
The anterior pituitary is often referred to as the “master gland” because, together with the hypothalamus, it orchestrates the complex regulatory functions of multiple other glands (Fig. 179-1). The anterior pituitary produces six major hormones: (1) prolactin (PRL); (2) growth hormone (GH); (3) adrenocorticotropin hormone (ACTH); (4) luteinizing hormone (LH); (5) follicle-stimulating hormone (FSH); and (6) thyroid-stimulating hormone (TSH). Pituitary hormones are secreted in a pulsatile manner, reflecting intermittent stimulation by specific hypothalamic-releasing factors. Each of these pituitary hormones elicits specific responses in peripheral target glands. The hormonal products of these peripheral glands, in turn, exert feedback control at the level of the hypothalamus and pituitary to modulate pituitary function. Disorders of the pituitary include neoplasms or other lesions (granulomas, hemorrhage) that lead to mass effects and clinical syndromes due to excess or deficiency of one or more pituitary hormones.
FIGURE 179-1 Diagram of pituitary axes. Hypothalamic hormones regulate anterior pituitary tropic hormones that, in turn, determine target gland secretion. Peripheral hormones feed back to regulate hypothalamic and pituitary hormones. GHRH, growth hormone-releasing hormone; SRIF, Somatostatin, somatotropin release-inhibiting factor; TRH, thyrotropin-releasing hormone; for other abbreviations, see text.
Pituitary adenomas are benign monoclonal tumors that arise from one of the five anterior pituitary cell types and may cause clinical effects from either overproduction of a pituitary hormone or compressive/destructive effects on surrounding structures, including the hypothalamus, pituitary, optic chiasm, and cavernous sinus. About one-third of all adenomas are clinically nonfunctioning and produce no distinct clinical hypersecretory syndrome. Among hormonally functioning neoplasms, tumors secreting prolactin are most common (~50%); they have a greater prevalence in women than in men. GH- and ACTH-secreting tumors each account for about 10–15% of functioning pituitary tumors. Adenomas are classified as microadenomas (<10 mm) or macroadenomas (≥10 mm). Pituitary adenomas (especially PRL- and GH-producing tumors) may be part of genetic familial syndromes such as MEN 1, Carney syndrome, or mutant aryl hydrocarbon receptor inhibitor protein (AIP) syndrome. Other entities that can present as a sellar mass include craniopharyngiomas, Rathke’s cleft cysts, sella chordomas, meningiomas, pituitary metastases, gliomas, and granulomatous disease (e.g., histiocytosis X, sarcoidosis).
Symptoms from mass effects include headache; visual loss through compression of the optic chiasm superiorly (classically a bitemporal hemianopia); and diplopia, ptosis, ophthalmoplegia, and decreased facial sensation from cranial nerve compression laterally. Pituitary stalk compression from the tumor may also result in mild hyperprolactinemia. Symptoms of hypopituitarism or hormonal excess may be present as well (see below).
Pituitary apoplexy, typically resulting from hemorrhage into a pre-existing adenoma or post-partum as Sheehan’s syndrome, is an endocrine emergency that typically presents with features that include severe headache, bilateral visual changes, ophthalmoplegia, and, in severe cases, cardiovascular collapse and loss of consciousness. It may result in hypotension, severe hypoglycemia, CNS hemorrhage, and death. Pts with no evident visual loss or impaired consciousness can usually be observed and managed conservatively with high-dose glucocorticoids; surgical decompression should be considered when visual or neurologic symptoms/signs are present.
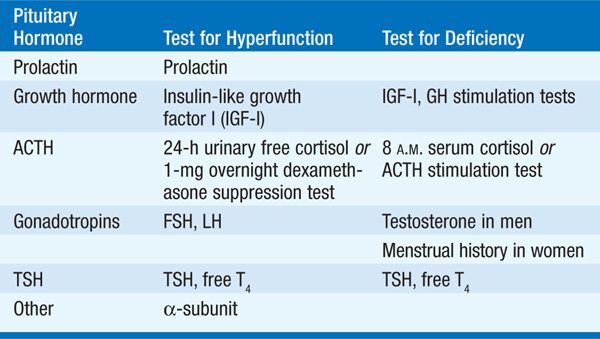
Sagittal and coronal T1-weighted MRI images with specific pituitary cuts should be obtained before and after administration of gadolinium. In pts with lesions close to the optic chiasm, visual field assessment that uses perimetry techniques should be performed. Initial hormonal evaluation is listed in Table 179-1.
