Chronic Diarrhea
Anat Guz-Mark, Raanan Shamir
Definition of Epidemiology
Chronic diarrhea is defined as stool volume of more than 10 g/kg/day in toddlers/infants and greater than 200 g/day in older children that lasts for 4 wk or more. Persistent diarrhea began acutely but lasts longer than 14 days. In practice, this usually means having loose or watery stools more than 3 times a day. Awakening at night to pass stool is often a sign of an organic cause of diarrhea. The epidemiology has 2 distinct patterns. In developing countries, chronic diarrhea is, in many cases, the result of an intestinal infection that persists longer than expected. This syndrome is often defined as protracted (persistent) diarrhea , but there is no clear distinction between protracted (persistent) and chronic diarrhea. In countries with higher socioeconomic conditions, chronic diarrhea is less frequent, and the etiology often varies with age. The outcome of diarrhea depends on the cause and ranges from benign, self-limited conditions, such as toddler's diarrhea, to severe congenital diseases, such as microvillus inclusion disease, that may lead to progressive intestinal failure.
Pathophysiology
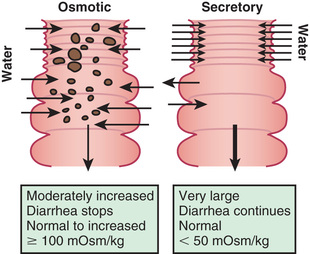
The mechanisms of diarrhea are generally divided into secretory and osmotic , but often diarrhea is a combination of both mechanisms . In addition, inflammation and motility disorders may contribute to diarrhea. Secretory diarrhea is usually associated with large volumes of watery stools and persists when oral feeding is withdrawn. Osmotic diarrhea is dependent on oral feeding, and stool volumes are usually not as massive as in secretory diarrhea (Fig. 367.1 ).
Secretory diarrhea is characterized by active electrolyte and water fluxes toward the intestinal lumen, resulting from either the inhibition of neutral NaCl absorption in villous enterocytes or an increase in electrogenic chloride secretion in secretory crypt cells as a result of the opening of the cystic fibrosis transmembrane regulator (CFTR) chloride channel or both. The result is more secretion from the crypts than absorption in the villous that persists during fasting. The other components of the enterocyte ion secretory machinery are (1) the Na-K 2Cl cotransporter for the electroneutral chloride entrance into the enterocyte; (2) the Na-K pump, which decreases the intracellular Na+ concentration, determining the driving gradient for further Na+ influx; and (3) the K+ selective channel, that enables K+ , once it has entered the cell together with Na+ , to return to the extracellular fluid.
Electrogenic secretion is induced by an increase of intracellular concentration of cyclic adenosine monophosphate, cyclic guanosine monophosphate, or calcium in response to microbial enterotoxins, or to endogenous endocrine or nonendocrine molecules, including inflammatory cytokines. Another mechanism of secretory diarrhea is the inhibition of the electroneutral NaCl-coupled pathway that involves the Na+ /H+ and the Cl− /HCO3 − exchangers. Defects in the genes of the Na+ /H+ and the Cl− /HCO3 − exchangers are responsible for congenital Na+ and Cl− diarrhea, respectively.
Osmotic diarrhea is caused by nonabsorbed nutrients in the intestinal lumen as a result of one or more of the following mechanisms: (1) intestinal damage (e.g., enteric infection); (2) reduced absorptive surface area (e.g., active celiac disease); (3) defective digestive enzyme or nutrient carrier (e.g., lactase deficiency); (4) decreased intestinal transit time (e.g., functional diarrhea); and (5) nutrient overload, exceeding the digestive capacity (e.g., overfeeding, sorbitol in fruit juice). Whatever the mechanism, the osmotic force generated by nonabsorbed solutes drives water into the intestinal lumen. A very common example of osmotic diarrhea is lactose intolerance. Lactose, if not absorbed in the small intestine, reaches the colon, where it is fermented to short-chain organic acids, releasing hydrogen that is detected in the lactose breath test, and generating an osmotic overload. Another risk for chronic osmotic diarrhea often noted in patients with diarrhea-associated irritable bowel syndrome are foods containing FODMAPs (fermentable oligo-di-monosaccharides and polyols).
In many children chronic diarrhea may be caused by the combination of multiple mechanisms.
Etiology
Table 367.1 summarizes the main etiologies of chronic diarrhea in infants and children.
Table 367.1
Infectious
Enteric infections are by far the most frequent cause of persistent or chronic diarrhea, both in developing and industrialized countries, however outcomes are often very different. In the former, comorbid conditions, such as HIV/AIDS, malaria, or tuberculosis, result in malnutrition that impairs the child's immune response, thereby potentiating the likelihood of prolonging diarrhea or acquiring another enteric infection. In children with HIV/AIDS, the viral infection itself impairs immune function and may trigger a vicious circle with malnutrition. Sequential infections with the same or different pathogens may also be responsible for chronic diarrhea. In developing countries, enteroadherent Escherichia coli and Giardia lamblia have been implicated in chronic diarrhea, whereas, in developed countries, chronic infectious diarrhea usually runs a more benign course and the etiology is often viral, with a major role of rotavirus and norovirus (Table 367.2 ).
Table 367.2
A Comparative List of Prevalent Agents and Conditions in Children With Persistent Infectious Diarrhea in Industrialized and Developing Countries
| AGENT/DISEASE | |
|---|---|
| INDUSTRIALIZED COUNTRIES | DEVELOPING COUNTRIES |
|
Astrovirus Norovirus Rotavirus* Small intestinal bacterial overgrowth (SIBO) Postenteritis diarrhea syndrome |
|
* More frequent in industrialized than in developing countries as agent of chronic diarrhea.
