AS WAS DISCUSSED IN CHAPTERS 5 AND 6 and will later be talked about in chapter 13, bacteria, plants, and animals have been genetically modified. This means that donor genes first had to be identified. Also, to properly diagnose diseases, it is necessary to pinpoint the defective gene. Identifying defective genes may then help with conventional therapy, as well as gene therapy. In order to accomplish these goals, genes first must be located, that is, their location on chromosomes be determined and their sequences deciphered.
We now know that higher organisms contain many thousands of genes. For example, the fruit fly harbors 16,000 genes, while humans have about 35,000. As a step in the identification of a gene, we need to determine the position of these genes in relation to other genes on the chromosome. We will now see how it is possible to map genes on chromosomes, thus locating and isolating genes of interest.
Chapters 2 and 3 described the inheritance pattern of genes located on the X chromosome. As we saw, it is the fact that sons always get their X chromosome from their mothers that allows the localization of several genes to that chromosome. Remember that we studied three such genes, the genes for hemophilia and color blindness in humans, as well as the gene for eye color in fruit flies. Very soon after Morgan discovered the behavior of sex-linked genes in fruit flies, he found that other genes were also located on the X chromosome. These included genes determining body color and wing size. Clearly, several genes are located on the X chromosome. This holds true for all the other chromosomes. Also, since the fruit fly has four pairs of chromosomes and 16,000 genes, one can calculate that each chromosome must carry thousands of genes. Humans have twenty-three pairs of chromosomes. Given that we have about 35,000 genes, the average number of genes per chromosome is about 1,500. Of course, long chromosomes are expected to carry many more genes than short ones.
Let us now consider two genes, located on different chromosomes, and see how these genes behave in a cross. This situation is a bit more complex than crosses involving a single gene existing in two forms, as we studied in chapters 2 and 3. To illustrate what happens, let us use traits that we mentioned already.
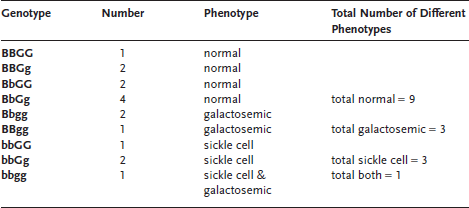
For example, let us see what genotypes of offspring a man heterozygous for sickle-cell anemia, who is also carrier of the galactosemia gene, can have with a woman who is also heterozygous for sickle-cell anemia and a carrier for galactosemia. Remember that both conditions are recessive so both parents are phenotypically normal. Let us call B the normal β-hemoglobin gene, and b its sickle cell counterpart. Further, let us use g for the galactosemia gene and G for the normal gene. The genotypes of both parents are thus BbGg. They will not show any sign of disease since they are heterozygous for both traits. We learned in chapter 2 that single sets of chromosomes are present in gametes. It is also known that the genes for sickle-cell anemia and galactosemia are located on different chromosomes. What kinds of gametes will these parents produce?
Each gamete has one set of chromosomes. So each gamete will have one copy of the hemoglobin gene and one copy of the “galactosemia gene,” either in its normal or abnormal form. Thus we can observe gametes with the gene combinations: BG, Bg, bG, and bg. These genes follow the third law of genetics, the law of independent assortment. This law states that genes located on different chromosomes assort independently. In other words, the B gene does not preferentially associate with the G gene. Rather, B can just as easily associate with g as G. In our example, the mother and the father will produce gametes having the same four chromosomal compositions; this is because they are both heterozygous for both traits. We can now build a Punnett square to see what kinds of offspring these two parents can have. In this case, since we have four possible gametes for each parent, the Punnett square will contain sixteen boxes. Remember that if only two contrasting genes are considered, the Punnett square contains only four boxes. Figure 9.1 shows that when two pairs of contrasting genes are studied, the situation becomes more complicated. Note that some gene combinations (genotypes) are represented more than once in this Punnett square. By careful counting, we see that nine different genotypes are possible (see table 9.1). Then, taking into account that B is dominant over b and that G is dominant over g, we can calculate the probability of individuals with various phenotypes. The individuals who are BBGG, BBGg, BbGG and BbGg will have the same phenotype: no symptoms of sickle-cell anemia and no symptoms of galactosemia. Next, the individuals who are BBgg and Bbgg will show no signs of sickle-cell anemia, but will be galactosemic. Then, those who are bbGG and bbGg will have sickle-cell anemia, but no galactosemia. Finally, the individuals with the bbgg genotype will unfortunately suffer from both diseases. Thus, crosses involving two pairs of contrasting genes produce nine different genotypic categories and four different phenotypic categories.
