Inge Russell
CONTENTS
8.2.6 Other Cytoplasmic Structures
8.3.8 Mass Mating and Genome Shuffling
8.4.3 Uptake and Metabolism of Wort Carbohydrates
8.4.4 Maltose and Maltotriose Uptake
8.4.5 Uptake and Metabolism of Wort Nitrogen
8.4.6 Uptake of Wort Minerals and Vitamins by Yeast
8.5.6 Diacetyl and 2,3-Pentanedione
8.7 Yeast Management in the Brewery
8.7.1 Glycogen and Trehalose—The Yeast’s Carbohydrate Storage Polymers
8.7.2.2 Multiple Cultures in a Plant Environment
8.7.2.3 Brettanomyces and Specialty Yeasts
8.9.1 Contamination of Cultures
8.9.2 Yeast Cropping from a Vertical Fermenter
8.11 Cell Viability/Vitality and Yeast Pitching
8.11.1 Microscopic Examination
8.11.2 Viability of Yeast and the Methylene Blue Test
8.11.5 Metabolic Activity Tests
8.11.7 Pitching Rate Calculations
8.1 TAXONOMY OF YEAST
Interest in brewing yeast centers around its different strains, and there are thousands of unique strains of Saccharomyces cerevisiae. These strains encompass brewing, baking, wine, distilling, and laboratory cultures. There is a problem classifying such strains in the brewing context; the minor differences among strains that the taxonomist dismisses can be of great technical importance to the brewer. Saccharomyces, Latin for sugar fungus, is the name first used for yeast in 1838 by Meyen, but it was the work of Hansen at the Carlsberg Laboratory in Denmark during the 1880s that gave us the species names of S. cerevisiae for head-forming yeast used in ale fermentations and S. carlsbergensis for nonhead-forming yeast associated with the lower temperature range of lager fermentations. Historically, lager yeast and ale yeast have been taxonomically distinguished on the basis of their ability to ferment the disaccharide melibiose. Strains of lager yeast possess the MEL genes, produce the extracellular enzyme α-galactosidase (melibiase), and are able to utilize melibiose, whereas ale strains that do not produce α-galactosidase are therefore unable to utilize melibiose. Traditionally, lager is produced by bottom-fermenting yeasts at fermentation temperatures between approximately 5°C and 15°C (maximum growth temperature is approximately 34°C) and, at the end of fermentation, these yeasts flocculate and collect at the bottom of the fermenter. Top-fermenting yeasts, used for the production of ale at fermentation temperatures between approximately 15°C and 26°C (maximum growth temperature is 37°C or higher), tend to be somewhat less flocculent, and loose clumps of cells adsorbed to carbon dioxide bubbles are carried to the surface of the fermenting wort. Consequently, top yeasts were traditionally collected by skimming from the surface of the fermenting wort, whereas bottom yeasts were collected, or cropped, from the fermenter bottom. The differentiation of lagers and ales on the basis of bottom and top cropping has become less distinct with the advent of vertical bottom fermenters and centrifuges. There is much greater diversity among ale strains than lager strains.
The taxonomy surrounding the yeast Saccharomyces has been confusing and ever-changing. Saccharomyces sensu stricto is a species complex that includes most of the yeast strains relevant in the fermentation industry as well as in basic science (i.e., S. bayanus, S. cerevisiae, S. paradoxus, and S. pastorianus). Names of species and single isolates have, and are still undergoing, name changes that cause confusion for yeast scientists and fermentation technologists.
There are three main reasons that taxonomists change yeast names. These are: (1) the yeast was incorrectly described or named; (2) new observations warrant such change; or (3) a prior published legitimate name is uncovered.1 Unfortunately, in the past, changes in nomenclature varied with changes in the criteria taxonomists believed should be used. These varied from phenotypic characteristics (such as microscopic appearance and ability to use certain substrates), nutritional characteristics (which could mutate), or the criteria of interfertility within strains of the same species. Newer techniques of molecular taxonomy and DNA relatedness are now being used for yeast classification. Recent findings have demonstrated that S. bayanus and S. pastorianus are not homogeneous and do not seem to be natural groups. As molecular biology methods become simpler and are marketed in inexpensive user-friendly kits and the cost of genome sequencing continues to drop in price, these newer identification methods are quickly becoming the methods of choice.2, 3
In 1970, taxonomists repositioned the lager yeast S. carlsbergensis as S. uvarum. In 1990, taxonomists repositioned S. uvarum as part of S. cerevisiae. Then, S. cerevisiae var. carlsbergensis was classified as S. pastorianus. The species was often written as S. pastorianus/carlsbergensis for clarity. There was an argument4 for keeping the name S. carlsbergensis for lager-brewing yeast rather than using the name S. pastorianus; however, today S. pastorianus is considered to be the correct taxonomic name of the species.
