Chapter 10. Running Single Workflows at Scale with Pipelines API
In Chapter 8, we started running workflows for the first time, working on a custom virtual machine in GCP. However, that single-machine setup didn’t allow us to take advantage of the biggest strength of the cloud: the availability of seemingly endless numbers of machines on demand! So in this chapter, we use a service offered by GCP called Genomics Pipelines API (PAPI), which functions as a sort of job scheduler for GCP Compute Engine instances, to do exactly that.
First, we try simply changing the Cromwell configuration on our VM to submit job execution to PAPI instead of the local machine. Then, we try out a tool called WDL_Runner that wraps Cromwell and manages submissions to PAPI, which makes it easier to “launch and forget” WDL executions. Both of these options, which we explore in the first half of this chapter, will open the door for us to run full-scale GATK pipelines that we could not have run on our single-VM setup in Chapter 9. Along the way, we also discuss important considerations such as runtime, cost, portability, and overall efficiency of running workflows in the cloud.
Introducing the GCP Genomics Pipelines API Service
The Genomics Pipelines API is a service operated by GCP that makes it easy to dispatch jobs for execution on the GCP Compute Engine without having to actually manage VMs directly. Despite its name, the Genomics Pipelines API is not at all specific to genomics, so it can be used for a lot of different workloads and use cases. In general, we simply refer to it as the Pipelines API, or PAPI. It is possible to use PAPI to execute specific analysis commands directly as described in the Google Cloud documentation, but in this chapter our goal is to use PAPI in the context of workflow execution with Cromwell, which is illustrated in Figure 10-1.
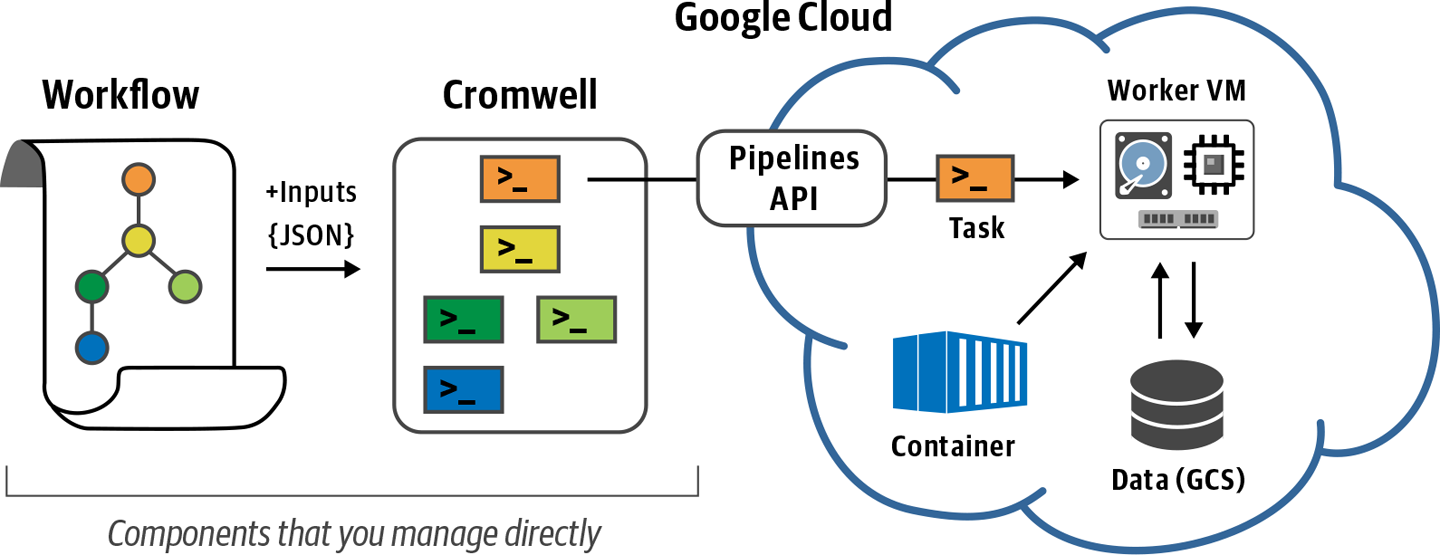
Figure 10-1. Overview of Cromwell + PAPI operation.
Just as in Chapter 8, we provide a WDL that describes our workflow to the Cromwell engine, which then interprets the workflow and generates the individual jobs that need to be executed. What’s new is that instead of having Cromwell hand the jobs over to the local executor of the machine it is itself running on, we’re going to point it to PAPI, as shown in Figure 10-1.
For every job that Cromwell sends to PAPI, the service will create a VM on Google Compute Engine with the specified runtime attributes (CPU, RAM, and storage), set up the Docker container specified in the WDL, copy input files to the VM’s local disk, run the command(s), copy any outputs and logs to their final location (typically a GCS bucket), and finally, delete the VM and free up any associated compute resources. This makes it phenomenally easy to rapidly marshal a fleet of custom VMs, execute workflow tasks, and then walk away with your results without having to worry about managing compute resources, because that’s all handled for you.
Note
As we wrap up this book, Google Cloud is rolling out an updated version of this service under a new name, “Life Sciences API.” Once we’ve had a chance to try it out, we’ll write a blog post about the new service and how to adapt the book exercises to use it. In the meantime, we expect that the Genomics Pipelines API will remain functional for the foreseeable future.
Enabling Genomics API and Related APIs in Your Google Cloud Project
To run the exercises in this chapter, you need to have three APIs enabled in your Google Cloud Project: Genomics API, Cloud Storage JSON API, and Compute Engine API. You can use the direct links that we’ve included, or you can go to the APIs & Services section of the Google Cloud console and click the “+ Enable APIs and Services” button. Clicking it will take you to the API Library, where you can search for each API by name. If you find yourself confused by APIs that have similar names in the search results, simply check the logos and descriptions shown in Figure 10-2.

Figure 10-2. Logos and descriptions for the three required APIs: Genomics API, Cloud Storage JSON API, and Compute Engine API.
On each API’s page, you will see a blue button that reads either Enable or Manage. The latter means that the API is already enabled in your project and you don’t need to do anything. If you see the Enable button, click it to enable the API. You might see a message that says “To use this API, you may need credentials. Click ‘Create credentials’ to get started.” but you can ignore it—GCP and Cromwell will handle authentication without requiring you to supply additional credentials.
Note
To use the Compute Engine API, you must have a means of payment set up in the Billing section even if you’re using the free credits. If you followed the instructions in Chapter 4, you should be all set; but if you skipped straight to here, you’ll need to go back and follow the billing setup in that chapter.
Directly Dispatching Cromwell Jobs to PAPI
In Chapter 8, we were running Cromwell without a configuration file, so by default it sent all jobs to the machine’s local executor. Now we’re going to provide a configuration file that points Cromwell to PAPI for execution.