TABLE 179-1 INITIAL HORMONAL EVALUATION OF PITUITARY ADENOMAS
In pituitary apoplexy, CT or MRI of the pituitary may reveal signs of sellar hemorrhage, with deviation of the pituitary stalk and compression of pituitary tissue.
TREATMENT Pituitary Tumors
Pituitary surgery is indicated for mass lesions that impinge on surrounding structures or to correct hormonal hypersecretion, except in the case of prolactinoma, where medical treatment is usually effective (see below). Transsphenoidal surgery, rather than transfrontal resection, is the desired surgical approach for most pts. The goal is selective resection of the pituitary mass lesion without damage to the normal pituitary tissue, to decrease the likelihood of hypopituitarism. Transient or permanent diabetes insipidus, hypopituitarism, CSF rhinorrhea, visual loss, and oculomotor palsy may occur postoperatively. Tumor invasion outside of the sella is rarely amenable to surgical cure, but debulking procedures may relieve tumor mass effects and reduce hormonal hypersecretion. Radiation may be used as an adjunct to surgery, but efficacy is delayed and >50% of pts develop hormonal deficiencies within 10 years, usually due to hypothalamic damage. GH-, and TSH-secreting tumors may also be amenable to medical therapy; in PRL-secreting tumors medical therapy is the initial treatment of choice.
Prolactin is unique among the pituitary hormones in that the predominant central control mechanism is inhibitory, reflecting dopamine-mediated suppression of prolactin release. Prolactin acts to induce and maintain lactation and decrease reproductive function and drive [via suppression of gonadotropin-releasing hormone (GnRH), gonadotropins, and gonadal steroidogenesis].
Physiologic elevation of prolactin occurs in pregnancy and lactation. Otherwise, prolactin-secreting pituitary adenomas (prolactinomas) are the most common cause of prolactin levels >100 μg/L. Less pronounced hyperprolactinemia is commonly caused by medications [risperidone, chlorpromazine, perphenazine, haloperidol, metoclopramide, opiates, H2 antagonists, amitriptyline, selective serotonin reuptake inhibitors (SSRIs), verapamil, estrogens], pituitary stalk damage (tumors, lymphocytic hypophysitis, granulomas, trauma, irradiation), primary hypothyroidism, or renal failure. Nipple stimulation may also cause acute prolactin increases.
In women, amenorrhea, galactorrhea, and infertility are the hallmarks of hyperprolactinemia. In men, symptoms of hypogonadism (Chap. 185) or mass effects are the usual presenting symptoms, and galactorrhea is rare.
Fasting, morning prolactin levels should be measured; when clinical suspicion is high, measurement of levels on several different occasions may be required. If hyperprolactinemia is present, nonneoplastic causes should be excluded (e.g., pregnancy test, hypothyroidism, medications).
If the pt is taking a medication that is known to cause hyperprolactinemia, the drug should be withdrawn, if possible. A pituitary MRI should be performed if the underlying cause of prolactin elevation is unknown. Resection of hypothalamic or sellar mass lesions can reverse hyperprolactinemia due to stalk compression. Medical therapy with a dopamine agonist is indicated in microprolactinomas for control of symptomatic galactorrhea, for restoration of gonadal function, or when fertility is desired. Alternatively, estrogen replacement may be indicated if fertility is not desired, but tumor size should be carefully monitored. Dopamine agonist therapy for macro-prolactinomas generally results in both adenoma shrinkage and reduction of prolactin levels. Cabergoline (initial dose 0.5 mg a week, usual dose 0.5–1 mg twice a week) or bromocriptine (initial dose 0.625–1.25 mg qhs, usual dose 2.5 PO three times a day) are the two most frequently used dopamine agonists. Cabergoline is the more effective and better-tolerated drug. These medications should initially be taken at bedtime with food, followed by gradual dose increases, to reduce the side effects of nausea and postural hypotension. Other side effects include constipation, nasal stuffiness, dry mouth, nightmares, insomnia, or vertigo; decreasing the dose usually alleviates these symptoms. Dopamine agonists may also precipitate or worsen underlying psychiatric conditions. Cabergoline in high doses can cause cardiac valvular disease. At doses typically used for prolactinoma treatment the risk for valvulopathy is small. Nevertheless, cardiac echo-cardiography should be performed before and 6–12 months after starting cabergoline therapy. In pts with microadenomas successfully treated (normal PRL, full tumors shrinkage), therapy may be withdrawn after 2 years, followed by careful monitoring for tumor recurrence. Spontaneous remission of microadenomas, presumably caused by infarction, occurs in some pts. Surgical debulking may be required for macroprolactinomas that do not adequately respond to medical therapy.
