Chapter 8. Automating Analysis Execution with Workflows
So far, we’ve been running individual commands manually in the terminal. However, the overwhelming majority of genomics work proper—the secondary analysis, in which we go from raw data to distilled information that we’ll then feed into downstream analysis to ultimately produce biological insight—involves running the same commands in the same order on all the data as it rolls off the sequencer. Look again at the GATK Best Practices workflows described in Chapters 6 and 7; imagine how tedious it would be to do all that manually for every sample. Newcomers to the field often find it beneficial to run through the commands involved step by step on test data, to gain an appreciation of the key steps, their requirements, and their quirks—that’s why we walked you through all that in Chapters 6 and 7—but at the end of the day, you’re going to want to automate all of that as much as possible. Automation not only reduces the amount of tedious manual work you need to do, but also increases the throughput of your analysis and reduces the opportunity for human error.
In this chapter, we cover that shift from individual commands or one-off scripts to repeatable workflows. We show you how to use a workflow management system (Cromwell), and language, WDL, that we selected primarily for their portability. We walk you through writing your first example workflow, executing it, and interpreting Cromwell’s output. Then, we do the same thing with a couple of more realistic workflows that run GATK and use scatter-gather parallelism.
Introducing WDL and Cromwell
You might remember that we introduced the concept of workflows in the technology primer (Chapter 3). As we noted at the time, many tooling options are available for writing and running workflows, with a variety of features, strengths, and weaknesses. We can’t tell you the best option overall, because much of that determination would depend on your particular backgrounds and needs. For the purposes of this book, we’ve selected an option, the combination of Cromwell and WDL, that is most suitable for the breadth of audience and scope of needs that we are targeting.
We already briefly introduced WDL at that time, as well, but let’s recap the main points given that it was half a book ago and a lot of water under the bridge. As just mentioned, its full name is Workflow Description Language, but it’s generally referred to as WDL (pronounced “widdle”) for short. It’s a DSL commonly used for genomics workflows that was originally developed at the Broad Institute and has since then evolved into a community-driven open source project under the auspices of a public group named OpenWDL. As a workflow language, it’s designed to be very accessible to bioinformatics newcomers who do not have any formal training in software programming while maximizing portability across different systems.
The workflow management system we’re going to use to actually run workflows written in WDL is Cromwell, an open source application, also developed at the Broad Institute. Cromwell is designed to be runnable in just about any modern Java-enabled computing environment, including popular HPC systems like Slurm and SGE, commercial clouds like GCP and AWS, and any laptop or desktop computer running on some flavor of Linux. This portability is a core feature of Cromwell, which is intended to facilitate running the same workflows at multiple institutions in order to maximize scientific collaboration and computational reproducibility in research. Another major portability-oriented feature of Cromwell is that it supports (but does not require) the use of containers for providing the code that you want to run within each component task of a given workflow. You’ll have the opportunity to try running both ways in the exercises in this chapter.
Finally, continuing in the spirit of collaboration and interoperability, Cromwell is also designed to support multiple workflow languages. Currently, it supports WDL as well as the CWL, another popular workflow language designed for portability and computational reproducibility.
Cromwell offers two modes of operation: the one-shot run mode and the server mode. The one-shot run mode is a simple way of running Cromwell that involves a single command line: you give it a workflow and a file that lists workflow inputs, and it will start running, execute the workflow, and finally shut down when the workflow is done. This is convenient for running workflows episodically with minimal hassle, and it’s what we use in the exercises in this chapter. The server mode of operation involves setting up a persistent server that is always running, and submitting workflow execution requests to that server via a REST API (a type of programmatic interface).
Starting the Cromwell server is quite easy. After it’s running, it offers functionality that is not available in the single-run mode, some of which we cover in Chapter 11. However, managing its operation securely on an ongoing basis requires a specialized skill set that most individual researchers or small groups without dedicated support staff do not possess. In Chapter 11, we introduce you to Terra, a managed system operated by the Broad Institute that provides access to a persistent Cromwell server through a GUI as well as an API. That will give you the opportunity to try out Cromwell in server mode without having to administer a server yourself.
Note
We won’t cover Cromwell server administration in this book, so see the Cromwell documentation if you’re interested in learning more.
Whether you run it as a one-shot or in server mode, Cromwell has interesting features that aim to promote efficiency and scalability—but no one wants to read a laundry list of features in a book like this, so let’s move on to the exercises and we’ll bring up those key features as they become relevant along the way.
Installing and Setting Up Cromwell
In this chapter, we examine and execute some workflows written in WDL to become acquainted with the basic structure of the language and learn how Cromwell manages inputs and outputs, logs, and so on. For continuity with the previous chapters, we run Cromwell on the GCP Compute Engine VM that we used earlier, in Chapters 4 and 5. However, we’re no longer running anything from within the GATK container. Instead, we install and run Cromwell directly in the VM environment.
You’ll need to run a few installation commands because Cromwell requires Java, which is not preinstalled on the VM we’re using. To do so, log back in to your VM via SSH, just as you did in earlier chapters. Remember that you can always find your list of VM instances in the GCP console by going directly to the Compute Engine, or, in the menu of GCP services on the left side of the console, click Compute Engine, if you’ve forgotten the URL.
In your VM, type java -version at the prompt. You should get the following output:
$ java -version Command ’java’ not found, but can be installed with: apt install openjdk-11-jre-headless # version 11.0.3+7-1ubuntu2~19.04.1, or apt install default-jre # version 2:1.11-71 apt install openjdk-8-jre-headless # version 8u212-b03-0ubuntu1.19.04.2 apt install openjdk-12-jre-headless # version 12.0.1+12-1 apt install openjdk-13-jre-headless # version 13~13-0ubunt1 Ask your administrator to install one of them.
Cromwell requires Java version 8, so let’s install the openjdk-8-jre-headless option, which is a lightweight environment sufficient for our needs:
$ sudo apt install openjdk-8-jre-headless Reading package lists... Done Building dependency tree Reading state information... Done [...] done.
This triggers the installation process, which should run to completion without error. You might see a few notifications but as long as you see that final done output, you should be fine. You can run the Java version check again to satisfy yourself that the installation was successful:
$ java -version openjdk version "1.8.0_222" OpenJDK Runtime Environment (build 1.8.0_222-8u222-b10-1ubuntu1~19.04.1-b10) OpenJDK 64-Bit Server VM (build 25.222-b10, mixed mode)
With Java installed, let’s set up Cromwell itself, which comes with a companion utility called Womtool that we use for syntax validation and creating input files. They are both distributed as compiled .jar files, and we’ve included a copy in the book bundle, so you don’t need to do anything fancy except point to where they are located. To keep our commands as short as possible, let’s set up an environment variable pointing to their location. Let’s call it BIN for binary, a term often used to refer to the compiled form of a program:
$ export BIN=~/book/bin
Let’s check that we can run Cromwell by asking for its help output, which presents a summary of the three commands you can give it: server and submit are part of the server mode we discussed earlier, and run is the one-shot mode that we use shortly:
$ java -jar $BIN/cromwell-48.jar --help
cromwell 48
Usage: java -jar /path/to/cromwell.jar [server|run|submit] [options] <args>...
--help Cromwell - Workflow Execution Engine
--version
Command: server
Starts a web server on port 8000. See the web server documentation for more
details about the API endpoints.
Command: run [options] workflow-source
Run the workflow and print out the outputs in JSON format.
workflow-source Workflow source file or workflow url.
--workflow-root <value> Workflow root.
-i, --inputs <value> Workflow inputs file.
-o, --options <value> Workflow options file.
-t, --type <value> Workflow type.
-v, --type-version <value>
Workflow type version.
-l, --labels <value> Workflow labels file.
-p, --imports <value> A directory or zipfile to search for workflow imports.
-m, --metadata-output <value>
An optional directory path to output metadata.
Command: submit [options] workflow-source
Submit the workflow to a Cromwell server.
workflow-source Workflow source file or workflow url.
--workflow-root <value> Workflow root.
-i, --inputs <value> Workflow inputs file.
-o, --options <value> Workflow options file.
-t, --type <value> Workflow type.
-v, --type-version <value>
Workflow type version.
-l, --labels <value> Workflow labels file.
-p, --imports <value> A directory or zipfile to search for workflow imports.
-h, --host <value> Cromwell server URL.
It’s also worth doing the same thing for Womtool, to get a sense of the various utility commands available:
$ java -jar $BIN/womtool-48.jar --help
Womtool 48
Usage: java -jar Womtool.jar
[validate|inputs|parse|highlight|graph|upgrade|womgraph] [options]
workflow-source
workflow-source Path to workflow file.
-i, --inputs <value> Workflow inputs file.
-h, --highlight-mode <value>
Highlighting mode, one of 'html', 'console'
(used only with 'highlight' command)
-o, --optional-inputs <value>
If set, optional inputs are also included in the
inputs set. Default is 'true' (used onl
y with the inputs command)
--help
--version
Command: validate
Validate a workflow source file. If inputs are provided then 'validate'
also checks that the inputs file is a valid set of inputs for the
workflow.
