CHAPTER 2
Neurotransmitters and Receptors in Psychiatric Disorders
Carolyn M. Drazinic, M.D., Ph.D.
Steven T. Szabo, M.D., Ph.D.
Todd D. Gould, M.D.
Husseini K. Manji, M.D., F.R.C.P.C.
This chapter serves as a primer on the recent advances in our understanding of neural function both in health and in disease. It is beyond the scope of this chapter to cover these important areas in extensive detail, and readers are referred to outstanding textbooks that are entirely devoted to the topic (Kandel 2013; Nestler et al. 2015; Squire 2013). Here, we focus on the principles of neurotransmission that are critical for an understanding of the biological bases of major psychiatric disorders, as well as the mechanisms by which effective treatments may exert their beneficial effects. In particular, our goal is to lay the groundwork for the subsequent chapters in this volume that focus on individual disorders and their treatments.
Although this chapter is intended to provide a general overview on neurotransmitter function, whenever possible, we emphasize the neuropsychiatric relevance of specific observations. We outline principles that are of fundamental importance to the study and practice of psychopharmacology. The figure legends contain additional details for the interested reader.
What Are Neurotransmitters?
Several criteria have been established for a neurotransmitter, including that 1) it is synthesized and released from neurons; 2) it is released from nerve terminals in a chemically or pharmacologically identifiable form; 3) it interacts with postsynaptic receptors and brings about the same effects as are seen with stimulation of the presynaptic neuron; 4) its interaction with the postsynaptic receptor displays a specific pharmacology; and 5) its actions are terminated by active processes (Kandel 2013; Nestler et al. 2015; Squire 2013). However, our growing appreciation of the complexity of the central nervous system (CNS) and of the existence of numerous molecules that exert neuromodulatory and neurohormonal effects has blurred the classical definition of neurotransmitters somewhat, and even well-known neurotransmitters do not meet all these criteria under certain situations (Cooper et al. 2003).
Most neuroactive compounds are small polar molecules that are synthesized in the CNS via local machinery or are able to permeate the blood–brain barrier. To date, more than 50 endogenous substances have been found to be present in the brain that appear to be capable of functioning as neurotransmitters. There are many plausible explanations for why the brain would need so many transmitters and receptor subtypes to transmit messages. Perhaps the simplest explanation is that the sheer complexity of the CNS results in many afferent nerve terminals impinging onto a single neuron. This requires a neuron to be able to distinguish the multiple information-conveying inputs. Although this can be accomplished partially by spatial segregation, it is accomplished in large part by chemical coding of the inputs—that is, different chemicals convey different information. Moreover, as we discuss in detail later, the evolution of multiple receptors for a single neurotransmitter means that the same chemical can convey different messages depending on the receptor subtypes it acts on. Additionally, the firing pattern of neurons is a means of conveying information; thus, the firing activities of neurons in the brain differ widely, and a single neuron firing at different frequencies can even release different neuroactive compounds depending on the firing rate (e.g., the release of peptides often occurs at higher firing rates than that which is required to release monoamines). These multiple mechanisms to enhance the diversity of responses—chemical coding, spatial coding, frequency coding—are undoubtedly critical in endowing the CNS with its complex repertoire of physiological and behavioral responses (Kandel 2013; Nestler et al. 2015). Finally, the existence of multiple neuroactive compounds also provides built-in safeguards to ensure that vital brain circuits are able to partially compensate for loss of function of particular neurotransmitters.
Receptors
An essential property of any living cell is its ability to recognize and respond to external stimuli. Cell surface receptors have two major functions: recognition of specific molecules (neurotransmitters, hormones, growth factors, and even sensory signals) and activation of “effectors.” Binding of the appropriate agonist (i.e., neurotransmitter or hormone) externally to the receptor alters the conformation (shape) of the protein. Cell surface receptors use a variety of membrane-transducing mechanisms to transform an agonist’s message into cellular responses. In neuronal systems, the most typical responses ultimately (in some cases rapidly, in others more slowly) involve changes in transmembrane voltage and hence neuronal changes in excitability. Collectively, the processes are referred to as transmembrane signaling or signal transduction mechanisms. This process is not restricted to neurons. For instance, astrocytes were once thought to be uninvolved in neurotransmission, but they have since been shown to possess volume-regulated Cl– anion channels, which work together with gap junction/hemichannels to permit efflux of amino acids such as taurine, glutamate, and aspartate in response to swelling due to brain injury (Mulligan and MacVicar 2006; Ye et al. 2009).
Interestingly, although increasing numbers of potential neuroactive compounds and receptors continue to be identified, it has become clear that translation of the extracellular signals (into a form that can be interpreted by the complex intracellular enzymatic machinery) is achieved through a relatively small number of cellular mechanisms. Generally speaking, these transmembrane signaling systems, and the receptors that use them, can be divided into four major groups (Figure 2–1):
- Those that are relatively self-contained in structure and whose message takes the form of transmembrane ion fluxes (ionotropic receptors)
- Those that are multicomponent in nature and generate intracellular second messengers (G protein–coupled receptors)
- Those that contain intrinsic enzymatic activity (receptor tyrosine kinases and phosphatases)
- Those that are cytoplasmic and translocate to the nucleus to directly regulate transcription (gene expression) after they are activated by lipophilic molecules (often hormones) that enter the cell (nuclear receptors)

FIGURE 2–1. Major receptor subtypes in the central nervous system.
See Plate 4 to view this figure in color.
This figure depicts the four major classes of receptors in the CNS. (A) Ionotropic receptors. These receptors comprise multiple protein subunits that are combined in such a way as to create a central membrane pore through this complex, allowing the flow of ions. This type of receptor has a very rapid response time (milliseconds). The consequences of receptor stimulation (i.e., excitatory or inhibitory) depend on the types of ions that the receptor specifically allows to enter the cell. Thus, for example, Na+ entry through the NMDA (N-methyl-D-aspartate) receptor depolarizes the neuron and brings about an excitatory response, whereas Cl– efflux through the γ-aminobutyric acid type A (GABAA) receptor hyperpolarizes the neuron and brings about an inhibitory response. Illustrated here is the NMDA receptor regulating a channel permeable to Ca2+, Na+, and K+ ions. The NMDA receptors also have binding sites for glycine, Zn2+, phencyclidine, MK801/ketamine, and Mg2+; these molecules are able to regulate the function of this receptor. (B) G protein–coupled receptors (GPCRs). The majority of neurotransmitters, hormones, and even sensory signals mediate their effects via seven transmembrane domain–spanning receptors that are G protein–coupled. The amino terminus of the G protein is on the outside of the cell and plays an important role in the recognition of specific ligands; the third intracellular loop and carboxy terminus of the receptor play an important role in coupling to G proteins and are sites of regulation of receptor function (e.g., by phosphorylation). All G proteins are heterotrimers (consisting of α, β, and γ subunits). The G proteins are attached to the membrane by isoprenoid moieties (fatty acid) via their γ subunits. Compared with the ionotropic receptors, GPCRs mediate a slower response (on the order of seconds). Detailed depiction of the activation of G protein–coupled receptors is given in Figure 2–2. Here we depict a receptor coupled to the G protein Gs (the s stands for stimulatory to the enzyme adenylyl cyclase [AC]). Activation of such a receptor produces activation of AC and increases in cyclic adenosine monophosphate (cAMP) levels. G protein–coupled pathways exhibit major amplification properties, and, for example, in model systems researchers have demonstrated a 10,000-fold amplification of the original signal. The effects of cAMP are mediated largely by activation of protein kinase A (PKA). One major downstream target of PKA is CREB (cAMP response element–binding protein), which may be important to the mechanism of action of antidepressants. (C) Receptor tyrosine kinases. These receptors are activated by neurotrophic factors and are able to bring about acute changes in synaptic function, as well as long-term effects on neuronal growth and survival. These receptors contain intrinsic tyrosine kinase activity. Binding of the ligand triggers receptor dimerization and transphosphorylation of tyrosine residues in its cytoplasmic domain, which then recruits cytoplasmic signaling and scaffolding proteins. The recruitment of effector molecules generally occurs via interaction of proteins with modular binding domains SH2 and SH3 (named after homology to the src oncogenes–src homology domains); SH2 domains are a stretch of about 100 amino acids that allow high-affinity interactions with certain phosphotyrosine motifs. The ability of multiple effectors to interact with phosphotyrosines is undoubtedly one of the keys to the pleiotropic effects that neurotrophins can exert. Shown here is a tyrosine kinase receptor type B (TrkB), which upon activation produces effects on the Raf, MEK (mitogen-activated protein kinase/ERK), extracellular response kinase (ERK), and ribosomal S6 kinase (RSK) signaling pathway. Some major downstream effects of RSK are CREB and stimulation of factors that bind to the AP-1 site (c-Fos and c-Jun). (D) Nuclear receptors. These receptors are transcription factors that regulate the expression of target genes in response to steroid hormones and other ligands. Many hormones (including glucocorticoids, gonadal steroids, and thyroid hormones) are able to rapidly penetrate into the lipid bilayer membrane, because of their lipophilic composition, and thereby directly interact with these cytoplasmic receptors inside the cell. Upon activation by a hormone, the nuclear receptor–ligand complex translocates to the nucleus, where it binds to specific DNA sequences, referred to as hormone responsive elements (HREs), and regulates gene transcription. Nuclear receptors often interact with a variety of coregulators that promote transcriptional activation when recruited (coactivators) and those that attenuate promoter activity (corepressors). However, nongenomic effects of neuroactive steroids have also been documented, with the majority of evidence suggesting modulation of ionotropic receptors. This figure illustrates both the genomic and the nongenomic effects. ATF1=activation transcription factor 1; BDNF=brain-derived neurotrophic factor; CaMKII=Ca2+/calmodulin–dependent protein kinase II; CREM=cyclic adenosine 5′-monophosphate response element modulator; D1=dopamine1 receptor; D5=dopamine5 receptor; ER=estrogen receptor; GR=glucocorticoid receptor; GRK=G protein–coupled receptor kinase; P=phosphorylation; PR=progesterone receptor.
A more extensive and continuously updated synopsis of these and many other receptors and ligands can be found at the International Union of Basic and Clinical Pharmacology/British Pharmacological Society Guide to Pharmacology Web site (www.guidetopharmacology.org) and associated publications (Alexander et al. 2013a, 2013b, 2013c, 2013d, 2013e, 2013f). We review the four major groups in the following subsections.
Ionotropic Receptors
The first class of receptors contains in their molecular complex an intrinsic ion channel. Receptors of this class include those for several amino acids, including glutamate (e.g., the NMDA [N-methyl-D-aspartate] receptor), GABA (γ-aminobutyric acid via the GABAA receptor), and the nicotinic acetylcholine (nACh) receptor and the serotonin type 3 (5-HT3) receptor. Ion channels are integral membrane proteins that are directly responsible for the electrical activity of the nervous system by virtue of their regulation of the movement of ions across membranes. Receptors containing intrinsic ion channels have been called ionotropic and are generally composed of four or five subunits that open transiently when a neurotransmitter binds, allowing ions to flow into (e.g., Na+, Ca2+, Cl–) or out of (e.g., K+) the neuron, thereby generating synaptic potential (see Figure 2–1).
Often, the ionotropic receptors are composed of different combinations of various subunits, thereby providing the system with considerable flexibility. For example, there is extensive research into the potential development of an anxiolytic that is devoid of sedative effects by targeting GABAA receptor subunits present in selected brain regions (Salerno et al. 2012; Taliani et al. 2009). In general, neurotransmission that is mediated by ionotropic receptors is very fast, with ion channels opening and closing within milliseconds, and these receptors regulate much of the tonic excitatory (e.g., glutamate-mediated) and inhibitory (e.g., GABA-mediated) activity in the CNS; as we discuss in the “Neurotransmitter and Neuropeptide Systems” section later in this chapter, many of the classic neurotransmitters (e.g., monoamines) exert their effects on a slower time scale and are therefore often considered to be modulatory in their effects.
G Protein–Coupled Receptors
Most receptors in the CNS do not have intrinsic ionic conductance channels within their structure but instead regulate cellular activity by the generation of various “second messengers.” Receptors of this class generally do not interact directly with the various second-messenger-generating enzymes but transmit information to the appropriate “effector” by the activation of interposed coupling proteins. These are the G protein–coupled receptor families, and they provide a slower onset, but longer duration of signaling, compared with ionotropic receptors (Squire 2013). The G protein–coupled receptors (GPCRs, which constitute more than 80% of all known receptors in the body and number about 800 in humans) all span the plasma membrane seven times and have three intracellular loops and three extracellular loops (see Figure 2–1) (Alexander et al. 2013b). G proteins are so named because of their ability to bind the guanine nucleotides guanosine triphosphate and guanosine diphosphate. Receptors coupled to G proteins include those for dopamine, serotonin, acetylcholine, various peptides, and even sensory signals such as light and odorants.
GPCRs have increasingly become the focus of extensive research in psychiatry (Catapano and Manji 2007). The amino terminus is on the outside of the cell and plays a critical role in recognition of the ligand, which can be a small molecule, peptide, or large protein; the carboxy terminus and third intracellular loop are inside the cell and regulate coupling to different G proteins, “cross-talk” between receptors, and desensitization (see Figure 2–1) (Alexander et al. 2013b). Although the bimodal model of ligands switching the GPCR “on” or “off” is appealing, an individual GPCR can actually assume many different conformations, which influences the nature of the ligand–receptor interaction and the predominant complex signal generated in a particular cell type; this concept is called ligand-induced selective signaling (Millar and Newton 2010). Differential oligomerization, differential phosphorylation, signaling through molecules other than G proteins, and second-messenger independent signaling together add even more complexity for future GPCR research (Kandel 2013; Millar and Newton 2010).
