big and diverse place, after all), it does suggest that the yield of the nucleoside would have been rather low at best. Fortunately, however, there may be a solution to this problem: in 2009, John Sutherland and his team, then of the University of Manchester, demonstrated a route to the formation of nucleosides that avoids this problem. And, as an added bonus, it produces a nucleotide—a nucleoside with a phosphate attached, like the ones used in Terrestrial biology.
Like those he followed, Sutherland had spent more than a decade thinking about the stepwise—and separate—synthesis of sugars and nucleobases, without ever demonstrating a route that could lead to them from start to finish. Finally, Sutherland’s group tried a radically different tack, one that scrambles together the cyanonitrile (−C≡N) chemistry that leads to the nucleobase and the aldehyde (−HC=O) chemistry that generates the sugar so as to avoid the problem associated with creating a bond between a preformed sugar and a preformed base (fig. 4.14). To do this, they added cyanimide, the first compound in the synthesis of the nucleobase, to glycolaldehyde, the first compound in the synthesis of the sugar, to form a molecule that was half a sugar bonded to half a nucleobase. Combining this with glyceraldehyde, the second component of the sugar, and cyanoacetylene, the second component of the nucleobase, they produced an intermediate containing, albeit hidden within it, both a ribose and a cytosine nucleobase. Treating this with phosphate (which serves as both a catalyst of the reaction and a reactant in it), they liberated the nucleoside of ribose and cytosine connected by the same glycosidic bond used in Earth’s biology. Better still, the phosphate remains attached to the ribose—thus the product is a nucleotide rather than the less useful nucleoside. A caveat of Sutherland’s postulated prebiotic synthesis, however, is that the various steps in the reaction pathway require varying conditions (e.g., changing pH), and thus he had to postulate that the environment that would have supported such a prebiotic synthesis would have had to fluctuate significantly with time. Still, the chemical principles that this work illustrates may yet prove more general, such that similar synthetic routes that rely less on the invocation of complex environmental fluctuations may exist.
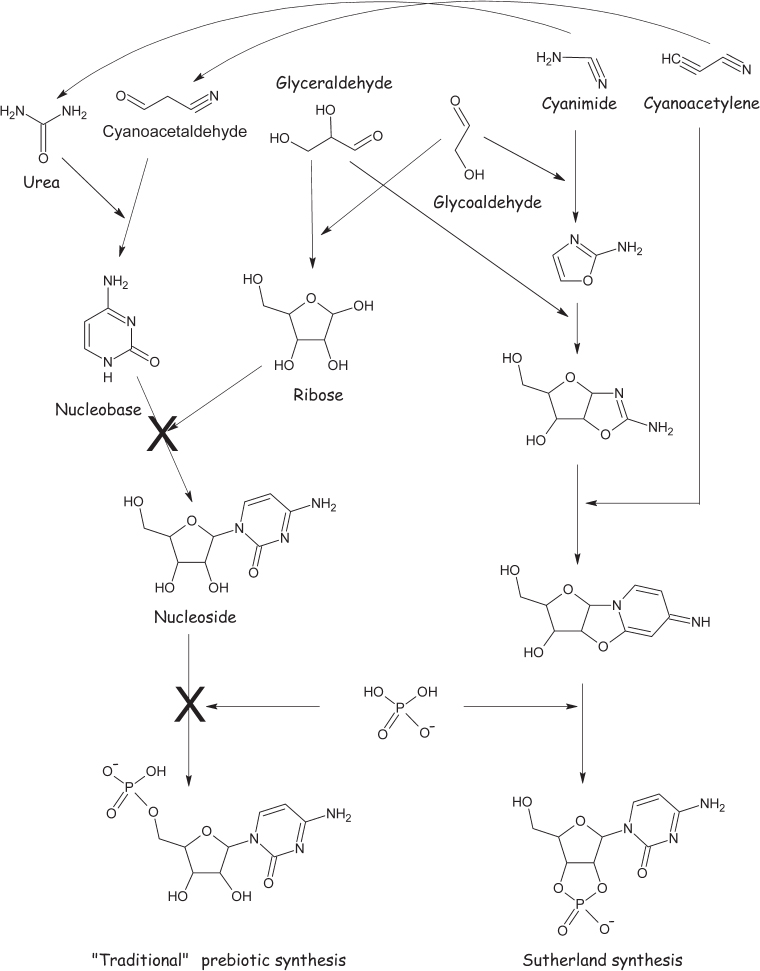
Figure 4.14 According to traditional thinking (illustrated on the left), ribose and the nucleobases were synthesized independently before being linked together with phosphate to form the nucleotides. Spontaneous, prebiotic bond formation between ribose and nucleobases, however, has not been demonstrated for any of the four nucleobases found in RNA. The addition of “unactivated” phosphate (last step) has likewise proved problematic. Fortunately, however, John Sutherland and coworkers demonstrated an alternative synthesis of cytidine phosphate that occurs spontaneously without utilizing either ribose or the nucleobase as intermediates. The addition and subtraction of water molecules, which occurs in several steps of the reaction, has been omitted for clarity.