Women with microprolactinomas who become pregnant should discontinue dopaminergic therapy, as the risk for significant tumor growth during pregnancy is low. In those with macroprolactinomas, visual field testing should be performed at each trimester. A pituitary MRI should be performed if severe headache and/or visual defects occur.
GH hypersecretion is primarily the result of pituitary somatotrope adenomas, mostly sporadic, but also in conjunction with MEN 1, Carney syndrome, McCune-Albright syndrome, and familial AIP mutations. Extrapituitary causes of acromegaly (ectopic GHRH production) are very rare.
The peak occurrence of acromegaly is at age 40–45. In children, GH hypersecretion prior to long bone epiphyseal closure results in gigantism. The presentation of acromegaly in adults is usually indolent, and diagnosis is typically delayed by up to a decade. Pts may note a change in facial features, widened teeth spacing, deepening of the voice, snoring, increased shoe or glove size, ring tightening, hyperhidrosis, oily skin, arthropathy, and carpal tunnel syndrome. Frontal bossing, mandibular enlargement with prognathism, macroglossia, an enlarged thyroid, skin tags, thick heel pads, and hypertension may be present on examination. Associated conditions include cardiomyopathy, left ventricular hypertrophy, diastolic dysfunction, sleep apnea, glucose intolerance, diabetes mellitus, colon polyps, and colonic malignancy. Overall mortality is increased approximately threefold.
Insulin-like growth factor type I (IGF-I) levels are a useful screening measure, with elevation suggesting acromegaly. Due to the pulsatility of GH, measurement of a single random GH level is not useful for screening. The diagnosis of acromegaly is confirmed by demonstrating the failure of GH suppression to <1 μg/L within 1–2 h of a 75-g oral glucose load. MRI of the pituitary usually reveals a macroadenoma.
TREATMENT Acromegaly
The primary treatment modality for acromegaly is transsphenoidal surgery. GH levels are not normalized by surgery alone in many pts with macroadenomas; in those, somatostatin analogues provide adjunctive medical therapy that suppresses GH secretion with modest to no effect on tumor size. Octreotide (50 μg SC three times a day) is used for initial therapy to determine response. Once a positive response and tolerance of side effects (nausea, abdominal discomfort, diarrhea, flatulence) is established, pts are changed to long-acting depot formulations (octreotide LAR 20–30 mg IM every 2–4 weeks or lanreotide autogel 90–120 mg IM once a month). Dopamine agonists (bromocriptine, cabergoline) can be used as adjunctive therapy but are generally not very effective. The GH receptor antagonist pegvisomant (10–30 mg SC daily) can be added in pts who do not respond to somatostatin analogues. Pegvisomant is highly effective in lowering IGF-I levels but does not lower GH levels or decrease tumor size. Pituitary irradiation may also be required as adjuvant therapy but has a slow therapeutic onset and a high rate of late hypopituitarism.
These tumors are the most common type of pituitary neoplasm and usually present with symptoms of one or more hormonal deficiencies or mass effect. They typically produce small amounts of intact gonadotropins (usually FSH) as well as uncombined α-subunit and LHβ and FSHβ subunits. Surgery is indicated for mass effects or hypopituitarism; asymptomatic small adenomas may be followed with regular MRI and visual field testing. Diagnosis is based on immunohistochemical analysis of resected tumor tissue. Medical therapy is usually ineffective in shrinking these tumors.
TSH-producing adenomas are rare but often large and locally invasive when they occur. Pts present with goiter and hyperthyroidism, and/or sella mass effects. Diagnosis is based on elevated serum free T4 levels in the setting of inappropriately normal or high TSH secretion and MRI evidence of a pituitary adenoma. Surgery is indicated and is usually followed by somatostatin analogue therapy to treat residual tumor. Somatostatin analogue therapy leads to normalization of TSH and euthyroidism in most and tumor shrinkage in 50–75% of pts. If necessary, thyroid ablation or antithyroid drugs can be used to reduce thyroid hormone levels.
