CHAPTER 6
Cardiovascular Pharmacology
Introduction
The cardiovascular system can be influenced by a variety of medications used in the operating room and intensive care units. Pharmacologic agents are commonly used to change the contractility of the heart (inotropy), heart rate (chronotropy), and conduction through the AV node (dromotropy). They also act as vasopressors when they constrict blood vessels or vasodilators when they cause blood vessels to relax. Most have their actions either by influencing receptors of the autonomic nervous system (alpha1, beta1, and beta2 receptors; see Chapter 13, Autonomic Nervous System) or by direct actions on the smooth muscle of the vascular wall.
Maintaining a normal heart rate and rhythm is essential for optimal cardiac function. Antiarrhythmic agents are commonly used in the perioperative period to accomplish this. Drugs impacting the cardiovascular system are used to overcome the sequelae of cardiovascular disease, the effects of cardiovascular-depressant drugs (e.g., anesthetic agents), physiologic reflexes, and/or any combination of these. This chapter introduces the anesthesia technician to the mechanism of action and uses of positive inotropic agents, vasoactive drugs, and antiarrhythmic agents. The cardiovascular actions of anesthetic drugs are described elsewhere.
Positive Inotropic Agents
Drugs that increase the force of myocardial contraction are called positive inotropic agents and include catecholamines, phosphodiesterase (PDE) inhibitors, and myofilament calcium sensitizers. Catecholamines and PDE inhibitors increase calcium concentration in the cytoplasm of cardiac muscle cells by different mechanisms to help generate a greater force of contraction. Myofilament calcium sensitizers enhance the interaction between the contractile proteins within myocardial cells without increasing intracellular calcium.
Stimulation of beta1-adrenergic receptors that are coupled to G proteins activates the enzyme adenylyl cyclase, which, in turn, forms cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (see Fig. 6.1). cAMP increases contractility (via increased intracellular calcium concentration) of cardiac muscle while also causing relaxation in smooth muscle (e.g., in blood vessels). It is metabolized by the enzyme PDE. PDE is targeted by a variety of drugs called PDE inhibitors that increase cAMP levels by inhibiting PDE and preventing breakdown of cAMP.
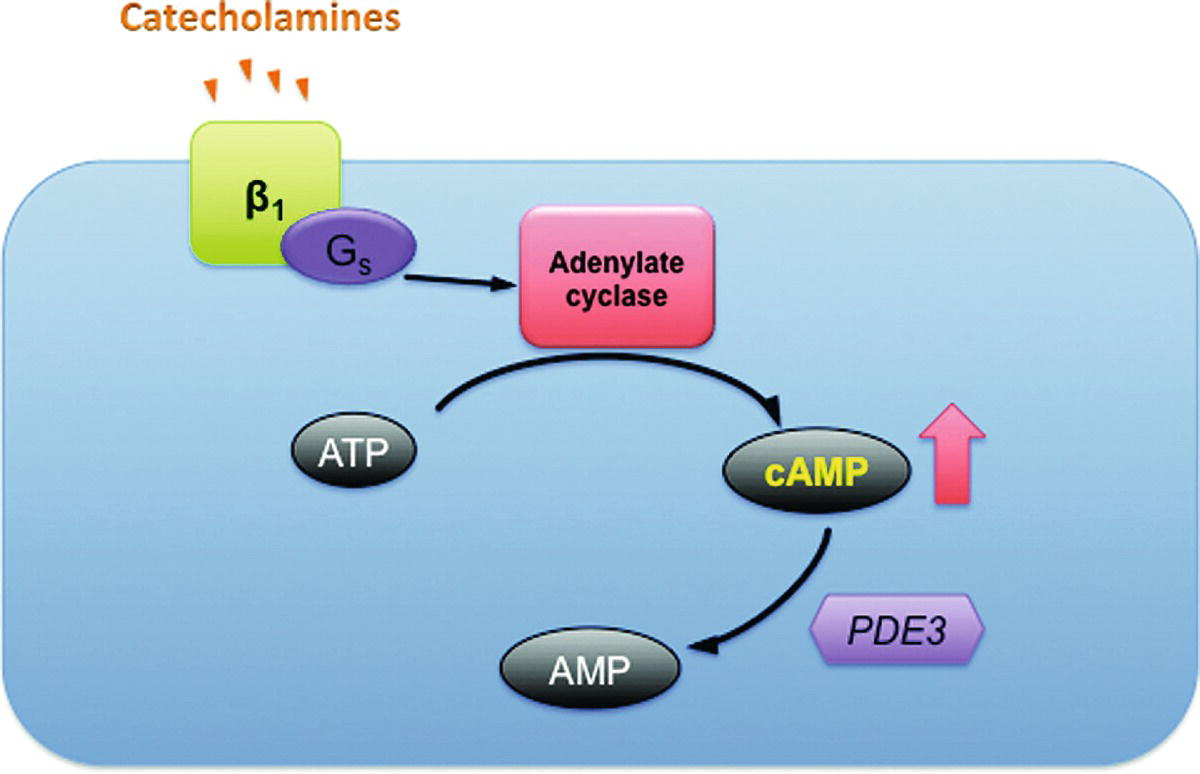
FIGURE 6.1. Function of catecholamines and phosphodiesterase inhibitors in the myocyte. Catecholamines increase cAMP through an energy-dependent pathway. In the presence of phosphodiesterase inhibitors, the breakdown of cAMP to AMP is slowed and the effect of catecholamines potentiated. AMP, adenosine monophosphate; ATP, adenosine triphosphate; β1, β1 adrenoreceptor; cAMP, cyclic adenosine monophosphate, Gs, Gs protein; PDE3, phosphodiesterase (isoenzyme 3). (Adapted from Klabunde RE. Cardiovascular Physiology Concepts. Philadelphia, PA: Lippincott Williams & Wilkins; 2005, with permission. Available from: www.cvpharmacology.com)
Thus, this same pathway is influenced by different classes of drugs as well as calcium; this becomes critically important when attempting to strengthen the heart to wean from cardiac bypass.
