Chapter 30
The Basal Ganglia
The basal ganglia and the cerebellum are large subcortical components of the motor system. They act in parallel, influencing both cortical and brainstem motor areas to ensure efficient, focused, and well-coordinated movements. Although they communicate with the same areas of cerebral cortex and with each other, they have distinct functions and mechanisms of action. They communicate with motor areas of cerebral cortex privately using separate thalamic nuclei. The outputs have opposite signs: the basal ganglia output is inhibitory and the cerebellar output is excitatory. The cerebellum receives direct input from spinal cord ascending pathways and from the vestibular system, whereas the basal ganglia receives the bulk of its sensory information from cerebral cortex. In human diseases, basal ganglia dysfunction results in changes in the slow voluntary movements or the development of involuntary movements; cerebellar dysfunction results in impaired coordination. In addition to their motor functions, both basal ganglia and cerebellum play roles in nonmotor behavior, including cognition, emotion, and possibly others. The basal ganglia are discussed in this chapter and the cerebellum in the next (Chapter 31).
The basal ganglia comprise several interconnected nuclei in the forebrain, diencephalon, and midbrain (Fig. 30.1). They are an important part of the motor system, and motor control is a large part of basal ganglia function: (1) the largest portion of basal ganglia inputs and outputs are connected with motor areas; (2) the discharge of many basal ganglia neurons correlates with movement; and (3) basal ganglia lesions cause severe movement abnormalities. In view of these features, this chapter concentrates on the motor functions of the basal ganglia. Participation of the basal ganglia in nonmotor functions is discussed after the basic framework of basal ganglia function is presented for motor control. The anatomy of the basal ganglia is discussed first, since it is anatomy that provides the infrastructure for the physiology. Next we will consider the activity of the individual components of the basal ganglia during movement. Then we will review the effect of placing a lesion in one basal ganglia component while leaving the rest of the nervous system intact. The combined strategy of measuring the activity of a structure during movement and then determining the effect of a selective lesion on that movement has been fruitful in studying the function of the motor system and understanding the effects of human basal ganglia diseases. After presentation of current models of basal ganglia motor function, the chapter concludes with a discussion of basal ganglia participation in cognitive function.
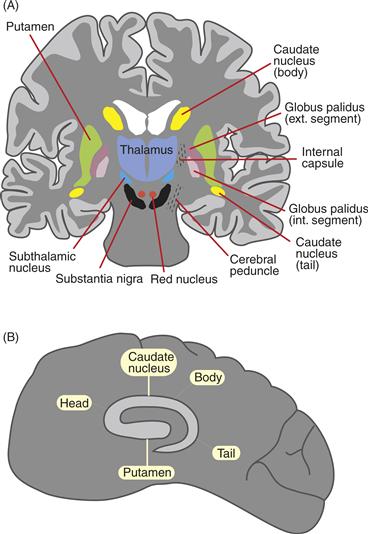
Figure 30.1 Location of basal ganglia in the human brain. (A) Coronal section. (B) Parasagittal section.
Anatomy of the Basal Ganglia
The basal ganglia receive a broad spectrum of cortical inputs. The information conveyed to the basal ganglia is processed to produce a focused output to areas of the frontal lobes and brainstem that are involved in the planning and production of movement. The basal ganglia output is inhibitory, which means that an increase in basal ganglia output leads to a reduction in the activity of its targets. The fact that the basal ganglia output is inhibitory to other motor mechanisms is key to understanding its normal function.
At first glance, the anatomy of the basal ganglia may seem confusing. There are several component nuclei at different levels in the brain, and two of the nuclei [substantia nigra (SN) and globus pallidus] are divided into functionally different components. The names of some structures are different in primates than in other mammals. Furthermore, the terminology has changed over the years so that historically an individual structure has been known by more than one name. However, with consistent use of modern terminology and a functional context in which to place the anatomy, the organization of basal ganglia can be learned readily. Once the organization is learned, many aspects of normal function and the response to injury become easier to understand.
The basal ganglia include the striatum (caudate, putamen, nucleus accumbens); the subthalamic nucleus (STN); the globus pallidus (internal segment—GPi; external segment—GPe; and ventral pallidum—VP); and the substantia nigra (pars compacta—SNpc; and pars reticulata—SNpr) (Fig. 30.2). The striatum and STN receive the majority of their inputs from outside the basal ganglia. Most of those inputs come from the cerebral cortex, but thalamic nuclei also provide strong inputs to striatum. There are no direct inputs from peripheral sensory or motor systems. The bulk of the outputs from basal ganglia arise from GPi and SNpr and are inhibitory to thalamic nuclei and to the brainstem. There are no direct outputs from basal ganglia to spinal motor circuitry.
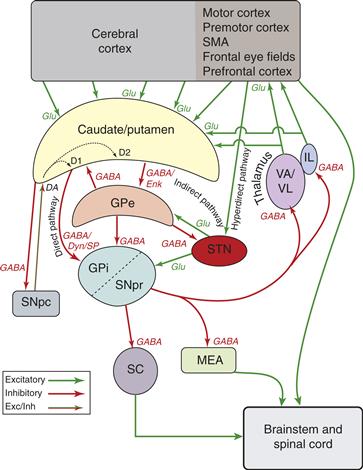
Figure 30.2 Simplified schematic diagram of basal ganglia circuitry. Excitatory connections are indicated by green arrows, inhibitory connections by red arrows, and the modulatory dopamine projection is indicated by a red and green arrow. GPe, globus pallidus, external segment; GPi, globus pallidus, internal segment; IL, intralaminar thalamic nuclei; MD, mediodorsal thalamic nucleus; MEA, midbrain extrapyramidal area; SC, superior colliculus; SNpc, substantia nigra pars compacta; SNpr, substantia nigra, pars reticulata; STN, subthalamic nucleus; VA, ventral anterior thalamic nucleus; VL, ventral lateral thalamic nucleus; DA, dopamine (with D1 and D2 receptor subtypes); Dyn, dynorphin; Enk, enkephalin; GABA, γ-aminobutyric acid; Glu, glutamate; SP, substance P.
The Striatum Receives Most of the Inputs to the Basal Ganglia
The striatum is located in the forebrain and comprises the caudate nucleus and putamen (neostriatum) and nucleus accumbens (ventral striatum). This section focuses on the neostriatum (see Box 30.3 and Chapter 41 for a discussion of the ventral system of the basal ganglia). Embryologically, the striatum develops from a basal region of the lateral telencephalic vesicle. It is named the striatum because axon fibers passing through the striatum give it a striped appearance. In rodents, the caudate and putamen form a single structure (caudatoputamen) with fibers of the internal capsule coursing through, but in carnivores and primates, the caudate and putamen are separated by the internal capsule (Fig. 30.1A).
Four major types of neurons have been described in the striatum, based on the size of the cell body, presence or absence of dendritic spines in individual neurons that have been stained with the Golgi technique or filled with horseradish peroxidase, and other staining properties. The first and by far the most numerous neuron type is the medium spiny neuron (Fig. 30.3). Medium spiny neurons make up 90 to 95% of the total number of striatal neurons. They utilize γ-aminobutyric acid (GABA) as a transmitter and constitute the output of the striatum, sending axonal projections to the GP and SN. Medium spiny neurons have large dendritic trees that span 200 to 500 μm. They also have extensive local axon collaterals that inhibit neighboring striatal neurons. The medium spiny neurons are morphologically homogeneous, but chemically heterogeneous, as discussed later. The second, large aspiny neurons, make up 1 to 2% of the striatal population. They are interneurons and use acetylcholine (ACh) as a neurotransmitter. They have extensive axon collaterals within the striatum that terminate on medium spiny neurons. The third type, small aspiny neurons, make up 2 to 5% of the striatal population. They stain positive for parvalbumin and use GABA as a neurotransmitter. The fourth type is the medium aspiny neuron. These are also interneurons and are thought to use somatostatin as a neurotransmitter.
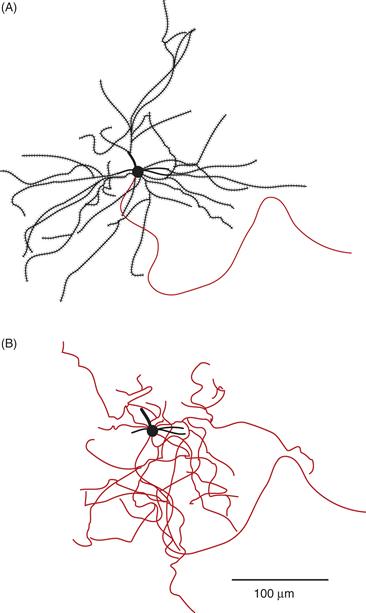
Figure 30.3 Two representations of a striatal medium spiny neuron that has been filled with HRP. (A) The soma and dendritic tree with numerous dendritic spines. The thin process is the axon, which has been drawn without its collaterals. (B) The same neuron drawn to show the axonal collaterals, which branch extensively within the same field as the dendritic tree.
From Wilson and Groves (1980).
The striatum receives excitatory input from nearly all of the cerebral cortex. The cortical input uses glutamate as its neurotransmitter and terminates largely on the heads of the dendritic spines of medium spiny neurons (Fig. 30.4). The projection from the cerebral cortex to the striatum has a roughly topographical organization. For example, the somatosensory and motor cortex project to the posterior putamen and the prefrontal cortex projects to the anterior caudate. Within the somatosensory and motor projection to the striatum, there is a preservation of somatotopy. It has been suggested that the topographic relationship between the cerebral cortex and the striatum provides a basis for the segregation of functionally different circuits in the basal ganglia (Kelly & Strick, 2004) (Fig. 30.5). These circuits include somatomotor, oculomotor, cognitive, and limbic connections. Within each circuit there appear to be subcircuits such that the primary motor cortex and premotor cortex have nonidentical connections with basal ganglia structures. Likewise, dorsolateral and orbitofrontal circuits have distinct connectivity patterns.
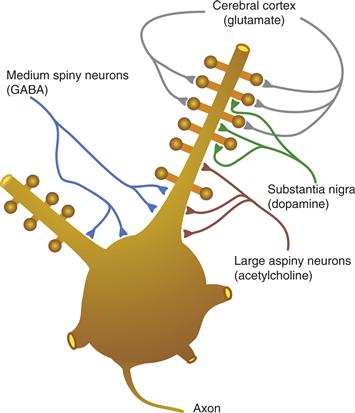
Figure 30.4 Pattern of termination of afferents on a medium spiny neuron. The soma and the proximal dendrites with their spines are shown.
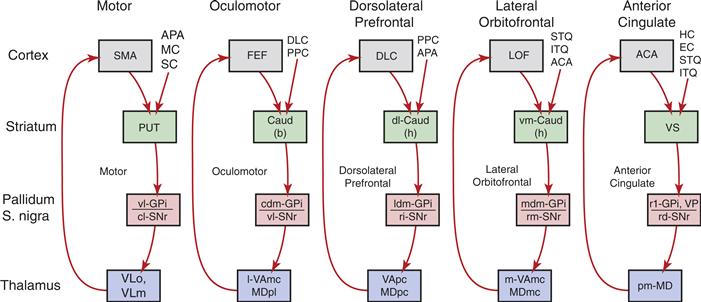
Figure 30.5 Hypothetical parallel segregated circuits connecting the basal ganglia, thalamus, and cerebral cortex. The five circuits are named according to the primary cortical target of the output from the basal ganglia: motor, oculomotor, dorsolateral prefrontal, lateral orbitofrontal, and anterior cingulate. ACA, anterior cingulate area; APA, arcuate premotor area; CAUD, caudate; b, body; h, head; DLC, dorsolateral prefrontal cortex; EC, entorhinal cortex; FEF, frontal eye fields; GPi, internal segment of globus pallidus; HC, hippocampal cortex; ITG, inferior temporal gyrus; LOF, lateral orbitofrontal cortex; MC, motor cortex; MDpl, medialis dorsalis pars paralarnellaris; MDme, medialis dorsalis pars magnocellularis; MDpc, medialis dorsalis pars parvocellularis; PPC, posterior parietal cortex; PUT, putamen; SC, somatosensory cortex; SMA, supplementary motor area; SNr, substantia nigra pars reticulate; STG, superior temporal gyrus; VAmc, ventralis anterior pars magnocellularis; Vapc, ventralis anterior pars parvocellularis; VLm, ventralis lateralis pars medialis; VLo, ventralis lateralis pars oralis; VP, ventral pallidum; VS, ventral striatum, cl, caudolateral; cdm, caudal dorsomedial; d1, dorsolateral; 1, lateral; 1dm, lateral dorsomedial; m, medial; mdm, medial dorsomedial; pm, posteromedial; rd, rostrodorsal; rl, rostrolateral; rm, rostromedial; vm, ventromedial; vl, ventrolateral.
