Renal Failure
Acute Kidney Injury
Prasad Devarajan
Acute kidney injury (AKI) has been traditionally defined as an abrupt loss of kidney function leading to a rapid decline in the glomerular filtration rate (GFR), accumulation of waste products such as blood urea nitrogen (BUN) and creatinine, and dysregulation of extracellular volume and electrolyte homeostasis. The term AKI has largely replaced acute renal failure (ARF), because the latter designation overemphasizes the discrete event of a failed kidney. AKI embodies a continuum of renal dysfunction that ranges from a small increase in serum creatinine to complete anuric renal failure. AKI is a common problem afflicting all ages, the leading reason to seek inpatient nephrology consultation, and associated with serious consequences and unsatisfactory therapeutic options. The incidence of AKI varies from 2–5% of all hospitalizations to > 25% in critically ill infants and children. The etiology of AKI varies widely according to age, geographic region, and clinical setting. Functional AKI induced by dehydration is usually reversible with early fluid therapy. However, the prognosis for patients with structural AKI in the intensive care setting with multiorgan failure remains guarded.
A classification system proposed by the Kidney Disease Improving Global Outcomes (KDIGO) AKI Consensus Conference takes both serum creatinine and urine output criteria into account to define and stage AKI (Table 550.1 ). Thus, AKI is defined as:
Table 550.1
- Increase in serum creatinine by ≥ 0.3 mg/dL from baseline within 48 hr; or
- Increase in serum creatinine to ≥ 1.5 times baseline within the prior 7 days; or
- Urine volume ≤ 0.5 mL/kg/hr for 6 hr
Pathogenesis
AKI has been conventionally classified into three categories: prerenal, intrinsic renal, and postrenal (Table 550.2 and Fig. 550.1 ).
Table 550.2
Common Causes of Acute Kidney Injury
| PRERENAL |
| INTRINSIC RENAL |
|
Postinfectious/poststreptococcal Anti–glomerular basement membrane Toxin and drugs (see Table 550.3 ) |
| POSTRENAL |
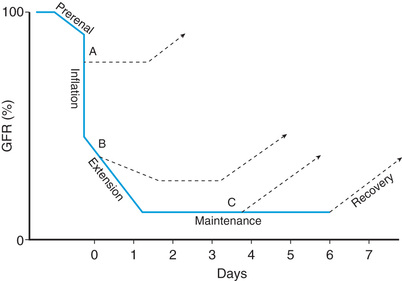
Prerenal AKI, also called prerenal azotemia, is characterized by a diminished effective circulating arterial volume, which leads to inadequate renal perfusion and a decreased GFR. Evidence of structural kidney damage is absent. Common causes of prerenal AKI include dehydration, sepsis, hemorrhage, severe hypoalbuminemia, and cardiac failure. If the underlying cause of the renal hypoperfusion is reversed promptly, renal function returns to normal. If hypoperfusion is sustained, intrinsic renal parenchymal damage can develop.
Intrinsic renal AKI includes a variety of disorders characterized by renal parenchymal damage, including sustained hypoperfusion and ischemia. Ischemic/hypoxic injury and nephrotoxic insults are the most common causes of intrinsic AKI in the United States and are more common with an underlying comorbid condition; most are associated with cardiac, oncologic, urologic, renal, and genetic disorders or prematurity (Table 550.3 ). Many forms of glomerulonephritis, including postinfectious glomerulonephritis, lupus nephritis, Henoch-Schönlein purpura nephritis, membranoproliferative glomerulonephritis, and anti–glomerular basement membrane nephritis, can also cause intrinsic AKI. Severe and prolonged ischemic/hypoxic injury and nephrotoxic insult lead to acute tubular necrosis (ATN), seen most often in critically ill infants and children. Mechanisms leading to ischemic AKI include hypotension/intravascular volume depletion (hemorrhage, third-space fluid losses, diarrhea), decreased effective intravascular volume (heart failure, cirrhosis, hepatorenal syndrome, peritonitis, abdominal compartment syndrome), vasodilation/vasoconstriction (sepsis, hepatorenal syndrome), renal artery obstruction (thrombosis, embolization, stenosis), intrarenal artery disease (vasculitis, hemolytic-uremic syndrome, sickle cell anemia, transplant rejection), and impaired renal blood flow (cyclosporine, tacrolimus, angiotensin-converting enzyme [ACE] inhibitors, angiotensin-receptor blocking agents, radiocontrast agents).
Table 550.3
Major Endogenous and Exogenous Toxins Causing Acute Tubular Injury
G6PD, Glucose-6-phosphate dehydrogenase; HGPRT, hypoxanthine-guanine phosphoribosyltransferase; HMG-CoA, 3-hydroxy-3-methylglutaryl–coenzyme A; PNH, paroxysmal nocturnal hemoglobinuria.
From Sharfuddin AA, Weisbord SD, Palevsky PM, Molitoris BA: Acute kidney injury. In Skorecki K, Chertow GM, Marsden PA, et al (eds): Brenner & Rector's the kidney, 10/e, Philadelphia, 2016, Elsevier, Tab 31-5.
The typical pathologic feature of ATN is tubular cell necrosis, although significant histologic changes are not consistently seen in patients with clinical ATN. The mechanisms of injury in ATN can include alterations in intrarenal hemodynamics, tubular obstruction, and passive backleak of the glomerular filtrate across injured tubular cells into the peritubular capillaries.
Tumor lysis syndrome is a specific form of AKI related to spontaneous or chemotherapy-induced cell lysis in patients with lymphoproliferative malignancies. This disorder is primarily caused by obstruction of the tubules by uric acid crystals (see Chapters 522 and 523 ). Acute interstitial nephritis is another common cause of AKI and is usually a result of a hypersensitivity reaction to a therapeutic agent or various infectious agents (see Chapter 539.2 ).
Postrenal AKI includes a variety of disorders characterized by obstruction of the urinary tract. In neonates and infants, congenital conditions, such as posterior urethral valves and bilateral ureteropelvic junction obstruction, account for the majority of cases of AKI. Other conditions, such as urolithiasis, tumor (intraabdominal lesion or within the urinary tract), hemorrhagic cystitis, and neurogenic bladder, can cause AKI in older children and adolescents. In a patient with two functioning kidneys, obstruction must be bilateral to result in AKI. Relief of the obstruction usually results in recovery of renal function, except in patients with associated renal dysplasia or prolonged urinary tract obstruction.
Clinical Manifestations and Diagnosis
A carefully taken history is critical in defining the cause of AKI. An infant with a 3-day history of vomiting and diarrhea most likely has prerenal AKI caused by volume depletion, but hemolytic-uremic syndrome (HUS) must also be a consideration. A 6 yr old child with a recent pharyngitis who presents with periorbital edema, hypertension, and gross hematuria most likely has intrinsic AKI related to acute postinfectious glomerulonephritis. A critically ill child with a history of protracted hypotension or with exposure to nephrotoxic medications most likely has ATN. A neonate with a history of hydronephrosis seen on prenatal ultrasound studies and a palpable bladder most likely has congenital urinary tract obstruction, probably related to posterior urethral valves.
The physical examination must be thorough, with careful attention to volume status. Tachycardia, dry mucous membranes, and poor peripheral perfusion suggest an inadequate circulating volume and the possibility of prerenal AKI. Hypertension, peripheral edema, rales, and a cardiac gallop suggest volume overload and the possibility of intrinsic AKI from glomerulonephritis or ATN. The presence of a rash and arthritis might indicate systemic lupus erythematosus (SLE) or Henoch-Schönlein purpura nephritis. Palpable flank masses may be seen with renal vein thrombosis, tumors, cystic disease, or urinary tract obstruction.
