Congenital Disorders of the Lung
Pulmonary Agenesis and Aplasia
Joshua A. Blatter, Jonathan D. Finder
Etiology and Pathology
Pulmonary agenesis differs from hypoplasia in that agenesis entails the complete absence of a lung. Agenesis differs from aplasia by the absence of a bronchial stump or carina that is seen in aplasia. Bilateral pulmonary agenesis is incompatible with life, manifesting as severe respiratory distress and failure. Pulmonary agenesis is thought to be an autosomal recessive trait, with an estimated incidence of 1 in 10,000-15,000 births.
Clinical Manifestations and Prognosis
Unilateral agenesis or hypoplasia can have few symptoms and nonspecific findings, resulting in only 33% of the cases being diagnosed while the patient is living. Symptoms tend to be associated with central airway complications of compression, stenosis, and/or tracheobronchomalacia. In patients in whom the right lung is absent, the aorta can compress the trachea and lead to symptoms of central airway compression. Right lung agenesis has a higher morbidity and mortality than left lung agenesis. Pulmonary agenesis is often seen in association with other congenital anomalies such as the VACTERL sequence (v ertebral anomalies, a nal atresia, c ongenital heart disease, t racheoesophageal fistula, r enal anomalies, and l imb anomalies), ipsilateral facial and skeletal malformations, and central nervous system and cardiac malformations. Compensatory growth of the remaining lung allows improved gas exchange, but the mediastinal shift can lead to scoliosis and airway compression. Scoliosis can result from unequal thoracic growth.
Diagnosis and Treatment
Chest radiographic findings of unilateral lung or lobar collapse with a shift of mediastinal structures toward the affected side can prompt referral for suspected foreign body aspiration, mucous plug occlusion, or other bronchial mass lesions. The diagnosis requires a high index of suspicion to avoid the unnecessary risks of bronchoscopy, including potential perforation of the rudimentary bronchus. CT of the chest is diagnostic, although the diagnosis may be suggested by chronic changes in the contralateral aspect of the chest wall and lung expansion on chest radiographs. Because pulmonary agenesis can be associated with a wide variety of congenital lesions, whole body MRI can be useful to determine whether other systems (e.g., cardiac, gastrointestinal) are affected. Conservative treatment is usually recommended, although surgery has offered benefit in selected cases.
Bibliography
Dillman JR, Sanchez R, Ladino-Torres MF, et al. Expanding upon the unilateral hyperlucent hemithorax in children. Radiographics . 2011;31(3):723–741.
Kim K, Min KH, Park SJ, et al. Right pulmonary agenesis in a 12-year-old girl. Am J Respir Crit Care Med . 2011;184(6):742–743.
Pierron C, Sigal-Cinqualbre A, Lambert V, et al. Left pulmonary artery sling with right lung aplasia. J Pediatr Surg . 2011;46(11):2190–2194.
Pulmonary Hypoplasia
Joshua A. Blatter, Jonathan D. Finder
Etiology and Pathology
Pulmonary hypoplasia involves a decrease in both the number of alveoli and the number of airway generations. The hypoplasia may be bilateral in the setting of bilateral lung constraint, as in oligohydramnios or thoracic dystrophy. Pulmonary hypoplasia is usually secondary to other intrauterine disorders that produce an impairment of normal lung development (see Chapter 122 ). Conditions such as deformities of the thoracic spine and rib cage (thoracic dystrophy), pleural effusions with fetal hydrops, congenital pulmonary airway malformation, and congenital diaphragmatic hernia physically constrain the developing lung. Any condition that produces oligohydramnios (fetal renal insufficiency or prolonged premature rupture of membranes) can also lead to diminished lung growth. In these conditions, airway and arterial branching are inhibited, thereby limiting the capillary surface area. Large unilateral lesions, such as congenital diaphragmatic hernia or pulmonary airway malformation, can displace the mediastinum and thereby produce a contralateral hypoplasia, although usually not as severe as that seen on the ipsilateral side.
Clinical Manifestations
Pulmonary hypoplasia is usually recognized in the newborn period, owing to either the respiratory insufficiency or the presentation of persistent pulmonary hypertension (see Chapter 122.7 ). Later presentation (tachypnea) with stress or respiratory viral infection can be seen in infants with mild pulmonary hypoplasia.