Catecholamines
Epinephrine, norepinephrine, and dopamine are all natural neurotransmitters and hormones that are also used as drugs. The naturally occurring catecholamines, epinephrine, norepinephrine, and dopamine, are produced in the medulla of the adrenal gland. Norepinephrine is also synthesized in adrenergic nerves and functions as a neurotransmitter in the sympathetic division of the autonomic nervous system (see Chapter 13). Epinephrine and norepinephrine are considered stress hormones when released into the bloodstream from the adrenal gland. The half-lives of catecholamines are short (minutes). They are deactivated by both reuptake into presynaptic neurons and metabolism by enzymes. The metabolites can be detected in the urine and are elevated in patients with catecholamine-producing tumors such as pheochromocytoma. The drugs stimulating alpha and beta adrenoceptors to produce their actions have proportionally different effects on heart, vascular, and other smooth muscles, depending on their affinity for receptor types (see Table 6.1).
Table 6.1. Pharmacology of Alpha- and Beta-Adrenergic Agonists
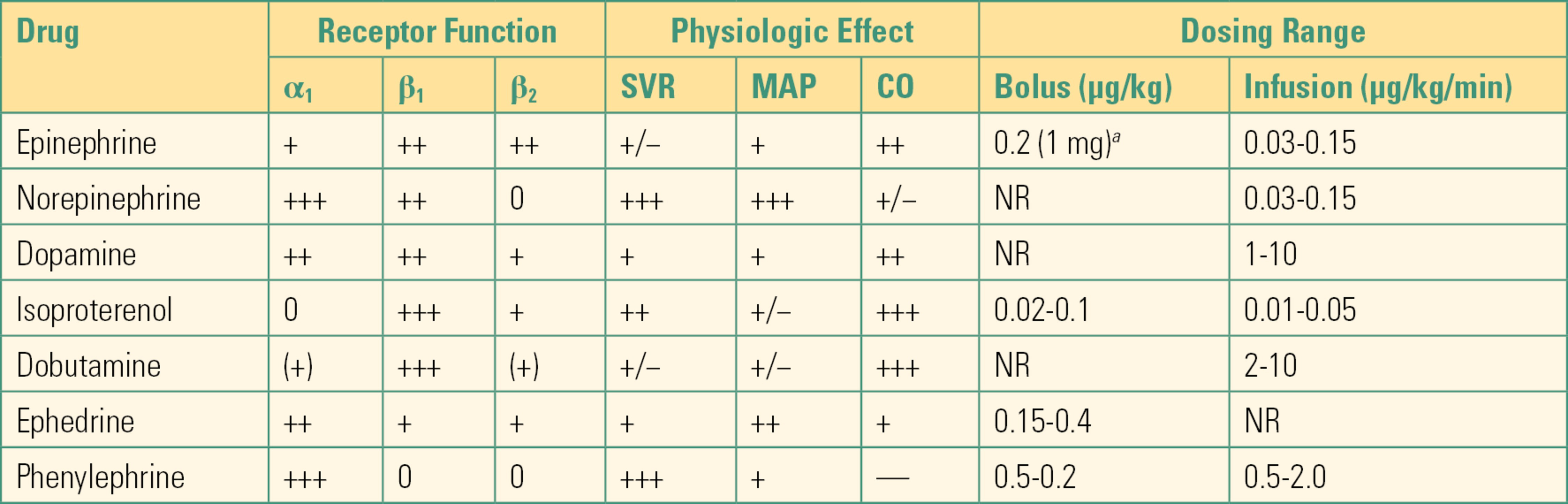
aFor cardiopulmonary resuscitation.
SVR, systemic vascular resistance; MAP, mean arterial pressure; CO, cardiac output; NR, not recommended.
Modified from Stoelting R, Hillier S, eds. Pharmacology & Physiology in Anesthetic Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006, with permission.
