Shock
David A. Turner, Ira M. Cheifetz
Shock is an acute process characterized by the body's inability to deliver adequate oxygen to meet the metabolic demands of vital organs and tissues. Insufficient oxygen at the tissue level is unable to support normal aerobic cellular metabolism, resulting in a shift to less efficient anaerobic metabolism. As shock progresses, increases in tissue oxygen extraction are unable to compensate for this deficiency in oxygen delivery, leading to progressive clinical deterioration and lactic acidosis. If inadequate tissue perfusion persists, adverse vascular, inflammatory, metabolic, cellular, endocrine, and systemic responses worsen physiologic instability.
Compensation for inadequate oxygen delivery involves a complex set of responses that attempt to preserve oxygenation of the vital organs (i.e., brain, heart, kidneys, liver) at the expense of other organs (i.e., skin, gastrointestinal tract, muscles). Of importance, the brain is especially sensitive to periods of poor oxygen supply given its lack of capacity for anaerobic metabolism. Initially, shock is often well compensated, but it may rapidly progress to an uncompensated state requiring more aggressive therapies to achieve clinical recovery. The combination of a continued presence of an inciting trigger and the body's exaggerated and potentially harmful neurohumoral, inflammatory, and cellular responses lead to the progression of shock. Irrespective of the underlying cause of shock, the specific pattern of response, pathophysiology, clinical manifestations, and treatment may vary significantly depending on the specific etiology (which may be unknown), the clinical circumstances, and an individual patient's biologic response to the shock state. Untreated shock causes irreversible tissue and organ injury (i.e., irreversible shock ) and, ultimately, death.
Epidemiology
Shock occurs in approximately 2% of all hospitalized infants, children, and adults in developed countries, and the mortality rate varies substantially depending on the etiology and clinical circumstances. Of patients who do not survive, most do not die in the acute hypotensive phase of shock, but rather as a result of associated complications and multiple-organ dysfunction syndrome (MODS). MODS is defined as any alteration of organ function that requires medical support for maintenance, and the presence of MODS in patients with shock substantially increases the probability of death. In pediatrics, educational efforts and the utilization of standardized management guidelines that emphasize early recognition and intervention along with the rapid transfer of critically ill patients to a pediatric intensive care unit (PICU) have led to decreases in the mortality rate for shock (Figs. 88.1 and 88.2 ).


Types of Shock
Shock classification systems generally define 5 major types of shock: hypovolemic, cardiogenic, distributive, obstructive, and septic (Table 88.1 ). Hypovolemic shock , the most common cause of shock in children worldwide, is most frequently caused by diarrhea, vomiting, or hemorrhage. Cardiogenic shock is seen in patients with congenital heart disease (before or after surgery, including heart transplantation) or those with congenital or acquired cardiomyopathies, including acute myocarditis. Obstructive shock stems from any lesion that creates a mechanical barrier that impedes adequate cardiac output, which includes pericardial tamponade, tension pneumothorax, pulmonary embolism, and ductus-dependent congenital heart lesions. Distributive shock is caused by inadequate vasomotor tone, which leads to capillary leak and maldistribution of fluid into the interstitium. Septic shock is often discussed synonymously with distributive shock, but the septic process usually involves a more complex interaction of distributive, hypovolemic, and cardiogenic shock.
Table 88.1
| HYPOVOLEMIC | CARDIOGENIC | DISTRIBUTIVE | SEPTIC | OBSTRUCTIVE |
|---|---|---|---|---|
| Decreased preload secondary to internal or external losses | Cardiac pump failure secondary to poor myocardial function | Abnormalities of vasomotor tone from loss of venous and arterial capacitance | Decreased cardiac output secondary to direct impediment to right- or left-sided heart outflow or restriction of all cardiac chambers | |
| POTENTIAL ETIOLOGIES | ||||
Pathophysiology
An initial insult triggers shock, leading to inadequate oxygen delivery to organs and tissues. Compensatory mechanisms attempt to maintain blood pressure (BP) by increasing cardiac output and systemic vascular resistance (SVR). The body also attempts to optimize oxygen delivery to the tissues by increasing oxygen extraction and redistributing blood flow to the brain, heart, and kidneys at the expense of the skin and gastrointestinal (GI) tract. These responses lead to an initial state of compensated shock in which BP is maintained. If treatment is not initiated or is inadequate during this period, decompensated shock develops, with hypotension and tissue damage that may lead to multisystem organ dysfunction and, ultimately, death (Tables 88.2 and 88.3 ).
