Chapter 9. Deciphering Real Genomics Workflows
In Chapter 8, we showed you how to string commands into workflows by using the WDL language, and we had you practice running those workflows on your cloud VM using Cromwell. Throughout that chapter, we used fairly simple example workflows in order to focus on the basic syntax and rules of WDL. In this chapter, we switch gears and tackle workflows that are more complex than what you’ve seen so far. Rest assured, we neither expect you to instantly master all of the intricacies involved nor to memorize the various code features that you’ll encounter.
Our main goal is to expose you to the logic, patterns, and strategies used in real genomics workflows and, in the process, present you with a methodology for deciphering new workflows of arbitrary complexity. To that end, we’ve selected two workflows from the gatk-workflows collection, but we won’t tell you up front what they do. Instead, for each mystery workflow, we tell you the functionality you’re going to learn about and then walk you through a series of steps to learn what the workflow does and how the functionality that we’re interested in is implemented. This won’t turn you into a WDL developer overnight, but it will equip you with the skills to decipher other people’s workflows and, with a bit of practice, learn to modify them to serve your own purposes if needed.
Mystery Workflow #1: Flexibility Through Conditionals
Our first mystery workflow gives you the opportunity to learn how to use conditional statements to control which tasks are run and under what conditions. It also shows you how to increase flexibility for controlling the workflow’s parameters and outputs. As we work through this case, you’ll encounter many familiar elements that you encountered in Chapter 8. However, unlike in that chapter, here we don’t immediately explain all of the code; in fact, there is some code that we won’t cover at all. Much of this exercise involves focusing on the more important parts and ignoring details that don’t matter for what we’re trying to figure out.
You’re going to work in your VM with the same setup as in Chapter 8, running commands from the home directory. To begin, let’s set up an environment variable in your VM shell to make the commands shorter:
$ export CASE1=~/book/code/workflows/mystery-1
Let’s also create a sandbox directory for storing outputs:
$ mkdir ~/sandbox-9
And with that, you’re ready to dive in.
Mapping Out the Workflow
When you encounter a new workflow, it can be tempting to dive straight into the WDL file and try to make sense of it by directly reading the code. However, unless you’re very familiar with the language (or the author did an unusually great job of documenting their work), this can end up being frustrating and overwhelming. We propose a two-step approach that can help you to figure out the overall purpose and structure of a workflow. Start by generating a graph diagram of the workflow tasks. Then, identify which parts of the code correspond to each of the main components in the graph. This will provide you with a map of the overall workflow and a starting point for digging deeper into its plumbing.
Generating the graph diagram
You may recall that in Chapter 8 we discussed how the various connections between tasks in a workflow form a directed graph. The workflow execution engine uses that graph to identify the dependencies between tasks and, from there, determine in what order it should run them. The good news is that we can also take advantage of that information, as we already did in Chapter 8 when we generated the graph diagram for the HaplotypeCaller workflow.
Let’s do the same thing now for our mystery workflow; run the womtool graph utility on the WDL file:
$ cat ~/sandbox-9/haplotypecaller-gvcf-gatk4.dot
$ java -jar $BIN/womtool-48.jar graph $CASE1/haplotypecaller-gvcf-gatk4.wdl \
> ~/sandbox-9/haplotypecaller-gvcf-gatk4.dot
This produces a DOT file that describes the structure of the workflow in JSON syntax. Open the DOT file with cat to see the contents, which should look like this:
digraphHaplotypeCallerGvcf_GATK4{#rankdir=LR;compound=true;# LinksCALL_HaplotypeCaller->CALL_MergeGVCFsSCATTER_1_VARIABLE_interval_file->CALL_HaplotypeCallerCALL_CramToBamTask->CALL_HaplotypeCaller# Nodessubgraphcluster_0{style="filled,dashed";fillcolor=white;CALL_CramToBamTask[label="call CramToBamTask"]CONDITIONAL_0_EXPRESSION[shape="hexagon"label="if (is_cram)"style="dashed"]}CALL_MergeGVCFs[label="call MergeGVCFs"]subgraphcluster_1{style="filled,solid";fillcolor=white;CALL_HaplotypeCaller[label="call HaplotypeCaller"]SCATTER_1_VARIABLE_interval_file[shape="hexagon"label="scatter over File as interval_file"]}}
This is a little difficult to directly interpret, so let’s use Graphviz to make a visual representation. Go to GraphvizOnline and then copy and paste the contents of the DOT file into the editor panel on the left. The panel on the right should refresh and display the visual rendering of the graph, as shown in Figure 9-1.