A variety of disorders may cause deficiencies of one or more pituitary hormones. These disorders may be genetic, congenital, traumatic (pituitary surgery, cranial irradiation, head injury), neoplastic (large pituitary adenoma, parasellar mass, craniopharyngioma, metastases, meningioma), infiltrative (hemochromatosis, lymphocytic hypophysitis, sarcoidosis, histiocytosis X), vascular (pituitary apoplexy, postpartum necrosis, sickle cell disease), or infectious (tuberculous, fungal, parasitic). The most common cause of hypopituitarism is neoplastic in origin (macroadenomatous destruction, or following hypophysectomy or radiation therapy). Pituitary hormone failure due to compression, destruction, or radiation therapy typically follows a sequential pattern: GH>FSH>LH>TSH>ACTH. Genetic causes of hypopituitarism may affect several hormones (e.g., pituitary dysplasia, PROP-1 and PIT-1 mutations) or be restricted to single pituitary hormones or axes (e.g., isolated GH deficiency, Kallmann syndrome, isolated ACTH deficiency). Hypopituitarism following cranial irradiation develops over 5–15 years. Varying degrees of partial to complete hormone deficiencies occur during evolution of pituitary destruction.
Each hormone deficiency is associated with specific findings:
• GH: growth disorders in children; increased intraabdominal fat, reduced lean body mass, hyperlipidemia, reduced bone mineral density, decreased stamina, and social isolation in adults
• FSH/LH: menstrual disorders and infertility in women (Chap. 186); hypogonadism in men (Chap. 185)
• ACTH: features of hypocortisolism (Chap. 182) without mineralocorticoid deficiency
• TSH: growth retardation in children, features of hypothyroidism in children and adults (Chap. 181)
• PRL: failure to lactate post-partum
Biochemical diagnosis of pituitary insufficiency is made by demonstrating low or inappropriately normal levels of pituitary hormones in the setting of low target hormone levels. Initial testing should include an 8 A.M. cortisol level, TSH and free T4, IGF-I, testosterone in men, assessment of menstrual cycles in women, and prolactin level. Provocative tests are required for definitive diagnosis of GH and ACTH deficiency. Adult GH deficiency is diagnosed by demonstrating a subnormal GH response to a standard provocative test (insulin tolerance test, L-arginine + GHRH). Acute ACTH deficiency may be diagnosed by a subnormal response in an insulin tolerance test, metyrapone test, or corticotropin-releasing hormone (CRH) stimulation test. Standard ACTH (cosyntropin) stimulation tests may be normal in acute ACTH deficiency; with adrenal atrophy, the cortisol response to cosyntropin is blunted.
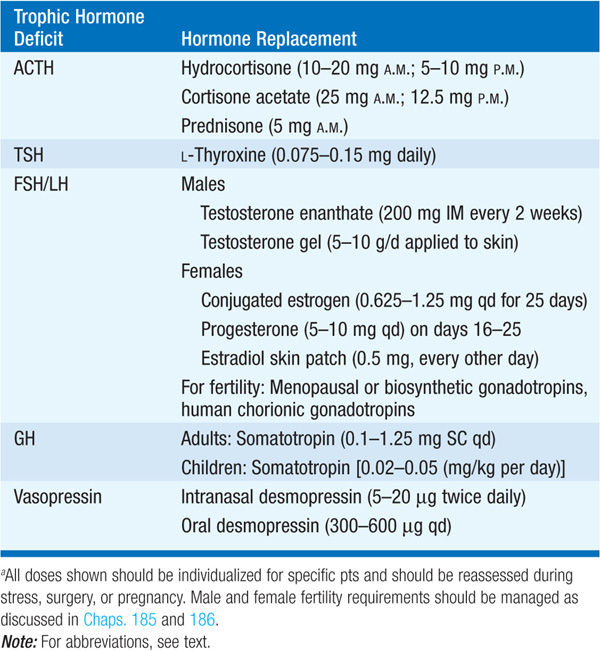
Hormonal replacement should aim to mimic physiologic hormone production. Effective dose schedules are outlined in Table 179-2. Doses should be individualized, particularly for GH, glucocorticoids and L-thyroxine. GH therapy, particularly when excessive, may be associated with fluid retention, joint pain, and carpal tunnel syndrome. Glucocorticoid replacement should always precede L-thyroxine therapy to avoid precipitation of adrenal crisis. Pts requiring glucocorticoid replacement should wear a medical alert bracelet and should be instructed to take additional doses during stressful events such as acute illness, dental procedures, trauma, and acute hospitalization.
TABLE 179-2 HORMONE REPLACEMENT THERAPY FOR ADULT HYPOPITUITARISMa

For a more detailed discussion, see Melmed S, Jameson JL: Disorders of the Anterior Pituitary and Hypothalamus, Chap. 339, p. 2876, in HPIM-18.