Chapter 41
Reward, Motivation, and Addiction
Reward and Motivation
Motivation is the process that mediates goal-directed responses to changes in the external or internal environment. Early explanations of motivated behavior focused on reductions of drives or needs, such as for sodium (salt) or water. Such purely homeostatic frameworks for motivation could plausibly account for ingestive and thermoregulatory behaviors and possibly even the drug-seeking behavior generated by the state of drug-withdrawal. Many behaviors, however, are triggered by external incentives which are perceived as appetizing, rewarding, or aversive. Thus, motivated behavior is determined both by biological needs and by learned associations between environmental cues and the value associated with the outcome of particular actions.
Behavioral goals, such as food or a sexual partner, are often not immediately available but must be searched for. Motivated behavior thus comprises both appetitive behavior and consummatory behaviors. Appetitive behavior, such as foraging for food or seeking a mate, tends to be adaptive, flexible, and learned through experience. In contrast, consummatory behavior, such as ingestion and copulation, tends to be reflexive or stereotyped and acquired early in development. Appetitive behavior occurs in conjunction with endocrine and autonomic responses, such as salivation and insulin secretion (see Chapter 34), which prepare the animal for efficient consummatory behavior.
The complexity of motivated behavior (Fig. 41.1), from the selection of learned voluntary actions to reflexive control of consummatory behavior, involves the coordination of several levels of neural control, including the prefrontal and cingulate cortices, the amygdala, the basal ganglia, the hypothalamus, and neuromodulatory nuclei in the brainstem.

Figure 41.1 Theory of incentive-motivation, as applied to feeding. Note that drive (internal stimuli) and food (external stimuli) separately affect central motivation to feed. Also, incentive (i.e., food) and discriminative (i.e., sensorimotor) influences exert their effects separately to control consummatory behavior (when food is contacted) and appetitive behavior (when food remains remote).
Modified with permission from Robbins (1986).
Conditioning and Learning
In the absence of immediate goals, such as food or sex, animals typically use what they have learned from past experiences to predict the outcome of particular actions. Two predominant forms of learning are critical for motivated behavior: classical conditioning (i.e., Pavlovian conditioning) and operant conditioning (the learned association between goal-directed, instrumental behaviors and their outcomes).
In operant conditioning, outcomes are used to strengthen preceding behaviors via either positive or negative reinforcement. Positive reinforcement increases the likelihood of a particular behavior by having an attractive stimulus, termed a positive reinforcer, follow a given behavior. Conversely, negative reinforcement strengthens a behavior by removing an aversive stimulus, or a negative reinforcer, after that behavior is performed. The addition of a positive reinforcer is thought to be rewarding for an animal, whereas the removal of a negative reinforcer is believed to be relieving. (Negative reinforcers are not to be confused with punishers, which serve to decrease, rather than increase, the probability of a response). Reinforcers can be stimuli such as food or drugs which are valuable or aversive in themselves (primary reinforcers), or stimuli that become valuable or aversive by association with primary reinforcers (conditioned reinforcers).
Because motivation depends on the contingency between a particular behavior and a reinforcer, as well as the strength of the reinforcer, researchers can employ operant conditioning paradigms to study an animal’s level of motivation. In the self-administration paradigm, animals learn arbitrary instrumental actions, such as a lever press, to gain access to positive reinforcers such as food and drugs (Fig. 41.2). Depending on the value of the reinforcer and the likelihood that their behavior will result in the reinforcer, animals will vary their level of activity (e.g., the amount they lever press). Thus, self-administration is a behavioral paradigm that measures an animal’s degree of motivation by assessing its capacity to work for a goal.

Figure 41.2 Intravenous self-administration by rats. (Top) Drawing illustrating the setup for intravenous self-administration of cocaine by rats. (Bottom) Event records for different unit doses of cocaine show a dose-response relationship relating dose of cocaine to number of infusions. Rats implanted with intravenous catheters and trained to self-administer cocaine with limited access (3 h per day) show stable and regular drug intake over each daily session. No obvious tolerance or dependence develops. Rats generally are maintained on a fixed-ratio (FR) schedule of drug infusion, such as FR-1 or FR-5. In an FR-1 schedule, one lever press is required to deliver an intravenous infusion of cocaine; in an FR-5 schedule, five lever presses are required to deliver an infusion of cocaine, and so on. With an FR schedule, rats appear to regulate the amount of drug self-administered. Lowering the dose from the training level of 0.75 mg/kg per injection increases the number of self-administered infusions. Raising the unit dose decreases the number of infusions. Pretreatment with the dopamine receptor antagonist SCH 23390 also increases the number of self-administered infusions.
Reprinted with permission from Caine et al. (1993).
Another measure of motivation involves assessing whether animals choose to execute particular behaviors when confronted with alternative choices. A commonly used experimental paradigm is conditioned place preference, in which a choice is made between two locations, only one of which is linked to a positive reinforcer (such as food or a drug). Following several learning trials, a test trial is conducted in the absence of the goal itself. In the test, the preference for that place (as measured by the time spent there) relative to another place without that association is taken as an index of the attractiveness, or incentive properties, of the goal.
The Hypothalamus Plays a Role in Motivation
Inspired by the discovery of brainstem centers for fundamental processes such as respiration, early investigators searched for a motivation center, conceptualized as a neural locus that could integrate internal and external stimuli and thereby coordinate neural circuits to produce adaptive behavioral, endocrine, and autonomic responses. The hypothalamus (Chapter 33) was thought to be ideal for such integrative functions, given its rich blood supply and exposure to cerebrospinal fluid (and thus access to a variety of chemical signals and the capacity also to deliver them), its control over the pituitary, and its neural connections (through brainstem and spinal centers) with visceral afferent information, autonomic and somatic motor outflow.
The hypothalamus is critical for a diverse range of motivated behaviors, including drinking, thermoregulation, aggression, and sex (see Chapter 33). For example, thermoregulatory behavior, such as reflexive shivering, panting, and grooming, is impaired by lesions of the preoptic region of the hypothalamus and is elicited by cooling or warming of this region (Satinoff, 1982). Defensive (affective) aggression and accompanying autonomic changes, such as piloerection, are produced by electrical stimulation of the ventromedial hypothalamus, whereas predatory (“quiet”) aggression is produced by electrical stimulation of the lateral hypothalamus (Flynn, Vanegas, Foote, & Edwards, 1970).
The role of the hypothalamus in sexual motivation has been particularly well studied. Some hypothalamic nuclei contain high concentrations of steroid receptors (e.g., for estradiol and testosterone), especially the medial preoptic area and ventromedial hypothalamic nucleus. In male rats, copulatory behavior is abolished by medial preoptic area lesions (Heimer & Larsson, 1966–1967), whereas electrical stimulation of this region elicits copulation. In female rats, lordosis is blocked (and aggression toward males increased) by lesions of the ventromedial hypothalamic nucleus. Lordosis is also induced by electrical stimulation of this region (see Pfaff, 1982). Most of these responses depend on hormones. In ovariectomized females, proceptive (“ear wiggling,” hopping, and darting) and receptive (lordosis) behaviors can be elicited by implanting estradiol into the ventromedial hypothalamic nucleus. Similarly, the sexual behavior of castrated males can be restored by implanting testosterone into the medial preoptic area. This hormone dependence underlines the interaction between external (environmental) and internal (plasma steroids) states in inducing motivated responses. Hormones influence the organization of sexually dimorphic brain development (e.g., development of the sexually dimorphic nuclei in the preoptic region), as well as sexually dimorphic behavior. Maternal behavior also is dependent on hormones and is impaired by lesions of the medial preoptic area.
Studies of the role of the hypothalamus in motivation that focus on consummatory, rather than learned instrumental, behavior can yield misleading conclusions. For example, when challenged with thermal stress, rats with preoptic lesions do not exhibit adaptive responses such as reflexive shivering, adjustments in food intake, nest building, or alterations in locomotion. They can, however, achieve thermoregulation by learning to press a lever for hot or cool air and remain motivated to do so (Carlisle, 1969). Similarly, male rats with preoptic area lesions exhibit locomotor excitement in the presence of a receptive female and will press levers to gain access to the female but cannot perform the consummatory behavior of copulation. Male monkeys with preoptic lesions continue to show motivated behavior (e.g., they will masturbate in the presence of females) but are unable to mount and intromit.
This dissociation between consummatory and instrumental behaviors is important because damage to the amygdala is known to produce the opposite pattern of effects—unimpaired mounting but reduced instrumental behavior (Everitt, 1990). These examples show that hypothalamic circuits predominantly control consummatory behaviors rather than learned, goal-directed instrumental behaviors. Behavioral flexibility, which requires learning, depends on the recruitment of additional neural systems, including the amygdala and prefrontal cortex, as described later (see Box 41.1).
Box 41.1 Logic of Methodology Used in Behavioral Neuroscience
Theorists (e.g., Hoebel, 1974) have defined principles for identifying the involvement of particular brain regions in motivational (and other behavioral) functions. For example, electrophysiological, neurochemical, or metabolic activity may be observed in a structure, correlated with a behavioral response of interest, which suggests an involvement of that structure with the function under study. However, this correlative relationship does not establish whether the functioning of that region is causally important for the behavior. This issue may be addressed by an intervention that disrupts (impairs or improves) the function under study. For example, if the function is lost following selective lesioning, this suggests that the brain structure is implicated in that function. Similarly, if the function is enhanced following a boosting of neurotransmission in that region (e.g., via mild levels of electrical or chemical stimulation through implanted electrodes or cannulae), this is similarly converging evidence of a causative role. However, it is important to realize that the use of these techniques is each associated with interpretative problems, and their effects must be critically appraised. For example, the designation of the lateral hypothalamic region as a “feeding center” was based on the following: (1) electrolytic lesions produced profound aphagia (abstention from eating); (2) electrical stimulation of the brain through electrodes implanted in the lateral hypothalamus induced eating in sated rats (Hoebel, 1974); (3) infusions of the neurotransmitter norepinephrine into the hypothalamus also induced eating in sated rats (Leibowitz, 1980); and (4) electrophysiological recording showed that when monkeys were deprived of food, individual neurons in the lateral hypothalamus were sensitive to the sight and taste of food (Rolls, 1985). While many of these experimental observations have remained valid, subsequent evidence has suggested that the lateral hypothalamus has a much more specific role in feeding motivation, as a result of using improved techniques (see main text). Moreover, many of the effects associated with diminishing and increasing activity within the lateral hypothalamus have been shown subsequently to depend upon coincidental influences on axons within the medial forebrain bundle which merely pass through the lateral hypothalamus (e.g., between midbrain and striatum or between prefrontal cortex and brainstem) and had little to do functionally with the lateral hypothalamus itself. Only through the use of several neurobiological techniques providing converging evidence can the relationships between brain and behavior become more certain.
Lesions of the Lateral Hypothalamus Disrupt Motivated Behavior and Dopaminergic Axons
Electrolytic lesions of the lateral hypothalamus cause dramatic deficits in food intake which were originally interpreted as a deficit in motivation. However, such lesions destroy far more than the lateral hypothalamic region itself, since they also interrupt ascending and descending fibers in the medial forebrain bundle, including dopaminergic axons (Fig. 41.3). Four major systems that are responsible for conveying dopamine to specific regions of the forebrain have been identified. The diencephalic incertohypothalamic dopamine system provides dopaminergic terminals to the hypothalamus, including the medial preoptic area responsible for sexual behavior (Pfaus, 2009). The tuberoinfundibular dopamine system controls hormone release from the anterior pituitary gland. The mesolimbic dopamine system conveys dopamine from neurons in the midbrain ventral tegmental area (VTA) to “limbic” regions of the forebrain, including the nucleus accumbens of the ventral striatum, amygdala, and medial prefrontal cortex. The nigrostriatal dopamine system originates in the substantia nigra and provides dopaminergic innervation to the caudate and putamen of the dorsal striatum.

Figure 41.3 Ascending monoamine neurotransmitter systems. The figure shows schematic sagittal (A–D) and coronal (E) sections through the lateral hypothalamus of a rat brain. (A) Origin and distribution of central noradrenergic pathways. Note noradrenergic cell groups A1–A7, including the locus coeruleus (A6). DNAB, dorsal noradrenergic ascending bundle; VNAB, ventral noradrenergic ascending bundle. (B) Origin and distribution of central dopamine pathways. Note dopaminergic cell groups A8–A10. (C) Origin and distribution of central cholinergic pathways. Note rostral cell groups. NBM, nucleus basalis magnocellularis (Meynert in primates); MS, medial septum; VDBB, vertical limb nucleus of the diagonal band of Broca; HDBB, horizontal limb nucleus of the diagonal band of Broca. (D) Origin and distribution of central serotonergic pathways. Note cell groups in the raphe nucleus, B4–B9. MFB, medial forebrain bundle; PFC, prefrontal cortex; VS, ventral striatum; DS, dorsal striatum. Based on Robbins and Everitt (1995). (E) Schematic coronal section through the rat brain at the level of the ventromedial hypothalamus. LH lesions could disrupt various ascending monoaminergic fibers. CC, corpus callosum; CP, caudate-putamen; DMH, dorsomedial hypothalamus; IC, internal capsule; GP, globus pallidus; PVH, paraventricular hypothalamus; LH, lateral hypothalamus; FX, fornix; VMH, ventromedial hypothalamus.
Modified from Robbins (1986), with permission.
Consistent with a critical role for dopamine in motivated behavior, injections of the catecholamine-selective neurotoxin 6-hydroxydopamine (6-OHDA) into the vicinity of the lateral hypothalamus reproduced many of the effects of electrolytic lesions (Ungerstedt, 1971a). The injected rats suffered large depletions (>90%) of dopamine from the striatum. They were cataleptic and had difficulty initiating head, limb, axial, and oral movements. Further experiments showed that the syndrome could be produced by injection of 6-OHDA at any of several points along the nigrostriatal dopamine pathway. Thus, dopamine depletion at sites far from the lateral hypothalamus reproduced much of the lateral hypothalamic syndrome. 6-OHDA-induced lesions of the mesolimbic pathway minimally affected ingestion but did disrupt other behaviors that can accompany feeding, including locomotion and exploration (Koob, Riley, Smith, & Robbins, 1978). These findings demonstrate that dopamine depletion impairs both consummatory and appetitive behaviors. Direct axonal projections from orexin neurons in the hypothalamus to dopamine neurons in the VTA provide a functional link between the homeostatic and incentive motivational components of ingestive behavior.