Fun fact: you can configure Cromwell to run jobs through PAPI from anywhere, whether you’re running Cromwell on your laptop, on a local server, or even on a VM on a different cloud platform. Here, we show you how to do it from your VM using GCP given that you already have it set up appropriately (you have Cromwell installed and you’re authenticated through GCP), but the basic procedure and requirements would be the same anywhere else.
Configuring Cromwell to Communicate with PAPI
The Cromwell documentation has a section on using GCP that includes a template configuration file for running on PAPI, which we’ve copied to the book bundle for your convenience. You can find it at ~/book/code/config/google.conf on your VM if you followed the instructions provided in Chapter 4. If you open up the google.conf file in a text editor, this is what it looks like:
$ cat ~/book/code/config/google.conf
# This is an example configuration file that directs Cromwell to execute
# workflow tasks via the Google Pipelines API backend and allows it to retrieve
# input files from GCS buckets. It is intended only as a relatively simple example
# and leaves out many options that are useful or important for production-scale
# work. See https://cromwell.readthedocs.io/en/stable/backends/Google/ for more
# complete documentation.
engine {
filesystems {
gcs {
auth = "application-default"
project = "<google-billing-project-id>"
}
}
}
backend {
default = PAPIv2
providers {
PAPIv2 {
actor-factory = "cromwell.backend.google.pipelines.v2alpha1.PipelinesApi
LifecycleActorFactory"
config {
# Google project
project = "<google-project-id>"
# Base bucket for workflow executions
root = "gs://<google-bucket-name>/cromwell-execution"
# Polling for completion backs-off gradually for slower-running jobs.
# This is the maximum polling interval (in seconds):
maximum-polling-interval = 600
# Optional Dockerhub Credentials. Can be used to access private docker images.
dockerhub {
# account = ""
# token = ""
}
# Number of workers to assign to PAPI requests
request-workers = 3
genomics {
# A reference to an auth defined in the `google` stanza at the top. This auth is used
# to create
# Pipelines and manipulate auth JSONs.
auth = "application-default"
# Endpoint for APIs, no reason to change this unless directed by Google.
endpoint-url = "https://genomics.googleapis.com/"
# Pipelines v2 only: specify the number of times localization and delocalization
# operations should be attempted
# There is no logic to determine if the error was transient or not, everything
# is retried upon failure
# Defaults to 3
localization-attempts = 3
}
filesystems {
gcs {
auth = "application-default"
project = "<google-billing-project-id>"
}
}
}
default-runtime-attributes {
cpu: 1
failOnStderr: false
continueOnReturnCode: 0
memory: "2048 MB"
bootDiskSizeGb: 10
# Allowed to be a String, or a list of Strings
disks: "local-disk 10 SSD"
noAddress: false
preemptible: 0
zones: ["us-east4-a", "us-east4-b"]
}
}
}
}
}
We know that’s a lot to take in if you’re not used to dealing with this sort of thing; the good news is that you can ignore almost all of it, and we’re going to walk you through the bits that you do need to care about. As a heads-up before we begin, be aware that you’re going to need to edit the file and save the modified version as my-google.conf in your sandbox directory for this chapter, which should be ~/sandbox-10. Go ahead and create that directory now. We recommend using the same procedure as you used in earlier chapters to edit text files:
$ mkdir ~/sandbox-10 $ cp ~/book/code/config/google.conf ~/sandbox-10/my-google.conf
Now set environment variables to point to your sandbox and related directories:
$ export CONF=~/sandbox-10 $ export BIN=~/book/bin $ export WF=~/book/code/workflows
In Chapter 4, we also defined a BUCKET environment variable. If you are working in a new terminal, make sure that you also redefine this variable because we use it a little later in this chapter. Replace my-bucket in the following command with the value you used in Chapter 4:
$ export BUCKET="gs://my-bucket"
Now that our environment is set up, we can get to work. First, let’s identify what makes this configuration file point to GCP and PAPI as opposed to another backend. Go ahead and open your copy of the configuration file now:
$ nano ~/sandbox-10/my-google.conf
You can see there are many references to Google throughout the file, but the key setting is happening here:
backend {
default = PAPIv2
providers {
PAPIv2 {
Here, PAPIv2 refers to the current name and version of the PAPI.
Scrolling deeper into that section of the file, you’ll find two settings, the project ID and the output bucket, which are very important because you must modify them in order to get this configuration file to work for you:
config {
// Google project
project = "<google-project-id>"
// Base bucket for workflow executions
root = "gs://<google-bucket-name>/cromwell-execution"
You need to replace google-project-id to specify the Google Project you’re working with, and replace google-bucket-name to specify the location in GCS that you want Cromwell to use for storing execution logs and outputs. For example, for the test project we’ve been using so far, it looks like this when it’s filled in:
config {
// Google project
project = "ferrous-layout-260200"
// Base bucket for workflow executions
root = "gs://my-bucket/cromwell-execution"
Finally, you also need to provide the project ID to charge when accessing data in GCS buckets that are set to the requester-pays mode, as discussed in Chapter 4. It appears in two places in this configuration file: once toward the beginning and once toward the end:
gcs {
auth = "application-default"
project = "<google-billing-project-id>"
}
Specifically, you need to replace google-billing-project-id to specify the billing project to use for that purpose. This setting allows you to use a different billing project from the one used for compute, for example if you’re using separate billing accounts to cover different kinds of cost. Here, we just use the same project as we just defined earlier:
gcs {
auth = "application-default"
project = "ferrous-layout-260200"
}
Make sure to edit both occurrences of this setting in the file.
When you’re done editing the file, be sure to save it as my-google.conf in your sandbox directory for this chapter, ~/sandbox-10. You can give it a different name and/or put it in a different location if you prefer, but then you’ll need to edit the name and path accordingly in the command line in the next section.
Running Scattered HaplotypeCaller via PAPI
You have one more step to do before you can actually launch the Cromwell run command to test this configuration: you need to generate a file of credentials that Cromwell will give to PAPI to use for authentication. To do so, run the following gcloud command:
$ gcloud auth application-default login
It’s similar to the gcloud init command you ran in Chapter 4 to set up your VM, up to and including the little lecture about security protocols. Follow the prompts to log in via your browser and copy the access code to finish the process. The credentials file, which the system refers to as Application Default Credentials (ADC), will be created in a standard location that the gcloud utilities can access. You don’t need to do anything more for it to work.