Command: inputs
Generate and output a new inputs JSON for this workflow.
Command: parse
(Deprecated; WDL draft 2 only) Print out the Hermes parser’s abstract
syntax tree for the source file.
Command: highlight
(Deprecated; WDL draft 2 only) Print out the Hermes parser’s abstract
syntax tree for the source file. Requires at least one of 'html' or 'console'
Command: graph
Generate and output a graph visualization of the workflow in .dot format
Command: upgrade
Automatically upgrade the WDL to version 1.0 and output the result.
Command: womgraph
(Advanced) Generate and output a graph visualization of Cromwell’s
internal Workflow Object Model structure for this workflow in .dot format
Of the functions just listed, you’ll have the opportunity to use inputs, validate, and graph in this chapter.
Now let’s check that you have all the workflow files that we provide for this chapter. If you followed the setup instructions in Chapter 4, you should have a code directory that you cloned from GitHub. Under ~/book/code, you’ll see a directory called workflows that contains all of the code and related files that you’ll use in this chapter (aside from the data, which came from the bucket). You’re going to run commands from the home directory (instead of moving into a subdirectory as we did in earlier chapters), so to keep paths short in the various commands, let’s set up an environment variable to point to where the workflow files reside:
$ export WF=~/book/code/workflows
Finally, let’s talk about text editors. In all but one of the exercises that follow, you’re simply going to view and run prewritten scripts that we provide, so for viewing you could just download or clone the files to your laptop and open them in your preferred text editor. In one exception, we suggest you modify a WDL to break it in order to see what Cromwell’s error messaging and handling behavior looks like, so you’ll need to actually edit the file. We show you how to do this using one of the shell’s built-in text editors, called nano, which is considered one of the most accessible for people who aren’t used to command-line text editors. You are of course welcome to use another shell editor like vi or emacs if you prefer; if so, it will be up to you to adapt the commands we provide accordingly.
Whatever you decide to use as a text editor, just make sure not to use a word processor like Microsoft Word or Google Docs. Those applications can introduce hidden characters and are therefore not appropriate for editing code files. With that all sorted out, let’s buckle up and tackle your very first WDL workflow.
Your First WDL: Hello World
We begin with the simplest possible working example of a WDL script: the quintessential HelloWorld. If you’re not familiar with this, it’s a common trope in the documentation of programming languages; in a nutshell, the idea is to provide an introductory example with the minimum amount of code that produces the phrase HelloWorld!. We’re actually going to run through three basic WDL workflows to demonstrate this level of functionality, starting with the absolute minimum example, and then adding on just enough code to show core functionality that is technically not required yet needed for realistic use.
Learning Basic WDL Syntax Through a Minimalist Example
Let’s pull up the simplest example by loading the hello-world.wdl workflow file into the nano editor:
$ nano $WF/hello-world/hello-world.wdl
As noted earlier, nano is a basic editor. You can use the arrow keys on your keyboard to move through the file. To exit the editor, press Ctrl+X.
This is what the minimalistic Hello World for WDL looks like:
version1.0workflowHelloWorld{callWriteGreeting}taskWriteGreeting{command{echo"Hello World"}output{Fileoutput_greeting=stdout()}}
First, let’s ignore everything except the one line that has the phrase HelloWorld in it, the one in which it’s in quotes. Do you recognize the command on that line? That’s right, it’s a simple echo command; you can run that line by itself right now in your terminal:
$ echo "Hello World" Hello World
So that’s the command at the heart of our script that performs the desired action, and everything else is wrapping to make it runnable in scripted form through our workflow management system.
Now let’s unpack that wrapping. At the highest level, we have just two distinct stanzas, or blocks of code: the one starting with workflow HelloWorld, and the one starting with task WriteGreeting, with several lines of code between the curly braces in each case (the original designer of WDL really liked curly braces; you’ll see a lot more of them). We can summarize them like this:
workflowHelloWorld{...}taskWriteGreeting{...}
This makes it really clear that our script is structured in two parts: the workflow block, which is where we call out the actions that we want the workflow to perform, and a task block, where we define the action details. Here we have only one task, which is not really typical given that most workflows consist of two or more tasks; we cover workflows with multiple tasks further in this section.
Let’s take a closer look at how the action—that is, the command—is defined in the WriteGreeting task:
taskWriteGreeting{command{echo"Hello World"}output{Fileoutput_greeting=stdout()}}
In the first line, we’re declaring that this is a task called WriteGreeting. Within the outermost curly braces, we can break the structure of the code into another two blocks of code: command {...} and output {...}. The command block is quite straightforward: it contains the echo "Hello World" command. So that’s pretty self-explanatory, right? In general, you can stick just about anything in there that you would run in your terminal shell, including pipes, multiline commands, and even blocks of “foreign” code like Python or R, provided that you wrap it in heredoc syntax. We provide examples of what that looks like in Chapter 9.
Meanwhile the output block is perhaps a bit less obvious. The goal here is to define the output of the command block we plan to run. We’re declaring that we expect the output will be a File, which we choose to call output_greeting (this name can be anything you want, except one of the reserved keywords, which are defined in the WDL specification). Then, in the slightly tricky bit, we’re stating that the output content itself will be whatever is emitted to stdout. If you’re not that familiar with command-line terminology, stdout is short for standard out, and refers to the text output to the terminal window, meaning it’s what you see displayed in the terminal when you run a command. By default, this content is also saved to a text file in the execution directory (which we examine shortly), so here we’re saying that we designate that text file as the output of our command. It’s not a terribly realistic thing to do in a genomics workflow (although you might be surprised…we’ve seen stranger things), but then that’s what a Hello World is like!
Anyway, that’s our task block explained away. Now, let’s look at the workflow block:
workflowHelloWorld{callWriteGreeting}
Well, that’s pretty simple. First, we declare that our workflow is called HelloWorld, and then, within the braces, we make a call statement to invoke the WriteGreeting task. This means that when we actually run the workflow through Cromwell, it will attempt to execute the WriteGreeting task. Let’s try that out.
Running a Simple WDL with Cromwell on Your Google VM
Exit the nano editor by pressing Ctrl+X and return to the shell of your VM. You’re going to launch the hello-world.wdl workflow using the Cromwell .jar file that resides in the ~/book/bin directory, which we aliased as $BIN during the setup part of this chapter. The command is straightforward Java:
$ java -jar $BIN/cromwell-48.jar run $WF/hello-world/hello-world.wdl
This command invokes Java to run Cromwell using its one-off (run) workflow execution mode, which we contrasted with the persistent server mode earlier in this chapter. So it’s just going to start up, run the workflow we provided as an input to the run command, and shut down when that’s done. For the moment, there is nothing else involved because our workflow is entirely self-contained; we cover how to parameterize the workflow to accept input files next.
Go ahead and run that command. If you have everything set up correctly, you should now see Cromwell begin to spit out a lot of output to the terminal. We show the most relevant parts of the output here, but we’ve omitted some blocks (indicated by [...]) that are of no interest for our immediate purposes:
[...]
[2018-09-08 10:40:34,69] [info] SingleWorkflowRunnerActor: Workflow submitted
b6d224b0-ccee-468f-83fa-ab2ce7e62ab7
[...]
Call-to-Backend assignments: HelloWorld.WriteGreeting -> Local
[2018-09-08 10:40:37,15] [info] WorkflowExecutionActor-b6d224b0-ccee-468f-83fa-
ab2ce7e62ab7 [b6d224b0]: Starting HelloWorld.WriteGreeting
[2018-09-08 10:40:38,08] [info] BackgroundConfigAsyncJobExecutionActor
[b6d224b0HelloWorld.WriteGreeting:NA:1]: echo "Hello World"
[2018-09-08 10:40:38,14] [info] BackgroundConfigAsyncJobExecutionActor
[...]
[2018-09-08 10:40:40,24] [info] WorkflowExecutionActor-b6d224b0-ccee-468f-83fa-
ab2ce7e62ab7 [b6d224b0]: Workflow HelloWorld complete. Final Outputs:
{
"HelloWorld.WriteGreeting.output_greeting": "/home/username/cromwell-
executions/HelloWorld/b6d224b0-ccee-468f-83fa-ab2ce7e62ab7/call-
WriteGreeting/execution/stdout"
}
[2018-09-08 10:40:40,28] [info] WorkflowManagerActor WorkflowActor-b6d224b0-ccee-
468f-83fa-ab2ce7e62ab7 is in a terminal state: WorkflowSucceededState
[2018-09-08 10:40:45,96] [info] SingleWorkflowRunnerActor workflow finished with
status ’Succeeded’.
[...]
[2018-09-08 10:40:48,85] [info] Shutdown finished.
As you can see, Cromwell’s standard output is a tad…well, verbose. Cromwell has been designed primarily for use as part of a suite of interconnected services, which we discuss in Chapter 11, where there is a dedicated interface for monitoring progress and output during routine use. The single-run mode is more commonly used for troubleshooting, so the development team has chosen to make the local execution mode very chatty to help with debugging. This can feel a bit overwhelming at first, but don’t worry: we’re here to show you how to decipher it all—or at least the parts that we care about.