Table 88.2
Criteria for Organ Dysfunction
BP, Blood pressure; FIO 2 , fraction of inspired oxygen; GCS, Glasgow Coma Scale; INR, international normalized ratio; PaCO 2 , arterial partial pressure of carbon dioxide; PaO 2 , partial pressure arterial oxygen; SD, standard deviations.
Table 88.3
| ORGAN SYSTEM | ↓ PERFUSION | ↓↓ PERFUSION | ↓↓↓ PERFUSION |
|---|---|---|---|
| Central nervous system | — | Restless, apathetic, anxious | Agitated/confused, stuporous, coma |
| Respiration | — | ↑ Ventilation | ↑↑ Ventilation |
| Metabolism | — | Compensated metabolic acidemia | Uncompensated metabolic acidemia |
| Gut | — | ↓ Motility | Ileus |
| Kidney | ↓ Urine volume | Oliguria (<0.5 mL/kg/hr) | Oliguria/anuria |
| ↑ Urinary specific gravity | |||
| Skin | Delayed capillary refill | Cool extremities | Mottled, cyanotic, cold extremities |
| Cardiovascular system | ↑ Heart rate | ↑↑ Heart rate | ↑↑ Heart rate |
| ↓ Peripheral pulses | ↓ Blood pressure, central pulses only |
In the early phases of shock, multiple compensatory physiologic mechanisms act to maintain BP and preserve tissue perfusion and oxygen delivery. Cardiovascular effects include increases in heart rate (HR), stroke volume, and vascular smooth muscle tone, which are regulated through sympathetic nervous system activation and neurohormonal responses. Respiratory compensation involves greater carbon dioxide (CO2 ) elimination in response to the metabolic acidosis and increased CO2 production from poor tissue perfusion. Renal excretion of hydrogen ions (H+ ) and retention of bicarbonate (HCO3 − ) also increase in an effort to maintain normal body pH (see Chapter 68.7 ). Maintenance of intravascular volume is facilitated via sodium regulation through the renin-angiotensin-aldosterone and atrial natriuretic factor axes, cortisol and catecholamine synthesis and release, and antidiuretic hormone secretion. Despite these compensatory mechanisms, the underlying shock and host response lead to vascular endothelial cell injury and significant leakage of intravascular fluids into the interstitial extracellular space.
Another important aspect of the initial pathophysiology of shock is the impact on cardiac output. All forms of shock affect cardiac output through several mechanisms, with changes in HR, preload, afterload, and myocardial contractility occurring separately or in combination (Table 88.4 ). Hypovolemic shock is characterized primarily by fluid loss and decreased preload. Tachycardia and an increase in SVR are the initial compensatory responses to maintain cardiac output and systemic BP. Without adequate volume replacement, hypotension develops, followed by tissue ischemia and further clinical deterioration. When there is preexisting low plasma oncotic pressure (caused by nephrotic syndrome, malnutrition, hepatic dysfunction, acute severe burns, etc.), even further volume loss and exacerbation of shock may result from endothelial breakdown and worsening capillary leak.
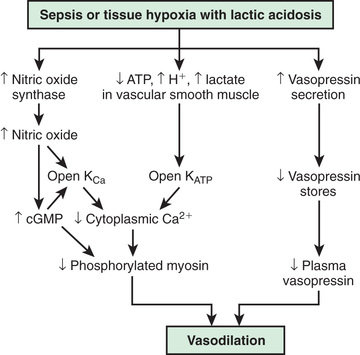
In contrast, the underlying pathophysiologic mechanism leading to distributive shock is a state of abnormal vasodilation and decreased SVR. Sepsis, hypoxia, poisoning, anaphylaxis, spinal cord injury, or mitochondrial dysfunction can cause vasodilatory shock (Fig. 88.3 ). The lowering of SVR is accompanied initially by a maldistribution of blood flow away from vital organs and a compensatory increase in cardiac output. This process leads to significant decreases in both preload and afterload. Therapies for distributive shock must address both these problems simultaneously.
Cardiogenic shock may be seen in patients with myocarditis, cardiomyopathy, arrhythmias and congenital heart disease (generally following cardiac surgery) (see Chapter 461 ). In these patients, myocardial contractility is affected, leading to systolic and/or diastolic dysfunction. The later phases of all forms of shock frequently have a negative impact on the myocardium, leading to development of a cardiogenic component to the initial shock state.