Given the difficulty in identifying solidly plausible prebiotic routes to the formation of the nucleotides in DNA and RNA, an increasing number of researchers have argued that the nucleobases, ribose, and phosphate that are the components of contemporary life on Earth might not have served as the foundation of its origins. The American chemist Nick Hud, for example, systematically surveyed more than 80 candidate pyrimidines and purines to show that 2,4,6-triaminopyrimidine, which, although it does not play any role in contemporary biology, is the most reactive with ribose and thus may have played a precursor role in our biology, being later supplanted by the current nucleobases. Likewise, the Swiss chemist Albert Eschenmoser has argued that the triazines, six-membered aromatic rings containing three nitrogen atoms, rather than the two contained in our pyrimidine nucleobases, are a perhaps intriguing alternative to the current nucleobases as they are readily synthesized under plausible prebiotic conditions. In parallel, alternative nucleic acids constructed from sugars other than ribose, including a number of four-, five-, and six-carbon sugars, have been demonstrated in the laboratory. This said, what, if any, roles these many alternatives might have played in the origins of life on Earth and, for that matter, whether they might support the creation of life elsewhere, remain open questions.
So far, so good. But free nucleosides and nucleotides are not the stuff upon which life is founded. Instead, life is built around polymers of nucleotides, long chains of nucleobases bound to ribose that are in turn linked, hydroxyl group to hydroxyl group, by intervening phosphate groups in what are known as phosphodiester bonds. These polymeric nucleic acids, called RNA (ribonucleic acid) or DNA (deoxyribonucleic acid), are higher in energy than monomers because the bonds that link the monomers together in these compounds are themselves unstable; they too are the products of dehydration reactions. A major question is thus: how do we generate “activated,” high-energy nucleotides whose chemical energy can be harnessed to drive polymerization? (Now might be a good time to check out sidebar 4.2, though, which touches on the inherent rareness of high-energy molecules.) Perhaps the most compelling answer to this is the use of high-energy polyphosphates, which is in fact the system Terrestrial biology uses today to store and shuttle energy.
The simplest nucleotides are the monophosphates, in which a single phosphate is linked to a hydroxyl group on the nucleoside’s sugar. And while Sutherland’s synthesis produces these by default, they can also be synthesized, in up to 60% yield, simply by heating a solution of nucleosides and phosphoric acid with urea, which acts as a catalyst. Unfortunately, however, nucleoside monophosphates are not the sort of high-energy compound we need to polymerize nucleotides. In contrast, a string of phosphates linked by oxygen bridges is high in energy because, at neutral pH, each phosphate takes on a negative charge, and the negative charges on adjacent phosphates repel one another. Contemporary Earth life utilizes the energy available from this repulsion to drive the polymerization of RNA (and DNA), not to mention countless other biochemical reactions within our cells. And it seems it just might be possible for nucleoside polyphosphates to be synthesized under reasonably plausible prebiotic conditions.