Interpreting the Important Parts of Cromwell’s Logging Output
First, let’s check that the output of our workflow is what we expected. Find this set of lines in the terminal output:
WorkflowExecutionActor-b6d224b0-ccee-468f-83fa-ab2ce7e62ab7 [b6d224b0]: Workflow
HelloWorld complete. Final Outputs:
{
"HelloWorld.WriteGreeting.output_greeting": "/home/username/cromwell-
executions/HelloWorld/b6d224b0-ccee-468f-83fa-ab2ce7e62ab7/call-
WriteGreeting/execution/stdout"
}
Without going into the details just yet, we see that this provides a list in JSON format of the output files that were produced; in this case, just the one file that captured the stdout of our one echo "Hello World" command. Cromwell gives us the fully qualified path, meaning it includes the directory structure above the working directory, which is really convenient because it allows us to use it in any command with a quick copy and paste. You can do that right now to look at the contents of the output file and verify that it contains what we expect:
Note
Keep in mind that in the command we show here, you need to replace the username and the execution directory hash. It might be easier to look for the equivalent line in your output than to customize our command.
$ cat ~/cromwell-executions/HelloWorld/b6d224b0-ccee-468f-83fa- ab2ce7e62ab7/call-WriteGreeting/execution/stdout Hello World
And there it is! So we know it worked.
Now let’s take a few minutes to walk through the information that Cromwell is giving us in all that log output to identify the most relevant nuggets:
SingleWorkflowRunnerActor: Workflow submitted b6d224b0-ccee-468f-83fa-ab2ce7e62ab7
= I’m looking at this one workflow and assigning it this unique identifier.
Cromwell assigns a randomly generated unique identifier to every run of every workflow and creates a directory with that identifier, within which all of the intermediate and final files will be written. We go over the details of the output directory structure in a little bit. For now, all you really need to know is that this is designed to ensure that you will never overwrite the results of a previous run of the same workflow or experience collisions between different workflows that have the same name:
Call-to-Backend assignments: HelloWorld.WriteGreeting -> Local
= I’m planning to send this to the local machine for execution (as opposed to a remote server).
By default, Cromwell runs workflows directly on your local machine; for example, your laptop. As we mentioned earlier, you can configure it to send jobs to a remote server or cloud service, instead; that’s what in Cromwell lingo is called a backend assignment (not to be confused with diaper duty):
Starting HelloWorld.WriteGreeting
= I’m executing the
WriteGreetingtask call from theHelloWorldworkflow now.
Cromwell treats each task call in the workflow as a separate job to execute, and will give you individual updates about each one accordingly. If the workflow involves multiple task calls, Cromwell will organize them in a queue and send each out for execution when appropriate. We discuss some aspects of how that works a little later. With regard to the status reporting aspect, you can imagine that as soon as we move to running more complex workflows, getting these reports through the standard out is rather impractical. This is where frontend software that provides an interface to parse and organize all of this information can really come in handy; you’ll have an opportunity to experience that in Chapter 11:
[b6d224b0HelloWorld.WriteGreeting:NA:1]: echo "Hello World"
= This is the actual command I’m running for this call.
It’s not obvious from this particular call because we didn’t include any variables in our minimal Hello World example, but what Cromwell outputs here is the real command that will be executed. In the parameterized example that comes next, you can see that if we include a variable in the script, the log output will show the form of the command in which the variable has been replaced by the input value that we provide. This fully interpreted command also is output to the execution directory for the record:
[b6d224b0]: Workflow HelloWorld complete. Final Outputs:
{
"HelloWorld.WriteGreeting.output_greeting": "/home/username/cromwell-
executions/HelloWorld/b6d224b0-ccee-468f-83fa-ab2ce7e62ab7/call-
WriteGreeting/execution/stdout"
}
= I’m done running this workflow. This is the full path to that output file(s) you wanted.
As noted earlier, this provides a list in JSON format of all the output files that were produced, identified by their full namespace. The namespace
HelloWorld.WriteGreeting.output_greeting
tells us that we are looking at the output_greeting output by the call to the WriteGreeting task belonging to the HelloWorld workflow.
The fully qualified path to the output file shows the entire directory structure; let’s unroll that and examine what each segment corresponds to:
~ (working directory)
cromwell-executions/ (Cromwell master directory)
HelloWorld (name of our workflow)
b6d224b0-ccee-468f-83fa-ab2ce7e62ab7 (unique identifier of the run)
call-WriteGreeting (name of our task call)
execution (directory of execution files)
The important piece in this structure is the nesting of workflow/identifier/calls. As you’ll see in the next exercise, any runs of a workflow with the same name will be added under the HelloWorld workflow directory, in a new directory with another unique identifier.
SingleWorkflowRunnerActor workflow finished with status 'Succeeded'.
= Yo, everything worked!
[2018-09-08 10:40:48,85] [info] Shutdown finished.
= I’m all done here. Buh-bye.
And that’s really all you need to care about at this point, concluding your first Cromwell workflow execution. Well done!
Adding a Variable and Providing Inputs via JSON
OK, but running a completely self-contained WDL is unrealistic, so let’s look at how we add variables to bring in some external input that can change from run to run. In the nano editor, go ahead and open the hello-world-var.wdl from the code directory:
$ nano $WF/hello-world/hello-world-var.wdl
What’s different? The workflow block is exactly the same, but now there’s a bit more going on in the WriteGreeting task block:
taskWriteGreeting{input{Stringgreeting}command{echo"${greeting}"}output{Fileoutput_greeting=stdout()}}
The Hello World input to the echo command has been replaced by ${greeting}, and we now have a new input block before the command block that contains the line String greeting. This line declares the variable called greeting and states that its value should be of type String; in other words, an alphanumeric sequence. This means that we have parameterized the greeting that will be echoed to the terminal; we’re going to be able to instruct Cromwell what to insert into the command on a run-by-run basis.
This leads to the next question: how do we provide Cromwell with that value? We definitely don’t want to have to give it directly on the command line, because although this particular case is simple, in the future we might need to run workflows that expect dozens of values, many of them more complex than a simple String.
Cromwell expects you to provide inputs in JavaScript Object Notation (JSON) text format. JSON has a key:value pair structure that allows us to assign a value to each variable. You can see an example of this in the $WF/hello-world/hello-world.inputs.json file that we provide:
{"HelloWorld.WriteGreeting.greeting":"Hello Variable World"}
In this simple inputs JSON file, we have defined the greeting variable from our HelloWorld workflow by its fully qualified name, which includes the name of the workflow itself (HelloWorld) and then the name of the task (WriteGreeting) because we declared the variable at the task level and then the name of the variable itself.
To provide the inputs JSON file to Cromwell, simply add it by using the -i argument (short for --input) to your Cromwell command, as follows:
$ java -jar $BIN/cromwell-48.jar run $WF/hello-world/hello-world-var.wdl \ -i $WF/hello-world/hello-world.inputs.json
Look for the output the same way you did earlier; you should see the message in the file output by the workflow match the text in the JSON file.
Cromwell enforces the use of fully qualified names at all levels, which makes it impossible to declare global variables. Although this might feel like a burdensome constraint, it is much safer than the alternative, because it means that you can have variables with the same name in different parts of a workflow without causing collisions. In simple workflows, it’s easy enough to keep track of variables and prevent such problems, but in more complex workflows with dozens of more variables, that can become quite difficult. That is especially the case when you use imports and subworkflows to facilitate code reuse, which we cover in Chapter 9 (ooh, spoilers). Note that you can declare a variable at the workflow level (and use the input naming syntax WorkflowName.variable in the inputs JSON file), but you’ll need to pass it explicitly to any task calls in which you want to use it. You’ll see an example of this in action later in this chapter.
Adding Another Task to Make It a Proper Workflow
Real-world workflows usually have more than one task, and some of their tasks are dependent on the outputs of others. In the nano editor, open hello-world-again.wdl:
$ nano $WF/hello-world/hello-world-again.wdl
Here’s our third iteration attempt at a Hello World example, showing two tasks chained into a proper workflow:
version1.0workflowHelloWorldAgain{callWriteGreetingcallReadItBackToMe{input:written_greeting=WriteGreeting.output_greeting}output{Fileoutfile=ReadItBackToMe.repeated_greeting}}taskWriteGreeting{input{Stringgreeting}command{echo"${greeting}"}output{Fileoutput_greeting=stdout()}}taskReadItBackToMe{input{String=read_string(written_greeting)}command{echo"${original_greeting} to you too"}output{Filerepeated_greeting=stdout()}}
You can see that the workflow block has quite a bit more going on now; it has an additional call statement pointing to a new task, ReadItBackToMe, and that call statement has some code in curly braces attached to it, which we’ll call the input block:
callReadItBackToMe{input:written_greeting=WriteGreeting.output_greeting}
The input block allows us to pass values from the workflow level to a particular task call. In this case, we’re referencing the output of the WriteGreeting task and assigning it to a variable called written_greeting for use within the ReadItBackToMe call.