In conclusion, many classes and subtypes of G proteins exist, playing key roles in amplifying and integrating signals.
Autoreceptors and Heteroreceptors
Autoreceptors are receptors located on neurons that produce the endogenous ligand for that particular receptor (e.g., a serotonergic receptor on a serotonergic neuron). By contrast, heteroreceptors are receptor subtypes that are present on neurons that do not contain an endogenous ligand for that particular receptor subtype (e.g., a serotonergic receptor located on a dopaminergic neuron).
Two major classes of autoreceptors play very important roles in fine-tuning neuronal activity. Somatodendritic autoreceptors are present on cell bodies and dendrites and exert critical roles in regulating the firing rate of neurons. In general, activation of somatodendritic autoreceptors (e.g., α2-adrenergic receptors for noradrenergic neurons, 5-HT1A receptors for serotonergic neurons, or dopamine type 2 [D2] receptors for dopaminergic neurons) inhibits the firing rate of the neurons by opening K+ channels and by reducing cyclic adenosine monophosphate (cAMP) levels, both of which may be important in psychiatric disease. For instance, TREK-1 is a background K+ channel regulator protein important in serotonin transmission and potentially in moodlike behavior regulation in mice (Heurteaux et al. 2006). This exemplifies how fundamental mechanisms of neuronal transmission such as K+ channels, which regulate membrane potentials, may relate to global alterations in brain functioning relevant to psychiatry.
The second major class of autoreceptors, nerve terminal autoreceptors, play an important role in regulating the amount of neurotransmitter released per nerve impulse, generally by closing nerve terminal Ca2+ channels. Both of these types of autoreceptors are typically members of the GPCR family. Neurotransmitter release is known to be triggered by influx and alterations of intracellular calcium, with functioning of three types of SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein [SNAP] receptor) proteins mediating a critical role. The distinct kinetics of neurotransmitter release modulators, such as botulinum and tetanus neurotoxins, induce prominent alterations in synaptobrevin and syntaxin, leading to calcium-independent mechanisms of neurotransmitter regulation (Sakaba et al. 2005). Most synapses are dependent on influx of Ca2+ through voltage-gated calcium channels for presynaptic neurotransmitter release; however, in the retina, this influx of calcium occurs via glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (Chávez et al. 2006). Beyond the receptor level, presynaptic SAD (synapses of amphids defective, originally identified as a regulator of neuronal polarity in the Caenorhabditis elegans worm model), an intracellular serine threonine kinase, is associated with the active zone cytomatrix that regulates neurotransmitter release, and SAD kinases in presynaptic neurons also control the maturation and arborization of synapses in the central and peripheral nervous systems (Inoue et al. 2006; Lilley et al. 2014). These data further exemplify the dynamic nature of basic processes involved in neurotransmitter regulation that may possibly aid in advancing treatment of psychopathology.
GPC Receptor Regulation and Trafficking
The mechanism by which GPCRs translate extracellular signals into cellular changes was once envisioned as a simple linear model. It is now known, however, that the activity of GPCRs is subject to at least three additional principal modes of regulation: desensitization, downregulation, and trafficking (Carman and Benovic 1998) (Figure 2–2). Desensitization, the process by which cells rapidly adapt to stimulation by agonists, is generally believed to occur by two major mechanisms: homologous and heterologous.
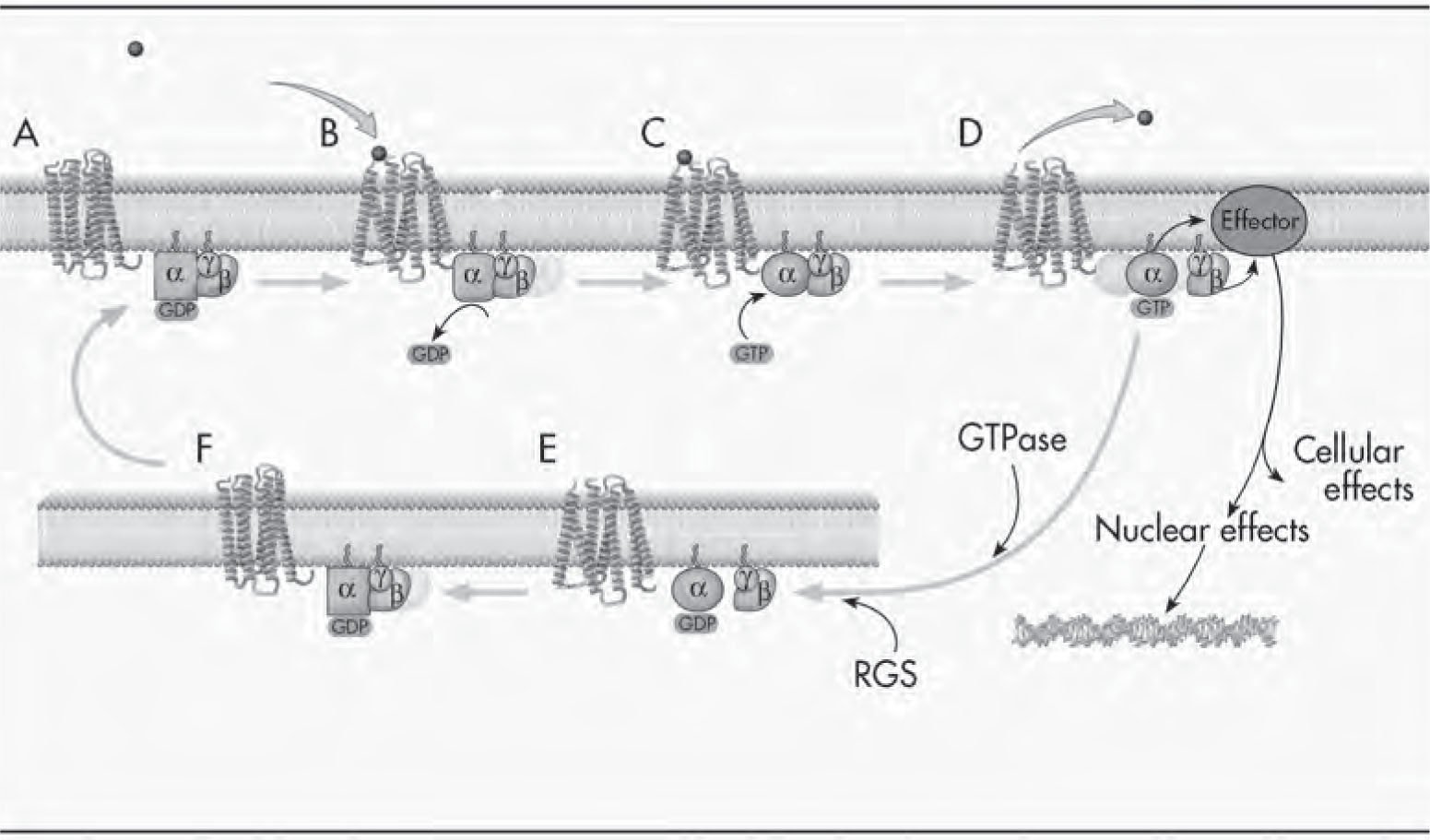
FIGURE 2–2. G protein–coupled receptors and G protein activation.
See Plate 5 to view this figure in color.
All G proteins are heterotrimers consisting of α, β, and γ subunits. The receptor shuttles between a low-affinity form that is not coupled to a G protein and a high-affinity form that is coupled to a G protein. (A) At rest, G proteins are largely in their inactive state, namely, as αβγ heterotrimers, which have GDP (guanosine diphosphate) bound to the α subunit. (B) When a receptor is activated by a neurotransmitter, it undergoes a conformational (shape) change, forming a transient state referred to as a high-affinity ternary complex, comprising the agonist, the receptor in a high-affinity state, and the G protein. A consequence of the receptor interaction with the G protein is that the GDP comes off the G protein α subunit, leaving a very transient empty guanine nucleotide binding domain. (C) Guanine nucleotides (generally GTP) quickly bind to this nucleotide binding domain; thus, one of the major consequences of active receptor–G protein interaction is to facilitate guanine nucleotide exchange—this is basically the “on switch” for the G protein cycle. (D) A family of GTPase-activating proteins for G protein–coupled receptors has been identified, and they are called regulators of G protein signaling (RGS) proteins. Since activating GTPase activity facilitates the “turn off” reaction, these RGS proteins are involved in dampening the signal. Binding of GTP to the α subunit of G proteins results in subunit dissociation, whereby the α-GTP dissociates from the βγ subunits. Although not covalently bound, the β and γ subunits remain tightly associated and generally function as dimers. The α-GTP and βγ subunits are now able to activate multiple diverse effectors, thereby propagating the signal. While they are in their active states, the G protein subunits can activate multiple effector molecules in a “hit and run” manner; this results in major signal amplification (i.e., one active G protein subunit can activate multiple effector molecules). The activated G protein subunits also dissociate from the receptor, converting the receptor to a low-affinity conformation and facilitating the dissociation of the agonist from the receptor. The agonist can now activate another receptor, and this also results in signal amplification. Together, these processes have been estimated to produce a 10,000-fold amplification of the signal in certain models. (E) Interestingly, the α subunit has intrinsic GTPase activity, which cleaves the third phosphate group from GTP (G-P-P-P) to GDP (G-P-P). Since α-GDP is an inactive state, the GTPase activity serves as a built-in timing mechanism, and this is the “turn off” reaction. (F) The reassociation of α-GDP with βγ is thermodynamically favored, and the reformation of the inactive heterotrimer (αβγ) completes the G protein cycle.
Homologous desensitization is receptor specific; that is, only the receptor actively being stimulated becomes desensitized. This form of desensitization occurs via a family of kinases known as G protein–coupled kinases. When a receptor activates a G protein and causes dissociation of the α subunit from the βγ subunits, the βγ subunits are able to provide an “anchoring surface” for the G protein–coupled kinases to allow them to come into the proximity of the activated receptor and phosphorylate it. This phosphorylation then recruits another family of proteins known as arrestins, which physically interfere with the coupling of the phosphorylated receptor and the G protein, thereby dampening the signal. This form of desensitization is very rapid and usually transient (i.e., the receptors get dephosphorylated and return to the baseline state). However, if the stimulation of the receptor is excessive and prolonged, it leads to an internalization of the receptor, and often its degradation, a process referred to as downregulation.
Heterologous desensitization is not receptor specific and is mediated by second-messenger kinases such as protein kinase A (PKA) and protein kinase C (PKC). Thus, when a receptor activates PKA, the activated PKA is capable of phosphorylating (and thereby desensitizing) not only that particular receptor but also other receptors that are present in proximity and have the correct phosphorylation motif, thereby producing heterologous desensitization.
After prolonged or repeated activation of receptors by agonist ligands, the process of receptor downregulation is observed. Downregulation is associated with a reduced number of receptors detected in cells or tissues, thereby leading to attenuation of cellular responses (Carman and Benovic 1998). The process of GPCR sequestration is mediated by a well-characterized endocytic pathway involving the concentration of receptors in clathrin-coated pits and subsequent internalization and recycling back to the plasma membrane (Tsao and von Zastrow 2000). Endocytosis can thus clearly serve as a primary mechanism to attenuate signaling by rapidly and reversibly removing receptors from the cell surface. However, endocytosis and receptor trafficking also mediate GPCR signaling by way of certain effector pathways, most notably mitogen-activated protein (MAP) kinase cascades. Evidence also indicates that endocytosis of GPCRs may be required for certain signal transduction pathways leading to the nucleus (Tsao and von Zastrow 2000). These diverse functions of GPCR endocytosis and trafficking lead to unexpected insights into the biochemical and functional properties of endocytic vesicles. Indeed, there is considerable excitement about our growing understanding of the diverse molecular mechanisms for signaling specificity and receptor trafficking and the possibility that this knowledge could lead to new selective therapeutics.
Receptor Tyrosine Kinases
The receptor tyrosine kinases, as their name implies, contain intrinsic tyrosine kinase activity and are generally used by growth factors, such as neurotrophic factors, and cytokines. Binding of an agonist initiates receptor dimerization and transphosphorylation of tyrosine residues in its cytoplasmic domain (Patapoutian and Reichardt 2001) (see Figure 2–1). The phosphotyrosine residues of the receptor function as binding sites for recruiting specific cytoplasmic signaling and scaffolding proteins. The ability of multiple effectors to interact with phosphotyrosines is undoubtedly one of the keys to the pleiotropic effects that neurotrophins can exert. These pleiotropic and yet distinct effects of growth factors are mediated by varying degrees of activation of three major signaling pathways: the MAP kinase pathway, the phosphoinositide-3 (PI3) kinase pathway, and the phospholipase C (PLC)–γ1 pathway. These pathways are part of complex secondary messaging systems of the cell, which are beyond the scope of this chapter.
Nuclear Receptors
Nuclear receptors are transcription factors that regulate the expression of target genes in response to steroid hormones and other ligands. Many hormones (including glucocorticoids, gonadal steroids, and thyroid hormones) are able to rapidly penetrate into the lipid bilayer membrane because of their lipophilic composition, and thereby directly interact with these cytoplasmic receptors inside the cell (see Figure 2–1). On activation by a hormone, the nuclear receptor–ligand complex translocates to the nucleus, where it binds to specific DNA sequences referred to as hormone-responsive elements, and subsequently regulates gene transcription (Mangelsdorf et al. 1995; Truss and Beato 1993). Nuclear receptors often interact with a variety of coregulators that promote transcriptional activation when recruited (coactivators) and those that attenuate promoter activity (corepressors). Numerous nuclear receptors have been identified, as reviewed elsewhere (Alexander et al. 2013e).