We provide a JSON file of inputs that is prefilled with the paths to test files (see $WF/scatter-hc/scatter-haplotypecaller.gcs.inputs.json). You can check what it contains; you’ll see the files are the same as you’ve been using locally, but this time we’re pointing Cromwell to their locations in GCS (in the book bucket):
$ cat $WF/scatter-hc/scatter-haplotypecaller.gcs.inputs.json
With that in hand, it’s time to run the following Cromwell command:
$ java -Dconfig.file=$CONF/my-google.conf -jar $BIN/cromwell-48.jar \
run $WF/scatter-hc/scatter-haplotypecaller.wdl \
-i $WF/scatter-hc/scatter-haplotypecaller.gcs.inputs.json
This command calls the same workflow that we last ran in Chapter 8, scatter-haplotypecaller.wdl, but this time we’re adding the -Dconfig.file argument to specify the configuration file, which will cause Cromwell to dispatch the work to PAPI instead of the local machine. PAPI in turn will orchestrate the creation of new VMs on Compute Engine to execute the work. It will also take care of pulling in any input files and saving the logs and any outputs to the storage bucket you specified in your configuration file. Finally, it will delete each VM after its work is done.
Monitoring Workflow Execution on Google Compute Engine
This all sounds great, but how can you know what is actually happening when you run that command? As usual Cromwell will output a lot of information to the terminal that is rather difficult to parse, so let’s walk through a few approaches that you can take to identify what is running where.
First, you should see in the early part of the log output that Cromwell has correctly identified that it needs to use PAPI. It’s not super obvious, but you’ll see a few lines like these that mention PAPI:
[2019-12-14 18:37:49,48] [info] PAPI request worker batch interval is 33333 milliseconds
Scrolling a bit farther down, you’ll find a line that references Call-to-Backend assignments, which lists the task calls from the workflow:
[2019-12-14 18:37:52,56] [info] MaterializeWorkflowDescriptorActor [68271de1]: Call-to-Backend assignments: ScatterHaplotypeCallerGVCF.HaplotypeCallerG VCF -> PAPIv2, ScatterHaplotypeCallerGVCF.MergeVCFs -> PAPIv2
The -> PAPIv2 bit means that each of these tasks was dispatched to PAPI (version 2) for execution. After that, past the usual detailed listing of the commands that Cromwell generated from the WDL and inputs JSON file, you’ll see many references to PipelinesApiAsyncBackendJobExecutionActor, which is another component of the PAPI system that is handling your workflow execution.
Of course, that tells you only that Cromwell is dispatching work to PAPI, and that PAPI is doing something in response, but how do you know what it’s doing? The most straightforward way is to go to the Compute Engine console, which lists VM instances running under your project. At the very least, you should see the VM that you have been using to work through the book exercises, whose name you should recognize. And if you catch them while they’re running, there can be up to four VM instances with obviously machine-generated names (google-pipelines-worker-xxxxx…), as shown in Figure 10-4. Those are the VMs created by PAPI in response to your Cromwell command.
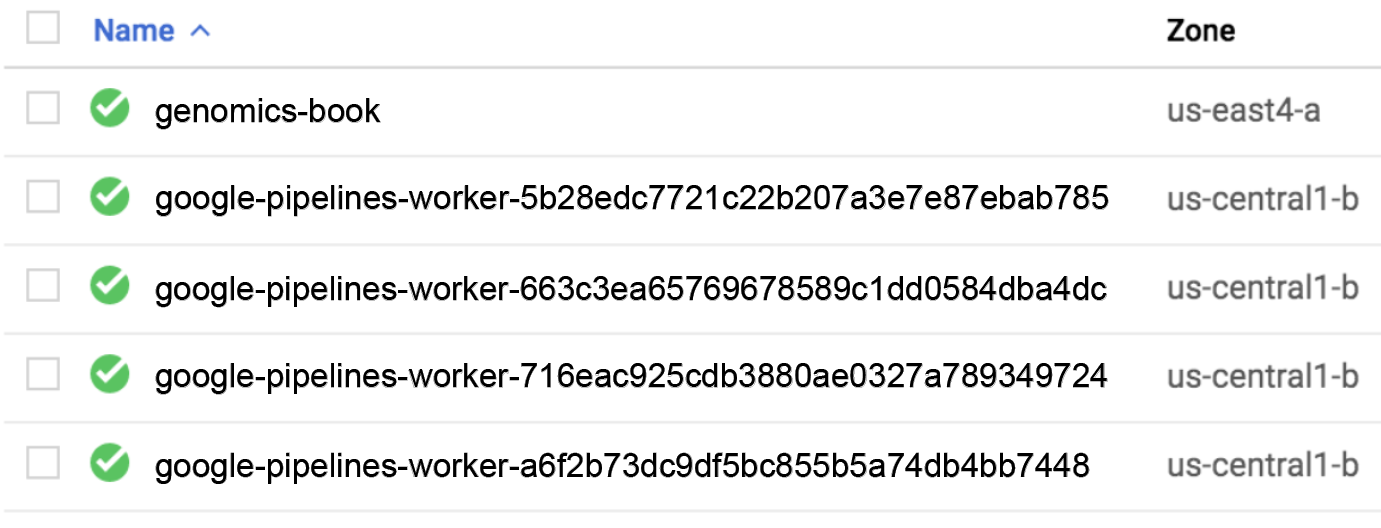
Figure 10-4. List of active VM instances
Why has PAPI created multiple VMs, you might ask? As a reminder, this workflow divides the work of calling variants into four separate jobs that will run on different genomic intervals. When Cromwell launched the first four jobs in parallel, PAPI created a separate VM for each of them. The only job Cromwell will have queued up for later is the merging task that collects the outputs of the four parallel jobs into a single task. That’s because the merging task must wait for all the others to be complete in order for its input dependencies to be satisfied. When that is the case, PAPI will once more do the honors to get the work done and corral the final output to your bucket.
Not to put too fine a point on it, but that right there is one of the huge advantages of using the cloud for this kind of analysis. As long as you can chop the work into independent segments, you can parallelize the execution to a much larger degree than you can typically afford to do on a local server or cluster with a limited number of nodes available at any given time. Even though the cloud is not actually infinite (sorry, the Tooth Fairy is also not real), it does have a rather impressive capacity for accommodating ridiculously large numbers of job submissions. The Broad Institute, for example, regularly runs workflows on Cromwell across thousands of nodes simultaneously.
Note
The zone of the PAPI-generated VMs may be different from the zone you set for the VM you created yourself, as it was in our case here. That’s because we didn’t explicitly specify a zone in the configuration file, so PAPI used a default value. There are several ways to control the zone where the work will be done, including at the workflow level or even at the task level within a workflow, as described in the Cromwell documentation.
You can also see an overview of Compute Engine activity in the Home Dashboard of your project, as shown in Figure 10-5. The advantage of the dashboard, even though it gives you only an aggregate view, is that it allows you to see past activity, not just whatever is running at the moment you’re looking.
