Obstruction of the Urinary Tract
Jack S. Elder
Most childhood obstructive lesions are congenital, although urinary tract obstruction can be caused by trauma, neoplasia, calculi, inflammatory processes, or surgical procedures. Obstructive lesions occur at any level from the urethral meatus to the calyceal infundibula (Table 555.1 ). The pathophysiologic effects of obstruction depend on its level, the extent of involvement, the child's age at onset, and whether it is acute or chronic.
Table 555.1
Etiology
Severe ureteral obstruction early in fetal life results in renal dysplasia, ranging from multicystic kidney, which is associated with ureteral or ureteropelvic junction (UPJ) atresia (see Fig. 552.3 in Chapter 552 ), to various degrees of histologic renal cortical dysplasia that are seen with less-severe obstruction. Chronic ureteral obstruction in late fetal life or after birth results in dilation of the ureter, renal pelvis, and calyces, with alterations of renal parenchyma ranging from minimal tubular changes to dilation of Bowman's space, glomerular fibrosis, and interstitial fibrosis. After birth, infection can complicate obstruction and worsen renal damage.
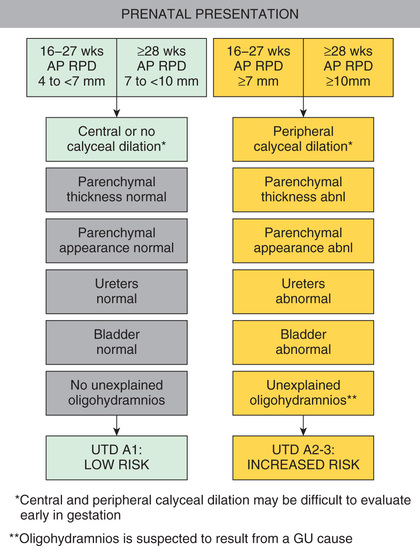
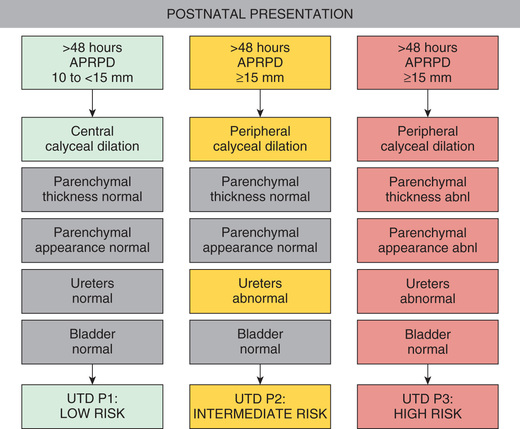
Prenatal screening with ultrasonography (US) may detect antenatal hydronephrosis (ANH), which is graded by the trimester and the anterior-posterior diameter of the renal pelvis (Table 555.2 ); most are mild. Table 555.3 notes the eventual etiology. Risk stratification for prenatal (Fig. 555.1 ) and postnatal (Fig. 555.2 ) urinary tract dilation helps plan for further evaluation and treatment.
Table 555.2
From Nguyen HT, Herndon CDA, Cooper C, et al: The Society for Fetal Urology consensus statement on the evaluation and management of antenatal hydronephrosis. J Pediatr Urol 6:212-231, 2010, Table 2, p. 215.
Table 555.3
From Nguyen HT, Herndon CDA, Cooper C, et al: The Society for Fetal Urology consensus statement on the evaluation and management of antenatal hydronephrosis. J Pediatr Urol 6:212-231, 2010, Table 5, p. 217.
Clinical Manifestations
Obstruction of the urinary tract generally causes hydronephrosis, which typically is asymptomatic in its early phases. An obstructed kidney secondary to a UPJ obstruction or ureterovesical junction obstruction can manifest as a unilateral mass or cause upper abdominal or flank pain on the affected side. Pyelonephritis can occur because of urinary stasis. An upper urinary tract stone can occur, causing abdominal and flank pain and hematuria. With bladder outlet obstruction, the urinary stream may be weak; urinary tract infection (UTI; see Chapter 553 ) is common. Many of these lesions are identified by antenatal US; an abnormality involving the genitourinary tract is suspected in as many as 1 in 50 fetuses (see Table 555.3 ).
Obstructive renal insufficiency can manifest itself by failure to thrive, vomiting, diarrhea, or other nonspecific signs and symptoms. In older children, infravesical obstruction can be associated with overflow urinary incontinence or a poor urine stream. Acute ureteral obstruction causes flank or abdominal pain; there may be nausea and vomiting. Chronic ureteral obstruction can be silent or can cause vague abdominal or typical flank pain with increased fluid intake.
Diagnosis
Urinary tract obstruction may be diagnosed prenatally by US, which typically shows hydronephrosis and occasionally a distended bladder. More complete evaluation, including imaging studies, should be undertaken in these children in the neonatal period.
In 2014, a multidisciplinary consensus conference that included pediatric urologists, pediatric nephrologists, pediatric radiologists, and maternal-fetal medicine specialists was convened to standardize the fetal evaluation and early postnatal management of babies with antenatal hydronephrosis (ANH). The US parameters include the anterior-posterior renal pelvic diameter (APRPD), calyceal dilation, whether the ANH involves the major and/or minor calyces, the parenchymal thickness and appearance, whether the ureter is normal or abnormal, and whether the bladder is normal or abnormal. Normal values for urinary tract dilation are the APRPD:
| Antenatal | 16-27 wk | <4 mm |
| ≥28 wk | <7 mm | |
| Postnatal | (>48 hr) | <10 mm |
Assuming there is no calyceal dilation, if the kidneys have a normal appearance and the ureter and bladder are normal, the study is considered normal.