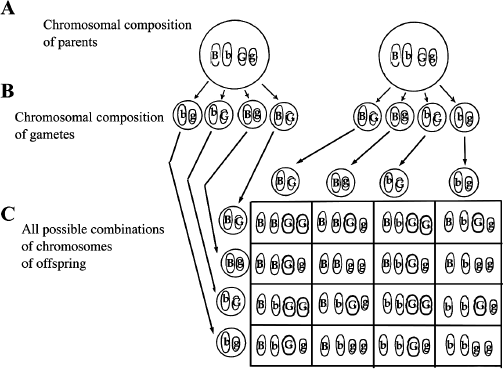
Figure 9.1 Independent Assortment of Two Pairs of Genes. The sixteen possible gene combinations observed in a cross involving two different pairs of genes located on separate chromosomes. A. The genotypes of the parents are shown with the genes located on separate chromosomes. Only the two pairs of chromosomes are shown for simplicity. B. The gene combinations of gametes are shown. C. A Punnett square shows all sixteen possible gene combinations of the cross.
Table 9.1 All Possible Genotype Combinations Produced by Parents with Genotype BbGg
By carefully counting the boxes in the Punnett square that contain different genotypes, we realize that the four phenotypic categories come in a 9:3:3:1 ratio. Nine out of sixteen boxes (~56 percent) represent the probability of being phenotypically normal for both traits. Three out of sixteen boxes (~19 percent) fall in the category where the first phenotype is normal (no sickle-cell anemia) but the other is not (galactosemia is present). Next, three out of sixteen boxes (~19 percent) represent the probability for an offspring to have sickle-cell anemia but not galactosemia. Finally, there is a 1 in 16 probability (~6 percent) that double heterozygous parents will have an offspring with both sickle-cell anemia and galactosemia.
This is different from a cross involving single genes (existing in two forms), where only three possible genotypes and two phenotypic categories are observed (see chapter 2). There, remember that the phenotypic ratio is 3:1. As you can well imagine, things get even more complicated if three pairs of genes are considered. In this case, the Punnett square contains sixty-four boxes, and there are eight possible phenotypic classes. In the human case, forty-six chromosomes (twenty-three pairs) segregate and assort independently during meiosis. This means that 223 possible phenotypic classes are potentially generated from each offspring, simply from the independent assortment of chromosomes into the gametes! You can now understand why each human is unique. Identical twins are an exception to this rule because they originate from the splitting of a single fertilized egg. For an example of independent assortment, see the “Try This at Home” at the end of this chapter.
We have already used the term “linked” to refer to genes located on the sex chromosome. These genes are called “sex-linked.” We saw at the beginning of this chapter that, given the large number of genes in an individual and given the limited number of chromosomes present in that individual’s cells, each chromosome must carry a large number of genes. When genes are located on the same chromosome, they are said to be linked. In the example above, which uses sickle-cell anemia and galactosemia as a pair of contrasting traits, the genes are on different chromosomes and are thus unlinked.
Gene linkage was elucidated in the laboratory of Thomas Hunt Morgan at Columbia University. As we explained before, Morgan generated many types of mutant fruit flies and crossed them. One cross involved flies with mutant wings, symbolized by r (the normal wing is represented by R), and black body indicated as b (B indicates again the dominant, normal body-color gene). Since mutant wings and black bodies are both recessive, these flies must be homozygous for the trait, and their genotype is thus rrbb. They were crossed with normal flies of genotype RRBB. Remember that normal is dominant over mutant. All the offspring of this cross are double heterozygotes, RrBb, and thus are phenotypically normal. Then, Morgan crossed these double heterozygous offspring. This cross, written RrBb x RrBb, involves two different pairs of traits, equivalent to the example with sickle-cell anemia and galactosemia in humans that we saw above.
Thus, in the offspring of his cross, Morgan should have observed four phenotypic classes: normal wings and body color, normal wings and black bodies, mutant wings with normal body color, and mutant wings with black bodies. Furthermore, these four categories should be produced in 9:3:3:1 proportions (56 percent, 19 percent, 19 percent, 6 percent) following the rule of independent assortment. Morgan did observe four categories, but the numbers of flies in each category were completely off! He observed 74 percent normal, only 1.5 percent each of just mutant wings or just black body, and 22 percent that had both mutant wings and black body. That is, he observed many more totally normal flies as well as flies with mutant wings and black body (rrbb) than expected. Further, he observed extremely low numbers of flies that had normal wings with a black body, or mutant wings with normal body color.
What can account for this gross deviation from the expected proportions of different phenotypes that we expect from the rules of independent assortment? Morgan correctly interpreted these results: he hypothesized that the R and B genes were on the same chromosome from one of the original parents. Similarly, the r and b genes were also located on the same chromosome, this time the one from the other original parent (figure 9.2). It is because the genes Morgan studied were not assorting independently (i.e., were not on different chromosomes) that a 9:3:3:1 ratio was not observed. Genes that do not assort independently must be on the same chromosome; they are linked.
If the two genes are on the same chromosome, figure 9.2 predicts that we should have gotten 75 percent flies with normal body color and normal wings (RRBB or RrBb), and 25 percent flies with mutant wings and black body (rrbb). We should not have gotten any flies that had just black bodies or mutant wings. So, how can we account for the small but significant numbers of flies that had just black bodies or mutant wings? The reason is that chromosomes can break and rejoin and, in the process, shuffle genes. This process is called “recombination.”
