72 | EPIGENETICS IN EARLY LIFE PROGRAMMING
TRACY L. BALE
Fetal antecedents such as maternal stress, infection, or dietary challenges have long been associated with an increased disease risk, capable of affecting multiple generations. The mechanisms through which such determinants contribute to disease development likely involve complex and dynamic relationships between the maternal environment, the endocrine placenta, and the epigenetic programming of
the developing embryo itself. Although an appreciation for the importance of the epigenome in offspring disease predisposition has evolved, the incredible variability in critical factors such as gestational timing of insults, sex of the fetus, and maternal genetics make clear interpretations difficult. However, animal models have proved highly informative in providing the best knowledge yet as to just how dynamically responsive the epigenome is, and in determining important mechanisms that shape and reprogram the developing brain. This chapter discusses the epidemiological and clinical evidence and supportive animal models related to environmental influences on neurodevelopmental and neuropsychiatric disease risk.
Historically, the term epigenetics has referred to heritable traits that are not mediated by changes in DNA sequence. More recently, epigenetics has been used more broadly to refer to any change in gene function not associated with sequence variation (Jiang et al., 2008) and has been embraced by the neuroscience community as a means by which we can integrate a role for the environment to influence or “program” gene expression or patterns that may or may not be heritable (Borrelli et al., 2008; Sweatt, 2009). Epigenetic mechanisms typically involve biochemical modifications of the DNA or histones such as methylation or acetylation, as well as noncoding RNAs, including microRNAs. Increasing evidence notes that numerous types of chromatin modifications, referred to as chromatin remodeling, are widespread in the brain and undergo dynamic regulation in both the developing and adult nervous system (Tsankova et al., 2007).
FETAL ANTECEDENTS AND PROGRAMMING IN NEURODEVELOPMENTAL DISORDERS: CLINICAL AND EPIDEMIOLOGICAL STUDIES
Although evidence points to a strong genetic component in risk factors for neurodevelopmental diseases, in which the concordance rate for schizophrenia in monozygotic twins is between 50% and 60%, and 40% to 90% of the variance in autism risk is attributed to genetic heritability, new studies have focused on the potential contributions of epigenetic factors (Brown, 2012). The interactions between the rapidly changing intrauterine environment and the genetic background of the developing fetus produce epigenetic programming changes at the level of transcription, ultimately shaping developmental trajectories. Birth cohort studies examining specific maternal exposures to stress, infection, and dietary challenges have provided the clearest evidence to date for the critical role such fetal antecedents play in disease risk, and the temporal specificity for exposure windows of susceptibility. These large registries allow prospective studies to be conducted, which greatly aids in the accuracy of exposure reporting and confirmation, and the collection of biological materials to be analyzed as potential biomarkers for genetic/epigenetic factors associated with susceptibility. In the following, evidence from clinical and epidemiological studies is described related to specific neurodevelopmental diseases and our current knowledge of fetal antecedents that increase risk for these disorders.
SCHIZOPHRENIA
Prenatal and early life events such as maternal stress and infection have been associated with an increased risk of schizophrenia (Brown, 2012). Studies from large birth cohorts in which clinical, neurocognitive, and neuroimaging measures have been obtained revealed strong associations between in utero exposure to stress, infections, hypoxia, starvation, and an increased risk for schizophrenia (Bale, 2011; Susser et al., 2008), including disturbances of executive function, working memory, verbal memory, and structural brain abnormalities among offspring with schizophrenia (Brown et al., 2009b). In support of a temporal specificity to the effects of fetal antecedents on long-term outcomes in neurodevelopmental disorders, a recent epidemiological study reported a significant association between maternal stress experienced during the first trimester of pregnancy with an increased risk of schizophrenia in males (Khashan et al., 2008). Prospective birth cohort studies have suggested that such stress exposures may act by altering brain developmental trajectories involving epigenetic modifications, the evidence for which in humans, however, is currently lacking.
Maternal infection and immune disturbances during pregnancy have a strong association with offspring schizophrenia risk. Influenza exposure during early or mid-gestation produced a three-fold increased risk for schizophrenia compared with controls (Brown, 2012). The presence of elevated IgG antibody to Toxoplasma gondii in maternal serum taken during pregnancy was also associated with a twofold increased risk of schizophrenia (Brown, 2012; Mortensen et al., 2007). Maternal genital reproductive infections around the time of conception have also been reported to have a fivefold increase in schizophrenia risk. Clinical studies examining neural end points have found an association of prenatal infection with deficits in executive function and morphological changes in schizophrenia patients (Brown, 2012). More specifically, neuroimaging findings indicated that prenatal infection was related to enlargement of the cavum septum pellucidum and diminished intracranial volume in these cases (Brown et al., 2009a).
Similar to the contributions made by maternal stress or infection to offspring development and programming, maternal diet directly regulates fetal growth, epigenetic programming, and disease risk. Epidemiological studies have repeatedly found an increased risk of schizophrenia in offspring from mothers exposed to severe caloric restriction or famine. In addition to the volume of epidemiological reports demonstrating intergenerational consequences of maternal starvation, obesity, and metabolic syndrome during pregnancy, the first data pointing to molecular mechanisms are now beginning to emerge. Periods of famine affecting human populations for well-defined durations afford the potential to track effects of undernutrition during pregnancy on offspring outcome. Findings stemming from the Dutch Hunger Winter of 1944–1945 reveal consequences ranging from glucose intolerance, increased coronary heart disease, altered stress responsiveness, and schizophrenia for the adult offspring of pregnancies occurring during a period of reduced caloric availability and increased psychological stress (Brown and Susser, 2008; Ravelli et al., 1976). These outcomes have been associated with changes detected in circulating growth factors and alterations in DNA methylation patterns of several genes, including the imprinted gene, insulin-like growth factor-2 (IGF-2) (Bale et al., 2010). These findings were detected 60 years following the famine exposure, supporting the long-lasting programming effects of the epigenetic marks. Interestingly, data from the Dutch Hunger Winter and the nineteenth century Swedish famine revealed that severe caloric restriction during pregnancy has transgenerational consequences in which even the second generation of offspring had increased neonatal adiposity, and overall lower quality health as adults when their mothers were exposed to famine (Brown and Susser, 2008), suggesting that epigenetic marks in germ cells must have occurred during the initial insult exposure.
Although most epidemiological studies have focused on offspring outcomes following periods of famine or malnutrition during pregnancy, our changing landscape is shifting the focus to effects of maternal overnutrition. Certainly, transgenerational epigenetics may be contributing to the rapid amplification of obesity rates and human height observed in recent generations (Bale et al., 2010). In addition, predisposing subsequent generations to these traits through epigenetic inheritance may compound the rate at which phenotypes progress. Maternal obesity has also been associated with a two- to threefold increased risk for schizophrenia in the offspring from two separate birth cohorts (Khandaker et al., 2012).