The consensus group then categorized ANH into antenatal and postnatal risk groups. For antenatal ANH, there are two risk groups: low risk and high risk (see Fig. 555.1 ). For postnatal ANH, there are three risk groups: low risk, intermediate risk, and high risk (see Fig. 555.2 ). The panel recommended that all seven urinary tract parameters be described in a written report.
For antenatal presentation, if the APRPD is 4-7 mm at 16-27 wk or 7-10 mm at ≥ 28 wk and there is central or no calyceal dilation, the fetus is categorized as having urinary tract dilation (UTD) A1 , Low Risk . In follow-up for UTD A1, the panel suggested one additional antenatal US at ≥ 32 wk, and after birth, a renal US at > 48 hr to 1 mo of age and a second renal US 6 mo later. Genetic screening is not indicated unless there are associated congenital malformations.
If the APRPD is ≥ 7 mm at 16-27 wk or ≥ 10 mm at ≥ 28 wk with any peripheral calyceal dilation or any other upper urinary tract abnormality, the fetus is classified as having UTD A2-3, or Increased Risk . The assigned risk is based on the most concerning feature. For UTD A2-3, the panel recommended a follow-up US in 4 to 6 wk, although with suspected posterior urethral valves (PUVs) or severe bilateral hydronephrosis, more frequent follow-up was recommended until delivery. Following delivery, a renal US after 48 hr but before 1 mo was suggested, again with more immediate evaluation if PUV is suspected or there is significant bilateral hydronephrosis. In addition, specialist consultation with pediatric urology or nephrology was recommended.
For postnatal presentation, at > 48 hr an APRPD < 10 mm is Normal. If the APRPD is 10-15 mm and there is central calyceal dilation but all other parameters are normal, the infant is classified as having UTD P1, Low Risk . Society of Fetal Urology (SFU) hydronephrosis grades 1 and 2 correspond to UTD P1. The panel recommends a follow-up renal US in 1 to 6 mo. A VCUG and antibiotic prophylaxis are optional, at the discretion of the clinician. A renal scan is not recommended.
If the postnatal APRPD is ≥ 15 mm and there is peripheral calyceal dilation and/or abnormal ureters, the infant is classified as having UTD P2, Intermediate Risk . SFU hydronephrosis grade 3 corresponds to UTD P2. The panel recommends a follow-up renal US in 1 to 3 mo. A VCUG, antibiotic prophylaxis, and a functional renal scan are optional, at the discretion of the clinician.
If the APRPD is ≥ 15 mm and there is peripheral calyceal dilation, abnormal parenchymal thickness, abnormal parenchymal appearance, abnormal ureters, and/or abnormal bladder, the infant is classified as having UTD P3, High Risk . SFU hydronephrosis grade 4 corresponds to UTD P3. The panel recommends a follow-up renal US in 1 mo. A VCUG and antibiotic prophylaxis are recommended. A functional renal scan is optional, at the discretion of the clinician (but is virtually always recommended).
Physical Findings
Urinary tract obstruction is often silent. In the newborn infant, a palpable abdominal mass most commonly is a hydronephrotic or multicystic dysplastic kidney. With PUVs, which constitutes an infravesical obstructive lesion in boys, a walnut-sized mass representing the bladder is palpable just above the pubic symphysis. A patent draining urachus also can suggest urethral obstruction. Urinary ascites in the newborn usually is caused by renal or bladder urinary extravasation secondary to PUVs. Infection and sepsis may be the first indications of an obstructive lesion of the urinary tract. The combination of infection and obstruction poses a serious threat to infants and children and generally requires parenteral administration of antibiotics and drainage of the obstructed kidney. Renal US should be performed in all children during the acute stage of an initial febrile UTI.
Imaging Studies
Renal Ultrasound
Hydronephrosis is the most common characteristic of obstruction (Fig. 555.3 ). Upper urinary tract dilation is not diagnostic of obstruction and often persists after surgical correction of a significant obstructive lesion. Dilation can result from vesicoureteral reflux, or it may be a manifestation of abnormal development of the urinary tract, even when there is no obstruction. Renal length, degree of caliectasis and parenchymal thickness, and presence or absence of ureteral dilation should be assessed. In addition to the UTD system, most pediatric urologists also grade the severity of hydronephrosis from 1-4 using the SFU grading scale (Table 555.4 ). The clinician should ascertain that the contralateral kidney is normal, and the bladder should be imaged to see whether the bladder wall is thickened, the lower ureter is dilated, and bladder emptying is complete. In acute or intermittent obstruction, the dilation of the collecting system may be minimal and US may be misleading.
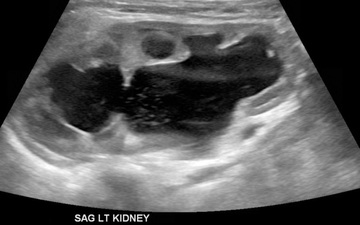
Table 555.4
Society for Fetal Urology Grading System for Hydronephrosis
| GRADE OF HYDRONEPHROSIS | RENAL IMAGE | |
|---|---|---|
| CENTRAL RENAL COMPLEX | RENAL PARENCHYMAL THICKNESS | |
| 0 | Intact | Normal |
| 1 | Slight splitting | Normal |
| 2 | Evident splitting, complex confined within renal border | Normal |
| 3 | Wide splitting, pelvis dilated outside renal border, calyces uniformly dilated | Normal |
| 4 | Further dilation of pelvis and calyces (calyces may appear convex) | Thin |
After Maizels M, Mitchell B, Kass E, et al: Outcome of nonspecific hydronephrosis in the infant: a report from the registry of the Society for Fetal Urology, J Urol 152:2324-2327, 1994.