During meiosis, chromosomes come into close physical contact. This can lead to chromosome breakage and rejoining. In this process, an overlapping chromosome breaks at the point of contact with another chromosome, and the two ends of the broken chromosomes can swap positions. Usually, chromosomes break and join at the same position so there is no gain or loss of genes in this process. But the result is that genes present on different copies of the same chromosomes can get shuffled. Let’s take the above example of the mutant wings and black bodies of fruit flies. First, figure 9.2 shows the result in a situation where chromosomes do not break and rejoin. Here, the R-B and r-b genes stay together and only two phenotypic classes are observed: 75 percent of the flies have normal wings and normal body color, whereas 25 percent of the flies have mutant wings and black body color. No recombinants are seen. Morgan did not obtain these numbers because he did observe recombinants. Let us now imagine that these chromosomes, one with the R and B genes, and one with the r and b genes, break and rejoin between R and B and between r and b (figure 9.3). You can see that the R gene, originally linked to the B gene, is now associated with the b gene. Similarly, r is now associated with B. These new gene combinations are due to chromosome recombination and the resultant chromosomes are called recombinants. Individuals with the recombinant chromosomes are also called recombinants.
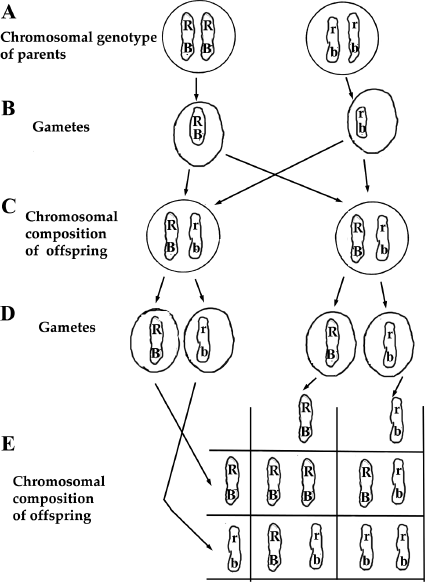
Figure 9.2 Inheritance of Two Pairs of Genes Located on the Same Chromosome. A. The genotype of double-homozygous parents are shown with both the eye color genes (R and r) and wing gene (B and b) on the same chromosome. B. Each parent produces only one type of gamete since they are homozygous. C. The offspring of the cross produces a double heterozygote, but note that the dominant form of the two genes are on the same chromosome and the recessive form of the two genes are also on the same chromosome. D. The double heterozygote offspring in this linked case produces only two types of gametes, not four types as seen in figure 9.1.E. There are only three different genotypes of offspring resulting from the double heterozygote parents if the genes are tightly linked on the chromosome.
How often does recombination occur? It is the distance between genes along the chromosome that determines how often they recombine. You can imagine that breakage anywhere between the R/r and B/b genes will result in the new combination shown in figure 9.3. Thus the frequency of recombination depends roughly on the distance between R/r and B/b genes. For example, if the two genes are at the opposite ends of the chromosome, the chances of recombination are high. In that case, there will be many recombinants. If, on the contrary, the two genes are very close together, chances of recombination between the two genes are small. In Morgan’s experiment, the distance between the two genes, R/r and B/b, was pretty close: he observed only 3 percent recombinants.
By determining the percentage recombination of linked genes in the offspring of a cross, we can map genes onto chromosomes. With fruit flies, if appropriate crosses are made, scientists can decide whether the genes are unlinked. This is the case when they observe a phenotypic ratio expected for independent assortment, for example a 9:3:3:1 ratio. If the ratio is significantly different from this expected ratio, they can conclude that the two genes are on the same chromosome, linked. Next, if two genes are linked, the proportion of individuals with phenotypes resulting from recombination is a measure of how far apart the genes are located on the chromosome. In fact, a standard unit of distances along chromosomes is the recombination percentage, named centimorgans after T. H. Morgan. We can thus establish a chromosome map giving the relative positions of many genes, their distances measured as the amount of recombination observed between them.
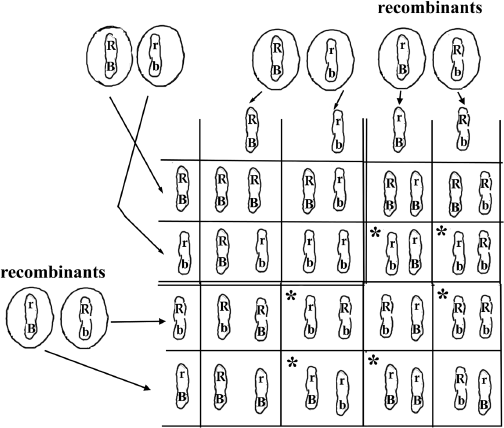
Figure 9.3 Breaking and Rejoining the Chromosomes Shown in Figure 9.2. The Punnett square shown in figure 9.2.E is expanded to include the chromosomes resulting from recombination. The original Punnett square is shown on the upper left enclosed by double lines. The recombinant chromosomes are added to the Punnett square. Those boxes marked by asterisk (*) represent the phenotypic classes not seen in the figure 9.2. Because the recombinant chromosomes represent only 6 percent of the chromosomes, the numbers of flies with the * phenotype are small.