Septic shock is generally a unique combination of distributive, hypovolemic, and cardiogenic shock. Hypovolemia from intravascular fluid losses occurs through capillary leak. Cardiogenic shock results from the myocardial-depressant effects of sepsis, and distributive shock is the result of decreased SVR. The degree to which a patient exhibits each of these responses varies, but there are frequently alterations in preload, afterload, and myocardial contractility.
In septic shock, it is important to distinguish between the inciting infection and the host inflammatory response. Normally, host immunity prevents the development of sepsis through activation of the reticular endothelial system along with the cellular and humoral immune systems. This host immune response produces an inflammatory cascade of toxic mediators, including hormones, cytokines, and enzymes. If this inflammatory cascade is uncontrolled, derangement of the microcirculatory system leads to subsequent organ and cellular dysfunction.
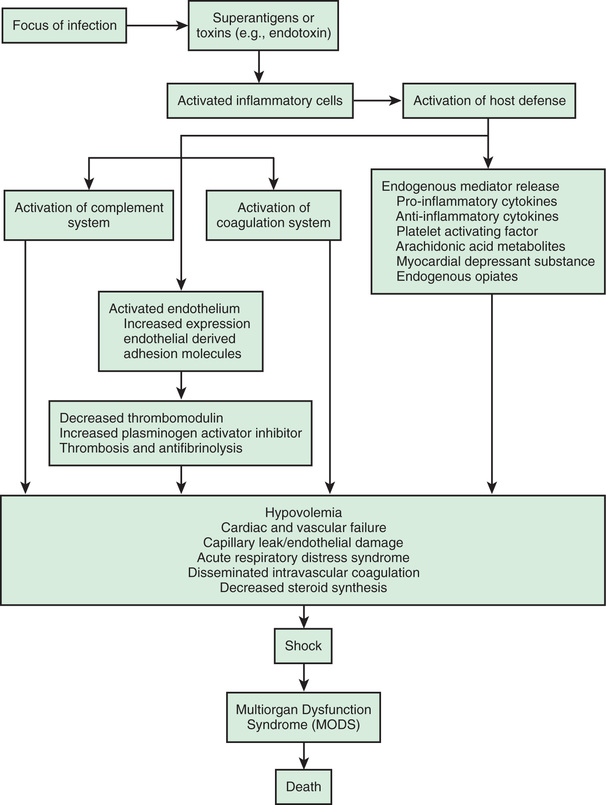
The systemic inflammatory response syndrome (SIRS ) is an inflammatory cascade that is initiated by the host response to an infectious or noninfectious trigger (Table 88.5 ). This inflammatory cascade is triggered when the host defense system does not adequately recognize and/or eliminate the triggering event. The inflammatory cascade initiated by shock can lead to hypovolemia, cardiac and vascular failure, acute respiratory distress syndrome (ARDS), insulin resistance, decreased cytochrome P450 activity (decreased steroid synthesis), coagulopathy, and unresolved or secondary infection. Tumor necrosis factor (TNF) and other inflammatory mediators increase vascular permeability, causing diffuse capillary leak, decreased vascular tone, and an imbalance between perfusion and metabolic demands of the tissues. TNF and interleukin (IL)-1 stimulate the release of proinflammatory and antiinflammatory mediators, causing fever and vasodilation. Proinflammatory mediators include IL-6, IL-12, interferon-γ, and macrophage migration inhibitory factor; antiinflammatory cytokines include IL-10, transforming growth factor-β, and IL-4. Arachidonic acid metabolites lead to the development of fever, tachypnea, ventilation-perfusion abnormalities, and lactic acidosis. Nitric oxide (NO), released from the endothelium or inflammatory cells, is a major contributor to hypotension. Myocardial depression is caused directly by myocardial-depressant factors, TNF, and some interleukins and is further depressed by depleted catecholamines, increased β-endorphin, and production of myocardial NO.
The inflammatory cascade is initiated by toxins or superantigens through macrophage binding or lymphocyte activation (Fig. 88.4 ). The vascular endothelium is both a target of tissue injury and a source of mediators that may cause further injury. Biochemical responses include the production of arachidonic acid metabolites, release of myocardial-depressant factors and endogenous opiates, activation of the complement system, and production and release of other mediators, which may be proinflammatory or antiinflammatory. The balance among these mediator groups for an individual patient contributes to the progression (and resolution) of disease and affects the prognosis.