Voiding Cystourethrogram
In neonates and infants with congenital grade 3 or 4 hydronephrosis and in any child with ureteral dilation, a contrast voiding cystourethrogram (VCUG) should be obtained, because the dilation is secondary to vesicoureteral reflux in 15% of cases. In males, the VCUG also is performed to rule out urethral obstruction, particularly in cases of suspected PUVs. In older children, the urinary flow rate can be measured noninvasively with a urinary flowmeter; decreased flow with a normal bladder contraction suggests infravesical obstruction (e.g., PUVs, urethral stricture). When the urethra cannot be catheterized to obtain a VCUG, the clinician should suspect a urethral stricture or an obstructive urethral lesion. Retrograde urethrography with contrast medium injected into the urethral meatus helps delineate the anatomy of the urethral obstruction.
Radioisotope Studies
Renal scintigraphy is used to assess renal anatomy and function. The two most commonly used radiopharmaceuticals are mercaptoacetyl triglycine (MAG-3) and technetium-99m-labeled dimercaptosuccinic acid (DMSA). MAG-3, which is excreted by renal tubular secretion, is used to assess differential renal function, and when furosemide is administered, drainage also can be measured. DMSA is a renal cortical imaging agent and is used to assess differential renal function and to demonstrate whether renal scarring is present. It is used infrequently in children with obstructive uropathy.
In a MAG-3 diuretic renogram, a small dose of technetium-labeled MAG-3 is injected intravenously (Figs. 555.4 and 555.5 ). During the first 2-3 min, renal parenchymal uptake is analyzed and compared, allowing computation of differential renal function.
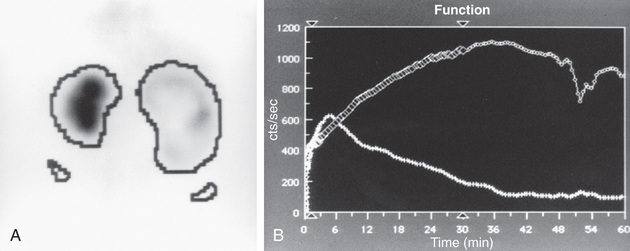
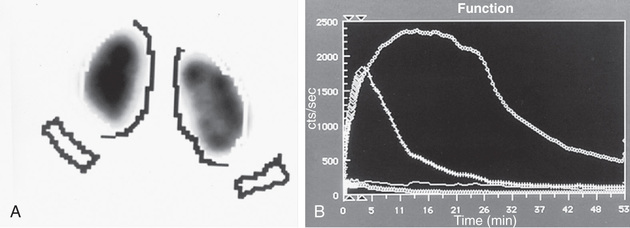
Subsequently, excretion is evaluated. After 20 min, furosemide 1 mg/kg is injected intravenously, and the rapidity and pattern of drainage from the kidneys to the bladder are analyzed. If no obstruction is present, half of the radionuclide should be cleared from the renal pelvis within 10-15 min, termed the half-time (t1/2 ). If there is significant upper tract obstruction, the t1/2 usually is longer than 20 min. A t1/2 of 15-20 min is indeterminate. An elevated t1/2 is suggestive but not diagnostic of obstruction. The images generated usually provide an accurate assessment of the site of obstruction. Numerous variables affect the outcome of the diuretic renogram. Newborn kidneys are functionally immature, and, in the first mo of life, normal kidneys might not demonstrate normal drainage after diuretic administration. Patient dehydration prolongs parenchymal transit and can blunt the diuretic response. Giving an insufficient dose of furosemide can result in slow drainage. If vesicoureteral reflux is present, continuous bladder drainage is mandatory to prevent the radionuclide from refluxing from the bladder into the dilated upper tract, which would prolong the washout phase.
Magnetic Resonance Urography
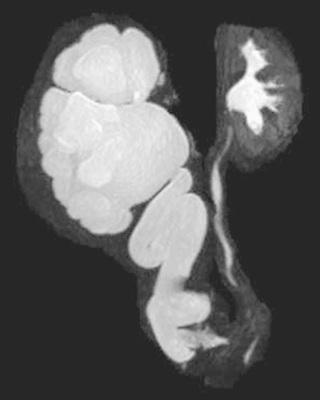
Magnetic resonance (MR) urography also is used to evaluate suspected upper urinary tract pathology. The child is hydrated and given intravenous furosemide. Gadolinium-diethylene tetrapentaacetic acid is injected, and routine T1-weighted and fat-suppressed fast spin-echo T2-weighted imaging is performed through the kidneys, ureters, and bladder. This study provides superb images of the pathology, and methodology permits assessment of differential renal function and drainage (Fig. 555.6 ). There is no radiation exposure; however, young children need sedation or anesthesia. It is used primarily when renal US and radionuclide imaging fail to delineate complex pathology.
Computed Tomography
In children with a suspected ureteral calculus (see Chapter 562 ), noncontrast low-dose spiral CT of the abdomen and pelvis is a standard method of demonstrating whether a calculus is present, its location, and whether there is significant proximal hydronephrosis. This study may be ordered when a renal/bladder US is inconclusive. The disadvantage of CT is the significant radiation exposure, and it should be used only when the results will direct management decisions.
Ancillary Studies
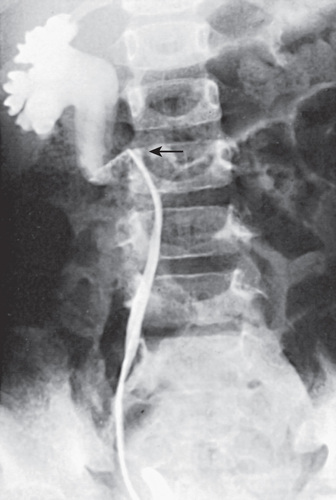
In unusual cases, an antegrade pyelogram (insertion of a percutaneous nephrostomy tube and injection of contrast agent) can be performed to assess the anatomy of the upper urinary tract. This procedure usually requires general anesthesia. In addition, an antegrade pressure-perfusion flow study (Whitaker test) may be performed, in which fluid is infused at a measured rate, usually 10 mL/min. The pressures in the renal pelvis and the bladder are monitored during this infusion, and pressure differences exceeding 20 cm H2 O suggest obstruction. In other cases, cystoscopy with retrograde pyelography provides excellent images of the upper urinary tract (Fig. 555.7 ).