Epinephrine
Epinephrine is a naturally occurring catecholamine that is produced from its precursor, norepinephrine, exclusively in the adrenal medulla. It has a wide range of physiologic effects including increasing heart rate, myocardial contractility, and conduction in the heart by stimulating primarily beta1-adrenergic receptors. Increased peripheral vascular tone (afterload) is mediated by the activation of alpha1-adrenergic receptors, while relaxing bronchial smooth muscle (bronchodilation) is caused by the activation of beta2 receptors. Beta2 receptors are also located on vascular smooth muscle cell membranes, and stimulation of these receptors by low doses of epinephrine produces vasodilation. At lower doses, the effects on beta1 and beta2 adrenoceptors (bronchodilation, inotropy, and chronotropy) usually predominate, while at high doses, epinephrine can cause profound vasoconstriction through alpha1 activation. Epinephrine also influences metabolism by increasing glycogenolysis in the liver and lipolysis in adipose tissue.
In anesthetic practice, epinephrine may be used intravenously in life-threatening situations including cardiac arrest, low cardiac output syndromes, anaphylaxis, or bronchospasm. Added to solutions of local anesthetics in a concentration of 1:200,000, it prolongs the action of the anesthetic by constricting surrounding blood vessels and decreasing systemic reabsorption.
In cardiopulmonary resuscitation, epinephrine is administered intravenously in 1 mg (0.02 mg/kg) increments every 3 to 5 minutes per advanced cardiac life support (ACLS) guidelines in a dilution of 1:10,000 (0.1 mg/mL) (see Chapter 58, Cardiac Arrest). At this high dose, epinephrine is primarily an arterial vasoconstrictor: it increases diastolic pressure, improving coronary artery perfusion. As a continuous infusion, epinephrine is usually administered between 0.03 and 0.15 μg/kg/min. Epinephrine can produce cardiac arrhythmias, especially in the presence of the anesthetic halothane. It produces dramatic increases in heart rate and, in patients with coronary artery disease, may cause myocardial ischemia. In general, the beta-adrenergic stimulant properties of epinephrine and other catecholamines will be diminished in patients taking beta-blocking drugs. There is a very high variability in the response between patients, and thus, this drug should always be titrated carefully to the desired effect. Infusion through a central venous line is recommended for higher concentrations as extravasation can cause tissue necrosis. In general, solutions containing catecholamines for infusion should be prepared in 5% glucose to avoid deactivation in alkaline solutions.
Norepinephrine
Norepinephrine is also a natural hormone secreted from the adrenal medulla and is the neurotransmitter released from postganglionic adrenergic nerve endings following sympathetic nervous system stimulation. In addition, norepinephrine plays a role in the stress response and is secreted from noradrenergic neurons in the central nervous system.
Norepinephrine must be infused intravenously (approximately 0.05-0.15 μg/kg/min) and has dose-dependent effects mediated via alpha1- and beta1-adrenergic receptors. In the lower dose range, increased heart rate and contractility through beta1 activation may result in a higher cardiac output. Unlike epinephrine, norepinephrine has little to no activity at beta2 receptors. Norepinephrine is a positive inotrope and is an excellent drug for the treatment of low-output, low–vascular resistance heart failure.
In higher doses, alpha1 activity predominates and norepinephrine causes a profound vasoconstriction in the vasculature of the kidney, liver, skeletal muscle, and skin. The resulting increase in systemic vascular resistance, reduced venous return, and increased arterial pressure can elicit reflex bradycardia (baroreceptor reflex), and cardiac output may actually decline. Norepinephrine is a drug of choice in shock states characterized by diminished peripheral vascular resistance such as septic shock. Like epinephrine, it reduces renal and intestinal perfusion and may contribute to organ dysfunction particularly if the patient is hypovolemic. This can lead to renal failure and mesenteric infarction. In the pulmonary vasculature, norepinephrine increases vascular resistance by stimulation of alpha1 receptors that may contribute to pulmonary hypertension and right heart failure. Patients should be carefully monitored while receiving norepinephrine, and due to its very short half-life (approximately 2 minutes), a continuous infusion is recommended. Like epinephrine and all other potent vasoconstrictors discussed here, norepinephrine can cause tissue necrosis if infused through a peripheral IV that infiltrates into surrounding tissue; a central line is recommended for infusion. Some providers will accept the risk of using norepinephrine, dopamine, or phenylephrine on a short-term basis (i.e., during a single anesthetic) through a reliable, visible peripheral intravenous line.
Dopamine
Dopamine is an endogenous catecholamine and an important neurotransmitter in the central nervous system. It is the direct precursor in the biosynthesis of norepinephrine and has a highly dose-dependent effect on cardiac, vascular, and endocrine functions. In addition to beta1 and alpha1 activity, dopamine has specific effects on a group of dopaminergic receptors. Due to its fast metabolism, dopamine has to be given as a continuous intravenous (IV) infusion. In lower dose ranges of approximately 1-3 μg/kg/min, dopamine activates mainly dopamine-1 (D1) receptors that cause vasodilation and increased blood flow in the coronary, renal, and intestinal vascular beds. Higher doses stimulate beta1 (3-10 μg/kg/min) and alpha1 adrenoceptors (>10 μg/kg/min). Dopamine at midrange doses will increase cardiac output by increasing stroke volume, but at higher doses, because of increased peripheral vasoconstriction, high pressure in the aorta may limit even a strong heart’s ability to eject. Like other catecholamines, dopamine has arrhythmogenic properties at high doses. Because of the individual variability of effects of dopamine, the dose should always be titrated to effect.