Dopamine Is Critical for Motivated Behavior
What is the nature of the contribution by dopaminergic neurons to motivation? Clues to answering this question were originally provided by experiments in which rats received unilateral 6-OHDA lesions of the nigrostriatal dopamine circuit. The rats rotated toward the side of the lesion following treatment with a dopamine-releasing drug, such as D-amphetamine (Ungerstedt, 1971b). Presumably, this rotation occurred because more dopamine is released on the intact side than on the lesioned side of the brain and because any output pathway from the striatum influences movements in the opposite side of the body. Of even greater interest, the direct dopamine receptor agonist apomorphine caused the rats to turn away from the side of the lesion, an effect explained by actions at supersensative dopamine receptors on the lesioned side.
Unilateral lesions of either the lateral hypothalamus or the nigrostriatal dopamine pathway produce polymodal sensorimotor neglect, measured by responses to contralateral somatic, auditory, and olfactory stimuli (Marshall & Teitelbaum, 1977). Although this neglect was originally attributed to sensory inattention, further analysis suggests that the fundamental deficit is a failure in initiating responses rather than in attention (see Box 41.2). Animals with lesions of the nigrostriatal dopamine system retain the capacity for movement but have great difficulty in initiating movement. Similarly, patients suffering from Parkinson’s disease have severe deficits in nigrostriatal dopamine with concommitant impairments in initiating movement. These considerations suggest that nigrostriatal dopamine is critical for activating or invigorating behavioral responses.
Box 41.2 Separation of Sensory and Response Factors in the Dopaminergic Syndrome of Neglect
Sensory and motor aspects of the neglect syndrome were separated in the following way (Carli et al., 1985). Rats were trained to detect brief flashes of light presented to either side of the head. In one case, they were trained to respond where the light was. In the alternate case, rats were trained to respond in a location away from the side of the light. Rats with dopamine depleted from one side of the striatum could not respond properly toward the contralateral side, whether or not the stimulus was presented on that side. Measurement of reaction time showed that the problem was in the initiation, rather than completion, of the movement. Consistent with these data, dopamine-depleted rats could detect a contralateral stimulus and respond to it with an unlateralized response. Thus, their deficit was in the initiation of movement in response to stimuli rather than in the detection of the stimuli.
Studies with molecular and genetic techniques have also implicated dopamine in behavioral activation (Palmiter, 2009). Dopamine production relies on the synthetic enzyme tyrosine hydroxylase, which is also critical for the production of norepinephrine. Genetic deletion of tyrosine hydroxylase specifically in dopaminergic neurons produces severe impairments in mice; they become hypoactive and aphagic, and they starve by 4 weeks of age. Daily treatment with L-DOPA restores activity and feeding, demonstrating the critical role of dopamine for goal-directed behaviors necessary for survival. Restoration of dopamine synthesis specifically to the dorsal striatum via stereotactic injections of a recombinant virus is sufficient to rescue deficits in feeding and the initiation of locomotory behavior, underscoring the critical role of dopaminergic innervation of striatal neurons for motivated behaviors.
Dopaminergic Neuron Firing Is Related to Incentive and Reward
Electrophysiological recordings from midbrain dopaminergic neurons in animals have provided a framework for understanding the role of dopamine in motivated behavior (Bromberg-Martin & Hikosaka, 2010; Schultz, Dayan, & Montague, 1997). Midbrain dopaminergic neurons fire tonically at low rates of about 5 Hz in awake animals. The resulting tonic release of dopamine is critical for the ongoing operation of downstream circuits. Stimuli that are either directly rewarding (such as juice) or predict rewards (such as a cue which has been paired with a subsequent reward) evoke phasic increases in firing which elevate dopamine concentration at synaptic terminals for a period of several seconds.
A critical insight into the function of dopamine neurons was revealed by the pioneering studies of Wolfram Schultz and colleagues (Schultz et al., 1997). Dopaminergic neurons in the VTA originally fire in response to the presentation of a reward, but if a sensory cue is provided which predicts the reward, dopamine neurons fire in response to the cue instead of the reward (Fig. 41.4). Remarkably, if a fully predicted reward fails to occur, dopamine neurons decrease rather than increase their firing rate. These and other findings have led to the hypothesis that dopamine neuronal firing encodes “reward prediction errors.” The dynamic changes in dopamine neuron firing as learning proceeds match quantitative predictions from reinforcement learning and have inspired researchers to borrow principles from economics to guide neuroscientific studies (Glimcher, 2011).

Figure 41.4 Responses of a dopamine neuron to unpredicted primary reward (top) and the transfer of this response to progressively earlier reward-predicting conditioned stimuli with training (middle). The bottom record shows a control baseline task when the reward is predicted by an earlier stimulus and not the light.
From Schultz et al. (1995), with permission.
Motivated behaviors can be directed toward rewarding stimuli and some dopamine neurons appear to encode the motivational salience of stimuli, responding to stimuli that are either rewarding or aversive (Matsumoto & Hikosaka, 2009). Signals encoding aversive stimuli (or the lack of rewarding stimuli) are thought to be conveyed to dopamine neurons indirectly from a nucleus in the diencephalon called the habenula (Hikosaka et al., 2010). Neurons in the habenula decrease their firing rate in response to rewarding stimuli and increase their firing rates in response to aversive stimuli and when expected rewards are absent. Dopaminergic neurons in the substantia nigra which fire in response to both rewarding and aversive stimuli are hypothesized to convey motivational signals to the superior colliculus and frontal cortex, brain regions responsible for orienting attention toward and evaluating potentially salient stimuli, as well as to the striatum.
Electrical Stimulation of the Brain Can Be a Positive Reinforcer
Olds and Milner (1954) discovered that rats learned new responses when their learning was positively reinforced by electrical stimulation of the brain, and the sites that supported robust electrical brain self-stimulation followed the trajectory of the medial forebrain bundle. That finding affirmed the importance of incentive factors in instrumental behavior. The best natural parallel appeared to be behavior in response to large incentives by animals in low states of drive (e.g., chocolate milk in nondeprived rats). Indeed, measurement of the strength of reinforcers in microamperes (Box 41.3 and Fig. 41.5) promised a pragmatic, as well as objective, new approach to the psychophysics of an operational hedonism. However, perhaps its greatest contribution was to enable the anatomical mapping of the central reward or reinforcement mechanisms of the brain (Phillips, 1984).
Box 41.3 Psychophysics of Hedonism as Measured by Brain Stimulation Reward
Electrical stimulation of the brain can be used as positive reinforcement of learned behaviors. An apparatus is set up so that when an animal presses a lever, its brain is stimulated. Alternatively, a threshold procedure can be used where a lever or wheel response is required. An advantage of using electrical stimulation to measure motivation is that the strength of the reward can be titrated easily by adjusting the amount of current delivered for each response. If the current is adjusted too low (i.e., below the threshold for reinforcement), then the animals will not press the lever. As current is increased, the relationship between the rate of lever pressing and the strength of current typically follows a sigmoidal curve called rate-intensity function. In the threshold procedure, a series of discrete trials are presented at different current intensities, and the responses within a certain cutoff period are recorded. Current levels are lowered and raised to capture the threshold current (Fig. 41.2). Pharmacologic manipulation of catecholamine neurotransmitter systems can lower thresholds (e.g., treatment with amphetamines or cocaine) or raise thresholds (e.g., treatment with the dopamine receptor antagonist pimozide). Such changes mean the amount of electrical current required to support lever presses varies, while the latency to respond is unchanged. Thus, these drugs affect reward processes, rather than motor function. The threshold of reinforcement also can be measured by allowing rats to adjust the strength of the current. In this setup, a process is employed in which rats press lever one to produce ICSS, but each lever-one press also reduces the intensity of the current. To reset current intensity, rats must press lever two. Such “self-titration” experiments yield conclusions similar to those based on threshold and rate-intensity functions.
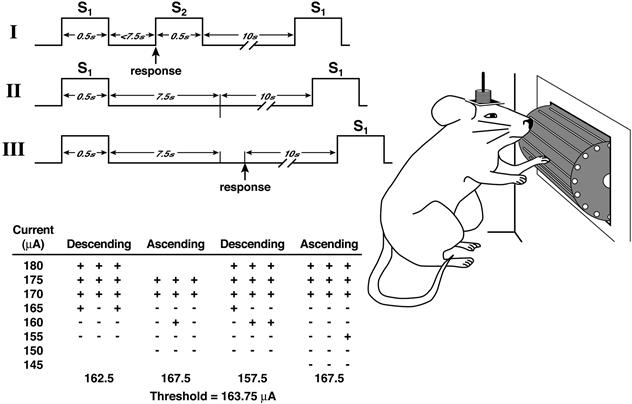
Figure 41.5 Intracranial self-stimulation threshold procedure. Panels I, II, and III illustrate the timing of events during three hypothetical discrete trials. Panel I shows a trial during which the rat responded within the 7.5 s following the delivery of the noncontingent stimulus (positive response). Panel II shows a trial during which the animal did not respond (negative response). Panel III shows a trial during which the animal responded during the intertrial interval (negative response). For demonstration purposes, the intertrial interval was set at 10 s. In reality, the interresponse interval had an average duration of 10 s and ranged from 7.5 to 12.5 s. The table depicts a hypothetical session and demonstrates how thresholds were defined for the four individual series. The threshold of the session is the mean of the four series’ thresholds.
Taken with permission from Markou and Koob (1992).
Catecholamine Hypothesis of Rewarding Brain Stimulation
Subsequent experiments indicated that animals can learn to engage in intracranial self-stimulation (ICSS) in several other brain regions (e.g., the locus coeruleus, midbrain central gray, cerebellum, trigeminal motor nucleus, substantia nigra, caudate nucleus, and nucleus accumbens) (Phillips, 1984). Although the diversity of these sites has not been fully elucidated, many of them are known to contain catecholamine-containing neurons, and while such neurons can express either norepinephrine or dopamine, most pharmacologic data can be interpreted in terms of dopaminergic mechanisms (Box 41.3 and Fig. 41.5). In particular, very high rates of ICSS were found in the ventral tegmental area, ventral striatum (including the nucleus accumbens to which dopamine neurons project), all key structures of the medial forebrain bundle. However, of several neural circuits that may subserve rewarding brain stimulation, only some are influenced directly by dopamine. The difficulty in interpreting the role of striatal dopamine in ICSS lies in distinguishing motor from reward effects (Box 41.3). The phenomenon of human drug dependence and the discovery in the early 1960s of drug self-administration in animals opened the way to more precisely analyze the neurochemical basis of reinforcement.
The Mesolimbic Dopamine System Plays a Key Role in Incentive Motivation
Wise (1982) proposed that natural reinforcers are perceived as rewards because they increase activity of the mesolimbic dopamine system. If this is correct, then the mesolimbic dopamine system should be important for behavioral responses to natural rewards, such as food and sex. In fact, depletion of dopamine in the nucleus accumbens does not impair consummatory behavior in rats (unlike dopamine depletion from the caudate-putamen), but it does reduce incentive-motivational responses. For example, proceptive behaviors (such as “ear wiggling,” hopping, and darting, all of which serve to solicit the attention of the male) are reduced in female rats with dopamine depletions. In addition, depletion of dopamine from the nucleus accumbens decreases locomotor excitement of hungry rats in the presence of food (Koob, Riley, Smith, & Robbins, 1978). Intriguingly, consummatory behavior, including eating of high incentive foods, appears to be mediated by distinct mechanisms within the nucleus accumbens that depend on opioid receptors (Hnasko, Sotak, & Palmiter, 2005; Kelley and Berridge, 2002).
Mesolimbic dopamine also may control behavior motivated by reward. For example, it was shown in studies by Robbins and Taylor that when a previously neutral light became a predictor of the occurrence of a primary reinforcer (water), rats learned a new behavior to turn on the light. Injection of amphetamine into the nucleus accumbens (but not the caudate) increased the frequency of the behavior, suggesting that amphetamine enhanced the motivational properties of this reward-related stimulus (i.e., the light, which was related to the reward, water). Amphetamine itself was well known to suppress eating and drinking; thus, the increased frequency of behavior was not due to increased thirst, for example. Furthermore, amphetamine did not increase the frequency of responding to a randomly paired stimulus. Finally, amphetamine-induced facilitation was completely blocked by dopamine depletion in the nucleus accumbens (but not by dopamine depletion in the caudate). These results indicated that the mesolimbic dopamine system enhances the motivational properties of stimuli predictive of natural rewards (Robbins, Taylor, Cador, & Everitt, 1989). Another way of describing this activational role of the dopamine system is that it confers “incentive salience” (Berridge & Robinson, 1998).
The Amygdala Also Has a Role in Appetitive Conditioning and Motivation
Growing evidence implicates the amygdala in evaluating both positive and negative stimuli (Morrison & Salzman, 2010). Excitotoxic, axon-sparing lesions of the amygdala diminish the capacity of stimuli associated with reward to motivate behavior. Such lesions also impair responses that are rewarded with access to female rats. However, mating per se is unaffected, in contrast with the effect of medial preoptic area lesions, (see above and Everitt, Cardinal, Hall, Parkinson, Robbins, 2000). This separation of control of consummatory and learned aspects of sexual behavior indicates that motivated sequences of behavior are constructed through the coordination of neural systems that are at least partly independent. Lesions of the basolateral amygdala also impair conditioned place preferences. In contrast, selective lesions of the central nucleus of the amygdala impair the learning of Pavlovian conditioned approach responses.
Motivated Behavior Translated to Habits
The ventral striatum, including the nucleus accumbens, receives afferents from the basolateral amygdala, hippocampal formation, and prefrontal cortex, and projects to the lateral hypothalamus and ventral pallidum (Fig. 41.6). Output from the ventral pallidum is routed in several different ways, including to brainstem motor regions and back to the prefrontal cortex through the mediodorsal thalamus. This circuit (prefrontal cortex-ventral striatum-ventral pallidum) appears to fit the requirements for “a neural mechanism by which motivation gets translated into action” (Mogenson, Jones, & Yim, 1980).