Chronic diarrhea in travelers to or expatriates from developing countries may depend on the country of origin. Nonetheless, common pathogens include giardia, E. coli , shigella, campylobacter, salmonella, and enteric viruses. Less common pathogens include amebiasis, strongyloides, and tropical spruce.
Opportunistic microorganisms induce diarrhea exclusively, more severely, or for more prolonged periods, in specific populations, such as immunocompromised children. Specific agents cause chronic diarrhea or exacerbate diarrhea in many chronic diseases. Clostridium difficile or cytomegalovirus act as opportunistic agents in oncologic patients as well as in patients with inflammatory bowel diseases. Cryptosporidium may induce severe and protracted diarrhea in AIDS patients.
Small intestinal bacterial overgrowth results in chronic diarrhea by either a direct interaction between the microorganism and the enterocyte, or the consequence of deconjugation and dihydroxylation of bile salts and hydroxylation of fatty acids due to an increased proliferation of bacteria in the proximal intestine.
Postenteritis diarrhea syndrome (Chapter 364.4 ) is a clinicopathologic condition in which small intestinal mucosal damage persists after acute gastroenteritis. Sensitization to food antigens, secondary disaccharidase deficiency, persistent infections, reinfection with an enteric pathogen, or side effects of medication may be responsible for causing postenteritis diarrhea syndrome, thought to be related to dysregulation of the intestinal microbiota. Functional diarrhea which may be related to the pathogenesis of irritable bowel syndrome may be caused by complications of an acute gastroenteritis.
Inflammatory/Immunologic
Celiac disease (Chapter 364.2 ) is a genetically determined permanent gluten intolerance that affects about 1 in 100 individuals, depending on geographic origin. In the genetically susceptible host, gliadin, the major protein of gluten, reacts with the immune system to cause villous atrophy. A reduction of intestinal absorptive surface is responsible for the diarrhea in celiac disease, which is reversible upon restriction of gluten from the diet.
Food allergy (mainly cow milk protein allergy Chapter 176 ) may present during infancy with chronic diarrhea. An abnormal immune response to food proteins can cause a proctitis/colitis or an enteropathy. Eosinophilic gastroenteritis is characterized by eosinophilic infiltration of the intestinal wall and is strongly associated with atopy. However, whereas diarrhea in food allergy responds to withdrawal of the responsible food, this does not always occur in eosinophilic gastroenteritis, in which immune suppression may be needed.
Inflammatory bowel diseases, including Crohn disease, ulcerative colitis, and inflammatory bowel disease–undetermined, cause chronic diarrhea that is often associated with abdominal pain, elevated inflammatory markers, and increased concentrations of fecal calprotectin or lactoferrin (see Chapter 362 ). The age of onset of inflammatory bowel disease is broad, with rare cases described in the 1st few mo of life, but the peak incidence in childhood occurs in adolescence. The severity of the symptoms is highly variable with a pattern characterized by long periods of well-being followed by exacerbations.
Autoimmune processes may target the intestinal epithelium, alone or in association with extraintestinal symptoms. Autoimmune enteropathy is associated with the production of antienterocyte and antigoblet cell antibodies, primarily immunoglobulin A, but also immunoglobulin G, directed against components of the enterocyte brush-border or cytoplasm and by a cell-mediated autoimmune response with mucosal T-cell activation. An X-linked immune-dysregulation, polyendocrinopathy, and enteropathy (IPEX syndrome ) is associated with variable gene mutations and phenotypes of chronic diarrhea (more on autoimmune enteropathy and IPEX syndrome is available on Chapter 364.3 ).
Immune deficiency can present as chronic diarrhea in children. In these cases (for example, severe combined immunodeficiency or AIDS) the child can be infected by an opportunistic pathogen; can exhibit a persistent diarrhea due to a pathogen usually causing an acute gastroenteritis; or be infected by multiple and recurrent different pathogens causing mucosal damage to the intestines. Other immunoregulatory defects, found in patients with agammaglobulinemia, isolated immunoglobulin A deficiency, and common variable immunodeficiency disorder, may result in mild persistent infectious diarrhea.
Pancreatic Deficiency
Chronic diarrhea may be the manifestation of maldigestion caused by exocrine pancreatic disorders (see Chapters 376 and 378.2 ). In most patients with cystic fibrosis , exocrine pancreatic insufficiency results in steatorrhea and protein malabsorption. In Shwachman-Diamond syndrome , exocrine pancreatic hypoplasia may be associated with neutropenia, bone changes, and intestinal protein-losing enteropathy. Specific isolated pancreatic enzyme defects, such as lipase deficiency, result in fat and/or protein malabsorption. Familial pancreatitis, associated with a mutation in the trypsinogen gene, may be associated with exocrine pancreatic insufficiency and chronic diarrhea. Mutations in CFTR , CTRC , PRSS1 , PRSS2 , SPINK 1 , and SPINK 5 are associated with hereditary pancreatitis.
Liver and Bile Acids Disorders
Liver disorders and cholestasis may lead to a reduction in the bile salts pool resulting in fat malabsorption causing chronic diarrhea in the form of steatorrhea. Bile acid loss may be associated with diseases affecting the terminal ileum, such as Crohn disease, or following ileal resection. In primary bile acid malabsorption , neonates and young infants present with chronic diarrhea and fat malabsorption caused by mutations of ileal bile transporter. In addition to the fat malabsorption, the bile acid loss from the intestinal lumen is a form of secretory diarrhea by itself (called cholorrheic diarrhea , which is usually associated with significant diaper dermatitis).