Diagnosis and Treatment
A variety of imaging techniques, including MRI and ultrasound, with estimation of oligohydramnios, can be helpful to identify hypoplasia but not to predict pulmonary function. Mechanical ventilation and oxygen may be required to support gas exchange. Specific therapy to control associated pulmonary hypertension, such as inhaled nitric oxide, may be useful. In cases of severe hypoplasia, the limited capacity of the lung for gas exchange may be inadequate to sustain life. Extracorporeal membrane oxygenation can provide gas exchange for a critical period of time and permit survival. Rib-expanding devices (vertically expansible prosthetic titanium ribs) can improve the survival of patients with thoracic dystrophies (see Chapter 720 ).
Bibliography
Parikh DH, Rasiah SV. Congenital lung lesions: postnatal management and outcome. Semin Pediatr Surg . 2015;24(4):160–167.
Puligandla PS, Laberge JM. Congenital lung lesions. Clin Perinatol . 2012;39(2):331–347.
Vergani P. Prenatal diagnosis of pulmonary hypoplasia. Curr Opin Obstet Gynecol . 2012;24(2):89–94.
Congenital Cystic Malformation (Congenital Pulmonary Airway Malformation)
Joshua A. Blatter, Jonathan D. Finder
Pathology
Congenital pulmonary airway malformation (CPAM) , formerly known as cystic adenomatoid malformation, consists of hamartomatous or dysplastic lung tissue mixed with more normal lung, generally confined to 1 lobe. This congenital pulmonary disorder occurs in approximately 1-4 in 100,000 births. Prenatal ultrasonographic findings are classified as macrocystic (single or multiple cysts >5 mm) or microcystic (echogenic cysts <5 mm). Five histologic patterns have been described. Type 0 (acinar dysplasia) is least common (<3%) and consists of microcystic disease throughout the lungs. The prognosis is poorest for this type, and infants die at birth. Type 1 (60%) is macrocystic and consists of a single or several large (>2 cm in diameter) cysts lined with ciliated pseudostratified epithelium; the lesion is localized involving only a part of 1 lobe. One third of cases have mucus-secreting cells. Presentation is in utero or in the newborn period. Cartilage is rarely seen in the wall of the cyst. This type has a good prognosis for survival. Type 2 (20%) is microcystic and consists of multiple small cysts with histology similar to that of the type 1 lesion. Type 2 is associated with other serious congenital anomalies (renal, cardiac, diaphragmatic hernia) and carries a poor prognosis. Type 3 (<10%) is seen mostly in males; the lesion is a mixture of microcysts and solid tissue with bronchiole-like structures lined with cuboidal ciliated epithelium and separated by areas of nonciliated cuboidal epithelium. The prognosis for this type, like type 0, is poor. Type 4 (10%) is commonly macrocystic and lacks mucus cells. It is associated with malignancy (pleuropulmonary blastoma) and can present either in childhood or in asymptomatic adults.
Etiology
The lesion probably results from an embryologic injury before the 35th day of gestation, with maldevelopment of terminal bronchiolar structures. Histologic examination reveals little normal lung and many glandular elements. Cysts are very common; cartilage is rare. The presence of cartilage might indicate a somewhat later embryologic insult, perhaps extending into the 10th-24th wk. Although growth factor interactions and signaling mechanisms have been implicated in altered lung-branching morphogenesis, the exact roles in the maldevelopment seen here remain obscure.
Diagnosis
Cystic airway malformations can be diagnosed in utero by ultrasonography (Fig. 423.1 ). Fetal cystic lung abnormalities can include CPAM (40%), pulmonary sequestration (14%) (see Chapter 423.4 ), or both (26%); the median age at diagnosis is usually 21 wk of gestation. In one series, only 7% had severe signs of fetal distress including hydrops, pleural effusion, polyhydramnios, ascites, or severe facial edema; 96% of the fetuses were born alive, 2 of whom died in the neonatal period. CPAM volume (i.e., CPAM volume ratio [CVR]) can be used to predict risk of hydrops. Lesions causing fetal hydrops have a poor prognosis. Large lesions, by compressing adjacent lung, can produce pulmonary hypoplasia in nonaffected lobes (see Chapter 423.2 ). Even lesions that appear large in early gestation can regress considerably or decrease in relative size and be associated with good pulmonary function in childhood. CT allows accurate diagnosis and sizing of the lesion and is indicated even in asymptomatic neonates.