We see that genes can be mapped on the chromosomes of Drosophila because we can do many crosses between flies with different genetic mutations. Then we can analyze hundreds of their offspring to see if the numbers correspond to ratios for independent assortment or not. In humans, we cannot do this. How do we measure linkage in humans?
Since gene mapping with crosses by linkage is generally not feasible in humans, how do we map human genes? One technique used to find the gene corresponding to a disease is called restriction fragment length polymorphism, or RFLP for short. The first word, restriction, refers to the first step in the process, which is to cut the DNA with restriction enzymes (see chapter 5). Let’s consider what happens to our DNA when we cut it with a restriction enzyme. A helpful analogy is to imagine a large document like an encyclopedia, though printed on a continuous roll of paper. Now, our “restriction enzyme” would be like using scissors to cut every place where there was a particular sequence of letters, for example the sequence of letters in “tem.” Then we would cut at words like temporary, stem, system, and so on. How many and what sizes of pieces do we get? In some articles of the encyclopedia, there may be many small pieces, but there may be long regions with no cuts and a wide range of sizes in between the small and the large pieces. With DNA, we would also get a range of sizes of fragments from small to large when we cut using a restriction enzyme that cuts DNA at a particular base sequence. So this is what FL in RFLP refers to, the fragment lengths created by cutting our DNA with restriction enzymes. Would a given restriction enzyme cut everybody’s DNA at always the same place and thus produce DNA pieces of the same size? The answer is no because individuals have differences in their DNA sequence. Therefore, when one cuts DNA with restriction enzymes, different individuals, even close relatives, produce different sizes of DNA pieces. The word polymorphism means “many forms,” in this case, many DNA fragment lengths. Thus we say that the lengths of our DNA fragments are polymorphic. But our genome contains billions of nucleotide base pairs, so when we cut our DNA with a restriction enzyme, there are hundreds of thousands of different sizes of DNA. If we now separate these DNA fragments using gel electrophoresis (recall that we used gel electrophoresis to separate DNA by size in chapter 1), we would not see bands of DNA of specific sizes but a smear of DNA running from the largest to the smallest fragment lengths. So even though different individuals have their DNA cut at different positions, because the DNA sequence is a little different between even closely related individuals, we would not be able to distinguish them by analyzing the cut DNA with gel electrophoresis alone.

Figure 9.4 A Portion of the β-hemoglobin Gene with Codes for Normal and Sickle-cell Traits. A. This portion of the normal hemoglobin gene contains a restriction enzyme site, which is underlined. B. The same region shown in A for the hemoglobin of an individual with sicklecell anemia. The asterisk shows the single nucleotide mutation. This mutation makes the restriction enzymes unable to recognize the site.
So how do we tell the difference between individuals? Remember from chapter 1 that DNA is a double helix held together by complementary base pairing. Recall also that we can now make single-stranded DNA molecules of a specific base sequence in the test tube and use them as primers for polymerase chain reaction. We can use these tools, restriction enzymes and DNA with specific sequences, to distinguish between the DNA from two different individuals. Let’s take the case of sickle-cell anemia. A short region of the DNA encoding normal β-hemoglobin is shown in figure 9.4.A. The underlined region indicates a restriction-enzyme site, a place that will be cut by a certain restriction enzyme. The mutation in sickle-cell anemia is a single nucleotide change indicated by an asterisk in figure 9.4.B. Because the base change occurs within the restriction enzyme site, the restriction enzyme no longer recognizes this sequence and so will not cut the mutant DNA at this position. The change in the cutting pattern of DNA is illustrated in figure 9.5.A. This is an example of a single-nucleotide polymorphism (abbreviated SNP) because only a single nucleotide was changed.
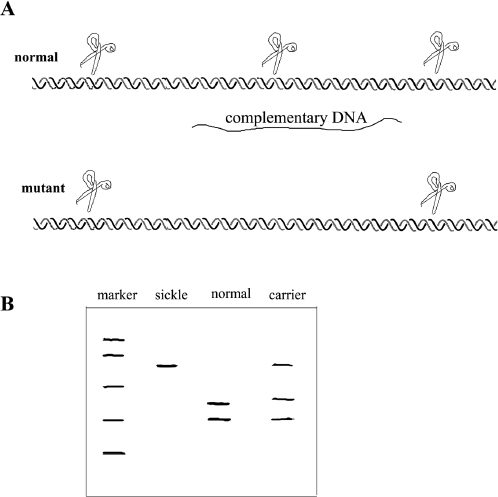
Figure 9.5 Restriction Fragment Length Polymorphism of Sickle-cell Anemia. A. Double strands of DNA are depicted with the positions of restriction enzyme cuts marked with scissors. The upper DNA shows a small portion of the normal β-hemoglobin gene from an individual with normal β-hemoglobin; below it is the same section from an individual with sickle-cell anemia. Note that a restriction site is now missing due to the mutation. The strand of complementary DNA is positioned between the two DNA double strands. B. A gel electrophoretic analysis of DNA from a sickle-cell anemia patient. The marker lane contains size markers; the sickle lane shows the long single fragment found in the mutant. The normal lane shows the two smaller bands of DNA corresponding to the two DNA fragments created by the restriction enzymes. The carrier lane, as expected, has bands representing both the mutant and the normal DNA.