Clinical Manifestations
Table 88.1 shows a classification system for shock. Categorization is important, but there may be significant overlap among these groups, especially in septic shock. The clinical presentation of shock depends in part on the underlying etiology, but if unrecognized and untreated, all forms of shock follow a common and untoward progression of clinical signs and pathophysiologic changes that may ultimately lead to irreversible organ injury and death.
Shock may initially manifest as only tachycardia, with or without tachypnea. Progression leads to decreased urine output, poor peripheral perfusion, respiratory distress or failure, alteration of mental status, and low BP (see Table 88.3 ). A significant misconception is that shock occurs only with low BP; hypotension is often a late finding and is not a criterion for the diagnosis of shock because of a complex set of compensatory mechanisms that attempt to preserve BP and peripheral perfusion. Hypotension reflects an advanced state of decompensated shock and is associated with increased morbidity and mortality.
Hypovolemic shock often manifests initially as orthostatic hypotension and is associated with dry mucous membranes, dry axillae, poor skin turgor, and decreased urine output. Depending on the degree of dehydration, the patient with hypovolemic shock may present with either normal or slightly cool distal extremities, and pulses may be normal, decreased, or absent depending on disease severity. The presenting signs of cardiogenic shock are tachypnea, cool extremities, delayed capillary filling time, poor peripheral and/or central pulses, declining mental status, and decreased urine output caused by the combination of decreased cardiac output and compensatory peripheral vasoconstriction (see Chapter 469.1 ). Obstructive shock often also manifests as inadequate cardiac output because of a physical restriction of forward blood flow, and the acute presentation may quickly progress to cardiac arrest. Distributive shock manifests initially as peripheral vasodilation and increased but inadequate cardiac output.
Regardless of etiology, uncompensated shock, with hypotension, high SVR, decreased cardiac output, respiratory failure, obtundation, and oliguria, occurs late in the progression of disease. Table 88.6 lists the hemodynamic findings in various shock states. Additional clinical findings in shock include cutaneous lesions such as petechiae, diffuse erythema, ecchymoses, ecthyma gangrenosum, and peripheral gangrene. Jaundice can be present either as a sign of infection or as a result of MODS.
Table 88.6
Hemodynamic Variables in Different Shock States
| TYPE OF SHOCK | CARDIAC OUTPUT | SYSTEMIC VASCULAR RESISTANCE | MEAN ARTERIAL PRESSURE | CAPILLARY WEDGE PRESSURE | CENTRAL VENOUS PRESSURE |
|---|---|---|---|---|---|
| Hypovolemic | ↓ | ↑ | ↔ or ↓ | ↓↓↓ | ↓↓↓ |
| Cardiogenic* | |||||
| Systolic | ↓↓ | ↑↑↑ | ↔ or ↓ | ↑↑ | ↑↑ |
| Diastolic | ↔ | ↑↑ | ↔ | ↑↑ | ↑ |
| Obstructive | ↓ | ↑ | ↔ or ↓ | ↑↑ † | ↑↑ † |
| Distributive | ↑↑ | ↓↓↓ | ↔ or ↓ | ↔ or ↓ | ↔ or ↓ |
| Septic | |||||
| Early | ↑↑↑ | ↓↓↓ | ↔ or ↓ ‡ | ↓ | ↓ |
| Late | ↓↓ | ↓↓ | ↓↓ | ↑ | ↑ or ↔ |
* Systolic or diastolic dysfunction.
† Wedge pressure, central venous pressure, and pulmonary artery diastolic pressures are equal.
‡ Wide pulse pressure.
Sepsis is defined as SIRS resulting from a suspected or proven infectious etiology. The clinical spectrum of sepsis begins when a systemic (e.g., bacteremia, rickettsial disease, fungemia, viremia) or localized (e.g., meningitis, pneumonia, pyelonephritis, peritonitis, necrotizing fasciitis) infection progresses from sepsis to severe sepsis (i.e., presence of sepsis combined with organ dysfunction). Further clinical deterioration leads to septic shock (severe sepsis plus the persistence of hypoperfusion or hypotension despite adequate fluid resuscitation or a requirement for vasoactive agents), MODS, and possibly death (Table 88.7 ). This is a complex spectrum of clinical problems that is a leading cause of mortality in children worldwide. Mortality can be mitigated and outcomes improved with early recognition and treatment.