Synthetic Catecholamines and Phosphodiesterase Inhibitors
Dobutamine
Dobutamine is a selective beta1 agonist and must be given by continuous infusion due to its short half-life. This drug has predominantly beta1 activity. It is used to treat ventricular failure and increases cardiac output by increasing myocardial contractility and heart rate. Doses between 2 and 10 μg/kg/min are commonly used. Lower doses have beta2 effects and can cause additional vasodilator actions, reducing systemic and pulmonary vascular resistance. Compared to dopamine, dobutamine is a coronary vasodilator improving blood flow to myocardium. Its use does not lead to significant increases in vascular resistance even at higher doses as does dopamine. Higher doses of dobutamine commonly cause extra ventricular beats and abnormal fast rhythms, especially in the presence of myocardial ischemia. Similar to other catecholamines, dobutamine will increase the oxygen demand of the heart and is often used in nonexercise cardiac stress testing. Dobutamine and other positive inotropic drugs can be combined with vasodilating drugs to further increase cardiac output.
Isoproterenol
The clinical value of isoproterenol for improving contractility has diminished with the introduction of other inotropic drugs. It is the most potent agonist of beta1 and beta2 adrenoceptors and is primarily used to increase heart rate to overcome heart block. Due to its short half-life (5 minutes), it must be given by continuous infusion (1-5 μg/min) but may be injected intramuscularly or subcutaneously (0.2 mg). A low-dose infusion should be started and titration slowly increased to the desired ventricular rate. Isoproterenol increases heart rate and myocardial contractility while decreasing arterial pressure. These hemodynamic effects can cause large increases in myocardial oxygen demand and decreases in oxygen supply resulting in myocardial ischemia in patients with coronary artery disease.
Milrinone
Milrinone is a PDE inhibitor that indirectly leads to increased intracellular cAMP concentrations in cardiac and vascular smooth muscle. This increases contractility of the heart and causes vasodilation in arterial blood vessels; both together improve output of blood from the heart. Milrinone is frequently used in cardiac surgery to improve ventricular pump function, particularly of the right heart, while simultaneously reducing pulmonary vascular resistance (and right ventricular afterload). In addition, this drug may have a positive effect on diastolic function. Milrinone is usually administered in a loading dose of 50 μg/kg followed by a maintenance infusion of 0.375-0.75 μg/kg/min. It is commonly used in combination with catecholamines. It will potentiate the effect of these agents by blocking the metabolism of cAMP, the concentration of which is increased by stimulation of beta adrenoceptors. Patients should be closely monitored during drug administration and the dose adjusted to hemodynamic or clinical end points, so as to avoid excessive hypotension and cardiac arrhythmias.
Myofilament Calcium Sensitizers
Levosimendan
Levosimendan is member of a new group of inotropes termed myofilament calcium sensitizers. It increases the contractility of muscle cells by binding to a regulatory protein. This allows actin and myosin filaments to contract more quickly and with greater force without increasing the intracellular calcium concentration. Levosimendan also opens adenosine triphosphate (ATP)-sensitive potassium channels in vascular smooth muscle and has PDE inhibition properties, which facilitate vasodilation of coronary, pulmonary, and systemic blood vessels, thereby reducing right and left ventricular afterload. This overall function leads to an increase in cardiac output while minimizing the risk of arrhythmias and with a more favorable balance of oxygen supply and demand as compared to catecholamines and PDE inhibitors. In clinical trials, this drug has been given as a loading dose of 6-12 μg/kg over 10 minutes followed by a continuous infusion of 0.05-0.2 μg/kg/min. Levosimendan is not yet FDA approved but has gained significant use in Europe and Asia.
Vasopressors
Vasopressin
Vasopressin (arginine vasopressin [AVP]) is a hypothalamic peptide hormone released from the posterior pituitary gland when the body senses dehydration. It regulates urine output in the kidney and is a very potent arterial vasopressor, which is mediated through receptors in blood vessel walls. Vasopressin has greater vasoconstrictive effects in the skeletal muscle and splanchnic vasculature and much less in coronary, cerebral, and pulmonary blood vessels. Vasopressin is used clinically to counteract the profound vasodilation in septic shock or catecholamine-resistant, postcardiopulmonary bypass shock. It is also used frequently in small (1-2 U) boluses to treat the dramatic drops in blood pressure that can be seen after induction of anesthesia in patients on some blood pressure medicines. High-dose vasopressin via bolus has now been removed from the 2015 AHA ACLS guidelines as an alternative to epinephrine in cardiopulmonary arrest.
Phenylephrine
Phenylephrine is an alpha1-adrenergic receptor agonist. Clinically, this drug has no positive inotropic activity, in contrast to norepinephrine. Pure vasoconstrictor agents like phenylephrine are best used to reverse hypotension caused by reduced peripheral resistance. This is why, as an anesthesia technician, you will see phenylephrine so often used by anesthesia providers; it counteracts the vasodilating effects of volatile anesthetics and of spinal and epidural anesthetics. Phenylephrine is less potent than norepinephrine, has a longer duration of action, and does not stimulate beta receptors to improve cardiac contractility. It causes greater vasoconstriction in veins than in arteries, and thus, it can increase cardiac output by increased venous return and stroke volume. Large increases in arterial pressure, however, can affect the increase in cardiac output by reflexively decreasing heart rate. All pure vasoconstrictors, including phenylephrine, when given in high doses can cause reduced blood flow to a variety of tissues, resulting in ischemia.
