The adrenal cortex produces three major classes of steroids: (1) glucocorticoids, (2) mineralocorticoids, and (3) adrenal androgens. Clinical syndromes may result from deficiencies or excesses of these hormones. The adrenal medulla produces catecholamines, with excess leading to pheochromocytoma (Chap. 126).
The most common cause of Cushing’s syndrome is iatrogenic, due to administration of glucocorticoids for therapeutic reasons. Endogenous Cushing’s syndrome results from production of excess cortisol (and other steroid hormones) by the adrenal cortex. The major cause is bilateral adrenal hyperplasia secondary to hypersecretion of adrenocorticotropic hormone (ACTH) by the pituitary (Cushing’s disease) or from ectopic sources such as small cell carcinoma of the lung; carcinoids of the bronchus, thymus, gut and ovary, medullary carcinoma of the thyroid; or pheochromocytoma. Adenomas or carcinoma of the adrenal gland account for about 15–20% of endogenous Cushing’s syndrome cases. There is a female preponderance in endogenous Cushing’s syndrome except for the ectopic ACTH syndrome.
Some common manifestations (central obesity, hypertension, osteoporosis, psychological disturbances, acne, hirsutism, amenorrhea, and diabetes mellitus) are relatively nonspecific. More specific findings include easy bruising, purple striae, proximal myopathy, fat deposition in the face and nuchal areas (moon facies and buffalo hump), and rarely virilization. Thin, fragile skin and plethoric moon facies also may be found. Hypokalemia and metabolic alkalosis are prominent, particularly with ectopic production of ACTH.
The diagnosis of Cushing’s syndrome requires demonstration of increased cortisol production and abnormal cortisol suppression in response to dexamethasone. For initial screening, measurement of 24-h urinary free cortisol, the 1-mg overnight dexamethasone test [8 A.M. plasma cortisol <1.8 μg/dL (50 nmol/L)], or late-night salivary cortisol measurement is appropriate. Repeat testing or performance of more than one screening test may be required. Definitive diagnosis is established in equivocal cases by inadequate suppression of urinary cortisol [<10 μg/d (25 nmol/d)] or plasma cortisol [<5 μg/dL (140 nmol/L)] after 0.5 mg dexamethasone every 6 h for 48 h. Once the diagnosis of Cushing’s syndrome is established, further biochemical testing is required to localize the source. This evaluation is best performed by an experienced endocrinologist. Low levels of plasma ACTH levels suggest an adrenal adenoma or carcinoma; inappropriately normal or high plasma ACTH levels suggest a pituitary or ectopic source. In 95% of ACTH-producing pituitary microadenomas, cortisol production is suppressed by high-dose dexamethasone (2 mg every 6 h for 48 h). MRI of the pituitary should be obtained but may not reveal a microadenoma because these tumors are typically very small. Furthermore, because up to 10% of ectopic sources of ACTH may also suppress after high-dose dexamethasone testing, inferior petrosal sinus sampling may be required to distinguish pituitary from peripheral sources of ACTH. Testing with corticotropin-releasing hormone (CRH) also may be helpful in determining the source of ACTH. Imaging of the chest and abdomen is required to localize the source of ectopic ACTH production; small bronchial carcinoids may escape detection by conventional CT. Pts with chronic alcoholism, depression, or obesity may have false-positive results in testing for Cushing’s syndrome—a condition named pseudo-Cushing’s syndrome. Similarly, pts with acute illness may have abnormal laboratory test results, since major stress alters the normal regulation of ACTH secretion.
TREATMENT Cushing’s Syndrome
Uncontrolled hypercorticism carries a poor prognosis, and treatment of Cushing’s syndrome is therefore necessary. Transsphenoidal surgery for pituitary ACTH-secreting microadenomas is curative in 70–80% when performed by a highly experienced surgeon, but long-term follow-up is required because these tumors may recur. Radiation therapy may be used when a surgical cure is not achieved (Chap. 179). Therapy of adrenal adenoma or carcinoma requires surgical excision; stress doses of glucocorticoids must be given pre- and postoperatively. Metastatic and unresectable adrenal carcinomas are treated with mitotane in doses gradually increased to 6 g/d in three or four divided doses. On occasion, debulking of lung carcinoma or resection of carcinoid tumors can result in remission of ectopic Cushing’s syndrome. If the source of ACTH cannot be resected, medical management with ketoconazole (600–1200 mg/d), metyrapone (2–3 g/d), or mitotane (2–3 mg/d) may relieve manifestations of cortisol excess. In some cases, bilateral total adrenalectomy is required to control hypercorticism. Pts with unresectable pituitary adenomas who have had bilateral adrenalectomy are at risk for Nelson’s syndrome (aggressive pituitary adenoma enlargement).