Specific Types of Urinary Tract Obstruction and Their Treatment
Hydrocalycosis
The term hydrocalycosis refers to a localized dilation of the calyx caused by obstruction of its infundibulum, termed infundibular stenosis. This condition can be developmental in origin or secondary to inflammatory processes, such as UTI. It usually is discovered during evaluation for pain or UTI. The diagnosis of infundibular stenosis is usually established by sonograph and CT scan or MR urography.
Ureteropelvic Junction Obstruction
UPJ obstruction is the most common obstructive lesion in childhood and usually is caused by intrinsic stenosis (see Figs. 555.3 to 555.5 ). An accessory artery to the lower pole of the kidney also can cause extrinsic obstruction. The typical appearance on US is grade 3 or 4 hydronephrosis without a dilated ureter. UPJ obstruction most commonly manifests on antenatal sonography revealing fetal hydronephrosis; as a palpable renal mass in a newborn or infant; as abdominal, flank, or back pain; as a febrile UTI; or as hematuria after minimal trauma. Approximately 60% of cases occur on the left side; the male:female ratio is 2 : 1. UPJ obstruction is bilateral in only 10% of cases . In kidneys with UPJ obstruction, renal function may be significantly impaired from pressure atrophy, but approximately half of affected kidneys have relatively normal glomerular function. The anomaly is corrected by performing a pyeloplasty, in which the stenotic segment is excised, and the normal ureter and renal pelvis are reattached. Success rates are 91–98%. Pyeloplasty can be performed using laparoscopic techniques, often robotic-assisted using the da Vinci robot.
Lesser degrees of UPJ narrowing might cause mild hydronephrosis, which usually is nonobstructive, and typically these kidneys function normally. The spectrum of UPJ abnormalities has been referred to as anomalous UPJ . Another cause of mild hydronephrosis is fetal folds of the upper ureter, which also are nonobstructive.
The diagnosis can be difficult to establish in an asymptomatic infant in whom dilation of the renal pelvis is found incidentally in a prenatal US. After birth, the sonographic study is repeated to confirm the prenatal finding. A VCUG is necessary because 10–15% of patients have ipsilateral vesicoureteral reflux. Because neonatal oliguria can cause temporary decompression of a dilated renal pelvis, it is ideal to perform the first postnatal US after the 3rd day of life. Delaying the US may be impractical. If no dilation is found on the initial US, a repeat study should be performed at 1 mo of age. If the kidney shows grade 1 or 2 hydronephrosis and the renal parenchyma appears normal, a period of observation usually is appropriate, with sequential renal US studies to monitor the severity of hydronephrosis, and the hydronephrosis usually disappears. Antibiotic prophylaxis is not indicated for children with mild hydronephrosis. If the hydronephrosis is grade 3 or 4, spontaneous resolution is less likely, and obstruction is more likely to be present, particularly if the renal pelvic diameter is 3 cm. A diuretic renogram with MAG-3 is performed at 4-6 wk of age. If there is poor upper tract drainage or the differential renal function is poor, pyeloplasty is recommended. After pyeloplasty the differential renal function often improves, and improved drainage with furosemide stimulation is expected.
If the differential function on renography is normal and drainage is satisfactory, the infant can be followed with serial US studies, even with grade 4 hydronephrosis. If the hydronephrosis remains severe with no improvement, a repeat diuretic renogram after 6-12 mo can help in the decision between continued observation and surgical repair. Prompt surgical repair is indicated in infants with an abdominal mass, bilateral severe hydronephrosis, a solitary kidney, or diminished function in the involved kidney. In unusual cases in which the differential renal function is < 10% but the kidney has some function, insertion of a percutaneous nephrostomy tube allows drainage of the hydronephrotic kidney for a few weeks to allow reassessment of renal function. In older children who present with symptoms, the diagnosis of UPJ obstruction usually is established by US and diuretic renography.
The following entities should be considered in the differential diagnosis: megacalycosis, a congenital nonobstructive dilation of the calyces without pelvic or ureteric dilation; vesicoureteral reflux with marked dilation and kinking of the ureter; midureteral or distal ureteral obstruction when the ureter is not well visualized on the urogram; and retrocaval ureter.
Midureteral Obstruction
Congenital ureteral stenosis or a ureteral valve in the midureter is rare. It is corrected by excision of the strictured segment and reanastomosis of the normal upper and lower ureteral segments. A retrocaval ureter is an anomaly in which the upper right ureter travels posterior to the inferior vena cava. In this anomaly, the vena cava can cause extrinsic compression and obstruction. A retrograde pyelogram or MR urogram shows the right ureter to be medially deviated at the level of the 3rd lumbar vertebra (see Fig. 555.7 ). Surgical treatment consists of transection of the upper ureter, moving it anterior to the vena cava, and reanastomosing the upper and lower segments. Repair is necessary only when obstruction is present. Retroperitoneal tumors, fibrosis caused by surgical procedures, inflammatory processes (as in chronic granulomatous disease), and radiation therapy can cause acquired midureteral obstruction.
Ectopic Ureter
A ureter that drains outside the bladder is referred to as an ectopic ureter. This anomaly is three times as common in females as in males and usually is detected prenatally. The ectopic ureter typically drains the upper pole of a duplex collecting system (two ureters).