The postulated reaction involves the triphosphate molecule trimetaphosphate, which consists of three molecules of phosphoric acid that have become linked together by dehydration reactions. On heating a dry mixture of nucleotide monophosphates with trimetaphosphate (a cyclic molecule comprised of three phosphate groups linked into a circle) in the presence of the catalyst magnesium (again, the heat drives off water, propelling the reaction forward), nucleoside polyphosphates are formed in good yield. When water is added, these compounds rapidly hydrolyze to form nucleoside triphosphates, which then only slowly hydrolyze to form di- and monophosphates. Unfortunately, however, the relevant phosphate compounds are quite rare on Earth; phosphate is usually the limiting mineral, for example, in freshwater ecosystems, and its high energy forms are not stable.
Of course, just because polyphosphates play the role of activator on contemporary Earth, this doesn’t mean that it’s the only way to activate nucleosides or, indeed, that it was the activation chemistry employed in the origins of life. For example, in the 1990s, Leslie Orgel proposed that the small, nitrogen-containing ring compound imidazole could react with phosphate to form a phosphorimidazolide containing a high-energy nitrogen-phosphorus bond that activates the phosphate for further reactions. Unfortunately, however, while phosphorimidazolides can be synthesized in the laboratory, no plausible prebiotic route to their synthesis has been described. And if some alternative activation chemistries were in play during the origins of life, they have not yet been identified.
The Missing Ingredient: Fats
Life on Earth is not built on amino acids and nucleotides alone. The dry weight of a bacterial cell, for example, is about 6% lipids, a word stemming from the Greek lipos, meaning “fat.” To a biochemist, though, lipids are anything that can be extracted from tissues by an organic solvent, and thus this family of molecules includes both fats and oils. Unfortunately, fats and oils are not readily produced by Miller-Urey chemistry, yet it seems likely that they played key roles in the origins of life. How, then, were appreciable amounts of these materials synthesized under prebiotic conditions? The answer to this question also remains unknown, but several seemingly plausible theories have been put forth.
Most lipids are long-chain molecules, and all consist almost entirely of carbon and hydrogen. Miller-Urey chemistry, in contrast, tends to produce only oxygen- and nitrogen-containing species, such as alcohols and amino acids. The only way long carbon chains can form via Miller-Urey chemistry is through radical-radical reactions in which methyl radicals add to a growing carbon chain. As the chances of the radical reacting with a water or ammonia molecule (to stop the growing chain) are reasonably large, purely carbon-hydrogen compounds become rarer with increasing chain length simply due to the improbability of one carbon radical reaction following another. Likewise, long-chain carbon compounds are not very volatile. Miller-Urey chemistry is gas-phase chemistry and, as we’ve mentioned, molecules with more than about three or four carbons fall out of the atmosphere as liquids or solids.

Figure 4.15 Shown are the wide variety of reduction reactions that can occur on the mineral troilite (FeS). Such reactions, which would probably have occurred as ocean water filtered past highly reduced rocks deep in the Earth’s primordial crust, could be a prebiotic source of lipids, which are critical for biology and are not produced by Miller-Urey chemistry.
Perhaps the most realistic theory described to date for the prebiotic formation of lipids is that they were synthesized by the reduction of that other class of large carbon-containing compounds: sugars. But again, these compounds are not volatile and thus the reduction cannot occur in the atmosphere. So where can this happen? Deep in the planetary crust, where reduction can be catalyzed by the iron mineral troilite, which is composed of iron sulfide (FeS). In the presence of the reducing gas hydrogen sulfide (remember: the crust is reduced, at least in this model of prebiotic chemistry), troilite is a strong reducing agent. In fact, it is a strong enough reductant to produce hydrogen; reduce alkenes (compounds with carbon-carbon double bonds), alkynes (with carbon-carbon triple bonds), and thiols (with −SH groups) to saturated hydrocarbons; and replace ketones (C=O groups) with thiols (fig. 4.15). And even today, more than 4 billion years after our planet formed, the Earth’s geology is still so active that deep-sea vents in the mid-ocean ridges recycle an entire ocean’s volume of water every 8 million years, and thus catalytic reduction deep within a planet’s crust is certainly a possibility.
