Congenital Anomalies and Dysgenesis of the Kidneys
Jack S. Elder
Embryonic and Fetal Development
The kidney is derived from interaction between the ureteral bud and the metanephric blastema. During the 5th wk of gestation, the ureteral bud arises from the mesonephric (wolffian) duct and penetrates the metanephric blastema, which is an area of undifferentiated mesenchyme on the nephrogenic ridge. The ureteral bud undergoes a series of approximately 15 generations of divisions and by wk 20 of gestation forms the entire collecting system: the ureter, renal pelvis, calyces, papillary ducts, and collecting tubules. Signals from the mesenchymal cells induce ureteric bud formation from the wolffian duct as well as ureteric bud branching. Reciprocal signals from the ureteric bud and, later, from its branching tips induce mesenchymal cells to condense, proliferate, and convert into epithelial cells. Under the inductive influence of the ureteral bud, nephron differentiation begins during the 7th wk of gestation. By the 20th wk of gestation, when the collecting system is developed, approximately 30% of the nephrons are present. Nephrogenesis continues at a nearly exponential rate and is complete by the 36th wk of gestation. During nephrogenesis, the kidneys ascend to a lumbar site just below the adrenal glands. At least 16 signaling agents have been identified that regulate renal development. Defects in any of the signaling activities could cause a kidney not to form (renal agenesis), or to differentiate abnormally (renal dysgenesis). Dysgenesis of the kidney includes aplasia, dysplasia, hypoplasia, and certain forms of renal cystic disease.
The fetal kidneys play a minor role in the maintenance of fetal salt and water homeostasis. The rate of urine production increases throughout gestation; at term, volumes have been reported to be 50 mL/hr. The glomerular filtration rate is 25 mL/min/1.73 m2 at term and triples by 3 mo postterm. The increase in the glomerular filtration rate is caused by a reduction in intrarenal vascular resistance and redistribution of intrarenal blood flow to the cortex, where more nephrons are located.
Renal Agenesis
Renal agenesis, or absent kidney development, can occur secondary to a defect of the wolffian duct, ureteric bud, or metanephric blastema. Unilateral renal agenesis has an incidence of 1 in 450-1,000 births. Unilateral renal agenesis often is discovered during the course of an evaluation for other congenital anomalies (VACTERL [vertebral defects, imperforate anus, congenital heart disease, tracheoesophageal fistula, renal and limb defects] syndrome; e.g., see Chapter 371 ). Its incidence is increased in newborns with a single umbilical artery. In true agenesis, the ureter and the ipsilateral bladder hemitrigone are absent. The contralateral kidney undergoes compensatory hypertrophy, to some degree prenatally but primarily after birth. Approximately 15% of these children have contralateral vesicoureteral reflux, and most males have an ipsilateral absent vas deferens because the wolffian duct is absent. Because the wolffian and müllerian ducts are contiguous, müllerian abnormalities in girls also are common. The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome (1 in 4,000 to 1 in 10,000 female births) is a group of associated findings that may include vaginal aplasia, uterine maldevelopment, and normal ovaries. Two types are described—type I and type II. In type I, only müllerian aplasia occurs, whereas in type II, there are associated anomalies, most commonly unilateral renal agenesis or a horseshoe kidney, and skeletal anomalies are present in 10% (see Chapter 569 ). Zinner syndrome is considered the male counterpart of MRKH syndrome (Fig. 552.1 ). In this condition, males with unilateral renal agenesis (or a regressed multicystic dysplastic kidney) have an ipsilateral seminal vesicle cyst, and a possible epididymal cyst and dilated distal ureteral segment. These patients typically present in adolescence.