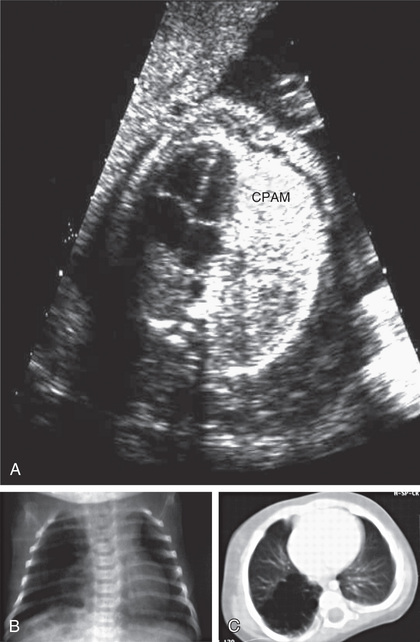
Clinical Manifestations
Patients can present in the newborn period or early infancy with respiratory distress, recurrent respiratory infection, and pneumothorax. The lesion may be confused with a diaphragmatic hernia (see Chapter 122.10 ). Patients with smaller lesions are usually asymptomatic until mid-childhood, when episodes of recurrent or persistent pulmonary infection or chest pain occur. Breath sounds may be diminished, with mediastinal shift away from the lesion on physical examination. Chest radiographs reveal a cystic mass, sometimes with mediastinal shift (Fig. 423.2 ). Occasionally, an air-fluid level suggests a lung abscess (see Chapter 431 ).
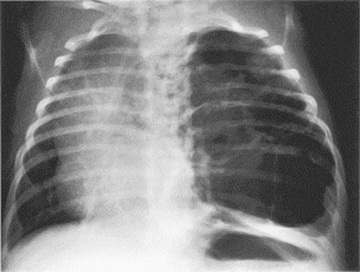
Treatment
Antenatal intervention in severely affected infants is controversial but can include excision of the affected lobe for microcystic lesions, aspiration of macrocystic lesions, and, rarely, open fetal surgery. In the postnatal period, surgery is indicated for symptomatic patients. Although surgery may be delayed for asymptomatic infants because postnatal resolution has been reported, true resolution appears to be very rare in that abnormalities usually remain detectable on CT or MRI. Sarcomatous and carcinomatous degeneration have been described in patients with CPAM, so surgical resection by 1 yr of age is recommended to limit malignant potential. The mortality rate is <10%. Another indication for surgery is to rule out pleuropulmonary blastoma (PPB) , a malignancy that can appear radiographically similar to type 1 CPAM. PPB is associated with germline mutations in DICER1 . In addition to the risk of malignancy, “asymptomatic” patients may have chronic inflammation with subtle systemic manifestations, which parents report resolves after the lesion is resected.
Bibliography
Baird R, Puligandla PS, Laberge JM. Congenital lung malformations: informing best practice. Semin Pediatr Surg . 2014;23(5):270–277.
Kotecha S, Barbato A, Bush A, et al. Antenatal and postnatal management of congenital cystic adenomatoid malformation. Paediatr Respir Rev . 2012;13(3):162–170 [quiz 170–171].
Singh R, Davenport M. The argument for operative approach to asymptomatic lung lesions. Semin Pediatr Surg . 2015;24(4):187–195.
Pulmonary Sequestration
Joshua A. Blatter, Jonathan D. Finder
Pulmonary sequestration is a congenital anomaly of lung development that can be intrapulmonary or extrapulmonary, according to the location within the visceral pleura. The majority of sequestrations are intrapulmonary.
Pathophysiology
The lung tissue in a sequestration does not connect to a bronchus and receives its arterial supply from the systemic arteries (commonly off the aorta) and returns its venous blood to the right side of the heart through the inferior vena cava (extralobar ) or pulmonary veins (intralobar ). The sequestration functions as a space-occupying lesion within the chest; it does not participate in gas exchange and does not lead to a left-to-right shunt or alveolar dead space. Communication with the airway can occur as the result of rupture of infected material into an adjacent airway. Collateral ventilation within intrapulmonary lesions via pores of Kohn can occur. Pulmonary sequestrations can arise through the same pathoembryologic mechanism as a remnant of a diverticular outgrowth of the esophagus. Some propose that intrapulmonary sequestration is an acquired lesion primarily caused by infection and inflammation; inflammation leads to cystic changes and hypertrophy of a feeding systemic artery. This is consistent with the rarity of this lesion in autopsy series of newborns. Gastric or pancreatic tissue may be found within the sequestration. Cysts also may be present. Other associated congenital anomalies, including CPAM (see Chapter 423.3 ), diaphragmatic hernia (see Chapter 122.10 ), and esophageal cysts, are not uncommon. Some believe that intrapulmonary sequestration is often a manifestation of CPAM and have questioned the existence of intrapulmonary sequestration as a separate entity.