Non-Miller-Urey Sources of Life’s Building Blocks
Given the possibility that primordial atmospheres are too oxidized to promote Miller-Urey chemistry, we should note that a small but vocal group within the astrobiology community has argued that the prebiotic precursors to life are not formed in situ but are delivered to terrestrial planets from space, where they can be synthesized under more reducing conditions.
Comets are known to contain ammonia, methane, and water (as solids, all of which are referred to as “ices” by planetary scientists), as are the asteroids that formed out beyond the snow line (discussed in chapter 3). Both types of objects are also subjected to significant radiation in the form of solar UV light and cosmic rays and therefore may contain Miller-Urey-type small molecules. Do they? The half dozen spacecraft that have visited comets have flown by at such high velocities that delicate molecules such as we are interested in here have difficulty surviving their encounter with the craft’s instruments. Nevertheless, in 2009 NASA announced the discovery of glycine by its Stardust spacecraft, which swept up dust particles while flying by comet Wild-2 at 6 km/s in 2004 and carried them back to Earth for analysis two years later. More detailed analysis of the organic inventory of a comet, however, had to wait until 2014, when the European Space Agency’s Rosetta mission, which dropped into orbit around comet 67P/Churyumov-Gerasimenko in 2014 (fig. 4.16), where it, too, found glycine in the dust the comet was spraying into space.

Figure 4.16 In 2014, the European Space Agency’s Rosetta spacecraft dropped into orbit around the comet 67P/Churyumov-Gerasimenko, a 4 km wide chunk of primordial ice and rock that was once a resident of the Kuiper belt. The spacecraft detected glycine in the jets of gas and dust seen here erupting from the comet’s surface. (Courtesy of the European Space Agency)
In contrast to comets, which we’ve had to sample via robotic spacecraft, meteoritic material is delivered to the Earth with some frequency, and thus it can be analyzed in the laboratory. Among the many classes of meteorites are the carbonaceous chondrites, which are water- and organic-containing stones thought to arise beyond the snow line, in the outer half of the asteroid belt. Because they consist of clays and other highly hydrated minerals, carbonaceous chondrites typically disintegrate soon after landing on our warm, wet planet, and thus they have been difficult to study. An important counterexample, however, arose in the fall of 1969 when a large carbonaceous chondrite exploded in the air over Murchison, Australia. The fragments of the Murchison meteorite were quickly collected under dry conditions, allowing their leisurely study in the laboratory.
Hot water extractions of the water-soluble organic material in the Murchison meteorite indicated that it contains a wide range of amino acids. In fact, some 70 different amino acids have been identified in the Murchison meteorite so far, including at least six of the 20 proteinogenic amino acids found in life on Earth (table 4.4). Probably not coincidentally, many of the amino acids most readily formed in the Miller-Urey chemistries are found in abundance in the Murchison material, suggesting that interstellar Strecker synthesis was occurring in the prestellar nebula. Consistent with these arguments, radio astronomers have identified the unambiguous spectral signatures of a number of small organic compounds, the most complex being the two-carbon, sugar-like molecule glycolaldehyde, in nebulae thousands of light-years from Earth.
Of interest to us, the organic inventory of carbonaceous chondrites isn’t just limited to amino acids. Dave Deamer at the University of California, Santa Cruz, for example, has extracted lipids from Murchison samples. When added to fresh water, these lipids spontaneously form hollow spheres, called vesicles, reminiscent of cell membranes. But Deamer’s experiments have led him to question the supposition that life originated in the ocean. It turns out that, in salt water, the lipids simply clump together without forming a hollow sphere, so Deamer has theorized that a freshwater pond would have been a more hospitable environment for life’s origins. Finally, in 2019, Tomoki Nakamura and coworkers in Sendai, Japan, reanalyzed Murchison samples and identified a number of sugars, including ribose, suggesting that this class of molecule can also be synthesized in prestellar nebulae or protoplanetary disks.