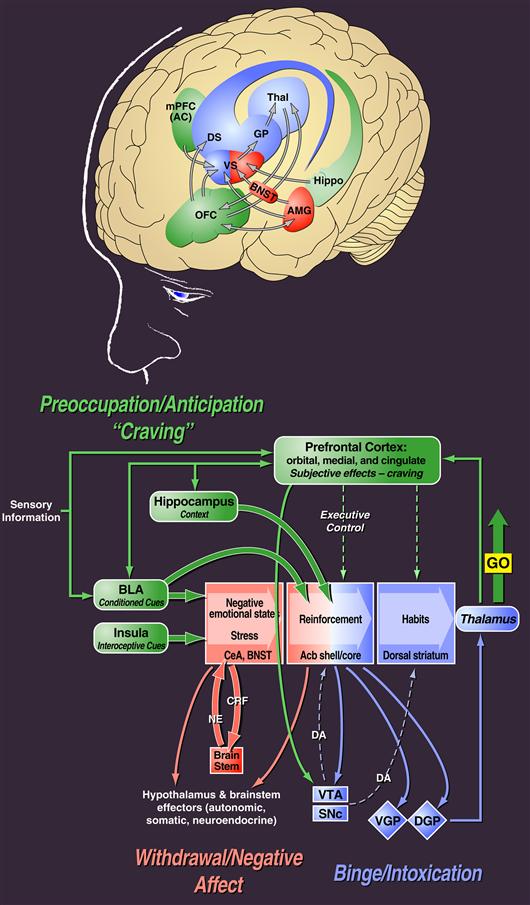
Figure 41.6 Neurocircuitry schematic illustrating the combination of neuroadaptations in the brain circuitry for the three stages of the addiction cycle that drive drug-seeking behavior in the addicted state. Note the activation of the ventral striatum/dorsal striatum/extended amygdala driven by cues via hippocampal and basolateral amygdala and stress via the insula. The frontal cortex system is compromised, producing deficits in executive function and contributing to the incentive salience of drugs compared to natural reinforcers. Dopamine systems are compromised, and brain stress systems such as corticotropin-releasing factor (CRF) are activated to reset further the salience of drugs and drug-related stimuli in the context of an aversive dysphoric state.
Modified with permission from Koob and Volkow (2010) and based on Everitt and Robbins (1995).
As described earlier, the basal and lateral amygdala convey associative information about stimuli that predict the occurrence of reinforcers. A link between the amygdala and ventral striatum is important in the translation of emotion (and motivational effects of stimuli) to behavioral output (Everitt et al., 2000). Glutamatergic inputs from the amgydala to ventral striatum work together with the ascending dopamine system to determine the output of ventral striatal γ-aminobutyric acid (GABA)-ergic medium spiny neurons that project to the globus pallidus (ventral pallidum) (see Chapter 30).
The nucleus accumbens is a heterogeneous structure. Its “shell” region is considered by some to be part of an “extended amygdala” encompassing the central nucleus of the amygdala and bed nucleus of the stria terminalis. The basolateral amygdala projects predominantly to the nucleus accumbens core subregion and specific parts of the dorsal striatum. These connections are paralleled by other cortico-striatal “loops” (see Chapter 30) (e.g., between different parts of the neocortex that project topographically to different sectors of the dorsal striatum). Moreover, there appears to be the possibility of cross-talk between these systems, arising from anatomical connections that potentially allow communication from the amygdala-ventral striatal circuitry to the cortico-dorsal striatal loops (Haber, Fudge, & McFarland, 2000). The ventral striatum has been linked to the initial acquisition of drug-seeking and drug-taking behavior and the subsequent cascade of loop circuitry involving the dorsal striatum to the gradual acquisition of stimulus-response habits that help to give the habits their “compulsive” quality (Belin, Jonkman, Dickinson, Robbins, & Everitt 2009; Everitt and Robbins, 2005).
Addiction
Epidemiology of Drug Abuse
Drug abuse is one of the world’s major public health problems. For example, statistics from the 2009 National Household Survey on Drug Abuse of the National Institute on Drug Abuse of the United States reveal that approximately 21.8 million Americans aged 12 and older were past-month users of illicit drugs, and 22.5 million could be classified with substance dependence or abuse. In addition, 15.4 million individuals were dependent on or abused alcohol, and 69.7 million were current smokers (Substance Abuse and Mental Health Services Administration, 2010). The cost of drug abuse to society is staggering. Total economic costs of alcohol abuse per year in the United States is listed as $166.5 billion dollars, smoking $157 billion dollars (Centers for Disease Control and Prevention, 2004), and illicit drugs $193 billion dollars (National Drug Intelligence Center, 2011). This totals over $500 billion dollars in lost productivity due to illness or death, health care expenditures, motor vehicle crashes, and social welfare, among others (Stein, 2001).
Addiction Cycle
Drug addiction, also known as substance dependence, is a chronically relapsing disorder characterized by (1) compulsion to seek and take the drug, (2) loss of control in limiting intake, and (3) emergence of a negative emotional state (e.g., dysphoria, anxiety, irritability) when access to the drug is prevented (Koob & Le Moal, 1997). Addiction and substance dependence (as currently defined by the Diagnostic and Statistical Manual of Mental Disorders, 4th edition; American Psychiatric Association, 1994) will be used interchangeably throughout this text and refer to a final stage of a usage process that moves from drug use to addiction. Clinically, the occasional but limited use of a drug with the potential for abuse or dependence is distinct from escalated drug use and the emergence of a chronic drug-dependent state (Table 41.1). An important goal of current neurobiological research is to understand the neuropharmacological and plastic mechanisms within specific neural circuits that mediate the transition from occasional, controlled drug use to the loss of behavioral control over drug-seeking and drug-taking that defines chronic addiction.
Table 41.1 DSM-IV and ICD-10 Diagnostic Criteria for Alcohol and Drug Dependence
| DSM-IV | ICD-10 | |
| Clustering criterion | A. A maladaptive pattern of substance use, leading to clinically significant impairment or distress as manifested by three or more of the following occurring at any time in the same 12-month period: | A. Three or more of the following have been experienced or exhibited at some time during the previous year: |
| Tolerance | 1. Need for markedly increased amounts of a substance to achieve intoxication or desired effect; or markedly diminished effect with continued use of the same amount of the substance. | 1. Evidence of tolerance, such that increased doses are required in order to achieve effects originally produced by lower doses. |
| Withdrawal | 2. The characteristic withdrawal syndrome for a substance or use of a substance (or a closely related substance) to relieve or avoid withdrawal symptoms. | 2. A physiological withdrawal state when substance use has ceased or been reduced as evidenced by: the characteristic substance withdrawal syndrome, or use of substance (or a closely related substance) to relieve or avoid withdrawal symptoms. |
| Impaired control | 3. Persistent desire or one or more unsuccessful efforts to cut down or control substance use. | 3. Difficulties in controlling substance use in terms of onset, termination, or levels of use. |
| 4. Substance used in larger amounts or over a longer period than the person intended. | ||
| Neglect of activities | 5. Important social, occupational, or recreational activities given up or reduced because of substance use. | 4. Progressive neglect of alternative pleasures or interests in favor of substance use; or |
| Time spent | 6. A great deal of time spent in activities necessary to obtain, to use, or to recover from the effects of substance used. | a great deal of time spent in activities necessary to obtain, to use, or to recover from the effects of substance use. |
| Inability to fulfill roles | None | None |
| Hazardous use | None | None |
| Continued use despite problems | 7. Continued substance use despite knowledge of having a persistent or recurrent physical or psychological problem that is likely to be caused or exacerbated by use. | 5. Continued substance use despite clear evidence of overtly harmful physical or psychological consequences. |
| Compulsive use | None | 6. A strong desire or sense of compulsion to use substance. |
| Duration criterion | B. No duration criterion separately specified. However, several dependence criteria must occur repeatedly as specified by duration qualifiers associated with criteria (e.g., “often,” “persistent,” “continued”). | B. No duration criterion separately specified. |
| Criterion for subtyping dependence | With physiological dependence: Evidence of tolerance or withdrawal (i.e., any of items A-1 or A-2 above are present). | None |
| Without physiological dependence: No evidence of tolerance or withdrawal (i.e., none of items A-1 or A-2 above are present). |
Addiction has been conceptualized as a chronic relapsing disorder with roots both in impulsivity and compulsivity and neurobiological mechanisms that change as an individual moves from one domain to the other (Hyman, Malenka, & Nestler, 2006). Subjects with impulse control disorders experience an increasing sense of tension or arousal before committing an impulsive act; pleasure, gratification, or relief at the time of committing the act; and finally, regret, self-reproach, or guilt following the act. In contrast, individuals with compulsive disorders experience anxiety and stress before committing a compulsive repetitive behavior and then relief from the stress by performing the compulsive behavior. In addiction, drug-taking behavior progresses from impulsivity to compulsivity in a three-stage cycle: binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation. As individuals move from an impulsive to a compulsive disorder, the drive for the drug-taking behavior shifts from positive to negative reinforcement (Fig. 41.7). Impulsivity persists, but compulsivity develops (Box 41.4). Impulsivity in general is an aberration of reward anticipation processes, mainly in the ventral striatum (plus possibly response preparation processes in the dorsal striatum), whereas compulsivity is in part the product of habit structures mediated by the dorsal striatum (plus possibly ventral striatal Pavlovian overmotivation; Everitt & Robbins, 2005). Compulsivity also involves the additional negative reinforcement recruited from sensitization of the brain stress systems (Koob & Le Moal, 2005).
Box 41.4 Impulsivity and Compulsivity
Impulsivity can be defined behaviorally as “actions which are poorly conceived, prematurely expressed, unduly risky, or inappropriate to the situation and that often result in undesirable consequences” (Durana and Barnes, 1993). However, compulsivity can be captured by a modification of the definition of impulsivity: actions inappropriate to the situation which persist, have no obvious relationship to the overall goal, and which often result in undesirable consequences (e.g., operationally defined as responding for a drug or alcohol in the face of adverse consequences (Wolffgramm & Heyne, 1995), responding in the face of punishment (Vanderschuren & Everitt, 2004), perseveration in reversal learning (Walker, Robbins, & Roberts, 2009), or responding for a drug or alcohol on a progressive-ratio schedule of reinforcement (Walker, Rasmussen, Raskind, Koob, 2008)).
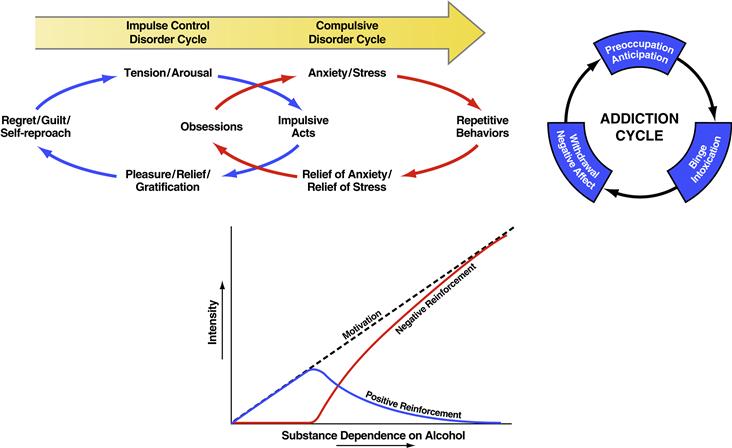
Figure 41.7 (Top) Diagram showing the stages of impulse control disorder and compulsive disorder cycles related to the sources of reinforcement. In impulse control disorders, an increasing tension and arousal occurs before the impulsive act, with pleasure, gratification, or relief during the act. Following the act, there may or may not be regret or guilt. In compulsive disorders, there are recurrent and persistent thoughts (obsessions) that cause marked anxiety and stress, followed by repetitive behaviors (compulsions) that are aimed at preventing or reducing distress (American Psychiatric Association, 1994). Positive reinforcement (pleasure/gratification) is more closely associated with impulse control disorders. Negative reinforcement (relief of anxiety or relief of stress) is more closely associated with compulsive disorders. (Bottom) Change in the relative contribution of positive and negative reinforcement constructs during the development of substance dependence on alcohol. Taken with permission from Koob, (2011). (Top right) Collapsing the cycles of impulsivity and compulsivity results in the addiction cycle, conceptualized as three major components: preoccupation/anticipation, binge/intoxication, and withdrawal/negative affect. Taken with permission from Koob (2008).
Much of the recent progress in understanding the neurobiology of addiction has derived from the study of animal models of addiction on specific drugs such as opioids, stimulants, and alcohol. While no animal model of addiction fully emulates the human condition, animal models do permit the investigation of specific elements of the process of drug addiction. Such elements can be defined by psychological constructs such as positive and negative reinforcement and different stages of the addiction cycle.
Neurobiological Substrates of Drug Reward
Positive Reinforcing Effects of Drugs Are Mediated by Multiple Systems Converging on Common Targets in the Basal Forebrain
As noted above, the medial forebrain bundle is involved in brain stimulation reward and natural rewards, and work in the neurobiology of addiction has led to an understanding of the neurochemical and neuroanatomical components of this system. The principal components of this system include the ventral tegmental area, basal forebrain (nucleus accumbens, olfactory tubercle, frontal cortex, and amygdala), and dopamine connection between the VTA and basal forebrain, called the mesolimbic dopamine system. Additional components are the opioid peptide, GABA, serotonin (5-hydroxytryptamine; 5-HT), and endocannabinoid systems that interact with the ventral tegmental area and the basal forebrain (Koob, 1992) (Fig. 41.8). The functional role of each component of the basal forebrain reward system is discussed in the following sections. The actions of several groups of drugs with respect to this system, including indirect sympathomimetics (cocaine and amphetamine), opioids (heroin), nicotine, sedative hypnotics (alcohol, barbiturates, and benzodiazepines), and Δ9-tetrahydrocannabinol (THC) also will be discussed.