Ephedrine
Ephedrine is an indirect-acting agent and commonly used by anesthesiologists to treat low blood pressure. It causes release of norepinephrine from adrenergic nerve terminals; norepinephrine then stimulates alpha and beta receptors, so that giving ephedrine results in actions very similar to those of norepinephrine. Bolus doses (5-10 mg IV) increase heart rate, blood pressure, and cardiac output similarly but of lesser magnitude as compared to giving epinephrine, and its duration of action is longer, approximately 10-15 minutes.
Antihypertensive Medications
Achieving hemodynamic stability of patients in the perioperative period can be challenging. Perioperative stress through anesthetic or surgical manipulation often contributes to high sympathetic nervous system activity with elevated arterial pressure and heart rate on the day of surgery. Large increases in blood pressures can cause significant morbidity and even mortality in conditions associated with cardiac and cerebrovascular events. Several classes of drugs have antihypertensive properties and can be used to manage blood pressure. These include direct vasodilators, beta-adrenergic blocking agents, and calcium antagonists.
Vasodilator Agents
Nitroprusside
Nitroprusside is a direct vasodilator with a very rapid onset (1 minute) and very short duration of action (1-2 minutes) and should only be given by continuous infusion. Nitroprusside reduces pulmonary as well as systemic vascular resistance and is frequently used when fast and reliable reduction of blood pressure is needed. Although this agent is used to reduce afterload to enhance left ventricular ejection, nitroprusside also reduces preload. Reduction of arterial pressure in chronically hypertensive patients should be done with caution, as a rapid decrease in pressure may lead to ischemia in brain, kidney, or heart. Nitroprusside may cause vascular steal by diverting blood flow away from ischemic areas of myocardium.
Doses from 0.1 to 2 μg/kg/min should be carefully titrated to the desired level of blood pressure and/or other hemodynamic parameter. Nitroprusside is deactivated by light, and thus, the drug infusion reservoir must be adequately protected from light to prevent degradation and loss of potency. The nitroprusside molecule contains cyanide, and cyanide toxicity can develop from the breakdown of the drug, particularly after long-term or high-dose infusions. Cyanide toxicity interrupts intracellular oxygen utilization, leading to tissue hypoxia and acidosis despite high oxygen saturations.
Nitroglycerin
Nitroglycerin directly relaxes vascular smooth muscle and has a greater effect on the venous vasculature compared to the arterial vasculature. This leads to pooling of blood in venous capacitance vessels and subsequent reduction in venous return to the heart with a decrease in right and left ventricular filling pressures. The latter results in a reduction in wall stress during systole and less energy consumption of the heart muscle. Nitroglycerin reduces pulmonary vascular resistance, increases coronary blood flow, and improves perfusion to ischemic regions of the heart. Stroke volume and cardiac output will decrease with lower preload in normal individuals, but in patients with myocardial ischemia, improvement in coronary perfusion can result in increased cardiac output. All these effects make nitroglycerin a drug of choice in the management of chronic heart failure (CHF) and acute myocardial infarction. Higher doses of nitroglycerin can cause a decrease in blood pressure by reducing systemic vascular resistance, which may compromise coronary perfusion. Low doses of this drug in hypovolemic patients can also cause profound decreases in pressure. The onset of action is rapid, and the half-life of nitroglycerin ranges from 1 to 3 minutes. IV bolus doses of 20-100 μg can be used to titrate to effect, while continuous infusions range between 0.1 and 7.0 μg/kg/min and often provide more stable hemodynamic conditions. Nitroglycerin also dilates the cerebral vasculature, which may lead to increased intracranial pressures through increases in intracranial blood volume.
Hydralazine
Hydralazine is a direct vasodilator and has a greater effect on arterial vessels than the venous system. It is mainly used in acute hypertension. Blood vessels in muscle and skin are less affected than coronary, cerebral, and renal vessels. Hydralazine can trigger sympathetic nervous system stimulation by the baroreflex with an increase in heart rate and cardiac output. Compared to other direct vasodilators, the onset of action is relatively slow (5-15 minutes), which may make treatment of acute hypertension more difficult. In addition, hydralazine has a longer duration of action, and therefore, moment-to-moment control of arterial pressure as with nitroprusside is impossible. Finally, the efficacy of hydralazine is considerably less than that of other vasodilators.
Fenoldopam
Fenoldopam is an agonist of dopamine D1 receptors (and to a lesser extent of alpha2-adrenergic receptors) and a rapidly acting vasodilator. It is approved for the management of severe hypertension when a rapid, easily reversible reduction in arterial pressure is warranted. It has strong vasodilator effects on the abdominal and renal arterial vessels and increases renal perfusion and diuresis. In the cerebral circulation, fenoldopam reduces global and regional blood flow.