Figure 9.5.B shows the gel electrophoresis pattern of several individuals, two of whom have at least one gene for sickle-cell anemia. The specific bands are differentiated from the background DNA smear because they can bind to the “complementary DNA” probe specific to a region of the β-hemoglobin gene. The complementary DNA can be labeled with a dye for easy detection. No other bands present in the DNA smear binds the complementary DNA, and so they remain invisible. Another example of this technique is shown in box 9.1.
In the sickle-cell anemia example, the defect is a specific change in the DNA sequence of a specific gene, the β-hemoglobin gene. This means that the band of DNA in RFLP is always associated with a sickle-cell defect. However, most often we do not know what gene or what defect in a gene is associated with a disease.
Restriction fragment length polymorphism allows us to identify the piece of DNA associated with a genetic disease. In this procedure, the DNA of affected and unaffected individuals are cut with the same restriction enzyme. If we arranged these pieces by size, we would get a large range, from small to large, and there would not be a distinct group of pieces of a particular size. Recall from chapter 1 that gel electrophoresis is used to separate pieces of DNA by size: if too many pieces of many different sizes are run together, the resulting gel would show the DNA spread out in a smear.
So how can we tell where a particular piece of DNA is in this smear? Recall that DNA has a double-helical structure held together by complementary base pairs. We can make a short piece of DNA with a specific sequence and label it with a dye. After the DNA is cut and put on a gel and separated by size, we can transfer these pieces of cut DNA onto a sheet that prevents them from floating away. By using appropriate chemicals, we can then separate the double strands of the cut DNA pieces. At that point, the specific short piece of labeled DNA is added and allowed to bind to the complementary sequence present somewhere among the cut pieces of DNA on the sheet. Recall from chapter 1 that short pieces of DNA find the complementary partner faster than large pieces. After that, we wash away any labeled DNA that has not bound to its complementary sequence. The band identified by the labeled DNA is a piece of DNA that has within its length a sequence complementary to the labeled DNA. The position of the labeled band determines the size of the DNA piece and how far it moved in the original gel electrophoresis.
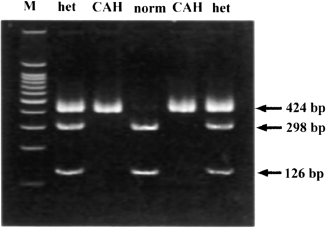
Figure B.9.1 RFLP to Identify Congenital Adrenal Hyperplasia. Gel electrophoresis of DNA from normal individuals (norm), those that are heterozygous (het) for congenital adrenal hyperplasia, and congenital adrenal hyperplasia patients (CAH). M provides size markers used to determine the size of the bands shown on the right as numbers of base pairs (bp). Note that both affected individuals (CAH) have a single band at 424 base pairs whereas normal individuals are missing that band and have instead two bands at 298 base pairs and 126 base pairs.
Now, we can treat this band of DNA on the gel as a genetic trait in the same way we view a person’s blood type. Figure B.9.1 shows an example of identifying the genetic defect for congenital adrenal hyperplasia. Congenital adrenal hyperplasia is a recessive disease caused by a defect in an enzyme important for salt balance. Infants with this disease can develop life-threatening dehydration or shock. This disease is treatable with hormones.
Even in cases where the disease mutation itself does not produce different banding patterns in RFLP, this technique can help track down the gene responsible for a disease. This is because a special banding pattern may be associated with a disease. Why might this be? This would be the case if a particular DNA sequence is closely linked to the disease gene, that is, located close to the disease gene on the chromosome. Recall that recombination between genes is caused by the breaking and rejoining of chromosomes. The recombination frequency depends upon the distance between the two genes. So we can consider a band on a gel as a gene. If the rate of recombination between a particular DNA sequence and the actual defective gene that causes the disease is low, the distance between them must be very short, that is, that piece of DNA must be close to the disease gene. By this logic, one can find a fragment of DNA that is close to the defective gene. The closer the piece of DNA represented by the restriction fragment, the more likely that piece will be inherited together with the defective gene. Since the restriction fragment can be detected by gel electrophoresis, we can locate the defective gene closely associated with it. Researchers analyze the DNA of large families in which the disease exists; affected family members’ DNA should show a higher chance of being associated with a specific DNA sequence close to the disease gene, whereas those of unaffected members should not. It is by this technique that scientists have closed in on genes responsible for many human genetic diseases.
