Anomalies of the Bladder
Jack S. Elder
Bladder Exstrophy
Exstrophy of the urinary bladder occurs in approximately 1 in 35,000-40,000 births. The male:female ratio is 2 : 1. The severity ranges from simple epispadias (in males) to complete exstrophy of the cloaca involving exposure of the entire hindgut and the bladder (termed cloacal exstrophy ).
Clinical Manifestations
Anomalies of the bladder are hypothesized to result when the mesoderm fails to invade the cephalad extension of the cloacal membrane; the extent of this failure determines the degree of the anomaly. In classic bladder exstrophy (Fig. 556.1 ), the bladder protrudes from the abdominal wall and its mucosa is exposed. The umbilicus is displaced downward, the pubic rami are widely separated in the midline, and the rectus muscles are separated. In males, there is complete epispadias with dorsal chordee, and the overall penile length is approximately half that of unaffected males. The scrotum typically is separated slightly from the penis and is wide and shallow. Undescended testes and inguinal hernias are common. Females also have epispadias, with separation of the two halves of the clitoris and wide separation of the labia. The anus is displaced anteriorly in both sexes, and there may be rectal prolapse. The pubic rami are widely separated. Persons with exstrophy tend to be shorter than normal.
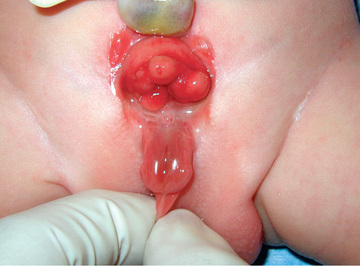
The consequences of untreated bladder exstrophy are total urinary incontinence and an increased incidence of bladder cancer, usually adenocarcinoma. The external and internal genital deformities cause sexual disability in both sexes, particularly in males. The wide separation of the pubic rami causes a characteristic broad-based gait but no significant disability. In classic bladder exstrophy, the upper urinary tracts usually are normal at birth.
Treatment
Management of bladder exstrophy should start at birth. The bladder should be covered with plastic wrap to keep the bladder mucosa moist. Application of gauze or petroleum-gauze to the bladder mucosa should be avoided, because significant inflammation will result. The infant should be transferred promptly to a center with pediatric urologic and pediatric anesthetic support for newborns with complex anomalies. These children are prone to latex allergy, and latex precautions should be practiced from birth, both in the nursery and in the operating room.
There are two surgical approaches: staged reconstruction and total single-stage reconstruction. Most babies also undergo bilateral iliac osteotomy, which allows the pubic symphysis to be approximated, which supports the bladder closure. In a staged reconstruction, the initial stage is bladder closure, the second stage (in males) is epispadias repair, and the final stage is bladder neck reconstruction. The single-stage reconstruction attempts to reconstruct the entire malformation in a single procedure. When this operation is performed in the newborn, there is an increased risk of intraoperative penile injury and postoperative hydronephrosis, compared with the staged reconstruction. The complication rate is high with both approaches and there is no consensus on which is better.
Although bladder closure within 48 hr has been the standard in the past, many centers of excellence now defer the procedure for 1-2 wk to be certain that the appropriate multidisciplinary surgical and anesthetic teams are available. During bladder exstrophy closure, the abdominal wall is mobilized and the pubic rami are brought together in the midline following pelvic osteotomy. Early bladder closure can be performed in almost all neonates with classic bladder exstrophy. Treatment should be deferred in selected situations when surgical therapy would be excessively risky or complex, as in a premature baby or when it would have to be performed by inexperienced surgeons. In the staged approach, in males, epispadias repair usually is performed at 1-2 yr of age. At this point the child has total urinary incontinence because there is no functional external urinary sphincter. Most infants with bladder exstrophy have vesicoureteral reflux and should receive antibiotic prophylaxis. Typically, the bladder capacity is monitored every 12-24 mo using cystoscopy under anesthesia. The final stage of reconstruction involves creation of a sphincter muscle for bladder control and correction of vesicoureteral reflux. At this point the child is 3-6 yr old, the bladder capacity should be at least 80-90 mL, and the child must have gained rectal sphincter control.
Total single-stage reconstruction includes newborn closure of the bladder and bladder neck narrowing, abdominal wall closure, and, in males, correction of epispadias using a technique of penile disassembly, in which the two corpora cavernosa and the midline urethra are mobilized separately into three parts. Postoperatively, the infant's upper urinary tract is monitored closely for possible development of hydronephrosis and infection. Comparison of outcomes between the multistage and single-stage approaches is ongoing.
At puberty, often the pubic hair is distributed to the sides of the external genitals. A monsplasty can performed to provide a normal escutcheon.
Long-Term Prognosis
Long-term management of individuals born with bladder exstrophy includes monitoring of upper urinary tract appearance and function, UTI, continence, erectile function, and, in adults, sexual function and fertility.
The previously described plan of treatment has yielded a continence rate of 60–70% in a few centers, with < 15% deterioration of the upper urinary tract. This continence rate reflects not only successful reconstruction but also the quality and size of the bladder. From a functional standpoint, the reconstructed bladder neck does not relax during voiding as in a normal child; instead the patient must void by Valsalva.
Children who remain incontinent for more than 1 yr after bladder neck reconstruction or those who are ineligible for bladder neck reconstruction because of a small bladder capacity are candidates for an alternative reconstructive procedure to achieve dryness. In selected cases, cystoscopic injection of dextranomer or polydimethylsiloxane microspheres into the bladder neck can provide sufficient bladder neck coaptation to establish continence. Alternatively, if the child is not a candidate for endoscopic therapy, options include:
- • Augmentation cystoplasty, in which the bladder is enlarged with a patch of small or large bowel to increase its capacity.