With this broad overview of neurotransmitters and receptor subtypes, we now turn to a discussion of selected individual neurotransmitters and their receptors.
Neurotransmitter and Neuropeptide Systems
Serotonergic System
Largely on the basis of the observation that most effective antidepressants and antipsychotics target these systems, the monoaminergic systems (e.g., serotonin, norepinephrine, dopamine) have been extensively studied. Serotonin was given that name because of its activity as an endogenous vasoconstrictor in blood serum (Rapport et al. 1948). It was later acknowledged as being the same molecule (secretine) that is found in the intestinal mucosa and that is “secreted” by chromaffin cells (Brodie 1900). Following these findings, serotonin later was characterized as a neurotransmitter in the CNS (Bogdanski et al. 1956).
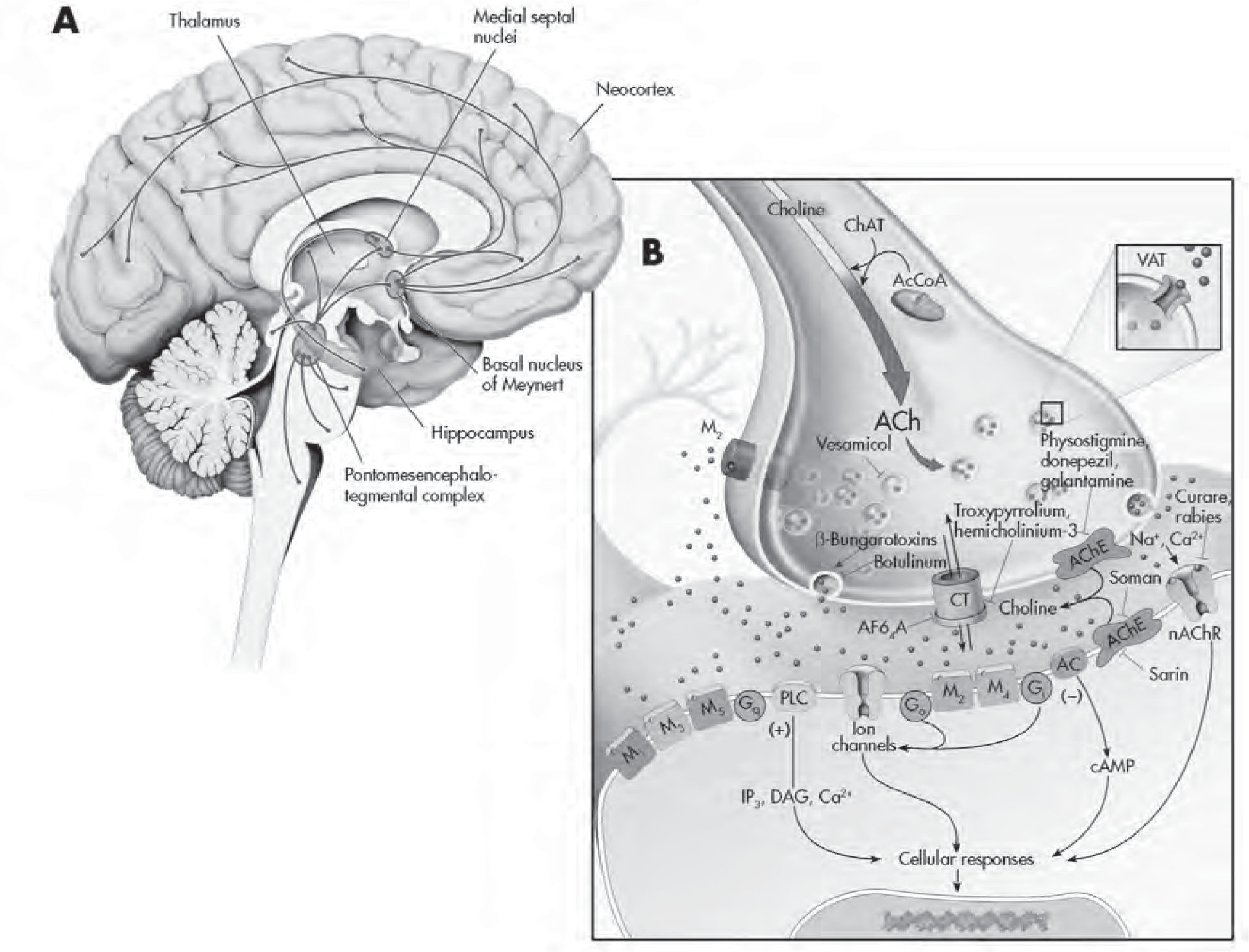
Serotonin-producing cell bodies in the brain are localized to the central gray, in the surrounding reticular formation, and in cell clusters located in the center, and thus the name raphe (from Latin, meaning “midline”) was adopted (Figure 2–3A). The dorsal raphe, the largest brain stem serotonin nucleus, contains approximately 50% of the total serotonin neurons in the mammalian CNS; in contrast, the medial raphe comprises 5% (Descarries et al. 1982; Wiklund and Björklund 1980). Serotonergic neurons project widely throughout the CNS rather than to discrete anatomical locations (as the dopaminergic neurons appear to do; see Figure 2–4A later in this chapter), leading to the suggestion that serotonin exerts a major modulatory role throughout the CNS (Reader 1980). Interestingly, evidence suggests that infralimbic and prelimbic regions of the ventromedial prefrontal cortex (vmPFC) in rats are responsible for detecting whether a stressor is under the organism’s control. When a stressor is controllable, stress-induced activation of the dorsal raphe nucleus is inhibited by the vmPFC, and the behavioral sequelae of the uncontrollable stress response are blocked (Amat et al. 2005). The ability to regulate serotonin neuron activity and function has been a major ongoing focus of psychiatric disorder research and treatments. Lysergic acid diethylamide, a hallucinogen, has a chemical structure similar to that of serotonin, and monoamine oxidase (MAO) inhibitors (MAOIs), which are classic antidepressants, increase the levels of monoamines such as serotonin in the synapse (Squire 2013).
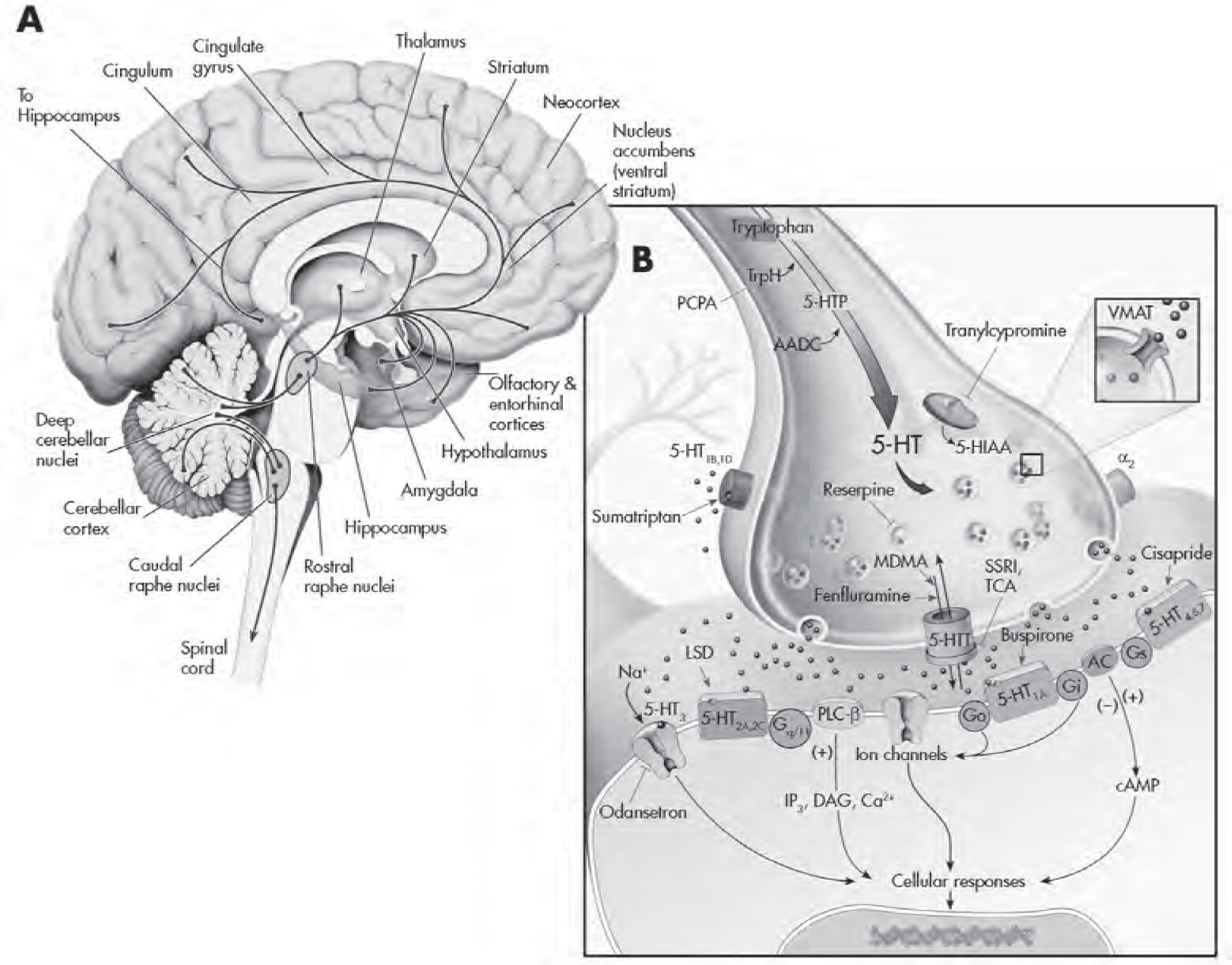
FIGURE 2–3. The serotonergic system.
See Plate 6 to view this figure in color.
This figure depicts the location of the major serotonin (5-HT)–producing cells (raphe nuclei) innervating brain structures (A), and various cellular regulatory processes involved in serotonergic neurotransmission (B). 5-HT neurons project widely throughout the CNS and innervate virtually every part of the neuroaxis. L-Tryptophan, an amino acid actively transported into presynaptic 5-HT-containing terminals, is the precursor for 5-HT. It is converted to 5-hydroxytryptophan (5-HTP) by the rate-limiting enzyme tryptophan hydroxylase (TrpH). This enzyme is effectively inhibited by the drug p-chlorophenylalanine (PCPA). Aromatic amino acid decarboxylase (AADC) converts 5-HTP to 5-HT. Once released from the presynaptic terminal, 5-HT can interact with a variety (15 different types) of presynaptic and postsynaptic receptors. Presynaptic regulation of 5-HT neuron firing activity and release occurs through somatodendritic 5-HT1A (not shown) and 5-HT1B,1D autoreceptors, respectively, located on nerve terminals. Sumatriptan is a 5-HT1B,1D receptor agonist. (The antimigraine effects of this agent are likely mediated by local activation of this receptor subtype on blood vessels, which results in their constriction.) Buspirone is a partial 5-HT1A receptor agonist that activates both pre- and postsynaptic receptors. Cisapride is a preferential 5-HT4 receptor agonist that is used to treat irritable bowel syndrome as well as nausea associated with antidepressants. The binding of 5-HT to G protein receptors (Go, Gi, etc.) that are coupled to adenylyl cyclase (AC) and phospholipase C–β (PLC-β) will result in the production of a cascade of second-messenger and cellular effects. Lysergic acid diethylamide (LSD) likely interacts with numerous 5-HT receptors to mediate its effects. Pharmacologically this ligand is often used as a 5-HT2 receptor agonist in receptor-binding experiments. Ondansetron is a 5-HT3 receptor antagonist that is marketed as an antiemetic agent for chemotherapy patients but is also given to counteract side effects produced by antidepressants in some patients. 5-HT has its action terminated in the synapse by rapidly being taken back into the presynaptic neuron through 5-HT transporters (5-HTTs). Once inside the neuron, it can either be repackaged into vesicles for reuse or undergo enzymatic catabolism. The selective 5-HT reuptake inhibitors (SSRIs) and older-generation tricyclic antidepressants (TCAs) are able to interfere/block the reuptake of 5-HT. 5-HT is then metabolized to 5-hydroxyindoleacetic acid (5-HIAA) by monoamine oxidase (MAO), located on the outer membrane of mitochondria or sequestered and stored in secretory vesicles by vesicular monoamine transporters (VMATs). Reserpine causes a depletion of 5-HT in vesicles by interfering with uptake and storage mechanisms (depressive-like symptoms have been reported with this agent). Tranylcypromine is an MAO inhibitor (MAOI) and an effective antidepressant. Fenfluramine (an anorectic agent) and 3,4-methylenedioxymethamphetamine (MDMA; “Ecstasy”) are able to facilitate 5-HT release by altering 5-HTT function. cAMP=cyclic adenosine monophosphate; DAG=diacylglycerol; IP3=inositol-1,4,5-triphosphate.
Source. Adapted from Cooper JR, Bloom FE, Roth RH: The Biochemical Basis of Neuropharmacology, 8th Edition. New York, Oxford University Press, 2001. Copyright 1970, 1974, 1978, 1982, 1986, 1991, 1996, 2001 by Oxford University Press, Inc. Used by permission of Oxford University Press, Inc. Modified from Nestler et al. 2001.