Aldosteronism is caused by hypersecretion of the adrenal mineralocorticoid aldosterone. Primary hyperaldosteronism refers to an adrenal cause and can be due to either an adrenal adenoma or bilateral adrenal hyperplasia. Rare causes include glucocorticoid-remediable hyperaldosteronism, some forms of congenital adrenal hyperplasia, and other disorders of true or apparent mineralocorticoid excess (see Table 342-3, HPIM-18). The term secondary hyperaldosteronism is used when an extraadrenal stimulus for renin secretion is present, as in renal artery stenosis, decompensated liver cirrhosis, or diuretic therapy.
Most pts with primary hyperaldosteronism have difficult to control hypertension (especially diastolic) and hypokalemia. Headaches are common. Edema is characteristically absent, unless congestive heart failure or renal disease is present. Hypokalemia, caused by urinary potassium losses, may cause muscle weakness, fatigue, and polyuria, although potassium levels may be normal in mild primary hyperaldosteronism. Metabolic alkalosis is a typical feature.
The diagnosis is suggested by treatment-resistant hypertension that is associated with persistent hypokalemia in a nonedematous pt who is not receiving potassium-wasting diuretics. In pts receiving potassium-wasting diuretics, the diuretic should be discontinued and potassium supplements should be administered for 1–2 weeks. If hypokalemia persists after supplementation, screening using a serum aldosterone and plasma renin activity should be performed. Ideally, antihypertensives should be stopped before testing, but that is often impractical. Aldosterone receptor antagonists, beta-adrenergic blockers, ACE inhibitors, and angiotensin receptor blockers interfere with testing and should be substituted for other antihypertensives if possible. A ratio of serum aldosterone (in ng/dL) to plasma renin activity (in ng/mL per hour) >30 and an absolute level of aldosterone >15 ng/dL suggest primary aldosteronism. Failure to suppress plasma aldosterone (to <5 ng/dL after 500 mL/h of normal saline × 4 h) or urinary aldosterone after saline or sodium loading (to <10 μg/d on day 3 of 200 mmol/d oral NaCl + fludrocortisone 0.2 mg twice daily × 3 days) confirms primary hyperaldosteronism. Caution should be used with sodium loading in a hypertensive pt. Localization should then be undertaken with a high-resolution CT scan of the adrenal glands. If the CT scan is negative, bilateral adrenal vein sampling may be required to diagnose a unilateral aldosterone-producing adenoma. Secondary hyperaldosteronism is associated with elevated plasma renin activity.
TREATMENT Hyperaldosteronism
Surgery can be curative in pts with adrenal adenoma but is not effective for adrenal hyperplasia, which is managed with sodium restriction and spironolactone (25–100 mg twice daily) or eplerenone (25–50 mg twice daily). The sodium channel blocker amiloride (5–10 mg twice a day) also can be used. Secondary hyperaldosteronism is treated with salt restriction and correction of the underlying cause.
See Chap. 186 for discussion of hirsutism and virilization.
Primary adrenal insufficiency is due to failure of the adrenal gland, whereas secondary adrenal insufficiency is due to failure of ACTH production or release.
Addison’s disease occurs when >90% of adrenal tissue is destroyed. The most common cause is autoimmune destruction (alone, or as part of type I or type II polyglandular autoimmune syndromes). Tuberculosis used to be the leading etiology. Other granulomatous diseases (histoplasmosis, coccidioidomycosis, cryptococcosis, sarcoidosis), bilateral adrenalectomy, bilateral tumor metastases, bilateral hemorrhage, CMV, HIV, amyloidosis, and congenital diseases (some types of congenital adrenal hypoplasia, adrenal hypoplasia congenita, and adrenoleukodystrophy) are additional etiologies.
Manifestations include fatigue, weakness, anorexia, nausea and vomiting, weight loss, abdominal pain, cutaneous and mucosal pigmentation, salt craving, hypotension (especially orthostatic), and, occasionally, hypoglycemia. Routine laboratory parameters may be normal, but typically serum Na is reduced and serum K increased. Extracellular fluid depletion accentuates hypotension. In secondary adrenal insufficiency, pigmentation is diminished and serum potassium is not elevated. Serum Na tends to be low because of hemodilution stemming from excess vasopressin secreted in the setting of cortisol deficiency.