Carbohydrate Malabsorption
Rare genetic mutations (Chapters 364.9 and 364.11 ) can cause carbohydrate malabsorption. More commonly, lactose intolerance is secondary to lactase deficiency caused by intestinal mucosal damage (usually as part of postenteritis syndrome, which is a self-limited process). Depending on ethnicity, a progressive, age-related, loss of lactase activity may begin around 7 yr of age and affects approximately 80% of the non-white population, and acquired hypolactasia may be responsible for chronic diarrhea in older children receiving cow’s milk (adult-type lactase deficiency).
Similarly, fructose malabsorption is common in Western countries with estimates as high as 40% of the population. These individuals cannot absorb fructose and often develop bloating, abdominal pain, diarrhea, and flatulence. Typically, they do not have liver disease. This is in contrast to hereditary fructose intolerance , a rare genetic disorder with incidence estimated to be 1 in 20,000-30,000. This disease is associated with mutations in the ASDOB gene that encodes for the aldolase B enzyme that is found primarily in the liver and is involved in the metabolism of fructose. Individuals with hereditary fructose intolerance may have nausea, abdominal pain/bloating, vomiting, diarrhea, and hypoglycemia. Continued ingestion of fructose results in hepatomegaly and eventually cirrhosis.
Protein-Losing Enteropathy
Chronic diarrhea can be the manifestation of obstructed intestinal lymphatic drainage, causing protein-losing enteropathy with steatorrhea, diarrhea, and lymphopenia. Besides intestinal lymphangiectasia , many diseases that cause intestinal mucosal injury can also result in protein-losing enteropathy, characterized by low serum protein levels and elevated fecal α1 -antitrypsin (see Chapter 364.3 ).
Motility Disorders
Disorders of intestinal motility include abnormal development and function of the enteric nervous system, such as in Hirschsprung disease and chronic intestinal pseudoobstruction (which encompass both the neurogenic and the myogenic forms). Other motility disorders may be secondary to extraintestinal disorders, such as in hyperthyroidism and scleroderma . Motility disorders are associated with either constipation or diarrhea or both, with the former usually dominating the clinical picture.
Short Bowel Syndrome
Short bowel syndrome is the single most frequent etiology of intestinal failure in children (Chapter 364.7 ). Many intestinal abnormalities such as stenosis, segmental atresia, gastroschisis, and malrotation may require surgical resection, but the most frequent primary cause of short bowel is necrotizing enterocolitis. Rarely, a child can be born with congenitally short small bowel. In these conditions, the residual intestine may be insufficient to carry on its digestive–absorptive functions, resulting in severe chronic diarrhea, malnutrition, and failure to thrive, requiring long-term treatment with parenteral nutrition.
Nonspecific Diarrhea, Including Toddler's Diarrhea
The most benign and common etiology of chronic diarrhea is nonspecific diarrhea that encompasses functional diarrhea (or toddler's diarrhea ) in children younger than 4 yr of age and irritable bowel syndrome in those 5 yr of age and older. It is the leading cause of chronic diarrhea in an otherwise well child. Toddler's diarrhea is defined by the daily painless recurrent passage of 4 or more large unformed stools, for 4 or more wk, with onset in infancy or preschool years. Nighttime defecation is usually absent. The child appears unperturbed by the diarrhea, there is no evidence of failure to thrive, and the symptoms resolve spontaneously by school age.
Diarrhea may also be the result of an excessive intake of fluid and nonabsorbable carbohydrate . If the child's fluid intake were > 150 mL/kg/24 hr, fluid intake should be reduced not to exceed 90 mL/kg/24 hr in order to decrease the stool frequency and volume. If the dietary history suggests that the child is ingesting significant amounts of fruit juice, especially apple juice, then the consumption of juice should be decreased. Sorbitol, which is a nonabsorbable sugar, is found in apple, pear, and prune juices, and often causes diarrhea in toddlers. Moreover, apple and pear juices contain higher amounts of fructose than glucose, a feature postulated to cause diarrhea in toddlers. In older children, irritable bowel syndrome is often associated with abdominal pain and may be related to anxiety, depression, and other psychological disturbances (Chapter 368 ). When the cause of the diarrhea remains undetermined and the clinical course is inconsistent with organic disorders, factitious disorder by proxy should be considered.
Congenital Diarrheal Disorders
The most severe etiology of chronic diarrhea includes a number of heterogeneous congenital conditions leading to syndromes often referred to as intractable or protracted diarrhea . This is the result of a permanent defect in the structure or function of the enterocyte, leading to progressive, potentially irreversible intestinal failure. The genetic and molecular basis of many causes of protracted diarrhea has been identified recently and a new classification of congenital diarrheal disorders (CDDs) has been proposed (Table 367.3 ). CDDs are a group of rare but severe enteropathies, with a similar clinical presentation despite a different pathogenesis and outcome. The diarrhea can be either secretory or osmotic, depending on the specific defect. Often severe diarrhea presents at birth or shortly thereafter, but in milder forms diarrhea may go unrecognized for years. CDDs can be classified in 4 groups: defects of digestion, absorption and transport of nutrients and electrolytes; defects of enterocyte differentiation and polarization; defects of enteroendocrine cell differentiation; and defects of modulation of intestinal immune response.