AUTISM SPECTRUM DISORDERS
Many factors have been examined as potential fetal antecedents related to an increased risk for autism spectrum disorders (ASD), including stress and infectious and immune disturbances (Brown, 2012). Although genome-wide association studies (GWAS) and other genetic analyses studies have identified candidate ASD susceptibility genes, more recent work in identical and fraternal twin pairs with autism showed that genetics accounted for less of the disease risk than did the shared environment (Angelidou et al., 2012). Such analyses support the critical importance for consideration of the interaction between genetic susceptibility and potential environmental insults that may produce epigenetic changes in neurodevelopment. Studies using the Danish Medical Birth Register found an association between viral infection resulting in hospitalization in the first trimester of pregnancy and a threefold increase in ASD risk. In a prospective study by the Early Markers for Autism, in which the presence of 17 cytokines was assayed in maternal serum and related to offspring ASD diagnosis, the authors found that specific cytokines, including IL-4, IL-5, and IFNγ, were increased at mid-gestation in mothers of children diagnosed with ASD. Prospective studies have also reported associations between amniotic fluid levels of specific cytokines such as TNFα, IL-4, and IL-5 with an increased risk for ASD (as reviewed in Brown, 2012).
Maternal stress has also been associated with an increased risk for ASD, including stress exposure at 21 to 32 weeks of gestation, and postnatal stressors such as the death of a relative in the first six months of life (Angelidou et al., 2012). However, in a metaanalysis by Gardner et al. on combined studies published since 2007, only four factors appear to consistently increase the risk for ASD: advanced maternal age, advanced paternal age, being first born, and having a mother born outside North America, Europe, or Australia (Gardener et al., 2009). A recent follow-up metaanalysis study from this group has added interactions of factors including umbilical cord complications, fetal distress, birth trauma, and a low five-minute Apgar score with increased risk for ASD (Gardener et al., 2011). What is clear is that more information and controlled prospective studies are still needed for confirming links between maternal stress or infection with ASD risk (Guinchat et al., 2012).
AFFECTIVE DISORDERS
There is growing evidence supporting an association between fetal risk factors and affective disorder predisposition, likely involving early epigenetic shifts in developmental trajectories. Birth cohort studies have identified prenatal conditions, including maternal immune and stress responses, as significant risk factors for major depressive disorder (MDD) (Bale et al., 2010). For instance, second trimester maternal exposure to type A2/Singapore influenza significantly increased the risk for unipolar and bipolar disorders in a cohort of Finnish and British adults. In addition, maternal exposure to famine during the second and third trimesters elevated offspring lifetime risk for MDD, supporting an important link between maternal nutrition and offspring neurodevelopment that may also relate to maternal stress resulting from insufficient food sources. Although maternal infection, stress, and undernutrition differentially affect the developing fetus, there are likely shared underlying mechanisms contributing to an increased vulnerability to MDD, including effects on the developing stress neurocircuitry (Bale et al., 2010).
As the brain continues to mature and develop well into adolescence, appreciation of the influences of the postnatal environment on programming of disease risk is necessary. Studies of the long-term consequences of adverse early childhood experience have unequivocally revealed that adults exposed to child abuse and/or neglect are at a greater risk for the lifetime development of affective disorders. In clinical studies, there is clear evidence for long-term neurobiological, neuroendocrine, and immune alterations following exposure to early life adverse events during critical periods in brain development (Bale, 2011). In addition, plasma stress hormone levels in response to relatively mild stressors are markedly increased in patients who have experienced early life trauma, including sexual or physical abuse.
In humans and non-human primates, hypersecretion of the stress hormone corticotropin-releasing factor (CRF), as detected in the cerebrospinal fluid (CSF), has been found years after the initial period of stress exposure during early life. Similar results were found in clinical studies in which women with a history of child abuse and/or neglect exhibit hyperactivity of the hypothalamic–pituitary–adrenal (HPA) axis in the Trier Social Stress test and increased CSF CRF concentrations. In addition, these patients exhibited decreased CSF concentrations of oxytocin, a peptide shown to be important in social biology and bonding, increased proinflammatory cytokines such as IL-6, and reduced hippocampal volume as measured by structural magnetic resonance imaging (Bale et al., 2010; Heim et al., 2009, 2010). Further supporting a gene x environment interaction for development of affective disorders is the growing evidence of a genetic predisposition that underlies a stress-sensitive phenotype, thereby increasing the likelihood for stress experience throughout life, and elevating the risk for disease presentation (Kendler, 1998).
What is clear from examination of the epidemiological literature is that although a relatively small number of fetal antecedents (e.g., stress, infection, dietary challenges) have been linked with the neuropsychiatric disorders discussed in the preceding, what distinguishes the outcomes is likely the timing of the exposure/insult combined with the genetic background and fetal sex of the individual. How these factors independently or combined can act to influence neurodevelopmental programming is best examined using animal models.
THE ANIMAL MODELS
The incredible complexity of neuropsychiatric diseases makes the generation of relevant and beneficial animal models difficult; but without these models, mechanistic studies are nearly impossible. Evaluation and diagnosis of a mental health disorder requires a clinical conversation with a patient so as to assess specific criteria. Obviously, this is not possible in an animal. Therefore, rather than producing models of diseases in their entirety, an alternate approach has been to focus on important aspects, or endophenotypes, of the disease that can be more clearly measured in animals (Table 72.1). Examples include stress responsivity (behavioral or physiological), cognitive performance, spatial learning, and social behaviors. Many of these outcomes have been pharmacologically validated, with human pharmaceuticals having known efficacies in mental health disorders. These studies provide a wealth of opportunities to manipulate the animals’ environment while controlling all other variables so as to identify important mechanisms involved in epigenetic programming and brain development.
TABLE 72.1. Summary of animal models being examined in relation to early life exposure and long-term outcomes of endophenotypes relevant to neuropsychiatric disease
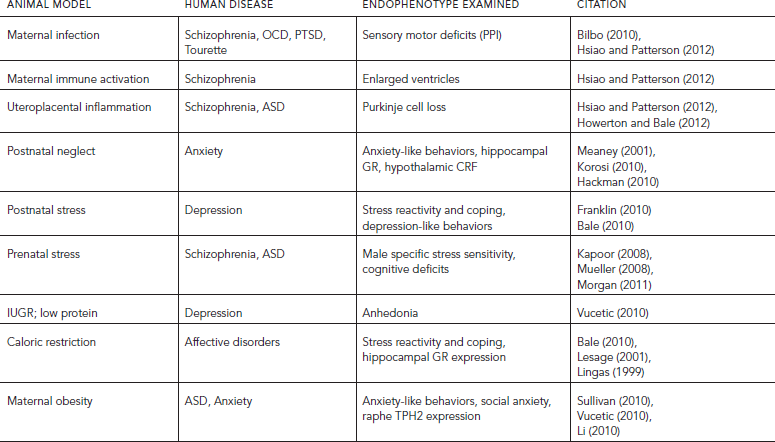
In examining the prenatal or intrauterine environment, animal studies have examined factors such as maternal stress exposure, infection, and dietary changes (high fat, or low protein) and their impact on offspring outcomes during specific developmental windows and as adults. Postnatal environments have also been studied for promoting changes in maternal care that are associated with long-term effects on offspring brain and behavior. Transgenerational effects have been examined in these models to determine potential epigenetic programming that involves the germ cells, potentially altering future generations’ disease susceptibility. An additional benefit provided by animal models is the ability to examine the temporal specificity for effects of fetal antecedents. Because fetal organ and tissue development occurs over the course of gestation, it follows that the impact of a given insult would depend on the stage at which it occurred. Thus, by more closely examining these specific periods of development, greater insight has been obtained as to points of increased vulnerability, as well as the potential for improved interventions and biomarker identification in predicting neurodevelopmental disorder risk. Animal models hold the greatest promise to accomplish this for their ability to control and study necessary variables such as genetic background and life experience, while manipulating the environment during specific windows of development.