Although the topography and somatotopy imply a certain degree of parallel organization, there are also convergence and divergence in the corticostriatal projection. The large dendritic fields of medium spiny neurons allow them to receive input from adjacent projections, which arise from different areas of cortex. Inputs from more than one cortical area overlap, and input from a single cortical area projects divergently to multiple striatal zones (Fig. 30.6). This convergent and divergent organization provides an anatomical framework for the integration and transformation of information from several areas of the cerebral cortex.
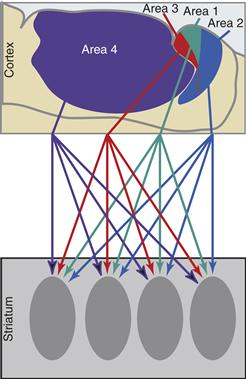
Figure 30.6 Schematic representation of the sensorimotor cortical projection to the striatum from arm areas in the somatosensory cortex (areas 1, 2, and 3) and motor cortex (area 4). Note that each cortical area projects to several striatal zones and several functionally related cortical areas project to a single striatal zone.
After Flaherty and Graybiel (1991).
In addition to cortical input, medium spiny striatal neurons receive a number of other inputs, including (1) excitatory glutamatergic inputs from intralaminar and ventrolateral nuclei of the thalamus; (2) cholinergic input from large aspiny neurons; (3) GABA, substance P, and enkephalin input from adjacent medium spiny striatal neurons; (4) GABA input from small interneurons; and (5) a large input from dopamine (DA)-containing neurons in the SNpc. The DA input is of particular interest because of its role in Parkinson disease (see Box 30.1).
Box 30.1 Parkinson’s Disease
Parkinson’s disease (PD) is the second most common progressive neurodegenerative disorder. Patients with PD experience slowness of movement, rigidity, a low-frequency rest tremor, and difficulty with balance. These major motoric features of PD are due to the degeneration of dopamine (DA) containing neurons in the substantia nigra pars compacta (SNC) with an accompanying loss of DA and its metabolites in the striatum. PD patients may exhibit other symptoms, including cognitive dysfunction, depression, anxiety, autonomic problems, and disturbances of sleep, which may be due to the degeneration of non-DA-containing neurons. PD is characterized pathologically by the cytoplasmic accumulation of aggregated proteins with a halo of radiating fibrils and a less defined core known as Lewy bodies. Measures of increased oxidative stress are also seen, including glutathione depletion, iron deposition, increased markers of lipid peroxidation, oxidative DNA damage and protein oxidation, and decreased expression and activity of mitochondrial complex 1 in the SNC. This mitochondrial complex 1 defect is specific to the SNC, as it has not been observed in other neurodegenerative diseases. Thus, in sporadic PD, oxidative stress and mitochondrial dysfunction appear to play prominent roles in the death of DA neurons. The majority of PD appears to be sporadic in nature, but an estimated 10% of cases are familial, with specific genetic defects. The identification of these rare genes and their functions has provided tremendous insight into the pathogenesis of PD and opened up new areas of investigation.
Several genes have been clearly linked to PD. The best studied are α-synuclein and parkin. α-Synuclein is a presynaptic protein and a primary component of Lewy bodies. Very rare missense mutations at A53T and A30P cause autosomal-dominant PD. How derangements in α-synuclein lead to dysfunction and death of neurons is not known. However, aggregation and fibrillization are thought to play a central role, perhaps leading to a dysfunction in protein handling by inhibiting the proteasome. The second “PD gene” parkin is an E3 ligase for the ubiquitination of proteins. Mutations in parkin include exonic deletions, insertions, and several missense mutations and result in autosomal recessive PD. Parkin mutations are now considered to be one of the major causes of familial PD. Substrates for parkin include the synaptic vesicle–associated protein CDCrel-1, which may regulate synaptic vesicle release in the nervous system. Whether CDCrel-1 is involved in the release of dopamine is not yet known. Synphilin-1 is a protein of unknown function that was identified as an α-synuclein interacting protein. It is a synaptic vesicle-enriched protein and is present in Lewy bodies. Synphilin-1 is a target of parkin, and expression of synphilin-1 with α-synuclein results in the formation of Lewy body–like aggregates that are ubiquitinated in the presence of parkin. Parkin is upregulated by unfolded protein stress, and expression of parkin suppresses unfolded protein stress-induced toxicity. The unfolded putative G-protein-coupled transmembrane receptor, the parkin-associated, endothelial-like receptor (Pael-R), is a parkin substrate. When over-expressed, Pael-R becomes unfolded, insoluble, and causes unfolded protein-induced cell death. Parkin ubiquitinates Pael-R, and coexpression of parkin results in protection against Pael-R-induced cell toxicity. Pael-R accumulates in the brains of autosomal-recessive Parkinson’s disease patients and thus may be an important parkin substrate.
Ultimately, it would be of interest to link all the multiple genes and the sporadic causes of PD into a common pathogenic biochemical pathway. Derangements in protein handling seem to be central in the pathogenesis of familial PD. Oxidative stress seems to play a prominent role in sporadic PD, and oxidative stress leads to synuclein aggregation and/or proteasomal dysfunction. Data suggest that there may be selective derangements in the proteasomal system in the substantia nigra of sporadic PD. Thus, protein mishandling may be central to the pathogenesis of PD.
In contrast to most other neurodegenerative disorders, there is effective temporary symptomatic treatment for PD consisting of DA replacement with levodopa or DA agonists and adjunctive medications or surgical approaches. Because the neurodegeneration in PD is progressive and there is no proven preventative, restorative, or regenerative therapy, patients eventually become quite disabled. Ultimately, to affect a true cure, the underlying mechanisms of neuronal cell death need to be understood, strategies for enhancing neuronal survival and regrowth need to be developed, and consideration needs to be made toward the replacement of cells lost during the degenerative process. If these goals can be obtained, a full recovery could be achieved.
Valina L. Dawson and Ted M. Dawson
1. Adapted from Dawson T. M. (2000). New animal models for parkinson’s disease. Cell, 101(2), 115–118.
2. Adapted from Zhang Y., Dawson V. L., & Dawson T. M. Oxidative stress and genetics in the pathogenesis of parkinson’s disease. Neurobiology of Disease, 7(4), 240–250.
The DA input to the striatum terminates largely on the shafts of the dendritic spines of medium spiny neurons (Fig. 30.4). The location of dopaminergic terminals puts them in a position to modulate transmission from the cerebral cortex to the striatum. The action of DA on striatal neurons depends on the type of DA receptor involved. Five types of G-protein-coupled DA receptors have been described (D1–D5). These have been grouped into two families based on their response to agonists. The D1 family includes D1 and D5 receptors, and the D2 family includes D2, D3, and D4 receptors. D1 receptors stimulate adenylate cyclase activity and potentiate the effect of cortical input to striatal neurons. D2 receptors inhibit adenylate cyclase activity and decrease the effect of cortical input to striatal neurons (see Chapters 5, 7, and 8).
Medium spiny neurons contain the inhibitory neurotransmitter GABA. In addition, medium spiny neurons have peptide neurotransmitters that are co-localized with GABA. Based on both the type of neurotransmitters and the type of DA receptor they contain, medium spiny neurons can be divided into two populations. One population contains GABA, dynorphin, and substance P and primarily expresses D1 receptors. These neurons send axons to GPi and to SNpr and constitute the “direct pathway” (Fig. 30.2). The predominant effect of these neurons is inhibition of their targets. The second population contains GABA and enkephalin and primarily expresses D2 receptors. These inhibitory neurons project to GPe and form the first limb of the “indirect pathway” (Fig. 30.2). The two populations of medium spiny neurons are morphologically indistinguishable and are not segregated topographically within the striatum (Fig. 30.7). This comingling of neurons suggests that they receive similar input and thus they may convey similar information to their respective targets. However, the different targets and transmitter types of the two populations provides the substrate for functional segregation within the striatum.
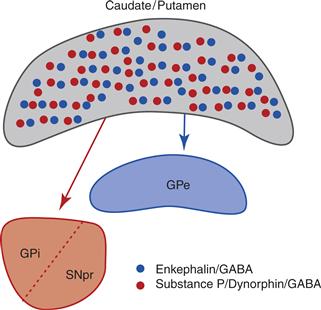
Figure 30.7 The two chemically different populations of striatal medium spiny neurons are intermixed. One population (blue) projects to the globus pallidus external segment (GPe) and contains GABA and enkephalin. The other population (red) projects to the globus pallidus internal segment (GPi) and substantia nigra pars reticulata (SNpr) and contains GABA, dynorphin, and substance P (red).
Although there are no apparent regional differences in the striatum based on cell morphology, an intricate internal organization has been revealed with special stains. When the striatum is stained for acetyl-cholinesterase (AChE), there is a patchy distribution of lightly staining regions within more heavily stained regions. The AChE-poor patches have been called striosomes and the AChE-rich areas have been called the extrastriosomal matrix. The matrix forms the bulk of the striatal volume and receives input from most areas of the cerebral cortex. Within the matrix are clusters of neurons with similar inputs that have been termed matrisomes. The bulk of the output from cells in the matrix goes to both segments of the GP and to SNpr. The striosomes receive input from the prefrontal cortex and send output to SNpc. Immuno-histochemical techniques have demonstrated that several chemicals, including substance P, dynorphin, and enkephalin, have a patchy distribution that may be partly or wholly in register with the striosomes. The striosome-matrix organization provides the substrate for another level of functional segregation within the striatum.
The Subthalamic Nucleus Receives Inputs from the Frontal Lobe
The STN is located at the junction of the diencephalon and midbrain, ventral to the thalamus and rostral and lateral to the red nucleus. Embryologically, it develops from the lateral hypothalamic cell column. The STN receives an excitatory, glutamatergic input from the frontal cortex with large contributions from motor areas, including the primary motor cortex, premotor and supplementary motor cortex, and frontal eye fields. The STN also receives an inhibitory GABA input from GPe. The output from the STN is excitatory and glutamatergic. STN projects to GPi and SNpr as well, projecting back to GPe.
Although the STN receives input from the neocortex and projects to both segments of GP and to SNpr, it is different from striatum in several ways. Unlike striatum, the cortical input to the STN is predominately from the frontal lobe. The output from STN is excitatory, whereas the output from striatum is inhibitory. Of the two routes from the cortex to GPi, GPe, and SNpr, the excitatory route through STN (5–8 ms) is faster than the inhibitory route through striatum (15–20 ms).
Globus Pallidus, Internal Segment and Substantia Nigra, and Pars Reticulata Are the Basal Ganglia Output Nuclei
GP lies medial to the putamen and rostral to the hypothalamus. Embryologically, it arises from the lateral hypothalamic cell column. In primates, GP is separated into an internal segment (GPi) and an external segment (GPe) by a fiber tract called the internal medullary lamina. In rodents and carnivores, the internal segment lies within the internal capsule and is called the entopeduncular nucleus.
The GPi is composed primarily of large neurons that project outside of the basal ganglia. The dendritic trees of GPi neurons radiate from the cell body in a disc-like distribution and are oriented so that the faces of the disc are perpendicular to incoming axons from the striatum. The dendrites of an individual GPi cell can span up to 1 mm in diameter, and therefore GPi neurons have the potential to receive a large number of converging inputs.