- • Creation of a neobladder out of small and large bowel with placement of a continent abdominal stoma through which clean intermittent catheterization can be performed.
- • Placement of an artificial urinary sphincter, with possible augmentation cystoplasty.
- • Ureterosigmoidostomy, in which the ureters are detached from the bladder and sutured to the sigmoid colon; individuals void urine and stool from the rectum and rely on their anal sphincter for continence.
- • Mainz II procedure, in which the sigmoid colon is reconfigured into a “bladder” into which the ureters are connected; the patient voids 3-6 times daily through the rectum, and the stool tends to be more solid.
Ureterosigmoidostomy carries a significant risk of chronic pyelonephritis (see Chapter 553 ), upper urinary tract damage, metabolic acidosis resulting from absorption of hydrogen ion and chloride in the intestine, and at least a 15% long-term risk of colon carcinoma. Patients from less-developed countries often undergo the Mainz II procedure because the continence rate is high and pyelonephritis and upper tract changes are uncommon.
Late follow-up has shown that although adult males with exstrophy have a penis that is half normal length, they usually experience satisfactory sexual function. Fertility has been low, possibly because of iatrogenic injury to the secondary sexual organs during reconstruction. With artificial reproductive technology, nearly all affected men can be fertile. In adult females, fertility is not affected, but uterine prolapse during pregnancy is a problem. In adult females who have undergone a continent urinary diversion, delivery by cesarean section may be necessary.
Other Exstrophy Anomalies
Children with more complex cases of cloacal exstrophy, which has an incidence of 1 in 400,000, have an omphalocele and severe abnormalities of the colon and the rectum and often have short bowel syndrome (see Chapter 364.7 ), the most devastating anomaly managed by pediatric urologists. Approximately 50% of patients have an upper urinary tract anomaly, and 50% have spina bifida (see Chapter 609.2 ). Children with cloacal exstrophy do not achieve normal urine or stool continence. Reconstructive techniques result in a satisfactory outcome in most patients with permanent urinary diversion (either ileal conduit or continent urinary diversion) and a colostomy. Because the penis in males with cloacal exstrophy usually is diminutive, genital reconstruction in males with cloacal exstrophy has been unsatisfactory. Until recently, many specialists recommended assigning a female gender to such infants, but currently there is debate as to whether these children, who have a 46,XY karyotype and brain androgen imprinting in utero, can have a satisfactory female gender identity (see Chapter 133 ). Many assume male gender characteristics by adolescence. Decisions regarding gender assignment should be made jointly by the physicians caring for the infant (surgical team, pediatric endocrinologist, child psychiatrist, and ethicist) and family.
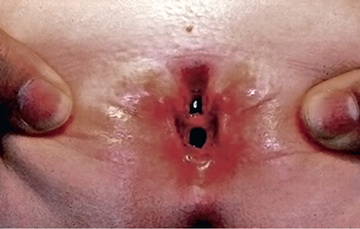
Epispadias is in the spectrum of exstrophy anomalies, affecting approximately 1 in 117,000 males and 1 in 480,000 females. In males, the diagnosis is obvious because the prepuce is distributed primarily on the ventral aspect of the penile shaft and the urethral meatus is on the dorsum of the penis. Distal epispadias in males (Fig. 556.2 ) usually is associated with normal urinary control and normal upper urinary tracts and should be repaired by 6-12 mo of age. In females, the clitoris is bifid, and the urethra is split dorsally (Fig. 556.3 ). In more severely affected males and in all females with epispadias, there is total urinary incontinence because the sphincter is incompletely formed, and there is wide separation of the pubic rami. These children require surgical reconstruction of the bladder neck, similar to the final management stage in children with classic bladder exstrophy.

Bladder Diverticula
Bladder diverticula develop as herniations of the bladder mucosa between defects of bladder smooth muscle fibers. Primary bladder diverticula usually develop at the ureterovesical junction and may be associated with vesicoureteral reflux, because the diverticulum interferes with the normal flap-valve attachment between the ureter and bladder. In rare circumstances, the diverticulum is so large that it interferes with normal micturition by obstructing the bladder neck. Bladder diverticula also commonly are associated with distal urethral obstructions such as posterior urethral valves or neurogenic bladder dysfunction. They occur commonly in children with connective tissue disorders, including Williams syndrome, Ehlers-Danlos syndrome, and Menkes syndrome (Fig. 556.4 ). Small diverticula require no treatment other than that of the primary disease, whereas large diverticula can contribute to inefficient voiding, residual urine, urinary stasis, and urinary tract infections and should be excised.

Urachal Anomalies
The urachus is an embryologic canal connecting the dome of the fetal bladder with the allantois, a structure that contributes to the formation of the umbilical cord. The lumen of the urachus is normally obliterated during embryonic development, transforming the urachus into a solid cord. Urachal abnormalities are more common in males than in females. A patent urachus can occur as an isolated anomaly or it may be associated with prune-belly syndrome or posterior urethral valves (see Chapter 555 ) (Fig. 556.5 ). A patent urachus results in continuous urinary drainage from the umbilicus. The tract should be excised. Another urachal anomaly is the urachal cyst, which can become infected. Typical symptoms and physical findings include suprapubic pain, fever, irritative voiding symptoms, and an infraumbilical mass, which can be erythematous. Diagnosis is made by ultrasonography or CT (Fig. 556.6 ). Treatment is intravenous antibiotic therapy and drainage and excision. Other urachal anomalies include the vesicourachal diverticulum, which is a diverticulum of the bladder dome, and umbilical–urachal sinus, which is a blind external sinus that opens at the umbilicus. These lesions should be excised.