PROGRAMMING EFFECTS OF EARLY STRESS EXPOSURE
PRENATAL STRESS
Stress pathway dysregulation is the most pervasive symptom in neuropsychiatric disease, and both clinical and basic research has identified factors important in the developmental programming and maturation of this system, as well as the sensitive periods during which perturbations may be disruptive. Animal models examining maternal stress have provided mechanistic insight into the long-term programming of important endophenotypes. As in humans, outcomes from maternal stress studies in animals have varied depending upon the stressors utilized, the outcomes examined, and timing of the stress event during pregnancy (Bale, 2011). Overall, in mice, rats, guinea pigs, and nonhuman primates, prenatal stress increases the sensitivity of the offspring HPA stress axis, anxiety-like and depressive-like behaviors, and cognitive deficits; all endophenotypes associated with neuropsychiatric disease.
Although prenatal stress has been broadly associated with offspring disease, the developing nervous system is unlikely to show uniform vulnerability to perturbations over the course of gestation. To compare maternal stress experience across early, mid, or late pregnancy on programming of offspring stress regulation, a chronic stress model in mice determined that early gestation (days 1 to 7 of the 20- to 21-day gestation in mice) resulted in male offspring with an increased stress-sensitive phenotype as adults (Mueller and Bale, 2008). These males had elevated amygdalar CRF, and reduced hippocampal glucocorticoid receptors (GR). Epigenetic analyses determined that DNA methylation changes of these genes correlated with their expression. Interestingly, similar outcomes have been reported following gestational glucocorticoid exposure on offspring GR expression and promoter methylation, suggesting potential common mechanisms and points of vulnerability that may relate to direct or indirect actions of glucocorticoids during brain development (Bale, 2011).
A prenatal stress model in the guinea pig has similarly explored the timing specificity of prenatal stress using short periods of daily flashing light as a maternal stress beginning on embryonic day E50 or E60 of the 70-day guinea pig gestation. Although stress beginning at E50 resulted in increased anxiety-like behaviors, elevated basal corticosterone levels, and lower hippocampal GR in adults, stress beginning at E60 failed to produce these outcomes (Kapoor et al., 2008). Overall, these results support epidemiological findings as to the importance of temporal specificity for maternal stress programming of offspring.
POSTNATAL STRESS
Rodent models examining the early postnatal window of brain development have produced a great deal of evidence for the effects of both enhanced and fragmented maternal care producing long-term effects on the developing brain. Meaney et al. have contributed a wealth of information to the field as to the epigenetic programming that occurs in early life related to the quality of maternal care (Hackman et al., 2010; Meaney, 2001). This group characterized a rat model in which dams had either high or low levels of early postnatal maternal care—defined as high or low licking and grooming. In this model, the postnatal interactions of the dam with her litter determined the behaviors of the offspring as adults, in which high licking and grooming offspring showed reduced levels of anxiety-like behaviors and diminished HPA stress axis activity. The epigenetics involved in these outcomes, at least in part, appeared related to the DNA methylation status of the GR in the hippocampus of these offspring. High licking and grooming mothers produced enhanced tactile stimulation of their pups, resulting in increased expression of specific transcription factors during brain development that ultimately dictate expression levels of GR. This is one example, then, of a gene that is epigenetically programmed during early development lasting into adulthood and having a potential impact on shaping offspring stress responsivity.
In contrast to models of stress resilience, chronic postnatal stress can promote cognitive decline via functional and structural programming of specific brain regions, including the hippocampus. These studies suggest that the magnitude of stress exposure may determine its long-lasting effects, such that modest postnatal stress may promote resilience, whereas severe or chronic stress may set in motion mechanisms that contribute to stress-related neurodevelopmental disease (Bale et al., 2010). For instance, in a model in which dams are provided very limited nesting and bedding material as a stressor in the postpartum environment, epigenetic changes were identified within the adult offspring hippocampus where the ability of a transcriptional repressor to bind to the silencing region of the stress gene, CRF, was found, thus altering the ability of these offspring to appropriately respond to and cope during stress experience as adults (Korosi et al., 2010).
Animal models have also examined transgenerational outcomes demonstrating germ cell programming from stress exposure in both the prenatal and postnatal environments. Prenatally, studies in mice have shown that males exposed to stress in utero and presenting with increased stress sensitivity themselves as adults, can pass on this phenotype to their male offspring (Morgan and Bale, 2011). In examination of the brain miRNA environment, it was found that there were significant differences in the expression of a number of these noncoding RNAs in these second-generation males, supporting an epigenetic target of maternal stress that was present into the second generation not requiring subsequent re-exposure to the insult. Similarly, in a mouse early postnatal stress model, offspring continued to present with a depressive-like phenotype through three generations, with comparable changes detected in DNA methylation in the germline of these mice (Franklin et al., 2010). These studies showing interactions of the environment with the genome at the level of placing long-lasting epigenetic marks in the chromatin that remain through cell divisions and across generations, support the susceptibility of the germline to external influences that could increase the disease susceptibility in subsequent generations.
MATERNAL INFECTION AND IMMUNE ACTIVATION
Numerous animal models of maternal infection show similar relationships with that reported in epidemiological studies in which maternal immune activation during pregnancy produced offspring endophenotypes associated with neurodevelopmental disorders, including ASD and schizophrenia (Spreckelmeyer et al., 2011). Such studies have utilized several models, including maternal influenza viral infection, injection of the pathogen mimics Poly I:C and lipopolysaccharide (LPS), and the proinflammatory cytokine IL-6 (Howerton and Bale, 2012). The most consistent phenotype produced in these models is a deficit in offspring pre-pulse inhibition (PPI) in response to an acoustic startle, a change that is reversed by the administration of the antipsychotic drugs haloperidol or clozapine. Sensorimotor gating deficiencies, phenotypes measured by PPI, are commonly associated with schizophrenia, and have also been demonstrated in numerous other psychiatric or affective disorders such as obsessive compulsive disorder, Tourette syndrome, and posttraumatic stress disorder, and thus are highly relevant endophenotypes in which potential epigenetic mechanisms can be explored. Again, highlighting the importance of gestational timing for the impact of fetal insults in producing long-term outcomes, a single administration of the proinflammatory cytokine, IL-6, at mid-gestation in mice was sufficient to produce significant deficits in both PPI and latent inhibition. Maternal immune activation in rodents using a low dose of LPS during mid-gestation also produced offspring with deficits in PPI, and these offspring also exhibited enlarged ventricles, a hallmark of schizophrenia (Hsiao and Patterson, 2012).