Let’s have a look at that new task definition:
taskReadItBackToMe{input{Filewritten_greeting}Stringgreeting=read_string(written_greeting)command{echo"${greeting} to you too"}output{Filerepeated_greeting=stdout()}
The read_string() bit is a function from the WDL standard library that reads in the contents of a text file and returns them in the form of a single string. So this task is meant to read in the contents of a file into a String variable and then use that variable to compose a new greeting and echo it to stdout.
In light of that, the extra code attached to the ReadItBackToMe call statement makes perfect sense. We’re calling the ReadItBackToMe task and specifying that the input file we used to compose the new greeting should be the output of the call to the WriteGreeting task.
Finally, let’s look at the last block of code we haven’t examined in this new version of the workflow:
output{Fileoutfile=ReadItBackToMe.repeated_greeting}
This workflow has an output block defined at the workflow level in addition to the individual task-level output blocks. This workflow-level output definition is entirely optional when the workflow is intended to be run by itself; it’s more a matter of convention than function. By defining a workflow-level output, we communicate which of the outputs produced by the workflow we care about. That being said, you can use this output definition for functional purposes; for example, when the workflow is going to be used as a nested subworkflow and we need to pass its output to further calls. You’ll see that in action in Chapter 9. For now, try running this workflow with the same input JSON as the previous workflow, then poke around the execution directories to see how the task directories relate to each other and where the outputs are located.
Your First GATK Workflow: Hello HaplotypeCaller
Now that you have a firm grasp of basic WDL syntax, let’s turn to a more realistic set of examples: actual GATK pipelines! We begin with a very simple workflow in order to build your familiarity with the language gradually. We want a workflow that runs GATK HaplotypeCaller linearly (no parallelization) in GVCF mode on a single sample BAM file, as illustrated in Figure 8-1.
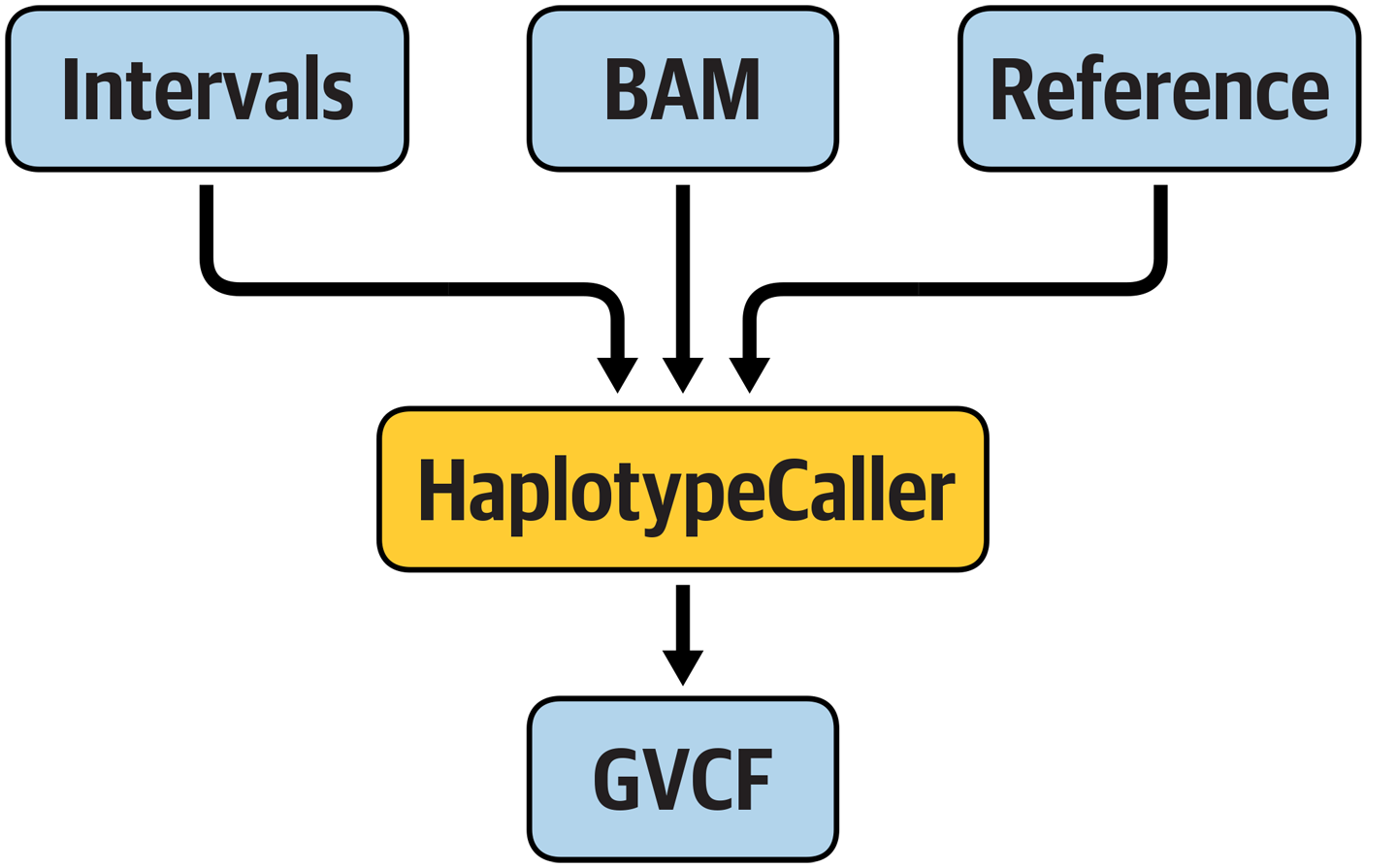
Figure 8-1. A hypothetical workflow that runs HaplotypeCaller.
The workflow should take the usual required files—the genome reference, input reads, and a file of intervals to analyze (technically optional as far as GATK is concerned, but here we made it required by the WDL)—and output a GVCF file named based on the input file.
Exploring the WDL
To illustrate all of that, we put together a WDL workflow that fulfills these requirements through a single task, HaplotypeCallerGVCF. Open it now in the nano editor:
$ nano $WF/hello-hc/hello-haplotypecaller.wdl
Let’s walk through the main sections of the script, recalling the structure of our HelloWorld example:
version1.0workflowHelloHaplotypeCaller{callHaplotypeCallerGVCF}taskHaplotypeCallerGVCF{input{Stringdocker_imageStringjava_optFileref_fastaFileref_indexFileref_dictFileinput_bamFileinput_bam_indexFileintervals}Stringgvcf_name=basename(input_bam,".bam")+".g.vcf"command{gatk--java-options${java_opt}HaplotypeCaller\-R${ref_fasta}\-I${input_bam}\-O${gvcf_name}\-L${intervals}\-ERCGVCF}output{Fileoutput_gvcf="${gvcf_name}"}runtime{docker:docker_image}}
Collapsing the task for clarity, you can see that it is indeed a single-task workflow, with only that single call and nothing else going on in the workflow block:
workflowHelloHaplotypeCaller{callHaplotypeCallerGVCF}taskHaplotypeCallerGVCF{…}
So let’s look at that HaplotypeCallerGVCF task in more detail, starting with the command block, because ultimately that’s where we’ll glean the most information about what the task actually does:
command{gatk--java-options${java_opt}HaplotypeCaller\-R${ref_fasta}\-I${input_bam}\-O${gvcf_name}\-L${intervals}\-ERCGVCF}
We see a classic GATK command that invokes HaplotypeCaller in GVCF mode. It uses placeholder variables for the expected input files as well as the output file. It also includes a placeholder variable for passing in Java options such as memory heap size, as described in Chapter 5. So far, that’s pretty straightforward.
Those variables should all be defined somewhere, so let’s look for them. Conventionally, we do this at the start of the task description, before the command block. This is what we see there:
input{Stringdocker_imageStringjava_optFileref_fastaFileref_indexFileref_dictFileinput_bamFileinput_bam_indexFileintervals}Stringgvcf_name=basename(input_bam,".bam")+".g.vcf"
Ignoring the String docker_image line for now, this shows that we declared all of the variables for the input files as well as the Java options, but we didn’t assign a value to them. So the task will expect to receive values for all of them at runtime. Not only that, but it will also expect values for all the accessory files we often take for granted: refIndex, refDict, and inputBamIndex, which refer to the indices and sequence dictionary. We don’t include those files in the command itself because GATK detects their presence automatically (as long as their names conform with their master file’s respective format conventions), but we do need to inform Cromwell that they exist so that it can make them available for execution at runtime.
There is one exception, though; for the output file, we see this line:
Stringgvcf_name=basename(input_bam,".bam")+".g.vcf"
The basename(input_bam, ".bam") piece is a convenience function from the WDL standard library that allows us to create a name for our output file based on the name of the input file. The basename() function takes the full path of the input file, strips off the part of the path that’s in front of the filename, and, optionally, strips off a given string from the end of the filename. In this case, we’re stripping off the expected .bam extension and then we’re using the + ".g.vcf" part of the line to add the new extension that will be appropriate for the output file.