Figure 41.8 Sagittal section through a representative rodent brain illustrating the pathways and receptor systems implicated in the acute reinforcing actions of drugs of abuse. Cocaine and amphetamines activate the release of dopamine in the nucleus accumbens and amygdala via direct actions on dopamine terminals. Opioids activate opioid receptors in the ventral tegmental area, nucleus accumbens, and amygdala via direct actions on interneurons. Opioids facilitate the release of dopamine in the nucleus accumbens via an action either in the ventral tegmental area or the nucleus accumbens (but also are hypothesized to activate elements independent of the dopamine system). Alcohol activates γ-aminobutyric acid-A (GABAA) receptors in the ventral tegmental area, nucleus accumbens, and amygdala via either direct actions at the GABAA receptor or through indirect release of GABA. Alcohol is hypothesized to facilitate the release of opioid peptides in the ventral tegmental area, nucleus accumbens, and central nucleus of the amygdala. Alcohol facilitates the release of dopamine in the nucleus accumbens via an action either in the ventral tegmental area or the nucleus accumbens. Nicotine activates nicotinic acetylcholine receptors in the ventral tegmental area, nucleus accumbens, and amygdala, either directly or indirectly, via actions on interneurons. Nicotine also may activate opioid peptide release in the nucleus accumbens or amygdala independent of the dopamine system. Cannabinoids activate CB1 receptors in the ventral tegmental area, nucleus accumbens, and amygdala via direct actions on interneurons. Cannabinoids facilitate the release of dopamine in the nucleus accumbens via an action either in the ventral tegmental area or the nucleus accumbens, but also are hypothesized to activate elements independent of the dopamine system. Endogenous cannabinoids may interact with postsynaptic elements in the nucleus accumbens involving dopamine and/or opioid peptide systems. The blue arrows represent the interactions within the extended amygdala system hypothesized to have a key role in drug reinforcement. AC, anterior commissure; AMG, amygdala; ARC, arcuate nucleus; BNST, bed nucleus of the stria terminalis; Cer, cerebellum; C-P, caudate-putamen; DMT, dorsomedial thalamus; FC, frontal cortex; Hippo, hippocampus; IF, inferior colliculus; LC, locus coeruleus; LH, lateral hypothalamus; N Acc., nucleus accumbens; OT, olfactory tract; PAG, periaqueductal gray; RPn, reticular pontine nucleus; SC, superior colliculus; SNr, substantia nigra pars reticulata; VP, ventral pallidum; VTA, ventral tegmental area.
Taken with permission from Koob (2005).
The Positive Reinforcing Effects of Cocaine and Other Indirect Sympathomimetics Depend Critically on the Mesolimbic Dopamine System
Amphetamine and cocaine are psychomotor stimulants that, in humans, have behavioral effects such as suppressing hunger and fatigue and inducing euphoria. In animals, these drugs increase locomotor activity, decrease food intake, stimulate operant behavior, enhance conditioned responding, and produce place preferences. Psychomotor stimulants also decrease thresholds for reinforcing brain stimulation and act as reinforcers for drug self-administration (Koob, 1992) (Fig. 41.8).
Psychomotor stimulants with a high potential for abuse have effects that lead to increases in the availability of monoamine neurotransmitters at synapses. By blocking reuptake or enhancing release, cocaine and amphetamine increase the synaptic availability of dopamine, norepinephrine, and serotonin. However, the acute reinforcing effects of these drugs depend critically on dopamine. Studies of intravenous self-administration have provided the most direct evidence implicating dopamine and, more specifically, the mesolimbic dopamine system in the reinforcing actions of cocaine and amphetamines. Low doses of dopamine receptor antagonists injected either systemically or centrally into the nucleus accumbens, amygdala, or bed nucleus of the stria terminalis block cocaine and amphetamine self-administration in rats. A specific role for the mesolimbic dopamine system in the reinforcing properties of cocaine and amphetamine was deduced from the observation that neurotoxin-induced lesions of the terminal regions of the mesolimbic dopamine system in the nucleus accumbens produce a significant and long-lasting decrease in self-administration of cocaine and amphetamine over days (Koob, 1992). 6-OHDA-induced lesions of the dopamine terminals in the nucleus accumbens even decrease the amount of work an animal will perform for cocaine without affecting work for many other reinforcers, such as food.
Neural Substrates of Sensitization Involve Changes in the Mesolimbic and Nigrostriatal Dopamine Systems
Under certain circumstances, repeated administration of stimulants and opioids results in increasingly strong motor activation, and these effects have been hypothesized to contribute to the development of the neuroadaptations associated with addiction, particularly those involving initial use and/or reinstatement of drug-taking following abstinence. This sensitization to the activating effects of stimulants involves activation of the mesolimbic and nigrostriatal dopamine system (Robinson & Berridge, 1993). Repeated microinjections of amphetamine into the somatodendritic region of the ventral tegmental dopamine cells, at doses that do not cause behavioral activation, are sufficient to sensitize the dopamine cells to later systemic injections of amphetamine. This sensitization can be manifested as increased locomotor activation or increased stereotyped-like behavior (see above). Thus, changes in the activity of these dopamine cells are sufficient to produce sensitization.
Multiple mechanisms for sensitization have been proposed, and a common theme is a time-dependent chain of adaptations that ultimately leads to long-lasting changes in the function of the mesolimbic dopamine system (Robinson & Berridge, 1993). For example, repeated administration of cocaine produces a decrease in the sensitivity of impulse-regulating somatodendritic dopamine D2 receptors on dopamine neurons (termed autoreceptors because the neuron is itself responding to the transmitter it releases). Because these autoreceptors normally exert an inhibitory influence on the activity of dopaminergic neurons, this change in sensitivity could translate into enhanced dopamine availability with subsequent injections of cocaine (Robinson & Berridge, 1993). However, the decrease in D2 receptor sensitivity lasts only 4–8 days, and behavioral sensitization can last for weeks; thus, other mechanisms also must be involved. N-methyl-D-aspartate (NMDA) glutamate receptor antagonists selectively block the development of sensitization to psychomotor stimulants, suggesting a role for brain glutamate systems and specific glutamate receptors in sensitization (Robinson & Berridge, 1993). In addition, an increased surface-to-intracellular ratio of glutamate-1 receptors (GluR1) has been observed 21 days after the last injection of cocaine, suggesting a slowly developing redistribution of AMPA receptors to the surface of nucleus accumbens neurons, particularly in those lacking GluR2 subunits (Boudreau & Wolf, 2005).
Stressors can cause sensitization to stimulant drugs, and both the stress axis and the extrahypothalamic corticotropin-releasing factor (CRF) system appear to be important in stress-induced sensitization (Koob, 1999). The hypothalamic-pituitary-adrenal stress axis (see Chapter 33) also may play an important role in sensitization. Sensitization may be hypothesized to play a key role in initial sensitivity to drugs and contribute to the high incentive salience attributed to drugs during the course of addiction. However, sensitization of the reward systems is not readily observed in individuals meeting the criteria for addiction, rather a profound tolerance and negative emotional state during withdrawal are the clinical norm.
Neurobiological Substrates for the Acute Reinforcing Effects of Opioids Involve Opioid Peptide Systems
Much like psychostimulants, opioid drugs such as heroin are readily self-administered intravenously by animals, and with limited access opioids do not produce dependence (Schuster & Thompson, 1969). Advances in opioid pharmacology, such as the identification of high specific binding for opioids in the brain and discovery of endogenous opioid peptides, provided insight into the brain systems responsible for the reinforcing effects of these drugs (Koob, 1992) (Box 41.5 and Figs. 41.7 and 41.10). The reinforcing actions of heroin and morphine appear to be mediated largely by the μ opioid receptor subtype. Antagonists at the μ receptor produce dose-dependent decreases in heroin reinforcement, and μ receptor knockout mice show loss of morphine-induced reward and loss of morphine-induced analgesia. Intracerebral opioid receptor antagonists block heroin self-administration in nondependent rats when these drugs are injected into the VTA or the region of the nucleus accumbens. Microinjections of opioids into the VTA also lower thresholds for ICSS and produce robust place preferences, and these effects appear to be dependent on mesolimbic dopamine function (Di Chiara & North, 1992). Rats will self-administer opioid peptides in the region of the nucleus accumbens. However, heroin self-administration is not blocked by dopamine receptor antagonists administered at doses that block cocaine self-administration or by large neurotoxin-induced lesions of the mesolimbic dopamine system. These results demonstrate that neural elements in the regions of the VTA and nucleus accumbens are responsible for the reinforcing properties of opioids, suggesting both dopamine-dependent and dopamine-independent opiate actions.
Box 41.5 Opioid Receptors
In 1978, Professors Solomon H. Snyder, John Hughes, and Hans W. Kosterlitz were awarded the Lasker award for their combined discovery of opioid receptors in the brain and for the even more amazing discovery of the existence of endogenous ligands for opioid receptors. The endogenous brain compounds were peptides, called enkephalins and endorphins, which shared all the physiologic properties of opioids. These observations opened a rich field of inquiry into the mechanism of action of opioid drugs, and these findings began a new outlook on neuropeptide neurotransmitters, an area of intense research. First, many new neuropeptides have been discovered with brain receptors potentially amenable to nonpeptidergic agonists and antagonists. Second, because neuropeptides are contained within neurons that also are more conventional amino acid or amine transmitters, the possibility of multiple mediators for any specific synapse had to be considered.
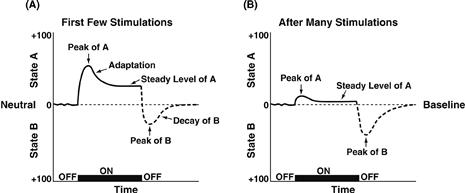
Figure 41.9 (A) The standard pattern of affective dynamics produced by a relatively novel unconditioned stimulus (nondependent drug intake). (B) The standard pattern of affective dynamics produced by a familiar, frequently repeated unconditioned stimulus (dependent drug intake). First, an unconditional arousing stimulus triggers a primary affective process, termed the a-process. An unconditional reaction that translates the intensity, quality, and duration of the stimulus (for example, the first injection of a drug). Second, as a consequence of the a-process, the b-process is evoked after a short delay, an opponent process. The two responses are consequently and temporarily linked (a triggers b) but are hypothesized to depend on different neurobiological mechanisms. The b-process has a longer latency but more inertia, slower recruitment, and more sluggish decay. At a given moment, the pattern of affect will be the algebraic sum of these opposite influences and the dynamics reveal, with the passage of time, the net product of the opponent process.
Taken with permission from Solomon (1980).
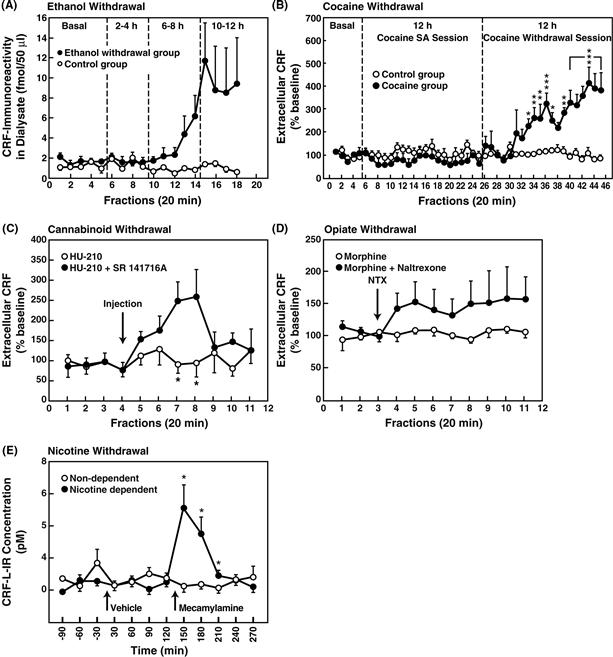
Figure 41.10 (A) Effects of ethanol withdrawal on CRF-like immunoreactivity (CRF-L-IR) in the rat amygdala determined by microdialysis. Dialysate was collected over four 2 h periods regularly alternated with nonsampling 2 h periods. The four sampling periods corresponded to the basal collection (before removal of ethanol), and 2–4 h, 6–8 h, and 10–12 h after withdrawal. Fractions were collected every 20 min. Data are expressed as mean ± SEM (n = 5 per group). Analysis of variance confirmed significant differences between the two groups over time (p < 0.05). Taken with permission from Merlo-Pich et al. (1995). (B) Mean (± SEM) dialysate CRF concentrations collected from the central nucleus of the amygdala of rats during baseline, 12 h cocaine self-administration, and a subsequent 12 h withdrawal period (cocaine group, n = 5). The control group consisted of rats with the same history of cocaine self-administration training and drug exposure but not given access to cocaine on the test day (n = 6). Data are expressed as percentages of basal CRF concentrations. Dialysates were collected over 2 h periods alternating with 1 h nonsampling periods shown by the timeline at the top. During cocaine self-administration, dialysate CRF concentrations in the cocaine group were decreased by about 25% compared with control animals. In contrast, termination of access to cocaine significantly increased CRF efflux that began approximately 5 h post-cocaine and reached about 400% of presession baseline levels at the end of the withdrawal session. *p < 0.05, **p < 0.01, ***p < 0.001, simple main effects after overall mixed-factorial analysis of variance. Taken with permission from Richter and Weiss (1999). (C) Effects of cannabinoid CB1 receptor antagonist SR 141716A (3 mg/kg) on CRF release from the central nucleus of the amygdala in rats pretreated for 14 days with the CB1 receptor agonist HU-210 (100 mg/kg). Cannabinoid withdrawal induced by SR 141716A was associated with increased CRF release (*p < 0.005, n = 5–8). Vehicle injections did not alter CRF efflux (n = 5–7). Data were standardized by transforming dialysate CRF concentrations into percentages of baseline values based on averages of the first four fractions. Taken with permission from Rodriguez de Fonseca, Carrera, Navarao, Koob, & Weiss, (1997). (D) Effects of morphine withdrawal on extracellular CRF in the central nucleus of the amygdala. Withdrawal was precipitated by administration of naltrexone (0.1 mg/kg) in rats prepared with chronic morphine pellet implants. Taken with permission from Weiss et al. (2001). (E) Effect of mecamylamine-precipitated (1.5 mg/kg, i.p.) nicotine withdrawal on extracellular levels of CRF-like immunoreactivity in the central nucleus of the amygdala measured by in vivo microdialysis in chronic nicotine pump-treated (nicotine-dependent, n = 7) and chronic saline pump-treated (nondependent, n = 6) rats. *p < 0.05, compared with nondependent. Taken with permission from George et al. (2007).