The half-life of fenoldopam is approximately 5 minutes, and infusion rates are commonly started at 0.05 μg/kg/min and subsequently titrated to the desired blood pressure response with doses up to 1.6 μg/kg/min. The diuretic effect is readily observed at lower doses. Compared to nitroprusside, fenoldopam does not carry as much risk of systemic toxicity especially at high doses.
Calcium Channel Blockers
The ability of cardiomyocytes and vascular smooth muscle cells to contract is directly related to the intracellular concentration of calcium. Calcium enters the cardiomyocyte through special Ca2+ channels, which triggers an additional boost of Ca2+ release from stores in the sarcoplasmic reticulum. During systole, the Ca2+ concentration increases, and during diastole, Ca2+ is pumped back into the sarcoplasmic reticulum enabling cardiac muscle to relax.
Calcium channel blockers reduce the flow of Ca2+ into the cell and in turn cause a much smaller release of Ca2+ from the sarcoplasmic reticulum. All calcium channel blockers produce vasodilation and reduce arterial pressure, which leads to a reduction in left ventricular afterload. Calcium antagonists are used to reduce peripheral resistance in the management of hypertension and to treat cerebral vasospasm after subarachnoid hemorrhage. They also slow conduction and impulse formation in areas of the heart and can be used as antiarrhythmic agents. The various calcium channel blockers show differences in their affinity for vascular smooth muscle and cardiac muscle cells. Nifedipine and nicardipine are much more effective vasodilators than myocardial depressants, while verapamil is used for its ability to slow conduction through the heart and has little effect on vascular muscle tone. Diltiazem has vasodilator action as well as antiarrhythmic effects. In patients with acute heart failure, calcium channel blockers should be avoided due to their negative inotropic effects.
Nicardipine
In clinical practice, nicardipine is a prototypical calcium channel blocker and is very effective in controlling perioperative hypertension, improving coronary perfusion in myocardial ischemia, or treating cerebral vasospasm after subarachnoid hemorrhage. It is associated with less rebound hypertension than other vasodilators, has a short half-life, and can be effectively titrated by continuous IV infusion (1-4 μg/kg/min).
Beta-Adrenergic Antagonists (Beta-Blockers)
Beta-adrenergic blocking agents have a variety of effects on the cardiovascular system including reduction in heart rate, contractility, and myocardial oxygen consumption. Beta-blockers play a role in the treatment of hypertension and have antiarrhythmic properties used mainly to treat atrial (supraventricular) arrhythmias. Due to their effect to slow conduction between atria and ventricles, these drugs can cause severe bradycardia and different severities of AV block (see Chapter 4, Cardiovascular Anatomy and Physiology.) By antagonizing the effects of endogenous catecholamines on beta2 receptors, bronchospasm may be triggered in susceptible patients. The use of beta-blockers has been shown to reduce mortality in a variety of medical conditions, including hypertension, postmyocardial infarction, and CHF, and in high-risk vascular surgery patients.
Beta-blockers differ in their affinity for beta-adrenoceptor subtypes (e.g., beta1 selective), duration of action, coactivity on alpha receptors (combined alpha- and beta-receptor antagonists), and intrinsic ability to stimulate as well as block receptors (partial agonist properties). Beta-blockers are commonly used perioperatively to reduce and/or prevent excessive increases in heart rate.
Metoprolol
Metoprolol is a beta-blocker commonly used in the management of hypertension and coronary artery disease. Like most beta-blockers, it undergoes extensive metabolism in the liver when given as an oral dose (first-pass effect) reducing drug bioavailability. Effective oral doses are much higher (100-200 mg/d) compared to the IV dosing at 2.5-5 mg every 5-10 minutes. With its high affinity for beta1 receptors, this drug carries a smaller risk for bronchospasm in patients with asthma—it is referred to as “cardioselective,” acting only on the heart.
Labetalol
Labetalol has nonselective competitive beta1-, beta2-, and selective competitive alpha1-adrenergic receptor blocking activity. Depending on oral or IV administration, the ratio of alpha to beta blockade is either 1:3 or 1:7, respectively, due to a profound first-pass effect. Labetalol produces a dose-dependent reduction in blood pressure without reflex tachycardia. Doses of 0.25-0.5 mg/kg decrease arterial pressure within 5 minutes, and subsequent doses can be repeated in 5- to 10-minute intervals until the target blood pressure is reached. Cumulative doses of more than 3 mg/kg may be necessary in severe hypertension. The duration of action of labetalol may be up to 18 hours.
Esmolol
Esmolol is a cardioselective beta1 antagonist that has to be administered intravenously. Its popularity is due to a very rapid onset (<2 minutes) and short duration of action (10-20 minutes). Esmolol can be administered by IV bolus or continuous infusion. It is used to suppress the increases in heart rate and blood pressure during intubation, surgical stimulus, or emergence from anesthesia with bolus doses of 50-100 mg (0.5 μg/kg). In the critical care setting, a continuous infusion (50-300 μg/kg/min) of esmolol facilitates rapid control of heart rate and blood pressure (i.e., in patients with aortic dissections or supraventricular tachycardias). The inactivation by red blood cell esterases leads to rapid loss of beta-blocker effect after discontinuation, which can be advantageous if undesirable side effects (hypotension, bradycardia) occur following drug administration. To avoid rebound hypertension and tachycardia, tapering of the dose is usually recommended.