Speaking of the output file, let’s now skip over to the task-level output block:
output{Fileoutput_gvcf="${gvcf_name}"}
This is also straightforward; we’re stating that the command will produce an output file that we care about, giving it a name for handling it within the workflow, and providing the corresponding placeholder variable so that Cromwell can identify the correct file after the command has run to completion.
Technically, that is all you need to have in the workflow and task definition if you’re planning to run this on a system with a local installation of the program that you want to run. However, in this chapter you’re working in your VM but outside the GATK container, and as a result, GATK is not available directly to your workflow. Fortunately, Cromwell is capable of utilizing Docker containers, so we just need to add a runtime block to the workflow in order to specify a container image:
runtime{docker:docker_image}
That’s why we had that String docker_image line in our task variables: we’re also using a placeholder variable for the container image. When we fill out the input JSON in the next step, we’ll specify the us.gcr.io/broad-gatk/gatk:4.1.3.0 image. Then, when we launch the workflow, Cromwell will spin up a new container from the image we specified and run GATK inside of it.
Technically we could hardcode the image name here, using double quotes (e.g., docker: "us.gcr.io/broad-gatk/gatk:4.1.3.0") but we don’t recommend doing that unless you really want to peg a particular script to a particular version, which reduces flexibility significantly. Some people use the latest tag to make their workflows always run with the latest available version of the program, but we consider that to be a bad practice with more downsides than upsides because you never know what might change in the latest version and break your workflow.
Generating the Inputs JSON
Alright, we’ve gone through all of the code in the workflow; now we need to determine how we’re going to provide the inputs to the workflow when we run it. In “Your First WDL: Hello World”, we were running an extremely simple workflow, first without any variable inputs and then with a single one. In the single-input case, we created a JSON file specifying that one input. Now we have eight inputs that we need to specify. We could proceed in the same way as before—create a JSON file and write in the name of every input expected by the HaplotypeCallerGVCF task—but there’s an easier way: we’re going to use the Womtool inputs command to create a template JSON.
First, because we’re going to be writing files that we care about for the first time in the chapter, let’s make a sandbox directory to keep our outputs organized:
$ mkdir ~/sandbox-8
Now, you can run the Womtool command that generates the inputs JSON template file:
$ java -jar $BIN/womtool-48.jar \
inputs $WF/hello-hc/hello-haplotypecaller.wdl \
> ~/sandbox-8/hello-haplotypecaller.inputs.json
Because we specify an output file on the last line of this command (which is actually optional), the command writes its output to that file. If everything goes smoothly, you shouldn’t see any output in the terminal. Let’s look at the contents of the file that we just created:
$ cat ~/sandbox-8/hello-haplotypecaller.inputs.json
{"HelloHaplotypeCaller.HaplotypeCallerGVCF.input_bam_index":"File","HelloHaplotypeCaller.HaplotypeCallerGVCF.input_bam":"File","HelloHaplotypeCaller.HaplotypeCallerGVCF.ref_fasta":"File","HelloHaplotypeCaller.HaplotypeCallerGVCF.ref_index":"File","HelloHaplotypeCaller.HaplotypeCallerGVCF.ref_dict":"File","HelloHaplotypeCaller.HaplotypeCallerGVCF.intervals":"File","HelloHaplotypeCaller.HaplotypeCallerGVCF.docker_image":"String","HelloHaplotypeCaller.HaplotypeCallerGVCF.java_opt":"String"}
There you go: all of the inputs that the HaplotypeCallerGVCF task expects are listed appropriately, with a placeholder value stating their type, in JSON format (although they might be in a different order in yours). Now we just need to fill in the values; those are the paths to the relevant files for the first six, and the runtime parameters (container image and Java options) for the last two. In the spirit of laziness, we provide a filled-out version that uses the snippet data that we used in Chapter 5, but you could also go through the exercise of filling in the inputs JSON with other inputs from the data bundle that you downloaded in Chapter 4 if you like. This is what the prefilled JSON looks like (paths are relative to the home directory):
$ cat $WF/hello-hc/hello-haplotypecaller.inputs.json
{"HelloHaplotypeCaller.HaplotypeCallerGVCF.input_bam_index":"book/data/germline/bams/mother.bai","HelloHaplotypeCaller.HaplotypeCallerGVCF.input_bam":"book/data/germline/bams/mother.bam","HelloHaplotypeCaller.HaplotypeCallerGVCF.ref_fasta":"book/data/germline/ref/ref.fasta","HelloHaplotypeCaller.HaplotypeCallerGVCF.ref_index":"book/data/germline/ref/ref.fasta.fai","HelloHaplotypeCaller.HaplotypeCallerGVCF.ref_dict":"book/data/germline/ref/ref.dict","HelloHaplotypeCaller.HaplotypeCallerGVCF.intervals":"book/data/germline/intervals/snippet-intervals-min.list","HelloHaplotypeCaller.HaplotypeCallerGVCF.docker_image":"us.gcr.io/broad-gatk/gatk:4.1.3.0","HelloHaplotypeCaller.HaplotypeCallerGVCF.java_opt":"-Xmx8G"}
Notice that all of the values are shown in double quotes, but this is a bit of an artifact because these values are all of String type. For other types such as numbers, Booleans, and arrays, you should not use double quotes. Except, of course, for arrays of strings, for which you should use double quotes around the strings, though not around the array itself, ["like","this"].
Running the Workflow
To run the workflow, we’re going to use the same command-line syntax as earlier. Make sure to execute this in your home directory so that the relative paths in the inputs JSON file match the location of the data files:
$ java -jar $BIN/cromwell-48.jar \
run $WF/hello-hc/hello-haplotypecaller.wdl \
-i $WF/hello-hc/hello-haplotypecaller.inputs.json
As mentioned earlier, Cromwell output is quite verbose. For this exercise, you’re looking for lines that look like these in the terminal output:
[2019-08-14 06:27:14,15] [info] BackgroundConfigAsyncJobExecutionActor
[9a6a9c97HelloHaplotypeCaller.HaplotypeCallerGVCF:NA:1]: Status change from
WaitingForReturnCode to Done
[2019-08-14 06:27:15,46] [info] WorkflowExecutionActor-9a6a9c97-7453-455c-8cd8-
be8af8cb6f7c [9a6a9c97]: Workflow HelloHaplotypeCaller complete. Final Outputs:
{
"HelloHaplotypeCaller.HaplotypeCallerGVCF.output_gvcf": "/home/username/cromwell-
executions/HelloHaplotypeCaller/9a6a9c97-7453-455c-8cd8-be8af8cb6f7c/call-
HaplotypeCallerGVCF/execution/mother.g.vcf"
}
[2019-08-14 06:27:15,51] [info] WorkflowManagerActor WorkflowActor-9a6a9c97-7453-
455c-8cd8-be8af8cb6f7c is in a terminal state: WorkflowSucceededState
[2019-08-14 06:27:21,31] [info] SingleWorkflowRunnerActor workflow
status ’Succeeded’.
{
"outputs": {
"HelloHaplotypeCaller.HaplotypeCallerGVCF.output_gvcf":
"/home/username/cromwell-executions/HelloHaplotypeCaller/9a6a9c97-7453-455c-8cd8-
be8af8cb6f7c/call-HaplotypeCallerGVCF/execution/mother.g.vcf"
workflow HelloHaplotypeCaller {
},
"id": "9a6a9c97-7453-455c-8cd8-be8af8cb6f7c"
}
The most exciting snippets here are Status change from WaitingForReturnCode to Done and finished with status 'Succeeded', which together mean that your workflow is done running and that all commands that were run stated that they were successful.
The other exciting part of the output is the path to the outputs. In the next section, we talk a bit about why they’re listed twice; for now, let’s just be happy that Cromwell tells us precisely where to find our output file so that we can easily peek into it. Of course, the output of this particular workflow is a GVCF file, so it’s not exactly pleasant to read through, but the point is that the file is there and its contents are what you expect to see.