In females, approximately 35% of these ureters enter the urethra at the bladder neck, 35% enter the urethrovaginal septum, 25% enter the vagina, and a few drain into the cervix, uterus, Gartner duct, or a urethral diverticulum. Often the terminal aspect of the ureter is narrowed, causing hydroureteronephrosis. With the exception of the ectopic ureter entering the bladder neck, in females an ectopic ureter causes continuous urinary incontinence from the affected renal moiety. UTI is common because of urinary stasis.
In males, ectopic ureters enter the posterior urethra (above the external sphincter) in 47%, the prostatic utricle in 10%, the seminal vesicle in 33%, the ejaculatory duct in 5%, and the vas deferens in 5%. Consequently, in males, an ectopic ureter does not cause incontinence, and most patients present with a UTI or epididymitis.
Evaluation includes a renal US, VCUG, and renal scan, which demonstrates whether the affected segment has significant function. The US shows the affected hydronephrotic kidney or dilated upper pole and ureter down to the bladder (Fig. 555.8 ). If the ectopic ureter drains into the bladder neck (female), a VCUG usually shows reflux into the ureter. Otherwise, there is no reflux into the ectopic ureter, but there may be reflux into the ipsilateral lower pole ureter or contralateral collecting system.
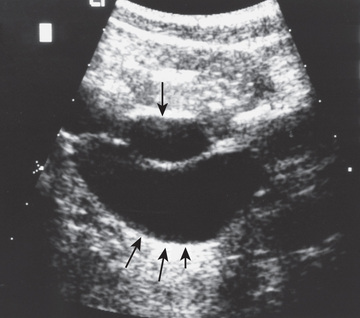
Treatment depends on the status of the renal unit drained by the ectopic ureter. If there is satisfactory function, ureteral reimplantation into the bladder or ureteroureterostomy (anastomosing the ectopic upper pole ureter into the normally inserting lower pole ureter) is indicated. If function is poor, partial or total nephrectomy is indicated. In many centers this procedure is done laparoscopically and often with robotic assistance using the da Vinci robot.
Ureterocele
A ureterocele is a cystic dilation of the terminal ureter and is obstructive because of a pinpoint ureteral orifice. Ureteroceles are much more common in females than in males. Affected children usually are discovered by prenatal US, but some present with a febrile UTI. Ureteroceles may be ectopic, in which case the cystic swelling extends through the bladder neck into the urethra, or orthotopic, in which case the ureterocele is entirely within the bladder. Both orthotopic and ectopic ureteroceles can be bilateral.
In females, ureteroceles nearly always are associated with ureteral duplication (Fig. 555.9 ), whereas in 50% of affected boys there is only one ureter. When associated with a duplication anomaly, the ureterocele drains the upper renal moiety, which commonly functions poorly or is dysplastic because of congenital obstruction. The lower pole ureter drains into the bladder superior and lateral to the upper pole ureter and may reflux.
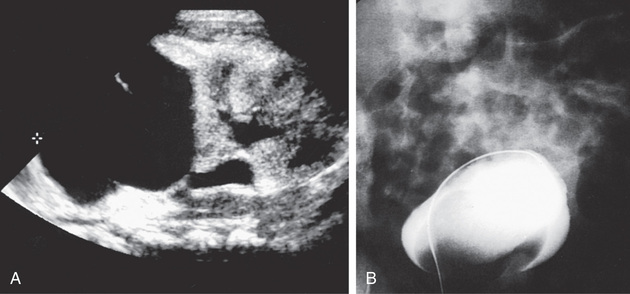
An ectopic ureterocele extends submucosally through the bladder neck into the urethra. Rarely, large ectopic ureteroceles can cause bladder outlet obstruction and retention of urine with bilateral hydronephrosis. In females, the ureterocele can prolapse from the urethral meatus. US is effective in demonstrating the ureterocele and whether the associated obstructed system is duplicated or single. VCUG usually shows a filling defect in the bladder, sometimes large, corresponding to the ureterocele, and it often shows reflux into the adjacent lower pole collecting system with typical findings of a “drooping lily” appearance to the kidney. Nuclear renal scintigraphy is most accurate in demonstrating whether the affected renal moiety has significant function.
Treatment of ectopic ureteroceles varies among different medical centers and depends on whether the upper pole functions on renal scan and whether there is reflux into the lower pole ureter. If there is nonfunction of the upper pole of the kidney and there is no reflux, treatment usually involves laparoscopic, robotic, or open excision of the obstructed upper pole and most of the associated ureter. If there is function in the upper pole or significant reflux into the lower pole ureter, or if the patient is septic from infection of the hydronephrotic kidney, then transurethral incision with cautery is appropriate initial therapy to decompress the ureterocele. Reflux into the incised ureterocele is common, and subsequent excision of the ureterocele and ureteral reimplantation usually is necessary. An alternative method is to perform an upper-to-lower ureteroureterostomy, allowing the obstructed upper pole ureter to drain through the normal lower ureter; this procedure often is performed with minimally invasive laparoscopic (robotic) technique or through a small incision.
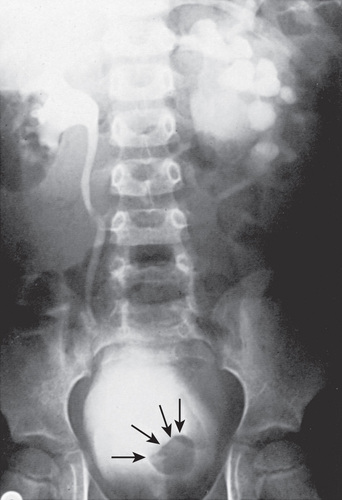
Orthotopic ureteroceles are associated with duplicated or single collecting systems, and the orifice is in the expected location in the bladder (Fig. 555.10 ). These anomalies usually are discovered during an investigation for prenatal hydronephrosis or a UTI. US is sensitive for detecting the ureterocele in the bladder and hydroureteronephrosis. Transurethral incision of the ureterocele effectively relieves the obstruction, but it can result in vesicoureteral reflux, necessitating ureteral reimplantation later. Some prefer open excision of the ureterocele and reimplantation as the initial form of treatment. Small, simple ureteroceles discovered incidentally without upper tract dilation generally do not require treatment.