The mechanism and potential epigenetic involvement in maternal immune activation programming of the offspring developing brain is not clear. Certainly, maternal cytokines have direct access to the placenta, the intermediary tissue that serves to protect the developing fetus, and have also been detected in the amniotic fluid following maternal immune challenge (Howerton and Bale, 2012). Therefore, there are likely direct as well as indirect downstream targets of these maternally produced cytokines that are involved in programming changes in the fetal brain. Of note, uteroplacental inflammation is sufficient to produce an increase in expression of apoptotic markers, including caspase-3 and -4 in Purkinje cells of the fetal ovine cerebellum, similar to the Purkinje cell loss reported in ASD and schizophrenia brains (Hsiao and Patterson, 2012). These results suggest that a contributing mechanism for maternal infection to alter brain development involves immune effectors at the level of the placenta. Interestingly, studies in mice and rats have clearly demonstrated that pregnant dams treated with viral “mimics” have reported features of schizophrenia in their offspring, supporting that a replicating virus is not required to transmit these programming outcomes, and that the array of pathogens that may promote inflammatory processes is great.
MATERNAL DIET: LOW PROTEIN AND HIGH FAT
Molecular and phenotypic evidence for programming effects of altered maternal nutrition including protein restriction, high fat diet, and methyl donor supplementation on brain development has been collected from extensive studies in animal models. In general, consequences of maternal dietary manipulations on first generation offspring reflect those observed in humans, although the extent of phenotypes, mechanisms, and sex-specific outcomes vary depending on the diet, timing of exposure, and model organism examined (Bale et al., 2010).
INTRAUTERINE GROWTH RESTRICTION
AND LOW PROTEIN MODELS
Rodent models have demonstrated an important link between the maternal nutritional availability and offspring brain development, especially for models of maternal low protein that typically induce an intrauterine growth restriction (IUGR) phenotype (Dunn et al., 2011). In mice, a protein deficient diet throughout pregnancy and lactation produced profound changes in offspring reward-related behaviors as adults in which IUGR mice showed a reduced preference for a palatable sucrose solution and hyperactivity in response to an acute cocaine injection, such reduced reward sensitivity or anhedonia is an endophenotype of affective disorders, including depression and anxiety. These behavioral changes were associated with programming differences in reward circuitry including increased expression and reduced DNA methylation of the promoter regions of key dopaminergic genes in the ventral tegmental area, prefrontal cortex, and nucleus accumbens (Vucetic et al., 2010). Mechanistically, one of the ways in which protein deficiency may produce changes in the offspring brain is via an intersection with immune activation. For instance, in a rat model of maternal low protein, increased levels of the proinflammatory cytokines TNFα and IL-6 were found in the placenta, again suggesting that common pathways may be involved in programming offspring disease risk.
It is also clear that pregnancy undernutrition is a stressor, and likely elevates maternal stress responsivity. In rats, maternal 50% food restriction during late pregnancy increased maternal plasma corticosterone levels and fetal adrenal weights. Offspring subsequently showed changes in hippocampal GRs and HPA axis sensitivity similar to that reported in maternal stress models (Lesage et al., 2001). Similar results were found in a guinea pig model of maternal nutrient restriction of 48 hours of food deprivation in which both maternal and fetal cortisol levels were elevated during late gestation, and GRs were reduced in the offspring hypothalamus and hippocampus as adults (Lingas et al., 1999).
MATERNAL HIGH-FAT DIET AND OBESITY MODELS
Because the organization of neural circuits controlling energy balance takes shape during perinatal life, considerable attention has focused on the developing hypothalamus, the source of neuroendocrine control, including that of stress regulation. Interestingly, the adipocyte hormone leptin, which is dramatically increased in the obese state, plays a vital role in directing development of hypothalamic projections in and around the arcuate nucleus. Moreover, secretion of leptin both within the fetus and from the placenta during development changes in response to the nutritional environment. Leptin signaling is thus poised to influence the developing brain regarding maternal over- or undernutrition via distinct actions on two functionally divergent neuronal populations, the anorexigenic pro-opiomelanocortin (POMC) and orexigenic neuropeptide Y (NPY) neurons (Bale et al., 2010). Target projections of these neurons include the paraventricular nucleus and the lateral hypothalamus, hypothalamic regions that exert widespread regulatory control over homeostatic functions throughout life. Such studies have perhaps identified specific modes of neurodevelopmental programming whereby the wiring of the hypothalamus is determined through endocrine signals. In addition, mice fed a high-fat diet throughout pregnancy produce offspring with altered DNA methylation patterns in dopaminergic and opioid genes and behaviors, suggesting that an obese maternal environment programs offspring with reward pathway dysregulation that may ultimately influence mood and affect (Li et al., 2010).
As mentioned, there is convincing evidence for a likely intersection between maternal dietary challenges and immune activation. In both rodents and nonhuman primates, studies have demonstrated increased inflammatory processes in important brain regions in offspring born to mothers on a high-fat or obesogenic diets. Rat offspring from a dam fed a high-saturated fat diet during pregnancy showed significant levels of activated microglia in their hippocampus at birth that remained high throughout life, consistent with an increased basal neuroinflammatory tone (Bilbo and Tsang, 2010). These offspring also displayed increased anxiety-like behaviors and reduced performance ability in a spatial learning and memory task, supporting a likely contribution of local immune factors such as proinflammatory cytokines in disrupting normal brain function and stress processing. Further, in the nonhuman primate, offspring from macaque mothers fed a high fat diet for four years before pregnancy showed substantial increases in proinflammatory cytokines and activated microglia in their hypothalamus during the third trimester (Grayson et al., 2010). Female offspring in these studies also demonstrated high levels of social anxiety and fear of novelty as infants (Sullivan et al., 2010). Results from these studies support a potential link between maternal nutritional intake throughout pregnancy and increased neuroinflammation in the developing brain and long-term changes in behaviors relevant to neurodevelopmental disease.
Epigenetic mechanisms, which exert lasting effects on gene expression and can be heritable, are a particularly intriguing target when examining links between the perinatal nutritional environment and offspring metabolic phenotype. For example, foods high in choline cause marked changes in DNA methylation, which, in turn, alter long-term gene expression. Studies have demonstrated that choline deficiency during pregnancy produced alterations in histone methylation and subsequent changes in gene expression in mice (Bale et al., 2010). Pregnant dams fed choline-deficient diets during late gestation produced offspring with diminished progenitor cell proliferation and increased fetal hippocampus apoptosis, altered hippocampal angiogenesis, insensitivity to long-term potentiation as adults and decreased visual-spatial and auditory memory. These changes in fetal brain development were associated with epigenetic alterations in DNA and histone methylation in the fetal hippocampus. Similarly, studies using changes in mouse coat color as a marker of dietary and nutrient influence on epigenetic regulation of the agouti locus demonstrated how relatively minor changes in DNA methylation can produce a profound phenotypic impact (Waterland, 2003). Such studies support the ultimate mechanistic impact for minor dietary and nutritional fluctuations during key points in development.