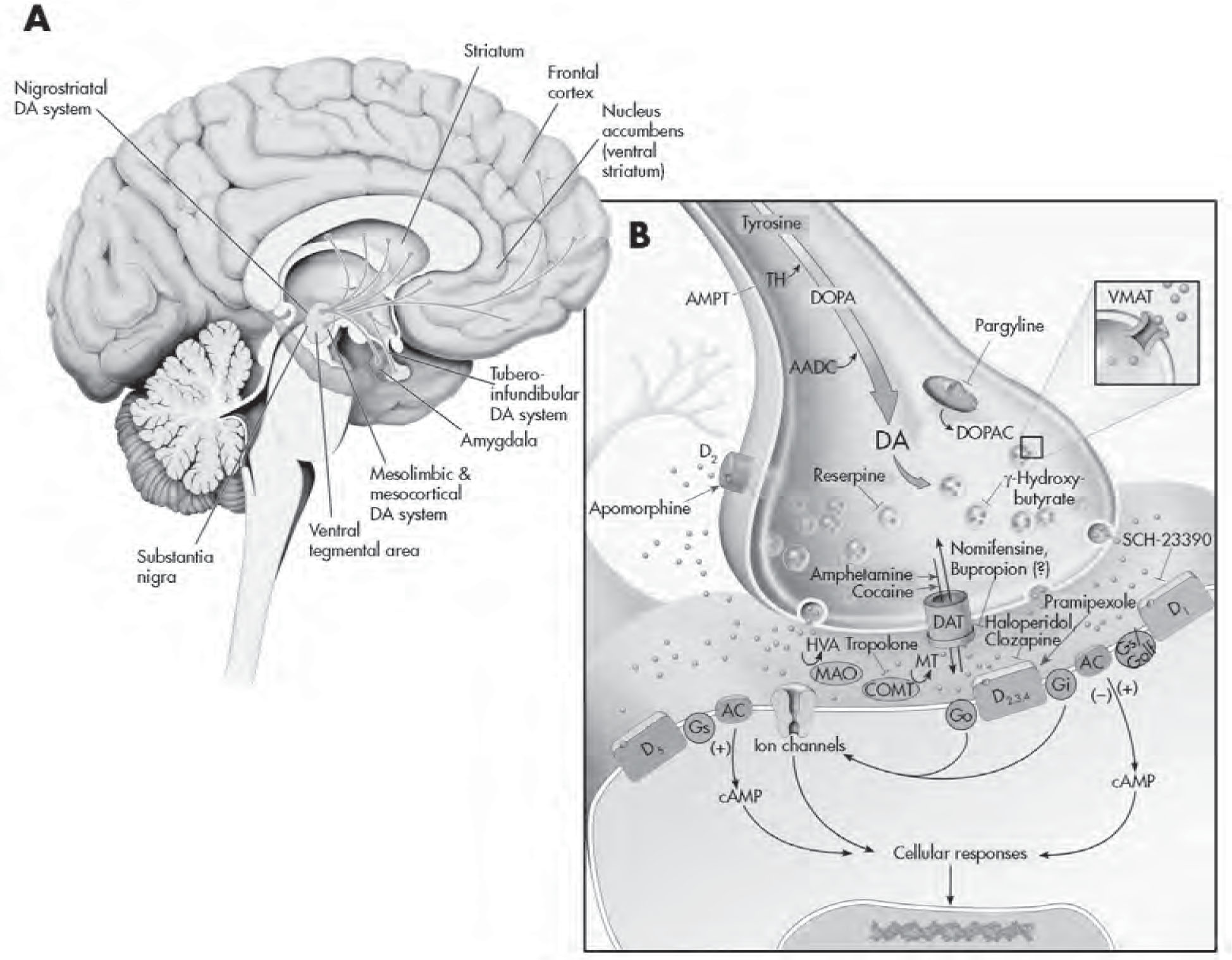
FIGURE 2–4. The dopaminergic system.
See Plate 7 to view this figure in color.
This figure depicts the dopaminergic projections throughout the brain (A) and various regulatory processes involved in dopaminergic neurotransmission (B). The amino acid L-tyrosine is actively transported into presynaptic dopamine (DA) nerve terminals, where it is ultimately converted into DA. The rate-limiting step is conversion of L-tyrosine to L-dihydroxyphenylalanine (L-dopa) by the enzyme tyrosine hydroxylase (TH). α-Methyl-p-tyrosine (AMPT) is a competitive inhibitor of tyrosine hydroxylase and has been used to assess the impact of reduced catecholaminergic function in clinical studies. The production of DA requires that L-aromatic amino acid decarboxylase (AADC) act on L-dopa. Thus, the administration of L-dopa to patients with Parkinson’s disease bypasses the rate-limiting step and is able to produce DA quite readily. DA has its action terminated in the synapse by rapidly being taken back into the presynaptic neuron through DA transporters (DATs). DA is then metabolized to dihydroxyphenylalanine (DOPAC) by intraneuronal monoamine oxidase (MAO; preferentially by the MAO-B subtype) located on the outer membrane of mitochondria, or is sequestered and stored in secretory vesicles by vesicular monoamine transporters (VMATs). Reserpine causes a depletion of DA in vesicles by interfering and irreversibly damaging uptake and storage mechanisms. γ-Hydroxybutyrate inhibits the release of DA by blocking impulse propagation in DA neurons. Pargyline inhibits MAO and may have efficacy in treating parkinsonian symptoms by augmenting DA levels through inhibition of DA catabolism. Other clinically used inhibitors of MAO are nonselective and thus likely elevate the levels of DA, norepinephrine, and serotonin. Once released from the presynaptic terminal (because of an action potential and calcium influx), DA can interact with five different G protein–coupled receptors (D1–D5), which belong to either the D1 or D2 receptor family. Presynaptic regulation of DA neuron firing activity and release occurs through somatodendritic (not shown) and nerve terminal D2 autoreceptors, respectively. Pramipexole is a D2/D3 receptor agonist and has been documented to have efficacy as an augmentation strategy in cases of treatment-resistant depression and in the management of Parkinson’s disease. The binding of DA to G protein receptors (Go, Gi, etc.) positively or negatively coupled to adenylyl cyclase (AC) results in the activation or inhibition of this enzyme, respectively, and the production of a cascade of second-messenger and cellular effects (see diagram). Apomorphine is a D1/D2 receptor agonist that has been used clinically to aid in the treatment of Parkinson’s disease. (SKF-82958 is a pharmacologically selective D1 receptor agonist.) SCH-23390 is a D1/D5 receptor antagonist. There are likely physiological differences between D1 and D5 receptors, but the current unavailability of selective pharmacological agents has precluded an adequate differentiation thus far. Haloperidol is a D2 receptor antagonist, and clozapine is a nonspecific D2/D4 receptor antagonist (both are effective antipsychotic agents). Once inside the neuron, DA can either be repackaged into vesicles for reuse or undergo enzymatic catabolism. Nomifensine is able to interfere/block the reuptake of DA. The antidepressant bupropion has affinity for the dopaminergic system, but it is not known whether this agent mediates its effects through DA or possibly by augmenting other monoamines. DA can be degraded to homovanillic acid (HVA) through the sequential action of catechol-O-methyltransferase (COMT) and MAO. Tropolone is an inhibitor of COMT. Evidence suggests that the COMT gene may be linked to schizophrenia (Akil et al. 2003). cAMP=cyclic adenosine monophosphate; DOPA=dihydroxyphenylalanine; MT=methoxytyramine;
Source. Adapted from Cooper JR, Bloom FE, Roth RH: The Biochemical Basis of Neuropharmacology, 8th Edition. New York, Oxford University Press, 2001. Copyright 1970, 1974, 1978, 1982, 1986, 1991, 1996, 2001 by Oxford University Press, Inc. Used by permission of Oxford University Press, Inc.
The precursor for serotonin synthesis is L-tryptophan, an amino acid that comes primarily from the diet and crosses the blood–brain barrier through a carrier for large neutral amino acids. Tryptophan hydroxylase is the rate-limiting enzyme in serotonin biosynthesis (Figure 2–3B), and polymorphisms in this enzyme have been extensively investigated in psychiatric disorders, with equivocal results. A more fruitful research strategy in humans has been tryptophan depletion via dietary restriction to study the role of serotonin in the pathophysiology and treatment of psychiatric disorders (Bell et al. 2001; van Donkelaar et al. 2011). These studies have indicated that acute tryptophan depletion (ATD) produces a rapid depressive relapse in patients taking selective serotonin reuptake inhibitors (SSRIs) but not in those taking norepinephrine reuptake inhibitors; the data suggesting induction of depressive symptoms in remitted patients or individuals with family histories of mood disorders are more equivocal (Bell et al. 2001). Findings similar to those from ATD depression studies also have been identified in numerous studies of panic disorder, whereby healthy subjects with no history of panic disorder, or patients with panic disorder in remission, were not very sensitive to ATD, but patients with unremitted panic disorder were extremely sensitive to carbon dioxide challenges (Sobczak and Schruers 2014). Some of the inconsistencies of the ATD findings in the literature may have been due to the various formulations of the tryptophan depletion amino acid cocktails used, but more surprising are suggestions that the classic ATD cocktails may not have been depleting extracellular synaptic serotonin levels after all but may have been working through other molecular mechanisms as described elsewhere (Sobczak and Schruers 2014; van Donkelaar et al. 2011). Nevertheless, numerous studies in the literature suggest that, in general, low levels of serotonin in the plasma and/or cerebrospinal fluid (CSF) correlate with depression, impulsivity, aggression, alcohol dependence, suicide attempts, completed suicides, and violent suicides (Bortolato et al. 2013; Moberg et al. 2011; Oo et al. 2016; Purselle and Nemeroff 2003; Ruljancic et al. 2011, 2013; Spreux-Varoquaux et al. 2001). Serotonin dysfunction–related endophenotypes that could predict patients at risk for suicide are being explored, including fenfluramine challenges leading to poor prolactin release, increased time spent in rapid eye movement (REM) sleep stages, increased loudness-dependent auditory evoked potentials, and increased frontal cortex P300 (Lee and Kim 2011). Furthermore, postmortem studies in depressed bipolar patients who completed suicide found lower serotonin and norepinephrine activity (Wiste et al. 2008).
More sophisticated molecular approaches targeting individual serotonin receptors and transporters are described in the following subsections.
Serotonin Transporters
As is the case for many classic neurotransmitters, termination of the effects of serotonin in the synaptic cleft is brought about in large part by an active reuptake process mediated by the serotonin transporter (5-HTT). Serotonin is taken up into the presynaptic terminals, where it is metabolized by the enzyme MAO or sequestered into secretory vesicles by the vesicle monoamine transporter (see Figure 2–3B). This presumably underlies the mechanism by which MAOIs initiate their therapeutic effects; that is, the blockade of monoamine breakdown results in an increase in the available pool for release when an action potential invades the nerve terminal. It is now well established that many tricyclic antidepressants and SSRIs exert their initial primary pharmacological effects by binding to the 5-HTT and blocking serotonin reuptake, thereby increasing the intrasynaptic levels of serotonin, which initiates a cascade of downstream effects (see Figure 2–3B for details). It has been hypothesized that the first step in serotonin transport involves the binding of serotonin to the 5-HTT and then a cotransport with Na+, and the second step involves the translocation of K+ across the membrane to the outside of the cell. SSRIs bind to the same site on the transporter as serotonin itself. Elegant biochemical and mutagenesis experiments have elucidated a leucine transporter from bacterial species, providing information that helped unravel the mechanism by which mammalian transporters couple ions and substrates to mediate neurotransmitter clearance. The crystal structure for sodium- and chloride-dependent neurotransmitter transporters (including transporters for serotonin [SERT], dopamine [DAT], norepinephrine [NET], glycine [GlyT1b], and GABA [GAT1]) with the L-leucine binding sites for Na+ ions also has been elucidated (Henry et al. 2006; Yamashita et al. 2005).
In the brain, 5-HTTs have been radiolabeled with [3H]-imipramine (Hrdina et al. 1985; Langer et al. 1980) and with SSRIs such as [3H]cyanoimipramine (Wolf and Bobik 1988), [3H]paroxetine (Habert et al. 1985), and [3H]citalopram (D’Amato et al. 1987). The regional distribution of 5-HTT corresponds to discrete regions of rat brain known to contain cell bodies of serotonin neurons and synaptic axon terminals, most notably the cerebral cortex, neostriatum, thalamus, and limbic areas (Cooper et al. 2003; Hrdina et al. 1985; Madden 2002). The specific cellular localization of 5-HTT in the CNS also has been accomplished by using site-specific antibodies (Lawrence et al. 1995a). Immunohistochemical studies that used antibodies against the serotonin carrier have reported both neuronal and glial staining in areas of the rat brain containing serotonin somata and terminals (i.e., dorsal raphe and hippocampus) (Lawrence et al. 1995b). Experimental alterations of 5-HTT in young mice for a brief period during early development indicate abnormal emotional behavior in the same mice later in life, similar to the phenotype in mice in which 5-HTT is deficient throughout life (Ansorge et al. 2004). This suggests the necessity of serotonin early in emotional development and provides a possible mechanism by which genetic changes in the 5-HTT system may lead to susceptibility to develop psychiatric diseases such as depression (Caspi et al. 2003). Furthermore, serotonin uptake ability has been documented in primary astrocyte cultures (Kimelberg and Katz 1985) and has been postulated to account for considerable serotonin uptake in the frontal cortex and periventricular region (Ravid et al. 1992).