Laboratory Findings
Laboratory abnormalities can include anemia (the anemia is usually dilutional or hemolytic, as in SLE, renal vein thrombosis, HUS); leukopenia (SLE, sepsis); thrombocytopenia (SLE, renal vein thrombosis, sepsis, HUS); hyponatremia (dilutional); metabolic acidosis; elevated serum concentrations of blood urea nitrogen, creatinine, uric acid, potassium, and phosphate (diminished renal function); and hypocalcemia (hyperphosphatemia).
The serum C3 level may be depressed (postinfectious glomerulonephritis, SLE, or membranoproliferative glomerulonephritis), and antibodies may be detected in the serum to streptococcal (poststreptococcal glomerulonephritis), nuclear (SLE), neutrophil cytoplasmic (granulomatosis with polyangiitis, microscopic polyarteritis), or glomerular basement membrane (Goodpasture disease) antigens.
The presence of hematuria, proteinuria, and red blood cell or granular urinary casts suggests intrinsic AKI, in particular glomerular disease and ATN. The presence of white blood cells and white blood cell casts with low-grade hematuria and proteinuria suggests tubulointerstitial disease. Urinary eosinophils may be present in some children with drug-induced tubulointerstitial nephritis.
Urinary indices may be useful in differentiating prerenal AKI from intrinsic AKI (Table 550.4 ). Patients whose urine shows an elevated specific gravity (>1.020), elevated urine osmolality (UOsm > 500 mOsm/kg), low urine sodium (UNa < 20 mEq/L), and fractional excretion of sodium < 1% (<2.5% in neonates) most likely have prerenal AKI. Those with a specific gravity of < 1.010, low urine osmolality (UOsm < 350 mOsm/kg), high urine sodium (UNa > 40 mEq/L), and fractional excretion of sodium > 2% (>10% in neonates) most likely have intrinsic AKI.
Table 550.4
Urinalysis, Urine Chemistries, and Osmolality in Acute Kidney Injury
| HYPOVOLEMIA | ACUTE TUBULAR NECROSIS | ACUTE INTERSTITIAL NEPHRITIS | GLOMERULONEPHRITIS | OBSTRUCTION | |
|---|---|---|---|---|---|
| Sediment | Bland, may have hyaline casts | Broad, brownish granular casts | White blood cells, eosinophils, cellular casts | Red blood cells, red blood cell casts | Bland or bloody |
| Protein | None or low | None or low | Minimal but may be increased with NSAIDs | Increased, > 100 mg/dL | Low |
| Urine sodium (mEq/L)* | <20 | >40 | >30 | <20 | <20 (acute) |
| >40 (few days) | |||||
| Urine osmolality (mOsm/kg) | >400 | <350 | <350 | >400 | <350 |
| Fractional excretion of sodium % † | <1 | >2 ‡ | Varies | <1 | <1 (acute) |
| >1 (few days) |
* The sensitivity and specificity of urine sodium of < 20 mEq/L in differentiating prerenal azotemia from acute tubular necrosis are 90% and 82%, respectively.
† Fractional excretion of sodium is the urine:plasma (U:P) ratio of sodium divided by U:P of creatinine ×100. The sensitivity and specificity of fractional excretion of sodium of < 1% in differentiating prerenal azotemia from acute tubular necrosis are 96% and 95%, respectively.
‡ The fractional excretion of sodium may be < 1% in acute tubular necrosis secondary to radiocontrast material or rhabdomyolysis.
NSAIDs, nonsteroidal antiinflammatory drugs.
From Singri N, Ahya SN, Levin ML: Acute renal failure, JAMA 289:747-751, 2003.
Chest radiography may reveal cardiomegaly, pulmonary congestion (fluid overload), or pleural effusions. Renal ultrasonography can reveal hydronephrosis and/or hydroureter, which suggest urinary tract obstruction, or nephromegaly, consistent with intrinsic renal disease. Renal biopsy can ultimately be required to determine the precise cause of AKI in patients who do not have clearly defined prerenal or postrenal AKI.
Although serum creatinine is used to measure kidney function, it is an insensitive and delayed measure of decreased kidney function following AKI. Other biomarkers under investigation include changes in plasma neutrophil gelatinase–associated lipocalin and cystatin C levels and urinary changes in neutrophil gelatinase-associated lipocalin, interleukin 18, and kidney injury molecule-1.
Treatment
Medical Management
Complications of acute kidney injury are noted in Table 550.5 . In infants and children with urinary tract obstruction, such as in a newborn with suspected posterior ureteral valves, a bladder catheter should be placed immediately to ensure adequate drainage of the urinary tract. The placement of a bladder catheter may also be considered in nonambulatory older children and adolescents to accurately monitor urine output during AKI; however, precautions to prevent iatrogenic infection should be taken.
Table 550.5
Common Complications of Acute Kidney Injury
| METABOLIC | CARDIOPULMONARY | GASTROINTESTINAL | NEUROLOGIC | HEMATOLOGIC | INFECTIOUS | OTHER |
|---|---|---|---|---|---|---|
| Hyperkalemia |
Pulmonary edema Arrhythmias Pericarditis Pericardial effusion Hypertension Myocardial infarction Pulmonary embolism |
Nausea |
Neuromuscular irritability Asterixis Seizures Mental status changes |
Anemia | Pneumonia |
Hiccups Elevated parathyroid hormone level |
|
Metabolic acidosis Hyponatremia Hypocalcemia Hyperphosphatemia Hypermagnesemia Hyperuricemia |
Vomiting Malnutrition Hemorrhage |
Bleeding |
Septicemia Urinary tract infection |
|||
| Low total triiodothyronine and thyroxine levels | ||||||
| Normal thyroxine level |
From Sharfuddin AA, Weisbord SD, Palevsky PM, Molitoris BA: Acute kidney injury. In Skorecki K, Chertow GM, Marsden PA, et al (eds): Brenner & Rector's the kidney, 10/e, Philadelphia, 2016, Elsevier, Tab 31-14.
Determination of the volume status is of critical importance when initially evaluating a patient with AKI. If there is no evidence of volume overload or cardiac failure, the intravascular volume should be expanded by intravenous administration of isotonic saline, 20 mL/kg over 30 min. In the absence of blood loss or hypoproteinemia, colloid-containing solutions are not required for volume expansion. Severe hypovolemia may require additional fluid boluses (see Chapters 69 , 70 , and 88 ). Determination of the central venous pressure may be helpful if adequacy of the blood volume is difficult to determine. After volume resuscitation, hypovolemic patients generally void within 2 hr; failure to do so suggests intrinsic or postrenal AKI. Hypotension caused by sepsis requires vigorous fluid resuscitation followed by a continuous infusion of vasopressors.
Diuretic therapy should be considered only after the adequacy of the circulating blood volume has been established. Furosemide (2-4 mg/kg) may be administered as a single intravenous dose. Bumetanide (0.1 mg/kg) may be given as an alternative to furosemide. If urine output is not improved, then a continuous diuretic infusion may be considered. To increase renal cortical blood flow, many clinicians administer dopamine (2-3 µg/kg/min) in conjunction with diuretic therapy, although no controlled data support this practice. There is little evidence that diuretics or dopamine can prevent AKI or hasten recovery. Mannitol may be effective in the prevention of pigment (myoglobin, hemoglobin)-induced renal failure. Atrial natriuretic peptide may be of value in preventing or treating AKI, although there is little pediatric evidence to support its use.