FETAL SEX AS A FACTOR IN EPIGENETIC PROGRAMMING
Epidemiological studies linking fetal antecedents with long-term disease risk have established gender as an important determinant in disease severity and onset. As an example, pregnant mothers exposed during their second trimester to the stress of the 1940 invasion of The Netherlands had male, but not female, offspring with an increased risk of schizophrenia (van van Os and Selten, 1998). Further, many neurodevelopmental disorders have a strong sex bias, including ASD with an overall sex ratio of 4.3:1 for boys-to-girls (as reviewed in Newschaffer et al., 2007). However, when further separated for cognitive impairment, this bias was even further increased for ASD without mental retardation to 5.5:1, suggesting that distinct underlying mechanisms or predisposing factors may be involved. As exposure to maternal stress before 32 weeks’ gestation has been suggested as a potential contributing factor to ASD (Beversdorf et al., 2005), understanding the role sex plays in the specificity of response to stress during development may provide unique insight as to disease etiology. A recent report detected a significant effect of maternal depression during pregnancy on offspring postnatal anxiety development, particularly in males (Gerardin et al., 2010). These studies support both a sex- and temporal-specificity in the association between maternal stress and offspring disease. Although there are many factors that likely contribute to sex differences in disease predisposition, sex-specific responses to fetal antecedents occurring during sensitive windows of development may promote long-term programming effects that underlie such disease biases. Along these lines, structural brain volume analyses using functional magnetic resonance imaging (fMRI) in male and female patients with schizophrenia have confirmed a disruption of the normal sexual dimorphism of the brain, including the dimorphic ratio of orbitofrontal cortex to amygdala where male schizophrenic patients showed a phenotypically more female pattern in these brain regions (Bale et al., 2010).
The brain develops in the face of combined and opposing forces of resiliency and vulnerability. The central factors important in resilience, ongoing neurogenesis, migration, myelination, differentiation, and synaptogenesis are the same processes subject to derailment, which can promote lasting consequences. The complex interplay of genetics, early experience, and later environment underlies the weak but consistent heritability of numerous neurodevelopmental disorders. The developing brain is organized by developmental hormone exposure, with males experiencing elevated testosterone levels during normal testes development. Aromatization of this testosterone to estradiol in the brain drives masculinization, an active process affecting cell differentiation and connectivity in the brain. Estrogenic involvement in cell death and cell birth in the developing nervous system is a critical component in programming the sexually dimorphic brain (McCarthy et al., 2009). The variety of mechanisms evoked by estradiol during development provides numerous avenues for disruption of the active process of masculinization. Programming of important regulatory brain regions, such as the neuroendocrine hypothalamus, via steroid hormone effects on cell migration patterns during early development may also contribute to sex differences in disease susceptibility (Bale et al., 2010).
In rodent models, studies have taken advantage of the ability to manipulate the early postnatal critical window to examine the organizational effects on long-term stress responsivity, resulting in an established sex difference in stress responsivity in adult mammals. Similar to outcomes reported with prenatal stress exposure, hippocampal GR expression was altered in female offspring masculinized at birth by a single injection of testosterone, supporting the importance of the male testosterone surge in normal wiring of these pathways (Bale, 2011). In addition, the early postnatal period is a sensitive window during which hormonal exposure produces organizational effects on maturation of the serotonin system, potentially leading to long-term changes in adult stress sensitivity. In addition to the early critical period of programming, the rise in testosterone beginning in puberty exerts modulatory actions on neurotransmitter systems critical in regulation of stress physiology and coping. Such systems affected included serotonergic and γ-aminobutyric acid (GABA)-ergic (Bitran et al., 1993). Therefore, the coordinated impact of masculinization at both time periods may interact with components of sex chromosomes to orchestrate a complete “normal” male phenotype. Neuropsychiatric disease predisposition may then involve a mismatch between brain organizational programming and activational hormones.
Similar to models of maternal stress, there is also evidence to support that offspring programming responses to maternal immune activation are also sex dependent. For example, male offspring of pregnant rats challenged with LPS late in gestation showed significant deficits in PPI, whereas females were unaffected by this insult (Howerton and Bale, 2012). Similarly, male but not female mice exposed during late gestation to the viral mimic Poly I:C displayed behavioral and cognitive inflexibility, key negative symptoms in schizophrenia. These sex-dependent programming effects were associated with significant reductions in glutamate content in the prefrontal cortex in the males exposed to the prenatal immune activation (Spreckelmeyer et al., 2011).
Little is currently understood regarding how offspring sex may influence the epigenetic response to a changing maternal environment. It is likely that sex-specific responses to fetal antecedents during sensitive windows of development contribute to these differences. Studies in animal models of prenatal stress have begun providing interesting clues as to the timing and mechanisms involved in how perturbations in the maternal milieu may have sex specificity in their effects on the developing brain (Bale et al., 2010). The disruption of sex-dependent processes is a common theme in the prenatal stress literature. Sex differences in neurodevelopment are the result of both genetic sex and circulating gonadal hormones. One of the first papers modeling prenatal stress in rats found that chronic restraint during the final week of gestation disrupted male sex behavior (Ward, 1972). Late gestational restraint stress has also been shown to disrupt the organizational perinatal testosterone surge, reduce adult testis size, shorten anogenital distance, and block the differentiation of sexually dimorphic brain nuclei in males (Bale, 2011). Similarly, male mice exposed to maternal stress during early gestation have reduced testis size and testosterone levels and a shortened anogenital distance as adults, supporting a disruption in their normal masculinization. Such outcomes demonstrate potential sex-specific mechanisms for changes in the programming of the sexually dimorphic brain despite a shared intrauterine environment.
There are many epigenetic mechanisms by which estrogen promotes and drives sex differences in the developing brain. During the critical window of brain sexually dimorphic brain programming, estradiol can broadly act to alter DNA methylation patterns, including that of its own estrogen receptor alpha (ERα), effectively shaping the landscape to be more or less responsive to the hormonal status of the animal (McCarthy et al., 2009). One mechanism by which estrogen is thought to modulate methylation is through its effects on DNMT gene expression and activity levels. These effects are brain region specific, providing yet another level of control for sex differences to progress and ultimately affect systems outcomes such as stress responsivity. Estradiol can also directly alter histone modifications, including actions on histone deacetylases (HDACs) (McCarthy et al., 2009). A single postnatal administration of an HDAC inhibitor in males during the critical window disrupted programming of the sexually dimorphic BNST, a brain region important in stress neurocircuitry. In addition, the broad epigenetic programming effect of estrogen has been shown for dramatic shifts in the miRNA environment in the developing brain. In the early postnatal mouse brain, males and females have significant differences in expression patterns of the 250 most abundant miRNAs. These sex differences were dependent on the conversion of testosterone to estradiol in males, as a single administration of an aromatase inhibitor on postnatal day 1 completely shifted the normal male pattern to that of females (Morgan and Bale, 2011). These studies provide substantial evidence for epigenetic mechanisms for steroid hormones to impact neurodevelopment, producing dramatic and important sex differences. However, these mechanisms are also vulnerable to perturbations in the environment, shifting “normal” developmental patterns and trajectories. As described, maternal stress and infection have both been associated with dysmasculinizing phenotypes, suggesting that a disruption at some level in male sexual differentiation or gonadal development occurs that alters testosterone production during the organizational period.