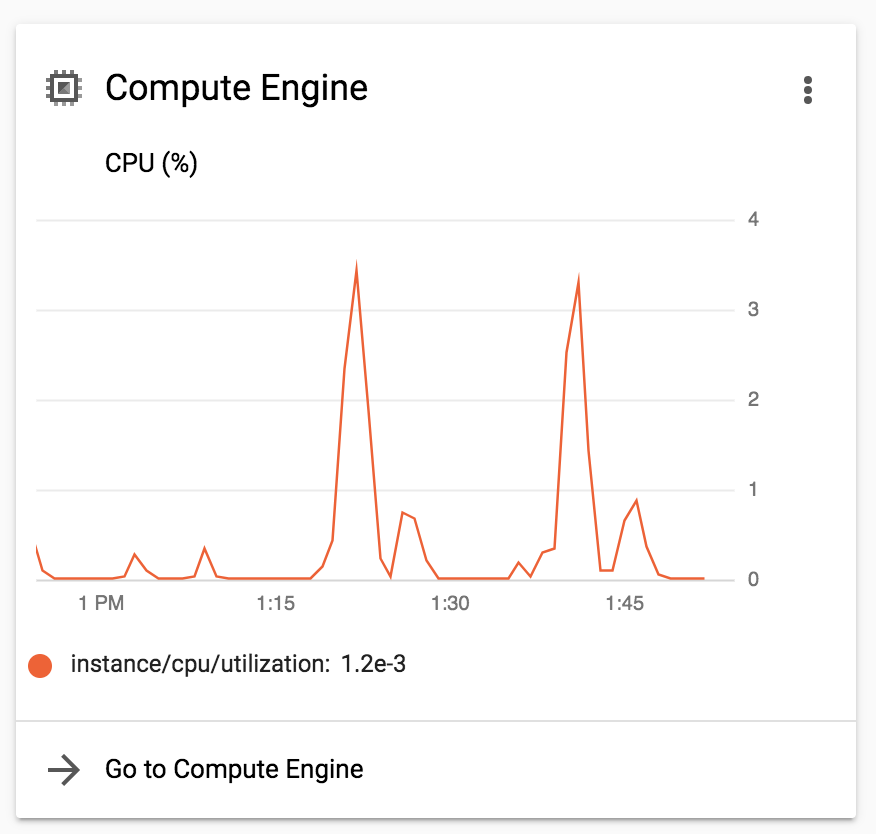
Figure 10-5. Overview of Compute Engine activity.
Finally, you can check the execution directory in the bucket that you specified in the config. You can do so either by navigating to the bucket in the Cloud Storage console, or by using gsutil, as we’ve previously described. Take a few minutes to explore that directory and see how the outputs are structured. It should look the same as in Chapter 8 when you ran this same workflow on the VM’s local system. However, keep in mind that the contents of the execution directory in the bucket don’t represent what is happening in real time on the VMs. A synchronization process updates the execution logs and copies any outputs as they become available, operating at intervals that start out very short and gradually increase to avoid overburdening the system in the case of long-running jobs. The maximum length of these intervals is customizable; you can see the relevant code in the configuration file we used earlier:
// Polling for completion backs-off gradually for slower-running jobs. // This is the maximum polling interval (in seconds): maximum-polling-interval = 600
The interval time is measured in seconds, so you can see that, by default, the maximum time between updates of the bucket is 10 minutes.
The workflow can take up to 10 minutes to complete. At that point, you’ll see the usual finished with status 'Succeeded' message, which is followed by the list of final outputs in JSON format:
[INFO] … SingleWorkflowRunnerActor workflow finished with status 'Succeeded'.
"outputs": {
"ScatterHaplotypeCallerGVCF.output_gvcf": "gs://genomics-book-test-99/cromwell-
execution/ScatterHaplotypeCallerGVCF/68271de1-4220-4818-bfaa-5694551cbe81/call-
MergeVCFs/mother.merged.g.vcf"
},
You should see that this time, the final output is located in the bucket that you specified in the configuration file. The path to the output has the same structure as we described for local execution.
Now, take a moment to think about what you just achieved: using a mostly prebaked configuration file and just one command line, you kicked off a process that involved marshaling sophisticated computational resources to parallelize the execution of a real (if simple) genomics workflow. You could use that exact same process to run a full-scale workflow, like the whole genome analysis pipeline we dissected in Chapter 9, or you could hold on to your hat while we roll out a few alternative options. Specifically, later in this chapter we show you two approaches that involve “wrapping” Cromwell in additional layers of tooling that increase both the ease of use and scalability of this system.
However, before we plunge into the verdant ecosystem of Cromwell-enabling add-ons, we’re going to take a bit of a side track to discuss the trade-offs and opportunities involved in running workflows on the cloud.
Understanding and Optimizing Workflow Efficiency
Did you pay attention to how much time it took to run the scatter-haplotypecaller.wdl workflow through PAPI? About 10 minutes, right? Do you remember how long it took to run just on your VM in Chapter 8? More like two minutes? So let’s get this straight: running the same workflow with parallelization on multiple machines took five times longer than just running the same jobs on a single machine. That sounds…kind of terrible?
The good news is that it’s mostly an artifact of the very small scale of the jobs we’re running. We’ve been using a set of intervals that cover only a tiny region of the genome, and HaplotypeCaller itself takes a trivially small amount of time to run on such short intervals. When you run the workflow locally on your VM, there’s not actually much work to do: the GATK container image and the files are already there, so all that really needs to happen is for Cromwell to read the WDL and launch the GATK commands, which, as we’ve mentioned, run quickly. In contrast, when you tell Cromwell to dispatch work to PAPI, you set into motion this massive machinery that involves creating VMs, retrieving container images, localizing files from GCS, and so on. That’s all overhead that shows up in the form of a longer runtime. So for short tasks, the actual amount of overall runtime spent on the “real work” is dwarfed by the time spent on behind-the-scenes setup. However, that setup time is roughly constant, so for longer-running tasks (e.g., if you ran this workflow on much larger genomic intervals), the setup time ends up being just a drop in the Google bucket.
With that example in mind, let’s take a stroll through some of the considerations you’ll need to keep in mind, whether you’re planning to develop your own workflows or simply using someone else’s on the cloud.
Granularity of Operations
The HaplotypeCaller workflow that we ran here is intended to run on much longer intervals that cover huge amounts of data, so it makes perfect sense to execute it through PAPI when processing full-scale datasets. But what if part of your workflow involves short-running operations like simple file format conversion and indexing? Well, that might be an opportunity to combine several operations into a single task in the workflow. We already saw an example of this in the second workflow we examined in Chapter 9, in the CramToBamTask command block:
command{set-eset-opipefail${samtools_path}view-h-T${ref_fasta}${input_cram}|${samtools_path}view-b-o${sample_name}.bam-${samtools_path}index-b${sample_name}.bammv${sample_name}.bam.bai${sample_name}.bai}
The workflow authors could have separated subsets of this command block into separate WDL tasks to maximize modularity. For example, having a standalone BAM indexing task might have been useful and reusable elsewhere. However, they correctly identified that doing so would increase the overhead when run through PAPI, so they traded off some potential modularity in favor of higher efficiency.