If there is no response to a diuretic challenge, diuretics should be discontinued and fluid restriction is essential. Patients with a relatively normal intravascular volume should initially be limited to 400 mL/m2 /24 hr (insensible losses) plus an amount of fluid equal to the urine output for that day. Extrarenal (blood, GI tract) fluid losses should be replaced, milliliter for milliliter, with appropriate fluids. Markedly hypervolemic patients can require further fluid restriction, omitting the replacement of insensible fluid losses, urine output, and extrarenal losses to diminish the expanded intravascular volume. Fluid intake, urine and stool output, body weight, and serum chemistries should be monitored on a daily basis.
In AKI, rapid development of hyperkalemia (serum potassium level > 6 mEq/L) can lead to cardiac arrhythmia, cardiac arrest, and death. The earliest electrocardiographic change seen in patients with developing hyperkalemia is the appearance of peaked T waves. This may be followed by widening of the QRS intervals, ST segment depression, ventricular arrhythmias, and cardiac arrest (see Chapter 450.2 ). Procedures to deplete body potassium stores should be initiated when the serum potassium value rises to > 6.0 mEq/L. Exogenous sources of potassium (dietary, intravenous fluids, total parenteral nutrition) should be eliminated. Sodium polystyrene sulfonate resin (Kayexalate), 1 g/kg, should be given orally or by retention enema. This resin exchanges sodium for potassium and can take several hours to take effect. A single dose of 1 g/kg can be expected to lower the serum potassium level by about 1 mEq/L. Resin therapy may be repeated every 2 hr, the frequency being limited primarily by the risk of sodium overload.
More severe elevations in serum potassium (>7 mEq/L), especially if accompanied by electrocardiographic changes, require emergency measures in addition to Kayexalate. The following agents should be administered:
- Calcium gluconate 10% solution, 100 mg/kg/dose (maximum 3000 mg/dose)
- Sodium bicarbonate, 1-2 mEq/kg intravenously, over 5-10 min
- Regular insulin, 0.1 units/kg, with glucose 50% solution, 1 mL/kg, over 1 hr
Calcium gluconate counteracts the potassium-induced increase in myocardial irritability but does not lower the serum potassium level. Administration of sodium bicarbonate, insulin, or glucose lowers the serum potassium level by shifting potassium from the extracellular to the intracellular compartment. A similar effect has been reported with the acute administration of β-adrenergic agonists in adults, but there are no controlled data in pediatric patients. Because the duration of action of these emergency measures is just a few hours, persistent hyperkalemia should be managed by dialysis.
Mild metabolic acidosis is common in AKI because of the retention of hydrogen ions, phosphate, and sulfate, but it rarely requires treatment. If acidosis is severe (arterial pH < 7.15; serum bicarbonate < 8 mEq/L) or contributes to significant hyperkalemia, treatment is indicated. The acidosis should be corrected partially by the intravenous route, generally by giving enough bicarbonate to raise the arterial pH to 7.20 (which approximates a serum bicarbonate level of 12 mEq/L). The remainder of the correction may be accomplished by oral administration of sodium bicarbonate after normalization of the serum calcium and phosphorus levels. Correction of metabolic acidosis with intravenous bicarbonate can precipitate tetany in patients with renal failure because rapid correction of acidosis reduces the ionized calcium concentration.
Hypocalcemia is primarily treated by lowering the serum phosphorus level. Calcium should not be given intravenously, except in cases of tetany, to avoid deposition of calcium salts into tissues. Patients should be instructed to follow a low-phosphorus diet, and phosphate binders should be orally administered to bind any ingested phosphate and increase the GI phosphate excretion. Common agents include sevelamer (Renagel), calcium carbonate (Tums tablets or Titralac suspension), and calcium acetate (PhosLo). Aluminum-based binders, commonly employed in the past, should be avoided because of the risk of aluminum toxicity.
Hyponatremia is most commonly a dilutional disturbance that must be corrected by fluid restriction rather than sodium chloride administration. Administration of hypertonic (3%) saline should be limited to patients with symptomatic hyponatremia (seizures, lethargy) or those with a serum sodium level < 120 mEq/L. Acute correction of the serum sodium to 125 mEq/L (mmol/L) should be accomplished using the following formula:
mEq sodium required =0.6×weight in kg×(125−serum sodium in mEq/L).
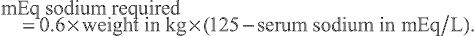
AKI patients are predisposed to GI bleeding because of uremic platelet dysfunction, increased stress, and heparin exposure if treated with hemodialysis or continuous renal replacement therapy. Oral or intravenous H2 blockers such as ranitidine are commonly administered to prevent this complication.
Hypertension can result from hyperreninemia associated with the primary disease process and/or expansion of the extracellular fluid volume and is most common in AKI patients with acute glomerulonephritis or HUS. Salt and water restriction is critical, and diuretic administration may be useful (see Chapter 472 ). Isradipine (0.05-0.15 mg/kg/dose, maximum dose 5 mg qid) may be administered for a relatively rapid reduction in blood pressure. Longer-acting oral agents such as calcium channel blockers (amlodipine, 0.1-0.6 mg/kg/24 hr qd or divided bid) or β blockers (labetalol, 4-40 mg/kg/24 hr divided bid or tid) may be helpful in maintaining control of the blood pressure. Children with severe symptomatic hypertension (hypertensive urgency or emergency) should be treated with continuous infusions of nicardipine (0.5-5.0 µg/kg/min), sodium nitroprusside (0.5-10.0 µg/kg/min), labetalol (0.25-3.0 mg/kg/hr), or esmolol (150-300 µg/kg/min) and converted to intermittently dosed antihypertensives when more stable.
Neurologic symptoms in AKI can include headache, seizures, lethargy, and confusion (encephalopathy). Potential etiologic factors include hypertensive encephalopathy, hyponatremia, hypocalcemia, cerebral hemorrhage, cerebral vasculitis, and the uremic state. Benzodiazepines are the most effective agents in acutely controlling seizures, and subsequent therapy should be directed toward the precipitating cause.
The anemia of AKI is generally mild (hemoglobin 9-10 g/dL) and primarily results from volume expansion (hemodilution). Children with HUS, SLE, active bleeding, or prolonged AKI can require transfusion of packed red blood cells if their hemoglobin level falls below 7 g/dL. In hypervolemic patients, blood transfusion carries the risk of further volume expansion, which can precipitate hypertension, heart failure, and pulmonary edema. Slow (4-6 hr) transfusion with packed red blood cells (10 mL/kg) diminishes the risk of hypervolemia. The use of fresh, washed red blood cells minimizes the acute risk of hyperkalemia, and the chronic risk of sensitization if the patient becomes a future candidate for renal replacement therapy. In the presence of severe hypervolemia or hyperkalemia, blood transfusions are most safely administered during dialysis or ultrafiltration.
Nutrition is of critical importance in children who develop AKI. In most cases, sodium, potassium, and phosphorus should be restricted. Protein intake should be moderately restricted while maximizing the caloric intake to minimize the accumulation of nitrogenous wastes. In critically ill patients with AKI, parenteral hyperalimentation with essential amino acids should be considered.