INTRICACIES OF THE PLACENTAL CONTRIBUTION TO FETAL PROGRAMMING
The developing placenta is an intriguing candidate tissue for mechanistic examination of epigenetic programming, as it is a rapidly developing and sex-specific endocrine tissue that continues to respond to the dynamic maternal milieu throughout pregnancy. The placenta serves as the critical messenger between the maternal and embryonic compartments, and therefore is poised to be influenced by perturbations occurring in pregnancy by a number of mechanisms, including alterations in: (1) nutrient and oxygen transport, (2) inflammatory responses, and (3) epigenetic programming (Bale et al., 2010) (Fig. 72.1). Tight control of placental and embryonic epigenetic machinery is critical during gestation when a wave of genome demethylation before de novo re-methylation by DNA methyltransferases, establishment of imprints, and sex determination occurs, identifying novel targets as highly vulnerable to maternal disturbances that could result in embryonic reprogramming. For instance, inflammatory cytokines can directly affect levels and activity of DNA methyltransferases (DNMT), as well as regulate placental receptors and transporters for folate, an important methyl donor shown to regulate levels of methylation of non-imprinted genes during pregnancy, thus shifting more broadly the epigenetic landscape (Bale, 2011).
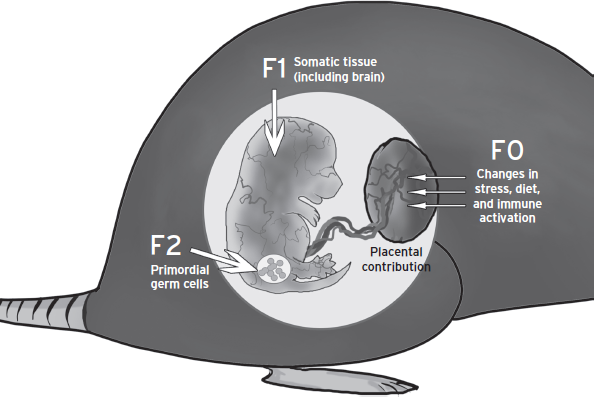
Figure 72.1 Intersection of prenatal insult and developmental timing to program tissue targets and affect subsequent generations. Information regarding maternal stress, infection, or diet perturbations, in the form of changes in hormonal milieu, is transmitted from the maternal compartment to the fetal compartment via the placenta. The dynamic process and trajectory of fetal development provides a temporal specificity for responses to fetal antecedents. Acute programming of somatic tissues can lead to changes in long-term health outcomes in the first generation. In addition, the primordial germ cells, which contribute genetic and epigenetic information to the second generation, are also present and can undergo reprogramming during embryonic development. Thus, a truly “transgenerational epigenetic” mark must be present into a third generation. In addition, embryo sex plays an important role in determining how an insult may become part of the epigenome and passed on to future generations.
Evidence from animal models of maternal stress supports an important regulatory role of the placenta in mediating effects between the maternal and fetal compartments. In early prenatal stress studies, a sex-specific effect of stress on placental epigenetic machinery was shown in which maternal stress increased expression of DNMT1 and the methyl binding protein, MeCP2, in males (Bale, 2011). Previous studies in mice have reported that regulation of placental methylation patterns is predictive of similar embryonic changes critical in neurodevelopment. Recent studies examining early life stress found similar changes for MeCP2 expression and methylation in the brains of first- and second-generation stressed mice (Bale, 2011). Further, maternal stress in mice has reported significant increases in the expression of genes important in growth and development including PPARα, IGFBP-1, GLUT4, and HIF3α in male but not female placentas (Bale et al., 2010). Mechanistically, the link between maternal stress and changes in placental gene expression may involve direct or indirect actions of stress hormones. As an example, stress-induced glucocorticoids increase expression of PPARα, and PPARα in turn drives expression of IGFBP-1, suggesting one mechanism whereby maternal stress could directly affect placental gene expression patterns directly relevant to embryo development. In addition, reductions in growth factors have been linked to affective and neurodevelopmental disorders, and IGFBP-1 is known to down-regulate genes involved in embryonic growth. Thus, as one example these studies suggest that an elevation in placental IGFBP-1 with a consequent decrease in available growth factors during critical developmental periods could impart a sex-specific effect on male fetal programming. Alterations in oxygen and nutrient availability have also been associated with inflammation, and there is an established association between placental inflammatory events and an increased risk for affective disorders, schizophrenia, and autism (Bale et al., 2010). How these placental outcomes are sex dependent remains unknown, but may be related to a protective or buffering mechanism of genes on the X chromosome. Because X inactivation occurs to a much lesser extent in the placenta, increased gene dosage in female placentas could underlie an altered response to the changing maternal environment.
The ability for maternal stress hormones (cortisol or corticosterone) to gain access to and affect fetal brain development is highly dependent on placental levels of 11β-hydroxysteroid dehydrogenase-2 (11β-HSD2), the placental barrier enzyme that converts glucocorticoids to their inactive metabolite. At the interface of the maternal:fetal circulation is the syncytiotrophoblastic cells that express high levels of 11β-HSD2, and function to exclude the majority of maternal active glucocorticoids before they reach the fetal compartment. Typically, in early-mid gestation, fetal blood has greater than 10-fold lower cortisol levels than maternal blood (O’O’Donnell et al., 2009). Environmental perturbations that would serve to decrease 11β-HSD2 expression could then expose the fetus to higher levels of active glucocorticoids. Such evidence has been reported in animal models in which pharmacological blockade of 11β-HSD2 was administered during pregnancy and resulted in significant increases in glucocorticoid receptors in the amygdala of the offspring as adults (Holmes et al., 2006). Genetic disruption of placental/fetal 11β-HSD2 to reduce its expression was also found to produce offspring with higher levels of anxiety-like behaviors. Perturbations in maternal diet are also known to affect placental 11β-HSD2 where a low protein diet during pregnancy significantly decreased its activity, supporting again a link between nutritionally deficiencies and stress axis changes (O’Donnell et al., 2009).
Fetal antecedents such as high fat diet or obesity and maternal stress have also been associated with increases in placental inflammation (O’O’Donnell et al., 2009; Frias et al., 2011). Cytokine production in the placenta can increase markers of immune activation in the developing fetus or disrupt overall placental function, producing long-term programming changes in the offspring. In macaques, maternal high-fat diet increased placental inflammatory cytokines and expression of Toll-like receptor-4 (Frias et al., 2011). Similarly, high-fat diets in rodents during pregnancy have also shown significant increases in placental inflammatory cytokines, supporting a change in appropriate placental function in response to an unhealthy environment. Timing of these effects is also important in predicting outcomes. For instance, maternal immune activation during early gestation can result in a failure for implantation to occur, whereas mid-gestation inflammation in the placenta has been linked to schizophrenia and ASD-like phenotypes (Hsiao and Patterson, 2012).