Antiarrhythmic Drugs
Cardiac arrhythmias are not uncommon in the perioperative period but usually only require treatment when they cause problems. Treatment with defibrillation or cardioversion should always be considered if arrhythmias cause the patient to become unstable. Normal conduction of the heart starts at the sinus node and passes through the atria to the AV node, where the speed of conduction is considerably slowed. This allows the atria to contract and relax before the ventricular contraction begins. The conduction continues through the bundle of His, splits into the right and left bundle branches, and spreads the impulse through the ventricle. Any portion of this pathway may be involved in the generation of arrhythmias, and the different classes of antiarrhythmics vary in their effects on this conduction system. The anesthesia provider will usually attempt to determine whether the patient is stable or unstable (i.e., are they supplying enough blood to their organs) and whether the arrhythmia is supraventricular or ventricular. Supraventricular arrhythmias start above the ventricle in the atria or in the AV node and usually (but not always) display normal conduction through the ventricle once the impulse arrives there; that is, the QRS complex, or main spike of the EKG, appears narrow, if it was narrow to begin with. These narrow-complex tachycardias can usually be managed by suppressing the conduction through the AV node with adenosine, beta-blockers, and calcium channel blockers. Ventricular arrhythmias start in the large muscle of the ventricle, below the AV node, and display a “wide-complex” bizarre form on the EKG. It is less common that patients with ventricular arrhythmias will be stable. Amiodarone is now the most common drug used to treat ventricular arrhythmias by reducing the excitability of myocytes.
Adenosine
Adenosine is a naturally occurring nucleotide that can produce total AV block. It is frequently used in the management of paroxysmal supraventricular tachycardias and can terminate the tachycardia or transiently slow the ventricular response. Adenosine can also help to differentiate the origin of narrow- or wide complex tachycardias. In the case of supraventricular origins, the ventricular response rate is reduced, but in tachyarrhythmias arising in the ventricle, the heart rate remains unaffected. Atrial fibrillation and atrial flutter are not AV node dependent, and they are not affected by adenosine.
The half-life of adenosine is extremely short, and it is administered in a bolus. For termination of supraventricular arrhythmias, 6 mg adenosine is injected intravenously. The onset of action is within 20 seconds and lasts for approximately 10 seconds. Conduction at the AV node halts, and there may be a long pause in the EKG, after which conduction resumes. This long pause is the expected response but can be alarming if you are not prepared for it. Awake patients often experience headache. A second dose of 12 mg can be given 1-2 minutes later if the first dose is ineffective.
Verapamil
Verapamil is a calcium channel blocker and is effective in controlling supraventricular tachycardia by slowing AV node conduction. It is also used to reduce the ventricular rate in atrial fibrillation or flutter. A slow IV injection of 75-150 μg/kg followed by a continuous infusion of 5 μg/kg/min is used for the management of supraventricular arrhythmias.
Amiodarone
Amiodarone plays a major role in the treatment of acute life-threatening supraventricular and ventricular arrhythmias but is also utilized in the chronic management of atrial fibrillation. It influences the cardiac conduction system in several ways. It can acutely cause vasodilation and myocardial depression, and vasopressor and/or inotropic support may be required during the initiation of therapy. Amiodarone is administered in a bolus dose of 150 mg (over 10-30 minutes), followed by a continuous infusion of 1 mg/min for 6 hours and 0.5 mg/min thereafter (1 g/d). An IV bolus dose of 300 mg is recommended in cardiac arrest with ventricular fibrillation that has not responded to defibrillation. Amiodarone accumulates extensively in tissue due to its high-lipid solubility, which leads to an elimination half-life of approximately 58 days. Patients with higher degrees of AV block or sinus bradycardia should not receive amiodarone unless they have an implanted pacemaker.
Lidocaine
Lidocaine is a local anesthetic that blocks sodium channels both in nerves and in the heart, including myocytes of the atrium, the ventricle, and the Purkinje pathway. Sodium channel blockade reduces the slope and amplitude of the cardiac action potential (phase 0; see Chapter 4, Cardiovascular Anatomy and Physiology). This ultimately leads to a reduction in conduction velocity throughout the myocardium (negative dromotropy) and limits reentry arrhythmias. Lidocaine is mainly used in the treatment of ventricular arrhythmias including hemodynamically significant premature ventricular contractions and ventricular fibrillation before weaning from cardiopulmonary bypass. It is administered as an IV bolus (1 mg/kg) followed by a continuous infusion (1-4 mg/min). For the treatment of refractory ventricular fibrillation, a second bolus can be administered although a cumulative dose of 3 mg/kg should not be exceeded. Compared to amiodarone, the routine use of lidocaine for ventricular fibrillation in the prehospital setting is associated with a higher mortality rate.