The principal inputs to GPi are from striatum and the STN. These two types of inputs differ in the neurotransmitters used, in their physiological action, and in their pattern of termination. As noted earlier, neurons projecting from striatum to GPi contain GABA, substance P, and dynorphin and are inhibitory. Each axon from the striatum enters GPi and sparsely contacts several neurons in passing before ensheathing a single neuron with a dense termination (Fig. 30.8). The excitatory, glutamate projection from STN to GPi is highly divergent such that each axon from the STN ensheathes many GPi neurons (Fig. 30.8). The cortico-STN-GPi pathway has been called the “hyperdirect” pathway. Thus, the striatal and STN inputs to GPi form a pattern of slow, focused, convergent inhibition from striatum (“direct pathway”) and fast, widespread, divergent excitation from STN (“hyperdirect pathway”) (Fig. 30.2).
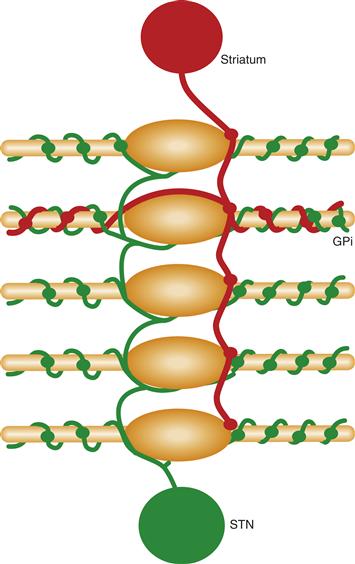
Figure 30.8 The two primary inputs to globus pallidus internal segment (GPi) have different patterns of termination. Axons from the subthalamic nucleus (green) are excitatory and terminate extensively on multiple GPi neurons. Axons from the striatum (red) contact several GPi neurons weakly in passing before terminating densely on a single neuron.
The output from GPi is inhibitory and uses GABA as its neurotransmitter. The majority (70%) of the GPi output is sent via collaterals to both the thalamus and the brainstem. In the thalamus, axons from GPi terminate in the oral part of the ventrolateral nucleus (VLo) and in the principal part of the ventral anterior nucleus (VApc). In turn, these thalamic nuclei project to the motor, premotor, supplementary motor, and prefrontal cortex. They also project back to striatum in a potential feedback loop. An individual GPi neuron sends output via the thalamus to just one area of the cortex (Hoover & Strick, 1993). Thus, just as there are parallel inputs from the cortex to striatum, there appear to be functionally parallel outputs from GPi.
Collaterals of the axons projecting from GPi to the thalamus project to an area at the junction of the midbrain and pons near the pedunculopontine nucleus, which has been termed the “midbrain extrapyramidal area.” The midbrain extrapyramidal area projects in turn to the reticulospinal motor system. Other GPi neurons (20%) project to intralaminar nuclei of the thalamus or to the lateral habenula. The specific role of these projections is not known.
Like the GP, the SN is divided into two segments. One is sparsely cellular and is called the pars reticulata (SNpr), and the other is a densely cellular region called the pars compacta (SNpc). SNpr is similar to GPi in many ways, including cell size, histochemistry, and connectional anatomy. The pars compacta contains DA cells and is not a principal output nucleus of the basal ganglia.
Like GPi, the SNpr contains large neurons that project outside of the basal ganglia. Like GPi, SNpr receives inhibitory inputs from striatum that contain GABA, SP, and dynorphin and excitatory inputs from STN that contain glutamate and provides inhibitory, GABAergic outputs. SNpr outputs are to the medial part of the ventro-lateral thalamus (VLm) and the magnocellular part of the ventral anterior thalamus (VAmc). These thalamic areas in turn project to the premotor and prefrontal cortex. Like the GPi output, SNpr sends collaterals to the midbrain extrapyramidal area and also has a projection to intralaminar nuclei of the thalamus. The primary difference in the outputs of GPi and SNpr is that the lateral portion of SNpr sends an inhibitory projection to the superior colliculus and to the paralaminar part of the medial dorsal thalamus (MDpl). MDpl projects in turn to the frontal eye fields. This lateral portion of SNpr is connected with cortical and brainstem areas which control eye movements (see Chapter 31).
GPe Is Connected to Several Other Basal Ganglia Nuclei
The GPe can be considered to be an intrinsic nucleus of the basal ganglia, as can the SNpc, which is considered in the next section. Both GPe and SNpc receive the bulk of their input from and send the bulk of their output to other basal ganglia nuclei.
The GPe is similar in some ways to GPi. Its inputs are an inhibitory projection from the striatum and an excitatory one from STN. The patterns of termination of the striatal and STN afferents are similar in that the striatal input is focused and convergent while the STN input is divergent (Parent & Hazrati, 1993). As noted earlier, GPe and GPi receive input from neighboring striatal neurons and thereby are likely to receive similar information. Unlike GPi, the striatal projection to GPe contains GABA and enkephalin but not substance P. However, the pathway taken by output of GPe is also quite different from that taken by the output of GPi, which undoubtedly has profound functional implications. The majority of the output of GPe projects to STN. The connections from striatum to GPe, from GPe to STN, and from STN to GPi thus are referred to as the “indirect” striatopallidal pathway to GPi (Alexander & Crutcher, 1990). In addition, there is a monosynaptic GABAergic inhibitory output from GPe directly to GPi and to SNpr and a recently described GABAergic projection back to striatum. Thus, GPe neurons are in a position to provide feedback inhibition to neurons in striatum and STN and feed-forward inhibition to neurons in GPi and SNpr. This circuitry suggests that GPe may act to oppose, limit, or focus the effect of the striatal and STN projections to GPi and SNpr, as well as focus activity in these output nuclei.
SNpc Provides DA Input to the Striatum
The SNpc is perhaps the most studied structure in the basal ganglia. The SNpc is made up of large DA-containing cells and it is these neurons that degenerate in PD (see later). DA neurons contain a substance called neuromelanin, which is a dark pigment that gives the SNpc a blackish appearance and appears to represent an oxidation product of DA. This is the basis for its name (substantia nigra = black substance). SNpc receives input from the striatum, specifically from the striosomes. This input is GABAergic and inhibitory. Other inputs to SNpc have been difficult to assess because the dendrites of SNpc and SNpr neurons overlap. The SNpc DA neurons project to all of caudate and putamen in a topographic manner. However, nigral DA neurons receive inputs from one striatal circuit and project back to the same and to adjacent circuits (Haber, Fudge, & McFarland, 2000). Thus, they appear to be in a position to modulate activity across circuits. The action of DA depends on the receptors located on target neurons, as was discussed earlier.
Summary of Basal Ganglia Circuitry
1. The striatum receives input from nearly all of the cerebral cortex such that several functionally related cortical areas project to overlapping striatal zones and an individual cortical area projects to several striatal zones. Cortical areas that are not functionally related project to separate zones of the striatum, although there may be some interaction between adjacent zones.
2. The striatum sends a focused and convergent inhibitory projection (“direct pathway”) to the basal ganglia output nuclei, GPi and SNpr.
3. The STN receives input from the frontal lobe, especially from the motor, premotor, and supplementary motor cortex and from the frontal eye fields.
4. The STN sends a fast divergent excitatory projection (“hyperdirect pathway”) to GPi and SNpr.
5. Reciprocal and loop-like connections among basal ganglia nuclei play a negative or positive feedback role and likely result in focusing of signals. These include the “indirect pathway” from striatum to GPe to STN to GPi and SNpr.
6. The output from the basal ganglia (GPi and SNpr) is inhibitory and projects to motor areas in the brainstem and thalamus.
The basal ganglia are sometimes viewed as a system with substantial convergence of information from the inputs to the outputs. In contrast, they have also been viewed as a system of multiple parallel functionally segregated loops through the basal ganglia, each with a separate output. In support of convergence are (1) the reduction of the number of neurons at each level from the cortex to striatum to GPi and SNpr, (2) the large number of synapses on each striatal neuron, (3) the large dendritic trees of striatal, GPi, and SNpr neurons, and (4) interaction across circuits mediated by SNpc and GPe neurons. In support of parallel segregated loops are (1) the preserved somatotopy with separate representations of the face, arm, and leg in the cortex, striatum, and GPi/SNpr; (2) the relative preservation of topography through the basal ganglia; and (3) the finding that separate groups of GPi neurons project via the thalamus to separate motor areas of the cortex. It is likely that both views are correct with parallel organization of function circuits, but with convergence within circuits and interaction between adjacent (and functionally related) circuits.
Signaling in Basal Ganglia
Although the anatomic organization may provide some clues as to what might be the function of basal ganglia circuits, inference of function from anatomy is speculative. One approach to studying the function of an area of the central nervous system is to record the electrical activity of individual neurons with an extracellular electrode in awake, behaving animals. Other approaches involve inferences of neuronal signaling from imaging studies of blood flow and metabolism, or of changes in gene expression. The activity of different regions can be correlated with performance of specific tasks. With electrophysiological studies, the timing of neural activity in one part can be compared to the timing of another and to the timing of the movement. This section will discuss movement-related signals in the basal ganglia that are correlated with the production or prevention of movement or with motor planning.
Striatal Output Neurons Have Low Spontaneous Activity That Increases With Movement
In awake animals, the majority of striatal neurons (80 to 90%) have low baseline discharge rates of 0.1 to 1 Hz (Fig. 30.9). These neurons project outside of the striatum and therefore are probably medium spiny neurons. In general, the activity of these neurons reflects the areas of cerebral cortex from which they receive inputs, though striatal neurons are less modality specific. Thus, neurons in areas of the putamen which receive input from the somatosensory and motor cortex have activity correlated with active and passive movement, but not with specific tactile modalities such as light touch, vibration, or joint position or with kinematic properties of movement such as amplitude, velocity, or force.

Figure 30.9 Representative neural discharge patterns from several basal ganglia nuclei. In the raster displays, each dot indicates the occurrence of an action potential. Each horizontal raster line represents a 1-s period of discharge. Several such periods are arranged vertically for each nucleus.
Striatal neurons related to movement have a distinct somatotopy with the face represented ventro-medially and the leg dorsolaterally. Neurons active in relation to eye movements are located in a longitudinal zone in the central part of the caudate nucleus. The striatum appears to be organized as clusters of neurons with similar discharge characteristics. These likely correspond to the anatomically defined matrisomes described earlier. Movement-related activity changes are typically increases above the very low baseline. On average, movement-related striatal neurons fire 20 msec prior to movement (Fig. 30.10). About half of these neurons fire in relation to the direction of movement. Some neurons fire in relation to the start of movement and others in relation to the stop. Some neurons in the putamen fire in relation to self-initiated movements, some in relation to stimulus-triggered movements, and some in relation to both (Romo, Scarnati, & Schultz, 1992). Neurons in the anterior part of the putamen and in the caudate nucleus that receive input from the premotor or prefrontal cortex have activity during the preparation for movement. This activity is time locked to instructional cues but not to the movement itself. Some signals correlate with the instruction of whether to move (“set”). Other signals correlate with the signal to move (“go”). Although individual neurons may have signals that preferentially correlate with specific tasks or task parameters, as a whole the striatum is not exclusively involved in any one task type or parameter.
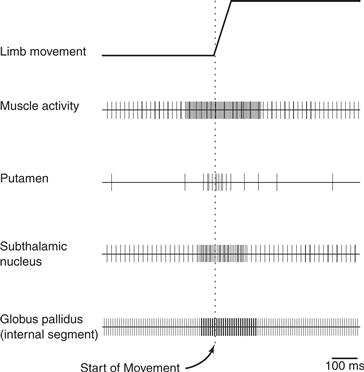
Figure 30.10 Schematic representation of the timing of neuronal activity in three nuclei of the basal ganglia in relation to limb movement and the muscle activity used to make the movement.