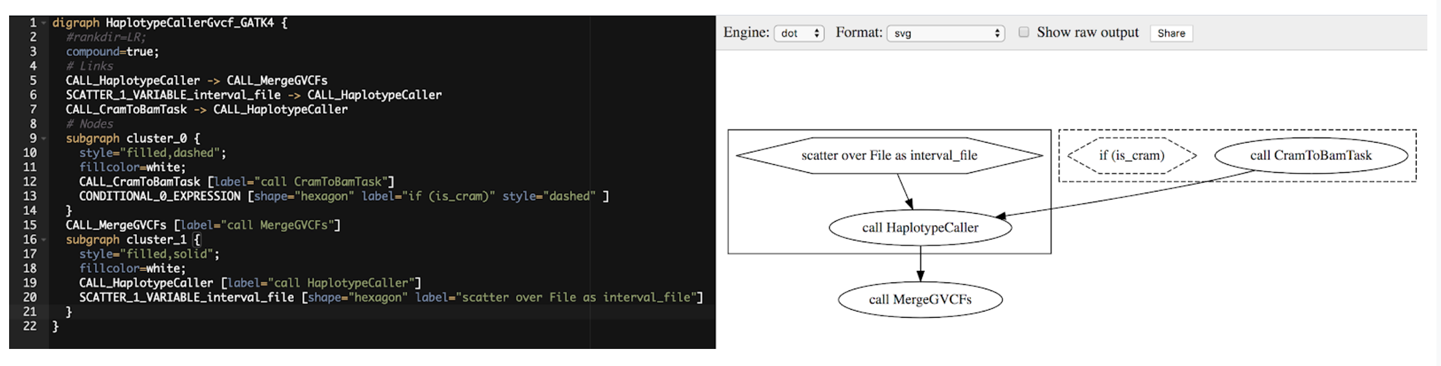
Figure 9-1. Graph description in JSON (left) and visual rendering (right).
Focus on the ovals first because they represent the calls to tasks in the workflow. We see that this workflow involves calls to three tasks: CramToBamTask, HaplotypeCaller, and MergeGVCFs. The first two calls are each in a box, which we know from Chapter 8 means there is some kind of control function that affects the call. Notice that the box around the call to CramToBamTask uses dotted lines, and that it also contains a hexagon labeled if (is_cram). Feel free to speculate and write down what you think that might do; you’ll find out if you were correct when we unpack that a little further along in the chapter. The other boxed call, to HaplotypeCaller, is under the control of a scatter function, which you should recognize from the last workflow we examined in Chapter 8. Indeed, if you ignore the CramToBamTask part, this workflow looks like the same scattered HaplotypeCaller example we reviewed then, which parallelized HaplotypeCaller over a list of intervals and then combined the per-interval GVCF outputs using MergeGVCFs to produce the final GVCF.
So, what’s up with that CramToBamTask call? Well, do you remember what CRAM is? As we mentioned in the genomics primer in Chapter 2, it’s a compression format for storing sequencing read data in smaller files than BAM format. Some tools like HaplotypeCaller are not yet able to work with CRAM files with absolute reliability, so if your input is in CRAM format, you need to convert it to BAM first. Based on the name of the task, it sounds like that’s exactly what CramToBamTask does. And we would want to run it only on actual CRAM files, so it makes sense to put it under the control of a conditional switch, if (is_cram), which asks: “Is this a CRAM file?” and calls the conversion task only if the answer is yes.
To summarize, based on the workflow graph, the names of tasks and variables, and some logical inferences, we can suppose that this mystery workflow is in fact very similar to the parallelized HaplotypeCaller workflow that you became familiar with in Chapter 8, but with a file format conversion step added at the beginning to handle CRAM files. That sounds reasonable, right? Let’s see if we can confirm this hypothesis and expand our understanding further.
Identifying the code that corresponds to the diagram components
Now we’re going to begin looking at the code, but in a very directed way. Rather than scrolling through all of it sequentially, we’re going to look up specific elements of code based on what we see in the graph diagram. Here’s an outline of the method:
-
List all tasks referenced in the graph (in ovals):
CramToBamTask HaplotypeCaller MergeGVCFs
-
Open the WDL file in your text editor and search for the
callstatements for each task:callCramToBamTask{line68callHaplotypeCaller{line84callMergeGVCFs{line100 -
For each call, capture the
callstatement and a few lines from the input definitions. If the task reference is shown inside a box in the graph, include the text that’s also in the box, likeif (is_cram)in Figure 9-1. Combine the code you captured into a table with screenshot slices of the graph diagram, as shown in Table 9-1.
| Calls in graph | Calls in workflow code |
|---|---|
 |
if ( is cram ) {
call CramToBamTask {
input:
input_cram = input_bam,
...
}
}
|
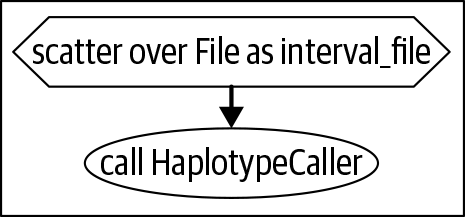 |
scatter (interval_file in
scattered_calling_intercals
) {
...
call HaplotypeCaller {}
input:
input_am =
select_first([CramToBamTask.output_bam,
input_bam]),
...
}
}
|
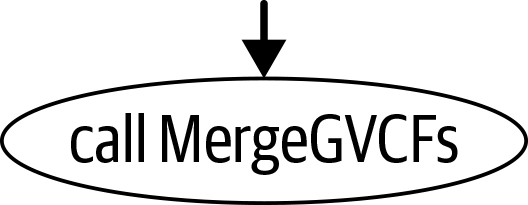 |
call MergeGVCFs {
input:
input_vcfs =
HaplotypeCalle.output_vcf,
...