Clinical Manifestations and Diagnosis
Physical findings in patients with sequestration include an area of dullness to percussion and decreased breath sounds over the lesion. During infection, crackles may also be present. A continuous or purely systolic murmur may be heard over the back. If findings on routine chest radiographs are consistent with the diagnosis, further delineation is indicated before surgical intervention (Fig. 423.3 ). CT with contrast can demonstrate both the extent of the lesion and its vascular supply. MR angiography is also useful. Ultrasonography can help to rule out a diaphragmatic hernia and demonstrate the systemic artery. Surgical removal is recommended. Identifying the blood supply before surgery avoids inadvertently severing its systemic artery. Coil embolization (transumbilical in neonates; arterial in older patients) has been successful in treating patients with sequestration.
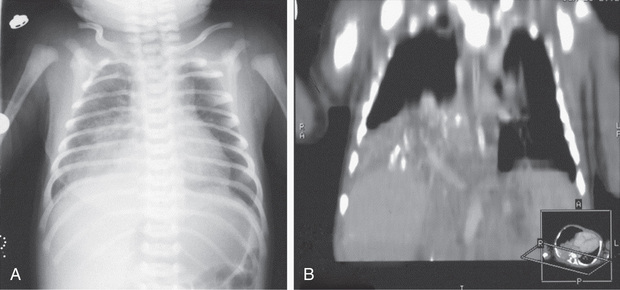
Intrapulmonary sequestration is generally found in a lower lobe and does not have its own pleura. Patients usually present with infection. In older patients, hemoptysis is common. A chest radiograph during a period when there is no active infection reveals a mass lesion; an air-fluid level may be present. During infection, the margins of the lesion may be blurred. There is no difference in the incidence of this lesion in each lung.
Extrapulmonary sequestration is much more common in boys and almost always involves the left lung. This lesion is enveloped by a pleural covering and is associated with diaphragmatic hernia and other abnormalities such as colonic duplication, vertebral abnormalities, and pulmonary hypoplasia. Many of these patients are asymptomatic when the mass is discovered by routine chest radiography. Other patients present with respiratory symptoms or heart failure. Subdiaphragmatic extrapulmonary sequestration can manifest as an abdominal mass on prenatal ultrasonography. The advent of prenatal ultrasonography has also enabled evidence that fetal pulmonary sequestrations can spontaneously regress.
Treatment
Treatment of intrapulmonary sequestration is surgical removal of the lesion, a procedure that usually requires excision of the entire involved lobe. Segmental resection occasionally suffices. Surgical resection of the involved area is often recommended for extrapulmonary sequestration as well, but observation can be considered for asymptomatic patients with small lesions. Coil embolization of the feeding artery has also been successful.
Bibliography
Abbey P, Das CJ, Pangtey GS, et al. Imaging in bronchopulmonary sequestration. J Med Imaging Radiat Oncol . 2009;53(1):22–31.
Adzick NS. Management of fetal lung lesions. Clin Perinatol . 2009;36(2):363–376.
Chien KJ, Huang TC, Lin CC, et al. Early and late outcomes of coil embolization of pulmonary sequestration in children. Circ J . 2009;73:938–942.
Lee BS, Kim JT, Kim EA, et al. Neonatal pulmonary sequestration: clinical experience with transumbilical arterial embolization. Pediatr Pulmonol . 2008;43:404–414.