Nicotine Activates Mesolimbic Dopamine
Nicotine has anti-fatigue, stimulant-like, and anti-anxiety effects and appears to improve cognitive performance in animals and humans. It has direct reinforcing actions, as measured by intravenous self-administration in animals and humans. Nicotine is a direct agonist at nicotinic acetylcholine receptors (see Chapter 8) and appears to activate nicotinic acetylcholine receptors in the mesolimbic dopamine system both at the level of the VTA and nucleus accumbens (Figs. 41.7 and 41.10). Knockout of the α4 and β2 nicotinic acetylcholine receptor subunit blocks nicotine self-administration (Picciotto et al., 1998). Blockade of dopaminergic systems with antagonists or lesions to the mesolimbic dopamine system can also block nicotine self-administration, suggesting a key role for dopamine in the acute reinforcing actions of nicotine.
Alcohol Has Multiple Neurochemical Substrates within Brain Reinforcement Systems
Alcohol (i.e., ethanol), barbiturates, and benzodiazepines all have measurable sedative–hypnotic actions, including euphoria, disinhibition, anxiety reduction, sedation, and hypnosis. All of these drugs also have anxiolytic (or antianxiety) effects, reflected in conflict situations as a reduction of behavior suppressed by punishment. The neurobiological basis of the anxiolytic properties of these sedative–hypnotic drugs has provided clues to their reinforcing properties and their abuse potential. For example, GABAergic receptor antagonists reverse many of the behavioral effects of alcohol, an observation that has led to the hypothesis that GABA has a role in alcohol’s intoxicating effects. Further support for a role of brain GABA in alcohol reinforcement is the observation that potent antagonists of GABA receptor function decrease alcohol reinforcement, and one particularly affected brain site is the central nucleus of the amygdala (Koob, 1992). Activation of opioid peptide systems has been implicated in alcohol reinforcement by numerous reports that the opiate antagonists naloxone and naltrexone reduce alcohol self-administration in several animal models (Tabakoff & Hoffman, 1992). The brain site most sensitive to opioid receptor antagonists are the VTA and central nucleus of the amygdala (Figs. 41.7 and 41.10).
Dopamine receptor antagonists also reduce lever pressing for alcohol in nondeprived rats (Koob, 1992), and extracellular dopamine levels increase in nondependent rats orally self-administering low doses of alcohol (Koob & Le Moal, 2001). Low doses of acetaldehyde are also self-administered by animals, and increasing evidence suggests that acetaldehyde may mediate some of ethanol’s effects on the mesolimbic dopamine system (Rodd et al., 2005). However, virtually complete destruction of dopamine terminals in the nucleus accumbens by 6-OHDA failed to alter voluntary responding for alcohol (Tabakoff & Hoffman, 1992). Combined with the pharmacologic data discussed earlier, these results suggest that although mesolimbic dopamine transmission may be associated with important aspects of alcohol reinforcement, it is not critical for the reinforcing properties of alcohol, and multiple neurotransmitters combine to orchestrate the reward profile of this drug (Engel, et al. 1987). For example, alcohol in a physiologic dose range may antagonize the actions of glutamate. In addition, increases and decreases in synaptic availability of serotonin (e.g., via blockade of serotonin reuptake or via administration of selective serotonin receptor antagonists) reduce the voluntary intake of alcohol.
Δ9-Tetrahydrocannabinol Has Effects Similar to Other Drugs of Abuse
THC shares effects in animal models of drug reinforcement similar to those of other drugs of abuse. Upon acute administration, THC decreases ICSS reward thresholds and produces a place preference in rats. It maintains self-administration behavior in squirrel monkeys, and a synthetic THC analog is self-administered intravenously in mice. THC binds to the cannabinoid CB1 receptor that is distributed widely throughout the brain, particularly in the striatum, and is thought to mediate some of the actions of the endogenous endocannabinoids. THC self-administration is blocked by both CB1 and opioid receptor antagonists (Justinova, Tanda, Munzar, & Goldberg, 2004; Justinova, Solinas, Tanda, Redhi, & Goldberg, 2005) (Figs. 41.7 and 41.10).
Neurobiological Substrates for the Withdrawal/Negative Affect State of Drug Dependence
Substance dependence is defined as compulsive, uncontrollable drug use; however, the etiology of that compulsive use is multidimensional. Although positive reinforcement is clearly necessary for the development of drug use, it may fall short in explaining the development of compulsive use. For example, tolerance or apparent tolerance develops to drug reward during chronic use, with positive reinforcement either being absent or subsumed by negative reinforcement. Indeed, this issue raises the question of what factors distinguish drug use from abuse and dependence or addiction.
Negative Affect Is a Common Result of Withdrawal from Chronic Administration of Drugs of Abuse
Withdrawal signs associated with the cessation of chronic drug administration usually are characterized by responses that are opposite to the initial effects of the drug. The physical signs (or somatic signs) of withdrawal are typically drug-specific. However, all drugs of abuse produce rewarding effects, and withdrawal commonly is associated with subjective symptoms of dysphoria, negative affect, and anxiety (Koob & Le Moal, 2001). In the case of opioid withdrawal, subjective symptoms such as dysphoria, anxiety, craving for drugs, and malaise are accompanied by both physical symptoms of extreme discomfort, including pupillary dilation, hot and cold flashes, goose bumps, and a flu-like state. In the case of alcohol withdrawal, humans show tremor and increases in heartbeat rate, blood pressure, and body temperature, as well as subjective signs, including dysphoria, anxiety, insomnia, and malaise. Indeed, whereas few physical signs of withdrawal usually are observed during cocaine withdrawal in humans, cessation of cocaine use often is characterized by severe depressive symptoms combined with irritability, anxiety, and anhedonia lasting several hours to several days (i.e., the “crash”) and may be a motivating factor in the maintenance of the cocaine dependence cycle. Withdrawal from nicotine is characterized by anxiety, negative affect, fatigue, irritability, sleep disturbances, and an intense craving for cigarettes. Withdrawal from THC resembles a combination of sedative–hypnotic or opioid withdrawal, with irritability, restlessness, hot flashes, sweating, disturbances in appetite and sleep, and low-level dysphoria. Withdrawal from benzodiazepines resembles that of alcohol withdrawal but generally is more prolonged in onset and less intense.
Chronic Drug Administration Compromises the Brain Reward Systems
In animal studies, withdrawal from chronic administration of stimulants has been studied using ICSS after repeated administration of cocaine or amphetamine over several weeks or 12–48 h of cocaine self-administration. Withdrawal from prolonged self-administration of cocaine in rats increases ICSS reward thresholds. Here, an increase in reward thresholds has been hypothesized to reflect a dysphoric-like state observed in humans. The increase in threshold dose is time-dependent and is an effect opposite to that of acute cocaine (Koob & Le Moal, 2001). Similar effects have been observed during withdrawal from chronic amphetamine and methamphetamine administration. Withdrawal from chronic alcohol, opioids, nicotine, and THC also produces pronounced increases in ICSS reward thresholds, suggesting that brain reward dysregulation is a common element of acute withdrawal from drugs of abuse (Table 41.2).
Table 41.2 Drug Effects on Thresholds of Rewarding Brain Stimulation
| Drug Class | Acute Administration | Withdrawal from Chronic Treatment |
| Psychostimulants (cocaine, amphetamines) | ↓ | ↑ |
| Opioids (morphine, heroin) | ↓ | ↑ |
| Nicotine | ↓ | ↑ |
| Sedative–hypnotics (ethanol) | ↓ | ↑ |
| Cannainoids | ↓ | ↑ |
Neural Circuitry Mediating Withdrawal/Negative Reinforcement
A brain system that mediates aversion was identified early on by a combination of anatomical and behavioral experiments. For example, mapping studies identified sites eliciting escape from brain stimulation, sites at which electrical brain stimulation elicits defensive responses, and sites at which fear or pain naturally evokes aggressive responses. One component of the neural circuitry that mediates aversion includes connections from the medial habenula to interpeduncular nucleus, which in turn projects to brainstem serotonergic neurons as well as projections from the lateral habenula to the rostral tegmental nucleus, which provides inhibitory inputs to dopaminergic neurons in the VTA and substantia nigra.
Aversion May Be Mediated by Opponent Motivational Processes
Animal learning theory has led to the concept of opponent motivational processes (one appetitive, one aversive). An influential model proposed which reinforcers produce affective and hedonic effects (a-processes) which are opposed by b-processes of opposite affective sign in a simple, dynamic control system for affect (Fig. 41.9) (Solomon & Corbit, 1974). The a-process follows the reinforcing event with short latency and then decays. Its size decreases with repeated presentation of the reinforcer. The b-process has a longer latency of onset, reaches its maximum only after repeated trials, and decays very slowly. These components of emotional responses are exhibited in response to many reinforcers, including not only drugs such as heroin, but also natural rewards that also may lead to different forms of “dependence,” such as attachment of offspring to a mother. So, for example, either heroin or the mother elicits a hedonic a-process, which is followed by a rebound negative or aversive b-process when the heroin wears off (or the mother leaves). These effects may get stronger with repeated experiences of heroin (or the mother at a critical period of development), leading to a form of dependence in each case. The opponent process also can work in reverse. For example, if tolerance develops for an initially aversive experience, then a rebound hedonic response can occur when this experience stops. Indeed, this has been suggested partly to explain the “pleasure” of jogging.
How might these opponent processes be mediated? The neuroadaptive mechanisms that reflect changes in reward function have been hypothesized to be alterations of the same neurotransmitters implicated in the acute reinforcing effects of drugs (Koob & Bloom, 1988). Examples of such homeostatic, “within-system” adaptations include decreases in dopaminergic and serotonergic transmission in the nucleus accumbens during drug withdrawal as measured by in vivo microdialysis (Parsons, Koob, & Weiss, 1995; Weiss, Markou, Lorang, & Koob, 1992), increased sensitivity of opioid receptor transduction mechanisms in the nucleus accumbens during opioid withdrawal (Shaw-Lutchman et al., 2002), and decreased GABAergic and increased NMDA glutamatergic transmission during ethanol withdrawal (Fitzgerald & Nestler, 1995; Roberts, Cole, & Koob, 1996; Weiss et al., 1996). However, recruitment of other neurotransmitter systems in the adaptive responses to drugs of abuse may involve neurotransmitter systems not linked to the acute reinforcing effects of the drug, or a “between-system” neuroadaptation (Koob & Bloom, 1988). An example of a between-system neuroadaptation is the activation of brain stress systems such as CRF and dysregulation of other stress-mediating neurotransmitter systems, such as dynorphin, during withdrawal (Koob & Le Moal, 2005) (see below).
Neurochemical Adaptation in Reward Neurotransmitters
Dopamine and Serotonin Systems Show Neuroadaptive Changes with Substance Dependence
Monoamines, including dopamine and serotonin, have been implicated in the reinforcing actions of each of the drugs discussed: psychostimulants, opioids, nicotine, alcohol, and THC. One way of assessing the prolonged effects of drug self-administration is to measure extracellular levels of a neurotransmitter during ongoing behaviors, such as intravenous self-administration of a drug or withdrawal from the drug after a period of chronic self-administration. Such measurements can be made by using the technique of in vivo microdialysis, which involves implanting into a specific site in the brain a guide cannula through which a probe with a semipermeable membrane at the tip perfuses and collects the neurotransmitter in extracellular fluid. Several studies using this technique have shown that extracellular dopamine and serotonin levels in the nucleus accumbens decrease during acute withdrawal from cocaine, opioids, and alcohol, an effect opposite to that of acute drug administration. Consistent with these results, many studies have shown that acute withdrawal from all drugs of abuse results in decreased firing of VTA dopamine neurons (Melis, Spiga, & Diana, 2005).
Brain Stress Systems: Corticotropin-Releasing Factor, Noradrenergic, and Dynorphin-κ Opioid Systems Show Neuroadaptive Changes with Substance Dependence
Anxiety, stress, and dysphoria are common elements of substance dependence and acute withdrawal from drugs of abuse in humans, and withdrawal from drugs of abuse produces stress-like behavior in animals (Sarnyai, Shaham, & Heinrichs, 2001). Stress can be defined simply as any challenge to or alteration in psychological homeostatic processes. Animal studies exploring the role of brain stress systems in the actions of alcohol, opioids, cocaine, nicotine, and THC have provided evidence of a neurochemical basis for the stress associated with abstinence following chronic drug administration. One major player is CRF. Not only is CRF a major hypothalamic releasing factor controlling the classic glucocorticoid stress response, but it also appears to have a neurotropic role in the central nervous system, modulating behavioral responses to stress. CRF itself produces stress-related behaviors, and CRF antagonists reverse a number of behavioral responses to stress. Rats treated repeatedly with drugs such as alcohol, opioids, and cocaine show significant stress-like responses in behavioral tests that follow the cessation of drug administration, and these responses are reversed by administration of a CRF antagonist directly into the brain. For example, injection of a CRF antagonist into the central nucleus of the amygdala reverses the stress-like effects of withdrawal from alcohol, cocaine, nicotine, opioid, and THC in rats. Withdrawal from alcohol, cocaine, nicotine, opioids, and THC is associated with an increase in the release of CRF into the amygdala, suggesting that drug withdrawal can activate CRF systems previously implicated in behavioral responses to stress (Fig. 41.10). Also, stress-induced reinstatement of drug self-administration following extinction can be reversed by CRF antagonists (Shalev et al., 2002).
Norepinephrine in the limbic forebrain also has been associated with the physical and motivational signs of opioid withdrawal. Projections to the bed nucleus of the stria terminalis—a critical part of the extended amygdala—may have a role in the motivational effects of opioid withdrawal (Delfs, Zhu, Druhan, & Aston-Jones, 2000). In addition, the bed nucleus of the stria terminalis is rich in CRF neurons and has been implicated in a feed-forward norepinephrine-CRF-norepinephrine system that may be involved in stress and drug dependence. In this hypothesized feed-forward system, CRF activates brainstem noradrenergic activity, which in turn activates forebrain CRF activity, effectively closing the loop. This mechanism could explain the potentiation of stress responses with repeated exposure that could lead to psychopathology.