Renal agenesis is distinguished from aplasia, in which a nubbin of nonfunctioning tissue is seen capping a normal or abnormal ureter. This distinction may be difficult but usually is clinically insignificant. Unilateral renal agenesis is diagnosed in some patients based on the finding of an absent kidney on ultrasonography or renal scintigraphy (renal scan). Some of these patients were born with a hypoplastic kidney or a multicystic dysplastic kidney that underwent complete cyst regression. Although the specific diagnosis is not critical, if the finding of an absent kidney is based on an ultrasonogram, a functional imaging study such as a renal scan should be considered because some of these patients have an ectopic kidney in the pelvis. If there is a normal contralateral kidney, long-term renal function usually remains normal.
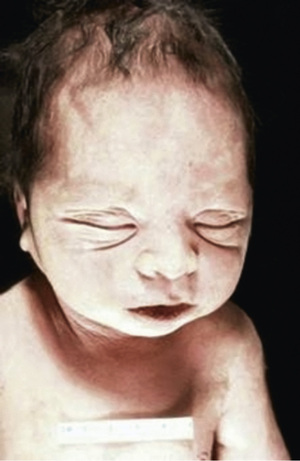
Bilateral renal agenesis is incompatible with extrauterine life and produces the Potter syndrome. Death occurs shortly after birth from pulmonary hypoplasia. The newborn has a characteristic facial appearance, termed Potter facies (Fig. 552.2 ). The eyes are widely separated with epicanthic folds, the ears are low set, the nose is broad and compressed flat, the chin is receding, and there are limb anomalies. Bilateral renal agenesis should be suspected when maternal ultrasonography demonstrates oligohydramnios, nonvisualization of the bladder, and absent kidneys. The incidence of this disorder is 1 in 3,000 births, with a male predominance, and represents 20% of newborns with the Potter phenotype. Other common causes of neonatal renal failure associated with the Potter phenotype include cystic renal dysplasia and obstructive uropathy. Less-common causes are autosomal recessive polycystic kidney disease (infantile), renal hypoplasia, and medullary dysplasia. Neonates with bilateral renal agenesis die of pulmonary insufficiency from pulmonary hypoplasia rather than renal failure (see Chapter 423 ).
The term familial renal adysplasia describes families in which renal agenesis, renal dysplasia, multicystic kidney (dysplasia), or a combination, occurs in a single family. This disorder has an autosomal dominant inheritance pattern with a penetrance of 50–90% and variable expression. Because of this association, some clinicians advise screening first-degree relatives of persons who have renal agenesis or dysplasia, but this is not standard practice.
Whether persons with a solitary kidney should avoid contact sports such as football and karate is unresolved. The arguments favoring participation are that there are other solitary organs (spleen, liver, and brain) that do not preclude participation in contact sports, and there have been only a few reports of persons losing a kidney from sports injuries. The arguments against such participation are that the contralateral normal kidney is hypertrophic and not as well protected by the ribs, and a serious renal injury could have serious lifelong consequences. The American Academy of Pediatrics recommends an “individual assessment for contact, collision, and limited-contact sports.”
Renal Dysgenesis: Dysplasia, Hypoplasia, and Cystic Anomalies
Renal dysgenesis refers to maldevelopment of the kidney that affects its size, shape, or structure. The three principal types of dysgenesis are dysplastic, hypoplastic, and cystic. Although dysplasia always is accompanied by a decreased number of nephrons (hypoplasia), the converse is not true: Hypoplasia can occur in isolation. When both conditions are present, the term hypodysplasia is preferred. The term dysplasia is technically a histologic diagnosis and refers to focal, diffuse, or segmentally arranged primitive structures, specifically primitive ductal structures, resulting from abnormal metanephric differentiation. Nonrenal elements, such as cartilage, also may be present. The condition can affect all or only part of the kidney. If cysts are present, the condition is termed cystic dysplasia. If the entire kidney is dysplastic with a preponderance of cysts, the kidney is referred to as a multicystic dysplastic kidney (MCDK) (Fig. 552.3 ).