Table 4.4
Amino acids observed in the Murchison meteorite
Isovaline |
β-Aminoisobutyric acid |
α-Aminoisobutyric acid |
Pipecolic acid |
Valine |
Glycine |
N-methylalanine |
β-Alanine |
α-Amino-n-butyric acid |
Proline |
Alanine |
γ-Aminobutyric acid |
N-methylglycine |
Aspartic acid |
N-ethylglycine |
Glutamic acid |
Norvaline |
|
Note: Proteinogenic amino acids shown in bold type.
So, it seems, meteorites and comets can deliver biologically relevant molecules from the outer reaches of a solar system to its rocky, inner planets. But the question remains: could they do so in sufficient quantity to be relevant to the origins of life? Jeffrey Bada, whom we met earlier in this chapter, long argued that the delivery of organic building blocks from space is insufficient. He arrived at this conclusion by searching for α-aminoisobutyric acid in places like Antarctica and Greenland, where meteoritic material falling on snow and ice can build up over significant time frames. This molecule is rare on Earth because, unlike the proteinogenic amino acids, it is not associated with life. But it is efficiently synthesized by Miller-Urey chemistry and is a major organic component of the Murchison meteorite. Bada found less than 0.05 mg of the compound per square centimeter of ice surface, leading him to argue that this extraterrestrial molecule—and by extrapolation, others—would not have built up to high enough concentrations to contribute much to the primordial soup.
More recently, however, Bada had to reevaluate this claim when he discovered that enormous quantities of “buckyballs” had arrived on Earth intact from outside the Solar System. Known more formally as fullerenes, buckyballs are large, spherical molecules of pure carbon that resemble geodesic domes, and thus were named after the inventor of said domes: Buckminster Fuller (1895–1983). But buckyballs are a common component of soot, so how did Bada know that the buckyballs he found were of extraterrestrial origin? The spherical structure of a buckyball allows it to trap atoms inside. With his associate Luann Becker, Bada explored a 2-billion-year-old impact site in Ontario, Canada, that contained about a million tons of carbon rich in buckyballs, many of which contained trapped helium. While helium is the second most common element in the Universe, it is rare on terrestrial planets (with its light weight and chemical inertness, it tends to be lost to space over geological time). It thus appears likely that these millions of tons of buckyballs, and presumably equally large amounts of other organic compounds, were indeed delivered from the outer Solar System by impacts. The consensus is now that both extraterrestrial delivery and in situ Miller-Urey chemistries contribute to the formation of the rich, prebiotic soup of organic materials necessary for life to form.
Prebiotic Polymerization
In our exploration of prebiotic chemistries, we still face a significant hurdle. While we understand where many of the monomers—individual amino acids and nucleobases—come from, monomers do not equal life. Even free nucleobases are only relevant to life in the context of polymers of nucleotides, the three-part, covalent combinations of sugars, bases, and phosphate we described above. The reactions that link such monomers into complex polymers are presumably critical steps in the origins of life, not only on Earth, but anywhere. But these reactions consume significant amounts of energy and thus are extremely unfavorable. How, then, might they have spontaneously occurred on a prebiotic planet? We don’t really know, but several (admittedly rather speculative) mechanisms have been proposed.