Intriguingly, activation of the dynorphin-κ opioid system has long been associated with the development of psychostimulant addiction. Activation of dopamine receptors in the nucleus accumbens shell stimulates a cascade of events that ultimately leads to cyclic adenosine monophosphate response element binding protein (CREB) phosphorylation and subsequent alterations in gene expression, notably the increased expression of protachykinin and prodynorphin mRNA. Subsequent activation of dynorphin systems has been hypothesized to result in decreased dopamine release, thereby contributing to the dysphoric syndrome associated with cocaine dependence (Nestler, 2001). Dynorphins produce aversive dysphoric-like effects in animals and humans and have been hypothesized to mediate negative emotional states by decreasing the functional activity of the mesolimbic dopamine system. However, such enhanced dynorphin action may also activate brain stress responses, such as CRF, or CRF in turn may activate dynorphin (McLaughlin, Marton-Poovici, & Chavkin, 2003; Land et al., 2008). Thus, activation of dynorphin systems by excessive drug use may drive both within-system (decreased reward) and between-system neuroadapatations (increased stress).
Addiction theories such as the opponent process theory postulate that the processes of tolerance to the positive affective state (hedonic tolerance) and subsequent development of the negative affective state (affective withdrawal) play important roles in the transition from drug use to drug dependence (Solomon, 1977). Whereas initial drug use may be motivated by the positive affective state produced by a drug and potentiated by sensitization, continued drug use leads to neuroadaptation to the presence of the drug, the development of a negative affective state, and ultimately self-medication of that affective state. Some theorists have gone so far as to argue that the presence of a negative affective state is the defining feature of addiction and that dysregulation of the residual reward system represents a state of allostasis, the process of obtaining stability through change (Box 41.6). The allostasis hypothesis suggests that both sensitization and opponent process mechanisms may contribute to changes in incentive motivation (or incentive salience) that convey the vulnerability to relapse for drug addiction.
Box 41.6 Allostasis
Counteradaptive processes, such as opponent process, that are part of normal homeostatic limitations of reward function can fail to return within the normal range and are hypothesized to contribute to the allostatic state of addiction. Allostasis from the addiction perspective is defined as the process of maintaining apparent reward function stability by changes in brain reward mechanisms. The allostatic state is fueled not only by dysregulation of reward circuits per se, but also by the activation of brain and hormonal stress systems (Koob & Le Moal, 2001). The following definitions apply: (1) allostasis, the process of achieving stability through change; (2) allostatic state, a state of chronic deviation of the regulatory system from its normal, or homeostatic, operating level; and (3) allostatic load, the cost to the brain and body of the deviation, accumulating over time, and reflecting in many cases pathological states and accumulation of damage.
Neuroadaptation, Prolonged Abstinence, and Relapse
An explanation for the chronic relapsing nature of drug addiction is that reinforcement can be derived from secondary sources that motivate continued drug use. Both positive and negative affective states can become associated with stimuli in the drug-taking environment through classical conditioning processes. When the drug taker is reexposed to these conditioned stimuli, compulsive drug use is reestablished (Box 41.7).
Box 41.7 Craving
Craving is a hypothetical construct that can be defined as the memory of the rewarding effects of a drug, superimposed on a negative motivational state (Markou, Kosten, & Koob, 1998). Human studies of craving have been fraught with difficulty in linking it to relapse per se in humans with drug addiction, but it is a ubiquitous construct that pervades the fabric of the clinical setting (Tiffany, Carter, & Singleton, 2000). Two types of craving can be conceptualized and linked to animal models.
Craving Type 1: Craving is induced by stimuli that have been paired with drug self-administration such as environmental cues. Termed conditioned positive reinforcement in experimental psychology, an animal model of Craving Type 1 is cue-induced reinstatement where a cue previously paired with access to drug reinstates responding for a lever that has been extinguished. Another example is drug-induced reinstatement where a small dose of the drug elicits responding for a lever that has been extinguished.
Craving Type 2: Craving is conceptualized as a state change characterized by anxiety and dysphoria or a residual negative affective state that combines with Craving Type 1 situations to produce relapse to drug seeking. Animal models of Craving Type 2 include stress-induced reinstatement of drug seeking after extinction or increased drug taking in animals during or after a prolonged deprivation. Craving Type 2 can be conceptualized as a residual negative emotional state or an acute stress-like state, both of which can elicit drug-seeking behavior.
Conditioning to the positive affective states induced by drugs has been demonstrated in animals. Stimuli associated with drugs of abuse can maintain responding in rats and monkeys when presented without the drug. It is also possible to demonstrate conditioned withdrawal. For example, when previously neutral stimuli are paired with naloxone-induced withdrawal, the neutral stimuli themselves elicit signs of opioid withdrawal. Conditioned withdrawal has been observed in opioid-dependent animals and humans.
Neurobiological substrates for conditioned drug effects have been investigated in some detail (Cardinal, Parkinson, Hall, & Everitt, 2002; Everitt, Cardinal, Hall, Parkinson, & Robbins, 2000). The enhancement of conditioned reinforcement produced by psychomotor stimulants appears to involve the basolateral amygdala and its glutamatergic projection to the nucleus accumbens with dopaminergic modulation (Robbins et al., 1989). The neurobiological substrates for conditioned withdrawal also may depend on the basolateral amygdala. In contrast, drug-induced reinstatement of drug-seeking appears to be mediated via a prefrontal cortex glutamatergic projection to the nucleus accumbens with dopaminergic modulation (Kalivas & McFarland, 2003). Another powerful source of relapse in human subjects is exposure to a stressor or the state of stress. Both conditions may involve activation of CRF, dynorphin, and noradrenergic systems in the central nucleus of the amygdala and bed nucleus of the stria terminalis. Another powerful source of neurobiological changes that leads to relapse is the loss of frontal cortical function in subregions that control impulsivity and delay of gratification. Damage to such circuits can perpetuate the vulnerability to relapse occasioned by impulsive acts and choices.
Imaging Brain Circuits Involved in Addiction
Brain imaging studies using positron emission tomography with ligands for measuring oxygen utilization or glucose metabolism or using functional magnetic resonance imaging techniques have provided important insights into changes in human brain circuits associated with the development, maintenance, and even vulnerability to addiction. These imaging results, overall, are consistent with circuitry identified in animal studies. Acute intoxication with drugs such as cocaine, methamphetamine, nicotine, alcohol, and THC produces changes in activation of the orbitofrontal cortex, prefrontal cortex, anterior cingulate cortex, amygdala, and ventral striatum. These changes are reflected both in functional magnetic resonance images and in oxygen utilization as measured by positron emission tomography. The changes evoked during acute intoxication often are accompanied by an increase in the availability of dopamine, particularly in striatal regions.
In contrast, acute and chronic withdrawal reverses these changes, resulting in decreases in metabolic activity in the orbitofrontal cortex, prefrontal cortex, and anterior cingulate cortex and decreases in basal dopamine activity in the striatum and prefrontal cortex as reflected in decreases in D2 receptor binding (Fig. 41.11). The decreases in dopamine activity have also been validated by challenge studies where cocaine- and alcohol-dependent individuals show blunted dopamine responses to psychostimulant challenge (Volkow et al., 1997b; Martinez et al., 2005).

Figure 41.11 (A) [18F]N-methylspiroperidol images in a normal control and in a cocaine abuser tested 1 month and 4 months after the last cocaine use. The images correspond to the four sequential planes where the basal ganglia are located. The color scale has been normalized to the injected dose. Notice the lower uptake of the tracer in the cocaine abuser compared to the normal control. Notice also the persistence of the decreased uptake even after 4 months of cocaine discontinuation. BNL, Brookhaven National Laboratory; SUNY, State University of New York. Taken with permission from Volkow et al. (1993). (B) Transaxial images illustrating the differential increase in relative regional cerebral blood flow (CBF) in the amygdala and anterior cingulate of a detoxified cocaine patient during a nondrug-related (nature) video and a cocaine-related video. Anatomical regions of interest first were localized on the patient’s magnetic resonance image (the first image in each row). Region templates were subsequently superimposed on [15O] positron emission tomography (PET) images, yielding radioactive count files for conversion to normalized (relative) regional CBF. The middle and final images in each row show relative regional CBF as measured by PET. The range on the arbitrary color scale is from 0.0 to 2.5 times background activity. Whole brain average regional CBF is 1.0 on the scale. Areas with greatest relative regional CBF are shown in red. Activity in the amygdala and in the anterior cingulate differentially increased during the cocaine video. Taken with permission from Childress et al. (1999).
Craving elicited by presenting cues associated with cocaine, alcohol, nicotine, and THC appears to activate the same circuits as acute intoxication, but with the reactivation of the prefrontal cortex, orbitofrontal cortex, and anterior cingulate cortex and the additional recruitment of activation in the amygdala (Childress et al., 1999) (Fig. 41.11).
Signal Transduction Mechanisms Show Neuroadaptative Changes with Substance Dependence
Chronic drug use not only increases transmitter release via presynaptic effects but also produces long-lasting alterations in postsynaptic receptor activation, signal transduction, and gene expression (Robison & Nestler, 2011). For example, whereas chronic opioid use has minimal effects on the binding of opioid receptors by opioid peptides, it dramatically alters signal transduction in the terminal regions of the mesolimbic dopamine system, such as the nucleus accumbens (Nestler, 2001). This occurs at least in part by increasing adenylate cyclase activity and CREB. CREB is a transcription factor that increases or decreases the transcription of genes. These changes can result in the expression of novel proteins and, consequently, changes in neuronal function that could explain clinical phenomena, such as protracted abstinence and vulnerability to relapse, in people formerly addicted to drugs (Fig. 41.12).
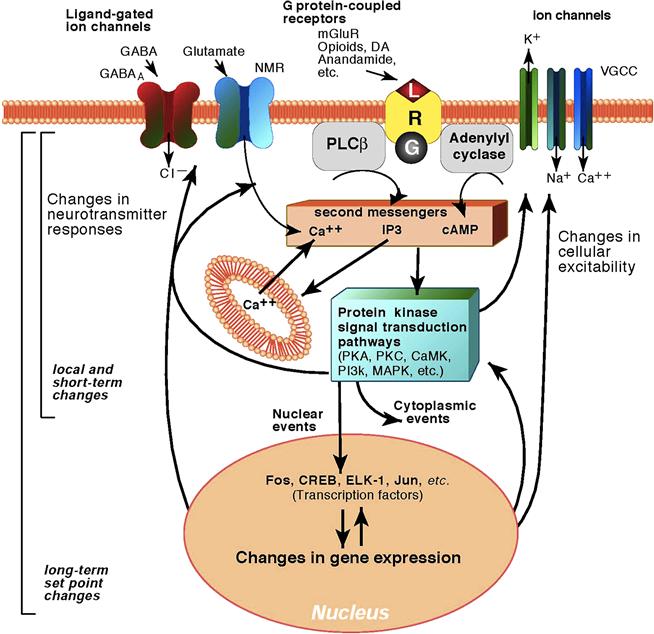
Figure 41.12 Molecular mechanisms of neuroadaptation. Cocaine and amphetamines, as indirect sympathomimetics, stimulate the release of dopamine which acts at G protein-coupled receptors, specifically D1, D2, D3, D4, and D5. These receptors modulate the levels of second-messengers like cyclic adenosine monophosphate (cAMP) and Ca2+, which in turn regulate the activity of protein kinase transducers. Such protein kinases affect the functions of proteins located in the cytoplasm, plasma membrane, and nucleus. Among membrane proteins affected are ligand-gated and voltage-gated ion channels (VGCC). Gi and Go proteins also can regulate K+ and Ca2+ channels directly through their βγ subunits. Protein kinase transduction pathways also affect the activities of transcription factors. Some of these factors, like cyclic adenosine monophosphate (cAMP) response element binding protein (CREB), are regulated posttranslationally by phosphorylation; others, like Fos, are regulated transcriptionally; still others, like Jun, are regulated both posttranslationally and/or transcriptionally. While membrane and cytoplasmic changes may be only local (e.g., dendritic domains or synaptic boutons), changes in the activity of transcription factors may result in long-term functional changes. These may include changes in gene expression of proteins involved in signal transduction and/or neurotransmission, resulting in altered neuronal responses. For example, chronic exposure to psychostimulants has been reported to increase levels of protein kinase A (PKA) and adenylyl cyclase in the nucleus accumbens and to decrease levels of Gαi. Chronic exposure to psychostimulants also alters the expression of transcription factors themselves. CREB expression, for instance, is depressed in the nucleus accumbens by chronic cocaine treatment. Chronic cocaine induces a transition from Fos induction to the induction of the much longer-lasting Fos-related antigens such as ΔFosB. Opioids, by acting on neurotransmitter systems, affect the phenotypic and functional properties of neurons through the general mechanisms outlined in the diagram. Shown are examples of ligand-gated ion channels such as the γ-aminobutyric acid-A (GABAA) and glutamate N-methyl-D-aspartate (NMDA) receptor (NMR) and G protein-coupled receptors such as opioid, dopamine (DA), or cannabinoid CB1 receptors, among others. These receptors modulate the levels of second messengers like cAMP and Ca2+, which in turn regulate the activity of protein kinase transducers. Chronic exposure to opioids has been reported to increase levels of PKA and adenylyl cyclase in the nucleus accumbens and to decrease levels of Gαi. Chronic exposure to opioids also alters the expression of transcription factors themselves. CREB expression, for instance, is depressed in the nucleus accumbens and increased in the locus coeruleus by chronic morphine treatment, while chronic opioid exposure activates Fos-related antigens such as ΔFosB. Alcohol, by acting on neurotransmitter systems, affects the phenotypic and functional properties of neurons through the general mechanisms outlined in the diagram. Shown are examples of ligand-gated ion channels such as the GABAA and the NMDA receptor and G protein-coupled receptors such as opioid, dopamine, or cannabinoid CB1 receptors, among others. The latter also are activated by endogenous cannabinoids such as anandamide. These receptors modulate the levels of second messengers such as cAMP and Ca2+, which in turn regulate the activity of protein kinase transducers. Such protein kinases affect the functions of proteins located in the cytoplasm, plasma membrane, and nucleus. Among membrane proteins affected are ligand-gated and VGCCs. Alcohol, for instance, has been proposed to affect the GABAA response via protein kinase C (PKC) phosphorylation. Gi and Go proteins also can regulate K+ and Ca2+ channels directly through their βγ subunits. Chronic exposure to alcohol has been reported to increase levels of PKA and adenylyl cyclase in the nucleus accumbens and to decrease levels of Giα. Moreover, chronic ethanol induces differential changes in subunit composition in the GABAA and glutamate inotropic receptors and increases expression of VGCCs. Chronic exposure to alcohol also alters the expression of transcription factors themselves. CREB expression, for instance, is increased in the nucleus accumbens and decreased in the amygdala by chronic alcohol treatment. Chronic alcohol induces a transition from Fos induction to the induction of the longer-lasting Fos-related antigens. Nicotine acts directly on ligand-gated ion channels. These receptors modulate the levels of Ca2+, which in turn regulate the activity of protein kinase transducers. Chronic exposure to nicotine has been reported to increase levels of PKA in the nucleus accumbens. Chronic exposure to nicotine also alters the expression of transcription factors themselves. CREB expression, for instance, is depressed in the amygdala and prefrontal cortex and increased in the nucleus accumbens and ventral tegmental area. Δ9-Tetrahydrocannabinol (THC), by acting on neurotransmitter systems, affects the phenotypic and functional properties of neurons through the general mechanisms outlined in the diagram. Cannabinoids act on the cannabinoid CB1 G protein-coupled receptor. The CB1 receptor also is activated by endogenous cannabinoids such as anandamide. This receptor modulates (inhibits) the levels of second messengers like cAMP and Ca2+, which in turn regulate the activity of protein kinase transducers. Chronic exposure to THC also alters the expression of transcription factors themselves. CaMK, Ca2+/calmodulin-dependent protein kinase; ELK-1, E-26-like protein 1; PLCβ, phosphlipase C β; IP3, inositol triphosphate; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; R, receptor. Modified with permission from Koob, Sanna, and Bloom, (1998).