The best screening test is the cortisol response 60 min after 250 μg ACTH (cosyntropin) IV or IM. Cortisol levels should exceed 18 μg/dL 30–60 min after the ACTH. If the response is abnormal, then primary and secondary deficiency may be distinguished by measurement of aldosterone from the same blood samples. In secondary, but not primary, adrenal insufficiency, the aldosterone increment from baseline will be normal (≥5 ng/dL). Furthermore, in primary adrenal insufficiency, plasma ACTH is elevated, whereas in secondary adrenal insufficiency, plasma ACTH values are low or inappropriately normal. Pts with recent onset or partial pituitary insufficiency may have a normal response to the rapid ACTH stimulation test. In these pts, alternative testing (metyrapone test or insulin tolerance testing) can be used for diagnosis.
TREATMENT Addison’s Disease
Hydrocortisone, at 15–25 mg/d divided into ⅔ in the morning and ⅓ in the afternoon, is the mainstay of glucocorticoid replacement. Some pts benefit from doses administered three times daily, and other glucocorticoids may be given at equivalent doses. Mineralocorticoid supplementation is usually needed for primary adrenal insufficiency, with administration of 0.05–0.1 mg fludrocortisone PO qd and maintenance of adequate Na intake. Doses should be titrated to normalize Na and K levels and to maintain normal blood pressure without postural changes. Measurement of plasma renin levels may also be useful in titrating the dose. Mineralocorticoid replacement is not needed in pts with secondary adrenal insufficiency. All pts with adrenal insufficiency should be instructed in the parenteral self-administration of steroids and should be registered with a medical alert system. During periods of intercurrent illness, the dose of hydrocortisone should be doubled. During adrenal crisis, high-dose hydrocortisone (10 mg/h continuous IV or 100-mg bolus IV three times a day) should be administered along with normal saline. Thereafter, if the pt is improving and is afebrile, the dose can be tapered by 20–30% daily to usual replacement doses.
Isolated aldosterone deficiency accompanied by normal cortisol production occurs with hyporeninism, as an inherited aldosterone synthase deficiency, postoperatively following removal of aldosterone-secreting adenomas (transient), and during protracted heparin therapy. Hyporeninemic hypoaldosteronism is seen most commonly in adults with diabetes mellitus and mild renal failure; it is characterized by mild to moderate hyperkalemia. This is usually a benign condition that can be managed by observation. If needed, oral fludrocortisone (0.05–0.15 mg/d PO) restores electrolyte balance if salt intake is adequate. In pts with hypertension, mild renal insufficiency, or congestive heart failure, an alternative approach is to reduce salt intake and to administer furosemide.
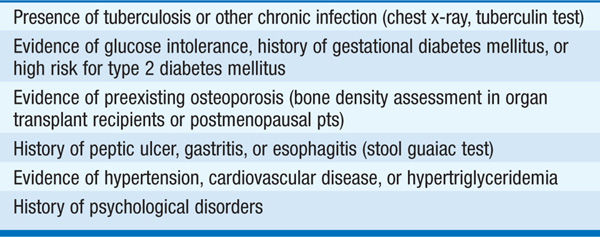
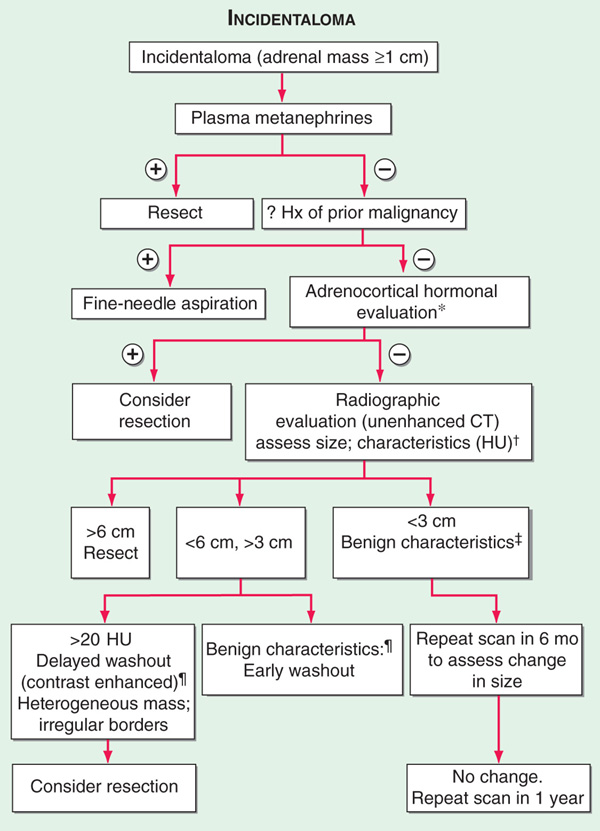