Because 5-HTT is transcribed from a single copy gene, abnormalities in platelet 5-HTT are thought to reflect CNS abnormalities (Owens and Nemeroff 1998). Several studies on platelet 5-HTT density have been undertaken using [3H]imipramine binding or [3H]paroxetine binding in mood disorders. Although the results of these studies are not entirely consistent, in total the results suggest that the receptor density value for platelet serotonin density is significantly lower in depressed subjects compared with healthy control subjects (Owens and Nemeroff 1998). The distribution of SERT (5-HTT) in the postmortem human brain was found to be highest in the thalamus, amygdala, putamen, globus pallidus externa, lateral geniculate body, hippocampus, and caudate, with the lowest amounts in the cerebral cortex and minimal levels in the cerebellum and white matter (Kish et al. 2005).
Numerous studies have examined suicide risk related to individual genetic polymorphisms in SERT. For example, the serotonin-transporter-linked polymorphic region (5-HTTLPR) short S allele, which decreases presynaptic 5-HTT expression and thereby decreases serotonin reuptake and has an interaction with childhood abuse to increase lifetime depression risk, has nevertheless yielded contradictory results related to suicide risk, likely because of other confounding polymorphisms (Purselle and Nemeroff 2003; Ressler et al. 2010). In patients with major depressive disorder (MDD) who attempted suicide, positron emission tomographic (PET) scanning with the [11C]N,N-dimethyl-2-(2-amino-4-cyanophenylthio) benzylamine ([11C]DASB) ligand indicated that SERT (5-HTT) levels were lower in the midbrain regions (Miller et al. 2013). In bipolar I patients with depression who completed suicide, a nearly 50% decrease in serotonin and norepinephrine activity was observed related to the locus coeruleus in postmortem brain studies, as compared with unipolar depressed patients who completed suicide and matched control subjects (Wiste et al. 2008). Three different antidepressants, sertraline (SSRI), citalopram (SSRI), and venlafaxine (serotonin–norepinephrine reuptake inhibitor), at clinical dosages in living patients were shown with PET ligand [11C]DASB to have 85% occupancy of SERT, effectively blocking SERT-mediated reuptake of free serotonin in the synapse and increasing serotonin synaptic levels (Voineskos et al. 2007). In summary, multiple mechanisms can lead to insufficient levels of serotonin in neuronal synapses, which contributes significantly to the risk for depression and for suicide.
Serotonin Receptors
In 1957, the existence of two separate serotonin receptors was first proposed, primarily because of the opposing phenomenon this neurotransmitter produces, in reference to cholinergic mediation of smooth muscle contraction (Gaddum and Picarelli 1957). Through the use of more precise molecular cloning and pharmacological and biochemical studies, seven distinct serotonin receptor families have been identified (5-HT1–7), many of which contain several subtypes. With the exception of the 5-HT3 receptor, which is an excitatory ionotropic receptor, all the other serotonin receptors are GPCRs. The 5-HT1A,B,D,E,F receptors are negatively coupled to adenylyl cyclase; the 5-HT2A,B,C subtypes are positively coupled to PLC; and the 5-HT4, 5-HT5, 5-HT6, and 5-HT7 subtypes are positively coupled to adenylyl cyclase (see Figure 2–3B) (Humphrey et al. 1993; Nestler et al. 2015). When all types and subtypes are counted, 13 serotonin receptors are identified in humans (Nestler et al. 2015).
5-HT1 receptors. 5-HT1A receptors are found in particularly high density in several limbic structures, including the hippocampus, septum, amygdala, and entorhinal cortex, as well as on serotonergic neuron cell bodies, where they serve as autoreceptors regulating serotonin neuronal firing rates (Blier et al. 1998; Cooper et al. 2003; Pazos et al. 1985). The highest density of labeling is found in the dorsal raphe, with lower densities observed in the remaining raphe nuclei (Pazos et al. 1985). The density and messenger RNA (mRNA) expression of 5-HT1A receptors appear insensitive to reductions in serotonin transmission associated with lesioning the raphe or administering the serotonin-depleting agent p-chlorophenylalanine. Similarly, elevation of serotonin transmission, resulting from chronic administration of an SSRI or MAOI, does not consistently alter 5-HT1A receptor density or mRNA in the cortex, hippocampus, amygdala, or hypothalamus. In contrast to the insensitivity to serotonin, 5-HT1A receptor density is downregulated by adrenal steroids. Postsynaptic 5-HT1A receptor gene expression is under tonic inhibition by adrenal steroids in the hippocampus and some other regions. Thus, in rodents, hippocampal 5-HT1A receptor mRNA expression is increased by adrenalectomy and decreased by corticosterone administration or chronic stress. The stress-induced downregulation of 5-HT1A receptor expression is prevented by adrenalectomy. Mineralocorticoid receptor stimulation has the most potent effect on downregulating 5-HT1A receptors, although glucocorticoid receptor stimulation also contributes to this effect.
In addition to being expressed on neurons, postsynaptic 5-HT1A receptors are also abundantly expressed by astrocytes and some other glia (Whitaker-Azmitia et al. 1990) (see Figure 2–7 later in this chapter). Stimulation of astrocyte-based 5-HT1A sites causes astrocytes to acquire a more mature morphology and to release the trophic factor S-100β, which promotes growth and arborization of serotonergic axons. Administration of 5-HT1A receptor antagonists, antibodies to S-100β, or agents that deplete serotonin produces similar losses of dendrites, spines, and/or synapses in adult and developing animals—effects that are blocked by administration of 5-HT1A receptor agonists or SSRIs. These observations have led to the hypothesis that a reduction of 5-HT1A receptor function may play an important role in mood disorders that are known to be associated with glial reductions (Manji et al. 2001). The use of conditional knockouts of the 5-HT1A receptor, in which gene expression is altered only in particular anatomical regions and/or during particular times, has illustrated the caution necessary in attributing complex behaviors to simple “too much” or “too little” neurotransmitter or receptor hypotheses. One report used a knockout/rescue approach with regional and temporal specificity to show that the anxiety-related effect of the 5-HT1A receptor knockout was actually developmental. Specifically, expression limited to the hippocampus and cortex during early postnatal development was sufficient to counteract the anxious phenotype of the mutant, even though the receptor was still absent in adulthood (Gross et al. 2002). As is discussed in the chapters on antidepressants (see Chapters 8–23), there is interest in the observation that antidepressants enhance hippocampal neurogenesis (Duman 2002; Malberg et al. 2000). It is noteworthy that data suggest that 5-HT1A receptor activation is required for SSRI-induced hippocampal neurogenesis in mice (Jacobs et al. 2000). Altering serotonin levels with the SSRI fluoxetine does not affect division of stem cells in the dentate gyrus, but rather increases symmetric divisions of an early progenitor cell class that exists after stem-cell division (Encinas et al. 2006).
5-HT1A receptors are now known to use a variety of signaling mechanisms to bring about their effects in distinct brain areas. Thus, somatodendritic 5-HT1A receptors appear to inhibit the firing of serotonergic neurons by opening a K+ channel through a pertussis toxin–sensitive G protein (likely Go, discussed later in the section on G proteins) (Andrade et al. 1986), as well as by reducing cAMP levels. Postsynaptic 5-HT1A receptors appear to exert many of their effects by inhibiting adenylyl cyclase via Gi (De Vivo and Maayani 1990) but also have been found to potentiate the activity of certain adenylyl cyclases (Bourne and Nicoll 1993) and to stimulate inositol-1,4,5-triphosphate (IP3) production and activate PKC (Liu and Albert 1991). Structurally, the 5-HT1A receptors are more related to D2 receptors than to other serotonin receptors (Squire 2013). Functional polymorphisms in the promotor of 5-HT1A receptors influencing 5-HT1A receptor expression did not lead to changes in binding of 5-HT1A receptor antagonists in healthy subjects based on two PET studies, suggesting that there are compensatory mechanisms in the brains of healthy living human subjects (Bortolato et al. 2013; David et al. 2005; Lothe et al. 2010). PET studies also reported that 5-HT1A receptors decrease with advancing age in men, which may potentially explain the increased risk of suicide in this population (Moses-Kolko et al. 2011). A functional promoter polymorphism, rs6295, for which the G allele causes decreased transcription of 5-HT1A, may have a role in increasing risk for depression and suicide, because it is overrepresented in patients with depression, although the mechanism is not clear, because postmortem brain samples from depressed individuals who committed suicide showed more equal expression of the G allele with the C allele (Donaldson et al. 2016; Savitz et al. 2009). Buspirone is a 5-HT1A receptor agonist indicated for treating generalized anxiety disorder (Nestler et al. 2015).
5-HT1D receptors are virtually absent in rodents but have been detected in guinea pigs and humans (Bruinvels et al. 1993). On the basis of an approximately 74% sequence homology, it has been proposed that 5-HT1B receptors are the rodent homologue of 5-HT1D receptors (Saxena et al. 1998). Furthermore, the distribution of the 5-HT1D receptors in guinea pigs and humans is approximately equivalent to that of the 5-HT1B receptors in rats (Bruinvels et al. 1993). Both 5-HT1B and 5-HT1D receptors have been proposed to represent the major nerve terminal autoreceptors regulating the amount of serotonin released per nerve impulse (Piñeyro and Blier 1999) (see Figure 2–3B). Like 5-HT1A receptors, 5-HT1B and 5-HT1D receptors inhibit cAMP formation and stimulate IP3 production and activate PKC (Schoeffter and Bobirnac 1995). This appears to be the case for many receptors coupled to Gi and Go (Table 2–1). The α subunits of the G protein (αi and αo) inhibit adenylyl cyclase and regulate ion channels, respectively, whereas the βγ subunits activate PLC isozymes to stimulate IP3 production and activate PKC. Examples of 5-HT1B/1D receptor agonists include the triptan family of medications used to treat migraines, which are incidentally contraindicated in patients who have comorbid coronary artery disease because of the presence of 5-HT1B receptors causing constriction of coronary arteries (Lambert 2005; Nestler et al. 2015). 5-HT1B receptor agonists have shown promise for decreasing reactive aggression and alcohol intake in animal models, but human polymorphism studies searching for a relationship between HTR1B polymorphisms and aggression or suicide risk have yielded inconsistent results (Bortolato et al. 2013).
G protein class |
Members |
Effector(s)/functions |
Examples of receptors |
αi |
Gαi1–3, Gαo |
AC (+) |
α2, D2, A1, μ, M2, 5-HT1A |
Ligand-type Ca2+ channels (+) |
Olfactory signals |
||
Gαz, Gαt1–2 |
K+ channels (+) |
||
Ca2+ channels (−)a |
GABAB |
||
cGMP |
Retinal rods, cones (rhodopsins) |
||
Phosphodiesterase (+) (Gαt1–2) |
|||
αq |
Gαq, Gα11, Gα14, Gα15, Gα16 |
PLC-β (+) |
TxA2, 5-HT2C, M1, M3, M5, α1 |
α12 |
Gα12, Gα13 |
RGS domain–containing rho exchange factors |
TxA2, thrombin |
βb |
β (×5) |
AC type I (−); AC types II, IV (potentiation) |
|
PLC (+) |
|||
Receptor kinases (+) |
|||
Inactivates αs |
|||
γ |
γ (×12) |
β γ subunits required for interaction of α subunit with receptor |
|
Note. AC=adenylyl cyclase; A1, A2=adenosine receptor subtypes; β1, α1, α2=adrenergic receptor subtypes; cGMP=cyclic guanosine monophosphate; D1, D2=dopamine receptor subtypes; Gαo=olfactory, but also found in limbic areas; Gαt=transducin; GABAB=γ-amino-butyric acid receptor subtype; 5-HT1A, 5-HT2C=serotonin receptor subtypes; M1, M2, M3, M5=muscarinic receptor subtypes; μ=opioid μ receptor; PLC=phospholipase C; RGS=regulators of G protein signaling; TxA2=thromboxane A2 receptor. aAlthough regulation of Na+/H+ exchange and Ca2+ channels by Gα1–2 and Gα1–3 has been demonstrated in artificial systems in vitro, these findings await definitive confirmation. bEffectors are regulated by β γ subunits as a dimer. |
|||
The 5-HT1C receptors have structural and transductional similarities to the 5-HT2 receptor class (Hoyer et al. 1986).
5-HT2 receptors. The three subtypes of 5-HT2 receptors are 5-HT2A, 5-HT2B, and 5-HT2C. The highest level of 5-HT2A binding sites and mRNA for these receptors exists in the cortex, and these receptors have been implicated in the psychotomimetic effects of agents such as lysergic acid diethylamide, as reviewed by Aghajanian and Marek (1999). In addition, lesioning serotonin neurons with 5,7-DHT does not reduce the 5-HT2 receptor density reported in brain regions (Hoyer et al. 1986), indicating that these receptors are primarily (if not exclusively) postsynaptic. Autoradiography performed with the potent and selective radioligand [3H]MDL 100,907 has localized 5-HT2A receptors to many similar brain regions in the rat and primate brain (López-Giménez et al. 1997). Hallucinogenesis is likely a result of cortical 5-HT2A receptor activation, because experiments in mice expressing 5-HT2A receptors in the cortex only incur receptor signaling and behavior changes to hallucinogenic drugs similar to that of genetically unaltered mice (González-Maeso et al. 2007). Competition studies with other radioligands (Westphal and Sanders-Bush 1994) and their mRNA distribution indicate that 5-HT2C receptors are considerably widespread throughout the CNS, with the highest density in the choroid plexus (Hoffman and Mezey 1989). 5-HT2B receptors are detected sparingly in the brain and are more prominently located in the fundus, gut, kidney, lungs, and heart (Hoyer et al. 1986).