Although septic shock is primarily distributive in nature, multiple other elements of pathophysiology are represented in this disease process. The initial signs and symptoms of sepsis include alterations in temperature regulation (hyperthermia or hypothermia), tachycardia, and tachypnea. In the early stages (hyperdynamic phase, low SVR, or warm shock), cardiac output increases to maintain adequate oxygen delivery and meet the greater metabolic demands of the organs and tissues. As septic shock progresses, cardiac output falls in response to the effects of numerous inflammatory mediators, leading to a compensatory elevation in SVR and the development of cold shock.
Diagnosis
Shock is a clinical diagnosis based on a thorough history and physical examination (see Tables 88.2 and 88.3 ). Septic shock has a specific consensus conference definition (see Table 88.7 ). In cases of suspected septic shock, an infectious etiology should be sought through culture of clinically appropriate specimens and prompt initiation of empirical antimicrobial therapy based on patient age, underlying disease, and geographic location, recognizing that time is necessary for incubation of cultures, and results often are not positive. Additional evidence for identifying an infectious etiology as the cause of SIRS includes physical examination findings, imaging, presence of white blood cells in normally sterile body fluids, and suggestive rashes such as petechiae and purpura. Affected children should be admitted to a PICU or other highly monitored environment as indicated by clinical status and the resources of the medical facility. These patients necessitate continuous monitoring, with a combination of noninvasive (e.g., pulse oximetry, capnography, near-infrared spectroscopy) and invasive (e.g., central venous pressure, arterial BP) techniques as clinically indicated.
Laboratory Findings
Laboratory findings often include evidence of hematologic abnormalities and electrolyte disturbances. Hematologic abnormalities may include thrombocytopenia, prolonged prothrombin and partial thromboplastin times, reduced serum fibrinogen level, elevation of fibrin split products, and anemia. Elevated neutrophil counts and increased immature forms (i.e., bands, myelocytes, promyelocytes), vacuolation of neutrophils, toxic granulations, and Döhle bodies can be seen with infection. Neutropenia or leukopenia may be an ominous sign of overwhelming sepsis.
Glucose dysregulation, a common stress response, may manifest as hyperglycemia or hypoglycemia. Other electrolyte abnormalities are hypocalcemia, hypoalbuminemia, and metabolic acidosis. Renal and/or hepatic function may also be abnormal. Patients with ARDS or pneumonia have impairment of oxygenation (decreased partial pressure of arterial oxygen [PaO 2 ]) as well as of ventilation (increased arterial partial pressure of carbon dioxide [PaCO 2 ]) in the later stages of lung injury (see Chapter 89 ).
The hallmark of uncompensated shock is an imbalance between oxygen delivery (DO 2 ) and oxygen consumption (VO 2 ). Oxygen delivery normally exceeds oxygen consumption by threefold. The oxygen extraction ratio is approximately 25%, thus producing a normal mixed venous oxygen saturation (SvO 2 ) of approximately 75%. A falling SvO 2 value, as measured by cooximetry, reflects an increasing oxygen extraction ratio and documents a decrease in oxygen delivery relative to consumption. This increase in oxygen extraction by the end organs is an attempt to maintain adequate oxygen delivery at the cellular level. This state is manifested clinically by increased lactic acid production (e.g., high anion gap, metabolic acidosis) caused by anaerobic metabolism and a compensatory increase in tissue oxygen extraction. The gold standard measurement of SvO 2 is from a pulmonary arterial catheter, but measurements from this location are often not clinically feasible. Sites such as the right ventricle, right atrium, superior vena cava (SvCO 2 ), or inferior vena cava can be as surrogate measures of mixed venous blood to follow the adequacy of oxygen delivery and effectiveness of therapeutic interventions. Elevated blood lactate levels reflect poor tissue oxygen delivery noted in all forms of shock.
Treatment
Initial Management
Early recognition and prompt intervention are extremely important in the management of all forms of shock (Tables 88.8 to 88.12 ; see Figs. 88.1 and 88.2 ). The vital sign targets and dose recommendations in Tables 88.9 to 88.12 should be adjusted to pediatric-size patients. Baseline mortality is much lower in pediatric shock than in adult shock, and further improvements in mortality are associated with early interventions (see Fig. 81.1 ). The initial assessment and treatment of the pediatric shock patient should include stabilization of airway, breathing, and circulation as established by the American Heart Association's pediatric advanced life support and neonatal advanced life support guidelines (see Chapter 81 ). Depending on the severity of shock, further airway intervention, including intubation and mechanical ventilation, may be necessary to lessen the work of breathing and decrease the body's overall metabolic demands.