The timing of neuronal activity related to limb or eye movements is important to understanding the function of those neurons in the production of movement. In the putamen, most movement-related activity occurs relatively late, before the onset of movement but after the onset of muscle activity responsible for producing the movement. When compared to activity in the cerebral cortex, movement-related putamen neurons fire after those in the overlying motor cortex and supplementary motor area. Putamen neurons related to movement preparation also fire substantially later than cortical neurons involved in movement preparation. Caudate neurons related to eye movements fire late relative to oculomotor areas of cortex. The late timing of striatal discharge during movement and movement preparation relative to discharge in cortical areas suggests that the striatum is not involved primarily in the initiation of movement. Instead, it receives information from cortical areas that are responsible for movement generation and may use that information for facilitation, gating, or scaling of cortically initiated movement.
Two additional subsets of striatal neurons represent fewer than 10% of striatal neurons but are critical for basal ganglia function. One subset called the tonically active neurons (TANs) have a relatively broad action potential, are distributed throughout the striatum, and fire tonically but have no discharge correlated with movement. TANs are thought to be cholinergic interneurons which fire in relation to certain sensory stimuli that are associated with reward (Aosaki et al., 1994). It has been suggested that these neurons signal aspects of tasks that are related to learning and reinforcement. The other subset is the fast-spiking interneurons (FSIs) that are thought to be GABAergic interneurons. They have narrow waveforms, and do discharge in relation to movements. In particular, they have been shown to fire in a coordinated manner throughout striatum when animals initiate one chosen action while suppressing a highly trained alternative (Gage, Stoetzner, Wiltschko, & Berke, 2010).
Functional imaging studies using functional Magnetic Resonance Imagining (fMRI) in human subjects have shown signals in striatum related to several aspects of task performance. In addition to activity correlated with motor performance, several studies have suggested an important role of striatum in specific types of procedural memory and motor learning.
STN Has Moderate Spontaneous Activity That Increases during Movement
Neurons in the STN are tonically active with average baseline discharge rates of about 20 Hz (Fig. 30.9). They are organized somatotopically and change activity in relation to eye or limb movement. For 90% of these neurons, the change is an increase, occurring on average 50 msec prior to the movement (Fig. 30.10). The majority of movement-related neurons in STN have signals related to movement direction, but little is known about their relationship to physical parameters of movement. Functional imaging studies in human studies have demonstrated signals in STN that appear to be correlated with the stopping of ongoing movement or preventing the initiation of unwanted movements (see Box 30.8).
GPi and SNpr Have High Spontaneous Activity That Increases or Decreases after Movement Initiation
As described earlier, GPi and SNpr form the output of the basal ganglia. Neurons in these output structures fire tonically at average rates of 60 to 80 Hz (Fig. 30.9). They are organized somatotopically with the leg and arm in GPi and the face and eyes in SNpr. Due to the somatotopy, studies of limb movements have focused on GPi and studies of eye movements have focused on SNpr.
The discharge of many GPi neurons is related to the direction of limb movement, but most GPi activity is not correlated with other physical parameters of movement, including joint position, force production, movement amplitude, or movement velocity. Few GPi neurons have activity correlated with the pattern of muscle activity. Like striatum, some GPi neurons have activity related to movement preparation. During a reaching movement, 25% of GPi neurons are related to movement preparation and 50% are related to movement, but many neurons have more than one type of response. Approximately 70% of arm movement–related GPi neurons increase activity and 30% decrease activity from this tonic baseline during movement. Because the output from GPi is inhibitory, the large proportion of activity increases during movement translates to broad inhibition of thalamic and brainstem targets with more restricted facilitation during movement. GPi neurons are more likely to decrease discharge in stimulus-triggered movement and increase discharge in memory-guided movement. Thus, movement-related discharge is highly influenced by behavioral context.
The timing of movement-related GPi activity is late compared to the activation of agonist muscles. The average onset of GPi activity is after the onset of EMG but before the onset of movement (Fig. 30.10). There is a tendency for GPi movement-related increases to occur earlier than decreases, which probably reflects the faster speed of the excitatory pathway from the cortex to GPi via STN compared with the slower inhibitory pathway from the cortex to GPi via striatum. The late timing of GPi movement-related activity suggests that the output of the basal ganglia is unlikely to initiate movement.
SNpr is the principal basal ganglia output for the control of eye movements, and most studies of SNpr activity have been in eye movement tasks (Hikosaka et al., 2000). Most saccade-related SNpr neurons fire in relation to the direction of the eye movements. Unlike GPi neurons during limb movements, virtually all saccade-related SNpr neurons decrease activity during the saccade. It is not known whether the fact that most GPi limb movement neurons increase and most SNpr saccade neurons decrease indicates a fundamental difference between eye and limb movement control or if it reflects task differences. Other differences between GPi limb movement neurons and SNpr saccade neurons have been noted. While GPi limb movement neurons have weak sensory responses, some SNpr neurons have strong responses to visual stimuli. For the visually responsive SNpr neurons, the spatial location of the stimulus seems to be the most salient feature.
Some SNpr neurons have their strongest relation to saccades made to remembered targets. If a visual target is presented and then removed and a monkey is trained to look to where the target had been, some SNpr cells will fire more in this condition as compared to when a saccade is made to a visible target. Thus, like GPi neurons, SNpr output neurons are strongly influenced by context.
GPe Has Irregular Activity That Increases or Decreases after Movement Initiation
Two types of neurons have been described in GPe based on their baseline activity patterns. Most fire at a high frequency (70 Hz) that is interrupted by long pauses. A smaller number fire at a low frequency (10 Hz on average) and have frequent spontaneous bursts of activity. Both types of neurons change activity in relation to limb movement and, for the majority, these changes are increases in activity. As has been described for GPi, GPe neurons weakly and inconsistently code for movement amplitude, velocity, muscle length, or force. Like the other structures of the basal ganglia, the activity of movement-related activity is late.
SNpc Has Low Spontaneous Activity That Does Not Change with Movement but Changes with Significant Environmental Stimuli
The activity of single neurons in the SNpc of trained animals is different from the activity of single neurons in the other basal ganglia structures. At baseline, SNpc neurons fire at a low rate (2 Hz on average). The activity of these neurons is not related to movement itself and there is no apparent somatotopy in SNpc. The neurons carry little specific information regarding sensory modality or spatial properties. The activity of SNpc neurons does change in relation to behaviorally significant events such as reward or the presentation of instructional cues. The responses to stimuli only occur if the stimulus is presented in the context of a movement task. Furthermore, the activity changes with conditioning. As an example, consider an SNpc neuron that fires in relation to an unexpected reward. If one records from this neuron over several trials while the reward is paired with a tone that precedes the reward, the SNpc neuron will gradually stop firing in relation to the reward and instead will begin to fire in relation to the tone that predicts the reward.
Thus, it appears that SNpc DA neurons can predict the occurrence of a behavioral event. In this way, SNpc neurons are similar to the TANs neurons described earlier. Remember that SNpc neurons also synapse extensively on medium spiny striatal neurons and that DA terminals are on the shafts of dendritic spines. It has been suggested that the DA input changes the sensitivity of striatal neurons to cortical inputs that terminate on the heads of dendritic spines. If so, the activity of SNpc neurons could modify the response of striatal neurons to cortical input that occurs in a specific behavioral context. Such influences may be related to the apparent ability of DA to mediate both long-term potentiation and long-term depression in striatal neurons, which in turn is likely to be an important mediator of learning and plasticity in the striatum.
Summary of Signaling in the Basal Ganglia
Several general statements about basal ganglia movement-related neuronal discharge can be made.
1. Movement-related neurons in the striatum, STN, GP, and SNpr are arranged somatotopically.
2. Medium spiny output neurons in the striatum are quiet at rest and increase during movement. Neurons in STN are tonically active and increase during movement. Recalling the anatomy, this means that GPi neurons receive a widespread, tonically active excitatory input and a focused, intermittent inhibitory input.
3. STN has increased activity when an ongoing movement is stopped suddenly or when initiation of movement is prevented.
4. Neurons in both GPe and GPi are tonically active. Most increase activity, but up to one-third decrease activity during limb movement.
5. Neurons in SNpr are also tonically active. Those that are related to saccadic eye movements decrease activity during the movement.
6. Neurons in SNpc discharge in relation to rewards and behaviorally relevant stimuli, but not to movement. They are likely to play a critical role in some types of motor learning.
7. Changes in the activity of basal ganglia occur at the onset of movement but after the muscles are already active. Thus, they are unlikely to initiate movement.
8. Movement-related activity throughout the basal ganglia is modulated by the behavioral context in which the movements are performed.
The Effect of Basal Ganglia Damage on Behavior
Valuable clues to the function of the basal ganglia have come from studying activity patterns during behavior. However, correlation of activity with an aspect of behavior does not necessarily mean that structure plays a causal role in that behavior. One approach to determining the precise roles of the basal ganglia in behavior is to selectively remove a specific component from an otherwise intact system. Certain human neurological diseases cause the loss of specific types of neuron in the basal ganglia. The movement disorders that result from basal ganglia damage are often dramatic. Depending on the site of the pathology, some basal ganglia diseases cause extreme slowness of movement and rigidity and others cause uncontrollable involuntary movements. Although human diseases are of great interest, they often affect more than one structure. This necessarily limits the power of functional models derived from the study of human basal ganglia diseases. In experimental animals, more selective lesions can be made with specific pharmacologic agents. More recently, mice have been genetically engineered to prevent development of specific neuron types.
Experimental Damage to the Striatum Causes Slow Voluntary Movements or Involuntary Postures and Movements
Lesions in the striatum produce variable results that depend on the location of the lesion, the lesion method, and what is measured. Many studies of unilateral striatal lesions have described only minimal deficits. If the putamen is inactivated pharmacologically with the GABA agonist muscimol unilaterally, the result is slightly slow movement of the contralateral limb. The slowed movement is associated with an increased activity of antagonist muscles. Despite slower movements after putamen lesions, reaction time is generally normal, indicating that movement initiation remains intact. Large bilateral lesions result in the paucity of movement, severe slowness of movement bilaterally, and postural abnormalities. In other studies, there appears to be little effect of bilateral electrolytic putamen lesions. The reason for this discrepancy is not clear, but it may be due to differences in lesion size.
Selective inactivation of striatal inhibitory interneurons (FSIs) results in abnormal involuntary movements (Gittis et al., 2011). The selective inactivation is thought to cause excessive firing of direct pathway striatal medium spiny neurons which, in turn, results in reduction of the inhibitory output from GPi and SNpr.
Huntington’s disease (HD) is a genetically based, degenerative disease in humans that results in disabling involuntary movements (see Box 30.4). These movements are called chorea (Greek for “dance”). They are frequent, brief, sudden, random twitch-like movements that involve all parts of the body and resemble fragments of normal voluntary movement. The pathologic hallmark of HD is a marked loss of neurons in the striatum. Studies have shown that not all striatal neurons are equally affected in HD (Albin, Young, & Penney, 1989). In early adult-onset HD, the enkephalin-containing neurons that project to GPe are lost first. Clinically, this is associated with the presence of chorea. In late adult-onset and in juvenile-onset HD, both the enkephalin-containing and the substance P-containing striatal neurons that project to GPi and SNpr are lost. Clinically, this loss is associated with the presence of rigidity and dystonia (sustained, abnormal postures). Interestingly, experimental destructive lesions of the striatum in monkeys do not result in chorea. This is probably due to the nonselective destruction of both GPe-and GPi projecting striatal neurons. More selective disruption of the striatal–GPe pathway with a GABA antagonist administered into the GPe does produce chorea. The suggested mechanism for chorea is that disinhibition of GPe neurons (pharmacologically, or as a result of selective striatal cell death) causes inhibition of STN and GPi. This results in abnormal overactivity of motor cortical and brainstem mechanisms, resulting in chorea.