Table 367.3
Classification of Congenital Diarrheal Disorders Based on Their Molecular Defect and Their Inheritance
| DEFECTS OF DIGESTION, ABSORPTION, AND TRANSPORT OF NUTRIENTS AND ELECTROLYTES | ||||
|---|---|---|---|---|
| DISEASE | GENE NAME | GENE LOCATION | TRANSMISSION AND INCIDENCE | MECHANISM |
| GENES ENCODING BRUSH-BORDER ENZYMES | ||||
| Congenital lactase deficiency (LD) | LCT | 2q21.3 | AR, 1 in 60,000 in Finland; lower in other ethnic groups | Osmotic |
| Congenital sucrase-isomaltase deficiency (SID) | SI | 3q26.1 | AR, 1 in 5,000; higher incidence in Greenland, Alaska, and Canada | Osmotic |
| Congenital maltase-glucoamylase deficiency (MGD) | Not defined | — | Few cases described | Osmotic |
| GENES ENCODING MEMBRANE CARRIERS | ||||
| Glucose-galactose malabsorption (GGM) | SLC5A1 | 22q13.1 | AR, few hundred cases described | Osmotic |
| Fructose malabsorption (FM) | Not defined | — | Up to 40% | Osmotic |
| Fanconi-Bickel syndrome (FBS) | SLC2A2 | 3q26.2 | AR, rare, higher frequency in consanguineous | Osmotic |
| Acrodermatitis enteropathica (ADE) | SLC39A4 | 8q24.3 | AR, 1 in 500,000 | Osmotic |
| Congenital chloride diarrhea (CCD, DIAR 1) | SLC26A3 | 7q31.1 | AR, sporadic; frequent in some ethnicities | Osmotic |
| Lysinuric protein intolerance (LPI) | SLC7A7 | 14q11.2 | AR, about 1 in 60,000 in Finland and Japan; rare in other ethnic groups | Osmotic |
| Primary bile acid malabsorption (PBAM) | SLC10A2 | 13q33.1 | AR | Secretory |
| Cystic fibrosis (CF) | CFTR | 7q31.2 | AR, 1 in 2,500 | Osmotic |
| GENES ENCODING PANCREATIC ENZYMES | ||||
| Enterokinase deficiency (EKD) | PRSS7 | 21q21 | AR | Osmotic |
| Hereditary pancreatitis (HP) |
PRSS1 PRSS2 SPINK1 CTRC |
7q34 7q34 5q32 1p36.21 |
AD, cases with compound mutations in different genes; SPINK1 mutations may also cause tropical pancreatitis | Osmotic |
| Congenital absence of pancreatic lipase (APL) | PNLIP | 10q25.3 | AR | Osmotic |
| GENES ENCODING PROTEINS OF LIPOPROTEIN METABOLISM | ||||
| Abetalipoproteinemia (ALP) | MTP | 4q27 | AR, about 100 cases described; higher frequency among Ashkenazi Jews | Osmotic |
| Hypobetalipoproteinemia (HLP) | APOB | 2p24.1 | Autosomal codominant | Osmotic |
| Chylomicron retention disease (CRD) | SAR1B | 5q31.1 | AR, about 40 cases described | Osmotic |
| GENES ENCODING OTHER TYPES OF PROTEINS | ||||
| Congenital sodium diarrhea (CSD) |
SPINT2 (only syndromic CSD) SLC9A3 |
19q13.2 5p15.33 |
AR | Osmotic |
| Shwachman-Diamond syndrome (SDS) | SBDS | 7q11 | AR | Osmotic |
| Activating guanylate cyclase-C mutation | GUCY2C | 12p12.3 | AD | Secretory |
| GENES ENCODING FOR OTHER ENZYMES | ||||
| Defect in triglyceride synthesis | DGAT1 | 8q24.3 | AR | Protein-losing enteropathy |
| DEFECTS OF ENTEROCYTE DIFFERENTIATION AND POLARIZATION | ||||
| Microvillous inclusion disease (MVID, DIAR 2) | MYO5B | 18q21.1 | AR; rare; higher frequency among Navajo | Secretory |
| Congenital tufting enteropathy (CTE, DIAR 5) | EPCAM | 2p21 | AR; 1 in 50,000-100,000; higher among Arabians | Secretory |
| Trichohepatoenteric syndrome (THE) |
TTC37 SKIV2L |
5q15 6p21.33 |
AR; 1 in 400,000 | Secretory |
| DEFECTS OF ENTEROENDOCRINE CELL DIFFERENTIATION | ||||
| Congenital malabsorptive diarrhea (CMD, DIAR 4) | NEUROG3 | 10q22.1 | AR; few cases described | Osmotic |
| Proprotein convertase 1/3 deficiency (PCD) | PCSK1 | 5q15 | AR | Osmotic |
| DEFECTS OF MODULATION OF INTESTINAL IMMUNE RESPONSE | ||||
| Autoimmune polyglandular syndrome type 1 (APS1) | AIRE | 21q22.3 | AR; AD (1 family) | Secretory |
| Immune dysfunction, polyendocrinopathy, X-linked (IPEX) | FOXP3 | Xp11.23 | X-linked (autosomal cases described), very rare | Secretory |
| IPEX-like syndrome | Not defined | — | Not X-linked | Secretory |
AD , autosomal dominant; AR , autosomal recessive.
Although CDDs are rare diseases, in most specific disorders the genetic defect and transmission are known. The incidence of genetic disorders associated with CDD can range from 1 in 2,500 for cystic fibrosis, 1 in 5,000 for sucrose-isomaltase deficiency, 1 in 60,000 for congenital lactase deficiency, to 1 in 400,000 for trichohepatoenteric syndrome. For most CDDs, such as IPEX syndrome or autoimmune polyglandular syndrome type 1, the clinical application of exome sequencing is likely to increase identification of more patients with these rare causes of chronic diarrhea. Selected CDDs are more frequent in ethnic groups where consanguineous marriages are common, or in some geographic areas because of founder effects. Congenital lactase deficiency is more common in Finland; lysinuric protein intolerance has a higher incidence either in Finland and in Japan because of founder effect, and a specific mutation is typically found in each of the 2 ethnic groups. A defect in the DGAT1 gene was identified using whole-exome sequencing in an Ashkenazi Jewish family and associated with the early onset of vomiting and nonbloody diarrhea with protein-losing enteropathy. For specific CDDs see Chapters 364.3 and 364.11 .