Table 88.8
Goal-Directed Therapy of Organ System Dysfunction in Shock
| SYSTEM | DISORDERS | GOALS | THERAPIES |
|---|---|---|---|
| Respiratory | |||
| Renal |
Prevent/treat: hypovolemia, hypervolemia, hyperkalemia, metabolic acidosis, hypernatremia/hyponatremia, and hypertension Monitor serum electrolytes |
||
| Hematologic | |||
| Gastrointestinal | |||
| Endocrine | Adrenal insufficiency, primary or secondary to chronic steroid therapy | Prevent/treat: adrenal crisis | |
| Metabolic | Metabolic acidosis |
Given the predominance of sepsis and hypovolemia as the most common causes of shock in the pediatric population, most therapeutic regimens are based on guidelines established in these settings. Immediately following establishment of intravenous (IV) or intraosseous (IO) access, aggressive, early goal-directed therapy should be initiated unless there are significant concerns for cardiogenic shock as an underlying pathophysiology. Rapid IV administration of 20 mL/kg isotonic fluid should be initiated to reverse the shock state. This bolus should be repeated quickly up to 60-80 mL/kg; it is not unusual for severely affected patients to require this volume within the 1st 3 hr of treatment.
Rapid fluid resuscitation totaling 60-80 mL/kg or more is associated with improved survival without an increased incidence of pulmonary edema. Fluid resuscitation in increments of 20 mL/kg should be titrated to normalize HR (according to age-based HRs), urine output (to 1 mL/kg/hr), capillary refill time (to <2 sec), and mental status. If shock remains refractory following 60-80 mL/kg of volume resuscitation, vasopressor therapy (e.g., norepinephrine, epinephrine) should be instituted while additional fluids are administered. Pediatric guidelines for septic shock unresponsive to fluid resuscitation suggest epinephrine (Fig. 88.2 ) or dopamine (Fig. 88.1 ), whereas adult guidelines recommend norepinephrine.
Fluid resuscitation may sometimes require as much as 200 mL/kg or greater. It must be stressed that hypotension is often a late and ominous finding, and BP normalization alone is not a reliable end-point for assessing the effectiveness of resuscitation. Although the type of fluid (crystalloid vs colloid) is an area of ongoing debate, fluid resuscitation (usually crystalloid) in the 1st hr is unquestionably essential to survival in septic shock, regardless of the fluid type administered.
Additional Early Considerations
In septic shock specifically, early (within 1 hr ) administration of broad-spectrum antimicrobial agents is associated with a reduction in mortality. The choice of antimicrobial agents depends on the predisposing risk factors and the clinical situation. Bacterial resistance patterns in the community and/or hospital should be considered in the selection of optimal antimicrobial therapy. Neonates should be treated with ampicillin plus cefepime and/or gentamicin. Acyclovir should be added if herpes simplex virus is suspected clinically. In infants and children, community-acquired infections with Neisseria meningitidis can initially be treated empirically with a third-generation cephalosporin (e.g., ceftriaxone, cefepime), as can Haemophilus influenzae infections. The prevalence of resistant Streptococcus pneumoniae requires the addition of vancomycin. Suspicion of community- or hospital-acquired, methicillin-resistant Staphylococcus aureus (MRSA) infection warrants coverage with vancomycin, depending on local resistance patterns. If an intraabdominal process is suspected, anaerobic coverage should be included with an agent such as metronidazole, clindamycin, or piperacillin-tazobactam.
Nosocomial sepsis should generally be treated with at least a third- or fourth-generation cephalosporin or a penicillin with an extended gram-negative spectrum (e.g., piperacillin-tazobactam). An aminoglycoside should be added as the clinical situation warrants. Vancomycin should be added to the regimen if the patient has an indwelling medical device (see Chapter 206 ), if gram-positive cocci are isolated from the blood, if MRSA infection is suspected, or as empirical coverage for S. pneumoniae in a patient with meningitis. Empirical coverage for fungal infections should be considered for selected immunocompromised patients (see Chapter 205 ). It should be noted that these are broad, generalized recommendations that must be tailored to the individual clinical scenario and to the local resistance patterns of the community and hospital.