}
|
This captures a lot of useful information about the key elements of code that determine how the tasks are connected and how their operation is controlled. In the next section, we dive deeper into these snippets of code as we attempt to reverse engineer how this workflow works.
You might need to tweak this approach and use your judgment regarding how many lines of input definitions or other code to include. For example, you might need to look up the task definitions to guide your choice of which inputs to include given that some are more informative than others. With practice, you will learn to reduce complex workflows to the key parts of their plumbing that matter.
Reverse Engineering the Conditional Switch
At this point, we’re reasonably confident that we understand what the workflow is doing overall. But we don’t yet understand how the conditional switch works and how the various tasks are wired together in practice. Let’s begin by looking at how the workflow logic is set up in the code.
How is the conditional logic set up?
In the match-up diagram in Table 9-1, we saw the box showing the call to CramToBamTask under the control of the presumed conditional switch. We identified the corresponding code by searching for call CramToBamTask and capturing the lines around it that are likely to be involved:
if(is_cram){callCramToBamTask{input:input_cram=input_bam,...}}
You can read this snippet of code as “If the is_cram condition is verified, call CramToBamTask and give it whatever was provided as input_bam.” This immediately brings up two important questions: how are we testing for that initial condition, and how are we handling the apparent contradiction in the input_cram = input_bam input assignment?
Let’s tackle the first question by scrolling up a bit in the workflow code until we find a reference to this mysterious is_cram variable. And here it is on line 59:
#is the input a cram file?Booleanis_cram=sub(basename(input_bam),".*\\.","")=="cram"
Can you believe our luck? There’s a note of documentation that confirms that the purpose of this line of code is to answer the question “Is the input a CRAM file?” All we need to do is determine how that second line is answering it. Let’s look at the left side first, before the equals sign:
Booleanis_cram
This means “I’m declaring the existence of this Boolean variable named is_cram.” Boolean refers to a type of variable that can have only one of two values: True or False. This variable type is named after English mathematician George Boole, who was a big fan of logic. Right there with you, George! Boolean variables are useful for expressing and testing conditions succinctly.
The equals sign is our value assignment statement, so whatever is on the other side is going to determine the value of the variable. Here’s what we see:
sub(basename(input_bam),".*\\.","")=="cram"
Try to ignore everything that’s in parentheses on the left side and just read this as something == "cram". The A == B syntax is a compact way of asking a question in code. You can ultimately read this as saying, “is A equal to B?” or bringing it back to this specific case, “is something equal to B?” So the full line now reads “My Boolean variable is_cram is True if something equals cram, and is False if something does not equal cram.”
So, what is this something that we’re testing? You might be able to guess it by now, but let’s keep decomposing the code methodically to get to the truth of the matter:
sub(basename(input_bam),".*\\.","")
Hopefully, you remember the basename() function from Chapter 8; it’s how we were able to name outputs based on the name of the input files. At some point we’re going to give this workflow some real inputs to run on, and the execution engine will match up the variable and the file path like this:
Fileinput_bam="gs://my-bucket/sample.bam"
When we just give the basename() function a file path, as in basename(input_bam), it produces the string sample.bam. If we also give it a substring, as in basename(input_bam, ".bam"), it will additionally clip that off to produce the string sample. This is convenient for naming an output the same thing as the input, but with a different file extension.
Here the workflow adds another layer of string manipulation by using the sub() function (also from the WDL standard library) to replace part of the string with something else. You use sub() like this:
sub("string to modify","substring or pattern we want to replace","replacement")
With that information, we can now interpret the full sub() command, as seen in the workflow:
-
Take the basename of our input file:
basename(input_bam) "sample.bam"
-
Separate the part up to the last period:
".*\\." "sample."+"bam"
-
Replace that part by nothing:
"" ""+"bam"
-
Output whatever is left:
"bam"
What is left? It’s the file extension! So finally, we can say for sure that that one line of code means “My Boolean variable is_cram is True if the input file extension equals cram, and is False if the input file extension does not equal cram.”
Note
You might argue that it would be worth adding a function to the WDL standard library to grab the file extension without having to jump through these string substitution hoops. If you feel very strongly about it, let the WDL maintainers know by posting an issue in the OpenWDL repository, and maybe they will add it! That’s how a community-driven language evolves: based on feedback from the people who use it.
Now that we know where the value of the is_cram variable comes from, we understand exactly how the flow is controlled: if we provide a file with a .cram extension as input to the workflow, the call to CramToBamTask will run on the file provided as input. If we provide anything else, that task will be skipped and the workflow will start at the next call, HaplotypeCaller. Switch on, switch off.
OK, what’s next? Well, before you go running off to the next feature, we have some more work to do to fully understand how the conditional switch affects the rest of the workflow. Conditionals are a really neat way to make workflows more flexible and multipurpose, but they tend to have a few side effects that you need to understand and watch out for. Let’s go over two important questions that we need to ask ourselves when we’re dealing with conditionals.
Does the conditional interfere with any assumptions we’re making anywhere else?
If we’re using a conditional to allow multiple file types to be used as input, we can’t know ahead of time what the extension might be for any given run. Yet a lot of genomics workflows, including this one, use basename() to name outputs based on the input name. That typically involves specifying what file extension to trim off the end of the input filename. The workflow must deal with that ambiguity explicitly.