Balance of Time Versus Money
It’s important to understand that these kinds of trade-offs will make a difference not just in the amount of time it takes to run your pipeline, but also its cost. This is probably not something you’re used to thinking about if you’ve worked mostly on local systems that have computational resources already paid for and your major limitation is how much quota you have been allotted. On the cloud, however, it’s almost entirely pay-as-you-go, so it’s worth thinking things through if you’re on a budget.
For example, here’s another trade-off to think about when moving workflows to the cloud: how widely should you parallelize the execution of your variant-calling workflow? You could simply parallelize it by chromosome, but those still create huge amounts of data, especially for whole genome samples, so it will take a very long time to process each, and you won’t really be taking advantage of the amazing capacity for parallelism of the cloud. In addition, in humans at least, the various chromosomes have enormously different sizes; for example, chromosome 1 is about five times longer than chromosome 22, so the latter will finish much faster, and its results will sit around waiting for the rest of the chromosomes to be done.
A more efficient approach is to chop the chromosomes themselves into subsets of intervals, preferably in areas of uncertainty where the reference sequence has stretches of N bases, meaning the content of those regions is unknown, so it’s OK to interrupt processing there. Then, you can balance the sizes of the intervals in such a way that most of them will take about the same amount of time to process. But that still leaves you a lot of leeway for deciding on the average length of the intervals when you design that list. The more you chop up the sequence, the sooner you can expect to have your results in hand, because you can (within reason) run all those intervals in parallel. However, the shorter the runtime of each individual interval, the more you will feel the pain—and more to the point, the cost—of the overhead involved in dispatching the work to separate VMs.
Not convinced? Suppose that you define three hundred intervals that take three hours each to analyze (these are made-up numbers, but should be in the ballpark of realistic). For each interval, you’ll pay up to 10 minutes of VM overhead time during the setup phase before the actual analysis begins (oh yeah, you get charged for that time). That’s 300 × 10 minutes; in other words, 50 hours’ worth of VM cost spent on overhead. Assuming that you’re using basic machines that cost about $0.03/hour, that amounts to $1.50 which is admittedly not the end of the world. Now let’s say you turn up the dial by a factor of 10 on the granularity of your intervals, producing 3,000 shorter intervals that take 18 minutes each to analyze. You’ll get your results considerably faster, but you’ll pay 3,000 × 10 minutes, or 500 hours’ worth of VM cost on overhead. Now you’re spending $15 per sample on overhead. Across a large number of samples, that might make a noticeable dent in your budget. But we’re not judging; the question is whether the speed is worth the money to you.
The takeaway here is that the cloud gives you a lot of freedom to find your happy point.
When the Broad Institute originally moved its entire genome analysis pipeline from its on-premises cluster to GCP, it cost about $45 to run per whole genome sample (at 30X coverage). Through a combination of workflow optimizations, and in collaboration with GCP engineers, the institute’s team was able to squeeze that down to $5 per whole genome sample. These phenomenal savings translated into lower production costs, which at the end of the day means more science per research dollar.
Admittedly, the cloud pipeline does take about 23 hours to run to completion on a single sample, which is quite a bit longer than the leading accelerated solutions that are now available to run GATK faster on specialized hardware, such as Illumina’s DRAGEN solution. For context, though, it typically takes about two days to prepare and sequence a sample on the wetlab side of the sequencing process, so unless you’re in the business of providing urgent diagnostic services, the additional day of analysis is usually not a source of concern.
Here’s the thing: that processing time does not monopolize computational resources that you could be using for something else. If you need more VMs, you just ask PAPI to summon them. Because there is practically no limit to the number of workflows that the operations team can launch concurrently, there is little risk of having a backlog build up when an order for a large cohort comes through—unless it’s on the scale of gnomAD, which contains more than 100,000 exome and 80,000 whole genome samples. Then, it takes a bit of advance planning and a courtesy email to your friendly GCP account manager so that they can tell their operations team to gird their loins. (We imagine the email: “Brace yourselves; gnomAD is coming.”)
Note
The cost of the Broad’s cloud-based whole genome analysis pipeline unfortunately increased to around $8 per sample following the discovery of two major computer security vulnerabilities, Spectre and Meltdown, that affect computer processors worldwide. The security patches that are required to make the machines safe to use add overhead that causes longer runtimes, which will apparently remain unavoidable until cloud providers switch out the affected hardware. Over time, though, the cost of storage and compute on the cloud continues to fall, so we expect the $8 price to decrease over time.
Suggested Cost-Saving Optimizations
If you’re curious to explore the workflow implementation that the Broad Institute team developed to achieve those substantial cost savings, look no further than back to Chapter 9. The optimized pipeline we’ve been talking about is none other than the second workflow we dissected in that chapter (the one with the subworkflows and task libraries).
Without going too far into the details, here’s a summary of the three most effective strategies for optimizing WDL workflows to run cheaply on Google Cloud. You can see these in action in the GermlineVariantDiscovery.wdl library of tasks that we looked at in Chapter 9.
Dynamic sizing for resource allocation
The more storage you request for a VM for a given task, the more it will cost you. You can keep that cost down by requesting only the bare minimum amount of disk storage that will fit the task, but how do you deal with variability in file input sizes without having to check them manually for every sample you need to process? Good news: there are WDL functions that allow you to evaluate the size of input files going into a task at runtime (but before the VM is requested). Then, you can apply some arithmetic (based on reasonable assumptions about what the task will produce) to calculate how much disk should be allocated. For example, the following code measures the total size of the reference genome files and then calculates the desired disk size based on the amount of space needed to account for a fraction of the input BAM file (more on that in a minute) plus the reference files, plus some padding to account for the output:
Floatref_size=size(ref_fasta,"GB")+size(ref_fasta_index,"GB")+size(ref_dict,"GB")Intdisk_size=ceil(((size(input_bam,"GB")+30)/hc_scatter)+ref_size)+20
See this blog post for a more detailed discussion of this approach.