Evidence from animal experiments in which cortical 5-HT2A receptors are disrupted indicates a specific role of these receptors in modulation of conflict anxiety without affecting fear conditioning and depression-like behaviors (Weisstaub et al. 2006). Furthermore, chronic administration of many antidepressants downregulates 5-HT2 receptors, suggesting that this effect may be important for their efficacy (Scott and Crews 1986). However, chronic electroconvulsive shock appears to upregulate 5-HT2 expression, precluding a simple mechanism for antidepressant efficacy. The obesity seen in 5-HT2C knockout animals suggests that in addition to histamine receptor blockade, 5-HT2C receptor blockade may play a role in the weight gain observed with certain psychotropic agents; this is an area of considerable research. In keeping, evidence suggests that the weight gain “orexigenic” properties of atypical antipsychotics are likely due to potent activation of hypothalamic AMP-kinase through histamine type 1 (H1) receptors (Kim et al. 2007). Aggressive and impulsive behaviors are also linked to the serotonin system, in which a neuropeptide Y (NPY) and serotonin interaction from studies in NPY knockout mice detected circuits responsible for aggressive behavior linked to feeding (Emeson and Morabito 2005). The regulation of 5-HT2 receptors is intriguing, as not only is it important in psychiatric disorders and therapeutic benefit, but also both agonists and antagonists appear to cause an internalization of the receptor. Moreover, data suggest that mRNA editing may play an important role in regulating the levels and activity of this receptor subtype (Niswender et al. 1998). All of the 5-HT2 receptor subtypes are linked to the phosphoinositide signaling system, and their activation produces IP3 and diacylglycerol (DAG), via PLC activation (Conn and Sanders-Bush 1987) (see Figure 2–3B).
A pharmacogenetic study searched for genetic predictors of treatment outcome in 1,953 patients with MDD who received the antidepressant citalopram in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study and were prospectively assessed (McMahon et al. 2006). In a split-sample design, a selection of 68 candidate genes was genotyped with 768 single-nucleotide-polymorphism markers chosen to detect common genetic variation. A significant and reproducible association was found between treatment outcome and a marker in HTR2A (P=1×10–6 to 3.7×10–5 in the total sample). The “A” allele (associated with better outcome) was six times more frequent in white than in black participants, for whom treatment was also less effective in this sample (McMahon et al. 2006). The A allele thus may contribute to racial differences in outcomes of antidepressant treatment. Taken together with prior neurobiological findings, these genetic data make a compelling case for a key role of HTR2A in the mechanism of antidepressant action.
A leading hypothesis for the mechanism of action of atypical antipsychotic agents suggests that the ratio of D2-to-5-HT2 blockade confers “atypicality” properties of many currently available antipsychotic agents (Meltzer 2002). Several antidepressants (e.g., mianserin, mirtazapine) and atypical antipsychotics (e.g., clozapine, risperidone, olanzapine) are antagonists of 5-HT2A receptors, raising the possibility that blockade of 5-HT2 receptors may play an important role in the therapeutic efficacy of these agents (Nestler et al. 2015). Studies comparing the various compounds and the state of 5-HT2A/2C receptors based on psychiatric phenotype would be enhanced by the development of a highly specific PET ligand, [18F]FECIMBI-36, which was confirmed in postmortem human brain tissue sections to bind at high levels to the prefrontal cortex (PFC), temporal cortex, and hippocampus for 5-HT2A receptors and to the choroid plexus for 5-HT2C receptors (Prabhakaran et al. 2015).
5-HT2C receptors are expressed on pro-opiomelanocortin (POMC) neurons of the arcuate nucleus of the hypothalamus (Nestler et al. 2015). The selective 5-HT2C receptor agonist lorcaserin is used for the treatment of obesity (Nestler et al. 2015). Many second-generation antipsychotic drugs cause weight gain through 5-HT2C receptor antagonism (Nestler et al. 2015).
5-HT3–7 receptors. Unlike the other serotonin receptors, 5-HT3 receptors are ligand-gated ion channels capable of mediating fast synaptic responses, although the opening of the channel is relatively slower compared with other ligand-gated ion channels (see Figure 2–3B) (Squire 2013; Yun and Rhim 2011). The cis–trans isomerization and molecular rearrangement at proline 8 is the structural mechanism that opens the 5-HT3 receptor protein pore (Lummis et al. 2005). 5-HT3 receptors are present in multiple brain areas, including the hippocampus, dorsal motor nucleus of the solitary tract, and area postrema (Laporte et al. 1992). The 5-HT3 receptor, located mostly in the peripheral nervous system, is effectively modulated by a variety of compounds, such as alcohols and anesthetics, and antagonists of this receptor are used as effective antiemetics in patients who are undergoing chemotherapy (e.g., ondansetron) (Squire 2013).
5-HT4, 5-HT6, and 5-HT7 are GPCRs that are preferentially coupled to Gs and activate adenylyl cyclases (see Figure 2–3B). 5-HT4 receptors are able to modulate the release of monoamines and GABA in the brain, appear to improve memory and learning in mice, and mediate pituitary prolactin release in the presence of estrogen (Darcet et al. 2016; Papageorgiou and Denef 2007). 5-HT4 receptor subtypes include 5-HT4A, 5-HT4B, 5-HT4C, 5-HT4D, 5-HT4E, 5-HT4F, 5-HT4G, 5-HT4H, and 5-HT4HB (Yun and Rhim 2011). 5-HT5 receptors are located in the hypothalamus, hippocampus, corpus callosum, fibra, cerebral ventricles, and glia (Hoyer et al. 2002). The 5-HT5A receptor is negatively coupled to adenylyl cyclase, whereas the 5-HT5B receptor is not functional because of stop codons (Grailhe et al. 2001). 5-HT6 receptors are located in the striatum, amygdala, nucleus accumbens, hippocampus, cortex, and olfactory tubercle (Hoyer et al. 2002). Of interest, many antipsychotic agents and antidepressants are high-affinity antagonists of 5-HT6 and 5-HT7 receptors, and 5-HT6 receptor antagonism is being specifically investigated for its ability to improve cognition in patients with Alzheimer’s disease (Parker et al. 2015; Roth et al. 1994; Yun and Rhim 2011). 5-HT7 receptors, made up of subtypes 5-HT7A, 5-HT7B, 5-HT7C, and 5-HT7D, have been localized to the cerebral cortex, medial thalamic nuclei, substantia nigra, central gray, and dorsal raphe nucleus (Hoyer et al. 2002; Yun and Rhim 2011). Chronic treatment with antidepressants downregulates 5-HT7 receptors, whereas acute stress has been reported to increase 5-HT7 expression (Sleight et al. 1995; Yau et al. 2001). Lurasidone and vortioxetine, agents used to treat schizophrenia and depression, respectively, are also nonselective 5-HT7 receptor antagonists that work through increased AMPA-mediated neurotransmission (Andreetta et al. 2016; Fountoulakis et al. 2015; Sanchez et al. 2015).
In the future, the subtle differences between the different serotonin receptors and their subtypes will be elucidated as specific PET ligands, genotype–phenotype correlations, and receptor-selective medications are developed (Kumar and Mann 2014).
Dopaminergic System
Dopamine was originally thought to be simply a precursor of norepinephrine and epinephrine synthesis, but the demonstration that its distribution in the brain was quite distinct from that of norepinephrine led to extensive research establishing its role as a critical, unique neurotransmitter. Dopamine synthesis requires transport of the amino acid L-tyrosine across the blood–brain barrier and into the cell. Once tyrosine enters the neuron, the rate-limiting step for dopamine synthesis is conversion of L-tyrosine to L-dihydroxyphenylalanine (L-dopa) by the enzyme tyrosine hydroxylase; L-dopa is readily converted to dopamine and, hence, is used as a precursor strategy to correct a dopamine deficiency in the treatment of Parkinson’s disease (Figure 2–4B). The activity of tyrosine hydroxylase can be regulated by many factors, including the activity of catecholamine neurons; furthermore, catecholamines function as end-product inhibitors of tyrosine hydroxylase by competing with a tetrahydrobiopterin cofactor (Cooper et al. 2003).
In contrast to the widespread serotonin and norepinephrine projections, dopamine neurons form more discrete circuits, with the nigrostriatal, mesolimbic, tuberoinfundibular, and tuberohypophysial pathways constituting the major CNS dopaminergic circuits (Figure 2–4A). The nigrostriatal circuit is composed of dopamine neurons from the mesencephalic reticular formation (region A8) and the pars compacta region of the substantia nigra (region A9) of the mesencephalon. These neurons give rise to axons that travel via the medial forebrain bundle to innervate the caudate nucleus and putamen (Andén et al. 1964; Ungerstedt 1971). The dopamine neurons that make up the nigrostriatal circuit have been assumed to be critical for maintaining normal motor control, because destruction of these neurons is associated with Parkinson’s disease; however, it is now clear that these projections subserve a variety of additional functions. For instance, evidence from human brain imaging studies indicates that drugs that modulate striatal dopamine receptor activation correlate with the subject’s ability to choose gradations of rewarding actions during instrumental learning tasks. This further implies that the dopamine reward pathway in the brain is likely convergent on many discrete brain circuits and neurotransmitter alterations and shows that striatal activity can also account for how the human brain proceeds toward making future decisions based on reward prediction (Pessiglione et al. 2006).
The mesolimbic dopamine circuit consists of dopamine neurons located in the midbrain just medial to the A9 cells in an area termed the ventral tegmental area (VTA) (Cooper et al. 2003; Nestler et al. 2015; Squire 2013). This circuit shares some similarities to the nigrostriatal circuit in that it is a parallel circuit consisting of axons that make up the medial forebrain bundle. However, these axons ascend through the lateral hypothalamus and project to the nucleus accumbens; olfactory tubercle; bed nucleus of the stria terminalis; lateral septum; and frontal, cingulate, and entorhinal regions of the cerebral cortex (Cooper et al. 2003). This circuit innervates many limbic structures known to play critical roles in motivational, motor, and reward pathways and has therefore been implicated in a variety of clinical conditions, including psychosis and drug abuse (Cooper et al. 2003). Data also suggest a potential role of dopamine—and, in particular, mesolimbic pathways—in the pathophysiology of bipolar mania, as well as bipolar and unipolar depression (Beaulieu et al. 2004; Dunlop and Nemeroff 2007; Goodwin et al. 2007; Roybal et al. 2007). It is perhaps surprising that the role of the dopaminergic system in the pathophysiology of mood disorders has not received greater study, because it represents a prime candidate on several theoretical grounds. The motoric changes in bipolar disorder are perhaps the most defining characteristics of the illness, ranging from near catatonic immobility to the hyperactivity of manic states. Similarly, loss of motivation is one of the central features of depression, whereas anhedonia and “hyperhedonic states” are among the most defining characteristics of bipolar depression and mania, respectively. In this context, it is noteworthy that the midbrain dopaminergic system is known to play a critical role in regulating not only motoric activity but also motivational and reward circuits. It is clear that motivation and motor function are closely linked and that motivational variables can influence motor output both qualitatively and quantitatively. Furthermore, considerable evidence indicates that the mesolimbic dopaminergic pathway plays a crucial role in the selection and orchestration of goal-directed behaviors, particularly those elicited by incentive stimuli (Goodwin et al. 2007).
The firing pattern of mesolimbic dopamine neurons appears to be an important regulatory mechanism; thus, in rats, electrical or glutamatergic stimulation of medial PFC elicits a burst firing pattern of dopaminergic cells in the VTA and increases dopamine release in the nucleus accumbens (Murase et al. 1993; Taber and Fibiger 1993). The burst firing of dopamine cell activity elicits more terminal dopamine release per action potential than the nonbursting, pacemaker firing pattern (Roth et al. 1987). The phasic, burst firing of dopamine neurons and accompanying rise in dopamine release normally occur in response to primary rewards (until they become fully predicted) and reward-predicting stimuli. Such a role also has been postulated to provide a neural mechanism by which PFC dysfunction could alter hedonic perceptions and motivated behavior in mood disorders (Drevets et al. 2002). Studies indicate that the amygdala has importance in the learning of new cocaine drug-seeking responses and its habit-forming properties (Lee et al. 2005). The supraphysiological levels of dopamine induced by cocaine and other drugs of abuse lead to powerful reinforcement of drug-seeking behavior, by co-opting the dopamine reward circuit of the brain, as reviewed elsewhere (Volkow and Morales 2015).