Megaureter
Table 555.5 presents a classification of megaureters (dilated ureter). Numerous disorders can cause ureteral dilation, and many are nonobstructive.
Table 555.5
| REFLUXING | OBSTRUCTED | NONREFLUXING AND NONOBSTRUCTED | |||
|---|---|---|---|---|---|
| PRIMARY | SECONDARY | PRIMARY | SECONDARY | PRIMARY | SECONDARY |
| Primary reflux | Neuropathic bladder | Intrinsic (primary obstructed megaureter) | Neuropathic bladder | Nonrefluxing, nonobstructive | Diabetes insipidus |
| Megacystic-megaureter syndrome | Hinman syndrome | Ureteral valve | Hinman syndrome | Infection | |
| Ectopic ureter | Posterior urethral valves | Ectopic ureter | Posterior urethral valves | Persistent after relief of obstruction | |
| Prune-belly syndrome | Bladder diverticulum | Ectopic ureterocele | Ureteral calculus | ||
| Postoperative | Extrinsic | ||||
| Postoperative | |||||
Megaureters usually are discovered during antenatal sonography, postnatal UTI, hematuria, or abdominal pain. A careful history, physical examination, and VCUG identify causes of secondary megaureters and refluxing megaureters, as well as the prune-belly syndrome. Primary obstructed megaureters and nonobstructed megaureters probably represent varying degrees of severity of the same anomaly.
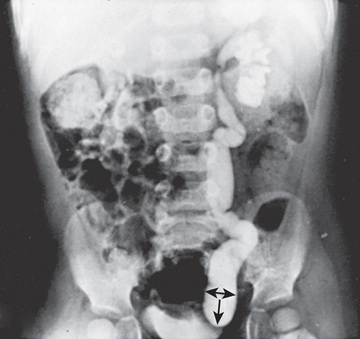
The primary obstructed nonrefluxing megaureter results from abnormal development of the distal ureter, with collagenous tissue replacing the muscle layer. Normal ureteral peristalsis is disrupted, and the proximal ureter widens. In most cases there is not a true stricture. On IVP or an MR urogram, the distal ureter is more dilated in its distal segment and tapers abruptly at or above the junction of the bladder (Fig. 555.11 ). The lesion may be unilateral or bilateral. Significant hydroureteronephrosis suggests obstruction. Megaureter predisposes to UTI, urinary stones, hematuria, and flank pain because of urinary stasis. In most cases, diuretic renography and sequential sonographic studies can reliably differentiate obstructed from nonobstructed megaureters. In most nonobstructed megaureters, the hydroureteronephrosis diminishes gradually (Fig. 555.12 ). Truly obstructed megaureters require surgical treatment, with excision of the narrowed segment, ureteral tapering, and reimplantation of the ureter. The results of surgical reconstruction usually are good, but the prognosis depends on preexisting renal function and whether complications develop.

If differential renal function is normal (>45%) and the child is asymptomatic, it is safe to manage the patient with observation with serial US and periodic diuretic renography to monitor renal function and drainage. In children with grade 4 hydroureteronephrosis, prophylactic antimicrobial therapy should be prescribed, as these children are prone to upper UTI. If renal function deteriorates, upper urinary tract drainage slows, or UTI occurs, ureteral reimplantation is recommended. Approximately 25% of children with a nonrefluxing megaureter undergo ureteral reimplantation.
Prune-Belly Syndrome
Prune-belly syndrome, also called triad syndrome or Eagle-Barrett syndrome, occurs in approximately 1 in 40,000 births; 95% of affected children are male. The characteristic association of deficient abdominal muscles, undescended testes, and urinary tract abnormalities probably results from severe urethral obstruction in fetal life (Fig. 555.13 ). Oligohydramnios and pulmonary hypoplasia are common complications in the perinatal period. Many affected infants are stillborn. Urinary tract abnormalities include massive dilation of the ureters and upper tracts and a very large bladder, with a patent urachus or a urachal diverticulum. Most patients have vesicoureteral reflux. The prostatic urethra usually is dilated, and the prostate is hypoplastic. The anterior urethra may be dilated, resulting in a megalourethra. Rarely, there is urethral stenosis or atresia. The kidneys usually show various degrees of dysplasia, and the testes usually are intraabdominal. Malrotation of the bowel often is present. Cardiac abnormalities occur in 10% of cases; >50% have abnormalities of the musculoskeletal system, including limb abnormalities and scoliosis. In females, anomalies of the urethra, uterus, and vagina usually are present.

Many neonates with prune-belly syndrome have difficulty with effective bladder emptying because the bladder musculature is poorly developed, and the urethra may be narrowed. When no obstruction is present, the goal of treatment is the prevention of UTI with antibiotic prophylaxis. When obstruction of the ureters or urethra is demonstrated, temporary drainage procedures, such as a vesicostomy, can help to preserve renal function until the child is old enough for surgical reconstruction. Some children with prune-belly syndrome have been found to have classic or atypical PUVs. UTIs occur often and should be treated promptly. Correction of the undescended testes by orchidopexy can be difficult in these children because the testes are located high in the abdomen and surgery is best accomplished in the first 6 mo of life. Reconstruction of the abdominal wall offers cosmetic and functional benefits.
The prognosis ultimately depends on the degree of pulmonary hypoplasia and renal dysplasia. One third of children with prune-belly syndrome are stillborn or die in the first few mo of life because of pulmonary hypoplasia. As many as 30% of the long-term survivors develop end-stage renal disease from dysplasia or complications of infection or reflux and eventually require renal transplantation. Renal transplantation in these children offers good results.