Activated nucleotides, as we’ve noted, are presumably the starting materials from which RNA polymers are synthesized. How such polymerization might take place has been the subject of extensive study. One possibility is simply the spontaneous polymerization of phosphorimidazolides (or other, still unknown activated nucleotides) in solution. This reaction, however, tends to be extremely inefficient. The problem is that each nucleotide has several free hydroxyl groups, each of which can form a phosphoester linkage (fig. 4.17). In fact, for the activated base adenosine phosphorimidazolide, incorrect linkages are six times more likely to form than the correct linkages found in the RNA polymers in life on Earth. Fortunately, mineral catalysts may provide a means of overcoming this problem. James Ferris (1932–2016) at Rensselaer Polytechnic Institute in New York State has shown, for example, that RNA polymers up to 55 monomers long can be synthesized (with mostly correct linkages) on the common clay montmorillonite; it seems that the surface of the clay binds the growing polymer and directs the addition of each new monomer. Alternatively, the polymerization could be template directed. RNA (like its relative DNA) can bind to a complementary sequence to form a double helix. This suggests that an existing RNA polymer could act as a template to direct the specific polymerization of a new strand of RNA. Using polycytidine (the polymer of the cytosine-containing nucleotide) as a template to direct the polymerization of activated guanosine (the guanine-containing nucleotide), Orgel found that the correct linkage is favored by 2:1 in the final polymerized product. The presence of zinc ions increases the yield of polymerized product without harming the yield of proper linkages. When the activator 2-methylimidazole is employed instead in the presence of magnesium, zinc, or iron ions, a template containing both uracil and cytosine can be efficiently copied as long as cytosine dominates the template’s composition. If the cytosine and uracil contents approach equality, the yield of polymerized material plummets. This limitation presents a serious obstacle to the formation of a self-replicating system from the simple, template-directed polymerization of RNA; as we discuss again in the next chapter, a cytosine-rich sequence that serves as an efficient template will produce cytosine-poor products that cannot, in turn, serve as new templates.
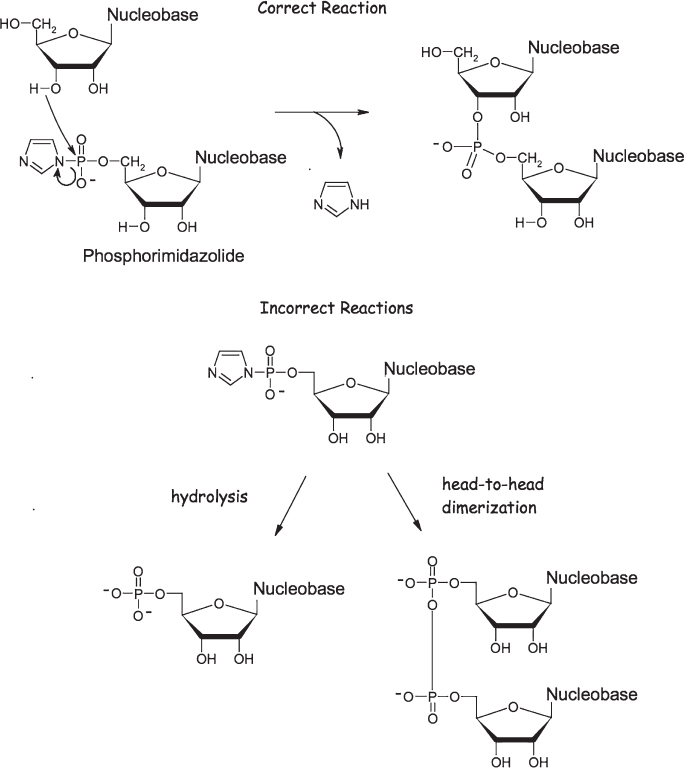
Figure 4.17 Activated nucleotides, such as the phosphorimidazolide shown here, readily polymerize in aqueous solution. Due to a multitude of side reactions, however, the yield of correctly linked RNA polymers produced by such polymerization is low.
With all these caveats and disclaimers and open questions, the prebiotic synthesis of RNA polymers is looking pretty bad, right? Sadly, the situation may be even worse than we’ve let on. Not only is the synthesis of nucleotide polymers an (energetically) uphill battle (see sidebar 4.2), but staying “uphill” is equally difficult. For example, the half-life of the phosphodiester linkages in unstructured, single-stranded RNAs is less than 1,000 years in water at the freezing point and less than 1 day at 35°C. And given that even a modest-length RNA polymer contains hundreds of phosphodiester bonds, the lifetime of an intact polymer is much, much less than the lifetime of any one linkage. This observation has, once again, led to the speculation that some as yet unknown polymer that shares RNA’s abilities to catalyze reactions and encode information and yet is more stable (and perhaps easier to synthesize prebiotically) was instead involved in the origins of life, a point that we’ll come back to a third time in the next chapter.