We use the head utility for this purpose; keep in mind that you’ll need to substitute the execution directory hash in the file path shown in the following example (9a6a9c97-7453-455c-8cd8-be8af8cb6f7c) to the one displayed in your output:
$ head ~/cromwell-executions/HelloHaplotypeCaller/9a6a9c97-7453-455c -8cd8-be8af8cb6f7c/call-HaplotypeCallerGVCF/execution/mother.g.vcf ##fileformat=VCFv4.2 ##ALT=<ID=NON_REF,Description="Represents any possible alternative allele at this location"> ##FILTER=<ID=LowQual,Description="Low quality"> ##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Allelic depths for the ref and alt alleles in the order listed"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Approximate read depth (reads with MQ=255 or with bad mates are filtered)"> ##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality"> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##FORMAT=<ID=MIN_DP,Number=1,Type=Integer,Description="Minimum DP observed within the GVCF block"> ##FORMAT=<ID=PGT,Number=1,Type=String,Description="Physical phasing haplotype information, describing how the alternate alleles are phased in relation to one another"> ##FORMAT=<ID=PID,Number=1,Type=String,Description="Physical phasing ID information, where each unique ID within a given sample (but not across samples) connects records within a phasing group">
This run should complete quickly because we’re using an intervals list that spans only a short region. Most of the time spent here is Cromwell getting started and spinning up the container, whereas GATK HaplotypeCaller itself runs for only the briefest of moments. As with the Hello World example, you might feel that this is an awful lot of work for such a small workload, and you’d be right; it’s like swatting a fly with a bazooka. For toy examples, the overhead of getting Cromwell going dwarfs the analysis itself. It’s when we get into proper full-scale analyses that a workflow management system like Cromwell really shows its value, which you’ll experience in Chapters 10 and 11.
Breaking the Workflow to Test Syntax Validation and Error Messaging
Hopefully, so far everything ran as expected for you, so you know what success looks like. But in reality, things occasionally go wrong, so now let’s look at what failure looks like. Specifically, we’re going to look at how Cromwell handles two common types of scripting error, WDL syntax and command block syntax errors, by introducing some errors in the WDL. First, let’s make a copy of the workflow file so that we can play freely:
$ cp $WF/hello-hc/hello-haplotypecaller.wdl ~/sandbox-8/hc-break1.wdl
Now, in your preferred text editor, open the new file and introduce an error in the WDL syntax. For example, you could mangle one of the variable names, one of the reserved keywords, or delete a curly brace to mess with the block structure. Here, we’ll be a bit sadistic and delete the second parenthesis in the basename() function call. It’s the kind of small error that is fatal yet really easy to overlook. Let’s see what happens when we run this:
$ java -jar $BIN/cromwell-48.jar \
run ~/sandbox-8/hc-break1.wdl \
-i $WF/hello-hc/hello-haplotypecaller.inputs.json
As expected, the workflow execution fails, and Cromwell serves us with some verbose error messaging:
[2019-08-14 07:30:49,55] [error] WorkflowManagerActor Workflow 0891bf2c-4539-498c-
a082-bab457150baf failed (during MaterializingWorkflowDescriptorState):
cromwell.engine.workflow.lifecycle.materialization.MaterializeWorkflowDescriptorAct
or$$anon$1: Workflow input processing failed:
ERROR: Unexpected symbol (line 20, col 2) when parsing ’_gen18’.
Expected rparen, got command .
command {
^
$e = :string
[stack trace]
[2019-08-14 07:30:49,57] [info] WorkflowManagerActor WorkflowActor-0891bf2c-4539-
498c-a082-bab457150baf is in a terminal state: WorkflowFailedState
There’s a lot in there that we don’t care about, such as the stack trace, which we’re not showing here. The really important piece is Workflow input processing failed: ERROR: Unexpected symbol. That is a dead giveaway that you have a syntax issue. Cromwell will try to give you more specifics indicating where the syntax error might lie; in this case, it’s pretty accurate—it expected but didn’t find the closing parenthesis (rparen for right parenthesis) on line 20—but be aware that sometimes it’s not as obvious.
When you’re actively developing a new workflow, you probably won’t want to have to launch the workflow through Cromwell each time you need to test the syntax of some new code. Good news: you can save time by using Womtool’s validate command instead. That’s what Cromwell actually runs under the hood, and it’s a very lightweight way to test your syntax. Try it now on your broken workflow:
$ java -jar $BIN/womtool-48.jar \
validate ~/sandbox-8/hc-break1.wdl
ERROR: Unexpected symbol (line 20, col 2) when parsing '_gen18'.
Expected rparen, got command .
command {
^
$e = :string
See? You get the important part of Cromwell’s output in a much shorter time frame—and you don’t even need to provide valid inputs to the workflow. As an additional exercise, try introducing other WDL syntax errors; for example, try deleting a variable declaration, changing a variable name in only one of its appearances, and misspelling a reserved keyword like workflow, command, or outputs. This will help you to recognize validation errors and interpret how they are reported by Womtool. In general, we heartily recommend using Womtool validate systematically on any new or updated WDLs (and yes, it works on CWL too).
That being said, it’s important to understand that Womtool’s WDL syntax validation will get you only so far: it can’t do anything about any other errors you might make that are outside its scope of expertise; for example, if you mess up the command syntax for the tool you want to run. To see what happens in that case, make another copy of the original workflow in your sandbox (call it hc-break2.wdl) and open it up to introduce an error in the GATK command this time; for example, by mangling the name of the tool, changing it to HaploCaller:
command {
gatk --java-options ${java_opt} HaploCaller \
-R ${refFasta} \
-I ${inputBam} \
-O ${gvcfName} \
-L ${intervals} \
-ERC GVCF
}
If you run Womtool validate, you’ll see this workflow sails right through validation; Womtool cheerfully reports Success! Yet if you actually run it through Cromwell, the workflow will most definitely fail:
$ java -jar $BIN/cromwell-48.jar \
run ~/sandbox-8/hc-break2.wdl \
-i $WF/hello-hc/hello-haplotypecaller.inputs.json
Scroll through the output to find the line showing the failure message:
[2019-08-14 07:09:52,12] [error] WorkflowManagerActor Workflow dd77316f-7c18-4eb1 -aa86-e307113c1668 failed (during ExecutingWorkflowState): Job HelloHaplotypeCaller.HaplotypeCallerGVCF:NA:1 exited with return code 2 which has not been declared as a valid return code. See ’continueOnReturnCode’ runtime attribute for more details. Check the content of stderr for potential additional information: /home/username/cromwell-executions/HelloHaplotypeCaller/dd77316f-7c18-4eb1-aa86- e307113c1668/call-HaplotypeCallerGVCF/execution/stderr. [First 300 bytes]:Picked up _JAVA_OPTIONS: -Djava.io.tmpdir=/cromwell- executions/HelloHaplotypeCaller/dd77316f-7c18-4eb1-aa86-e307113c1668/call- HaplotypeCallerGVCF/tmp .e6f08f65 USAGE: <program name> [-h] Available Programs:
The log line that says Job HelloHaplotypeCaller.HaplotypeCallerGVCF:NA:1 exited with return code 2 means that HaplotypeCallerGVCF was the task that failed and, specifically, that the command it was running reported an exit code of 2. This typically indicates that the tool you were trying to run choked on something—a syntax issue, unsatisfied input requirements, formatting errors, insufficient memory, and so on.
The error message goes on to point out that you can find out more by looking at the standard error (stderr) output produced by the command, and it helpfully includes the full file path so that you easily can peek inside it. It also includes the first few lines of the stderr log for convenience, which is sometimes enough if the tool’s error output was very brief. The stderr output produced by GATK is of the more verbose variety. So here we’ll need to check out the full stderr to find out what went wrong. Again, make sure to substitute the username and execution directory hash shown here (dd77316f-7c18-4eb1-aa86-e307113c1668) with the ones in your output:
$ cat /home/username/cromwell-executions/HelloHaplotypeCaller/dd77316f-7c18-4eb1
-aa86-e307113c1668/call-HaplotypeCallerGVCF/execution/stderr
(...)
***********************************************************************
A USER ERROR has occurred: 'HaploCaller' is not a valid command.
Did you mean this?
HaplotypeCaller
***********************************************************************
(...)
Ah, look at that! We wrote the name of the tool wrong; who knew? Props to GATK for suggesting a correction, by the way; that’s new in GATK4.
Note
All command-line tools output a return code when they finish running as a concise way to report their status. Depending on the tool, the return code can be more or less meaningful. Conventionally, a return code of 0 indicates success, and anything else is a failure. In some cases, a nonzero code can mean the run was successful; for example, the Picard tool ValidateSamFile reports nonzero codes when it ran successfully but found format validation errors in the files it examined.
Other things can go wrong that we haven’t covered here, such as if you have the wrong paths for input files, or if you forgot to wrap string inputs in double quotes in the inputs JSON. Again, we recommend that you experiment by making those errors on purpose, because it will help you learn to diagnose issues more quickly.
Introducing Scatter-Gather Parallelism
We’re going to go over one more workflow example in this chapter to round out your first exposure to WDL and Cromwell, because we really want you to get a taste of the power of parallelization if you haven’t experienced that previously. So now we’re going to look at a workflow that parallelizes the operation of the HaplotypeCaller task we ran previously through a scatter() function and then merges the outputs of the parallel jobs in a subsequent step, as illustrated in Figure 8-2.