At this point, you could do a text search for “basename” to find the code we need to investigate, but in our case, what we’re looking for is on the very next line of code, after the is_cram variable assignment:
Stringsample_basename=ifis_cramthenbasename(input_bam,".cram")elsebasename(input_bam,".bam")
This is another variable declaration, stating the variable’s type (String) and name (sample_basename) on the left side of the equals sign, with a value assignment on the right. Let’s look at that value assignment in more detail:
ifis_cramthenbasename(input_bam,".cram")elsebasename(input_bam,".bam")
If you read that out loud, it’s almost reasonable English, and it takes only a few additional words of interpretation to make complete sense:
“If the input is a CRAM file, run the basename function with the .cram extension; otherwise, run it with the .bam extension.”
Problem solved! It’s not the only way to achieve that result in WDL but it’s probably the most readily understandable. In fact, if you look carefully at the next few lines in the workflow, you should be able to find and interpret another (unrelated) conditional variable assignment that’s doing something very similar, though that one comes into play at the other end of the pipeline.
How does the next task know what to run on?
Depending on the format of the input file, HaplotypeCaller will be either the first or the second task called in the workflow. If it’s the first, it should run on the original input file; if it’s the second, it should take the output of the first task. How does the workflow code handle that?
Have another look at Table 9-1, which showed how the code maps to the graph diagram. This was the code we highlighted at the time for the call to HaplotypeCaller:
scatter(...){...callHaplotypeCaller{input:input_bam=select_first([CramToBamTask.output_bam,input_bam]),...}}
And there it is: in the input section of the call statement, we see that the input_bam is being set with yet another function, select_first(). The name suggests it’s in charge of selecting something that comes first, and we see it’s reading in an array of values, but it’s not super obvious what that means, is it? If we just wanted it to use the first element in the array, why give it that array in the first place?
Yep, this is a bit of an odd one. The subtlety is that select_first() is designed to tolerate missing values, which is necessary when you’re dealing with conditionals: depending on what path you follow, you might generate some outputs but not others. Most WDL functions will choke and fail if you try to give them an output that doesn’t exist. In contrast, select_first() will simply ask: what else do you have that might be available?
So, in light of that information, let’s look at that value assignment again in more detail. Here’s a handy way to represent how the code can be broken into segments of meaning:
input_bam = select_first([ |
CramToBamTask.output_bam |
, input_bam]) |
The input file for HaplotypeCaller should be... |
...the output from CramToBamTask... |
...but if that doesn’t exist, just use the original input file. |
At the moment in time when the workflow execution engine is evaluating what tasks need to be run and on what inputs/outputs, the output of the first task doesn’t exist yet. However, that output might be created at a later point, so the engine can’t just start running HaplotypeCaller willy-nilly on what might be a CRAM file. It must check the input file format to resolve the ambiguity, determine what tasks will run, and what outputs to expect. Having done that, the engine is able to evaluate the result of the select_first() function. If it determines that the CramToBamTask will run, it will hold off on running the HaplotypeCaller task until the CramToBamTask.output_bam has been generated. Otherwise, it can go ahead with the original input.
Hopefully, that makes more sense now. Though it might prompt a new question in your mind...
Can we use conditionals to manage default settings?
Yes, yes you can! If you skimmed through the workflow-level inputs, you might have noticed pairs of lines like this that look unusually complex for what should be straightforward variable declarations:
String?gatk_docker_overrideStringgatk_docker=select_first([gatk_docker_override,"us.gcr.io/broad-gatk/gatk:4.1.0.0"])
The first line in the pair simply declares a variable named gatk_docker_override, which was marked as optional by adding the question mark (?) after the variable type (String). Based on its name, it sounds like this variable would allow us to override the choice of Docker container image used to run GATK tasks, presumably by specifying one in the JSON file we hand to Cromwell at runtime. This implies that there’s a preset value somewhere, but we haven’t seen a variable declaration for that yet.
We find it in the second line in the pair, which declares a variable named gatk_docker and assigns it a value using the select_first() function. Let’s try breaking down that line the same way we did earlier, with a few adaptations based on context:
String gatk_docker = select_first([ |
gatk_docker_override |
, "us.gcr.io/broad-gatk/gatk:4.1.0.0"]) |
| The GATK Docker image should be... | ...what we provide in the JSON file of inputs... | ...but if we didn’t provide one, just use us.gcr.io/broad-gatk/gatk:4.1.0.0 by default. |
Isn’t that neat? This is not the only way to set and manage default values in WDL, and it’s admittedly not the most readily understandable if you’re not familiar with the select_first() function. However, it’s a very convenient way for the workflow author to peg their workflow to specific versions of software, environment expectations, resource allocations, and so on, while still allowing a lot of flexibility for using alternative settings without having to modify the code. You will encounter this pattern frequently in most up-to-date GATK workflows produced by the Broad Institute.
That wraps up our exploration of Mystery Workflow #1, which turned out to be a rather garden-variety HaplotypeCaller workflow hopped up with a few conditional statements. It had a few twists that mostly had to do with the consequences of using conditionals; but overall, the complexity was moderate. Let’s see if Mystery Workflow #2 can take us a little bit further up the scale of WDL complexity.