Most cases of protracted diarrhea syndrome are not easily treated. The natural history of protracted diarrhea is related to the primary intestinal disease and the specific defect in nutrient absorption. Treatment is more favorable for motility disorders and autoimmune enteropathy than for structural enterocyte defects. Children with motility disorders may have persistent symptoms, but they are rarely fatal, whereas children with structural enterocyte defects have a more severe course, poorer prognosis, and are more likely to be candidates for intestinal transplantation (see Chapter 365 ). Some late-onset CDDs may be relatively mild and are recognized only later in life.
Evaluation of Patients
Because of the spectrum of etiologies, the medical approach should be based on diagnostic algorithms that begin with assessment for infectious causes, and then consider the age of the child, growth, and clinical and epidemiologic factors. Early onset in the neonatal period is rare and may suggest a congenital or severe condition (see also Table 364.3 ), however infections and food allergy are more frequent in this age group, and together with gastrointestinal (GI) malformations should be high on the differential diagnosis. In later infancy and up to 2 yr of age, infections and allergies are the most common; inflammatory diseases are more frequent in older children and adolescents. Celiac disease as well as functional nonspecific diarrhea should always be considered independently of age because of their relatively high frequency at all ages.
Specific clues in the family and personal history may provide useful indications, suggesting a congenital, allergic, or inflammatory etiology. A history of polyhydramnios is consistent with congenital chloride-sodium diarrhea (where a typical sonographic finding of dilated fetal bowel loops is present), cystic fibrosis, and other CDDs, as well as a family history of a chronic or intractable diarrhea in a relative presenting in the 1st mo of life, as well as consanguinity . An acute onset of diarrhea that runs a protracted course suggests postenteritis diarrhea, secondary lactase deficiency, small intestinal overgrowth, or the onset of chronic nonspecific diarrhea (toddler's diarrhea). The association of diarrhea with specific foods may indicate a nutrient basis, such as intolerance to selected nutrients (fructose). Anthropometric evaluation is essential to understand if diarrhea has affected weight gain and growth, providing estimation of the severity of diarrhea. Normal weight and growth strongly support functional diarrhea that may respond to simple dietary management. It should be noted that a child with functional diarrhea may be inappropriately “treated” with a diluted hypocaloric diet in an effort to reduce the diarrhea, resulting in impaired growth.
Initial clinical examination should include the evaluation of general and nutritional status. Dehydration, marasmus, or kwashiorkor require prompt supportive interventions to stabilize the patient. Nutritional evaluation should start with the evaluation of the weight and height curves, and of the weight-for-height index to determine the impact of diarrhea on growth. Weight is generally impaired before height, but with time, linear growth also becomes affected, and both parameters may be equally abnormal in the long term. Assessment of nutritional status includes a dietary history, physical examination, and biochemical testing including nutritional investigations. Caloric intake should be quantitatively determined, energy requirements determined, and the relationship between weight modifications and energy intake should be carefully considered. Assessment of body composition may be performed by measuring mid-arm circumference and triceps skinfold thickness or by bioelectrical impedance analysis, dual-emission x-ray absorptiometry scans, or air plethysmography. Biochemical markers including albumin, prealbumin, retinol binding protein, serum iron, and transferrin may assist in grading malnutrition, as the half-life of serum proteins may distinguish between short- and long-term malnutrition. Evaluation of micronutrient concentrations should always be considered. Zinc, magnesium, vitamin A, and folate deficiency are associated with chronic diarrhea and should be provided if needed.
In infants with chronic diarrhea, feeding history must be carefully obtained, providing clues for allergy or specific food intolerance, such as cow's-milk protein allergy or sucrose-isomaltase deficiency. Associated symptoms and selected investigations provide important diagnostic clues. Signs of general inflammation such as fever, mucoid or bloody stools, and abdominal pain may suggest inflammatory bowel disease. The presence of eczema or asthma is associated with an allergic disorder, whereas specific extraintestinal manifestations (arthritis, diabetes, thrombocytopenia, etc.) may suggest an autoimmune disease. Specific skin lesions may be suggestive of acrodermatitis enteropathica that might respond to zinc supplementation. Typical facial abnormalities and woolly hair are associated with phenotypic diarrhea.
Investigations
Microbiologic investigation should include a thorough list of intestinal bacterial, viral, and protozoan pathogens. Proximal intestinal bacterial overgrowth may be determined using the lactulose hydrogen breath test, but false-positive tests are common (see Chapter 364.4 ).
Initial investigations of a child with chronic diarrhea should always include an assessment of intestinal inflammation using fecal calprotectin or lactoferrin, and serology for celiac disease (see Chapter 364.2 ). The role of a mucosal biopsy is determined by the noninvasive diagnostic evaluation in consultation with a pediatric gastroenterologist.
Noninvasive assessment of digestive-absorptive function and of intestinal inflammation plays a key role in the diagnostic workup (Table 367.4 ). Abnormalities in the digestive-absorptive function tests suggest small bowel involvement, whereas intestinal inflammation, as demonstrated by increased fecal calprotectin or lactoferrin, supports colitis.
Table 367.4
Determining the osmotic versus secretory nature of the diarrhea in neonates and infants with protracted diarrhea is especially important. The stool osmolar gap , sometimes called stool ion gap, is calculated as 290 mOsm/kg (or measured stool osmolality) minus [2 × (stool Na + stool K)]. If the osmolar gap is above 100 mOsm/kg, fecal osmolality is derived from ingested or nonabsorbed osmotically active solutes or nonmeasured ions. In contrast, a low gap (<50 mOsm/kg) is typically observed in secretory diarrhea. It is also important to measure Cl− concentration in the stool to rule out CCD, which is characterized by low osmolar gap due to high Cl− fecal loss (>90 mmol/L).