File streaming to GATK4 tools
This is another great way to reduce the amount of disk space you need to request for your task VMs. Normally, Cromwell and PAPI localize all input files to the VM as a prerequisite for task execution. However, GATK (starting in version 4.0) is capable of streaming data directly from GCS, so if you’re running a GATK tool on a genomic interval, you can instruct Cromwell not to localize the file and just let GATK handle it, which it will do by retrieving only the subset of data specified in the interval. This is especially useful, for example, if you’re running HaplotypeCaller on a 300 Gb BAM file but you’re parallelizing its operation across many intervals to run on segments that are orders of magnitude smaller. In the previous code example, that’s why the size of the input BAM file is divided by the width of the scatter (i.e., the number of intervals). To indicate in your WDL that an input file can be streamed, simply add the following to your task definition, for the relevant input variable:
parameter_meta{input_bam:{localization_optional:true}}
Note that this currently works only for files in GCS and is not available to Picard tools bundled in GATK.
Preemptible VM instances
This involves using a category of VMs, called preemptible, that are much cheaper to use than the normal pricing (currently 20% of list price). These discounted VMs come with all the same specifications as normal VMs, but here’s the catch: Google can take them away from you at any time. The idea is that it’s a pool of spare VMs that you can use normally as long as there’s no shortage of resources. If somebody out there suddenly requests a large number of machines of the same type as you’re using and there aren’t enough available, some or all of the VMs that you are using will be reallocated to them, and your job will be aborted.
To head off the two most common questions, no, there is no (built-in) way to save whatever progress had been made before the interruption; and yes, you will get charged for the time you used them. On the bright side, PAPI will inform Cromwell that your jobs were preempted, and Cromwell will try to restart them automatically on new VMs. Specifically, Cromwell will try to use preemptible VMs again, up to a customizable number of attempts (three attempts by default in most Broad Institute workflows); then, it will fall back to using regular (full-price) VMs. To control the number of preemptible attempts that you’re willing to allow for a given task, simply set the preemptible: property in the runtime block. To disable the use of preemptibles, set it to 0 or remove that line altogether. In the following example, the preemptible count is set using a preemptible_tries variable so that it can be easily customized on demand, as are the Docker container and the disk size:
runtime{docker:gatk_dockerpreemptible:preemptible_triesmemory:"6.5 GiB"cpu:"2"disks:"local-disk "+disk_size+" HDD"}
The bottom line? Using preemptible VMs is generally worthwhile for short-running jobs because the chance of them getting preempted before they finish is typically very small, but the longer the job, the less favorable the odds become.
Platform-Specific Optimization Versus Portability
All of these optimizations are very specific to running on the cloud—and for some of them, not just any cloud—so now we need to talk about their impact on portability. As we discussed when we introduced workflow systems in Chapter 8, one of the key goals for the development of Cromwell and WDL was portability: the idea that you could take the same workflow and run it anywhere to get the same results, without having to deal with a whole lot of software dependencies or hardware requirements. The benefits of computational portability range from making it easier to collaborate with other teams, to enabling any researcher across the globe to reproduce and build on your work after you’ve published it.
Unfortunately, the process of optimizing a workflow to run efficiently on a given platform can make it less portable. For example, earlier versions of WDL did not include the localization_optional: true idiom, so to use the GATK file-streaming capability, workflow authors had to trick Cromwell into not localizing files by declaring those input variables as String types instead of File types. The problem? The resulting workflows could not be run on local systems without changing the relevant variable declarations. Instant portability fail.
The introduction of localization_optional: true was a major step forward because it functions as a “hint” to the workflow management system. Basically, it means, “Hey, you don’t have to localize this file if you’re in a situation that supports streaming, but if you’re not, please do localize it.” As a result, you can run the same workflow in different places, enjoy the optimizations where you can, and rest assured that the workflow will still work everywhere else. Portability win!
The other optimizations we showed you have always had this harmless “hint” status, in the sense that their presence does not affect portability. Aside from the docker property, which we discussed in Chapter 8, Cromwell will happily ignore any properties specified in the runtime block that are not applicable to the running environment, so you can safely have preemptible, disk, and so on specified even if you are just running workflows on your laptop.
Where things might get tricky is that other cloud providers such as Microsoft Azure and AWS have their own equivalents of features like preemptible VM instances (on AWS, the closest equivalents are called Spot Instances), but they don’t behave exactly the same way. So, inevitably the question will arise as to whether Cromwell should use the preemptible property to control Spot Instances when running on AWS, or provide a separate property for that purpose. If it’s the latter, where does it stop? Should every runtime property exist in as many flavors as there are cloud platforms?
Oh, no, we don’t have an answer to that. We’ll be over here heating up some popcorn and watching the developers battle it out. The best way to keep track of what a given Cromwell backend currently can and can’t do is to take a look at the online backend documentation. For now, let’s get back to talking about the most convenient and scalable ways to run Cromwell on GCP.
Wrapping Cromwell and PAPI Execution with WDL Runner
It’s worth repeating that you can make Cromwell submit workflows to PAPI from just about anywhere, as long as you can run Cromwell and connect to the internet. As mentioned earlier, we’re making you use a GCP VM because it minimizes the amount of setup required to get to the good parts (running big workflows!), and it reduces the risk of things not working immediately for you. But you could absolutely do the same thing from your laptop or, if you’re feeling a bit cheeky, from a VM on AWS or Azure. The downside of the laptop option, however, is that you would need to make sure that your laptop stays on (powered and connected) during the entire time that your workflow is running. For a short-running workflow that’s probably fine; but for full-scale work like a whole genome analysis, you really want to be able to fire off a workflow and then pack up your machine for the night without interrupting the execution.
Enter the WDL Runner. This open source toolkit acts as a lightweight wrapper for Cromwell and PAPI. With a single command line from you, WDL Runner creates a VM in GCP, sets up a container with Cromwell configured to use PAPI, and submits your workflow to be executed via PAPI, as shown in Figure 10-6. Does any of that sound familiar? Aside from a few details, this is pretty much what you did in earlier chapters: create a VM instance, put Cromwell on it, configure it to communicate with PAPI, and run a Cromwell command to launch a workflow.