Bladder Neck Obstruction
Bladder neck obstruction usually is secondary to ectopic ureterocele, bladder calculi, or a tumor of the prostate (rhabdomyosarcoma). The manifestations include difficulty voiding, urinary retention, UTI, and bladder distention with overflow incontinence. Apparent bladder neck obstruction is common in cases of PUVs, but it seldom has any functional significance. Primary bladder neck obstruction is extremely rare.
Posterior Urethral Valves
The most common cause of severe obstructive uropathy in children is PUVs, affecting 1 in 8,000 males. The urethral valves are tissue leaflets fanning distally from the prostatic urethra to the external urinary sphincter. A slit-like opening usually separates the leaflets. Valves are of unclear embryologic origin and cause varying degrees of obstruction. Approximately 30% of patients experience end-stage renal disease or chronic renal insufficiency. The prostatic urethra dilates, and the bladder muscle undergoes hypertrophy. Vesicoureteral reflux occurs in 50% of patients, and distal ureteral obstruction can result from a chronically distended bladder or bladder muscle hypertrophy. The renal changes range from mild hydronephrosis to severe renal dysplasia; their severity probably depends on the severity of the obstruction and its time of onset during fetal development. As in other cases of obstruction or renal dysplasia, there may be oligohydramnios and pulmonary hypoplasia.
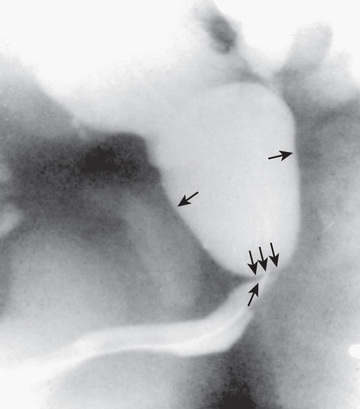
Affected males with PUVs often are discovered prenatally when maternal US reveals bilateral hydronephrosis, a distended bladder, and, if the obstruction is severe, oligohydramnios. Prenatal bladder decompression by percutaneous vesicoamniotic shunt or open fetal surgery has been reported. Experimental and clinical evidence of the possible benefits of fetal intervention is lacking, and few affected fetuses are candidates. Prenatally diagnosed PUVs, particularly when discovered in the second trimester, carry a poorer prognosis than those detected in the third trimester following a normal second-trimester fetal US. In the male neonate, PUVs are suspected when there is a palpably distended bladder and the urinary stream is weak. If the obstruction is severe and goes unrecognized during the neonatal period, infants can present later in life with failure to thrive because of uremia or sepsis caused by infection in the obstructed urinary tract. With lesser degrees of obstruction, children present later in life with difficulty in achieving diurnal urinary continence or with UTI. The diagnosis is established with a VCUG (Fig. 555.14 ) or by perineal US.
After the diagnosis is established, renal function and the anatomy of the upper urinary tract should be carefully evaluated. In the healthy neonate, a small polyethylene feeding tube (No. 5 or No. 8 French) is inserted in the bladder and left for several days. Passing the feeding tube may be difficult because the tip of the tube can coil in the prostatic urethra. A sign of this problem is that urine drains around the catheter rather than through it. A Foley (balloon) catheter should not be used , because the balloon can cause severe bladder spasm, which can produce severe ureteral obstruction.
If the serum creatinine level remains normal or returns to normal, treatment consists of transurethral ablation of the valve leaflets, which is performed endoscopically under general anesthesia. If the urethra is too small for transurethral ablation, temporary vesicostomy is preferred, in which the dome of the bladder is exteriorized on the lower abdominal wall. When the child is older, the valves may be ablated and the vesicostomy closed.
If the serum creatinine level remains high or increases despite bladder drainage by a small catheter, secondary ureteral obstruction, irreversible renal damage, or renal dysplasia should be suspected. In such cases, a vesicostomy should be considered. Cutaneous pyelostomy rarely affords better drainage when compared with cutaneous vesicostomy, and the latter also allows continued bladder growth and gradual improvement in bladder wall compliance.
In the septic and uremic infant, lifesaving measures must include prompt correction of the electrolyte imbalance and control of the infection by appropriate antibiotics. Drainage of the upper tracts by percutaneous nephrostomy and hemodialysis may be necessary. After the patient's condition becomes stable, evaluation and treatment may be undertaken. PUVs are diagnosed in some older males because of a poor stream, diurnal incontinence, or a UTI; these males generally are treated by primary valve ablation.
Favorable prognostic factors include a normal prenatal US between 18 and 24 wk of gestation, a serum creatinine level < 0.8-1.0 mg/dL after bladder decompression, and visualization of the corticomedullary junction on renal sonography. In several situations, a “popoff valve” can occur during urinary tract development, which preserves the integrity of one or both kidneys. For example, 15% of males with PUVs have unilateral reflux into a nonfunctioning dysplastic kidney, termed the VURD syndrome (valves, unilateral reflux, dysplasia). In these males, the high bladder pressure is dissipated into the nonfunctioning kidney, allowing normal development of the contralateral kidney. In newborn males with urinary ascites, the urine generally leaks out from the obstructed collecting system through the renal fornices, allowing normal development of the kidneys. Unfavorable prognostic factors include the presence of oligohydramnios in utero, identification of hydronephrosis before 24 wk of gestation, a serum creatinine level > 1.0 mg/dL after bladder decompression, identification of cortical cysts in both kidneys, and persistence of diurnal incontinence beyond 5 yr of age.