Dialysis
Indications for dialysis in AKI include the following:
- Anuria/oliguria
- Volume overload with evidence of hypertension and/or pulmonary edema refractory to diuretic therapy
- Persistent hyperkalemia
- Severe metabolic acidosis unresponsive to medical management
- Uremia (encephalopathy, pericarditis, neuropathy)
- Calcium:phosphorus imbalance, with hypocalcemic tetany that cannot be controlled by other measures
An additional indication for dialysis is the inability to provide adequate nutritional intake because of the need for severe fluid restriction. In patients with AKI, dialysis support may be necessary for days or for up to 12 wk. Many patients with AKI require dialysis support for 1-3 wk. Table 550.6 lists the advantages and disadvantages of the three types of dialysis.
Table 550.6
Comparison of Peritoneal Dialysis, Intermittent Hemodialysis, and Continual Renal Replacement Therapy
| PD | IHD | CRRT | |
|---|---|---|---|
| BENEFITS | |||
| Fluid removal | + | ++ | ++ |
| Urea and creatinine clearance | + | ++ | + |
| Potassium clearance | ++ | ++ | + |
| Toxin clearance | + | ++ | + |
| COMPLICATIONS | |||
| Abdominal pain | + | − | − |
| Bleeding | − | + | + |
| Dysequilibrium | − | + | − |
| Electrolyte imbalance | + | + | + |
| Need for heparinization | − | + | +/− |
| Hyperglycemia | + | − | − |
| Hypotension | + | ++ | + |
| Hypothermia | − | − | + |
| Central line infection | − | + | + |
| Inguinal or abdominal hernia | + | − | − |
| Peritonitis | + | − | − |
| Protein loss | + | − | − |
| Respiratory compromise | + | − | − |
| Vessel thrombosis | − | + | + |
PD, peritoneal dialysis; IHD, intermittent hemodialysis; CRRT, continual renal replacement therapy.
Adapted from Rogers MC: Textbook of pediatric intensive care, Baltimore, 1992, Williams & Wilkins.
Intermittent hemodialysis is useful in patients with a relatively stable hemodynamic status. This highly efficient process accomplishes both fluid and electrolyte removal in sessions of 3-4 hr using a pump-driven extracorporeal circuit and large central venous catheter. Intermittent hemodialysis may be performed 3-7 times per week based on the patient's fluid and electrolyte balance.
Peritoneal dialysis is most commonly employed in neonates and infants with AKI, although this modality may be used in children and adolescents of all ages. Hyperosmolar dialysate is infused into the peritoneal cavity via a surgically or percutaneously placed peritoneal dialysis catheter. The fluid is allowed to dwell for 45-60 min and is then drained from the patient by gravity (manually or with the use of machine-driven cycling), accomplishing fluid and electrolyte removal. Cycles are repeated for 8-24 hr/day based on the patient's fluid and electrolyte balance. Anticoagulation is not necessary. Peritoneal dialysis is contraindicated in patients with significant abdominal pathology.
Continuous renal replacement therapy (CRRT) is useful in patients with an unstable hemodynamic status, concomitant sepsis, or multiorgan failure in the intensive care setting. CRRT is an extracorporeal therapy in which fluid, electrolytes, and small- and medium-size solutes are continuously removed from the blood (24 hr/day) using a specialized pump-driven machine. Usually, a double-lumen catheter is placed into the internal jugular or femoral vein. The patient is then connected to the pump-driven CRRT circuit, which continuously passes the patient's blood across a highly permeable filter.
CRRT may be performed in three basic fashions. In continuous venovenous hemofiltration, a large volume of fluid is driven by systemic or pump-assisted pressure across the filter, bringing with it by convection other molecules, such as urea, creatinine, phosphorus, and uric acid. The blood volume is reconstituted by an intravenous infusion of a replacement fluid having a desirable electrolyte composition similar to that of blood. Continuous venovenous hemofiltration dialysis uses the principle of diffusion by circulating dialysate in a countercurrent direction on the ultrafiltrate side of the membrane. No replacement fluid is used. Continuous hemodiafiltration employs both replacement fluid and dialysate, offering the most effective solute removal of all forms of CRRT.
Table 550.6 compares the relative risks and benefits of the various renal replacement therapies.
Prognosis
The mortality rate in children with AKI is variable and depends entirely on the nature of the underlying disease process rather than on the renal failure itself. Children with AKI caused by a renal-limited condition such as postinfectious glomerulonephritis have a very low mortality rate (<1%); those with AKI related to multiorgan failure have a very high mortality rate (>50%).
The prognosis for recovery of renal function depends on the disorder that precipitated AKI. Recovery of renal function is likely after AKI resulting from prerenal causes, ATN, acute interstitial nephritis, or tumor lysis syndrome. Complete recovery of renal function is unusual when AKI results from most types of rapidly progressive glomerulonephritis, bilateral renal vein thrombosis, or bilateral cortical necrosis. Medical management may be necessary for a prolonged period to treat the sequelae of AKI, including chronic renal insufficiency, hypertension, renal tubular acidosis, and urinary concentrating defect.
Bibliography
Barasch J, Zager R, Bonventre JV. Acute kidney injury: a problem of definition. Lancet . 2017;389:779–781.
Ciccia E, Devarajan P. Pediatric acute kidney injury: prevalence, impact and management challenges. Int J Nephrol Renovasc Dis . 2017;10:77–84.
Devarajan P. Acute kidney injury: still misunderstood and misdiagnosed. Nat Rev Nephrol . 2017;13(3):137–138.
Kaddourah A, Basu RK, Bagshaw SM, et al. Epidemiology of acute kidney injury in critically ill children and young adults. N Engl J Med . 2017;376(1):11–20.
Kellum JA, Chawla LS, Keener C, et al. The effects of alternative resuscitation strategies on acute kidney injury in patients with septic shock. Am J Respir Crit Care Med . 2016;193(3):281–287.
McCaffrey J, Dhakal AK, Milford DV, et al. Recent developments in the detection and management of acute kidney injury. Arch Dis Child . 2017;102(1):91–96.
Selewski DT, Charlton JR, Jetton JG, et al. Neonatal acute kidney injury. Pediatrics . 2015;136(2):e463–e473.
Vanmassenhove J, Kielstein J, Jörres A, Van Biesen W. Intensive care medicine and renal transplantation 1—management of patients at risk of acute kidney injury. Lancet . 2017;389:2139–2148.
Zarbock A, Kellum JA, Schmidt C, et al. Effect of early vs delayed initiation of renal replacement therapy on mortality in critically ill patients with acute kidney injury. JAMA . 2016;315(20):2190–2199.
Chronic Kidney Disease
Donna J. Claes, Mark Mitsnefes
Chronic kidney disease (CKD) is determined by the presence of kidney damage and level (or severity) of kidney function (glomerular filtration rate, or GFR; Tables 550.7 and 550.8 ). End-stage renal disease (ESRD) is an administrative term in the United States; it is used to define all patients who are treated with dialysis or kidney transplantation, and is a subset of patients with stage 5 CKD.
Table 550.7
Criteria for Definition of Chronic Kidney Disease (NKF KDOQI Guidelines)
NKF KDOQI, National Kidney Foundation Kidney Disease Outcomes Quality Initiative.
Table 550.8
GFR, glomerular filtration rate; NKF KDOQI, National Kidney Foundation Kidney Disease Outcomes Quality Initiative.
The pediatric CKD prevalence is approximately 18 per 1 million children. The prognosis for the infant, child, or adolescent with CKD has improved dramatically since the 1970s, mostly because of improved medical management, dialysis techniques, and kidney transplantation. Yet, childhood-onset ESRD still carries significant morbidity and a 30-fold increased mortality rate as compared with healthy peers, with cardiovascular and infectious diseases as the leading causes of death.