Whereas most etiologies of chronic diarrhea can be exaggerated by feeding and have osmotic or mixed nature to the stool, secretory diarrhea necessitates investigation for congenital defects in enterocytes, defects in the intestinal immune response (IPEX and autoimmune enteropathy), and disorders of bile acid malabsorption. Because of the overlap between secretory and osmotic features of the diarrhea in many diseases, a classification based on the response to bowel rest was also introduced. Severe diarrhea that persists at bowel rest is characteristic of congenital enteropathies (microvillous inclusion disease, tufting enteropathy, syndromic diarrhea). Diarrhea that disappears at bowel rest can imply carbohydrate or fat malabsorptive syndromes, as well as defects in enteroendocrine cells. In most other etiologies the diarrhea can decrease significantly, but not disappear, in response to bowel rest, including some congenital diseases as well as acquired inflammatory and other enteropathies.
Histology is important in establishing mucosal involvement, noting changes in the epithelial cells, or in identifying specific intracellular inclusion bodies caused by pathogens, such as cytomegalovirus, or the presence of parasites. Electron microscopy is essential to detect subcellular structural abnormalities such as microvillous inclusion disease. Immunohistochemistry allows the study of mucosal immunity as well as of other cell types (smooth muscle cells and enteric neuronal cells).
Imaging has a major role in the diagnostic approach. Abdominal ultrasound may help in detecting liver and pancreatic abnormalities or an increase in distal ileal wall thickness that suggests inflammatory bowel disease. A preliminary plain abdominal x-ray is useful for detection of abdominal distention, suggestive of intestinal obstruction, or increased retention of colonic feces. Intramural or portal gas may be seen in necrotizing enterocolitis or intussusception. Structural abnormalities such as diverticula, malrotation, stenosis, blind loop, and congenital short bowel, as well as motility disorders, may be investigated through a barium meal and a small bowel follow-through. Capsule endoscopy can be done when the patient weighs more than 10 kg and allows the exploration of the entire intestinal tract searching for structural changes, inflammation, or bleeding; the new SmartPill measures pressure, pH, and temperature as it moves through the GI tract, assessing motility.
Specific investigations should be carried out for specific diagnostic indications. Prick and patch test may support a diagnosis of food allergy. However, an elimination diet with withdrawal of the suspected harmful food from the diet and subsequent challenge is the most reliable strategy by which to establish a diagnosis. Bile malabsorption may be explored by the retention of the bile acid analog 75 Se-homocholic acid-taurine (75 SeHCAT) in the enterohepatic circulation. A scintigraphic examination, with radio-labeled octreotide is indicated in suspected APUD cell neoplastic proliferation. In other diseases, specific imaging techniques such as computed tomography, or nuclear magnetic resonance endoscopic retrograde cholangiopancreatography and magnetic resonance cholangiopancreatography, may have important diagnostic value.
Once infectious agents have been excluded and nutritional assessment performed, a stepwise approach to the child with chronic diarrhea may be applied. The main causes of chronic diarrhea should be investigated, based on the features of the diarrhea and the specific nutrient(s) that is (are) affected. The use of whole-exome sequencing or specific molecular analysis may be especially essential in children suspected of having CDD. A step-by-step diagnostic approach is important to minimize the unnecessary use of invasive procedures as well as the cost, while optimizing the yield of the diagnostic evaluation (Table 367.5 ).
Table 367.5
Stepwise Diagnostic Approach to Children and Infants With Chronic Diarrhea
| INITIAL EVALUATION | |
| ⇩ | |
| LABORATORY TESTS | |
| ⇩ | |
| IMAGING | |
| ⇩ | |
| ENDOSCOPIES AND INTESTINAL HISTOLOGY | |
|
Endoscopy and standard jejunal/colonic histology* ; morphometry; PAS staining; intestinal immunohistochemistry; electron microscopy |
|
| ⇩ | |
| GENETIC INVESTIGATION | |
| ⇩ | |
| OTHER SPECIAL INVESTIGATIONS | |
* The decision to perform an upper or a lower endoscopy may be supported by noninvasive tests. PAS , Periodic acid–Schiff; 75 SeHCAT , 75 Se-homocholic acid-taurine.
Treatment
Chronic diarrhea associated with impaired nutritional status should always be considered a serious disease, and therapy should be started promptly. Treatment includes general supportive measures, nutritional rehabilitation, elimination diet, and medications. The latter include therapies for specific etiologies as well as interventions aimed at counteracting fluid secretion and/or promoting restoration of disrupted intestinal epithelium. Because death in most instances is caused by dehydration, replacement of fluid and electrolyte losses is the most important early intervention.
Nutritional rehabilitation is often essential and is based on clinical and biochemical assessment. In moderate to severe malnutrition, caloric intake should be carefully advanced to avoid the development of refeeding syndrome and may be progressively increased to 50% or more above the recommended dietary allowances. The intestinal absorptive capacity should be monitored by digestive function tests. In children with steatorrhea, medium-chain triglycerides may be the main source of lipids. A lactose-free diet should be started in all children with chronic diarrhea and is recommended by the World Health Organization. Lactose is generally replaced by maltodextrin or a combination of complex carbohydrates. A sucrose-free formula is indicated in sucrase-isomaltase deficiency. Semi-elemental or elemental diets have the dual purpose of overcoming food intolerance, which may be the primary cause of chronic diarrhea, particularly in infancy and early childhood, and facilitating nutrient absorption. The sequence of elimination should begin from less to more restricted diets, that is, cow's-milk protein hydrolysate to amino-acid–based formulas, depending on the child's situation. In severely compromised infants, it may be prudent to start with amino-acid–based feeding.