Conclusions
We’ve come a long way in the half century following the discoveries of the double helix and prebiotic chemistry, but the latter did not advance as fast as molecular biology, and perhaps not as far as Miller would have hoped while marveling over his results back in 1953. Thanks to his work and that of those who followed, we understand in detail how quite a few of the key molecular elements of life as we know it might have been created in the prebiotic world. But equally many questions about prebiotic chemistry remain.
And what happened to the pioneering protagonists of our story? In the early 1960s, Harold Urey went on to head the Chemistry Department at the newly founded University of California, San Diego, and he brought along his protégé Stanley Miller. Miller spent the next 40 years pursuing origins-of-life research, publishing several hundred papers and numerous books on the topic and spawning a whole community of scientists pursuing the same elusive goal, although progressing only slowly. Indeed, when Miller died in 2007, the unfulfilled promise of his 1953 breakthrough was a key theme of many of his obituaries. However, the legacy of his pioneering experiments lives on. For example, when his former student Jeffrey Bada inherited the contents of Miller’s lab, he discovered samples of experiments conducted in 1958, which were clearly labeled and referenced in Miller’s notebooks. The previously unreported 1958 experiment was the first Miller had conducted that included hydrogen sulfide (H2S). Reanalyzing these samples using modern high-performance liquid chromatography and mass spectrometry, Bada found that they contained the oxidation products of the sulfur-containing amino acids cysteine and methionine, which Miller himself didn’t report making until the 1970s. Give them a few million years more, and Miller’s samples may come up with life after all.
Further Reading
Prebiotic Chemistry
Cleaves, H. James, II. “Prebiotic Chemistry: What We Know, What We Don’t.” Evolution: Education and Outreach 5 (2012): 342–60.
Miller, S. L. “A Production of Amino Acids under Possible Primitive Earth Conditions.” Science 15, no. 3046 (1953): 528–29.
Orgel, Leslie E. “Prebiotic Chemistry and the Origin of the RNA World.” Critical Reviews in Biochemistry and Molecular Biology 39, no. 2 (2004): 99–123.
Parker, Eric T., Henderson J. Cleaves, Jason P. Dworkin, Daniel P. Glavin, Michael Callahan, Andrew Aubrey, Antonio Lazcano, and Jeffrey L. Bada. “Primordial Synthesis of Amines and Amino Acids in a 1958 Miller H2S-Rich Spark Discharge Experiment.” Proceedings of the National Academy of Sciences USA 108, no. 14 (2011): 5526–31.
Sutherland, John D. “Studies on the Origin of Life — the End of the Beginning.” Nature Reviews Chemistry 1 (2017): 0012.
Alternatives to RNA in the Origins of Life
Hud, Nicholas V., Brian J. Cafferty, Ramanarayanan Krishnamurthy, and Loren Dean Williams. “The Origin of RNA and ‘My Grandfather’s Axe’.” Chemistry & Biochemistry 20, no. 4 (2013): 466–74.
- * W. M. Garrison et al., “Reduction of Carbon Dioxide in Aqueous Solutions by Ionizing Radiation,” Science 114, no. 2964 (1951): 416–18.
- * Haldane is also known for his succinct and illuminating description of the four stages of acceptance that any new scientific theory goes through: (1) this is worthless nonsense, (2) this is an interesting, but perverse, point of view, (3) this is true, but quite unimportant, (4) I always said so.
- ** J. B. S. Haldane, “The Origin of Life,” Rationalist Annual 148 (1929): 3–10.
- * As a former student of Miller’s, Bada is the intellectual “grandchild” of Urey.
- * Consistent with this, the Galileo entry probe failed to detect molecules containing more than three carbon atoms when it sampled Jupiter’s highly reduced, lightning-filled atmosphere. Thus our earlier argument that hydrogen-filled “blimpoids” are unlikely to be found living in the atmospheres of gas giant planets.