The lager yeast S. pastorianus (syn. Saccharomyces carlsbergensis) is a natural allopolyploid hybrid of Saccharomyces eubayanus and S. cerevisiae. This hybrid yeast was created by the fusion of a Saccharomyces cerevisiae ale yeast with a cryotolerant Saccharomyces species, Saccharomyces eubayanus sp. nov., which resembles Saccharomyces bayanus (a complex hybrid of S. eubayanus, Saccharomyces uvarum, and S. cerevisiae, found only in the brewing environment).5
S. eubayanus is a species that until recently had eluded identification, and there is debate as to its geographical origin (Patagonia, Tibetan Plateau, China, or some other yet unidentified region) and it is the part of the hybrid that endows the yeast with its capacity to ferment at relatively cool temperatures, while the S. cerevisiae part of the hybrid contributes the ability to ferment maltotriose.6 The complete genome sequence of S. eubayanus has now been published,7 and it is 99.5% identical to the non- S. cerevisiae portion of the S. pastorianus genome sequence and suggests specific changes in sugar and sulfite metabolism occurred during domestication in the lager-brewing environment.
Gallone et al.8 recently examined the history and domestication of ale-brewing yeast using genomic and phenotypic analyses by sequencing 157 S. cerevisiae yeasts. They concluded that there are five sub-lineages that separate the brewing yeast from wild strains and that these originated from only a few ancestors. They found industry-specific selection for stress tolerance, sugar utilization such as maltotriose, and flavor production. Krogerus et al.9 in a recent review explore this topic of natural hybrids and the creation of new hybrids and further attempt to explain the complex history of brewing yeast and discuss future potential opportunities.
For simplicity, brewing yeast will be referred to as S. cerevisiae throughout this chapter unless distinct reference is made to lager yeast.
8.2 STRUCTURE OF YEAST
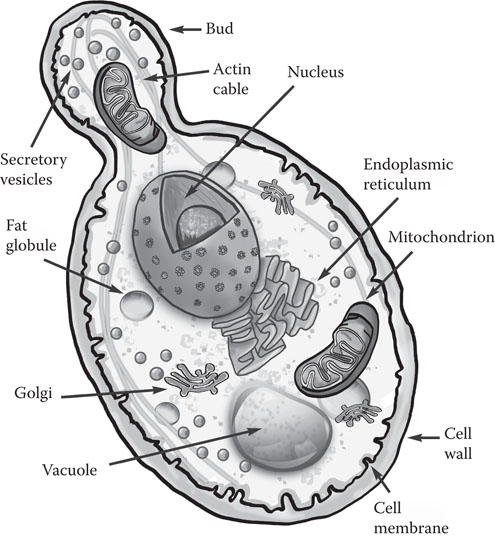
The group of microorganisms known as “yeast” is by traditional agreement limited to fungi in which the unicellular form is predominant.10 Figure 8.1 shows the features of a typical yeast cell, and Figure 8.2 is an electron micrograph of a budding yeast cell.
Figure 8.1 Main features of a typical yeast cell.
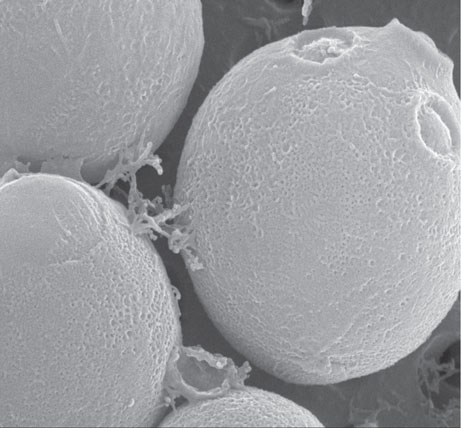
Figure 8.2 Electron micrograph of budding yeast cell. (Courtesy of Alex Speers, Heriot-Watt University.)
Yeasts vary in size, from roughly 5 μm to 10 μm in length to a breadth of 5 μm to 7 μm. The mean cell size varies with the growth cycle stage, fermentation conditions, and cell age (older cells are larger in size).