When oral nutrition is not feasible or fails, enteral or parenteral nutrition should be considered. Enteral nutrition may be provided via nasogastric or gastrostomy tube and is indicated in a child who is not able to be fed orally, either because of inability to tolerate nutrient requirements or because of extreme weakness. In extreme wasting and in cases of significant intestinal mucosal damage or dysfunction, enteral nutrition may not be tolerated, and parenteral nutrition is required.
Micronutrient and vitamin supplementation are part of nutritional rehabilitation, especially in malnourished children in developing countries. Zinc supplementation is important in both prevention and therapy of chronic diarrhea, since it promotes ion absorption, restores epithelial proliferation, and stimulates immune response. Nutritional rehabilitation has a general beneficial effect on the patient's general condition, intestinal function, and immune response.
Functional diarrhea in children may benefit from a diet based on the “4 F” principles (reduce fructose and fluids, increase fat and fiber). The use of probiotics in persistent infectious and postinfectious diarrhea in children appear to hold promise as adjunctive therapy with reduction in symptom duration, but there is still insufficient evidence to recommend their routine use.
Pharmacologic therapy includes, based on the etiology, anti-infectious drugs, immune suppression, and drugs that may inhibit fluid loss and promote cell growth. If a bacterial agent is detected, specific antibiotics should be prescribed. Empiric antibiotic therapy may be used in children with either small bowel bacterial overgrowth or with suspected infectious diarrhea. Table 367.6 summarizes the antimicrobial treatment of infectious persistent diarrhea. Immune suppression should be considered in selected conditions such as autoimmune enteropathy and inflammatory bowel disease.
Table 367.6
Antimicrobial Treatment for Persistent Diarrhea
| DRUG | INDICATIONS | DOSAGE | DURATION | |
|---|---|---|---|---|
| Antibiotics | Trimethoprim-sulfamethoxazole | Salmonella spp., Shigella spp. | 6-12 mg/kg/day (of Trimethoprim) in 2 divided doses–daily os | 5-7 days |
| Azithromycin | Shigella spp., Campylobacter |
1 day: 12 mg/kg/day once–daily os 2-5 days: 6 mg/kg/day once–daily os *alternative: 10 mg/kg/day once–daily os, for 3 days |
5 days | |
| Ciprofloxacin | Shigella spp. | 20-30 mg/kg/day in 2 divided doses–os or IV | 3 days | |
| Ceftriaxone | Shigella spp. | 50-100 mg/kg/day once–IM or IV | 2-5 days | |
| Metronidazole | Giardia, Amebiasis , Blastocystis , Clostridium difficile | 15-35 mg/kg/day in 2-3 divided doses–os | 7-10 days | |
| Paromomycin | Amebiasis | 25-35 mg/kg/day in 3 divided doses–os | 7 days | |
| Vancomycin | Clostridium difficile | 40 mg/kg/day in 4 divided doses–os | 10 days | |
| Antiparasitic | Nitazoxanide | Amebiasis , Giardiasis , Blastocystis, Cryptosporidiosis |
100 mg every 12 hr for children ages 12-47 mo 200 mg every 12 hr for children ages 4-11 yr 500 mg every 12 hr for children older than 11 yr |
3 days |
| Albendazole | Ascaris, Hookworm, and Pinworm infection | 400 mg | Once |
Depends on local susceptibility profile. IM , intramuscular; IV , intravenous; os , by mouth.
Treatment may be also directed at modifying specific pathophysiologic processes. Secretion of ions may be reduced by antisecretory agents, such as the enkephalinase inhibitor racecadotril. Some benefit from absorbents, such as diosmectite, has been described, with reduction of diarrhea duration in infectious diarrhea. In diarrhea caused by neuroendocrine tumors (NETs), microvillus inclusion disease and enterotoxin-induced severe diarrhea, a trial of somatostatin analog octreotide may be considered. Zinc promotes both enterocyte growth and ion absorption and may be effective when intestinal atrophy and ion secretion are associated. However, when therapeutic attempts and other nutritional support have failed, the only option to treat children with intestinal failure, while maintaining adequate growth and development, may be long-term parenteral nutrition or eventually intestinal transplantation.
Diarrhea From Neuroendocrine Tumors
Shimon Reif, Raanan Shamir
The incidence of neuroendocrine tumors (NET) originating in the gastrointestinal (GI) tract is increasing globally. The commonly perceived notion of NETs is of slow-growing malignancies with a benign course. Indeed, well-differentiated GI-NETs may exhibit indolent clinical behavior, but recent studies indicate that they are frequently already metastatic at diagnosis. The most common tumor in children is carcinoid and mostly it is a low-grade tumor especially when it is small, that is <1 cm. It is equally distributed between the small and large intestine and can commonly be found in the appendix. Most carcinoids are found incidentally and are asymptomatic, especially those that are located in the appendix. Some NET patients (around 10%) will develop secretory diarrhea requiring symptom control to optimize quality of life and clinical outcomes. Such patients are defined as having carcinoid syndrome, characterized by excessive production of one or more peptides, which, when released into the circulation, exert their endocrine effects and can be measured by radioimmunologic methods (in the plasma or as their urinary metabolites). These peptides therefore also act as tumor markers. In clinically functioning tumors, the secreted peptides cause a recognizable syndrome that can include watery diarrhea. Compared to carcinoid, vasoactive intestinal polypeptide (VIP)omas are much less frequent. Because it secretes VIP, a more potent vasoactive peptide, it induces more profuse diarrhea, with up to 70% of patients having volumes greater than 3 L/day. Though rare as a cause of watery diarrhea, a NET should be considered in the differential diagnosis when diarrhea is unusually severe or takes a chronic course (resulting in electrolyte and fluid depletion). GI-NETs may be associated with flushing, palpitations, or bronchospasm. Furthermore, patients may have a positive family history of multiple endocrine neoplasia MEN 1 or 2 syndromes. (Table 367.7 ).