The pathogenesis of dysplasia is multifactorial. The “bud” theory proposes that if the ureteral bud arises in an abnormal location, such as an ectopic ureter, there is abnormal penetration and induction of the metanephric blastema, which causes abnormal kidney differentiation, resulting in dysplasia. Renal dysplasia also can occur with severe obstructive uropathy early in gestation, as with the most severe cases of posterior urethral valves or in an MCDK, in which a portion of the ureter is absent or atretic.
MCDK is a congenital condition in which the kidney is replaced by cysts and does not function; it can result from ureteral atresia. Kidney size is highly variable. The incidence is approximately 1 in 2,000. Some clinicians incorrectly use the terms multicystic kidney and polycystic kidney interchangeably. However, polycystic kidney disease is an inherited disorder that may be autosomal recessive or autosomal dominant and affects both kidneys (see Chapter 541 ). MCDK usually is unilateral and generally is not inherited. Bilateral MCDKs are incompatible with life.
MCDK is the most common cause of an abdominal mass in the newborn, but the vast majority are nonpalpable at birth. In most cases, it is discovered incidentally during prenatal sonography. In some patients, the cysts are identified prenatally, but the cysts regress in utero and no kidney is identified on imaging at birth. Contralateral hydronephrosis is present in 5–10% of patients. Sonography shows the characteristic appearance of a kidney replaced by multiple cysts of varying sizes that do not communicate, and no identifiable parenchyma is present. In the past, most cases were confirmed with a renal scan, which should demonstrate nonfunction. However, presently the diagnosis of MCDK is usually straightforward based on the renal ultrasound, and a scan generally is unnecessary. In some patients, usually boys, a small nonobstructing ureterocele is present in the bladder (see Chapter 555 ). Although 15% have contralateral vesicoureteral reflux, it is usually low grade, and obtaining a voiding cystourethrogram also is unnecessary, unless there is significant contralateral hydronephrosis or the child develops an upper urinary tract infection. Management is controversial. Complete cyst regression occurs in nearly half of MCDKs by age 7 yr. The risk of associated hypertension is 0.2–1.2%, and the risk of Wilms tumor arising from an MCDK is approximately 1 in 1,200. Because neoplasms arise from the stromal rather than the cystic component, even if the cysts regress completely, the likelihood that the kidney could develop a neoplasm is not altered.
Because of the occult nature of these potential problems, some clinicians advise annual follow-up with sonography and blood pressure measurement. The most important aspect of follow-up is being certain that the solitary kidney is functioning normally. If there is an abdominal mass, the cysts enlarge, the stromal core increases in size, or hypertension develops, nephrectomy is recommended. In lieu of follow-up screening, laparoscopic nephrectomy may be performed.
Renal hypoplasia refers to a small nondysplastic kidney that has fewer than the normal number of calyces and nephrons. The term encompasses a group of conditions with an abnormally small kidney and should be distinguished from aplasia, in which the kidney is rudimentary. If the condition is unilateral, the diagnosis usually is made incidentally during evaluation for another urinary tract problem or hypertension. Bilateral hypoplasia usually manifests with signs and symptoms of chronic renal failure and is a leading cause of end-stage renal disease during the first decade of life. A history of polyuria and polydipsia is common. Urinalysis results may be normal. In a rare form of bilateral hypoplasia called oligomeganephronia, the number of nephrons is markedly reduced and those present are markedly hypertrophied.
The Ask-Upmark kidney, also termed segmental hypoplasia, refers to small kidneys, usually weighing not more than 35 g, with one or more deep grooves on the lateral convexity, underneath which the parenchyma consists of tubules resembling those in the thyroid gland. It is unclear whether the lesion is congenital or acquired. Most patients are 10 yr or older at diagnosis and have severe hypertension. Nephrectomy usually controls the hypertension.