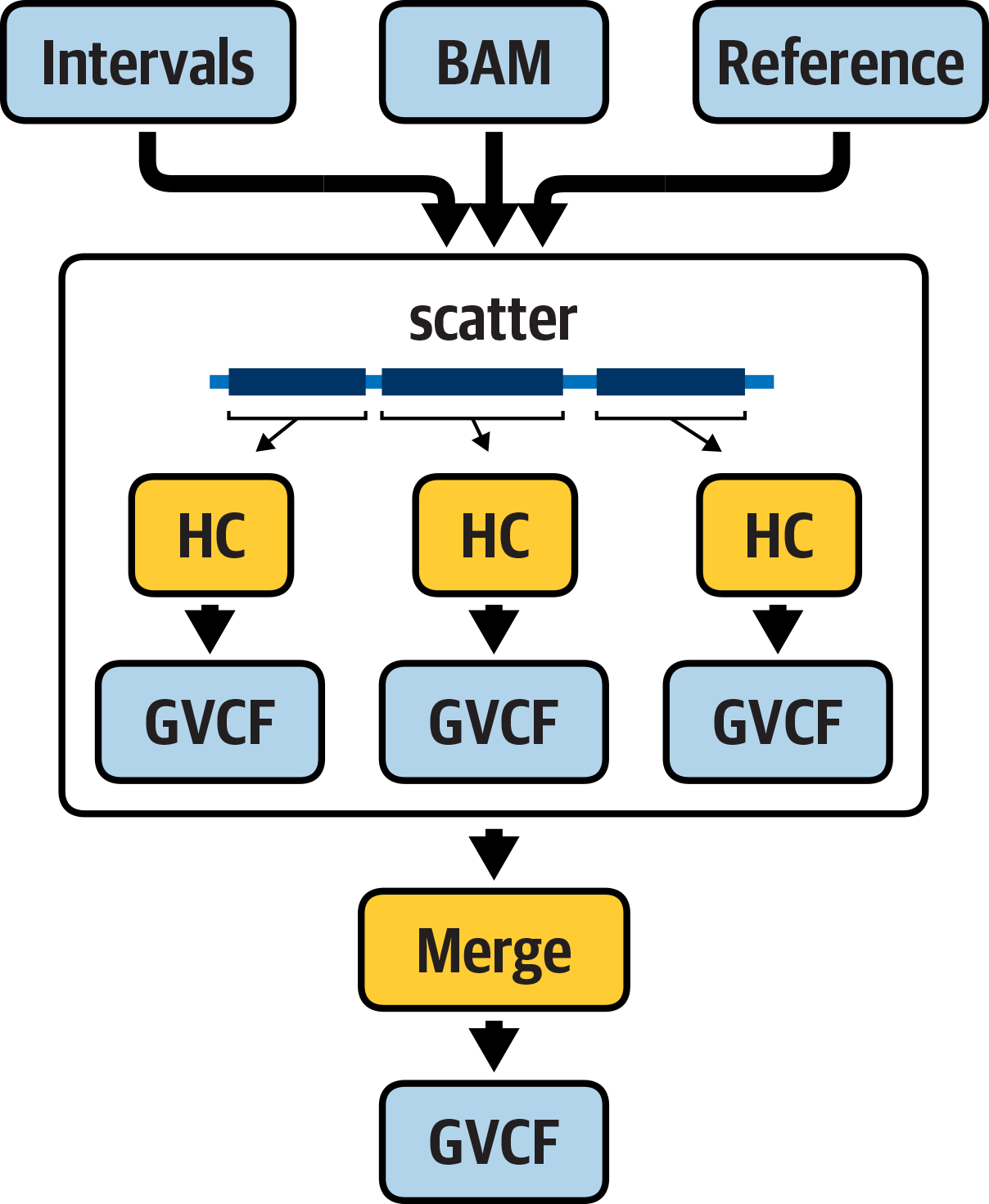
Figure 8-2. A workflow that parallelizes the execution of HaplotypeCaller.
This workflow will expose you to the magic of scatter-gather parallelism, which is a staple of genomics workflows, especially in the cloud. It will also give you an opportunity to dig into the mechanics of stringing multiple tasks together based on their inputs and outputs in a bit more detail than we covered earlier.
Exploring the WDL
Here’s a full WDL that parallelizes the operation of HaplotypeCallerGVCF over subsets of intervals. In the nano editor, open it as usual:
$ nano $WF/scatter-hc/scatter-haplotypecaller.wdl
Let’s walk through the main sections of the script, calling out what has changed compared to the linear implementation of this workflow:
version1.0workflowScatterHaplotypeCallerGVCF{input{Fileinput_bamFileinput_bam_indexFileintervals_list}Stringoutput_basename=basename(input_bam,".bam")Array[String]calling_intervals=read_lines(intervals_list)scatter(intervalincalling_intervals){callHaplotypeCallerGVCF{input:input_bam=input_bam,input_bam_index=input_bam_index,intervals=interval,gvcf_name=output_basename+".scatter.g.vcf"}}callMergeVCFs{input:vcfs=HaplotypeCallerGVCF.output_gvcf,merged_vcf_name=output_basename+".merged.g.vcf"}output{Fileoutput_gvcf=MergeVCFs.mergedGVCF}}taskHaplotypeCallerGVCF{input{Stringdocker_imageStringjava_optFileref_fastaFileref_indexFileref_dictFileinput_bamFileinput_bam_indexStringintervalsStringgvcf_name}command{gatk--java-options${java_opt}HaplotypeCaller\-R${ref_fasta}\-I${input_bam}\-O${gvcf_name}\-L${intervals}\-ERCGVCF}output{Fileoutput_gvcf="${gvcf_name}"}runtime{docker:docker_image}}taskMergeVCFs{input{Stringdocker_imageStringjava_optArray[File]vcfsStringmerged_vcf_name}command{gatk--java-options${java_opt}MergeVcfs\-I${sep=’-I’vcfs}\-O${merged_vcf_name}}output{Filemerged_vcf="${merged_vcf_name}"}runtime{docker:docker_image}}
The most obvious difference is that now a lot more is happening in the workflow block. The core of the action is this subset (collapsing the two call blocks for readability):
scatter(intervalsincalling_intervals){callHaplotypeCallerGVCF{...}}callMergeVCFs{...}
Previously, we were simply making one call to the HaplotypeCallerGVCF task, and that was it. Now, you see that the call to HaplotypeCallerGVCF is subordinated to a higher-level action, under this line, which opens a scatter block:
scatter(intervalsincalling_intervals){
For such a short line, it does a lot of work: this is how we parallelize the execution of HaplotypeCaller over subsets of intervals, which are specified in the parentheses. The calling_intervals variable refers to a list of intervals, and the HaplotypeCallerGVCF task will be run as a separate invocation on each interval provided in that file. This might look a lot like a for loop, if you’re familiar with that scripting construct, and indeed it is similar in the sense that its purpose is to apply the same operation to each element in a list. However, the scatter is specifically designed to allow independent execution of each operation, whereas a for loop leads to linear execution in which each operation can be run only after the previous one (if any) has run to completion.
So now that you understand what the scatter() instruction does, let’s look into the HaplotypeCallerGVCF task itself, starting with how it’s being called:
callHaplotypeCallerGVCF{input:input_bam=input_bam,input_bam_index=input_bam_index,intervals=intervals,gvcf_name=output_basename+".scatter.g.vcf"}
In the previous version of the workflow, the call to the task was just that: the call keyword followed by the task name. In this version, the statement includes an input block that specifies values for some of the variables that are required by the task: the input BAM file and its index, the intervals file, and the name of the GVCF output file. Speaking of which, you’ll notice that the basename function call that we use to generate a name for the output is now happening within this input block instead of happening within the task. If you compare the task definitions for HaplotypeCallerGVCF between this version of the workflow and the previous one, that’s the only difference. Let’s make a mental note of that because it will come up again in a few minutes.
Now let’s talk about what follows the scatter block containing the HaplotypeCallerGVCF call: a call to a new task named MergeVCFs. To be clear, this call statement is outside of the scatter block, so it will be run only once:
callMergeVCFs{...}
You probably already have a pretty good idea of what this task is for based on its name, but let’s pretend it’s not that obvious and follow the logical path for deciphering the structure of the workflow. Peeking ahead into the MergeVCFs task definition, we see that the command it runs is a GATK command that invokes a tool called MergeVcfs (actually, a Picard tool bundled into GATK4). As input, it takes one or more VCF files (of which GVCFs are a subtype) and outputs a single merged file:
command{gatk--java-options${java_opt}MergeVcfs\-I${sep='-I'vcfs}\-O${vcf_name}}
We can infer this from the command arguments and the variable names, and confirm it by looking up the MergeVcfs tool documentation on the GATK website.
Note
One novel point of WDL syntax to note here: the -I ${sep=' -I' vcfs} formulation is how we deal with having a list of items of arbitrary length that we need to put into the command line with the same argument for each item. Given a list of files [FileA, FileB, FileC], the preceding code would generate the following portion of the command line: -I FileA -I FileB -I FileC.
The MergeVCFs task definition also tells us that this task expects a list of files (technically expressed as Array[File]) as its main input. Let’s look at how the task is called in the workflow block:
callMergeVCFs{input:vcfs=HaplotypeCallerGVCF.output_gvcf,merged_vcf_name=output_basename+".merged.g.vcf"}
This might feel familiar if you remember the ReadItBackToMe task in the HelloWorldAgain workflow. Much as we did then, we’re assigning the vcfs input variable by referencing the output of the HaplotypeCallerGVCF task. The difference is that in that case, we were passing a single-file output to a single-file input. In contrast, here we’re referencing the output of a task call that resides inside a scatter block. What does that even look like?
