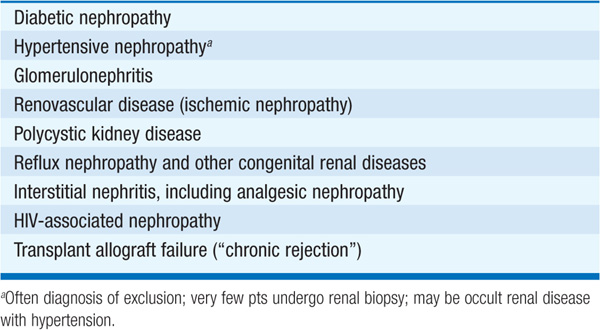
The prevalence of chronic kidney disease (CKD), generally defined as a longstanding, irreversible impairment of kidney function, is substantially greater than the number of pts with end-stage renal disease (ESRD), now ≥500,000 in the United States. There is a spectrum of disease related to decrements in renal function; clinical and therapeutic issues differ greatly depending on whether the glomerular filtration rate (GFR) reduction is moderate (stage 3 CKD, 30–59 mL/min per 1.73 m2) (Table 52-1), severe (stage 4 CKD, 15–29 mL/min per 1.73 m2), or “end-stage renal disease” (stage 5 CKD, <15 mL/min per 1.73 m2). Dialysis is usually required once GFR <10 mL/min per 1.73 m2. Common causes of CKD are outlined in Table 149-1.
TABLE 149-1 COMMON CAUSES OF CHRONIC RENAL FAILURE
The first step in the differential diagnosis of CKD is establishing its chronicity, i.e., disproving a major acute component. The two most common means of determining disease chronicity are the history and prior laboratory data (if available) and the renal ultrasound, which is used to measure kidney size. In general, kidneys that have shrunk (<10–11.5 cm, depending on body size) are more likely affected by chronic disease. While reasonably specific (few false positives), reduced kidney size is only a moderately sensitive marker for CKD, i.e., there are several relatively common conditions in which kidney disease may be chronic without any reduction in renal size. Diabetic nephropathy, HIV-associated nephropathy, and infiltrative diseases such as multiple myeloma may in fact be associated with relatively large kidneys despite chronicity. Renal biopsy, although rarely performed in pts with CKD, is a more reliable means of proving chronicity; a predominance of glomerulosclerosis or interstitial fibrosis argues strongly for chronic disease. Hyperphosphatemia, anemia, and other laboratory abnormalities are not reliable indicators in distinguishing acute from chronic disease.
Once chronicity has been established, clues from the physical exam, laboratory panel, and urine sediment evaluation can be used to determine etiology. A detailed history (Hx) will identify important comorbid conditions, such as diabetes, HIV seropositivity, or peripheral vascular disease. The family Hx is paramount in the workup of autosomal dominant polycystic kidney disease or hereditary nephritis (Alport’s syndrome). An occupational Hx may reveal exposure to environmental toxins or culprit drugs (including over-the-counter agents, such as analgesics or Chinese herbs).
Physical exam may demonstrate abdominal masses (i.e., polycystic kidneys), diminished pulses or femoral/carotid bruits (i.e., atherosclerotic peripheral vascular disease), or abdominal or femoral bruits (i.e., renovascular disease). The Hx and exam may also yield important data regarding severity of disease. Excoriations (uremic pruritus), pallor (anemia), muscle wasting, and a nitrogenous fetor are all signs of advanced CKD, as are pericarditis, pleuritis, and asterixis, complications of particular concern that usually prompt the initiation of dialysis.