Renal Cysts in Children
Although uncommon, there are many renal cystic disorders in children (Table 552.1 ). The most common is the simple renal cyst . The mean incidence is 0.22%; they are usually discovered incidentally during urinary tract imaging. Most are small and asymptomatic and do not require treatment, although follow-up imaging is recommended. If there are septations, irregular margins, calcifications, or a cluster of cysts, further evaluation may be indicated, however. The Bosniak classification of simple and complex renal cysts places various cystic lesions into four risk categories, and helps guide a decision on whether removal of a lesion is necessary. A calyceal diverticulum is an outpouching of the collecting system into the corticomedullary region of the kidney, and it usually arises from the fornix of a calyx, typically in the upper or lower pole. Typically, the infundibulum between the diverticulum and renal pelvis is narrow. Occasionally, calculi form within the lesion or it causes symptoms of flank pain, necessitating removal of the diverticulum.
Table 552.1
From Glassberg KI, Stephens FD, Lebowitz RL, et al: Renal dysgenesis and cystic disease of the kidney: a Report of the Committee on Terminology, Nomenclature and Classification, Section on Urology, American Academy of Pediatrics, J Urol 138[4]:1085-1092, 1987, Tab 2.
A multilocular cyst (multilocular cystic nephroma) is a lesion in the kidney that falls in a spectrum of diseases, along with multilocular cyst with partially differentiated Wilms tumor, multilocular cyst with nodules of Wilms tumor, or cystic Wilms tumor. The multilocular cyst is considered benign and is unrelated to the multicystic dysplastic kidney. More than 95% occur in children < 4 yr, and most are discovered during evaluation for an abdominal or flank mass. The lesion should be removed.
Anomalies in Shape and Position
During renal development, the kidneys normally ascend from the pelvis into their normal position behind the ribs. The normal process of ascent and rotation of the kidney may be incomplete, resulting in renal ectopia or nonrotation. The ectopic kidney may be in a pelvic, iliac, thoracic, or contralateral position. If the ectopia is bilateral, in 90% of persons the two kidneys fuse. The incidence of renal ectopia is approximately 1 in 900 (Fig. 552.4 ).
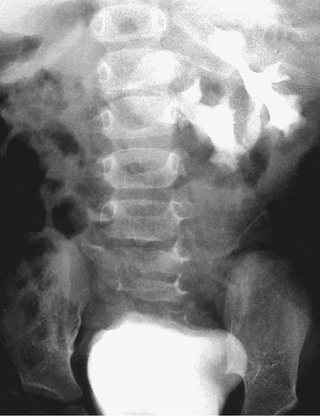
Renal fusion anomalies are more common. The lower poles of the kidneys can fuse in the midline, resulting in a horseshoe kidney (Fig. 552.5 ); the fused portion is termed the isthmus and may be thick functioning parenchyma or a thin fibrous strand. Horseshoe kidneys occur in 1 in 400-500 births and are seen in 7% of patients with Turner syndrome. Horseshoe kidney is one of the many renal anomalies that occur in 30% of patients with Turner syndrome (see Chapter 604 ). Wilms tumors are four times more common in children with horseshoe kidneys than in the general population. Stone disease and hydronephrosis secondary to ureteropelvic junction obstruction are other potential late complications. The incidence of MCDK affecting one of the two sides of a horseshoe kidney also is increased. With crossed fused ectopia, one kidney crosses over to the other side and the parenchyma of the two kidneys is fused. Renal function usually is normal. Most commonly, the left kidney crosses over and fuses with the lower pole of the right kidney. The insertion of the ureter to the bladder does not change, and the adrenal glands remain in their normal positions. The clinical significance of this anomaly is that if renal surgery is necessary, the blood supply is variable and can make partial nephrectomy more difficult.

Associated Physical Findings
Upper urinary tract anomalies are more common in children with certain physical findings. The incidence of renal anomalies is increased if there is a single umbilical artery and an abnormality of another organ system (congenital heart disease). External ear anomalies (particularly if the child has multiple congenital anomalies), imperforate anus, and scoliosis are associated with renal anomalies. Infants with these physical findings should undergo a renal sonogram.