Adrenal masses are common findings on abdominal CT or MRI scans (1–7% prevalence with increasing age). The majority (70–80%) of such “incidentalomas” are clinically nonfunctional, and the probability of an adrenal carcinoma is low (<0.01%). Genetic syndromes such as MEN 1, MEN 2, Carney syndrome, and McCune-Albright syndrome are all associated with adrenal masses. The first step in evaluation is to determine the functional status by measurement of plasma free metanephrines to screen for pheochromocytoma (Fig. 182-1). In a pt with a known extraadrenal malignancy, there is a 30–50% chance that the incidentaloma is a metastasis. Additional hormonal evaluation should include overnight 1-mg dexamethasone suppression testing in all pts, plasma renin activity/aldosterone ratio in hypertensives, DHEAS in women with signs of androgen excess, and estradiol in males with feminization. Fine-needle aspiration is rarely indicated and absolutely contraindicated if a pheochromocytoma is suspected. Adrenocortical cancer is suggested by large size (>4–6 cm), irregular margins, tumor inhomogeneity, soft tissue calcifications, and high unenhanced CT attenuation values (>10 HU).
FIGURE 182-1 Incidentaloma. *Adrenocortical hormonal evaluation: Dexamethasone suppression test in all pts; plasma renin activity/aldosterone ratio for hypertensives; sex steroid (DHEA sulfate, estradiol) for clinical signs in females and males, respectively. †Hounsfield units (HU): a measurement of x-ray attenuation or lipid content of neoplasms. A lipid-rich mass (<10 HU) is diagnostic of a benign cortical adenoma.  Benign characteristics: homogeneous mass, smooth borders,
Benign characteristics: homogeneous mass, smooth borders, 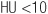 . §Benign adrenal adenomas are also characterized by earlier washout of contrast enhancement than other neoplasms.
. §Benign adrenal adenomas are also characterized by earlier washout of contrast enhancement than other neoplasms.
Glucocorticoids are pharmacologic agents used for a variety of disorders such as asthma, rheumatoid arthritis, and psoriasis. The almost certain development of complications (weight gain, hypertension, Cushingoid facies, diabetes mellitus, osteoporosis, myopathy, increased intraocular pressure, ischemic bone necrosis, infection, and hyperlipidemia) must be weighed against the potential therapeutic benefits of glucocorticoid therapy. These side effects can be minimized by a careful choice of steroid preparations (Table 182-1), dose minimization, and alternate-day or interrupted therapy; the use of topical steroids, i.e., inhaled, intranasal, or dermal, whenever possible; the judicious use of nonsteroid therapies; monitoring of caloric intake; and instituting measures to minimize bone loss. Pts should be evaluated for the risk of complications before the initiation of glucocorticoid therapy (Table 182-2). Higher doses of glucocorticoids may be required during periods of stress, since the hypothalamo-pituitary-adrenal axis is suppressed and the adrenal gland may atrophy in the setting of exogenous glucocorticoids. In addition, following long-term use, glucocorticoids should be tapered with the dual goals of allowing the pituitary-adrenal axis to recover and the avoidance of underlying disease flare.
TABLE 182-1 GLUCOCORTICOID PREPARATIONS
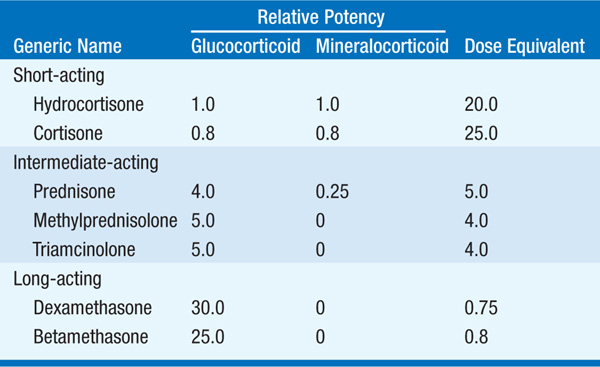
TABLE 182-2 A CHECKLIST FOR USE PRIOR TO THE ADMINISTRATION OF GLUCOCORTICOIDS IN PHARMACOLOGIC DOSES