The most interesting and important genes, such as disease-causing genes in humans or disease-resistance genes in plants, are often the most difficult to identify because we do not know what proteins they code for or what their function might be. Cloning a gene is the first step toward learning how it functions and using it to suit our needs.
Stem rust is a fungal disease in barley that routinely reduced crop yields until the early 1940s. In a particularly bad epidemic of the disease in 1935, Sam Lykken, a farmer from Kindred, North Dakota, identified a single healthy plant in his field, which was otherwise totally decimated by stem rust. This single healthy plant was saved and became the source of resistance genes in barley strains. It turns out that this strain of barley contained the dominant stem rust–resistance gene that has since been given the name “Rpg1.” The presence of this gene has prevented any significant losses due to this disease since then.
To learn more about the stem rust–resistance gene in barley, a team of scientists led by Andris Kleinhofs at Washington State University in Pullman, Washington, set out to clone this gene. How can one identify this gene or other genes that are known only by their phenotype? We can look for DNA markers or bands on gels linked to the trait. The barley genome is huge; thus a gene can be hundreds of thousands of base pairs away from the DNA marker to which it is linked! So the scientists put large pieces of the barley genome into bacteria in such a way that each bacterium contained a portion of the barley genome. Recombinant DNA molecules like these are called bacterial artificial chromosomes, or BAC clones. One can identify the BAC clone of interest with the DNA markers linked to Rpg1. Because the BAC clones contain random pieces of the genome, if one has enough of them overlapping pieces can be found. Chromosome walking is a process used to follow the sequence of DNA in overlapping pieces of DNA like BAC clones. Once a number of overlapping DNA clones including the gene of interest are found, they are sequenced to identify potential genes and candidates for the gene of interest (figure B.9.2.A)
We say candidate genes at this point in the process because we still do not know which gene is the one responsible for disease resistance. Searching among the candidate genes to identify the resistance gene is difficult. The DNA sequences must be compared between resistant and susceptible strains to identify which gene is consistently different between them. If a particular gene always shows a difference, this is strong evidence, but not proof, that the correct gene has been identified. Kleinhofs’s team used this method to identify the Rpg1 gene, but they also transformed barley with the Rpg1 gene. If a gene is indeed the one that confers resistance to stem rust, the transformed barley should be resistant. Indeed the transformation of a susceptible barley strain with the Rpg1 gene conferred resistance to the stem-rust pathogen! Thus Rpg1 is truly the stem rust–resistance gene! Interestingly, the transgenic plants were more resistant to stem rust than the original resistant strain from which the gene was isolated (figure B.9.2.B). Though this was an arduous process that took years, the identification of the resistance gene by the Kleinhofs’s team allows researchers to understand disease resistance better and to use this knowledge and this gene for agricultural purposes.
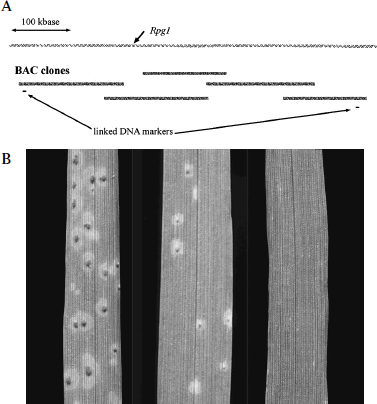
Figure B.9.2 Identifying the Disease-Resistance Gene in Barley. A. A diagram illustrating map-based cloning. Over half a million base pairs of the barley genome containing Rpg1, the stem rust–resistance gene, are shown at the top. The two DNA markers linked to Rpg1 are shown below the bacterial artificial chromosomes (BAC clones) that contain this DNA sequence. Additional contiguous BAC clones used to “walk” to the disease resistance gene are shown in dark mottled lines. B. Photographs of barley leaves treated with spores of the fungus that causes stem-rust disease. On the left is the susceptible strain, in the middle is the resistant strain, and on the right, a susceptible strain transformed with the resistance gene Rpg1. Note that the transformed strain shows no evidence of rust, and is thus more resistant than the original resistant strain. Photo courtesy David Hansen.
The procedure outlined above for mapping a disease gene on a chromosome is quite involved. Because of this, a major international scientific collaboration to sequence the whole human genome was set up. Sequencing the 3.15 billion base pairs of the human genome was accomplished in 2001. With this information in hand, finding and cloning the gene for a disease should be greatly facilitated. Indeed, after sequencing the DNA fragment associated with the disease gene, powerful software can scan the whole human DNA sequence and precisely position that fragment. By so doing, researchers can determine the sequence of the disease gene located next to the fragment used to probe the DNA.