Critical epigenetic processes occur within the placenta as part of the regulation for normal development and function, and disruptions in programmatic changes in the chromatin have been linked to increased disease risk for the offspring (Hsiao and Patterson, 2012). Chromatin remodeling complexes and DNA methyltransferases (DNMTs) are necessary for epigenetic reprogramming that occurs in pre-implantation development. This highly regulated process produces competent embryos and the trophoblastic cells of the placenta. The same epigenetic marks found in fetal tissue including DNA methylation, miRNAs, and histone modifications have also been reported in placental tissue. Studies have established unique roles for DNMT isoforms during pre-implantation in the placenta to maintain uniform methylation imprints in all embryonic and extraembryonic cells, including the placenta (Ackerman et al., 2012). Recent studies have identified miRNAs that are specific to the placenta, where their expression pattern appears to dynamically change across pregnancy in humans and rodents. As these miRNAs are also released from the placenta and are detectable in the maternal serum encased in microvesicles, there is potential to classify them as biomarkers of placental function. The necessity of miRNA expression in placental development has been established, in which mice deficient in the Argonaut-2 gene, a protein component of the RNA-induced silencing complex (RISC), show abnormal placenta function resulting in embryonic lethality (Ackerman et al., 2012). The placental miRNA environment also appears to be susceptible to perturbations resulting from such insults as hypoxia, immune activation, and dietary challenges, supporting a likely role for miRNAs in programmatic changes that are poised to alter fetal development.
CONCLUSIONS
In examination of the epigenetic mechanisms that may contribute to neurodevelopmental disease risk, the necessity for and value in studies using valid animal models in which variables such as environmental conditions, temporal specificity, and offspring sex can be evaluated. The transmission of epigenetic marks to future generations through reprogramming of the germ cells adds to the complexity in establishing causal links between disease presentation and fetal antecedents in clinical and epidemiological studies. In looking at offspring phenotypic changes, it is clear that although some phenotypes terminate with the first generation, others will be transmitted across multiple generations (Dunn et al., 2011). The degree to which there is true transgenerational “epigenetic” involvement in a given end point is determined by the ability for that mark to be erased and re-established outside of a re-exposure or continuation of the insult. Further, the sex-specificity of transgenerational epigenetic programming requires examination at both the level of transmission of an effect as well as the inheritance of it. In other words, can both mom and dad pass on the trait? And do both male and female offspring inherit it? Maternal exposure to aversive environments such as maternal stress, infection, or obesity results in a variety of phenotypes that have different transmission characteristics as they pass through the generations. Information about maternal experience, in the form of changes in hormonal milieu, can be transmitted via the placenta to the fetal compartment producing a direct action on the developing embryo (Dunn et al., 2011) (see Fig. 72.1).
In addition to ascertaining the transmission and generational passage of epigenetic marks, understanding the cell type specificity and involvement is also helpful in determining in what tissue an effect might occur as windows of developmental vulnerability to environmental challenges may be tissue specific (Fig. 72.2). Somatic tissues may be acutely programmed, leading to changes in health outcomes in the first generation. However, if prenatal insults do not directly affect the germline, the phenotype will terminate with this generation. Phenotypes that persist through the second generation but not a third may relate to the programming of the primordial germ cells during the initial maternal insult, and acquired transient epigenetic marks only capable of passing through a single generation. Thus, transmission of an epigenetic trait does not demonstrate a fully integrated, transgenerational germline-based mechanism until it reaches the third generation. However, if stable inheritance is only observed through the female line it may be caused by the presence of a maternal behavioral or uterine effect. Examination of transgenerational transmission through the paternal line avoids many of these confounding variables.
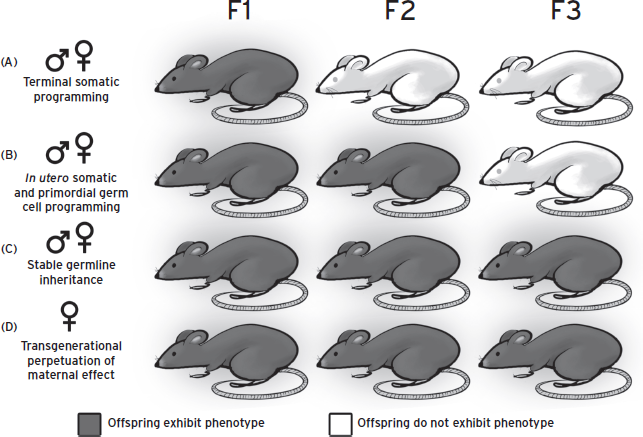
Figure 72.2 Modes of transgenerational epigenetic transmission. Several mechanisms for transgenerational epigenetic transmission are possible, as exemplified here in dark gray. Dark gray mice represent a changed phenotype, whereas white mice represent the “normal” end point, indicating that the phenotype has not been transmitted to that generation. The sex symbols at left indicate the maternal or paternal lineage capable of transmitting phenotypes in the indicated manner. (A) In utero somatic programming may acutely affect tissue and brain development in the fetus, initiating a developmental trajectory that results in an adult phenotype that is not transmitted into the next generation. (B) In addition to in utero somatic programming, changes in the maternal environment can acutely program primordial germ cells, resulting in transmission of the phenotype into the second generation. In this scenario, germ cells must be directly exposed to a maternal insult. (C) In utero exposure is sufficient to stably alter the germline of offspring, resulting in many generations of transmission without the need for re-exposure to the original maternal insult. (D) Transgenerational phenotypes may occur through a maternal effect wherein programming during gestation or through exposure to altered maternal behavior results in an adult phenotype in female offspring. The presence of this phenotype during pregnancy in the mother can in turn acutely program the next generation, perpetuating the transgenerational trait.
Our understanding of the mechanisms contributing to transgenerational epigenetic programming by fetal antecedents such as maternal stress and diet is likely to dramatically increase in the next decade. Prospective epidemiological studies are underway that will utilize banked tissue samples from birth cohorts to identify potential biomarkers predictive of disease. Such insight will no doubt yield great advances in disease etiology and provide novel targets and therapies in disease prevention and treatment. Studies examining effects across the maternal, placental, and embryonic compartments will be able to identify temporal and sex-specific markers that may provide our greatest insight into both direct and indirect gene targets. Necessary in this pursuit is the dissection of the complex role sex plays on both sides of the proverbial epigenetic coin for the transmission as well as the inheritance of given traits.
DISCLOSURE
Dr. Bale has no conflicts of interest to disclose. She is funded by NIMH only. Grant Support: R01MH073030, R01MH087597, R01MH091258, and P50MH099910.
REFERENCES
Ackerman, W.E., Bulmer, J.N., et al. (2012). IFPA Meeting 2011 workshop report III: placental immunology; epigenetic and microRNA-dependent gene regulation; comparative placentation; trophoblast differentiation; stem cells. Placenta 33(Suppl):S15–S22.
Angelidou, A., Asadi, S., et al. (2012). Perinatal stress, brain inflammation and risk of autism: review and proposal. BMC Pediatr. 12:89.
Bale, T.L. (2011). Sex differences in prenatal epigenetic programming of stress pathways. Stress 14:348–356.
Bale, T.L., Baram, T.Z., et al. (2010). Early life programming and neurodevelopmental disorders. Biol. Psychiatry 68:314–319.
Beversdorf, D.Q., Manning, S.E., et al. (2005). Timing of prenatal stressors and autism. J. Autism Dev. Disord. 35:471–478.
Bilbo, S.D., and Tsang, V. (2010). Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 24:2104–2115.