Atropine and Glycopyrrolate
Atropine and glycopyrrolate are competitive antagonists of the muscarinic acetylcholine receptor and belong to the anticholinergic drug class. Increased activity of the parasympathetic nervous system (see Chapter 13, Autonomic Nervous System) is one of the primary causes of intraoperative bradycardia, and resting activity of the vagal nerve is particularly high in young adults, resulting both in bradycardia and in heart rate variability. The parasympathetic blocking properties of atropine make it very effective in the treatment of bradycardia caused by perioperative vagal responses. The ACLS protocol for treatment of pulseless electrical activity (PEA) and asystole has recently been revised, and atropine was removed from the guidelines as it had not been shown to be effective. Atropine increases the activity and firing rate of the sinoatrial node and accelerates conduction through the AV node, increasing heart rate. The dose for IV atropine is usually 0.5-1 mg, which may be repeated every 3 minutes up to a total dose of 3 mg. Glycopyrrolate has the same mechanism of action but is not an emergency drug as it has a much slower onset of action (2-4 minutes). It is often used to treat asymptomatic bradycardia intraoperatively. Both drugs will increase myocardial oxygen consumption due to increased heart rate, which limits their use in patients with coronary artery disease.
Summary
Anesthesia providers often use medications to optimize the function of the cardiovascular system. Medications are most often used to reduce or increase afterload (vasodilators and vasopressors), increase inotropy, or treat arrhythmias. Anesthesia technicians are often called upon to assist with the setup and administration of these medications. This chapter provides a brief introduction to the properties of common cardiovascular medications used in the operating room.
Review Questions
1. Drugs that increase the force of myocardial contraction are called
A) Local anesthetics
B) Beta-blockers
C) Antiarrhythmic agents
D) Positive inotropic agents
E) None of the above
Answer: D
Drugs that increase the force of myocardial contraction are called positive inotropic agents and include catecholamines, PDE inhibitors, and myofilament calcium sensitizers. Stimulation of beta receptors increase inotropy, increase heart rate, and cause bronchodilation. Blockade of these receptors can decrease inotropy (beta-blockers are negative inotropes). Local anesthetics are used to block nerve conduction, but they can have antiarrhythmic properties (suppress abnormal heart rhythms).
2. Epinephrine may be used in anesthesia for all of the following effects EXCEPT:
A) Constrict bronchial smooth muscle
B) Constrict blood vessels
C) Increase heart rate
D) Increase myocardial contractility
E) Prolong the action of local anesthetics
F) Treat cardiac arrest
Answer: A
Epinephrine is a powerful alpha- and beta-receptor agonist. The beta2 receptors on smooth muscle cause relaxation. Epinephrine relaxes bronchial smooth muscle and can be used to treat bronchoconstriction. The beta receptors on the heart cause increased contractility and heart rate. The alpha receptors in vascular smooth muscle cause contraction (vasoconstriction); thus, epinephrine increases blood pressure. When injected in peripheral tissues, epinephrine causes local vasoconstriction and can prolong the action of local anesthetics by decreasing their uptake back into the circulation. Epinephrine is the first drug for use in the ACLS algorithm after CPR and defibrillation, regardless of the suspected cause of cardiac arrest.
3. Use of dobutamine can increase oxygen demand of the heart.
A) True
B) False
Answer: A
4. Vasopressors
A) Cause vasoconstriction
B) Include phenylephrine, ephedrine, and vasopressin
C) Cause vasodilation
D) Include nitroprusside, nitroglycerin, and nicardipine
E) A and B
Answer: E
Vasopressors cause vasoconstriction and include phenylephrine, ephedrine, and vasopressin. Vasodilators include nitroprusside, nitroglycerin, and nicardipine.
5. Beta-blockers can be used to treat
A) Tachycardia
B) Low blood pressure
C) Bronchospasm
D) Acute heart failure
E) All of the above
Answer: A
Beta-blockers can block beta1, beta2, or both types of receptors. Blockade of these receptors will slow the heart rate and would be useful in the treatment of tachycardia. Because beta blockade decreases contractility and slows heart rate, beta-blockers can decrease blood pressure and would not be used to treat low blood pressure or severe acute heart failure.
SUGGESTED READINGS
De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362(9):779-789.
Dellinger RP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36(1):296-327.
Dyer RA, Reed AR, van Dyk D, et al. Hemodynamic effects of ephedrine, phenylephrine and phenylephrine co-administered with oxytocin, during spinal anesthesia for elective cesarean delivery. Anesthesiology. 2009;111:753-765.
Hensley FA, Martin DE, Gravlee GP. A Practical Approach to Cardiac Anesthesia. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006.
Klabunde RE. Cardiovascular Physiology Concepts. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
Neumar RW, Shuster M, Callaway CW, et al. Part 1: executive summary: 2015 American Heart Association guidelines update for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2015;132 S315-S367.
Pagel PS, Haikala H, Pentikainen PJ, et al. Pharmacology of levosimendan: a new myofilament calcium sensitizer. Cardiovasc Drug Rev. 1996;14:286-316.
Petersen JW, Felker GM. Inotropes in the management of acute heart failure. Crit Care Med. 2008;36(1 suppl):S106-S111.
Rodriguez MA, Kumar SK, DeCaro M. Hypertensive crisis. Cardiol Rev. 2010;18:102-107.
Stoelting K, Hillier S, eds. Pharmacology & Physiology in Anesthetic Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006.
Thompson A, Balser JR. Perioperative cardiac arrhythmias. Br J Anaesth. 2004;93(1):86-94.