Mystery Workflow #2: Modularity and Code Reuse
Oh yes, now we’re in the big leagues: without spoiling the story, we can tell you that this one is a major pipeline that runs in production at the Broad Institute. It does a ton of work and involves many tools. It’s going to teach us a lot about how to build modular pipelines and minimize duplication of code. Let’s break it down.
First, make an environment variable pointing to the code, just as you did for the first workflow:
$ export CASE2=~/book/code/workflows/mystery-2
We can use the same sandbox as the first workflow, ~/sandbox-9/.
Mapping Out the Workflow
Let’s see if we can apply the same approach as for the previous workflow: generate the graph diagram and then map the call statements in the code back to the task bubbles in the diagram.
Generating the graph diagram
You know the drill: first, let’s whip out womtool graph to make the DOT file:
$ java -jar $BIN/womtool-48.jar graph $CASE2/WholeGenomeGermlineSingleSample.wdl \
> ~/sandbox-9/WholeGenomeGermlineSingleSample.dot
Next, open the DOT file; does it look any longer than the other one to you?
$cat~/sandbox-9/WholeGenomeGermlineSingleSample.dotdigraphWholeGenomeGermlineSingleSample{#rankdir=LR;compound=true;# LinksCALL_UnmappedBamToAlignedBam->CALL_BamToCramCALL_UnmappedBamToAlignedBam->CALL_CollectRawWgsMetricsCALL_UnmappedBamToAlignedBam->CALL_CollectWgsMetricsCALL_UnmappedBamToAlignedBam->CALL_AggregatedBamQCCALL_UnmappedBamToAlignedBam->CALL_BamToGvcfCALL_AggregatedBamQC->CALL_BamToCram# NodesCALL_AggregatedBamQC[label="call AggregatedBamQC";shape="oval";peripheries=2]CALL_BamToGvcf[label="call BamToGvcf";shape="oval";peripheries=2]CALL_UnmappedBamToAlignedBam[label="callUnmappedBamToAlignedBam";shape="oval";peripheries=2]CALL_BamToCram[label="call BamToCram";shape="oval";peripheries=2]CALL_CollectRawWgsMetrics[label="call CollectRawWgsMetrics"]CALL_CollectWgsMetrics[label="call CollectWgsMetrics"]}
Hmm, that doesn’t seem all that long. Considering we told you it involves a lot of tools, the graph file seems rather underwhelming. Let’s visualize it: copy the contents of the graph file into the panel on the left side in the online Graphviz app, as you’ve done before to produce the graph diagram, as depicted in Figure 9-2.
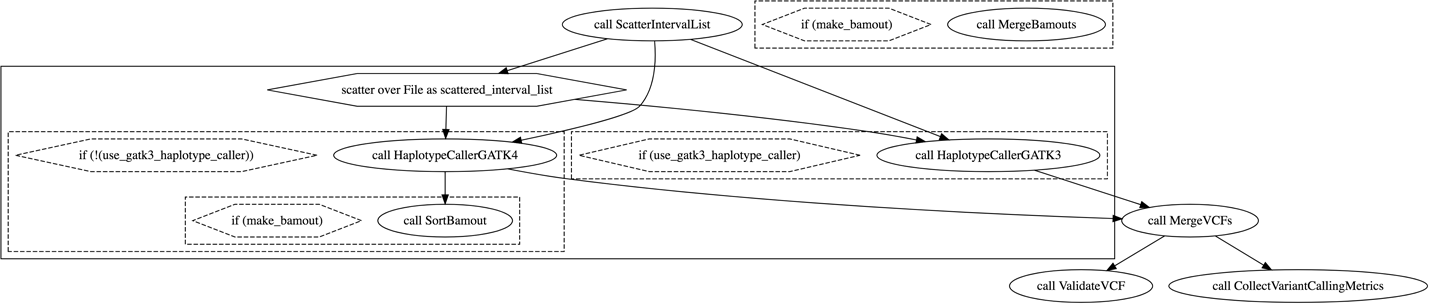
Figure 9-2. Visual rendering of the workflow graph.
What’s the first thing that is visually different compared to the previous workflow diagrams we’ve looked at?
There are no boxes, and some of the ovals representing the calls to tasks have a double outline. We don’t have any grounding yet for guessing what the double outline signifies, so let’s make a note of that as something to solve. However, we do know from the two previous workflows we looked at that scatter blocks show up as boxes, so the absence of boxes in this diagram suggests that, at the very least, this workflow does not have any parallelized steps. Does that seem likely to you given how much we’ve been talking up the parallelization capabilities of the cloud? (Yes; we know that’s called leading the witness.)
Just as we did for the first workflow, let’s try to form a hypothesis about what the workflow does based on the names of the task calls and their relative positions in the graph diagram.
The very first oval is labeled call UnmappedBamToAlignedBam, which suggests a transformation from the unmapped BAM file format that we referenced (ever so briefly) in Chapters 2 and 6 to an aligned BAM file—or as we’ll call it for consistency, a mapped BAM file. Based on what you learned of the data preprocessing pipeline in Chapter 6, that sounds like it could correspond to the step that applies the mapping to the raw unmapped data.