Dopamine Transporters
As with serotonin, the dopamine signal in the synaptic cleft is terminated primarily by reuptake into the presynaptic terminal. The DAT comprises 12 transmembrane domains and is located somatodendritically as well as on dopamine nerve terminals (see Figure 2–4B). Like other monoamine transporters, the DAT functions as a Na+/K+ pump to clear dopamine from the synaptic cleft on its release. However, data suggest that many drugs of abuse are capable of altering the function of these transporters. Thus, the amphetamines are thought to mediate their effects, in part, by reversing the direction of the transporter so that it releases dopamine. Cocaine is capable of blocking the reuptake of DAT, leading to an increase in dopamine in the synaptic cleft. Of interest, altered neuronal long-term potentiation in the VTA in response to chronic cocaine exposure has been linked to drug-associated memory and likely contributes to the powerful addictive potential of this drug of abuse (Liu et al. 2005). Dopamine in the medial frontal cortex is taken up predominantly by the norepinephrine transporter, which goes against the dogma of transporters being able to selectively take up only their respective neurotransmitter. Furthermore, this provides a mechanism by which norepinephrine reuptake–inhibiting antidepressants also may increase synaptic levels of dopamine in the frontal cortex, an effect that may be therapeutically very important. Interestingly, a meta-analysis of single photon emission computed tomography (SPECT) scans examining the DAT gene SLC6A3 variable-number tandem repeat (VNTR) polymorphism did not find significant changes in the levels of the dopamine transporters in the brains of patients with schizophrenia, attention-deficit/hyperactivity disorder (ADHD), and even Parkinson’s disease (Costa et al. 2011). However, another DAT polymorphism of note, a VNTR in the nontranslated 3′ end of exon 15, causes a 25% decreased density of DAT in humans with a VNTR of 9 repeats instead of the more common 10 repeats (Lacerda-Pinheiro et al. 2014). SPECT scanning of the brain with the presynaptic DAT radioligand is now being used to distinguish Parkinson’s disease syndromes from other causes of parkinsonism such as antipsychotic-induced parkinsonism, the latter of which does not show a deficit in DAT binding in the caudate and putamen (Tatsch and Poepperl 2013).
In patients with MDD and bipolar disorder, SPECT studies showed that DAT availability is higher—and, by inference, synaptic dopamine is lower—in patients with depression (Camardese et al. 2014a, 2014b). PET studies identified decreased binding potential of the DAT PET ligand to the dopamine transporter in the striatum in MDD patients and more specifically in the caudate in bipolar patients (Anand et al. 2011; Meyer et al. 2001; Savitz and Drevets 2013). Postmortem studies in patients with MDD showed lower DAT levels in the amygdala and higher D2 and D3 receptor levels (no change in D1 receptors), consistent with similar observations in rats with dopamine depletion (Klimek et al. 2002).
To treat refractory depression, the new triple transporter reuptake inhibitor compound BMS-820836, reported by PET studies to specifically inhibit monoamine transporters DAT, SERT, and NET, was developed for the goal of increasing synaptic levels of the respective monoamines dopamine, serotonin, and norepinephrine and thereby decreasing depressive symptoms (Risinger et al. 2014). Moderate to severe MDD or atypical MDD (with reversed neurovegetative symptoms) was found by PET studies to be associated with higher levels of MAO-A, an enzyme that catabolizes and thus decreases synaptic dopamine, serotonin, and norepinephrine, presumably increasing depressive symptoms (Chiuccariello et al. 2014). Classic MAOIs increase all three monoamines in the synapse by blocking the catabolizing enzyme MAO (both A and B enzyme subtypes).
Bupropion is widely believed to increase synaptic dopamine by blocking the DAT-mediated reuptake, but a PET study showed that it has at most only 22% occupancy of the DAT (Meyer et al. 2002). Given the observation of a decreased binding potential of 15% of DAT in patients with MDD, it remains unknown whether bupropion occupies the same binding site on DAT as the PET ligand, whether 22% occupancy of DAT is sufficient for bupropion to work (as compared with 80% binding to SERT for SSRIs), and whether bupropion works by another uncharacterized mechanism to achieve its antidepressant properties (Meyer et al. 2001, 2002).
Dopaminergic Receptors
The existence of two subtypes of dopamine receptors, D1 and D2, was initially established with classic pharmacological techniques in the 1970s (Stoof and Kebabian 1984). Subsequent molecular biological studies have shown that the D1 family contains both the D1 and the D5 receptors, whereas the D2 family contains the D2, D3, and D4 receptors (Cooper et al. 2003). D1 receptor family members were originally defined solely on the ability to stimulate adenylyl cyclase, whereas the D2 family inhibited the enzyme. Interestingly, dopamine receptors complexed with subunits from other subclasses of dopamine receptors within a receptor family are able to form distinct hetero-oligomeric receptors also termed kissing cousin receptors. Notably, hetero-oligomeric D1–D2 receptor complexes in the brain require binding to active sites of both receptor subtypes to induce activation of the hetero-oligomeric receptor complex. These receptors have been shown to use traditional D1 receptor intracellular signaling components of Gq/11 and Ca2+/calmodulin–dependent protein kinase II (CaMKII) second-messenger activation as seen in the nucleus accumbens (Rashid et al. 2007). This opens up possibilities for the brain to use different receptor subunit proportions to further fine-tune brain neurophysiology. Similar to the DAT exon 15 VNTR, the D2 dopamine receptor gene (DRD2) has a TaqIA polymorphism (allele A1+) in the 3′ nontranslated region that reduces D2 receptor density (Lacerda-Pinheiro et al. 2014).
D1 and D5 receptors. The D1 and D5 receptors stimulate adenylyl cyclase activity via the activation of Gs or Golf (a G protein originally thought to be present exclusively in olfactory tissue but now known to be abundantly present in limbic areas) (see Figure 2–4B). Other second-messenger pathways also have been reported to be activated by D1 receptors, an effect that may play a role in the reported D1–D2 cross-talk (Clark and White 1987). The frontal cortex contains almost exclusively D1 receptors (Clark and White 1987), suggesting that this receptor may play an important role in higher cognitive function and perhaps in the actions of medications such as methylphenidate. A D1/D5 receptor agonist, dihydrexidine, has been developed and tested for its ability to improve cognition and working memory in patients with schizophrenia but was prematurely terminated because of hypotension and potentially increased seizure risk (Arnsten et al. 2017). Although the D5 receptor is a neuron-specific receptor located primarily in limbic areas of the brain, no compounds have been developed so far that pharmacologically distinguish it from the homologous D1 receptor (Arnsten et al. 2017). Drugs such as cocaine and methylphenidate cause peak dopamine activation of D1 receptors on striatal GABAergic medium spiny neurons, which plays a key role in the direct striatal pathway mediating expectations of reward (Volkow and Morales 2015).
D2 receptors. Four types of D2 receptors have been identified: two subtypes of D2 receptors (the short and long splice variants, D2S and D2L, respectively) and D3 and D4 receptors. Although a seemingly identical pharmacological profile for these receptors exists, there are undoubtedly physiological differences between the two subtypes. D2 receptors mediate their cellular effects via the Gi/Go proteins and thereby several effectors (see Figure 2–4B). In addition to the well-characterized inhibition of adenylyl cyclase, D2 receptors in different brain areas also regulate PLC, bring changes in K+ and Ca2+ currents, and possibly regulate phospholipase A2. D2 receptors are located on cell bodies and nerve terminals of dopamine neurons and function as autoreceptors. Thus, activation of somatodendritic D2 receptors reduces dopamine neuron firing activity, likely via the opening of K+ channels, whereas activation of nerve-terminal D2 autoreceptors reduces the amount of dopamine released per nerve impulse, in large part by closing voltage-gated Ca2+ channels. As discussed extensively elsewhere in this volume (see Chapter 24, “Classic Antipsychotic Medications,” by Nasrallah and Tandon), D2 receptors have long been implicated in the pathophysiology and treatment of schizophrenia. In one study, transgenic mice overexpressing D2 receptors in the striatum had many phenotypic hallmarks of schizophrenia (Kellendonk et al. 2006). However, the potential role of D2 receptors in contributing to depression is a comparatively more recent recognition, confirmed by the observation that D2 receptor binding is increased by PET ligand [11C]raclopride in the putamen of patients with MDD and psychomotor retardation, reflecting decreased dopamine levels in the synapse (Meyer et al. 2006).
The typical antipsychotics are generally defined by their high-affinity antagonism or blockade of the D2 receptor, which substantially prevents synaptic dopamine from activating the postsynaptic receptor but also has a significant risk for extrapyramidal side effects (EPS) such as dystonia and akathisia in the short term and parkinsonism and tardive dyskinesia in the longer term (see Figure 2–4B). Medications with high anticholinergic properties, such as diphenhydramine and benztropine, are frequently used to avoid development of EPS, but tardive dyskinesia can develop nevertheless after many years of antipsychotic exposure. Cocaine and methylphenidate stimulation of D2 receptors in the indirect, inhibitory striatal pathway, which mediates punishment, leads to sustained motivation to procure these drugs in the future (Volkow and Morales 2015).
D3 receptors. D3 receptors possess a different anatomical distribution than D2 receptors and, because of their preferential limbic expression, have been postulated to represent an important target for antipsychotic drugs. Numerous studies have investigated the position association of a polymorphism in the coding sequence of the D3 receptor with schizophrenia, with equivocal results. It has been suggested that brain-derived neurotrophic factor (BDNF) may regulate behavioral sensitization via its effects on D3 receptor expression (Guillin et al. 2001). Cariprazine, an antipsychotic with possible efficacy in treating acute mania, has antagonist/partial agonist binding to D2 and D3 receptors, with a much higher affinity for D3 receptors compared with other antipsychotics, but it still has significant D2-related EPS (Altinbaş et al. 2013). Buspirone, a 5-HT1a receptor partial agonist, also has D3/D4 receptor antagonist activity, so it is being considered for treating methamphetamine and cocaine dependence, given the observation of increased brain D3 receptors by PET studies in addicted individuals and the attenuation of self-administration in animals given D3 receptor antagonists (Paterson et al. 2014).
D4 receptors. The D4 receptor has received much interest in psychopharmacological research because of the fact that clozapine has a high affinity for this receptor. More selective D4 antagonists are being explored as adjunctive agents in the treatment of schizophrenia. Furthermore, considerable attention has focused on the possibility that genetic D4 variants may be associated with thrill-seeking behavior (Zuckerman 1985), ADHD (Roman et al. 2001), and responsiveness to clozapine (Van Tol et al. 1992). A potent, selective antagonist of the D4 receptor called ML398 has been developed that is still being investigated (Berry et al. 2014).
Noradrenergic System
Named sympathine because it was initially encountered as being released by sympathetic nerve terminals, the molecule was later given the name norepinephrine after meeting the criteria for a neurotransmitter in the CNS (Cooper et al. 2003). Norepinephrine is produced from the amino acid precursor L-tyrosine found in neurons in the brain, chromaffin cells, sympathetic nerves, and ganglia. The enzyme dopamine β-hydroxylase converts dopamine to norepinephrine, and as is the case for dopamine synthesis, tyrosine hydroxylase is the rate-limiting enzyme for norepinephrine synthesis (Figure 2–5B). The dietary depletion of tyrosine and α-methyl-p-tyrosine (a tyrosine hydroxylase inhibitor) has played an important part in efforts aimed at delineating the role of catecholamines in the pathophysiology and treatment of mood and anxiety disorders (Coupland et al. 2001; McCann et al. 1995).
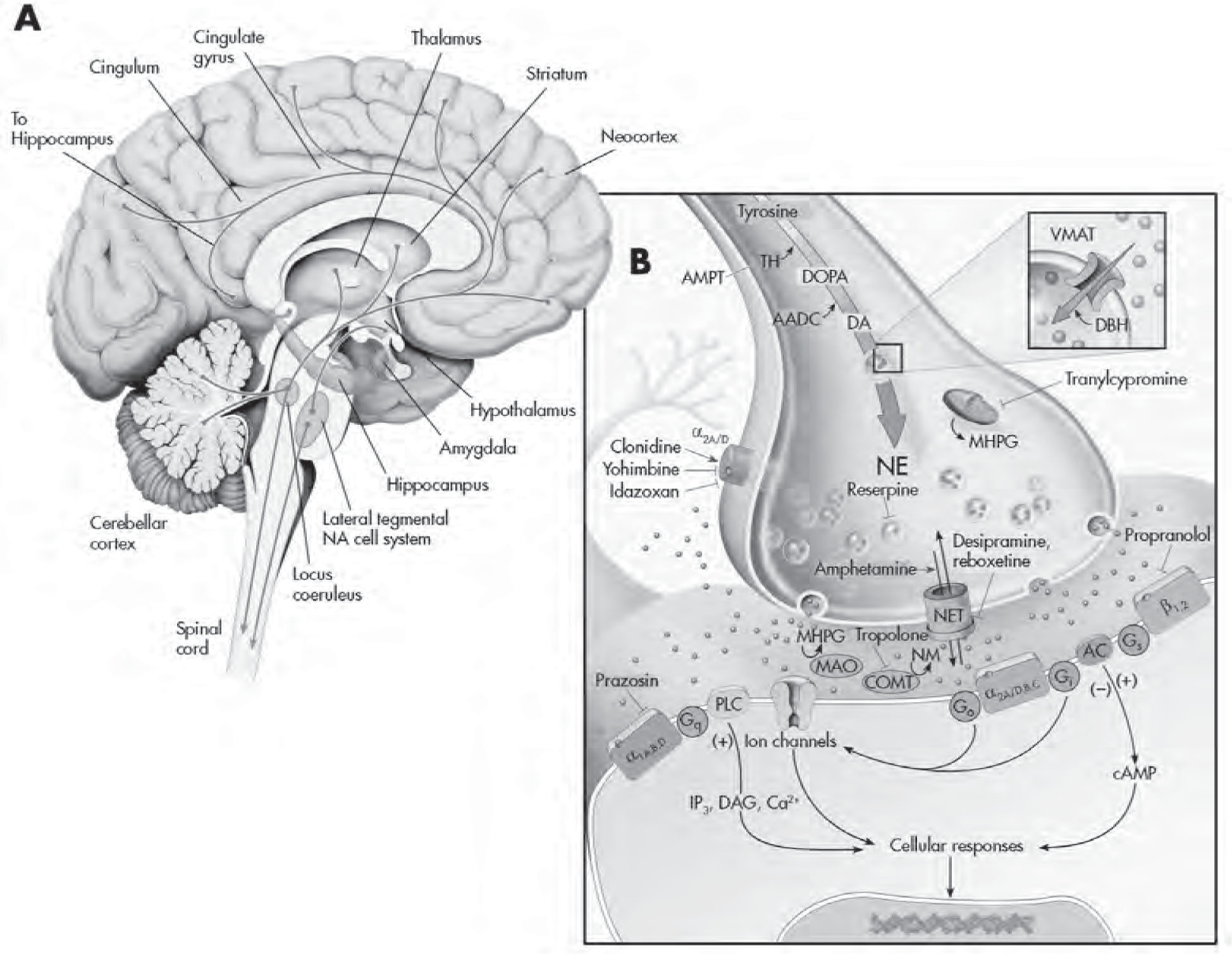
FIGURE 2–5. The noradrenergic system.