Etiology
The etiology of pediatric CKD may be the result of congenital, acquired, inherited, or metabolic renal disease; causes of kidney disease in children are typically subdivided as being either nonglomerular or glomerular in origin (Table 550.9 ). The underlying cause correlates with the age at the time of diagnosis. In children < 5 yr of age, CKD is most commonly a result of congenital abnormalities of the kidney and urinary tract (i.e., renal hypoplasia, dysplasia, or obstructive uropathy) and is often diagnosed with prenatal ultrasonography. In children older than 5 yr of age, acquired or inherited forms of glomerulonephritis predominate.
Table 550.9
Etiologies of Pediatric Chronic Kidney Disease
Pathogenesis
In addition to progressive injury with ongoing structural or metabolic genetic diseases, renal injury can progress despite removal of the original insult.
Hyperfiltration injury may be an important final common pathway of glomerular destruction, independent of the underlying cause of renal injury. As nephrons are lost, the remaining nephrons undergo structural and functional hypertrophy characterized by an increase in glomerular blood flow. The driving force for glomerular filtration is thereby increased in the surviving nephrons. Although this compensatory hyperfiltration temporarily preserves total renal function, it can cause progressive damage to the surviving glomeruli, possibly by a direct effect of the elevated hydrostatic pressure on the integrity of the capillary wall and/or the toxic effect of increased protein traffic across the capillary wall. Over time, the remaining nephrons suffer an increased excretory burden, resulting in a vicious cycle of increasing glomerular blood flow and hyperfiltration injury.
Other pathologic etiologies of chronic kidney disease include proteinuria, hypertension, hyperphosphatemia, and hyperlipidemia. Proteinuria itself can contribute to renal functional decline. Proteins that traverse the glomerular capillary wall can exert a direct toxic effect on tubular cells and recruit monocytes and macrophages, enhancing the process of glomerular sclerosis and tubulointerstitial fibrosis. Uncontrolled hypertension can exacerbate the disease progression by causing arteriolar nephrosclerosis and by increasing the hyperfiltration injury. Hyperphosphatemia can increase the progression of disease by leading to calcium phosphate deposition in the renal interstitium and blood vessels. Hyperlipidemia, a common condition in CKD patients, can adversely affect glomerular function through oxidant-mediated injury.
CKD is viewed as a continuum of disease, with increasing biochemical and clinical manifestations as renal function deteriorates. Regardless of the etiology, the progression of tubulointerstitial fibrosis is the primary determinant of CKD progression.
Clinical Manifestations
Table 550.10 outlines the pathophysiologic manifestations of CKD. The clinical presentation of CKD is varied and depends on the underlying etiology and CKD stage (Fig. 550.2 ). CAKUT and some genetic forms of renal disease (i.e., familial nephronophthisis) demonstrate growth failure, vomiting, and polyuria with associated polydipsia. Urinary tract infection can also be common in those with urologic abnormalities. Glomerular forms of CKD often present with edema, hypertension, hematuria, and proteinuria; in severe forms of glomerulonephritis, malnutrition can be seen. As renal deterioration advances in severity, patients can develop uremic symptoms (i.e., worsening fatigue, weakness, nausea, vomiting, anorexia, and poor sleep patterns), as well as edema, hypertension, and other findings of fluid overload, regardless of the cause of CKD.
Table 550.10
Pathophysiology of Chronic Kidney Disease
| MANIFESTATION | MECHANISMS |
|---|---|
| Accumulation of nitrogenous waste products | Decrease in glomerular filtration rate |
| Acidosis | |
| Sodium wasting | |
| Urinary concentrating defect | |
| Hyperkalemia | |
| Renal osteodystrophy | |
| Growth retardation | |
| Anemia | |
| Bleeding tendency | Defective platelet function |
| Infection | |
| Decreased academic achievement, attention regulation, or executive functioning | |
| Gastrointestinal symptoms (feeding intolerance, abdominal pain) | |
| Hypertension | |
| Hyperlipidemia | |
| Cardiomyopathy | |
| Glucose intolerance | Tissue insulin resistance |
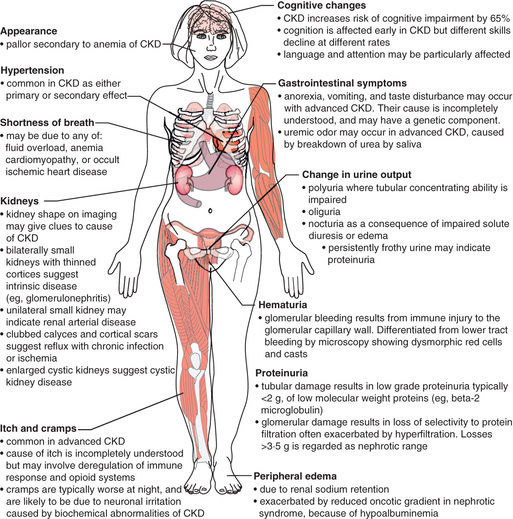
Physical examination in CKD should focus on overall growth and development, with special attention and/or evaluation of the blood pressure, as well as the skin (pallor) and the extremities (edema; bony abnormalities of rickets seen in untreated renal osteodystrophy).
Laboratory Findings
Laboratory findings can include elevations in blood urea nitrogen and serum creatinine in addition to hyperkalemia, hyponatremia (secondary to either renal salt wasting versus volume overload), hypernatremia (loss of free water), acidosis, hypocalcemia, hyperphosphatemia, and an elevation in uric acid. Patients with heavy proteinuria can have hypoalbuminemia. A complete blood cell count may show a normochromic, normocytic anemia. Dyslipidemia is commonly seen. In children with glomerulonephritis, the urinalysis (UA) shows hematuria and proteinuria, whereas in children with congenital lesions such as renal dysplasia, the UA often has a low specific gravity with minimal other abnormalities.
Renal function can be measured or estimated by the GFR. Inulin clearance is the gold standard to measure the GFR, but it is no longer readily available. Other methods for measuring the GFR in clinical practice include using iohexol or various radioisotopes (99m Tc-DTPA, 51 Cr-EDTA, or 125 Iothalamate). However, estimating the GFR by endogenous markers (e.g., creatinine and/or cystatin C) is the most utilized method to understand the severity of renal disease. A new bedside creatinine-based estimating equation of estimated GFR (mL/min/1.73 m2 ) = 0.43 × height (cm)/serum creatinine (mg/dL) has been validated in a pediatric CKD population of children aged 1-16 yr and whose GFR was between 15 and 90 mL/min/1.73 m2 .
Treatment and Management
CKD treatment is supportive, with an aim to screen for and treat various metabolic complications of CKD in hopes to improve the quality of life and potentially slow the progression of renal dysfunction. Children with CKD should be treated at a pediatric center capable of supplying multidisciplinary services, including medical, nursing, social service, nutritional, and psychological support.
CKD management requires close monitoring of blood studies, urine studies (including quantitative measurement for proteinuria using either a spot urine protein/urine creatinine ratio or 24 hr urine collection), and overall clinical symptomatology. Ambulatory blood pressure monitoring (ABPM) over 24 hr, the gold standard of blood pressure evaluation, is recommended in patients with renal disease to diagnose and treat hypertension, especially masked hypertension. Masked hypertension (defined as a normal office blood pressure but abnormal ABPM) is seen in up to 35% of pediatric predialysis CKD patients and carries a 4-fold increased risk of having left ventricular hypertrophy (LVH).