That first oval then connects to all five other ovals in the diagram, including the last one, which seems mighty strange but OK. Four of those ovals are on the same level, one step down from the first, and seem to be completely independent of one another. The two on the left have names that suggest they collect metrics related to WGS data (call CollectWgsMetrics and call CollectRawWgsMetrics), which would make sense as a quality-control step applied to evaluate the mapped BAM produced by the first call. In addition, they are both end nodes on their respective branches and have a single-line outline, so there’s probably not much more to them than meets the eye.
The third oval from the left, labeled call AggregatedBamQC, also evidently involves quality control, but what is the “aggregated BAM” to which it is applied? If the original data was divided among several read groups as recommended in the GATK Best Practices, it would make sense to have an “aggregated BAM” after the read group data has been merged per sample. However…do you remember from Chapter 6 when the merge step happens in the Best Practices pipeline? It happens after the mapping, during the duplicate marking step. Unless we’re totally off base in our interpretation of these task names, this would suggest that the duplicate marking and merging is happening within the same task as the mapping. That is technically feasible with a WDL workflow but generally not advisable from the point of view of performance: the mapping can be massively parallelized, but the duplicate marking cannot, so it would be a bad idea to bundle both of them for execution within the same task. Hold on to that thought.
Interestingly, the call AggregatedBamQC task does have a child node one more level down; it links to the oval labeled call BamToCram, which is also dependent on the first oval. BamToCram by itself sounds like the exact opposite of the CramToBamTask that you encountered in the previous workflow, so let’s venture to guess that this one allows you to create a CRAM file for archival purposes based on the mapped BAM created by the first step. Presumably, if the data in that file is considered ready for archival, it must have already been fully preprocessed, which lends further support to the idea that that first step is doing much more than just mapping the reads to the reference. As to the contribution of the AggregatedBamQC task, let’s suppose for now that there is something that affects the cramming step that is decided based on the results of the quality control analysis.
Finally, that just leaves the rightmost oval on the second level, an end node that is labeled call BamToGvcf. That sounds very familiar indeed; would you care to take a guess? Are you sick of seeing HaplotypeCaller everywhere yet?
So just based on the graph diagram, the names of the calls, and the way they connect to one another, we’ve provisionally deduced that this pipeline takes in raw unmapped sequencing data (which is probably whole genome sequence), maps it, applies some quality control, creates a CRAM archive, and generates a GVCF of variant calls that will be suitable for joint calling. That’s a lot of work for such an apparently minimalistic pipeline; we should probably dig into that.
Identifying the code that corresponds to the diagram components
The book says that we must match the code with the diagram now, and you can’t argue with a book, so in your text editor, go ahead and open the WDL file and follow the same steps as you did for the first workflow. List all tasks referenced in the graph (in ovals) and then, for each task, search for the call statement. For each call, capture the call statement and a few lines from the input definitions. Combine the code you captured into a table with screenshot slices of the graph diagram, as shown in Table 9-2.
| Workflow calls in the graph | Workflow calls in WDL code |
|---|---|
 |
call ToBam.UnmappedGamToAlignedBam {
input:
sample_and_unmapped_bams =
sample_and_unmapped_bams,
...
}
|
 |
call AggregatedQC.AggregatedBamQC {
input:
base_recalibrated_bam =
UnmappedBamToAlignedBam.output_bam,
...
}
|
 |
call ToCram.BamToCram as BamToCram {
input:
input_bam =
UnmappedBamToAlignedBam.output_bam,
...
}
|
 |
#QC the sample WGS metrics (stringent thresholds)
call QC.CollectWgsMetrics as
CollectWgsMetrics{
input:
input_bam =
UnmappedBamToAlignedBam.output_bam,
...
}
|
 |
#QC the sample WGS metrics (common thresholds)
call QC.CollectRawWgsMetrics as
CollectWgsMetrics{
input:
input_bam =
UnmappedBamToAlignedBam.output_bam,
...
}
|
 |
call ToGvcf.VariantCalling as BamToGvcf {
input:
...
input_bam =
UnmappedBamToAlignedBam.output_bam,
...
}
|
Looking at the code for these calls, something odd pops right up. The task names look a bit weird. Check out the first one:
callToBam.UnmappedBamToAlignedBam
Did you know periods are allowed in task names in WDL? (Spoiler: they’re not.)
You can try searching for ToBam.UnmappedBamToAlignedBam to find the task definition; usually that can be helpful for figuring what’s going on here. But here’s a twist: you’re not going to find it in this file. Go ahead, scroll down as much as you want; this WDL workflow does not contain any task definitions. (What?)