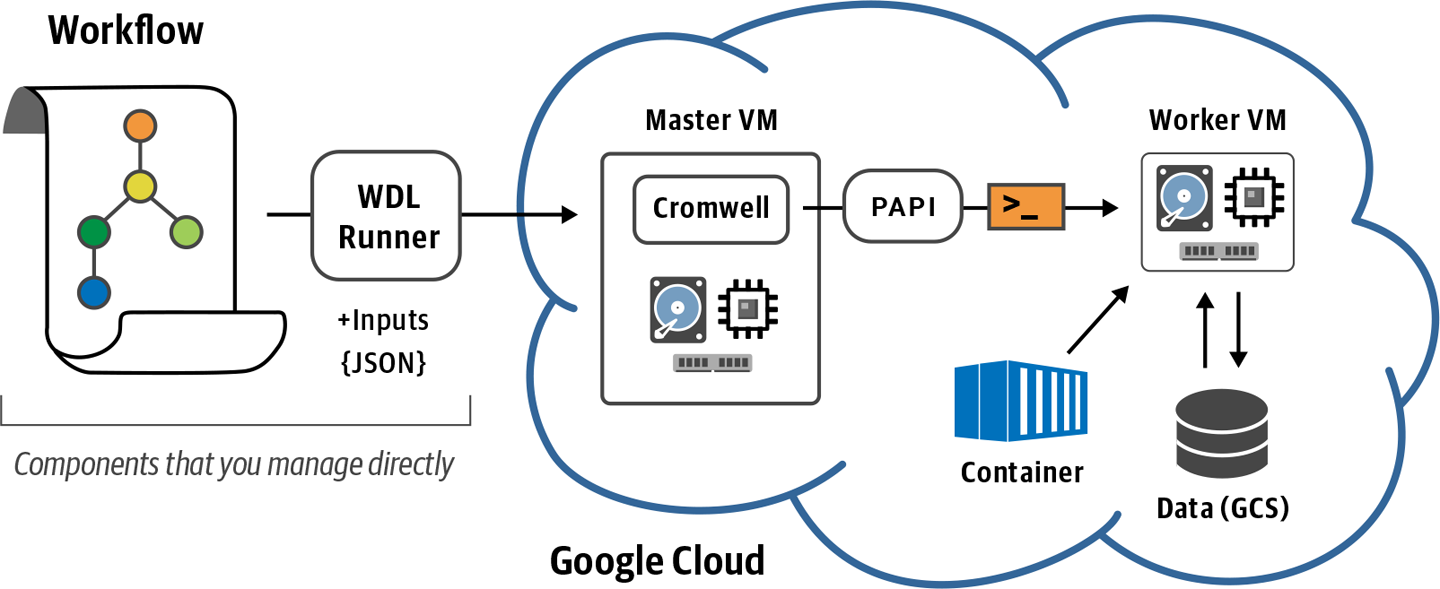
Figure 10-6. Overview of WDL Runner operation.
So at a basic level, you can view WDL Runner as a way to outsource all of that work and achieve the same result without having to manually manage a VM. But there’s more: when the workflow completes, WDL Runner will transfer the execution logs to a location of your choosing and delete the VM, so it won’t keep costing you money if you don’t immediately turn it off. The cherry on the cake is that WDL Runner will also copy the final outputs to a location of your choosing, so you don’t need to go dig through Cromwell’s (annoyingly deep) execution directories to find the result you care about. Oh, and here’s another cherry: WDL Runner also comes with monitoring tools that allow you to get a status summary for your workflow.
Needless to say, you can run WDL Runner from anywhere, but we’re going to have you run it from your VM because it’s right there and it has everything you need on it. Feel free to try it out from your laptop, as well, of course.
Setting Up WDL Runner
Good news: there’s nothing to set up! We’ve included a copy of the WDL Runner code, which is available in GitHub, in the book bundle that you already copied to your VM. WDL Runner’s main requirements are Python 2.7, Java 8, and gcloud, which are all available on your VM already.
To make the paths easier to manage, we are once again going to use environment variables. Two of them are variables that you should already have defined earlier in the chapter, $WF pointing to the workflows directory and $BUCKET pointing to your Google bucket. In addition, you are going to create a $WR_CONF variable pointing to the config directory and a $WR_PIPE variable pointing to the location of the WDL_Runner pipeline configuration file, wdl_pipeline.yaml, as follows:
$ export WR_CONF=~/book/code/config $ export WR_PIPE=~/book/wdl_runner/wdl_runner
That’s not a mistake, by the way; there are two nested directories that are both called wdl_runner; the lower-level one refers to code that is specific to wdl_runner whereas the other one refers to the overall package. If you poke around in the latter, you’ll see that beside the other directory called wdl_runner, there is as a directory called monitoring_tools. We’ll give you three guesses as to what’s in there, and the first two don’t count.
Running the Scattered HaplotypeCaller Workflow with WDL Runner
This is going to be reasonably straightforward. As a heads-up, you’ll notice that the command line for launching a workflow with WDL Runner is a bit longer than the one for launching it directly with Cromwell itself. That’s because WDL Runner gives you a few more options for where to store outputs and so on without having to mess around with a configuration file. Here it is:
$ gcloud alpha genomics pipelines run \
--pipeline-file $WR_PIPE/wdl_pipeline.yaml \
--regions us-east4 \
--inputs-from-file WDL=$WF/scatter-hc/scatter-haplotypecaller.wdl,\
WORKFLOW_INPUTS=$WF/scatter-hc/scatter-haplotypecaller.gcs.inputs.json,\
WORKFLOW_OPTIONS=$WR_CONF/empty.options.json \
--env-vars WORKSPACE=$BUCKET/wdl_runner/test/work,\
OUTPUTS=$BUCKET/wdl_runner/test/output \
--logging $BUCKET/wdl_runner/test/logging
But don’t run it yet! Let’s walk through it briefly so that you know what to customize versus what to leave alone.
The first line tells you that this command is primarily calling gcloud, which puts an interesting twist on the situation. Previously, we were running Cromwell and instructing it to communicate with Google Cloud things. Now, we’re calling a Google Cloud thing to have it run Cromwell and make it communicate with Google Cloud things. You might not be entirely surprised to learn that WDL Runner was originally written by a team at GCP.
The --pipeline-file argument takes the path to a sort of configuration file that comes prebaked with the WDL Runner code; you would need to modify the path if you wanted to run WDL Runner from a different working directory.
The --regions argument, unsurprisingly, controls the region where you want the VMs to reside. Funny thing: the WDL Runner seems to require this, but it conflicts with the default gcloud settings that were set on your VM when you originally authenticated yourself with gcloud. Specifically, the command fails and returns this error message:
ERROR: (gcloud.alpha.genomics.pipelines.run) INVALID_ARGUMENT: Error: validating pipeline: zones and regions cannot be specified together
To work around this issue, you need to unset the default zone by running the following gcloud command:
$ gcloud config set compute/zone ""
The --inputs-from-file argument lists the workflow WDL file, inputs JSON file, and options file. The options file allows you to specify runtime configuration settings that will apply to the entire workflow. We don’t need to specify anything, but because WDL Runner requires that we provide one, we included a stub file that doesn’t actually contain any settings. As an aside, make sure there is absolutely no whitespace between the key:value pairs that specify the files (even around the commas), or the command will fail with a weird error about unrecognized arguments. Yes, we try to experience all the failures so you don’t have to.
The --env-vars argument lists your desired locations for storing the execution directory and the final outputs; these can be anything you want, as long as they are valid GCS paths starting with gs://. Here, we’re using the $BUCKET environment variable that we originally set up in Chapter 4, and we’re using a directory structure that we find convenient for managing outputs and logs. Feel free to chart your own paths, as it were. The --logging argument does the same thing as the previous argument but for the execution logs.