Serum and urine laboratory findings typically provide additional information useful in determining the etiology and severity of CKD; serial studies determine the pace of progression and/or whether the renal failure is in fact acute. Heavy proteinuria (>3.5 g/d), hypoalbuminemia, hypercholesterolemia, and edema suggest nephrotic syndrome (Chap. 152). Diabetic nephropathy, membranous nephropathy, focal segmental glomerulosclerosis, minimal change disease, amyloid, and HIV-associated nephropathy are principal causes. Proteinuria may decrease slightly with decreasing GFR, but rarely to normal levels. Hyperkalemia and metabolic acidosis may complicate all forms of CKD eventually, but can be more prominent in pts with interstitial renal diseases. Serum and urine protein electrophoresis, in addition to serum free light chains, should be obtained in all pts >35 years of age with CKD to exclude paraproteinemia-associated renal disease. If underlying glomerulonephritis is suspected, autoimmune disorders such as lupus and infectious etiologies such as hepatitis B and C should be assessed. Serum concentrations of calcium, phosphate, vitamin D, and PTH should be measured to evaluate metabolic bone disease. Hemoglobin, vitamin B12, folate, and iron studies should be measured to evaluate anemia.
The culprit toxin(s) responsible for the uremic syndrome remain elusive. The serum creatinine (Cr) is the most common laboratory surrogate of renal function. GFR can be estimated using serum Cr–based equations derived from the Modification of Diet in Renal Disease Study. This “eGFR” is now reported with serum Cr by most clinical laboratories in the United States and is the basis for the National Kidney Foundation classification of chronic kidney disease (Table 52-1).
Uremic symptoms tend to develop with serum Cr >530–710 μmol/L (>6–8 mg/dL) or CrCl <10 mL/min, although these values vary widely. Uremia is thus a clinical diagnosis made in pts with CKD. Symptoms of advanced uremia include anorexia, weight loss, dyspnea, fatigue, pruritus, sleep and taste disturbance, and confusion and other forms of encephalopathy. Key findings on physical exam include hypertension, jugular venous distention, pericardial and/or pleural friction rub, muscle wasting, asterixis, excoriations, and ecchymoses. Laboratory abnormalities may include hyper-kalemia, hyperphosphatemia, metabolic acidosis, hypocalcemia, hyperuricemia, anemia, and hypoalbuminemia. Most of these abnormalities eventually resolve with initiation of dialysis or renal transplantation (Chaps. 150 and 151) or with appropriate drug therapies (see below).
TREATMENT Chronic Kidney Disease and Uremia
Hypertension complicates many forms of CKD and warrants aggressive treatment to reduce the risk of stroke and potentially to slow the progression of CKD (see below). Volume overload contributes to hypertension in many cases, and potent diuretic agents are frequently required. Anemia can be ameliorated with recombinant human erythropoietin (rHuEPO); current practice is to target a hemoglobin concentration of 100–110 g/L. Iron deficiency and/or other causes of anemia can reduce the response to rHuEPO and should be investigated if present. Iron supplementation is often required; many pts require parenteral iron therapy, since intestinal iron absorption is reduced in CKD.
Hyperphosphatemia can be controlled with judicious restriction of dietary phosphorus and the use of postprandial phosphate binders, either calcium-based salts (calcium carbonate or acetate) or nonabsorbed agents (e.g., sevelamer). Hyperkalemia should be controlled with dietary potassium restriction. Dialysis should be considered if the potassium is >6 mmol/L on repeated occasions. If these conditions cannot be conservatively controlled, dialysis should be instituted (Chap. 150). It is also advisable to begin dialysis if severe anorexia, weight loss, and/or hypoalbuminemia develop, as it has been definitively shown that outcomes for dialysis pts with malnutrition are particularly poor.
SLOWING PROGRESSION OF RENAL DISEASE Prospective clinical trials have explored the roles of blood pressure control and dietary protein restriction on the rate of progression of renal failure. Control of hypertension is of benefit, although angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) may exert unique beneficial effects, most likely due to their effects on intrarenal hemodynamics. The effects of ACE inhibitors and ARBs are most pronounced in pts with diabetic nephropathy and in those without diabetes but with significant proteinuria (>1 g/d). Diuretics and other antihypertensive agents are often required, in addition to ACE inhibitors and ARBs, to optimize hypertension control and attenuate disease progression; diuretics may also help control serum [K+].