The persistence of changes in drug reinforcement mechanisms that characterize drug addiction suggests that the underlying molecular mechanisms are long-lasting, and considerable attention has been given to the drug regulation of gene expression. Current research focuses on several types of transcription factors, including CREB and novel Fos-like proteins termed chronic Fos-related antigens. FosB, a member of the Fos family of transcription factors that dimerizes with a member of the Jun family to form activator protein-1 transcription factor complexes, has been implicated in the adaptation to chronic exposure to drugs of abuse. Activator protein-1 transcription factor complexes bind to sites in the regulatory regions of many genes. Acute administration of several drugs of abuse activates c-fos and Fos-related antigens, but chronic administration of drugs of abuse eliminates these activations and produces a gradual accumulation of FosB. This highly stable protein is hypothesized to function as a sustained molecular switch contributing to vulnerability to relapse after prolonged periods of abstinence (Nestler, 2001). These transcription factors may be possible mediators of chronic drug action. However, it has not yet been possible to relate regulation of a specific transcription factor to specific features of drug reinforcement or addiction.
Genetic and molecular genetic animal models have provided a convergence of data to provide hypotheses regarding vulnerability to addiction in some of the neuropharmacological substrates identified in neurocircuitry studies. High alcohol-preferring rats have been bred that show high voluntary consumption of alcohol, increased anxiety-like responses, and numerous neuropharmacological phenotypes such as decreased dopaminergic activity, increased CRF activity, and decreased neuropeptide Y activity, some of which have support from genetic studies of chromosomal linkage (Carr et al., 1998; McBride, Murphy, Lumeng, & Li, 1990; Murphy et al., 2002).
Box 41.8 Major Depressive Disorders
Feeling sad and blue are common and universal moods experienced in response to transient loss, stress, or disappointment. The clinical diagnosis of major depressive disorder (MDD) involves a persistent symptom complex that includes some combination of depressed mood, diminished interest or pleasure, changes in sleep and appetite, excessive guilt, agitation or retardation, loss of energy, diminished ability to think or concentrate, thoughts of death, and thoughts of suicide. These symptoms are recurrent and persistent and produce impairment.
Major depression is highly prevalent in all cultures and countries studied, and it is the most common psychiatric condition in medical patients. The community lifetime prevalence is approximately 12 to 15%, with the lowest rates in Asian countries and the highest in the Americas, Europe, and Australia. Even in countries with lower rates, it is not rare. The rates are two- to threefold higher in women than men. MDD is uncommon before puberty, with a marked increase in females during adolescence and following childbirth, and a decrease in first onset after menopause. Major depression with a first onset in the 60s or later is often associated with dementia or a medical condition.
Epidemiologic studies from the United States and elsewhere have documented higher lifetime rates of MDD in younger age groups. An analysis by birth cohort showed that rates of depression rose in cohorts born after World War II (the baby-boom generation). Numerous hypotheses noting the social changes that have occurred since that period have been considered, with no confirmed explanation. Follow-up epidemiologic studies carried out a decade or more later, in the 2000s, found the highest prevalence among the middle-age group (30–64 years), suggesting that the cohort born after WWII were carrying forth their high rates as they aged.
Different subtypes of major depression have been identified, including depression with psychotic features and a typical depression. Efforts to understand the heterogeneity of MDD by subtyping symptoms, however, are inherently difficult, as patients’ symptoms may change with different episodes. Major depression in the context of a history of mania (bipolar disorder) has the most severe course and requires treatment that prevents both the depressive and the manic symptoms.
Depression is an extremely disabling disorder. About 75% of patients have recurrent episodes. Ten to 30% recover incompletely, with persistent residual symptoms. MDD also complicates the course of cardiovascular illness, diabetes, hypertension, and other chronic medical conditions. Suicide, accidents, and the risk of death from heart disease are highest among the depressed. Because onset is early, recurrence rates are high, and the years lived with disability are high, MDD is considered, according to the World Health Organization, to be among the most disabling medical conditions in the world, similar to ischemic heart disease.
Rates of MDD are highest in persons who are divorced, separated, or widowed, and lowest in unmarried or never-married persons. These differences vary by country and are confounded by the difficulty in determining the sequence of divorce, separation, and depression. The one clear finding is that the ending of marriage, when it occurs, is associated with an increase in MDD.
The increased risk of MDD in first-degree relatives of a depressed patient is approximately two- to sixfold, depending on the severity of the disorder. This pattern of transmission is found across cultures and countries. Early-onset recurrent MDD has the highest relative risk, with estimates as high as eightfold. The best replicated findings are the high rates of MDD, anxiety disorders, and conduct disorder found in the biological young offspring of depressed as compared to nondepressed parents. These studies have been carried out in three generations and show that grandchildren with two generations affected are at extremely high risk of developing MDD at a young age.
The high familial rate of MDD across generations is supported by twin studies showing higher rates of depression in monozygotic twins, and an estimated heritability of 37 to 43%. These estimates are similar to those described for Type 2 diabetes. Several large-scale molecular genetic studies of MDD have been undertaken; most have focused on early-onset (<30 years) recurrent MDD, since this subtype has the highest familial rate. DNA and deidentified clinical information from these studies are stored in the National Institute of Mental Health (NIMH)’s national repositories for use by qualified scientists. As of 2012, a large Genome-Wide Association (GWA) study with participation by numerous investigators across the globe is underway. Many sequence variations in several genes have been reported, but no consistently replicated findings are yet available.
Availability of families at high risk for MDD (by virtue of having a FDR with the disorder) and the numerous positive clinical trials of drugs for MDD have led to a search for biomarkers or “endophenotypes” for MDD. By identifying subjects at risk for depression but not yet ill, this approach allows the disentangling of processes that present prior to the illness and are therefore more likely to be true risk factors. One study using magnetic resonance imaging (MRI) found 30% cortical thinning in the second and third generations of high-risk, as compared to low-risk, families that was independent of presence of MDD in the subject. Cortical thinning in areas that regulate emotion and cognitive stress response may thus be a familial trait marker for vulnerability to MDD. The first large-scale study to develop a platform for biomarkers using MRI, electroencephalography, behavioral tasks, and genetics to predict treatment response to MDD using selective serotonin reuptake inhibitors was initiated in 2011. Other promising areas for depression research include studies of circulatory levels of peripheral/serum growth factors and cytokines, which are altered in patients with MDD and which may be normalized or reversed with antidepressant treatment. There is agreement that a single biomarker approach may not be useful. The development of biomarker panels that profile a diverse array of peripheral/serum growth factors, cytokines, hormones, and metabolic markers may provide better coverage of the biologic abnormalities in MDD.
There are numerous studies on the efficacy of both medications and psychotherapy for depression. Treatment can hasten remission, may reduce suicide risk, and, if continued, can prevent relapse. Many of the pharmacologic agents with effective antidepressant action amplify serotonin or neurepinephrine signaling by inhibiting reuptake. There are also several evidence-based psychotherapies developed for MDD. They target the distorted thinking of depression (cognitive therapy) or stressful life events (grief, disputes, transitions) that trigger an episode (interpersonal psychotherapy). Drugs and psychotherapy in combination have been shown usually to have the best outcome, but many patients do not have access to, cannot afford, or do not want both. Both classes of treatment need to deal with the delayed response rate (3 to 4 weeks) and the high recurrence rate. Continuation or maintenance therapy (drugs or psychotherapy) is usually indicated.
Myrna M. Weissman and Ardesheer Talati
Advances in molecular biology have led to the ability to systematically inactivate the genes that control the expression of proteins that make up receptors or neurotransmitter/neuromodulators in the central nervous system using the gene knockout approach. Notable positive results with gene knockout studies in mice have focused on knockout of the μ opioid receptor, which eliminates opioid, nicotine, alcohol, and cannabinoid reward in mice (Contet, Kieffer, & Befort, 2004). Selective deletion of the genes for expression of the dopamine D1 and D2 receptor subtypes and the dopamine transporter has revealed significant decreases in activation and reward produced by psychomotor stimulants. Although developmental factors must be taken into account for the compensatory effect of deleting any one or a combination of genes, it is clear that D1 and D2 receptors and the dopamine transporter play important roles in the actions of psychomotor stimulants (Caine, Gabriel, Berkowitz, Zhang, & Xu, 2002).
References
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders 4th ed. Washington, DC: American Psychiatric Press; 1994.
2. Belin D, Jonkman S, Dickinson A, Robbins TW, Everitt BJ. Parallel and interactive learning processes within the basal ganglia: Relevance for the understanding of addiction. Behavioural Brain Research. 2009;199:89–102.
3. Berridge KC, Robinson TE. What is the role of dopamine in reward: Hedonic impact Reward learning, or incentive salience?. Brain Research Reviews. 1998;28:309–369.
4. Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. Journal of Neuroscience. 2005;25:9144–9151.
5. Bromberg-Martin ES, Hikosaka O. Dopamine in motivational control: Rewarding, aversive, and alerting. Neuron. 2010;68:815–834.
6. Caine SB, Gabriel KI, Berkowitz JS, Zhang J, Xu M. Decreased cocaine self-administration in dopamine D-1 receptor knockout mice. Drug and Alcohol Dependence. 2002;66:s25.
7. Caine SB, Lintz R, Koob GF. Intravenous drug self-administration techniques in animals. In: Sahgal A, ed. New York, NY: Oxford University Press; 1993:117–143. Behavioral neuro-science: A practical approach. Vol. 2.
8. Cardinal RN, Parkinson JA, Hall J, Everitt BJ. Emotion and motivation: The role of the amygdala, ventral striatum and prefrontal cortex. Neuroscience and Biobehavioral Reviews. 2002;26:321–352.
9. Carli M, Evenden JL, Robbins TW. Depletion of unilateral striatal dopamine impairs initiation of contralateral actions and not sensory attention. Nature (Lond.). 1985;313:679–682.
10. Carlisle HJ. The effects of preoptic and anterior hypothalamic lesions on behavioral thermoregulation in the cold. Journal of Comparative and Physiological Psychology. 1969;69:391–402.
11. Carr LG, Foroud T, Bice P, et al. A quantitative trait locus for alcohol consumption in selectively bred rat lines. Alcoholism, Clinical and Experimental Research. 1998;22:884–887.
12. Centers for Disease Control and Prevention. The health consequences of smoking: A report of the Surgeon General Atlanta, GA: US Department of Health and Human Services, CDC; 2004.
13. Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP. Limbic activation during cue-induced cocaine craving. American Journal of Psychiatry. 1999;156:11–18.
14. Contet C, Kieffer BL, Befort K. Mu opioid receptor: A gateway to drug addiction. Current Opinion in Neurobiology. 2004;14:370–378.
15. Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434.
16. Di Chiara G, North RA. Neurobiology of opiate abuse. Trends in Pharmacological Sciences. 1992;13:185–193.
17. Durana JH, Barnes PA. A neurodevelopmental view of impulsivity and its relationship to the superfactors of personality. In: McCown WG, Johnson J, Shure MB, eds. The impulsive client: Theory, research, and treatment. Washington, DC: American Psychological Association; 1993;23–37.
18. Engel J, Oreland L, Ingvar DH, Pernow B, Rossner S, Pellborn LA. Brain reward systems and abuse New York, NY: Raven Press; 1987.
19. Everitt BJ. Sexual motivation: A neural and behavioral analysis of the mechanisms underlying appetitive copulatory responses of male rats. Neuroscience and Biobehavioral Reviews. 1990;14:217–232.
20. Everitt BJ, Cardinal RN, Hall J, Parkinson JA, Robbins TW. Differential involvement of amygdala sub-systems in appetitive conditioning and drug addiction. In: Aggleton JP, ed. The amygdala: A functional analysis. Oxford: Oxford Universiry Press; 2000;353–390.
21. Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nature Neuroscience. 2005;8:1481–1489.
22. Fitzgerald LW, Nestler EJ. Molecular and cellular adaptations in signal transduction pathways following ethanol exposure. Clinical Neuroscience. 1995;3:165–173.
23. Flynn JP, Vanegas H, Foote W, Edwards S. Neural mechanisms involved in a cat’s attack on a rat. In: Whalen R, Thompson RF, Verzeano M, Weinberger NM, eds. The neural control of behaviour. New York, NY: Academic Press; 1970;135–173.