8.2.1 Cell Wall
The yeast cell wall is a multifunctional organelle of protection, shape, cell interaction, reception, attachment, and specialized enzymic activity. The cell wall, which is 100 nm to 200 nm thick, constitutes 15% to 25% of the dry weight of the cell and consists primarily of equal amounts of phosphomannan (31%) and glucans (29%). There are three glucans present in the wall (Figure 8.3).
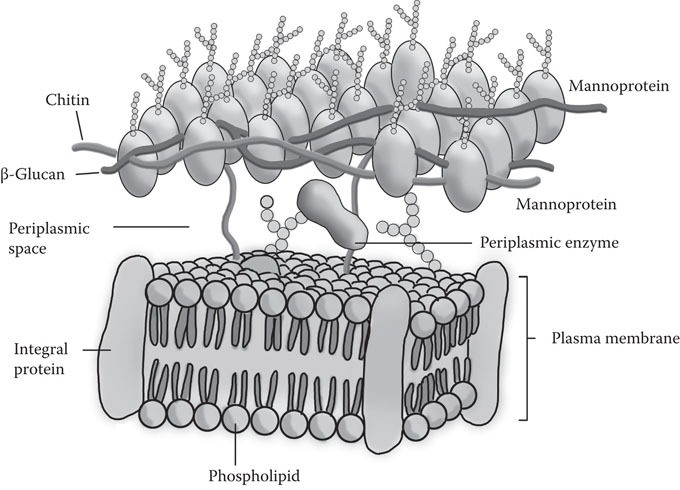
Figure 8.3 Simplified structure of the yeast cell wall and plasma membrane.
The major component of the wall is an alkali-insoluble, acid-insoluble β-1,3-linked polymer, which helps the wall maintain its rigidity. There is an alkali-soluble branched glucan, with predominantly β-1,3-linkages, but with some β-1,6-linkages as well. The cell wall also contains a small portion of predominantly β-1,6-linked glucan. Chitin, a polymer of N-acetyl glucosamine, is present in small quantities (2% to 4%) and is almost always restricted to the bud scar. Mannans are present as an α-1,6-linked inner core with α-1,2- and α-1,3-side chains. Lipids are present at about 8.5% and proteins at about 13%. The carbohydrate portion of the mannoprotein on the yeast cell surface determines the immunochemical properties of the cell. The exact composition of the cell wall is dependent on growth conditions, age of the culture, and the specific yeast strain. The construction and structures of the yeast cell wall have been reviewed in detail by Klis et al.11
8.2.2 Plasma Membrane
The plasma membrane acts as a barrier to separate the aqueous interior of the cell from its aqueous exterior. It consists of lipids and proteins, in more or less equal amounts, together with a small amount of carbohydrate. It is 8 nm to 10 nm thick, with occasional invaginations protruding into the cytoplasm (Figure 8.3).
The carbohydrate portions of the membrane-bound glycoproteins are believed to extend only from the external surface of the membrane. The plasma membrane has a role in regulating the uptake of nutrients and in the excretion of metabolites. It is also the site of cell wall synthesis and assembly, and secretion of extracellular enzymes.
The intrinsic membrane proteins are inserted in a lipid bilayer consisting primarily of phospholipids and sterols. The principal phospholipids are phosphatidylcholine and phosphatidylethanolamine, and their primary function is believed to be the maintenance of a barrier between the external and internal cell environment. The fluidity of the plasma membrane is regulated by phospholipid unsaturation and is an important factor in ethanol tolerance. The sterols in the plasma membrane are ergosterol, 24(28)-dehydro-ergosterol, zymosterol, and smaller quantities of fecosterol and lanosterol. They confer integrity and rigidity on the membrane.
The main function of the plasma membrane is to dictate what enters and what leaves the cytoplasm. The extrinsic proteins, those that interact with membrane lipids and proteins by polar binding, cover part of the bilayer surface, and a large group of these proteins is involved in solute transport.12 Brewer’s yeast requires initial oxygenation, prior to fermentation, to ensure correct plasma membrane synthesis as oxygen is absolutely required for the synthesis of the unsaturated fatty acids and sterols.
8.2.3 The Periplasmic Space
This is the thin area between the outer surface of the plasma membrane and the inner surface of the cell wall. Secreted proteins, which are unable to permeate the cell wall, are located here. This includes enzymes such as invertase, acid phosphatase, and melibiase. For example, sucrose is broken down in the periplasmic space by the enzyme invertase to fructose and glucose.