When you’re happy with the look of your command, run it and then sit back and enjoy the sight of software at work. (Yes, saying this does feel a bit like tempting fate, doesn’t it.) If everything is copacetic, you should get a simple one-line response that looks like this:
Running [projects/ferrous-layout-260200/operations/8124090577921753814].
The string of numbers (and sometimes letters) that follows operations/ is a unique identifier, the operations ID, that the service will use to track the status of your submission. Be sure to store that ID somewhere because it will be essential for the monitoring step, which is coming up next.
Monitoring WDL Runner Execution
And here we are, ready to monitor the status of our workflow submission. Remember the not-so-mysterious directory called monitoring_tools? Yep, we’re going to use that now. Specifically, we’re going to use a shell script called monitor_wdl_pipeline.sh that is located in that directory. The script itself is fairly typical sysadmin fare, meaning that it’s really painful to decipher if you’re not familiar with that type of code. Yet, in a stroke of poetic justice, it’s quite easy to use: you simply give the script the operations ID that you recorded in the previous step, and it will let you know how the workflow execution is going.
Before you jump in, however, we recommend that you open a new SSH window to your VM so that you can leave the script running. It’s designed to continue to run, polling the gcloud service at regular intervals, until it receives word that the workflow execution is finished (whether successfully or not). As a reminder, you can open any number of SSH windows to your VM from the Compute Engine console.
When you’re in the new SSH window, move into the first wdl_runner directory and run the following command, substituting the operations ID you saved from the previous step:
$ cd ~/book/wdl_runner $ bash monitoring_tools/monitor_wdl_pipeline.sh 7973899330424684165 Logging: Workspace: Outputs: 2019-12-15 09:21:19: operation not complete No operations logs found. There are 1 output files Sleeping 60 seconds
This is the early stage output that you would see if you start the monitoring script immediately after launching the WDL Runner command. As time progresses, the script will produce new updates, which will become increasingly more detailed.
If you’re not satisfied by the content of the monitoring script’s output, you can always go to the Compute Engine console as we did earlier to view the list of VMs that are working on your workflow, as shown in Figure 10-7. This time, you’ll see up to five new VMs with machine-generated names (in addition to the VM you created manually).
Figure 10-7. List of active VM instances (WDL Runner submission).
But hang on, why would there be five VMs this time instead of four? If you’re confused, have a quick look back at Figure 10-6 to refresh your memory of how this system works. This should remind you that the fifth wheel in this case is quite useful: it’s the VM instance with Cromwell that is controlling the work.
Getting back to the monitoring script, you should eventually see an update that starts with operation complete:
2019-12-15 09:42:47: operation complete
There’s a huge amount of output below that, though, so how do you know whether it was successful? It’s the next few lines that you need to look out for. Either you see something like this and you breathe a happy sigh
Completed operation status information done: true metadata: events: - description: Worker released
or you see see the dreaded error: line immediately following done: true, and you take a deep breath before diving into the error message details:
Completed operation status information done: true error: code: 9 message: ’Execution failed: while running "WDL_Runner": unexpected exit status 1 was not ignored’
We’re not going to sugarcoat this: troubleshooting Cromwell and PAPI errors can be rough. If something went wrong with your workflow submission, you can find information about the error both in the output from the monitor_wdl_pipeline.sh script if you’re using WDL Runner, and in the files saved to the bucket, which we take a look at after this.
Finally, let’s take a look at the bucket to see the output produced by WDL Runner, shown in Figure 10-8.
Figure 10-8. Output from the WDL Runner submission.
The logging output is a text file that contains the Cromwell log output. If you peek inside it, you’ll see that it looks different from the log output you saw earlier when you directly ran Cromwell. That’s because this instance of Cromwell was running in server mode! This is exciting: you ran a Cromwell server and didn’t even know you were doing it. Not that you were really taking advantage of the server features—but it’s a start. The downside is that the Cromwell server logs are extra unreadable for standard-issue humans, so we’re not even going to try to read them here.
The work directory is simply a copy of the Cromwell execution directory, so it has the same rabbit-hole-like structure and contents as you’ve come to expect by now.
Finally, the output directory is one of the features we really love about the WDL Runner: this directory contains a copy of whatever file(s) were identified in the WDL as being the final output(s) of the workflow. This means that if you’re happy with the result, you can just delete the working directory outright without needing to trawl through it to save outputs you care about. When you have workflows that produce a lot of large intermediate outputs, this can save you a lot of time—or money from not paying storage costs for those intermediates.
At the end of the day, we find WDL Runner really convenient for testing workflows quickly with little overhead while still being able to “fire and forget”; that is, launch the workflows, close your laptop, and walk away (as long as you record those operations IDs!). On the downside, it hasn’t been updated in a while and is out of step with WDL features; for example, it neither supports subworkflows nor task imports because it lacks a way to package more than one WDL file. In addition, you need to be very disciplined with keeping track of those operations IDs if you find yourself launching a lot of workflows.
Here we showed you how to use WDL Runner as an example of how a project can wrap Cromwell, not as a fully-baked solution, and in the next chapter we’re going to move on to a system that we like better. However, if you like the concept of wdl_runner, don’t hesitate to make your voice heard on the project GitHub page to motivate further development.
Note
What time is it? Time to stop your VM again, and this time it’s especially important to do so because we won’t use it in any of the book’s remaining exercises. Feel free to delete your VM if you don’t anticipate coming back to previous tutorials.
Wrap-Up and Next Steps
In this chapter, we showed you how to take advantage of the cloud’s scaling power by running your workflows through the GCP Genomics PAPI service, first directly from the Cromwell command line and then from a light wrapper called WDL Runner. However, although these two approaches gave you the ability to run individual workflows of arbitrary complexity in a scalable way in GCP, the overhead involved in spinning up and shutting down a new instance of Cromwell every time you want to run a new workflow is highly inefficient and will scale poorly if you plan to run a lot of workflows.
For truly scalable work, we want to use a proper Cromwell server that can receive and execute any number of workflows on demand, and can take advantage of sophisticated features like call caching, which allows Cromwell to resume failed or interrupted workflows from the point of failure. Call caching requires a connection to a database and is not supported by WDL Runner. Yet, setting up and administering a server and database—not to mention operating them securely in a cloud environment—involves more complexity and tech support burden than most of us are equipped to handle.
That is why, in the next chapter, we’re going to show you an alternative path to enjoying the power of a Cromwell server without shouldering the maintenance burden. Specifically, we’re going to introduce you to Terra, a scalable platform for biomedical research built and operated on top of GCP by the Broad Institute in collaboration with Verily. Terra includes a Cromwell server that you’ll use to run GATK Best Practices workflows at full scale for the first time.