Bronchogenic Cysts
Joshua A. Blatter, Jonathan D. Finder
Etiology and Pathology
Bronchogenic cysts arise from abnormal budding of the tracheal diverticulum of the foregut before the 16th wk of gestation and are originally lined with ciliated epithelium. They are more commonly found on the right and near a midline structure (trachea, esophagus, carina), but peripheral lower lobe and perihilar intrapulmonary cysts are not infrequent. Diagnosis may be precipitated by enlargement of the cyst, which causes symptoms by pressure on an adjacent airway. When the diagnosis is delayed until an infection occurs, the ciliated epithelium may be lost, and accurate pathologic diagnosis is then impossible. Cysts are rarely demonstrable at birth. Later, some cysts become symptomatic by becoming infected or by enlarging and compromising the function of an adjacent airway.
Clinical Manifestations and Treatment
Fever, chest pain, and productive cough are the most common presenting symptoms. Dysphagia may be present; some bronchogenic cysts are asymptomatic. A chest radiograph reveals the cyst, which can contain an air-fluid level (Fig. 423.4 ). CT scan or MRI is obtained in most cases to better demonstrate anatomy and extent of lesion before surgical resection. Treatment of symptomatic cysts is surgical excision after appropriate antibiotic management. Asymptomatic cysts are generally excised in view of the high rate of infection.
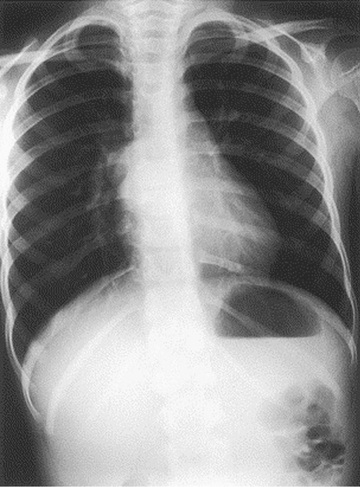
Bibliography
Chowdhury MM, Chakraborty S. Imaging of congenital lung malformations. Semin Pediatr Surg . 2015;24(4):168–175.
Fievet L, D'Journo XB, Guys JM, et al. Bronchogenic cyst: best time for surgery? Ann Thorac Surg . 2012;94(5):1695–1699.
Maurin S, Hery G, Bourliere B, et al. Bronchogenic cyst: clinical course from antenatal diagnosis to postnatal thoracoscopic resection. J Minim Access Surg . 2013;9(1):25–28.
Congenital Pulmonary Lymphangiectasia
Joshua A. Blatter, Jonathan D. Finder
Etiology and Pathology
Congenital pulmonary lymphangiectasia is characterized by greatly dilated lymphatic ducts throughout the lung. It can occur in three pathologic circumstances: pulmonary venous obstruction that produces an elevated transvascular pressure and engorges the pulmonary lymphatics; generalized lymphangiectasia , as a generalized disease of several organ systems, including lymphedema, lungs, and the intestines either associated with other syndromes (Noonan, Hennekam, yellow nail, trisomy 21) or nonsyndromic. Gorham-Stout disease (vanishing bone disease) presents with pulmonary and abdominal chylous effusions, destructive bone cysts, and multiple lymphangiomas; and primary lymphangiectasia limited to the lung as a manifestation of an abnormality in lymphatic development.
Clinical Manifestations and Treatment
Children with pulmonary venous obstruction or severe pulmonary lymphangiectasia present with dyspnea and cyanosis in the newborn period. Hydrops fetalis may be diagnosed antenatally. Chest radiographs reveal diffuse, dense, reticular densities with prominence of Kerley B lines. Pleural effusions are common; thoracentesis will reveal chylothorax in this setting. If the lung is not completely involved, the spared areas appear hyperlucent. Respiration is compromised because of impaired diffusion and decreased pulmonary compliance. The diagnosis can be suggested by CT scan and/or cardiac catheterization; definitive diagnosis requires lymphangiography or lung biopsy (either thoracoscopic or open) (Fig. 423.5 ).
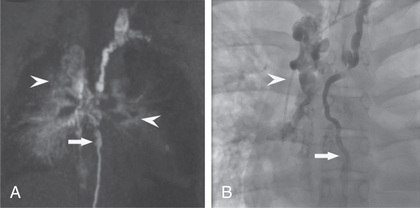
Treatment is supportive and includes administration of oxygen, mechanical ventilation, nutritional support (including gastrostomy placement and use of feedings containing medium-chain triglycerides), and careful fluid management with diuretics. Octreotide, the somatostatin analog, can reduce chylous effusion in some patients. Primary pulmonary lymphangiectasia in the neonate can produce severe pulmonary dysfunction that can require long-term mechanical ventilation; long-term survival and resolution of respiratory insufficiency are possible even in severe cases, especially if the chylous effusions can be managed. Occasionally, the pulmonary venous obstruction is secondary to left-sided cardiac lesions; relief of the latter can produce improvement in pulmonary dysfunction. Generalized lymphangiectasia produces milder pulmonary dysfunction, and survival to mid-childhood and beyond is not unusual.