However, searching for either the first part of the name, ToBam, or the second, UnmappedBamToAlignedBam, does bring up this line in a stack of other similar lines that we ignored when we originally started looking at the workflow:
import"tasks/UnmappedBamToAlignedBam.wdl"asToBam
Aha. That’s new; it looks like we’re referencing other WDL files. In the preceding line, the import part points to another WDL file called UnmappedBamToAlignedBam.wdl, located in a subdirectory called tasks. Now things are starting to make sense: when the workflow engine reads that, it’s going to import whatever code is in that file and consider it part of the overall workflow. The as ToBam part of this import statement assigns an alias so that we can refer to content from the imported file without having to reference the full file path. For example, this was the call we encountered earlier:
callToBam.UnmappedBamToAlignedBam
We can interpret this as saying, “From the ToBam code, call UnmappedBamToAlignedBam.” So what is UnmappedBamToAlignedBam? Is it a task? Have a quick peek inside the WDL file referenced in the import statement and then take a deep breath. It’s a full workflow in its own right, called UnmappedBamToAlignedBam:
# WORKFLOW DEFINITIONworkflowUnmappedBamToAlignedBam{
We’re dealing with a workflow of workflows? Voltron, assemble!
This is actually a happy surprise. We suspected coming in that this was going to be a rather complex pipeline, even though the graph diagram initially gave us the wrong impression. So the discovery that it uses imports to tie together several simpler workflows into a mega-workflow is a good thing because it’s going to allow us to investigate the pipeline in smaller chunks at a time. In effect, the workflow authors have already done the hard work of identifying the individual segments that can be logically and functionally separated from the others.
Now we need to see how the workflow nesting works, beyond just the import statements. Surely there’s some additional wiring that we need to understand, like how are outputs passed around from one subworkflow to the next?
Unpacking the Nesting Dolls
We begin by digging down one level, into one of the subworkflows, to better understand how importing works and how the subworkflow connects to the master workflow. Which subworkflow should we pick? The VariantCalling workflow is a great option because it’s likely to be the most comfortably familiar. That way we can focus on understanding the wiring rather than a plethora of new tools and operations. But before we delve any further into the code, let’s map this subworkflow at a high level.
What is the structure of a subworkflow?
The good news is that subworkflows are, in fact, perfectly normal in terms of their WDL structure and syntax, so you can run all the usual womtool commands on them, and you can even run them on their own. Once more, womtool graph comes in handy:
$ java -jar $BIN/womtool-48.jar graph $CASE2/tasks/VariantCalling.wdl \
> ~/sandbox-9/VariantCalling.dot
Copy the DOT file contents into the online Graphviz website to generate the graph diagram. As you can see in Figure 9-3, this workflow looks a lot like the now-classic scattered HaplotypeCaller workflow that we’ve been exploring from all angles, with a few additions. In particular, it has a few conditional switches that you can investigate in detail if you’d like to get more practice with conditionals.
Figure 9-3. Graph diagram of the VariantCalling.wdl workflow.
Now open up the VariantCalling.wdl file in your text editor and scroll through it briefly. It has a workflow definition section, workflow VariantCalling {...}. If you’re up for it, you can run through the exercise of mapping the code for call statements to their position in the diagram; we won’t do it explicitly here. We’re more interested in the fact that this subworkflow has import statements of its own. Still no task definitions, though, except for one all the way at the bottom. Does that mean there’s going to be yet another level of nested workflows? We’ve got to find out where those task definitions are hiding.
Where are the tasks defined?
Let’s pick the first call statement we come across and hunt down whatever it points to. Scrolling down from the top, we come to this call:
callUtils.ScatterIntervalListasScatterIntervalList
You should recognize the pattern from the top-level workflow: this means that we need to look for an import statement aliased to Utils. Sure enough, we find this toward the top of the file:
import"tasks/Utilities.wdl"asUtils
In your text editor, open the Utilities.wdl file, and you should see the following (some lines omitted):
version1.0## Copyright Broad Institute, 2018#### This WDL defines utility tasks used for processing of sequencing data.##...# Generate sets of intervals for scatter-gathering over chromosomestaskCreateSequenceGroupingTSV{input{Fileref_dict...
Wait, it’s just a bunch of tasks? No workflow definition at all? That’s…well, kind of a relief. We seem to have found the deepest level of our nested structure, at least on this branch. To confirm, search for the ScatterIntervalList task in the Utilities.wdl file, and, sure enough, there is its task definition starting on line 77:
taskScatterIntervalList{input{Fileinterval_listIntscatter_countIntbreak_bands_at_multiples_of}command<<<...
Note
This task’s command block is pretty wild—first it runs a Picard tool via a Java command and then some bulk Python code that generates lists of intervals on the fly. We won’t analyze its syntax in detail; just note that it’s the sort of thing you can do within a WDL task.
The takeaway here is that this Utilities.wdl file functions as a library of tasks. Think for a moment about the implications: this means that you can store task definitions for commonly used tasks in a library and simply import them into individual workflows with a one-line statement, rather than rewriting or copying the code over each time. Then, when you need to fix or update a task definition, you need to do it only once rather than having to update all the copies that exist in different workflows. To be clear though, you’re not obligated to use subworkflows in order to use imports for tasks. For an example of non-nested workflows that share a task library, have a look at the workflows on GitHub.
Hey, look at how far we’ve come; this chapter is almost over. You should feel pretty good about yourself. You’ve worked through almost everything you need in order to understand imports and use them in your own work. But there’s one more thing we want to make sure is really clear, and it has to do with connecting inputs and outputs between subworkflows.
How is the subworkflow wired up?