Damage to STN Causes Large-Scale Involuntary Movements
In monkeys and humans, lesions in the STN cause dramatic involuntary flinging movements of the contralateral arm and leg. These movements have been called hemiballismus. They resemble chorea in their brief, random, and sudden nature, but they tend to be much larger in amplitude. After a lesion in STN, the hemiballism lasts for days to weeks before gradually resolving. Animals and humans with hemiballism can still make voluntary movements and often the voluntary movements appear to be quite normal. It has been suggested that the mechanism underlying the hemiballism is a loss of excitatory input to GPi, resulting in decreased GPi activity and ultimately in the disinhibition of cortical and brainstem motor mechanisms. Several lines of evidence support this. First, blocking the excitatory transmission from STN to GPi with a glutamate antagonist administered into GPi causes involuntary movements. Second, metabolic activity patterns as shown with 2–deoxyglucose uptake indicate decreased GPi output in hemiballism. Third, neuronal recording during stereotaxic surgical treatment of movement disorders has shown decreased GPi activity in patients with chorea or hemiballism. However, it is likely that chorea or hemiballism is due to abnormal patterns of basal ganglia output signals and not just tonically decreased activity. Thus, if hemiballism or chorea is produced by an STN lesion in monkeys, a second lesion placed in GPi abolishes the involuntary movements. Furthermore, in monkeys with STN lesions, the level of GPi activity is reduced even after the involuntary movements resolve. Finally, destruction of GPi in humans with hemiballism or chorea abolishes the involuntary movements and experimental lesions of GPi in monkeys do not cause chorea. Therefore, it seems likely that chorea and hemiballism reflect an abnormal fluctuating or bursting output from the basal ganglia rather than a simple reduction of output.
In rodent studies, involuntary movements are uncommon but STN lesions cause animals to respond prematurely to cues and impair the ability to prevent unwanted movements. Similar difficulties with impulse control occur in people with Parkinson disease who have been treated with Deep Brain Stimulation of STN (see Box 30.5).
Damage to GP Causes Slow Voluntary Movements and Involuntary Postures and Does Not Delay Movement Initiation
Lesions of the GPi result in slowing of movement but normal movement initiation. Because of their close proximity to each other, it is difficult to lesion GPi without including part of GPe or vice versa. Therefore, caution must be used in interpreting results of GP lesions. However, in most cases, combined GPe and GPi lesions have a similar effect to the lesion of GPi alone. Unilateral lesions of GPe and GPi result in slowness of movement, with abnormal cocontraction of agonist and antagonist muscles, but normal initiation of movement. Bilateral lesions of GPe and GPi result in even more severely abnormal flexed postures with an apparent inability to move out of them. Electrolytic lesions producing this severe abnormality have tended to be large, involving both GPe and GPi and part of the internal capsule. Similar abnormalities are seen with carbon monoxide or carbon disulfide poisoning, which results in neuron death in GPi and SNpr.
Lesions restricted to GPi result in slowness of movement of the contralateral limbs. After GPi lesions, reaction time is normal, which indicates that mechanisms involved in the initiation of movement are intact. In some studies, the slowness of movement is accompanied by a cocontraction of agonist and antagonist muscles, which results in abnormal postures similar to those that occur in dystonia, a basal ganglia movement disorder. In other studies, the slowing may depend on the context of the task. Monkeys with GPi lesions are more impaired when movements are made by turning off active muscles than when movements are made by further turning on active muscles (Fig. 30.11). Thus, lesions that remove the inhibitory output of the basal ganglia appear to interfere with the ability to turn off unwanted muscle activity.
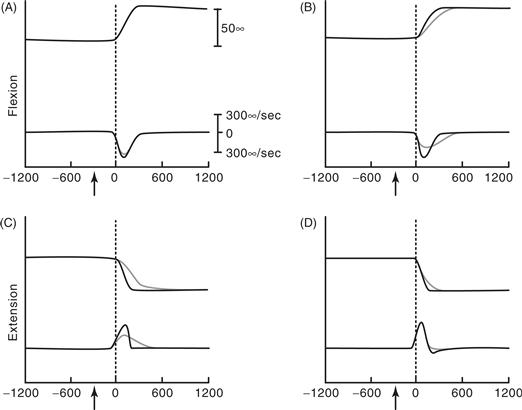
Figure 30.11 Wrist position and velocity in visually guided wrist movements before (black traces) and after (blue traces) a lesion of the globus pallidus internal segment. In each graph, the top traces represent wrist position, the lower traces represent wrist velocity. (A) Flexion with the flexor muscles loaded (movement made by further activating the loaded muscles). (B) Flexion with extensor muscles loaded (movement made by turning off the loaded muscles). (C) Extension with flexor muscles loaded (movement made by turning off the loaded muscles). (D) Extension with extensor muscles loaded (movement made by further activating the loaded muscles). After the lesion, the peak velocity was slower when the movements were made by decreasing the activity of the loaded muscle than when made by increasing the activity of the loaded muscle.
From Mink and Thach (1991).
Damage to SNpr Causes Involuntary Eye Movements
Due to its proximity to SNpc, it is difficult to produce electrolytic lesions exclusively in SNpr. However, with the use of the GABA agonist muscimol, it has been possible to inactivate neurons focally in SNpr. Injection of muscimol into the lateral SNpr inactivates neurons that are normally involved in saccadic eye movements. Inactivation in this area results in an inability to maintain visual fixation because of involuntary saccades (Fig. 30.12). The inability to suppress involuntary saccades appears to result from disinhibition of the superior colliculus. Injection of the GABA antagonist bicuculline into the superior colliculus mimics the effects of muscimol in SNpr. Thus, just as GPi inactivation results in abnormal excess limb and trunk muscle activity, SNpr inactivation results in abnormal excess eye movements.
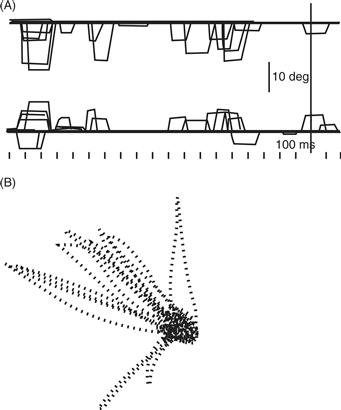
Figure 30.12 After inactivation of substantia nigra pars reticulata, monkeys are unable to maintain fixation of gaze because of involuntary contraction of the eye muscles. (A) The top line represents vertical eye position, and the bottom line represents horizontal eye position during attempted visual fixation. (B) Lines represent the trajectory of the involuntary eye movement when the monkey was instructed to maintain its gaze in the center dot.
Damage to SNpc Causes Symptoms of Parkinson’s Disease
Lesions of SNpc are of particular interest because the DA neurons in this nucleus die in Parkinson’s disease (PD). PD is a neurological disease that affects older adults (see Box 30.1). The main symptoms of PD are (1) tremor at rest that decreases during movement, (2) slowness of movement (bradykinesia), (3) paucity of movement (akinesia), (4) muscular rigidity, and (5) unstable posture. The primary pathology in PD is a progressive degeneration of neurons in the SNpc. Additionally, there is some degree of loss of DA neurons in the ventral tegmental area and of norepinephrine neurons in the locus coeruleus. Although PD has been recognized for over a century, it has only been since the early 1980s that a complete animal model has been available. As noted earlier, the interdigitation of SNpc and SNpr has made selective electrolytic lesions of SNpc or SNpr difficult to produce. In the 1960s, it was found that 6-hydroxydopamine (6-OHDA) produces specific lesions of catecholamine neurons. Therefore, injection of 6-OHDA into the rat SN bilaterally produces a selective loss of SNpc DA neurons. Lesions made in this way result in some of the abnormalities of PD—namely, the slowness of movement and rigidity, but they did not produce tremor. In the 1980s, a street drug contaminant called MPTP (see Box 30.2) was found to produce all of the symptoms of PD in humans and in certain species of monkeys. The MPTP monkey has proven an extremely valuable model with which to explore this disease.
Box 30.2 The MPTP Story
Until the early 1980s, the quest for a complete animal model of Parkinson’s disease (PD) was largely unsuccessful. Although some of the abnormalities of PD could be caused by electrolytic lesion of SNpc or by injecting the neurotoxin 6-hydroxydopamine into SNpc, neither of these methods produced the full syndrome of PD. Then, in 1982 an unfortunate, but fortuitous, accident happened. Four young drug users in northern California developed symptoms of PD. Because PD is highly unusual in young adults, the neurologists caring for these patients began a search for the cause (Langston, Ballard, Tetrud, & Irwin, 1983). They discovered that each of the patients had recently obtained a “new” synthetic heroin. This “new heroin” contained an analog of the narcotic meperidine, 1-methyl-4-phenyl-proprionoxy-piperidine (MPPP), and a contaminant, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP). It turned out that MPTP was the agent responsible for the parkinsonism. MPTP is oxidized in the brain to MPP+ by monoamine oxidase. MPP+ is taken up by DA neurons where it inhibits oxidative metabolism in the mitochondria and ultimately leads to cell death.
Subsequently, it has been found that MPTP produces a Parkinson’s-like syndrome in several species of monkeys. Unlike previous models, monkeys given MPTP had not only slowness of movement, paucity of movement, and rigidity, but also tremor. The Parkinson’s-like syndrome in monkeys given MPTP is associated with a nearly complete degeneration of DA neurons in SNpc and the ventral tegmental area and variable degeneration in the locus coeruleus. MPTP monkeys improve when given the DA precursor L-DOPA and have side effects of chorea when they are given too much L-DOPA, similar to what happens in people with PD. Hence, by behavioral, pharmacological, and pathological measures, the MPTP monkey is an excellent model of human PD. It is currently used to study the pathophysiology and pharmacology of PD and it has lead to new ideas about possible causes of PD. One such idea is that environmental toxins may play a role in the etiology of PD. In this regard, it is important to note that paraquat and rotenone, two compounds that are used commonly as pesticides and/or herbicides, both have MPTP-like structures and have also been shown to damage DA neurons.
Jonathan W. Mink
Reference
1. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980.
Despite extensive investigation, the fundamental mechanism of the tremor in PD is not known. Some evidence suggests that it results from abnormal bursting of neurons in the thalamus. The slowness of movement in PD has been associated with a reduced magnitude and duration of muscle activity during movement. In monkeys that have been given 6-OHDA or MPTP, the activity of motor cortex neurons during arm movements is also reduced compared with normal monkeys. In the MPTP monkey model of PD, some animals have abnormally increased activity of STN and GPi and decreased activity of GPi. GPi neurons in the MPTP monkey have abnormal bursting activity and abnormally increased responses to somatosensory stimuli. It has been suggested that the increased activity of GPi causes an excess inhibition of motor mechanisms in the cortex and brainstem and that this excess inhibition causes movements to be slow (Albin et al., 1989). However, it has been argued that the abnormal patterns of GPi activity may be more important than the more modest increase in tonic activity.
The rigidity of PD has been attributed in part to hyperactivity of the transcortical stretch reflex. Normally, the transcortical stretch reflex is active to resist displacement from an actively held posture. It is inhibited when subjects are instructed not to resist the displacement. People with PD have abnormally increased transcortical stretch reflexes and are unable to suppress them in response to instruction. The inability to inhibit long-loop reflexes may also account for the postural instability of PD. Patients with PD have an inappropriate cocontraction of leg and back muscles in response to perturbation from an upright stance. When the same subjects perturbed from a sitting position, they are not able to inhibit the postural reflexes that were active during stance. This suggests that the mechanism of rigidity and postural instability may be similar and that they reflect an inability to suppress unwanted reflex activity.
Paradoxically, it is known that lesions of GPi or STN can ameliorate many or all of the signs and symptoms of PD without producing the abnormalities associated with lesions in these areas in normal animals or humans. This is particularly paradoxical for GPi lesions that can produce the signs of PD (flexed posture, slow movement, rigidity) in normal monkeys and in some rare human diseases, but can reduce or eliminate those signs in monkeys and humans with preexisting lesions of the DA system. Why this is the case is not known, but it may relate to long-term changes in basal ganglia physiology that result from chronic DA depletion. Chronic, high-frequency stimulation of the STN or GPi has been found to be effective for the treatment of symptoms in advanced PD. This has been called deep brain stimulation (DBS; see Box 30.5). The mechanisms by which DBS works are not well understood, but it is thought that high-frequency stimulation disrupts neural signaling and thus mimics a lesion in the stimulated area. The growth of neurosurgical treatment of PD is a direct result of knowledge about basal ganglia circuitry.