Distributive shock that is not secondary to sepsis is caused by a primary abnormality in vascular tone. Cardiac output in affected patients is usually maintained and may initially be supranormal. These patients may benefit temporarily from volume resuscitation, but the early initiation of a vasoconstrictive agent to increase SVR is an important element of clinical care. Patients with spinal cord injury and spinal shock may benefit from either phenylephrine or vasopressin to increase SVR; epinephrine is the treatment of choice for patients with anaphylaxis (Table 88.13 ). Epinephrine has peripheral α-adrenergic as well as inotropic effects that may improve the myocardial depression seen with anaphylaxis and its associated inflammatory response (see Chapter 174 ).
Table 88.13
Cardiovascular Drug Treatment of Shock
| DRUG | EFFECT(S) | DOSING RANGE | COMMENT(S) |
|---|---|---|---|
| Dopamine | 3-20 µg/kg/min | ↑ Risk of arrhythmias at high doses | |
| Epinephrine | 0.05-3.0 µg/kg/min | ||
| Dobutamine | 1-10 µg/kg/min | — | |
| Norepinephrine | 0.05-1.5 µg/kg/min | ||
| Phenylephrine | Potent vasoconstriction | 0.5-2.0 µg/kg/min | |
Patients with cardiogenic shock have poor cardiac output secondary to systolic and/or diastolic myocardial depression, often with a compensatory elevation in SVR. These patients may show poor response to fluid resuscitation and may decompensate quickly when fluids are administered. Smaller boluses of fluid (5-10 mL/kg) should be given in cardiogenic shock to replace deficits and maintain preload. In any patient with shock whose clinical status deteriorates with fluid resuscitation, a cardiogenic etiology should be considered, and further administration of IV fluids should be provided judiciously. Early initiation of myocardial support with epinephrine or dopamine to improve cardiac output is important in this context, and early consideration should be given to administration of an inodilator, such as milrinone.
Despite adequate cardiac output with the support of inotropic agents, a high SVR with poor peripheral perfusion and acidosis may persist in cardiogenic shock. Therefore, if not already started, milrinone therapy may improve systolic function and decrease SVR without causing a significant increase in HR. Furthermore, this agent has the added benefit of enhancing diastolic relaxation. Dobutamine or other vasodilating agents, such as nitroprusside, may also be considered in this setting (Table 88.14 ). Titration of these agents should target clinical end-points, including increased urine output, improved peripheral perfusion, resolution of acidosis, and normalization of mental status. Even though they may be beneficial in other forms of shock, agents that improve BP by increasing SVR, such as norepinephrine and vasopressin, should generally be avoided in patients with cardiogenic shock. These agents may cause further decompensation and potentially precipitate cardiac arrest as a result of the increased afterload and additional work imposed on the myocardium. The combination of inotropic and vasoactive agents must be tailored to the pathophysiology of the individual patient with close and frequent reassessment of the patient's cardiovascular status.
Table 88.14
Vasodilators/Afterload Reducers in Treatment of Shock
| DRUG | EFFECT(S) | DOSING RANGE | COMMENT(S) |
|---|---|---|---|
| Nitroprusside | Vasodilator (mainly arterial) | 0.5-4.0 µg/kg/min | |
| Nitroglycerin | Vasodilator (mainly venous) | 1-20 µg/kg/min | |
| Prostaglandin E1 | 0.01-0.2 µg/kg/min | ||
| Milrinone | Phosphodiesterase inhibitor—slows cyclic adenosine monophosphate breakdown | ||
For patients with obstructive shock , fluid resuscitation may be briefly temporizing in maintaining cardiac output, but the primary insult must be immediately addressed. Examples of lifesaving therapeutic interventions for such patients are pericardiocentesis for pericardial effusion, pleurocentesis or chest tube placement for pneumothorax, thrombectomy/thrombolysis for pulmonary embolism, and the initiation of a prostaglandin infusion for ductus-dependent cardiac lesions. There is often a last-drop phenomenon associated with some obstructive lesions, in that small additional amounts of intravascular volume depletion may lead to a rapid deterioration, including cardiac arrest, if the obstructive lesion is not corrected.