See Plate 8 to view this figure in color.
This figure depicts the noradrenergic projections throughout the brain (A) and the various regulatory processes involved in norepinephrine (NE) neurotransmission (B). NE neurons innervate nearly all parts of the neuroaxis, with neurons in the locus coeruleus being responsible for most of the NE in the brain (90% of NE in the forebrain and 70% of total NE in the brain). The amino acid L-tyrosine is actively transported into presynaptic NE nerve terminals, where it is ultimately converted into NE. The rate-limiting step is conversion of L-tyrosine to L-dihydroxyphenylalanine (L-dopa) by the enzyme tyrosine hydroxylase (TH). α-Methyl-p-tyrosine (AMPT) is a competitive inhibitor of tyrosine hydroxylase and has been used to assess the impact of reduced catecholaminergic function in clinical studies. Aromatic amino acid decarboxylase (AADC) converts L-dopa to dopamine (DA). L-dopa then becomes decarboxylated by decarboxylase to form DA. DA is then taken up from the cytoplasm into vesicles, by vesicular monoamine transporters (VMATs), and hydroxylated by dopamine β-hydroxylase (DBH) in the presence of O2 and ascorbate to form NE. Normetanephrine (NM), which is formed by the action of catechol-O-methyltransferase (COMT) on NE, can be further metabolized by monoamine oxidase (MAO) and aldehyde reductase to 3-methoxy-4-hydroxyphenylglycol (MHPG). Reserpine causes a depletion of NE in vesicles by interfering with uptake and storage mechanisms (depressive-like symptoms have been reported with this hypertension). Once released from the presynaptic terminal, NE can interact with a variety of presynaptic and postsynaptic receptors. Presynaptic regulation of NE neuron firing activity and release occurs through somatodendritic (not shown) and nerve-terminal α2 adrenoreceptors, respectively. Yohimbine potentiates NE neuronal firing and NE release by blocking these α2 adrenoreceptors, thereby disinhibiting these neurons from a negative feedback influence. Conversely, clonidine attenuates NE neuron firing and release by activating these receptors. Idazoxan is a relatively selective α2 adrenoreceptor antagonist primarily used for pharmacological purposes. The binding of NE to G protein receptors (Go, Gi, etc.) that are coupled to adenylyl cyclase (AC) and phospholipase C–β (PLC-β) produces a cascade of second-messenger and cellular effects (see diagram and later sections of the text). NE has its action terminated in the synapse by rapidly being taken back into the presynaptic neuron via NE transporters (NETs). Once inside the neuron, it can either be repackaged into vesicles for reuse or undergo enzymatic degradation. The selective NE reuptake inhibitor and antidepressant reboxetine and older-generation tricyclic antidepressant desipramine are able to interfere/block the reuptake of NE. On the other hand, amphetamine is able to facilitate NE release by altering NET function. Green spheres represent DA neurotransmitters; blue spheres represent NE neurotransmitters. cAMP=cyclic adenosine monophosphate; DAG=diacylglycerol; DOPA=dihydroxyphenylalanine; IP3=inositol-1,4,5-triphosphate; NA=nucleus accumbens.
Source. Adapted from Cooper JR, Bloom FE, Roth RH: The Biochemical Basis of Neuropharmacology, 8th Edition. New York, Oxford University Press, 2001. Copyright 1970, 1974, 1978, 1982, 1986, 1991, 1996, 2001 by Oxford University Press, Inc. Used by permission of Oxford University Press, Inc. Modified from Nestler et al. 2001.
The mammalian CNS has seven norepinephrine cell groups, designated A1 through A7. In the brain stem, these are the lateral tegmental neurons (A5 and A7) and the locus coeruleus neurons (A6) (Dahlström 1971) (see Figure 2–5B). In general, the projections from A5 and A7 are more restricted to brain stem areas and do not interact with those of A6. The term locus coeruleus (LC) was derived from the Greek because of its saddle shape and its “blueish color” (caeruleum). The LC is the most widely projecting CNS nucleus known (Foote et al. 1983), responsible for approximately 90% of the norepinephrine innervation of the forebrain and 70% of the total norepinephrine in the brain (Figure 2–5A). Indeed, the LC norepinephrine neurons, although small in number, constitute a diffuse system of projections to widespread brain areas via highly branched axons. The extensive efferent innervation suggests that the LC plays a modulatory and integrative role rather than a role in specific sensory or motor processing (Foote et al. 1983).
Several physiological roles have been ascribed to the LC, notably in the control of vigilance and the initiation of adaptive behavioral responses (Foote et al. 1983). Considerable data support the hypothesis that norepinephrine neurons in the LC constitute a CNS response or defense system, because the neurons are activated by “challenges” in both the behavioral/environmental and the physiological domains (Jacobs et al. 1991). Thus, various sensory stimuli are capable of increasing LC activity, but noxious or stressful stimuli are particularly potent in this regard. Moreover, considerable evidence also supports a role for LC norepinephrine neurons in the learning of aversively motivated tasks and in the conditioned response to stressful stimuli (Rasmussen and Jacobs 1986; Rasmussen et al. 1986), with obvious implications for a variety of psychiatric conditions (Gould et al. 2002; Szabo and Blier 2001). Indeed, tonic activation of the LC occurs preferentially in the response to stressful stimuli, in contrast to stimuli limited to simply evoking activation or arousal (Rasmussen and Jacobs 1986; Rasmussen et al. 1986). Preclinical studies have found increased sensitivity of the LC norepinephrine system in females compared with males, which may help explain the increased incidence of posttraumatic stress disorder (PTSD) and depression in human females (Bangasser et al. 2016). Of note, the LC also releases neuropeptides such as galanin, NPY, cocaine- and amphetamine-related transcript, and BDNF; galanin specifically co-localizes with norepinephrine in LC neurons as they project to the PFC, hippocampus, and VTA and mediate stress responses and addictive behaviors (Weinshenker and Holmes 2016). Increased norepinephrine and activation of the LC may be involved in both fear and anger responses, and excessive anger may be a “vent” for fear of the unexpected, as suggested in one review (Gu et al. 2016).
Norepinephrine Transporter
NET, the first of the monoamine transporters to be cloned in humans, transports norepinephrine from the synaptic cleft back into the neuron (Pacholczyk et al. 1991). Like other monoamine transporters, the NET comprises 12 putative transmembrane domains, and autoradiography with various norepinephrine reuptake inhibitors has been used to determine the brain distribution of NET. A high level of NET is found in the LC, with moderate to high levels found in the dentate gyrus, raphe nuclei, and hippocampus (Tejani-Butt and Ordway 1992; Tejani-Butt et al. 1990). This pattern of expression is consistent with the norepinephrine innervation to these structures. The NET is expressed mainly on norepinephrine terminals, as shown by a drastic reduction in labeling following norepinephrine destruction with the neurotoxin 6-hydroxydopamine or N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) (Tejani-Butt and Ordway 1992; Tejani-Butt et al. 1990).
The NET is dependent on extracellular Na+ to mediate norepinephrine reuptake and the effectiveness of norepinephrine reuptake inhibitors in inhibiting norepinephrine reuptake (Brüss et al. 1997, 1999; Harder and Bönisch 1985). The uptake of norepinephrine is chloride dependent, meaning that the electrogenic process of norepinephrine transport is Na+ and Cl– driven (Harder and Bönisch 1985). In addition to the electrogenic process, the NET has properties of a channel-like pore, in that it can transport norepinephrine showing an infinite stoichiometry that can be blocked by cocaine and desipramine (Galli et al. 1995, 1996). Several studies suggest that the NET can be regulated by diverse stimuli, neuronal activity, and peptide hormones, as well as protein kinases. Indeed, studies have shown that all monoaminergic transporters (5-HTT, DAT, and NET) are rapidly regulated by direct or receptor-mediated activation of cellular kinases, particularly PKC (Bauman et al. 2000). PKC activation results in an activity-dependent transporter phosphorylation and sequestration. Protein phosphatase–1/2A (PP-1/PP-2A) inhibitors, such as okadaic acid and calyculin A, also promote monoaminergic transporter phosphorylation and functional downregulation (Bauman et al. 2000). These phenomena that occur beyond the receptor level may well be important in the long-term actions of psychotropic drugs known to regulate protein kinases (Chen et al. 1999; Manji and Lenox 1999). Given that norepinephrine neurons co-localize and release orexins, it is of interest that these neuropeptides have been implicated in sleep disorders and hypoglycemia through the glucose-sensing tandem membrane receptor K+ channels (K2P type) affecting coordinated arousal (Scott et al. 2006). It is also interesting that enkephalin, an endogenous opioid peptide, co-localizes with NET on the axon terminals of the basolateral amygdala, as indicated by transmission electron microscopy and immunohistochemistry (Zhang and McDonald 2016). The latest PET radiotracer developed for human brain NET showed high uptake in the LC as well as the raphe nucleus, red nucleus, and thalamus, which could aid in monitoring disease progression of various conditions with NET dysregulation, including Parkinson’s disease, Alzheimer’s disease, epilepsy, ADHD, depression, and anxiety (Adhikarla et al. 2016). Atomoxetine, a selective NET inhibitor and the first nonstimulant medication approved for the treatment of ADHD in humans, was found to occupy both the NET and the SERT at clinically relevant doses when tested in rhesus monkeys (Ding et al. 2014).
Adrenergic Receptors
The α and β catecholamine receptors were first discovered more than 50 years ago (Ahlquist 1948) and were later subdivided further into α1, α2, β1, β2, and β3 adrenoreceptors—all of which are GPCRs—on the basis of molecular cloning and pharmacological and biochemical studies (see Figure 2–5B). The crystal structures of these receptors were subsequently solved, along with those of other GPCRs, as reviewed elsewhere (Millar and Newton 2010).
α Receptors. The three subtypes of α1 receptors are denoted 1A, 1B, and 1D; they are all positively coupled to PLC and possibly phospholipase A2 (see Figure 2–5B). The α2 family comprises the 2A/D, 2B, and 2C subtypes, which couple negatively to adenylyl cyclase and regulate K+ and Ca2+ channels (see Figure 2–5B). The 2A, 2B, and 2C adrenoceptors correspond to the human genes ADRA2A, ADRA2B, and ADRA2C, respectively. The bovine, guinea pig, rat, and mouse α2D adrenoceptor is thought to be a species homologue or variant of the human α2A adrenoceptor (Bylund et al. 1994) and is often referred to as α2A/D. The α2 receptors represent autoreceptors for norepinephrine neurons, and blockade of these autoreceptors results in increased norepinephrine release—a biochemical effect that has been postulated to play a role in the mechanisms of action of selected antidepressants (e.g., mianserin, mirtazapine) and antipsychotics (e.g., clozapine). In the LC, α2-adrenergic receptors converge onto similar K+ channels as μ opioid receptors, and this convergence has been postulated to represent a mechanism for the efficacy of clonidine (an α2 agonist) in attenuating some of the physical symptoms of opioid withdrawal. In addition, clonidine and another α2 receptor agonist, guanfacine, have both been approved as nonstimulant medications to treat ADHD (Chan et al. 2016). The α2 receptor antagonist yohimbine, which robustly increases norepinephrine neuron firing and norepinephrine release, has been used as a provocative challenge in clinical studies of anxiety disorders and as an antidepressant-potentiating agent. Yohimbine also has been used to explore the role of α2 adrenoceptors in modulating different types of pain in healthy humans given high-frequency electrical stimulation (Vo and Drummond 2016).
β Receptors. The β receptor family comprises β1, β2, and β3 adrenoceptors, which are all positively coupled to adenylyl cyclase (Bylund et al. 1994) (see Figure 2–5B). As is discussed in greater detail in the chapters on antidepressants (see Chapters 8–23), most effective antidepressants produce a downregulation/desensitization of β1 receptors in rat forebrain, leading to the suggestion that these effects may play a role in their therapeutic efficacy. Interestingly, β receptors also have been shown to play a role in regulating emotional memories, leading to the proposal that β antagonists may have utility in the treatment of PTSD (Cahill et al. 1994; Przybyslawski et al. 1999). Propranolol, a combined β1–β2 receptor antagonist, has been used to try to prevent consolidation of traumatic memories in people who had just experienced trauma, such as rape victims or soldiers, but a meta-analysis suggested that it was not effective (Argolo et al. 2015). More recently, propranolol is being proposed to mitigate the development of intensive care unit–induced PTSD, which commonly stems from the trauma of hospitalization in an intensive care unit (Gardner and Griffiths 2014). β3 receptors are not believed to be present in the CNS but are abundantly expressed on brown adipose tissue (BAT), where they exert lipolytic and thermogenic effects. Not surprisingly, active researchers are attempting to develop selective β3 agonists for the treatment of obesity, and they are now called Class I BAT activators (Mukherjee et al. 2016).