Nutrition
Nutritional management by a dietician experienced in pediatric renal patients is recommended by the National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI). Patients should receive 100% of the estimated energy requirement for age, individually adjusted for the physical activity level, body mass index, and response in the rate of weight gain or loss. When oral supplemental nutrition with increased calories or fluid volume is insufficient, tube feeding (by nasogastric tubes or gastrostomy tubes) should be considered. Calories should be balanced between carbohydrate, unsaturated fat in physiologic ranges (per dietary reference intake [DRI]), and protein. Dietary protein restriction is not suggested for children with CKD because of the concern about adverse effects on growth and development; in fact, the recommended protein intake is often 100% (or more for those receiving dialysis) of the DRI for ideal weight for children. Children with CKD stages 2-5 should receive 100% of the DRI of vitamins and trace elements; water-soluble vitamin supplements are often required for patients receiving dialysis.
CKD Mineral and Bone Disorder (CKD-MBD)
Chronic kidney disease is characterized by systemic disorders of calcium, phosphorus, PTH, and vitamin D metabolism that can not only lead to bone disorders (renal osteodystrophy) but also vascular and soft tissue calcification (Fig. 550.3 ). Efforts have focused on the role of the hormone fibroblast growth factor 23 (FGF23) and its cofactor, Klotho, in CKD-MBD. An elevated FGF23 results in increased urinary phosphate excretion and suppression of 1-α-hydroxylase activity, leading to reduced 1,25-dihydroxycholecalciferol (1,25OH2 D) values and increased PTH secretion. Elevated FGF23 is the first sign of altered osteocyte function in pediatric and adult CKD, is seen as early as CKD stage 2 (GFR 60-90 mL/min/1.73 m2), and occurs despite normal calcium, phosphorus, PTH, and 1,25OH2 D levels. With a continued loss of renal function, further FGF23 elevation results in the development of secondary hyperparathyroidism (low 1,25OH2 D, with hypocalcemia, hyperphosphatemia, and elevated PTH values).
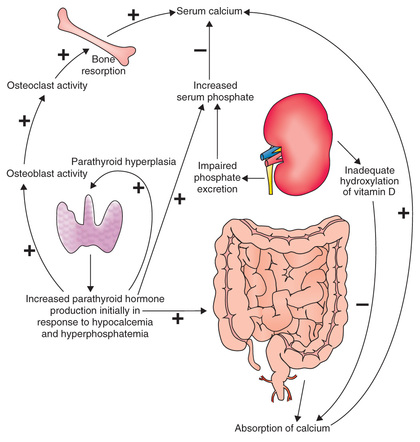
Renal osteodystrophy is characterized by abnormalities in bone turnover (high versus low), mineralization, and bone volume. High-turnover bone disease, or osteitis fibrosa cystica, is the most common condition seen in advanced pediatric CKD, with characteristic laboratory findings (hypocalcemia, hyperphosphatemia, and elevated alkaline phosphatase and PTH values) and radiographic findings (subperiosteal bone resorption, metaphyseal widening). Clinical manifestations may include bone pain, fractures with minor trauma, and various bony abnormalities (rachitic changes, varus and valgus deformities of the long bones, and slipped capital femoral epiphyses, or SCFE). In contrast, low-turnover bone disease (adynamic renal osteodystrophy) is associated with PTH oversuppression, hypercalcemia, and low alkaline phosphatase activity; it is more commonly seen in pediatric dialysis patients receiving treatment for secondary hyperparathyroidism. Defective bone mineralization occurs in states of either high bone turnover (mixed lesion) or low to normal bone turnover (osteomalacia). In terms of bone volume, most pediatric CKD patients have normal to high bone volume on bone histomorphometry unless they were exposed to prolonged corticosteroid use.
Vascular calcification in CKD-MBD typically occurs within the vascular media, which is in contrast to the atherosclerotic plaques that form within the vascular intima in patients without renal disease but with traditional cardiovascular risk factors (hypertension, diabetes/obesity, cigarette smoking, and dyslipidemia). Vascular calcification in CKD has been associated with hypercalcemia, hyperphosphatemia, and an elevated calcium-phosphorus product (Ca×PO4 ;); yet, studies of adult and pediatric patients with mild to moderate CKD have noted findings of vascular calcification despite normal serum calcium and phosphorus values. The cause of vascular calcification in CKD is not completely understood and is being actively studied; the proposed pathophysiologic etiology involves the transition of vascular smooth muscle cells to osteoblast-like cells in response to trigger(s) that are currently unknown.
Treatment for CKD-MBD is guided by the clinical assessment of calcium, phosphorus, 25OH Vitamin D, and PTH. The goals of treatment are to normalize mineral metabolism with the goal of improving growth, reducing bone deformities and fragility, and reducing vascular and other soft tissue calcification. This is typically accomplished with reduced phosphorus intake, normalization of 25OH vitamin D, and use of active vitamin D sterol agents.
CKD patients of all ages should typically follow a low-phosphorus diet with the goal of maintaining age-appropriate serum phosphorus values. Infants should be provided with a low-phosphorus formula (Similac PM 60/40). Phosphate binders (given with meals) are used to enhance GI phosphate excretion, and at present are recommended to be started at the onset of hyperphosphatemia. Phosphate binders should be adjusted to maintain normal serum calcium and phosphorus levels, and to ensure that the recommended total daily intake of calcium is not exceeded. Phosphate binders can be either calcium-based (calcium carbonate, calcium acetate) or non–calcium-based (sevelamer and ferric citrate). Because aluminum may be absorbed from the GI tract and can lead to aluminum toxicity, aluminum-based binders should be avoided.
Correcting 25OH Vitamin D insufficiency can delay the onset of secondary hyperparathyroidism in predialysis CKD patients, and it improves bone mineralization. 25OH vitamin D provides a substrate for the formation of 1,25OH2 D, and has been shown to directly suppress PTH production at the level of the parathyroid gland. US-based pediatric CKD treatment guidelines define 25OH vitamin D sufficiency as a serum value of ≥ 30 ng/mL; ergocalciferol or cholecalciferol are typically recommended to treat insufficient 25OH vitamin D.
Active vitamin D sterols have been traditionally indicated when (1) 1,25OH2 D levels fall below the established goal range for the child's particular stage of CKD, (2) PTH levels increase above the established goal range for CKD stage (after correcting for insufficient 25OH vitamin D), or (3) in patients with elevated PTH levels and hypocalcemia. Vitamin D sterols increase calcium and phosphorus absorption from the GI tract and are effective in reducing PTH values. Calcitriol is the most well-known and studied active vitamin D sterol; newer agents such as paricalcitol and doxercalciferol have less intestinal calcium and phosphorus reabsorption and are used in CKD patients predisposed to hypercalcemia. The ideal PTH target at which to initiate and monitor active vitamin D sterol therapy is debated, particularly in the predialysis CKD population.
Fluid and Electrolyte Management
Infants and children with renal dysplasia may be polyuric, with significant urinary sodium and free water losses. These children benefit from high-volume, low-caloric-density feedings with sodium supplementation. Children with high blood pressure or edema benefit from sodium restriction and diuretic therapy. Fluid restriction is necessary in severe cases of nephrotic syndrome or when renal function worsens to the point of requiring dialysis.