Regardless of the etiology of shock, metabolic status should be meticulously maintained (see Table 88.8 ). Electrolyte levels should be monitored closely and corrected as needed. Hypoglycemia is common and should be promptly treated. Neonates and infants in particular may have profound glucose dysregulation in association with shock. Glucose levels should be checked routinely and treated appropriately, especially early in the course of illness. Hypocalcemia, which may contribute to myocardial dysfunction, should be treated with a goal of normalizing the ionized calcium concentration. There is no evidence that supranormal calcium levels benefit the myocardium, and hypercalcemia may be associated with increased myocardial toxicity.
Adrenal function is another important consideration in shock, and hydrocortisone replacement may be beneficial. Up to 50% of critically ill patients may have absolute or relative adrenal insufficiency. Patients at risk for adrenal insufficiency include those with congenial adrenal hypoplasia, abnormalities of the hypothalamic-pituitary axis, and recent therapy with corticosteroids (including those with asthma, rheumatic diseases, malignancies, and inflammatory bowel disease). These patients are at high risk for adrenal dysfunction and should receive stress doses of hydrocortisone. Corticosteroids may also be considered in patients with shock that is unresponsive to fluid resuscitation and catecholamines. Although a subset of pediatric septic shock patients may benefit from treatment with hydrocortisone, currently available pediatric data do not demonstrate an overall survival benefit in patients with shock treated with hydrocortisone. Determination of baseline cortisol levels before corticosteroid administration may be beneficial in guiding therapy, although this approach remains controversial.
Considerations for Continued Therapy
After the 1st hr of therapy and attempts at early reversal of shock, focus on goal-directed end-points should continue in an intensive care setting (see Figs. 88.1 and 88.2 and Table 88.8 ). Clinical end-points serve as global markers for organ perfusion and oxygenation. Laboratory parameters such as SvO 2 (or ScvO2 ), serum lactate concentration, cardiac index, and hemoglobin serve as adjunctive measures of tissue oxygen delivery. Hemoglobin should be generally maintained at 10 g/dL, SvO 2 (or ScvO2 ) >70%, and cardiac index at 3.3-6.0 L/min/m2 to optimize oxygen delivery in the acute phase of shock. It is important to note that cardiac index is rarely monitored in the clinical setting because of the limited use of pulmonary artery catheters and lack of accurate noninvasive cardiac output monitors for infants and children. Blood lactate levels and calculation of base deficit from arterial blood gas values are very useful markers for the adequacy of oxygen delivery. These traditional markers are indicators of global oxygen utilization and delivery. There is increasing use of measures of local tissue oxygenation, including near-infrared spectroscopy of the cerebrum, flank, or abdomen.
Respiratory support should be used as clinically appropriate. When shock leads to ARDS requiring mechanical ventilation, lung-protective strategies to keep plateau pressure <30 cm H2 O and maintain tidal volume at 6 mL/kg have been shown to improve mortality in adult patients (see Chapter 89 ). These data are extrapolated to pediatric patients because of the lack of definitive pediatric studies in this area. Additionally, after the initial shock state has been reversed, data demonstrate that judicious fluid administration, renal replacement therapy, and fluid removal may also be useful in children with anuria or oliguria and fluid overload (see Chapter 550 ). Other interventions include correction of coagulopathy with fresh-frozen plasma or cryoprecipitate and platelet transfusions as necessary, especially in the presence of active bleeding.
If shock remains refractory despite maximal therapeutic interventions, mechanical support with extracorporeal membrane oxygenation (ECMO) or a ventricular assist device (VAD) may be indicated. ECMO may be lifesaving in cases of refractory shock regardless of underlying etiology. Similarly, a VAD may be indicated for refractory cardiogenic shock in the setting of cardiomyopathy or recent cardiac surgery. Systemic anticoagulation, which is required while patients are receiving mechanical support, may be difficult, given the significant coagulopathy often encountered in refractory shock, especially when the underlying etiology is sepsis. Mechanical support in refractory shock poses substantial risks but can improve survival in specific populations of patients.
Prognosis
In septic shock, mortality rates are as low as 3% in previously healthy children and 6–9% in children with chronic illness (compared with 25–30% in adults). With early recognition and therapy, the mortality rate for pediatric shock continues to improve, but shock and MODS remain one of the leading causes of death in infants and children. The risk of death involves a complex interaction of factors, including the underlying etiology, presence of chronic illness, host immune response, and timing of recognition and therapy.