As you might recall, this subworkflow is imported as follows in the master workflow:
import"tasks/VariantCalling.wdl"asToGvcf
As noted previously, this makes the code within the imported file available to the workflow interpreter and assigns the ToGvcf alias to that code so that we can refer to it elsewhere. We see this in the following call statement:
callToGvcf.VariantCallingasBamToGvcf{input:...input_bam=UnmappedBamToAlignedBam.output_bam,
Now that we know VariantCalling is a workflow defined within of the imported WDL, we can read the call like this in plain English:
From the pile of imported code named ToGvcf, call the workflow defined as VariantCalling, but name it BamToGvcf within the scope of this master workflow. Also, for the input BAM file it expects, give it the output from the UnmappedBamToAlignedBam workflow.
You might notice that there’s an interesting difference between how the UnmappedBamToAlignedBam workflow and the VariantCalling workflow are treated: the latter is assigned an alias, whereas the former is not. Instead, UnmappedBamToAlignedBam retains the original name given in the WDL where it is defined. Based on the naming patterns we’ve observed so far, our guess is that the authors of the workflow wanted to identify the high-level segments by the file formats of the main inputs and outputs at each stage. This is a very common pattern in genomics; it’s not the most robust strategy, but it certainly is concise and does not require too much domain understanding to decipher beyond the file formats themselves.
Anyway, the practical consequence is that we point to the outputs of the subworkflow by either its original name or by its alias if it has one. To be clear, however, we can point only to outputs that were declared as workflow-level outputs in the subworkflow. If any tasks in the subworkflow were not declared at the workflow level, but only at the task level, they will be invisible to the master workflow and to any of its other subworkflows.
Note
Aliases are a convenient feature of WDL that are useful beyond dealing with imports; they can also be used to call the same task in different ways within the same workflow.
As we’ve already observed, the import wiring system also allows us to call individual tasks from an imported WDL by referring to the alias given to the WDL file, as in this call statement to one of the tasks in the Utility.wdl library:
callUtils.ScatterIntervalListasScatterIntervalList
To reprise our model for loose translation to plain English, this reads as follows:
From the pile of imported code named Utils, call the task defined as ScatterIntervalList, but name it ScatterIntervalList instead of Utils.ScatterIntervalList within the scope of this master workflow.
The twist here is that if we don’t provide an alias, we must refer to the task as Utils.ScatterIntervalList everywhere. When we define this alias, we can just refer to the task directly as ScatterIntervalList. This is a subtle but important difference compared to the way workflow imports work. Note that we could also use a different name from the original if we wanted, like ScatterList for short, or Samantha if we’re actively trying to mess with anyone who might read our code (but please don’t do that).
Finally, remember that some of the ovals in the top-level workflow have a double outline, including the one labeled UnmappedBamToAlignedBam, which happens to be a workflow—do you think that could possibly be related? Let’s check out the other calls. The AggregatedBamQC oval had a double outline, so look up the import statement to identify the corresponding WDL file (tasks/AggregatedBamQC.wdl) and peek inside. Sure enough, it’s another workflow. Now look up the import statement for one of the ovals that had a single outline, for example CollectWgsMetrics, and peek inside the file (tasks/Qc.wdl). Ooh, no workflow declaration, just tasks. It seems we’ve identified what the double outline means: it denotes that the call points to a nested workflow, or subworkflow. Now that’s really useful.
Note
It would be great to also be able to recognize in the diagram whether an oval with a single outline is a true task or whether it’s pointing to a library of tasks. Something to request from the Cromwell development team.
To recap, we elucidated the structure of this complex pipeline, which implements the full GATK Best Practices for germline short variant discovery in whole genome sequencing data. It combines data preprocesssing (read alignment, duplicate marking, and base recalibration) with the scattered HaplotypeCaller we’ve been working with previously, and adds on a layer of quality control.
As it happens, the original form of this pipeline was a massive, single-WDL implementation, but it later was split into multiple WDL files to be more readable and manageable. The current pipeline utilizes import statements with several levels of nesting of both workflows and tasks. The result is a high level of code modularity and reusability that make it a good model for developing new pipelines, especially complex ones.
If you’re looking for more practice analyzing the use of imports with nested workflows and task libraries, try to map out the complete structure of this monster pipeline, including the preprocessing and QC workflows. When you’re done with that, take a look at the exome version of this pipeline, which follows the same model and, in fact, reuses some of the same code! See if you can combine them into a single codebase that maximizes code reuse.
Wrap-Up and Next Steps
In this chapter, we explored real genomics workflows that apply advanced WDL features to do some seriously interesting things. We began with an exploration of how you can use conditionals to control the flow of a WDL workflow, branching the analysis based on an input file type. We then looked at a rather complex production workflow from the Broad Institute that uses subworkflows, structures, and task libraries to break the workflow into more manageable (and reusable) pieces. Along the way, we focused on the skills and techniques you need to take a complex workflow, pick it apart, and understand it component by component. This will help not only in understanding the complex production Best Practices workflows that we’ve just started to examine, but it will serve you well when building your own workflows. In this chapter, we didn’t actually run the workflows because our focus was on developing your skills as a workflow detective. In Chapter 10, we dive into the execution of complex workflows but, this time, we show you how to take advantage of the full power of the cloud instead of just a single VM.