Bitran, D., Kellogg, C.K., et al. (1993). Treatment with an anabolic-androgenic steroid affects anxiety-related behavior and alters the sensitivity of cortical GABAA receptors in the rat. Horm. Behav. 27:568–583.
Borrelli, E., Nestler, E.J., et al. (2008). Decoding the epigenetic language of neuronal plasticity. Neuron 60:961–974.
Brown, A.S. (2012). Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev. Neurobiol. 72(10):1272–1276.
Brown, A.S., Deicken, R.F., et al. (2009a). Prenatal infection and cavum septum pellucidum in adult schizophrenia. Schizophrenia Res. 108:285–287.
Brown, A.S., and Susser, E.S. (2008). Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophrenia Bull. 34:1054–1063.
Brown, A.S., Vinogradov, S., et al. (2009b) Prenatal exposure to maternal infection and executive dysfunction in adult schizophrenia. Am. J. Psychiatr. 166:683–690.
Dunn, G.A., Morgan, C.P., et al. (2011). Sex-specificity in transgenerational epigenetic programming. Horm. Behav. 59:290–295.
Franklin, T.B., Russig, H., et al. (2010). Epigenetic transmission of the impact of early stress across generations. Biol. Psychiatry 68:408–415.
Frias, A.E., Morgan, T.K., et al. (2011). Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 152:2456–2464.
Gardener, H., Spiegelman, D., et al. (2011). Perinatal and neonatal risk factors for autism: a comprehensive meta-analysis. Pediatrics 128:344–355.
Gardener, H., Spiegelman, D., et al. (2009). Prenatal risk factors for autism: comprehensive meta-analysis. Br. J. Psychiatry 195:7–14.
Gerardin, P., Wendland, J., et al. (2010). Depression during pregnancy: is the developmental impact earlier in boys? A prospective case–control study. J. Clin. Psychiatry 72(3):378–387.
Grayson, B.E., Levasseur, P.R., et al. (2010). Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology 151:1622–1632.
Guinchat, V., Thorsen, P., et al. (2012). Pre-, peri- and neonatal risk factors for autism. Acta obstetricia et gynecologica Scandinavica 91:287–300.
Hackman, D.A., Farah, M.J., et al. (2010). Socioeconomic status and the brain: mechanistic insights from human and animal research. Nat. Rev. Neurosci. 11:651–659.
Heim, C., Bradley, B., et al. (2009). Effect of childhood trauma on adult depression and neuroendocrine function: sex-specific moderation by CRH receptor 1 gene. Frontiers Behav. Neurosci. 3:41.
Heim, C., Shugart, M., et al. (2010). Neurobiological and psychiatric consequences of child abuse and neglect. Dev. Psychobiol. 52:671–690.
Holmes, M.C., Abrahamsen, C.T., et al. (2006). The mother or the fetus? 11beta-hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J. Neurosci. 26:3840–3844.
Howerton, C.L., and Bale, T.L. (2012). Prenatal programing: at the intersection of maternal stress and immune activation. Horm. Behav. 62(3):237–242.
Hsiao, E.Y., and Patterson, P.H. (2012). Placental regulation of maternal-fetal interactions and brain development. Dev. Neurobiol. 72:1317–1326.
Jiang, Y., Langley, B., et al. (2008). Epigenetics in the nervous system. J. Neurosci. 28:11753–11759.
Kapoor, A., Leen, J., et al. (2008). Molecular regulation of the hypothalamic–pituitary–adrenal axis in adult male guinea pigs after prenatal stress at different stages of gestation. J. Physiol. 586:4317–4326.
Kendler, K.S. (1998). Anna-Monika-Prize paper: major depression and the environment: a psychiatric genetic perspective. Pharmacopsychiatry 31:5–9.
Khandaker, G.M., Dibben, C.R., et al. (2012). Does maternal body mass index during pregnancy influence risk of schizophrenia in the adult offspring? Obesity Rev. 13:518–527.
Khashan, A.S., Abel, K.M., et al. (2008). Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch. Gen. Psychiatry 65:146–152.
Korosi, A., Shanabrough, M., et al. (2010). Early-life experience reduces excitation to stress-responsive hypothalamic neurons and reprograms the expression of corticotropin-releasing hormone. J. Neurosci. 30:703–713.
Lesage, J., Blondeau, B., et al. (2001). Maternal undernutrition during late gestation induces fetal overexposure to glucocorticoids and intrauterine growth retardation, and disturbs the hypothalamo-pituitary adrenal axis in the newborn rat. Endocrinology 142:1692–1702.
Li, C.C., Maloney, C.A., et al. (2010). Epigenetic programming by maternal nutrition: shaping future generations. Epigenomics 2:539–549.
Lingas, R., Dean, F., et al. (1999). Maternal nutrient restriction (48 h) modifies brain corticosteroid receptor expression and endocrine function in the fetal guinea pig. Brain Res. 846:236–242.
McCarthy, M.M., Auger, A.P., et al. (2009). The epigenetics of sex differences in the brain. J. Neurosci. 29:12815–12823.
Meaney, M.J. (2001). Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu. Rev. Neurosci. 24:1161–1192.
Meyer, U., Feldon, J., et al. (2011). Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr. Res. 69:26R–33R.
Morgan, C.P., and Bale, T.L. (2011). Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J. Neurosci. 31:11748–11755.
Mortensen, P.B., Norgaard-Pedersen, B., et al. (2007). Toxoplasma gondii as a risk factor for early-onset schizophrenia: analysis of filter paper blood samples obtained at birth. Biol. Psychiatry 61:688–693.
Mueller, B.R., and Bale, T.L. (2008). Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 28:9055–9065.
Newschaffer, C.J., Croen, L.A., et al. (2007). The epidemiology of autism spectrum disorders. Annu. Rev. Public Health 28:235–258.
O’Donnell, K., O’Connor, T.G., et al. (2009). Prenatal stress and neurodevelopment of the child: focus on the HPA axis and role of the placenta. Dev. Neurosci. 31:285–292.
Ravelli, G.P., Stein, Z.A., et al. (1976). Obesity in young men after famine exposure in utero and early infancy. NEJM 295:349–353.
Sullivan, E.L., Grayson, B., et al. (2010). Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J. Neurosci. 30:3826–3830.
Susser, E., St Clair, D., et al. (2008). Latent effects of prenatal malnutrition on adult health: the example of schizophrenia. Ann. NY Acad. Sci. 1136:185–192.
Sweatt, J.D. (2009). Experience-dependent epigenetic modifications in the central nervous system. Biol. Psychiatry 65:191–197.
Tsankova, N., Renthal, W., et al. (2007). Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8:355–367.
van Os, J., and Selten, J.P. (1998). Prenatal exposure to maternal stress and subsequent schizophrenia: the May 1940 invasion of The Netherlands. Br. J. Psychiatry 172:324–326.
Vucetic, Z., Kimmel, J., et al. (2010). Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology 151:4756–4764.
Ward, I.L. (1972). Prenatal stress feminizes and demasculinizes the behavior of males. Science 175:82–84.
Waterland, R.A. (2003). Do maternal methyl supplements in mice affect DNA methylation of offspring? J. Nutr. 133:238; author reply 239.