Bibliography
Boland JM, Tazelaar HD, Colby TV, et al. Diffuse pulmonary lymphatic disease presenting as interstitial lung disease in adulthood: report of 3 cases. Am J Surg Pathol . 2012;36(10):1548–1554.
Mêlée P, Sridhar S. Congenital pulmonary lymphangiectasia: an unusual presentation of nonimmune hydrops in a preterm infant. Adv Neonatal Care . 2012;12(3):166–171.
Yuan SM. Congenital pulmonary lymphangiectasia. J Perinat Med . 2017 [Epub ahead of print].
Lung Hernia
Joshua A. Blatter, Jonathan D. Finder
Etiology and Pathology
A lung hernia is a protrusion of the lung beyond its normal thoracic boundaries. Approximately 20% are congenital, with the remainder being noted after chest trauma or thoracic surgery or in patients with pulmonary diseases such as cystic fibrosis (see Chapter 432 ) or asthma (see Chapter 169 ), which cause frequent cough and generate high intrathoracic pressure. A congenital weakness of the suprapleural membrane (Sibson fascia) or musculature of the neck can play a role in the appearance of a lung hernia. More than half of congenital lung hernias and almost all acquired hernias are cervical. Congenital cervical hernias usually occur anteriorly through a gap between the scalenus anterior and sternocleidomastoid muscles. Cervical herniation is usually prevented by the trapezius muscle (posteriorly, at the thoracic inlet) and by the 3 scalene muscles (laterally).
Clinical Manifestations and Treatment
The presenting sign of a cervical hernia (Sibson hernia) is usually a neck mass noticed while straining or coughing. Some lesions are asymptomatic and detected only when a chest film is taken for another reason. Findings on physical examination are normal except during Valsalva maneuver, when a soft bulge may be noticed in the neck. In most cases, no treatment is necessary, although these hernias can cause problems during attempts to place a central venous catheter through the jugular or subclavian veins. They can resolve spontaneously.
Paravertebral or parasternal hernias are usually associated with rib anomalies. Intercostal hernias usually occur parasternally, where the external intercostal muscle is absent. Posteriorly, despite the seemingly inadequate internal intercostal muscle, the paraspinal muscles usually prevent herniation. Straining, coughing, or playing a musical instrument can have a role in causing intercostal hernias, but in most cases, there is probably a preexisting defect in the thoracic wall.
Surgical treatment for lung hernia is occasionally justified for cosmetic reasons. In patients with severe chronic pulmonary disease and chronic cough and for whom cough suppression is contraindicated, permanent correction might not be achieved.
Other Congenital Malformations of the Lung
Joshua A. Blatter, Jonathan D. Finder
Congenital Lobar Emphysema and Pulmonary Cysts
See Chapter 420 .
Pulmonary Arteriovenous Malformation
See Chapters 459 and 471 .
Bronchobiliary Fistula
A bronchobiliary fistula consists of a fistulous connection between the right middle lobe bronchus and the left hepatic ductal system. Although diagnosis can be delayed until adulthood, this rare anomaly typically manifests with life-threatening bronchopulmonary infections in early infancy. Girls are more commonly affected. Definitive diagnosis requires endoscopy or exploratory surgery. Treatment includes surgical excision of the entire intrathoracic portion of the fistula. If the hepatic portion of the fistula does not communicate with the biliary system or duodenum, the involved segment might also have to be resected. Bronchobiliary communications also occur as acquired lesions resulting from hepatic disease complicated by infection.
Bibliography
Kim MJ, Kim SH, Hwang IK, et al. Presence of bilirubin in bronchobiliary fistula easily confirmed with urinary dipstick test and treated with embolization. Korean J Intern Med . 2017;32(1):182–185.
Sachdev A, Chugh K, Krishana A, et al. Congenital tracheobiliary fistula: a case report with review of literature. Pediatr Surg Int . 2011;27(8):899–905.