Summary
1. Damage to any basal ganglia structure may cause slowness of voluntary movement, involuntary movements, involuntary postures, or a combination of these.
2. Damage to the striatum causes voluntary movements to be slow and may produce involuntary movements or postures depending on the mechanism of damage.
3. Damage to the STN causes large amplitude involuntary limb movements.
4. Damage to the GP causes slowness of movement, abnormal postures, and difficulty relaxing muscles, but does not delay movement initiation.
5. Damage to SNpr causes abnormal eye movements, but does not delay the initiation of eye movements.
6. Damage to SNpc causes tremor at rest, slowness of movement, rigidity, and postural instability, which are the main features of PD.
Principles of Basal Ganglia Operation for Motor Control
How do the basal ganglia participate in motor control? Several hypotheses have been advanced over the past century. A growing consensus suggests that models based on human disease states may not be sufficient to explain normal basal ganglia function. Nonetheless, the models based on human disease have been useful in developing new treatments for movement disorders and for the development of testable hypotheses relating to basal ganglia function.
An old model of basal ganglia function is that the basal ganglia initiate movement. This model was based in large part on the manifestation of basal ganglia diseases. The paucity and slowness of movement in PD were attributed to an inability to initiate movements, whereas the involuntary movements of chorea and hemiballism were attributed to a release of normal motor systems from basal ganglia control. This model gained support from the fact that much of the output from the basal ganglia goes to parts of the thalamus that project to the premotor and motor cortex. It was argued that motor programs are stored in the basal ganglia and are called up and sent to the motor cortex for execution. This model is no longer widely accepted because it is now apparent that basal ganglia are active relatively late in relation to movement and to the activation of those brain mechanisms that are known to be involved in initiation. Furthermore, lesions of basal ganglia output do not delay the initiation of movement.
If basal ganglia do not initiate movement, what do they do? We consider three current hypotheses. One hypothesis states that although basal ganglia do not initiate movement, they contribute to the automatic execution of movement sequences. This hypothesis suggests that other mechanisms initiate the first component in a sequence, but that basal ganglia contain the programs for completion of the sequence. The second hypothesis states that the basal ganglia circuitry is made up of opposing parallel pathways that adjust the magnitude of the inhibitory GPi output in order to increase or decrease movement. According to this hypothesis, increased GPi output slows movements and decreased GPi output increases movement. The third hypothesis states that basal ganglia act to permit desired movements and to inhibit unwanted competing movements.
Do Basal Ganglia Automatically Generate Learned Movement Sequences?
A popular hypothesis states that basal ganglia are responsible for the automatic execution of learned movement sequences (Marsden, 1987). It has been pointed out that patients with PD have difficulty moving several body parts simultaneously or sequentially, and that this difficulty is more than one would expect from a simple addition of the deficits of each component of the movement. An example of an apparent problem with sequential movements in PD is the phenomenon of micrographia (or small writing). A patient begins to write a sentence with nearly normal-sized writing, but within several letters, the writing begins to get smaller so that by the end of the sentence, it may be illegible. It has been emphasized that the early components of the sequence are larger and faster than are the subsequent components. One experiment compared the performance of elbow flexion and hand grip individually or in sequence (Benecke, Rothwell, Dick, Day, & Marsden, 1986). Patients with PD performed each movement more slowly than normal subjects. However, when the movement was part of a sequence, it was slowed to an even greater degree than when it was performed separately. Another experiment involved recording the activity of GPi neurons in monkeys trained to perform two successive prompt wrist movements. It was found that some GPi neurons fired after the first component of the movement but before the second component. Proponents of the sequencing hypothesis speculate that the loss of this GPi output signal in PD is responsible for the relatively greater difficulty in producing sequential movements than in producing individual movements.
Do Basal Ganglia Produce or Prevent Movement by Using Opposing Direct and Indirect Parallel Pathways?
This hypothesis emphasizes the two major paths of information flow from the striatum to GPi and SNpr that were described earlier (Fig. 30.2). To recapitulate, one is an inhibitory “direct” pathway from striatum to GPi/SNpr and the other is a net excitatory “indirect” pathway from striatum to GPe (inhibitory), from GPe to STN (inhibitory), and from STN to GPi/SNpr (excitatory). In this hypothesis, the two pathways are in balance in such a way that increased activity in the “direct” pathway causes decreased GPi/SNpr output and increased activity in the “indirect” pathway causes increased GPi/SNpr output. By adjusting the balance, cortical targets of the basal ganglia can be facilitated or inhibited. The hypothesis predicts that abnormally decreased output results in excessive movements (chorea) and abnormally increased output results in a decreased movement (PD). The “direct/indirect pathway” model has been useful for understanding and developing certain treatments of movement disorders, but it has several shortcomings when it comes to explaining normal basal ganglia function and the mechanism of treatments for involuntary movements. Despite the shortcomings, this has been a central hypothesis in the field of basal ganglia research for over 20 years. If the success of a model can be measured by the amount of research it stimulates, this one has been highly successful.
Do Basal Ganglia Select and Inhibit Competing Motor Patterns?
The output of the basal ganglia is inhibitory to posture and movement pattern generators in the cerebral cortex (via thalamus) and in the brainstem. The inhibitory output neurons fire tonically at high frequencies. In this hypothesis, the motor output of the basal ganglia is analogous to a brake (Mink, 1996). The hypothesis states that when a movement is initiated by a particular motor pattern generator, GPi neurons projecting to that generator decrease their discharge, thereby removing tonic inhibition and “releasing the brake” on that generator. GPi neurons projecting to other movement pattern generators increase their firing rate, thereby increasing inhibition and applying a “brake” on those generators. Thus, other postures and movements are prevented from interfering with the one selected.
How might this mechanism work (Fig. 30.13)? When one makes a voluntary movement, that movement is initiated by the prefrontal, premotor, and motor cortex and by the cerebellum. The premotor and motor cortex send a corollary signal to STN, exciting it. STN projects to GPi in a widespread pattern and excites GPi. In parallel, signals are sent from the cortex to the striatum, which inhibits GPi focally via a direct pathway. Striatum can also disinhibit GPi via two indirect pathways (striatum → GPe → GPi and striatum → GPe → STN → GPi). The indirect pathways further focus the effects of the fast excitatory cortico-STN pathway and the slower inhibitory cortico-striatal pathway to GPi. The net result is to release the “brake” from the selected voluntary movement pattern generator and to apply the “brake” to potentially competing posture-holding pattern generators (transcortical, vestibular, tonic neck, and other postural reflexes; see Chapter 28). The result is the focused selection of desired motor patterns and surround inhibition of competing patterns. Disruption of the ability to facilitate desired movements and inhibit unwanted movements results in slow voluntary movements (parkinsonism), abnormal involuntary movement (chorea, dystonia, tics), or both (Mink, 2003).

Figure 30.13 Relationship of proposed center-surround organization of GPi to inputs from the striatum and subthalamic nucleus. During voluntary movement, excitatory subthalamo-pallidal neurons increase the activity of the pallidal neurons in the surround territory. Inhibitory striatopallidal neurons inhibit the functional center in a focused manner. Pallidal activity changes are conveyed to the targets in the thalamus and midbrain brainstem, causing disinhibition of neurons involved in the desired motor program and inhibition of surrounding neurons involved in competing motor programs. Excitatory projections are indicated with green arrows; inhibitory projections are indicated with red arrows. The relative magnitude of activity is represented by line thickness.
Basal Ganglia Participation in Nonmotor Functions
Although the focus of this chapter has been on motor control, it has become increasingly clear that the basal ganglia participate in a variety of nonmotor functions. These include functions of the limbic system (see Box 30.3) and cognitive functions. The anatomy of basal ganglia circuits has revealed outputs to all areas of frontal cortex, placing the basal ganglia in a position to influence a wide variety of behaviors. As discussed earlier, specific areas of the basal ganglia are connected preferentially with specific areas of the cerebral cortex. This provides the anatomic substrate for the localization of different functions in the basal ganglia circuits. It is known that focal lesions of the striatum tend to produce deficits similar to those seen after lesions of afferent areas of the cortex. Thus, lesions of posterior putamen cause movement deficits, lesions of inferior caudate produce deficits similar to lesions of the orbitofrontal cortex, and lesions of the dorsolateral caudate produce deficits similar to lesions of dorsolateral prefrontal cortex. The basal ganglia have been implicated in a variety of nonmotor disorders, including depression, obsessive-compulsive disorder (see Box 30.6), attention deficit hyperactivity disorder, and schizophrenia. It is becoming clear that the basic underlying principles of basal ganglia function are similar. Thus, the intrinsic circuitry is the same for cognitive and motor parts of the basal ganglia, the basic neurophysiology appears to be the same, and the effects of lesions are analogous. For example, chorea is characterized by excessive involuntary movements, and obsessive-compulsive disorder is characterized by excessive involuntary thoughts and complex behaviors. The hypothesis of focused selection and surround inhibition has been applied to non-motor basal ganglia functions (Redgrave, Prescott, & Gurney, 1999) and data support each hypothesis from cognitive science and psychiatric research.
Box 30.3 Ventral System of the Basal Ganglia
There are nuclei in the brain that historically have not been included with the basal ganglia, but that are analogous to the traditional components. These lie ventral to the neostriatum and GP and are called the ventral striatum (nucleus accumbens and olfactory tubercle) and the ventral pallidum. The ventral striatum receives input from limbic and olfactory areas of the cortex, including the amygdala and hippocampus. Like the neostriatum, the ventral striatum receives a DA input, but it is from the ventral tegmental area (VTA), which lies medial to the SN. The ventral striatum sends a projection back to VTA and to the adjacent SNpr. The ventral pallidum is analogous to both GPe and GPi. It receives input from the ventral striatum and possibly from STN. Unlike the GP, the ventral pallidum receives direct input from the amygdala. The output of the ventral pallidum projects to the dorsomedial nucleus of the thalamus (DM) and from there to limbic areas of the cortex. By virtue of its inputs and outputs, the ventral system is closely linked to the limbic system. This linkage has led to the suggestion that the ventral system is involved to some degree in motivation and emotion (see Chapter 41). The exact nature of this role is not known, but it may be analogous to the motor role of the basal ganglia, with the inhibitory output of the ventral pallidum acting to suppress or select potentially competing limbic mechanisms.
Lesions of the ventral pallidum lead to an inability to inhibit incorrect responses in an odor discrimination task, supporting the hypothesis.
Jonathan W. Mink
Increasing evidence points to a role for the basal ganglia in procedural learning. Much of the research has focused on the learning of tasks or of sequential behavior. There is evidence for a role of the basal ganglia in procedural learning that leads to the formation of habits and the performance of behavioral routines once they are learned (Jog, Kubota, Connally, Hillegart, & Graybiel, 1999). As described earlier, TANs and SNpc DA neurons fire in relation to behaviorally significant events. The activity patterns of these neurons change as the task becomes learned and when novel stimuli or events are introduced. There is also growing evidence for DA-mediated long-term potentiation and long-term depression in striatal neurons that is likely to play an important role in learning. It has been shown that non-TAN striatal neurons also change activity in relation to learning. In rats performing a T-maze task, striatal neurons changed activity patterns as the task became learned and more automatic (Jog et al., 1999). In monkeys performing a discrimination learning task, striatal neurons changed activity as the animal learned new associations between stimuli and reward (Tremblay, Hollerman, & Schultz, 1998). Functional imaging studies have also shown basal ganglia activity correlated with the learning of new tasks. Striatal lesions or focal striatal DA depletion impairs the learning of new movement sequences, and procedural learning has been shown to be impaired in people with PD. These findings together support a role for the basal ganglia in certain types of procedural learning. However, other brain structures have also been implicated in procedural learning, including the cerebellum (see Chapter 31). Just as the cerebellum and basal ganglia play different roles in motor control, they are likely to play different, but complementary, roles in procedural learning. The exact nature of those different roles is the subject of current research.