The prognosis in the newborn is related to the child's degree of pulmonary hypoplasia and potential for recovery of renal function. Severely affected infants often are stillborn. Of those who survive the neonatal period, approximately 30% eventually require kidney transplantation and 15% have renal insufficiency. In some series, kidney transplantation in children with PUVs has a lower success rate than does transplantation in children with normal bladders, presumably because of the adverse influence of altered bladder function on graft function and survival.
After valve ablation, antimicrobial prophylaxis is beneficial in preventing UTI, because hydronephrosis to some degree often persists for many years. These males should be evaluated annually with a renal US, physical examination including assessment of somatic growth and blood pressure, urinalysis, and determination of serum levels of electrolytes. Many males have significant polyuria resulting from a concentrating defect secondary to prolonged obstructive uropathy. If these children acquire a systemic illness with vomiting and/or diarrhea, urine output cannot be used to assess their hydration status. They can become dehydrated quickly, and there should be a low threshold for hospital admission for intravenous rehydration. Some of these patients have renal tubular acidosis, requiring oral bicarbonate therapy. If there is any significant degree of renal dysfunction, growth impairment, or hypertension, the child should be followed closely by a pediatric nephrologist. When vesicoureteral reflux is present, expectant treatment and prophylactic doses of antibacterial drugs are advisable. If breakthrough UTI occurs, surgical correction should be undertaken.
After treatment, males with urethral valves often do not achieve diurnal urinary continence as early as other males. Incontinence can result from a combination of factors, including uninhibited bladder contractions, poor bladder compliance, bladder atonia, bladder neck dyssynergia, or polyuria. Often these males require urodynamic evaluation with urodynamics or videourodynamics to plan therapy. Males with a poorly compliant bladder are at significant risk for ongoing renal damage, even in the absence of infection. Overnight catheter drainage has been shown to be beneficial in males with polyuria and can help preserve renal function. Urinary incontinence usually improves with age, particularly after puberty. Meticulous attention to bladder compliance, emptying, and infection can improve results in the future.
Urethral Atresia
The most severe form of obstructive uropathy in males is urethral atresia, a rare condition. In utero there is a distended bladder, bilateral hydroureteronephrosis, and oligohydramnios. In most cases, these infants are stillborn or succumb to pulmonary hypoplasia. Some males with prune-belly syndrome also have urethral atresia. If the urachus is patent, oligohydramnios is unlikely, and the infant usually survives. Urethral reconstruction is difficult, and most patients are managed with continent urinary diversion.
Urethral Hypoplasia
Urethral hypoplasia is a rare form of obstructive uropathy in males that is less severe than urethral atresia. In urethral hypoplasia, the urethral lumen is extremely small. Neonates with urethral hypoplasia typically have bilateral hydronephrosis and a distended bladder. Passage of a small pediatric feeding tube through the urethra is difficult or impossible. Usually a cutaneous vesicostomy must be performed to relieve upper urinary tract obstruction, and the severity of renal insufficiency is variable. The most severely affected males have end-stage renal disease. Treatment includes urethral reconstruction, gradual urethral dilation, or continent urinary diversion.
Urethral Stricture
Urethral strictures in males usually result from urethral trauma, either iatrogenic (catheterization, endoscopic procedures, previous urethral reconstruction) or accidental (straddle injuries, pelvic fractures). Because these lesions can develop gradually, the decrease in force of the urinary stream is seldom noticed by the child or the parents. More commonly, the obstruction causes symptoms of bladder instability, hematuria, or dysuria. Catheterization of the bladder usually is impossible. The diagnosis is made by a retrograde urethrogram, in which contrast is injected toward the bladder through a catheter inserted into the distal urethra. US also has been used to diagnose urethral strictures. Endoscopy is confirmatory. Endoscopic treatment of short strictures by direct-vision urethrotomy is often successful initially and results in a profoundly improved urinary stream, but often the stricture recurs. Longer strictures surrounded by periurethral fibrosis often require urethroplasty. Repeated endoscopic procedures generally should be avoided, because they can cause additional urethral damage. Noninvasive measurement of the urinary flow rate and pattern is useful for diagnosis and follow-up.
In females, true urethral strictures are rare because the female urethra is protected from trauma, particularly in childhood. In the past it was thought that a distal urethral ring commonly caused obstruction of the female urethra and UTI and that affected females benefited from urethral dilation. The diagnosis was suspected when a “spinning-top” deformity of the urethra was found in the VCUG (see Fig. 558.3 in Chapter 558 ) and was confirmed by urethral calibration. There is no correlation between the radiologic appearance of the urethra in the VCUG and the urethral caliber and no significant difference in urethral caliber between females with recurrent cystitis and normal age-matched controls. The finding usually is secondary to detrusor–sphincter discoordination. Consequently, urethral dilation in females rarely is indicated.
Anterior Urethral Valves and Urethral Diverticula in the Male
Anterior urethral valves are rare. The obstruction is not obstructing valve leaflets, as occurs in the posterior urethra. Rather, it is a urethral diverticulum in the penile urethra that expands during voiding. Distal extension of the diverticulum causes extrinsic compression of the distal penile urethra, causing urethral obstruction. There is usually a soft mass on the ventral surface of the penis at the penoscrotal junction. In addition, the urinary stream often is weak, and the physical findings associated with PUVs often are present. The diverticulum may be small and minimally obstructive, or, in other cases, may be severely obstructive and cause renal insufficiency. The diagnosis is suspected on physical examination and is confirmed by the VCUG. Treatment involves open excision of the diverticulum or transurethral excision of the distal urethral cusp. Urethral diverticulas occasionally occur after extensive hypospadias repair.
Fusiform dilation of the urethra or megalourethra can result from underdevelopment of the corpus spongiosum and support structures of the urethra. This condition is commonly associated with the prune-belly syndrome.
Male Urethral Meatal Stenosis
See Chapter 559 for information on urethral meatal stenosis in males.