Hyperkalemia can develop with severe deterioration in renal function, as well as in patients with moderate renal insufficiency who have excessive dietary potassium intake, severe acidosis, or hyporeninemic hypoaldosteronism (related to destruction of the renin-secreting juxtaglomerular apparatus). Hyperkalemia may be treated by the restriction of dietary potassium intake, administration of oral alkalinizing agents, and/or use of Kayexalate. Sodium zirconium and patiromer are additional oral agents used to treat hyperkalemia in adults.
Metabolic acidosis develops because of a decreased net acid excretion by the failing kidneys. Either Bicitra (1 mEq sodium citrate/mL) or sodium bicarbonate tablets (650 mg = 8 mEq of base) may be used to maintain the serum bicarbonate level ≥ 22 mEq/L.
Linear Growth
Short stature is a significant long-term sequela of childhood CKD. CKD results in an apparent growth hormone–resistant state, with elevated growth hormone levels but decreased insulin-like growth factor 1 levels and abnormalities of insulin-like growth factor–binding proteins.
Children with CKD who remain less than −2 SD for height despite optimal medical support (adequate caloric intake and effective treatment of renal osteodystrophy, anemia, and metabolic acidosis) may benefit from treatment with recombinant human growth hormone (rHuGH). rHuGH is given by daily subcutaneous injections and continues until the patient reaches the 50th percentile for midparental height, achieves a final adult height, or undergoes kidney transplantation. Long-term rHuGH treatment significantly improves the final adult height and induces persistent catch-up growth; some patients are able to achieve normal adult height.
Anemia
Anemia in patients with CKD is primarily the result of inadequate erythropoietin production by the failing kidneys and typically manifests when renal function falls below 40 mL/min/1.73 m2 . Other contributory factors for anemia in CKD include iron, folic acid, and/or vitamin B12 deficiency, and decreased erythrocyte survival secondary to uremia.
Anemia in pediatric CKD patients is defined when the hemoglobin falls to < 5% for age and gender; alternatively, anemia can be defined when the hemoglobin falls to < 11g/dL (ages 0.5-5 yr of age), < 11.5 g/dL (5-12 yr of age), < 12 g/dL (females > 12 yr of age, males 12-15 yr of age), and < 13 g/dL (males > 15 yr of age). Once anemia is diagnosed, the recommendation is to investigate for deficiencies in iron and/or other vitamins (i.e., vitamin B12 , folate). Iron supplementation (oral or intravenous) is recommended for patients who demonstrate a transferrin saturation (TSAT) ≤ 20% and ferritin ≤ 100 ng/mL.
Erythropoiesis-stimulating agents (ESAs) have decreased the need for transfusion in CKD patients, especially those receiving hemodialysis. Erythropoietin and darbepoetin alfa are common prescribed ESAs. All patients receiving ESA therapy should be provided with either oral or intravenous iron supplementation. Patients who appear to be resistant to ESA should be evaluated for iron deficiency, occult blood loss, a chronic infection or inflammatory state, vitamin B12 or folate deficiency, or bone marrow fibrosis related to secondary hyperparathyroidism.
Hypertension and Proteinuria
Hypertension in pediatric CKD can be secondary to volume overload and/or excessive renin production due to glomerular disease. Both hypertension and proteinuria have been independently associated with more rapid CKD progression in various pediatric CKD observational studies. The ESCAPE trial demonstrated that more aggressive blood pressure control delays CKD progression. In this study, participants with 24 hr mean arterial pressure (MAP) < 50th percentile for age and sex by ABPM had a 35% risk reduction of reaching the composite outcome (doubling of serum creatinine, eGFR of < 10 mL/min/1.73 m2 , or need for dialysis or kidney transplantation) as compared with those randomized to a conventional blood pressure target (MAP of 50–95% by ABPM); this effect was more notable in those with significant proteinuria.
Therapy for hypertension involves both dietary interventions and, often, pharmacologic agents. Dietary sodium restriction (< 2 g of sodium/24 hr) and lifestyle modifications that promote achieving a healthy weight are both important aspects of achieving good blood pressure control. Treatment guidelines recommend initiating pharmacologic antihypertensive therapy when systolic or diastolic blood pressures are > 90% for age, gender, and height. Once therapy is started, it is recommended to titrate medications to achieve a systolic and diastolic blood pressure < 50% for age, gender, and height, especially for those patients with proteinuria. ACE inhibitors (ACE; e.g., enalapril or lisinopril) and angiotensin II receptor blockers (ARB; e.g., losartan) are the antihypertensive medications of choice in all children with pediatric CKD, irrespective of the level of proteinuric renal disease, because of their potential ability to slow CKD and their superiority in controlling blood pressure as noted in various observational and research studies. It is important to closely monitor the renal function and electrolyte balance while using ACEs or ARBs, particularly in those with advanced CKD. Thiazide (hydrochlorothiazide, chlorothiazide) or loop diuretics (furosemide) can be helpful in controlling hypertension related to salt and fluid retention. Thiazides become ineffective when a patient's estimated GFR falls below 30 mL/min/1.73 m2 . Calcium channel blockers (amlodipine), β-blockers (propranolol, atenolol), and centrally acting agents (clonidine) may be useful as adjunctive agents in children with CKD whose blood pressure cannot be controlled using dietary sodium restriction, ACE inhibitors, and diuretics.
Immunizations
Children with CKD should receive all standard immunizations according to the schedule used for healthy children, with an exception to withhold live virus vaccines (such as measles, mumps, rubella, varicella) from those receiving immunosuppressive medications (i.e., kidney transplant recipients and some patients with glomerulonephritis). It is critical to make every attempt to administer live virus vaccines before kidney transplantation. All children with CKD should receive a yearly influenza vaccine. Data from a number of studies suggest that children with CKD might respond suboptimally to immunizations.
Adjustment in Drug Dose
Drugs excreted by the kidneys might need to be dose adjusted in CKD patients to maximize their effectiveness and minimize the risk of toxicity. Strategies in dosage adjustment include lengthening of the interval between doses or decreasing the absolute dose, or both.
Progression of Disease
The timing of CKD progression from minimal renal injury to the onset of ESRD is variable. The median loss of GFR in children enrolled in the Chronic Kidney Disease in Children (CKiD) study is 1.5 mL/min/1.73 m2 /yr (nonglomerular CKD etiology) versus 4.3 mL/min/1.73 m2 /yr (glomerular CKD etiology). Nonmodifiable risk factors associated with more rapid CKD progression include older age, glomerular etiology of renal disease, CKD severity, and onset of puberty. In terms of potential modifiable risk factors (in addition to an elevated blood pressure), persistent nephrotic range proteinuria, anemia, and dyslipidemia, as well as no ACE/ARB use, were important predictors of CKD progression.
In addition to addressing and treating the risk factors as noted above, prompt treatment of infectious complications and episodes of dehydration can minimize additional loss of renal parenchyma. Other potentially beneficial recommendations include tobacco avoidance, prevention of obesity, and avoidance of potential nephrotoxic medications (which includes nonsteroidal antiinflammatory medicines, various illegal street drugs, and herbal and/or homeopathic medications or supplements).
Bibliography
ESCAPE Trial Group. Strict blood pressure control and progression of renal failure in children. N Engl J Med . 2009;361:1639–1650.
Harambat J, van Stralen KJ, Kim JJ, et al. Epidemiology of chronic kidney disease in children. Pediatr Nephrol . 2012;27:363–373.
KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney Int Suppl . 2012;2:280–335.