Box 30.4 Huntington’s Disease
Huntington’s disease (HD) is an inherited progressive neurodegenerative disorder that affects about 1 in 10,000 people. Symptoms include abnormal movements (comprising both involuntary movements termed chorea or dystonia and motor incoordination), cognitive difficulties, and emotional difficulties, including depression, apathy, and irritability. Onset is usually in midlife, but can range between childhood and old age. HD is generally fatal within 15 to 20 years after onset. The disease is caused by an expanded CAG repeat coding for polyglutamine in the HD gene product, termed the “huntingtin” protein. CAG repeat lengths between about 10 and 25 are normal, whereas those above 36 cause HD. Within the expanded range, the longer the repeat, the earlier the age of onset. The normal function of huntingtin is poorly understood, but it may be involved with cytoskeletal function, or cellular transport.
Pathologically, HD is characterized by selective neuronal vulnerability. The caudate and putamen of the corpus striatum are most affected in early stages, but as the disease progresses, other areas of the brain also become affected. Within the striatum, medium spiny GABA neurons are severely affected, with up to 95% loss in advanced cases, whereas large interneurons are relatively spared. In addition, there are intranuclear inclusion bodies and perinuclear and neuritic aggregates of huntingtin in HD neurons.
The pathogenesis of HD is still incompletely understood and represents a challenge for neuroscience. The huntingtin protein is widely expressed throughout the brain and is expressed at lower levels in other regions of the body as well. HD, like other neurodegenerative diseases, involves abnormal protein folding and aggregation. The inclusions themselves appear not to be directly toxic, but they likely represent a late stage of an abnormal process, whose earlier steps are pathogenic. The study of the disease has been facilitated by the generation of cell models, invertebrate models, and mouse models. Chaperone proteins can ameliorate the pathology, perhaps by refolding abnormally folded huntingtin. Huntingtin may normally be degraded by intracellular proteolytic machinery, termed the proteasome, and inhibition of the proteosome may be one of the toxic effects of the abnormally expanded polyglutamine.
A number of lines of evidence have pointed to a possible role for abnormal gene transcription in HD. Several studies have suggested that targeting of huntingtin to the nucleus enhances toxicity. Huntingtin may normally have a role in regulation of gene transcription, although this is poorly understood. Expression array studies show that mouse and cell models have an alteration of normal gene expression patterns. Abnormal intereactions with coactivator and corepressor complexes may contribute.
Whatever the initial mechanisms, symptoms appear to be caused by both cell dysfunction and cell death, with cell death correlating best with functional disability. Thus, preventing cell death is a major goal of experimental therapeutics in HD and other neurodegenerative disorders. Cell death may partly involve apoptotic mechanisms with activation of caspases and may also involve mechanisms such as excitotoxicity, metabolic, or free radical stress. Therapeutic strategies under investigation include protection of neurons with neurotrophins, altering abnormal gene transcription patterns, and reducing cell death with inhibitors of caspase activation or blockers of excitotoxicity.
Christopher A. Ross
Box 30.5 Deep Brain Stimulation
Ablative neurosurgery has been used for treating movement disorders, epilepsy, and pain for over 80 years. In ablative procedures, a lesion is placed in the brain in order to disrupt a dysfunctional circuit. Ablative neurosurgery is still used, but increasingly deep brain stimulation (DBS) is replacing ablations to treat disorders associated with neural circuit dysfunction. This is particularly true for movement disorders related to basal ganglia dysfunction.
The idea of treating neurologic disorders with chronic electrical stimulation emerged in the 1960s. By the 1990s, technological advances provided the possibility of a system that could be implanted with no external components that could provide continuous stimulation for years without battery replacement. DBS involves stereotaxic surgical placement of electrodes into specific brain targets, fixing the electrodes to the skull, passing the electrode wires under the skin and ultimately connecting to a pacemaker-like device that is implanted under the skin in the chest, back, or abdomen. The pacemaker device can be programmed telemetrically to deliver desired stimulation by adjusting voltage, pulse width, frequency, and electrode configuration.
DBS was used initially for treating tremor associated with Essential Tremor or Parkinson disease, with electrodes placed in the cerebellar-receiving thalamic nucleus (ventral intermediate nucleus; VIM). By the end of the 1990s, DBS was used increasingly for treating other symptoms of Parkinson disease including bradykinesia and rigidity (targets: STN or GPi) and for treating dystonia (target: GPi). DBS can be dramatically effective for these movement disorders.
The mechanism by which DBS works is a subject of investigation. Although the effects are similar to the effect of ablation of the brain target in which the electrodes are placed, DBS does not appear to work simply by inactivating the target nucleus. In fact, there is evidence that DBS activates the efferent axons from that target. Regardless of the mechanism, there is no controversy regarding the efficacy of DBS for specific disorders. Investigations are under way to determine whether other neuropsychiatric disorders such as depression, OCD, and Tourette Syndrome, can be treated safely and effectively with DBS.
Jonathan W. Mink
Suggested Readings
1. Perlmutter JS, Mink JW. Deep brain stimulation. Annual Review of Neuroscience. 2006;29:229–257.
2. Wichmann T, Delong MR. Deep-brain stimulation for basal ganglia disorders. Basal Ganglia. 2011;1:65–77.
Box 30.6 Obsessive-Compulsive Disorder
Obsessive-compulsive disorder (OCD) is a chronic disorder characterized by recurrent intrusive thoughts and ritualistic behaviors that consume much of the afflicted individual’s attentional and goal-directed processes. Among the more agonizing illnesses in clinical medicine, OCD is classified as an anxiety disorder because of the marked tension and distress produced by resisting the obsessions and compulsions. Common obsessions involve thoughts of harming oneself or others, of being afflicted by various illnesses, or of being contaminated by germs. Compulsions may be a response to obsessive thoughts (e.g., repetitive hand washing following skin contact with any object due to obsessive worries of having contacted germs) or may instead reflect cognitive-behavioral rituals performed according to stereotyped rules (e.g., counting one’s footsteps to avoid ending on certain numbers). Although individuals with OCD recognize that such thoughts and behaviors are irrational, they feel irresistibly compelled to engage in them.
The onset of OCD usually occurs between late childhood and early adulthood. Without treatment, OCD is often disabling. However, chronic treatment with drugs that potently inhibit serotonin reuptake can reduce the amount of time engaged in obsessions and compulsions and the magnitude of the associated anxiety. In addition, behavioral therapy involving repeated in vivo exposure and response prevention (e.g., having patients touch a toilet seat and subsequently preventing them from hand washing) may facilitate extinction of the anxiety responses generated by resisting obsessive thoughts and compulsive behaviors.
The etiology and pathophysiology of OCD are unknown. Converging evidence from analysis of the lesions that result in obsessive-compulsive symptoms, functional neuroimaging studies of OCD, and observations regarding the neurosurgical interventions that can ameliorate OCD implicate prefrontal cortical-striatal circuits in the pathogenesis of obsessions and compulsions. The neurological conditions associated with the development of secondary obsessions and compulsions include lesions of the globus pallidus and putamen, Sydenham chorea (a poststreptococcal autoimmune disorder associated with neuronal atrophy in the caudate and putamen), Tourette disorder (an idiopathic syndrome characterized by motoric and phonic tics that may have a genetic relationship with OCD, see Box 30.7), chronic motor tic disorder, and lesions of the ventromedial prefrontal cortex. Several of these conditions are associated with complex motor tics (repetitive, coordinated, involuntary movements occurring in patterned sequences in a spontaneous, unpredictable, and transient manner). Complex tics and obsessive thoughts may thus both reflect aberrant neural processes originating in distinct portions of the cortical-striatal-pallidal-thalamic circuitry that are manifested within the motor and cognitive-behavioral domains, respectively. Compatible with this hypothesis, neurosurgical procedures that interrupt the white matter tracts carrying projections between the frontal lobe, basal ganglia, and thalamus are effective at reducing obsessive-compulsive symptoms in OCD cases that prove intractable to other treatments.
Functional neuroimaging studies have also implicated prefrontal cortical-striatal circuits in the pathophysiology of OCD. In primary OCD, “resting” cerebral blood flow and metabolism are increased abnormally in the orbitofrontal cortex and the caudate nucleus, and increase further during symptom provocation in these areas, as well as in the putamen, thalamus, and anterior cingulate cortex. During effective pharmacotherapy, orbital metabolism decreases toward normal, and both drug treatment and behavioral therapy are associated with a reduction of caudate metabolism. In contrast, imaging studies of obsessive-compulsive syndromes arising in the setting of Tourette syndrome or basal ganglia lesions have found reduced metabolism in the orbital cortex in such subjects relative to controls. Differences in the functional anatomical correlates of primary versus secondary OCD suggest that dysfunction arising at multiple points within the ventral prefrontal cortical-striatal-pallidal-thalamic circuitry may result in pathological obsessions and compulsions.
This circuitry in general appears to be involved in the organization of internally guided behavior toward a reward, switching of response strategies, habit formation, and stereotypic behavior. Electrophysiological and lesion analysis studies indicate that parts of the orbitofrontal cortex are specifically involved in the correction of behavioral responses that become inappropriate as reinforcement contingencies change. Such functions could potentially become disturbed at the level of this cortex itself or within the circuits conveying projections from the ventral PFC through the basal ganglia, conceivably giving rise to the perseverative patterns of nonreinforced thought and behavior that characterize the cognitive-behavioral state of OCD.
Wayne C. Drevets
Suggested Readings
1. Baxter LR. Neuroimaging studies of human anxiety disorders. In: Bloom FE, Kupfer DJ, eds. Psychopharmacology: The fourth generation of progress. New York, NY: Raven Press; 1995; (Chap. 80, pp. 921–932).
2. LaPlane D, Levasseur M, Pillon B, et al. Obsessive-compulsive and other behavioural changes with bilateral basal ganglia lesions. Brain. 1989;112:69–725.
3. McDougle CJ. The neurobiology and treatment of obsessive-compulsive disorder. In: Charney DS, Nestler EJ, Bunney BJ, eds. Neurobiology of mental illness. New York, NY: Oxford Univ. Press; 1999:518–533.
Box 30.7 Tourette Syndrome
A “tic” is a sudden, rapid, recurrent, nonrhythmic, stereotyped movement or sound. Individuals with Tourette syndrome (TS) exhibit prominent and persistent motor tics and at least one phonic tic, beginning in childhood. Current estimates of TS prevalence range between 0.1 and 1.0%, with a 3:1 male:female ratio. Symptoms wax and wane; roughly half of all TS patients experience a significant symptom decline in their early 20s, whereas others experience lifelong symptoms.
Tics differ in complexity (simple, complex) and domain of expression (motor, phonic). In addition to their outward manifestations, a variety of sensory and mental states are associated with tics. “Simple” sensory tics are rapid, recurrent, and stereotyped, and are sensations at or near the skin. “Premonitory urges” are more complex phenomena, which often include both sensory and psychic discomfort that may be momentarily relieved by a tic. The full elaboration of tics, therefore, can include a sequence: (1) a sensory event or premonitory urge, (2) a complex state of inner conflict over if and when to yield to the urge, (3) the motor or phonic production, and (4) a transient sensation of relief.
Functional impairment in TS often results from comorbid conditions. More than 40% of individuals with TS experience symptoms of obsessive-compulsive disorder (OCD), and a comparable number have symptoms of attentional deficits and hyperactivity (ADHD).
Limited neuropathological evidence suggests four locations of potential pathology within cortico-striato-pallido-thalamic (CSPT) circuitry in TS: (1) intrinsic striatal neurons (increased neuronal packing density); (2) diminished striato-pallidal “direct” output (reduced dynorphin-like immunoreactivity in the lenticular nuclei); (3) increased dopaminergic innervation of the striatum (increased density of dopamine transporter sites); and (4) reduced glutamatergic output from the STN (reduced lenticular glutamate content).