Table 367.7
Diarrhea Caused by Neuroendocrine Tumors
| TUMOR AND CELL TYPE | SITE | MARKERS | SIGNS OF HORMONE HYPERSECRETION | THERAPY |
|---|---|---|---|---|
| Carcinoid | Intestinal argentaffin cells, typically midgut, also foregut and hindgut, ectopic bronchial tree |
Serotonin (5-HT), urine 5-HIAA * (diagnostic) Also produce substance P, neuropeptide K, somatostatin, VIP Chromogranin A |
Secretory diarrhea, crampy abdominal pain, flushing, wheezing (and cardiac valve damage if foregut site) |
Resection Somatostatin analog, (palliative) Genetic MEN-1 |
| Gastrinoma, Zollinger-Ellison syndrome | Pancreas, small bowel, liver, and spleen | Gastrin | Multiple peptic ulcers, secretory diarrhea |
H2 -blockers, PPI, tumor resection, (gastrectomy) Genetic MEN-1 |
| Mastocytoma | Cutaneous, intestine, liver, spleen | Histamine , VIP |
Pruritus, flushing, apnea If VIP, diarrhea |
H1 - and H2 -blockers, steroids, resection if solitary |
| Medullary carcinoma | Thyroid C-cells | Calcitonin , VIP , prostaglandins | Secretory diarrhea | Radical thyroidectomy ± lymphadenectomy (genetic MEN-2A/B, familial MTC) |
| Ganglioneuroma, pheochromocytoma, ganglioneuroblastoma, neuroblastoma | Chromaffin cells; abdominal > other sites; extraadrenal or adrenal |
Metanephrines and catecholamines , VIP VMA, HMA in neuroblastoma |
Hypertension, tachycardia, paroxysmal palpitations, sweating, anxiety, watery diarrhea † |
Perioperative α-adrenergic (BP) and β-adrenergic blockade with volume support tumor resection Genetic MEN-2 (RET gene), VHL, NF-1, SDH |
| Somatostatinoma | Pancreas | Somatostatin | Secretory diarrhea, steatorrhea, cholelithiasis, diabetes |
Resection Genetic MEN-1 |
| VIPoma | Pancreas | VIP , prostaglandins | Secretory diarrhea, achlorhydria, hypokalemia |
Somatostatin analogs, resection Genetic MEN-1 |
* Bold indicates major markers.
† Diarrhea has been reported only in adult patients with pheochromocytoma.
BP , blood pressure; H1 , histamine receptor type 1; H2 , histamine receptor type 2; HMA , homovanillic acid; MEN-1 , multiple endocrine neoplasia type 1; MTC , medullary thyroid carcinoma; NF-1 , neurofibromatosis type 1; PPI , proton pump inhibitor; SDH , succinate dehydrogenase; VHL , von Hippel-Lindau disease; VIP , vasoactive intestinal polypeptide; VMA , vanillylmandelic acid.
(Modified from Spoudeas HA, editor: Paediatric endocrine tumors. A multidisciplinary consensus statement of best practice from a working group convened under the auspices of the British Society of Paediatric Endocrinology and Diabetes (BSPED) and the United Kingdom Children's Cancer Study Group (UKCCSG) , Crawley, West Sussex, 2005, Novo Nordisk.)
Baseline tests should include plasma chromogranin A and urinary 5-hydroxyindoloacetic acid (metabolite of serotonin) and other specific biochemistry being guided by the suspected syndrome (see Table 367.7 ). Localization of any NET is best achieved using a multimodality approach. Whole-body CT, MRI, and somatostatin receptor scintigraphy may be required (because nearly all NETs express membrane receptors for small peptides, e.g., somatostatin), with gallium-68 positron emission tomography. Therapeutic interventions to be considered include surgical, pharmacological, and radioisotope therapy. Details to be considered when making therapeutic decisions include disease extent and location, tumor grade, pace of disease progression, symptoms, and co-morbidities.
Tumor resection is the treatment of choice when the tumor is small and localized. However, resection is potentially hazardous as it can precipitate life-threatening adrenergic crises. When arising in the appendix, carcinoid tumors less than 2 cm in size can be managed by simple appendectomy. When greater than 2 cm in size or arising from the base of the appendix, a right hemicolectomy is indicated. Fortunately, in pediatric patients, metastases (most frequently to the liver) are rare. Tumor histochemistry will confirm the NET type and classification. Pharmacologic treatment may include the use of long-acting somatostatin analogues. This usually results in a pronounced improvement of symptoms including diarrhea. However, the improvement is mostly temporary with many patients becoming resistant to somatostatin. An oral medication, Everolimus, a more specific target of Rapamycin (mTOR)-inhibitor, has been reported as add-on treatment to octreotide mainly in adult patients. Data suggest a positive effect of ondansetron, a serotonin-3-receptor antagonist, on diarrhea. Peptide receptor radioisotope therapy also has been reported as a therapeutic modality.
The diagnosis of NET in children should prompt a genetic referral to exclude a familial tumor predisposition syndrome.
Bibliography
Singh S, Asa SL, Dey C, et al. Diagnosis and management of gastrointestinal neuroendocrine tumors: An evidence-based Canadian consensus. Cancer Treat Rev . 2016;47:32–45.
Iqbal CW. Whaof DC: Diagnosis and management of neuroendocrine tumor in children. Curr Opin Pediatr . 2009;21:379–385.