DNA sequencing is largely automated today. First, DNA is isolated from an organism, broken into small fragments, and cloned in a plasmid vector. The sequences of these fragments are determined using chemicals that react with the four bases, and the sequences of the fragments are arranged in a linear fashion using powerful computer software. Bacterial genomes that consist of a few million base pairs can be assembled in a few months. Of course, it takes longer to sequence larger genomes. An interesting finding of the Human Genome Project was that most of the human DNA does not contain genes. Indeed, over 95 percent of human DNA does not code for proteins. This also holds true for other animals whose DNA has been sequenced. The function of noncoding DNA is not entirely clear. Much of it consists of simple sequences repeated thousands of times. It goes without saying that spotting human genes in a mass of DNA is not an easy task. This work is still in progress. To date, it is estimated that humans have about 35,000 genes, compared to 16,000 in the fruit fly and 19,000 in the simple nematode worm Caenorhabditis elegans.
Other genome projects aim at sequencing the fruit fly, mouse, rat, and chimpanzee genomes. Examples of plant genomes sequenced or with sequencing in progress are rice and Arabidopsis, a small plant with a small genome. Genomes of medical importance, such as that of the malaria parasite have been sequenced. Comparison of genes sequenced in humans and fruit flies have identified hundreds of genes that are so similar between them that scientists can use fruit flies to investigate genes implicated in human genetic diseases. Mice and rats are often used as model systems for the study of human diseases. Therefore, knowing their DNA sequence will facilitate the study of disease genes in animal models. Knowledge of the chimpanzee genome may tell us what genes make us different from our closest cousins.
One of the goals of the Human Genome Project is to determine which genes make us prone to disease. This does not mean the types of genetic diseases that are well understood (such as PKU and others) that we described in chapter 3. Rather, we are referring to genes that make some of us more susceptible to certain diseases, such as bacterial infection, high blood pressure, and cancer. These diseases and many others involve many genes as well as environmental factors.
Now that the human genome has been deciphered, it should be possible to identify disease-susceptibility genes and attribute a function to them. This process is called “annotation.” It is not enough to know the 3.15-billion base-pair sequence of the human genome if the genes contained in this sequence cannot be identified. Annotation is not an easy task, especially when you know that more than 95 percent of the human genome does not contain any genes. How does one discover genes in all that sequenced DNA?
One way to go about this is to compare the human genome with other, similar sequenced genomes. Mice have been used for a long time in genetic research, and many mutants, including behavioral and neurological mutants, have been created. In many cases, these mutations have been mapped precisely, and the genes involved have been located. Thus, once the mouse genome is completed, researchers will be able to annotate human genes based on similar locations on chromosomes and similar genes in mice. However, there may be better model than the mouse for annotating human genes.
Quite recently, investigators have sequenced the genome of the puffer fish, Fugu rubripes. It turns out that the genome of this fish contains only 365 million base pairs, one-ninth the number found in humans. It also appears that Fugu has about the same number of genes as humans, because it does not contain the enormous amount of non-coding DNA that humans have. But, you might think, how could fish genes help annotate the human genome? Thus it might surprise you that over 1,000 human genes have already been identified by their similarity to the Fugu genes.
But what about disease genes? Here too, researchers are optimistic that the Fugu sequence will help them identify genes in humans. The puffer fish evolved a very long time before humans did. Therefore, its genes have had a chance to change a lot more than human genes. Thus, if one finds in Fugu a gene that closely corresponds to a gene in humans, this means that this particular gene may have a fundamental role in survival. In fact, some researchers think that the Fugu sequence will be more important in that respect than the mouse sequence. This is because mice and humans are more closely related than either is to Fugu. In this case, annotation will not be easy because humans and mice are evolutionarily too close to each other, and it will be more difficult to decide which genes in both mice and humans are critical for survival.
We have studied the techniques used by geneticists to determine whether genes are located on the same chromosome, that is, whether they are linked. Phenotypic ratios of offspring from crosses with linked genes differ from the expected ratios for unlinked genes. This is because genes on the same chromosome tend to be inherited together. However, chromosomes can break and rejoin, resulting in recombinant chromosomes. The percentage of offspring with recombinant chromosomes gives us a measure of the distance between linked genes on the chromosome. In humans, where extensive, controlled crosses cannot be performed, other techniques are needed to determine gene linkage. One of these techniques consists in determining the close association of RFLP DNA fragments with a diseased phenotype. The full sequencing of the human and other genomes will allow efficient identification and study of disease genes.
This game will allow you to directly see how the independent assortment of chromosomes results in new gene combinations. In order to make the game not take too long, we’ll consider an organism with only three pairs of chromosomes. Remember we actually have twenty-three pairs of chromosomes.