KDIGO clinical practice guideline for the management of blood pressure in chronic kidney disease: blood pressure management in children with CKD ND. Kidney Int Suppl . 2012;2:372–376.
Levey AS, Coresh J. Chronic kidney disease. Lancet . 2012;379:165–178.
Mitsnefes MM, Laskin BL, Dahhou M, et al. Mortality risk among children initially treated with dialysis for end-stage kidney disease, 1990-2010. JAMA . 2013;309(18):1921–1928.
Mitsnefes MM. Cardiovascular disease in children with chronic kidney disease. J Am Soc Nephrol . 2012;23:578–585.
National Kidney Foundation. KDOQI clinical practice guideline and clinical practice recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis . 2007;50(3):471–530.
National Kidney Foundation. KDOQI clinical practice guideline for nutrition in children with CKD: 2008 update. Am J Kidney Dis . 2009;53(3 Suppl 2):S1–S124.
National Kidney Foundation, KDOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis . 2002;39(2 Suppl 1):s1–s266.
Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med . 2015;372(3):222–232.
Schaefer B, Wuhl E. Progression in chronic kidney disease and prevention strategies. Eur J Pediatr . 2012;1717(11):1579–1588.
The Medical Letter. Ferric citrate (auryxia) for hyperphosphatemia. Med Lett Drugs Ther . 2015;57(1483):166–167.
Warady BA, Abraham AG, Schwartz GJ, et al. Predictors of rapid progression of glomerular and non-glomerular kidney disease in children: the CKiD cohort. Am J Kidney Dis . 2015;65:878–888.
Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet . 2017;389:1238–1252.
Weir MR, Bakris GL, Bushinsky DA, et al. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med . 2015;372(3):21–220.
Wesseling-Perry K, Salusky IB. Chronic kidney disease: mineral and bone disorder in children. Semin Nephrol . 2013;33:169–179.
Wong CJ, Moxey-Mims M, Jerry-Fluker J, et al. CKiD (CKD in children) prospective cohort study: a review of current findings. Am J Kidney Dis . 2012;60:1002–1011.
Wong CS, et al. Association of proteinuria with race, cause of chronic kidney disease, and glomerular filtration rate in the chronic kidney disease in children study. Clin J Am Soc Nephrol . 2009;4:812–819.
End-Stage Renal Disease
Donna J. Claes, Stuart L. Goldstein
ESRD represents the state in which a patient's renal dysfunction has progressed to the point at which homeostasis and survival can no longer be sustained by maximal medical management. At this point, renal replacement therapy (dialysis or renal transplantation) becomes necessary. The ultimate goal for children with ESRD is successful kidney transplantation (see Chapter 551 ) because it provides the most normal lifestyle and improved mortality and morbidity rates.
In the United States, 75% of children with ESRD require dialysis prior to transplantation. It is recommended that plans for renal replacement therapy (RRT) be initiated when a child reaches stage 4 CKD (GFR < 30 mL/min/1.73 m2 ). Indications for initiating maintenance dialysis include diuretic-resistant fluid overload, severe fluid restrictions that inhibit the ability to provide appropriate nutrition sufficient for linear growth, uncontrolled electrolyte abnormalities (hyperkalemia, hyperphosphatemia, metabolic acidosis), and subjective findings of uremia (fatigue, weakness, nausea, vomiting, anorexia, and poor sleep patterns), especially if these symptoms are negatively affecting academic performance. Dialysis initiation should be considered as the GFR approaches 10-15 mL/min/1.73 m2 . Most nephrologists attempt to initiate dialysis early enough to prevent the development of severe fluid and electrolyte abnormalities, malnutrition, and uremic symptoms.
The dialysis modality selection must be individualized to fit the needs of each child.
In the United States, peritoneal dialysis is still the most utilized dialysis modality (~55%) as compared with hemodialysis (~44%); however, there is a temporal trend toward greater use of hemodialysis as the initial maintenance dialysis therapy. Age is a defining factor in dialysis modality selection: 85% of infants and children from birth to 5 yr of age initiate maintenance dialysis treatment using peritoneal dialysis, whereas 50% of children ≥ 13 yr of age initiate maintenance dialysis treatment with hemodialysis.
Peritoneal dialysis utilizes the patient's peritoneal membrane to transport fluid and solutes. Excess body water is removed by an osmotic gradient created by the relatively high dextrose concentration in the dialysis fluid; wastes are removed by diffusion from the peritoneal capillaries into the dialysis fluid. Access to the peritoneal cavity is achieved by a surgically inserted, tunneled catheter. Peritoneal dialysis may be provided either as continuous ambulatory peritoneal dialysis (CAPD) or as an automated therapy using a cycler (APD), which allows exchanges of peritoneal fluid to be performed automatically during sleep by a cycler machine. APD is the PD modality of choice in countries without cost restraints. Cycler-driven peritoneal dialysis therapy allows the child and family an uninterrupted day of activities (including decreased school interruption), a reduction in the number of dialysis catheter connections and disconnections (which decreases the risk of peritonitis), often less strict fluid and dietary restrictions, and a reduction in the time required by patients and parents to perform dialysis, reducing the risk of caregiver fatigue and burnout. Because peritoneal dialysis is not as efficient as hemodialysis, it must be performed at least 6 times per week. Contraindications to peritoneal dialysis use include anatomic abnormalities (e.g., significant surgical adhesions, omphalocele, gastroschisis, or bladder exstrophy), peritoneal injury (including injury secondary to previous severe peritoneal infections), or lack of an appropriate caregiver who can reliably perform peritoneal dialysis in the home.
Hemodialysis, unlike peritoneal dialysis, is usually performed in a hospital or outpatient clinic setting; home pediatric hemodialysis programs or programs that provide intensified hemodialysis are available but uncommon. Access to the child's circulation is achieved by a surgically created arteriovenous fistula (AVF), arteriovenous graft (AVG), or tunneled dual-lumen catheter. The internal jugular vein is the preferred catheter site; indwelling subclavian catheters can cause subclavian stenosis that limits that ability to utilize a future AVF or AVG in the ipsilateral arm. Each hemodialysis treatment is typically prescribed to provide appropriate solute clearance and fluid removal. Hemodialysis has historically been provided 3 times per week; however, more frequent dialysis treatments (up to 4-5 times per week) are seen in the United States. Intensified hemodialysis programs (such as short daily hemodialysis, intermittent nocturnal hemodialysis, and daily nocturnal hemodialysis) have demonstrated improved control of blood pressure, fluid overload, phosphorus, anemia, and improved growth. Many pediatric dialysis centers work with schools or have hospital-based teachers that can help hemodialysis patients stay on track academically. Contraindications to hemodialysis include inadequate vascular access.
Dialysis-associated infections (peritonitis, hemodialysis-related bloodstream infections) are the leading causes of hospitalization and the second-leading cause of death in pediatric dialysis patients.
Bibliography
Muller D, Goldstein SL. Hemodialysis in children with end-stage renal disease. Nat Rev Nephrol . 2011;7:650–658.
North American Pediatric Renal Transplant Cooperative Study (NAPRTCS). 2011 Annual report. Rockville, MD . [Available from] http://www.naprtcs.org .
Schaefer F, Warady BA. Peritoneal dialysis in children with end-stage renal disease. Nat Rev Nephrol . 2011;7:659–668.
Warady BA, Neu AM, Schaefer F. Optimal care of the infant, child, and adolescent on dialysis: 2014 update. Am J Kidney Dis . 2014;64:128–142.