Volumetric neuroimaging studies report an enlarged corpus callosum, reduced caudate volume, or diminished striatal and lenticular asymmetry; these changes are subtle and are not replicated uniformly. Metabolic neuroimaging studies in TS report a reduced glucose uptake in the orbitofrontal cortex, caudate, parahippocampus, and midbrain regions and reduced blood flow in the caudate nucleus, anterior cingulate cortex, and temporal lobes. Regional glucose uptake patterns suggest distributed CSPT dysfunction, based on covariate relationships between reduced glucose uptake in striatal, pallidal, thalamic, and hippocampal regions. Tic suppression is associated with increased right caudate activation, measured by functional MRI, and by bilaterally diminished activity in the putamen, globus pallidus, and thalamus. Neurochemical imaging studies have reported relatively subtle abnormalities in the levels of dopamine receptors, dopamine release, DOPA decarboxylase, and dopamine transporter in the striatum of some individuals with TS. A report of greater caudate D2 receptor binding among more symptomatic TS identical twins may be relevant to factors contributing to heterogeneity of the TS phenotype.
There are a number of reported subtle abnormalities in the levels of major neurotransmitters, precursors, metabolites, biogenic amines, and hormones in blood, cerebrospinal fluid (CSF), and urine of TS subjects compared to controls. One qualitatively different finding is that of approximately 40% elevations of serum antiputamen antibodies in TS children. This finding may have particular importance, based on growing evidence for autoimmune contributions to at least some forms of TS.
Converging evidence for CSPT pathology in TS comes from neuropsychological and psychophysiological studies. Some forms of TS appear to be accompanied by abnormalities in sensorimotor gating, oculomotor functions, and visuospatial priming consistent with mild corticostriatal dysfunction.
Treatments for TS focus on education, comorbid conditions, and direct tic suppression. Education is a critically important intervention for both family members and affected individuals. Treatments for comorbid conditions, particularly OCD and ADHD, are highly effective and can provide significant relief. Dopamine antagonists, particularly high potency, D2 preferential blockers, are the most potent and rapid acting tic-suppressing agents, but have important, undesirable side effects. Newer, “atypical” antipsychotics are better tolerated, and some are effective in suppressing tics. The DA depleter tetrabenazine also offers significant tic suppression. α2-Adrenergic agonists are often used as anti-tic agents; compared to dopamine antagonists, these drugs have relatively weaker anti-tic abilities and their benefit generally evolves more gradually. New therapeutic avenues for TS are being explored in controlled studies, including dopamine D1 antagonists, Δ9-tetrahydro-cannabinol, and deep brain stimulation. Intractable, localized tics have also been treated successfully with injections of botulinum toxic. An effective nonpharmacologic therapy, habit reversal therapy, involves the application of cognitive and behavioral therapy principles to TS, analogous to the successful use of these therapies in the treatment of OCD. Behavioral therapy for tics is as effective as most currently employed pharmacologic therapies.
Neal R. Swerdlow and Jonathan W. Mink
Suggested Readings
1. Albin RL, Mink JW. Recent advances in Tourette syndrome research. Trends in Neuroscience. 2006;29:175–182.
2. McNaught KS, Mink JW. Advances in understanding and treatment of Tourette Syndrome. Nature Reviews Neurology. 2011;8:667–676.
3. Piacentini J, Woods DW, Scahill L, et al. Behavior therapy for children with Tourette disorder: A randomized controlled trial. JAMA. 2010;303:1929–1937.
Box 30.8 The Role of the Subthalamic Nucleus in Stopping
You are sitting astride your bicycle at an intersection and just about to press down on the pedal when all of a sudden a motorist runs the light. This requires the rapid cancellation of an initiated action. Recent studies suggest that rapid stopping of this kind is implemented by a “hyperdirect” pathway between the frontal cortex and the subthalamic nucleus.
The broader sequence of events that engages this pathway is as follows. Sensory information about the stop signal (in this case, the car) is quickly relayed to the prefrontal cortex, where the stopping command must be generated. Two broad regions of the prefrontal cortex are apparently critical for stopping behavior: the right inferior frontal cortex and the dorsomedial frontal cortex (especially the presupplementary motor area). These two regions appear to work together to send a stop command to intercept the Go process, via the subthalamic nucleus. To appreciate how, first consider the Go process (the initiated pedal press). This is likely generated by premotor areas that project via the direct pathway of the basal ganglia (through striatum, pallidum, and thalamus), eventually exciting primary motor cortex and generating corticospinal volleys to the foot. An important point of interaction for Stop and Go processes could be the globus pallidus. Whereas the Go process activates the striatum and inhibits part of the globus pallidus pars interna, the Stop process activates the subthalamic nucleus which then excites the globus pallidus pars interna, which reduces thalamocortical excitability, leading to increased GABAergic inhibition in primary motor cortex. The consequence is a cessation of the corticospinal volleys for the Go process.
Interestingly, recent studies show that rapidly stopping the hand leads to suppression of foot motor representations and rapidly stopping the voice leads to suppression of hand motor representations. This shows that stopping has very broad effects on the motor system (a “global stop command”). While not proven, this could relate to the fact that the subthalamic nucleus appears to have a very broad effect on multiple neurons in the globus pallidus pars interna (Fig. 30.8).
Although rapid stopping has only so far been explored in the action domain, note that the subthalamic nucleus has different territories, including sensorimotor, associative and limbic, which connect to motor/premotor cortical, lateral prefrontal and cingulate/orbital frontal regions, respectively. It is an open question whether the kind of stopping discussed here could extend, for example, to the limbic domain of the subthalamic nucleus to enable emotional and motivational control. Notably, the limbic domain of the subthalamic nucleus is now a target for deep brain stimulation in obsessive compulsive disorder, and some studies have reported that inadvertent stimulation of this area in Parkinson’s patients induces hypomania.
Other research points to the importance of neural oscillations in the beta frequency band for the stopping system. Successfully stopping action is associated with increased beta power in the subthalamic nucleus, as well as the presupplementary motor area and the right inferior frontal cortex. It is possible that stopping is implemented via long-distance synchronization in the beta band in the overall cortico-basal ganglia network.
Adam R. Aron
Suggested Readings
1. Aron AR, Durston S, Eagle DM, Logan GD, Stinear CM, Stuphorn V. Converging evidence for a fronto-basal-ganglia network for inhibitory control of action and cognition. Journal of Neuroscience. 2007;27:11860–11864.
2. Chambers CD, Garavan H, Bellgrove MA. Insights into the neural basis of response inhibition from cognitive and clinical neuroscience. Neuroscience and Biobehavioral Reviews. 2009;33:631–646.
3. Ray N, Brittain J, Holland P, Joundi R, Stein J. The role of the subthalamic nucleus in response inhibition: Evidence from local field potential recordings in the human subthalamic nucleus. Neuroimage. 2012;60(1):271–278.
Summary
It is appropriate to consider the basal ganglia as part of the motor system because the largest portion of the basal ganglia is devoted to motor control and the most prominent deficits resulting from basal ganglia damage are motor. However, it has become clear that the basal ganglia play substantial roles in nonmotor function and in more cognitive aspects of movement. These roles are the subject of active research. Ultimately there may be a unifying theory of basal ganglia function that will encompass all of the subdivisions, but at this point one can only say that it is likely that the different circuits employ similar mechanisms.
References
1. Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends in Neurosciences. 1989;12(10):366–375.
2. Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: Neural substrates of parallel processing. Trends in Neurosciences. 1990;13(7):266–271.
3. Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M. Responses of tonically active neurons in the primate’s striatum undergo systematic changes during behavioral sensorimotor conditioning. Journal of Neuroscience. 1994;14(6):3969–3984.
4. Benecke R, Rothwell JC, Dick JP, Day BL, Marsden CD. Performance of simultaneous movements in patients with Parkinson’s disease. Brain. 1986;109:739–757.
5. Flaherty AW, Graybiel AM. Corticostriatal transformations in the primate somatosensory system: Projections from phsyiologically mapped body-part representations. Journal of Neurophysiology. 1991;66(4):1249–1263.
6. Gage GJ, Stoetzner CR, Wiltschko AB, Berke JD. Selective activation of striatal fast-spiking interneurons during choice execution. Neuron. 2010;67:466–479.
7. Gittis AH, Leventhal DK, Fensterheim BA, Pettibone JR, Berke JD, Kreitzer AC. Selective inhibition of striatal fast-spiking interneurons causes dyskinesias. Journal of Neuroscience. 2011;31:15727–15731.
8. Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorso-lateral striatum. Journal of Neuroscience. 2000;20:2369–2382.
9. Hikosaka O, Takikawa Y, Kawagoe R. Role of the basal ganglia in the control of purposive saccadic eye movements. Physiological Reviews. 2000;80:953–978.
10. Hikosaka O, Wurtz RH. Modification of saccadic eye movements by GABA-related substances II Effects of muscimol in monkey substantia nigra pars reticulata. Journal of Neurophysiology. 1985;53:292–308.
11. Hoover JE, Strick PL. Multiple output channels in the basal ganglia. Science. 1993;259:819–821.
12. Jog M, Kubota Y, Connally CI, Hillegart V, Graybiel AM. Building neural representations of habits. Science. 1999;286:1745–1749.
13. Kelly R, Strick PL. Macro-architecture of basal ganglia loops with the cerebral cortex: Use of rabies virus to reveal multisynaptic circuits. Progress in Brain Research. 2004;143:449–459.
14. Marsden CD. What do the basal ganglia tell premotor cortical areas?. Ciba Foundation Symposium. 1987;132:282–300.
15. Mink JW. The basal ganglia and involuntary movements: Impaired inhibition of competing motor patterns. Archives of Neurology. 2003;60:1365–1368.
16. Mink JW, Thach WT. Basal ganglia motor control III Pallidal ablation: normal reaction time, muscle cocontraction, and slow movement. Journal of Neurophysiology. 1991;65:330–351.
17. Parent A, Hazrati L-N. Anatomical aspects of information processing in primate basal ganglia. Trends in Neurosciences. 1993;16(3):111–116.
18. Redgrave P, Prescott T, Gurney K. The basal ganglia: A vertebrate solution to the selection problem?. Neuroscience. 1999;89:1009–1023.
19. Romo R, Scarnati E, Schultz W. Role of primate basal ganglia and frontal cortex in the internal generation of movements II: Movement-related activity in the anterior striatum. Experimental Brain Research. 1992;91:385–395.
20. Tremblay L, Hollerman J, Schultz W. Modifications of reward expectation-related neuronal activity during learning in primate striatum. Journal of Neurophysiology. 1998;80:964–977.
21. Wilson CJ, Groves PM. Fine structure and synaptic connections of the common spiny neuron of the rat neostriatum: A study employing intracellular injection of horseradish peroxidase. The Journal of Comparative Neurology. 1980;194:599–614.
Suggested Readings
1. Bolam JP, Hanley JJ, Booth PA, Bevan MD. Synaptic organisation of the basal ganglia. Journal of Anatomy. 2000;196:527–542.
2. Graybiel A. The basal ganglia and chunking of action repertoires. Neurobiology of Learning and Memory. 1998;70:119–136.
3. Houk JC, Davis JL, Beiser DG, eds. Models of information processing in the basal ganglia: Computational neuroscience. Cambridge, MA: MIT Press; 1995.
4. Kreitzer AC. Physiology and pharmacology of striatal neurons. Annual Review of Neuroscience. 2009;32:127–147.
5. Middleton F, Strick P. Basal ganglia output and cognition: Evidence from anatomical, behavioral, and clinical studies. Brain and Cognition. 2000;42:183–200.
6. Mink JW. The basal ganglia: Focused selection and inhibition of competing motor programs. Progress in Neurobiology. 1996;50:381–425.
7. Penney JB, Young AB. Speculations on the functional anatomy of basal ganglia disorders. Annual Review of Neuroscience. 1983;6:73–94.
8. Watts RL, Roller WC. Movement disorders: Neurologic principles and practice New York, NY: McGraw-Hill; 1997.