For each pair of chromosomes, we’ll have two different types that we’ll call heads (H) and tails (T) since we’ll flip a coin to determine which we pick. Thus we have chromosome 1 heads (1 H) and chromosome 1 tails (1 T), chromosome 2 heads (2 H) and chromosome 2 tails (2 T), and chromosome 3 heads (3 H) and chromosome 3 tails (3 T). We begin with a father and a mother, each with three pairs of chromosomes. We will indicate the chromosomes from the father with f and the chromosome from the mother with m. So the chromosomal compositions of mother and father are:
Father: 1 f H and 1 f T, 2 f H and 2 f T, 3 f H and 3 f T
Mother: 1 m H and 1 m T, 2 m H and 2 m T, 3 m H and 3 m T
Each team of two has a "mother" and a "father," with each person representing either the father or the mother. To begin the game, each person flips a coin three times to determine which of the pairs of chromosome they contribute to the next generation. The first flip determines whether chromosome 1 H or T is contributed, the second flip determines which chromosome 2 is contributed, and so forth. Then the team writes down the chromosome composition of their "child." If done properly, you will see that there is one f and one m chromosome for each pair of chromosomes. Now compare the chromosome composition of your "child" with that of other teams. Are there any with the identical chromosome composition? What is the chance that we would get two children with the same chromosome composition?
There is an explosion of genetic information being accumulated in databases around the world. Much of the information is available to anyone with Internet access. We will introduce you to some of that huge amount of information in this exercise to help you get acquainted with the databases available for genetic studies.
In this exercise, you will first find a gene associated with a human genetic disease. Then we will search the DNA database for the gene. Finally, we’ll use one of many tools available to analyze DNA sequences, called BLAST (Basic Local Alignment Search Tool). We will use the example of Marfan syndrome that we encountered in chapter 3, but we encourage you to follow along in this exercise with another disease or issue of interest to you.
First, open an Internet browser and go to the Entrez search and retrieval system of the National Center for Biotechnology Information of the National Library of Medicine at the National Institutes of Health site at http://www.ncbi.nlm.nih.gov/Entrez/. At this site you will see a link called PubMed, which links to the professional biomedical literature database, another labeled Nucleotide, which links to the DNA-sequence database GenBank, and many others.
We will first look up a genetic disease in OMIM, the Online Mendelian Inheritance in Man, by clicking on "OMIM." Then, in the space provided, type in a disease or an organ of the body that you are interested in or even a characteristic of interest like left-handedness. As an example, we will type in Marfan and click "Go," which gives us a listing of OMIM entry numbers. Each of these listings provides authoritative information about the disease. Genes can be identified with a set of upper case letters sometimes followed by a number. When we click on #154700, the listing gives information about Marfan syndrome. At the beginning it states that "all cases of the true Marfan syndrome appear to be due to mutation in the fibrillin-1 gene (134797)." Clicking on that number gives us the name for the gene and its acronym, FBN1. In the column on the left, the light orange box labeled "R" provides a link to the reference-gene sequence. The default display is a short summary of the sequence. If you want more detailed information, you can choose GenBank from the pull down menu and then click "Display" to show the information. For our purpose, however, we want to get the simple DNA sequence, so choose the FASTA format from the same menu and click "Display." Because this gene is huge, for demonstration purposes, select the first few lines of the sequence and copy it.
Now we will use BLAST, the Basic Local Alignment Search Tool, to find other related sequences. To do this, go to the BLAST page at http://www.ncbi.nlm.nih.gov/BLAST/. You will see the many types of BLAST searches that can be performed. There is a BLAST tutorial page at http://www.ncbi.nlm.nih.gov/BLASTinfo/informa-tion3.html. For our example, we will use the Translated BLAST search. This search takes a DNA sequence and translates it into protein sequences. The program can search through the protein database (blastx) or through the DNA database that is translated into protein (tblastx). We will do the latter by clicking on that line and then using the first few lines of our FBN1 gene that we copied by pasting it into the box next to "search." We can leave everything else at the default settings and click on "BLAST." You should now get a box with your request ID and an estimate of the time it will take to do this search. Depending upon how crowded internet traffic is in general and at the NIH site in particular, and depending on the size of your search (which includes both the query DNA sequence length and the database that you are searching), the search can take from seconds to many minutes, and sometimes hours. You can shorten the time needed for your search by using a shorter DNA sequence for the query and limiting your search to a subset of organisms. There are millions of people using this search tool throughout the world, searching through an ever-increasing database. Thus, even the few minutes that a search may take is quite fast!
A BLAST search seeks to find DNA sequences that are similar to the query sequence. The results display the closest match, first in a color-coded graph that shows the region of similarity, ranging from red being the closest match to black being the least similar. The higher the "Score" value, the better the match. The "E "value provides a statistical measure of how likely it is to have found this match by random chance. The smaller this value, the more likely it is that the match is meaningful. This tool is commonly used when one first sequences a piece of DNA in order to find the closest match and help identify the gene sequenced. The E value can also find similar genes in closely related or even distantly related organisms. For our example, not surprisingly, the best matches to our Marfan gene piece are to human fibrillin genes. But further down the list, one finds Bos taurus, cow, Sus domesticus, pig, Mus musculus, mouse, and Rattus norvicus, rat genes. If one goes down further, we can even find that the fruit fly, Drosophila melanogaster, has a gene similar to human fibrillin, admittedly with less similarity than the mammals listed above.