24. George O, Ghozland S, Azar MR, et al. CRF-CRF1 system activation mediates withdrawal-induced increases in nicotine self-administration in nicotine-dependent rats. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17198–17203.
25. Glimcher PW. Understanding dopamine and reinforcement learning: the dopamine reward prediction error hypothesis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15647–15654.
26. Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. Journal of Neuroscience. 2000;20:2369–2382.
27. Heimer L, Larsson K. Impairment of mating behaviour in male rats following lesions in the preoptic-anterior hypothalamic contimuum. Brain Research. 1966–1967;3:248–263.
28. Hikosaka O, Sesack SR, Lecourtier L, Shepard PD. Habenula: Crossroad between the basal ganglia and the limbic system. Journal of Neuroscience. 2008;28:11825–11829.
29. Hnasko TS, Sotak BN, Palmiter RD. Morphine reward in dopamine-deficient mice. Nature. 2005;438:854–857.
30. Hoebel BG. Brain reward and aversion systems in the control of feeding and sexual behavior. In: Cole J, Sonderegger T, eds. Nebraska Symposium on Motivation. Lincoln: University of Nebraska Press; 1974;49–112.
31. Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annual Review Neuroscience. 2006;29:565–598.
32. Justinova Z, Solinas M, Tanda G, Redhi GH, Goldberg SR. The endogenous cannabinoid anandamide and its synthetic analog R(+)-methanandamide are intravenously self-administered by squirrel monkeys. Journal of Neuroscience. 2005;25:5645–5650.
33. Justinova Z, Tanda G, Munzar P, Goldberg SR. The opioid antagonist naltrexone reduces the reinforcing effects of delta 9 tetrahydrocannabinol (THC) in squirrel monkeys. Psychopharmacology. 2004;173:186–194.
34. Kalivas PW, McFarland K. Brain circuitry and the reinstatement of cocaine-seeking behavior. Psychopharmacology. 2003;168:44–56.
35. Kelley AE, Berridge KC. The neuroscience of natural rewards: Relevance to addictive drugs. Journal of Neuroscience. 2002;22:3309–3311.
36. Koob GF. Drugs of abuse: Anatomy, pharmacology, and function of reward pathways. Trends in Pharmacological Sciences. 1992;13:177–184.
37. Koob GF. Corticotropin-releasing factor, norepinephrine and stress. Biological Psychiatry. 1999;46:1167–1180.
38. Koob GF. Neurobiology of addiction. In: Galanter M, Kleber HD, eds. Textbook of substance abuse treatment. 4th ed. Washington, DC: American Psychiatric Publishing; 2008;3–16.
39. Koob GF. Theoretical frameworks and mechanistic aspects of alcohol addiction: Alcohol addiction as a reward deficit disorder. In: Spanagel R, Sommer W, eds. Behavioral neurobiology of alcohol addiction. New York, NY: Springer; 2011; in press.
40. Koob GF, Bartfai T, Roberts AJ. The use of molecular genetic approaches in the neuropharmacology of corticotropin-releasing factor. International Journal of Comparative Psychology. 2001;14:90–110.
41. Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988;242:715–723.
42. Koob GF, Kandel D, Volkow ND. Pathophysiology of addiction. In: Tasman A, Kay J, Lieberman JA, First MB, Maj M, eds. 3rd ed. Chichester: Wiley; 2008;354–378. Psychiatry. Vol. 1.
43. Koob GF, Le Moal M. Drug abuse: Hedonic homeostatic dysregulation. Science. 1997;278:52–58.
44. Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129.
45. Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nature Neuroscience. 2005;8:1442–1444.
46. Koob GF, Riley S, Smith SC, Robbins TW. Effects of 6-hydroxydopamine lesions to nucleus accumbens septi and olfactory tubercle on food intake, locomotor activity and amphetamine anorexia in the rat. Journal of Comparative and Physiological Psychology. 1978;92:917–927.
47. Koob GF, Sanna PP, Bloom FE. Neuroscience of addiction. Neuron. 1998;21:467–476.
48. Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacol Reviews. 2010;35:217–238.
49. Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin κ-opioid system. Journal of Neuroscience. 2008;28:407–414.
50. Leibowitz SF. Neurochemical systems of the hypothalamus: Control of feeding and drinking behaviour and water-electrolyte excretion. In: Morgan PJ, Panksepp J, eds. New York: NY: Raven Press; 1980;299–437. Handbook of the hypothalamus. Vol. 3, Part A.
51. Markou A, Koob GF. Construct validity of a self-stimulation threshold paradigm: Effects of reward and performance manipulations. Physiology and Behavior. 1992;51:111–119.
52. Markou A, Kosten TR, Koob GF. Neurobiological similarities in depression and drug dependence: A self-medication hypothesis. Neuropsychopharmacology. 1998;18:135–174.
53. Marshall JF, Teitelbaum P. New considerations in the neuropsychology of motivated behaviors. In: Iversen LL, Iversen SD, Snyder SH, eds. New York, NY: Plenum; 1977;201–229. Handbook of psychopharmacology. Vol. 7.
54. Martinez D, Gil R, Slifstein M, Hwang DR, Huang Y, Perez A. Alcohol dependence is associated with blunted dopamine transmission in the ventral striatum. Biological Psychiatry. 2005;58:779–786.
55. Matsumoto M, Hikosaka O. Two types of dopamine neurons distinctly convey positive and negative signals. Nature. 2009;459 837–431.
56. McBride WJ, Murphy JM, Lumeng L, Li TK. Serotonin, dopamine and GABA involvement in alcohol drinking of selectively bred rats. Alcohol. 1990;7:199–205.
57. McLaughlin JP, Marton-Popovici M, Chavkin C. κ Opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. Journal of Neuroscience. 2003;23:5674–5683.
58. Melis M, Spiga S, Diana M. The dopamine hypothesis of drug addiction: hypodopaminergic state. International Review of Neurobiology. 2005;63:101–154.
59. Merlo-Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Koob GF, Weiss F. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and alcohol withdrawal as measured by microdialysis. Journal of Neuroscience. 1995;15:5439–5447.
60. Mogenson GJ, Jones DL, Yim CY. From motivation to action: Functional interface between the limbic system and the motor system. Progress in Neurobiology. 1980;14:69–97.
61. Morrison SE, Salzman CD. Re-valuing the amygdala. Current Opinion in Neurobiology. 2010;20:221–230.
62. Murphy JM, Stewart RB, Bell RL, et al. Phenotypic and genotypic characterization of the Indiana University rat lines selectively bred for high and low alcohol preference. Behavior Genetics. 2002;32:363–388.
63. National Drug Intelligence Center. The Economic impact of illicit drug use on American society [Product No 2011-Q0317-002] Washington DC: U.S. Department of Justice; 2011.
64. Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nature Reviews Neuroscience. 2001;2:119–128.
65. Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. Journal of Comparative and Physiological Psychology. 1954;47:419–427.
66. Parsons LH, Koob GF, Weiss F. Serotonin dysfunction in the nucleus accumbens of rats during withdrawal after unlimited access to intravenous cocaine. The Journal of Pharmacology and Experimental Therapeutics. 1995;274:1182–1191.
67. Pfaff DW. The physiological mechanisms of motivation New York, NY: Springer-Verlag; 1982; (pp. 217–251).
68. Pfaus JG. Pathways of sexual desire. Journal of Sexual Medicine. 2009;6:1506–1533.
69. Phillips AG. Brain reward circuitry: A case for separate systems. Brain Research Bulletin. 1984;12:195–201.
70. Picciotto MR, Zoli M, Rimondini R, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177.
71. Richter RM, Weiss F. In vivo CRF release in rat amygdala is increased during cocaine withdrawal in self-administering rats. Synapse. 1999;32:254–261.
72. Robbins TW. Hunger. In: Lightman SL, Everitt BJ, eds. Neuroendocrinology. Oxford: Blackwell; 1986;252–303.
73. Robbins TW, Everitt BJ. Arousal systems and attention. In: Gazzaniga MS, ed. The cognitive neurosciences. Cambridge, MA: MIT Press; 1995;703–720.
74. Robbins TW, Taylor JR, Cador M, Everitt BJ. Limbic-striatal interactions and reward-related processes. Neuroscience and Biobehavioral Reviews. 1989;13:155–162.
75. Roberts AJ, Cole M, Koob GF. Intra-amygdala muscimol decreases operant ethanol self-administration in dependent rats. Alcoholism, Clinical and Experimental Research. 1996;20:1289–1298.
76. Robinson TE, Berridge KC. The neural basis of drug craving: An incentive-sensitization theory of addiction. Brain Research Reviews. 1993;18:247–291.
77. Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nature Review Neuroscience. 2011;12:623–637.
78. Rodd ZA, Bell RL, Zhang Y, et al. Regional heterogeneity for the intracranial self-administration of ethanol and acetaldehyde within the ventral tegmental area of alcohol-preferring (P) rats: Involvement of dopamine and serotonin. Neuropsychopharmacology. 2005;30:330–338.
79. Rodriguez de Fonseca F, Carrera MRA, Navarro M, Koob GF, Weiss F. Activation of corticotropin-releasing factor in the limbic system during cannabinoid withdrawal. Science. 1997;276:2050–2054.
80. Rolls ET. The neurophysiology of feeding. In: Sandler M, Silverstone T, eds. Psychopharmacology and food. Oxford: Oxford University Press; 1985;1–16.
81. Sarnyai Z, Shaham Y, Heinrichs SC. The role of corticotropin-releasing factor in drug addiction. Pharmacological Reviews. 2001;53:209–243.
82. Satinoff E. Are there similarities between thermoregulation and sexual behaviour?. In: Pfaff DW, ed. The physiological mechanisms of motivation. New York, NY: Springer-Verlag; 1982;217–251.
83. Schultz W, Romo R, Ljungberg T, Mirenowicz J, Hollerman JR, Dickinson A. Reward-related signals carried by dopamine neurons. In: Houk JR, Davis JL, Beiser D, eds. Models of information processing in the basal ganglia. Cambridge, MA: MIT Press; 1995;233–248.
84. Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599.
85. Schuster CR, Thompson T. Self administration and behavioral dependence on drugs. Annual Review of Pharmacology and Toxicology. 1969;9:483–502.
86. Shalev U, Grimm JW, Shaham Y. Neurobiology of relapse to heroin and cocaine seeking: A review. Pharmacological Reviews. 2002;54:1–42.
87. Shaw-Lutchman TZ, Barrot M, Wallace T, et al. Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. Journal of Neuroscience. 2002;22:3663–3672.
88. Solomon RL. The opponent-process theory of acquired motivation: The affective dynamics of addiction. In: Maser JD, Seligman MEP, eds. Psychopathology: Experimental models. San Francisco: Freeman; 1977;124–145.
89. Solomon RL. The opponent-process theory of acquired motivation: The costs of pleasure and the benefits of pain. American Psychology. 1980;35:691–712.
90. Solomon RL, Corbit JD. An opponent-process theory of motivation: I Temporal dynamics of affect. Psychological Reviews. 1974;81:119–145.
91. Stein JJ. Substance abuse: The nation’s number one health problem Princeton NJ: Schneider Institute for Health Care Policy, Robert Wood Johnson Foundation; 2001.
92. Substance Abuse and Mental Health Services Administration (2010). Results from the 2009 National Survey on Drug Use and Health: Volume I. Summary of National Findings (Office of Applied Studies, NSDUH Series H-38A, HHS Publication No. SMA 10-4856). Rockville, MD.
93. Tabakoff B, Hoffman PL. Alcohol: Neurobiology. In: Lowenstein JH, Ruiz P, Millman RB, eds. Substance Abuse: A Comprehensive Textbook. 2nd Ed Baltimore, MD: Williams & Wilkins; 1992;152–185.
94. Tiffany ST, Carter BL, Singleton EG. Challenges in the manipulation, assessment and interpretation of craving relevant variables. Addiction. 2000;95:s177–s187.
95. Ungerstedt U. Adipsia and aphagia after 6-hydroxydopamine induced degeneration of the nigrostriatal dopamine system. Acta physiologica Scandinavica Supplementum. 1971a;367:95–122.
96. Ungerstedt U. Striatal dopamine release after amphetamine or nerve degeneration revealed by rotational behavior. Acta physiologica Scandinavica Supplementum. 1971b;367:49–68.
97. Vanderschuren LJ, Everitt BJ. Drug seeking becomes compulsive after prolonged cocaine self-administration. Science. 2004;305:1017–1019.
98. Volkow ND, Fowler JS, Wang GJ, et al. Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse. 1993;14:169–177.
99. Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, Hitzemann R. Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature. 1997;386:830–833.
100. Walker BM, Rasmussen DD, Raskind MA, Koob GF. α1-Noradrenergic receptor antagonism blocks dependence-induced increases in responding for ethanol. Alcohol. 2008;42:91–97.
101. Walker SC, Robbins TW, Roberts AC. Response disengagement on a spatial self-ordered sequencing task: Effects of regionally selective excitotoxic lesions and serotonin depletion within the prefrontal cortex. Journal of Neuroscience. 2009;29:6033–6041.
102. Weiss F, Ciccocioppo R, Parsons LH, et al. Compulsive drug-seeking behavior and relapse: neuroadaptation, stress, and conditioning factors. In: Quinones-Jenab V, ed. New York, NY: New York Academy of Sciences; 2001;1–26. The biological basis of cocaine addiction. Vol. 937.
103. Weiss F, Markou A, Lorang MT, Koob GF. Basal extracellular dopamine levels in the nucleus accumbens are decreased during cocaine withdrawal after unlimited-access self-administration. Brain Research. 1992;593:314–318.
104. Weiss F, Parsons LH, Schulteis G, et al. Ethanol self-administration restores withdrawal-associated deficiencies in accumbal dopamine and 5-hydroxytryptamine release in dependent rats. Journal of Neuroscience. 1996;16:3474–3485.
105. Wise R. Neuroleptics and operant behavior: The anhedonia hypothesis. Behavioral and Brain Sciences. 1982;5:39–87.
106. Wolffgramm J, Heyne A. From controlled drug intake to loss of control: The irreversible development of drug addiction in the rat. Behavioural Brain Research. 1995;70:77–94.