Excellent question. By definition, each separate invocation of the task made within the scatter block generates its own separate output. The neat thing about the scatter construct is that those separate outputs are automatically collected into a list (technically an array) under the name of the output variable specified by the task. So here, although the output value of a single invocation of the HaplotypeCallerGVCF task, named HaplotypeCallerGVCF.output_gvcf, is a single GVCF file, the value of HaplotypeCallerGVCF.output_gvcf referenced in the workflow block is a list of the GVCF files generated within the scatter block. When we pass that reference to the next task call, we’re effectively providing the full list of files as an array.
Let’s finish this exploration by calling out a few more details. First, you might notice that we explicitly declare the output of the MergeVCFs call as the final output of the workflow in the workflow-level output block. This is technically not required, but it is good practice. Second, the variable declarations for the BAM file, its index, and the intervals file were all pushed up to the workflow level. In the case of the BAM file and its index, this move allows us to generate the names of the output files in the input blocks of both task calls, which among other advantages gives us more flexibility if we want to put our task definitions into a common library. For something like that, we want the tasks to be as generic as possible, and leave details like file-naming conventions up to the workflow implementations. As for the intervals file, we need to have it available at the workflow level in order to implement the scatter block.
Finally, you should now be able to generate the inputs JSON, fill it out based on the previous exercise, and run the workflow using the same setup as previously. We included a prefilled JSON if you want to save yourself the hassle of filling in file paths; just make sure to use the .local.inputs.json version rather than the .gcs.inputs.json, which we use in Chapter 10.
Here’s the Cromwell command using the prefilled local inputs JSON. Make sure to execute this in your home directory so that the relative paths in the inputs JSON file match the location of the data files:
$ java -jar $BIN/cromwell-48.jar \
run $WF/scatter-hc/scatter-haplotypecaller.wdl \
-i $WF/scatter-hc/scatter-haplotypecaller.local.inputs.json
When you run this command, you might notice the scatter jobs running in parallel on your VM. This is great because it means jobs will finish sooner. However, what happens if you’re trying to run five hundred scatter calls across a full genome? Running these all in parallel is going to cause problems; if it doesn’t outright crash, it will at least grind your VM to a halt as it swaps RAM to disk frantically. The good news is there are a couple solutions for this. First, you can control the level of parallelism allowed by the “local” backend for Cromwell, as described in the online documentation. Alternatively, you can use a different backend that is designed to handle this kind of situation gracefully. In Chapter 10, we show you how to use the Google backend to automatically send parallel jobs to multiple VMs.
Generating a Graph Diagram for Visualization
As a coda to this exercise, let’s learn to apply one more Womtool utility: the graph command. The workflows we’ve looked at so far have been quite simple in terms of the number of steps and overall plumbing. In the next chapter (and out in the real world), you will encounter more complex workflows, for which it might be difficult to build a mental model based on the code alone. That’s where it can be incredibly helpful to be able to generate a visualization of the workflow. The Womtool graph command allows you to generate a graph file in .dot format and visualize it using generic graph visualization tools:
$ java -jar $BIN/womtool-48.jar \
graph $WF/scatter-hc/scatter-haplotypecaller.wdl \
> ~/sandbox-8/scatter-haplotypecaller.dot
The .dot file is a plain-text file, so you can view it in the terminal; for example:
$cat~/sandbox-8/scatter-haplotypecaller.dotdigraphScatterHaplotypeCallerGVCF{#rankdir=LR;compound=true;# LinksCALL_HaplotypeCallerGVCF->CALL_MergeVCFsSCATTER_0_VARIABLE_interval->CALL_HaplotypeCallerGVCF# NodesCALL_MergeVCFs[label="call MergeVCFs"]subgraphcluster_0{style="filled,solid";fillcolor=white;CALL_HaplotypeCallerGVCF[label="call HaplotypeCallerGVCF"]SCATTER_0_VARIABLE_interval[shape="hexagon"label="scatter over String as interval"]}}
However, that’s not terribly visual, so let’s load it into a graph viewer. There are many available options, including the very popular open source package Graphviz. You can either install the package on your local machine or use it through one of its many online implementations. To do so, simply copy the contents of the .dot file into the text window of the visualizer app, and it will generate the graph diagram for you, as shown in Figure 8-3.
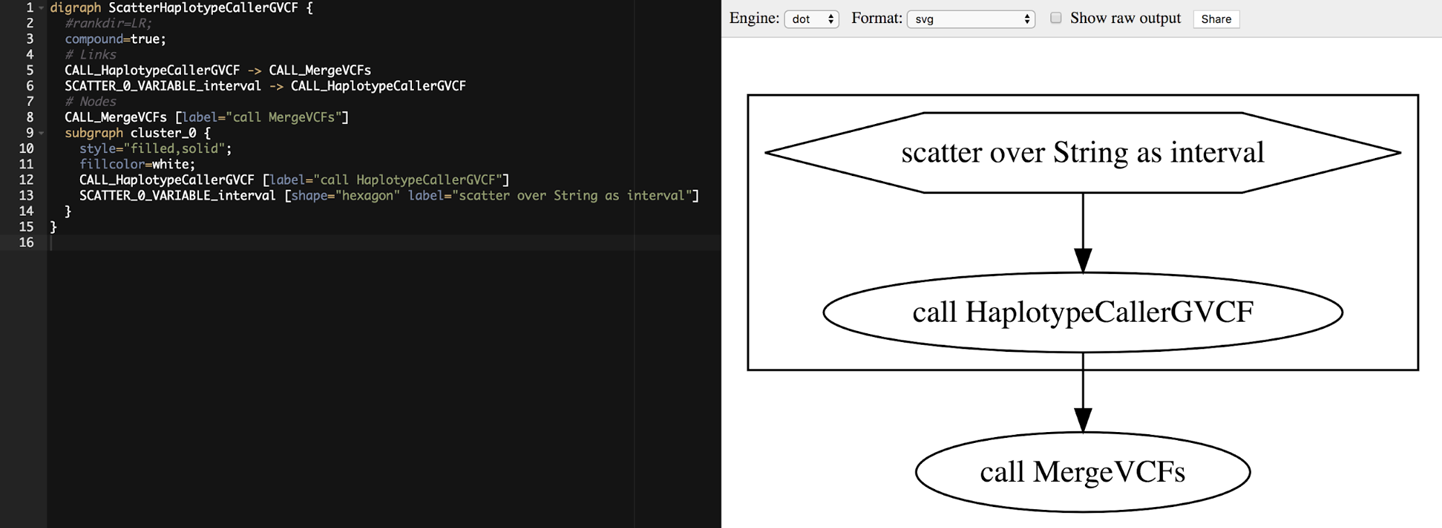
Figure 8-3. Visualizing the workflow graph in an online Graphviz application.
When we visualize a workflow graph like this, we typically look at the ovals first, which represent calls to tasks in the workflow. Here you can see that the two task calls in our workflow are indeed present, HaplotypeCallerGVCF and MergeVCFs. The direction of the arrows connecting the ovals indicates the flow of execution, so in Figure 8-3, you can see that the output of HaplotypeCallerGVCF is going to be the input to MergeVCFs.
Interestingly, the call to HaplotypeCallerGVCF is displayed with a box around it, which means it is under the control of a modifying function. The modifier function is represented as a hexagon, and here you can see the hexagon is labeled “scatter over String as interval.” That all makes sense because we just discussed how in this workflow the execution of the HaplotypeCaller task is scattered over a list of intervals.
In this case, the workflow was straightforward enough that the graph visualization didn’t really tell us anything we didn’t already know, but when we tackle more complex workflows in the next chapter, graph visualization is going to be a crucial part of our toolkit.
Note
This concludes the exercises in this chapter, so don’t forget to stop your VM; otherwise, you’ll be paying just to have it idly ponder the futility of existence.
Wrap-Up and Next Steps
In this chapter, we looked at how to string individual commands into a simple Hello World workflow, and we executed it using Cromwell in one-shot run mode on the single VM that we had set up in Chapter 4. We covered the basics of interpreting Cromwell’s terminal output and finding output files. We iterated on the original HelloWorld workflow, adding variables and an additional task. Then, we moved on to examine more realistic workflows that run real GATK commands and use scatter-gather parallelism, though still at a fairly small scale. Along the way, we exercised key utilities for generating JSON templates, validating WDL syntax, testing error handling, and generating graph visualizations.
However, we only scratched the surface of WDL’s capabilities as a workflow language, so now it’s time to move on to some more sophisticated workflows. In Chapter 9, we examine two mystery workflows and try to reverse engineer what they do, which will give you the opportunity to hone your detective skills and also learn some useful patterns that are used in real genomics analysis workflows.