8.2.4 Nucleus
The cell nucleus is roughly spherical, about 2 μm in diameter, and is visible with phase contrast microscopy. In resting cells, it is usually situated next to a prominent vacuole. The nucleus consists primarily of DNA and protein and is surrounded by the nuclear membrane. Saccharomyces contains 16 linear chromosomes. The nuclear membrane, which is perforated at intervals with pores, remains intact throughout the cell cycle.
8.2.5 Mitochondria
Electron micrographs of yeast cells reveal round or elongated mitochondrial structures composed of two distinct membranes, the outer and the inner, within the cytoplasm. The cristae within the mitochondria are formed by the folding of the inner membrane. Specific enzymes are associated within four distinct locations: the outer membrane, the intermembrane space, the inner membrane, and the matrix. Most of the enzymes of the tricarboxylic acid cycle are present in the matrix of the mitochondrion. The enzymes involved in electron transport and oxidative phosphorylation are associated with the inner membrane. When yeast is grown aerobically on a nonfermentable carbon source, the mitochondria of the fully respiring cells are rich in the cristae structures. Under aerobic conditions, yeast mitochondria are primarily involved in adenosine triphosphate (ATP) synthesis during respiration. In glucose-repressed cells, only a few mitochondria with poorly developed cristae can be seen. Mitochondrial DNA only comprises approximately 0.1 % of total cellular DNA. Stewart13 recently reviewed the influence of mitochondria on brewer’s yeast fermentations. Jensen et al.14 and Mishra and Chan15 provide in-depth reviews on overall yeast mitochondrial dynamics.
8.2.6 Other Cytoplasmic Structures
8.2.6.1 Vacuoles
Vacuoles are a part of an intra-membranous system that includes the endoplasmic reticulum and are easily seen under the light microscope. They serve as dynamic stores of nutrients, and they provide a site for the breakdown of macromolecules. They are also key in intracellular protein trafficking in yeasts. The form and size of the vacuoles change during the cell cycle, and they can occupy up to one-third of the volume of a cell. Mature cells contain large vacuoles, which fragment into small vesicles when bud formation is initiated. Later in the cell cycle, the small vacuoles fuse again to produce a single vacuole in the mother and daughter cells. The vacuoles contain proteases and hydrolases, and they also store metabolites such as amino acids. Vacuoles are bounded by a single membrane called the tonoplast. The secretory pathway in yeast is believed to work in the following fashion: endoplasmic reticulum → Golgi complex → vesicle → cell surface or vacuolar compartment. This sequence allows the transport of soluble and membrane-bound proteins, both for extracellular secretion and for assembly of the vacuole.16 Further details in chapter 14.
8.2.6.2 Peroxisomes
Peroxisomes are simple organelles up to 1 nm in size. They are sealed vesicles surrounded by a single membrane. Yeast peroxisomes perform a variety of functions and are also the sites of fatty acid degradation. The number and volume of the peroxisomes change depending on the external conditions. Dedicated membrane proteins are required to allow communication across the peroxisomal membrane.17
8.2.6.3 Endoplasmic Reticulum
The endoplasmic reticulum (ER) is a large organelle formed as a network of tubules and sacs that extends from the nucleus. The whole ER is surrounded by a continuous membrane. The ER is divided into two parts: the rough ER on which the ribosomes are attached and the smooth ER. Secreted proteins are synthesized by the membrane-bound ribosomes and transported into the ER, where they are processed and then either retained or transported further to the Golgi apparatus. English and Voeltz18 review ER structure and its interconnections with other organelles.
8.2.6.4 Golgi Complex
The Golgi complex is an organelle built of several flattened, membrane-enclosed sacs and is part of the cellular endomembrane system. Secretory proteins exit from the ER in coated vesicles and then progress through the Golgi complex for processing before delivery to their final destination.19
8.2.6.5 Cytosol
The cytosol (cytoplasmic matrix) is the liquid matrix around the organelles in the cell and is separated into compartments by membranes. It is the prime site for protein synthesis and degradation. The cytosol is contained within the plasma membrane and surrounds the nucleus. It makes up more than half of the cell volume and consists of, among other things, all the free ribosomes and proteasomes. Proteasomes are responsible for the digestion of proteins that may be detrimental to the cell. The cytosol together with the rest of the cell content, except the nucleus, constitute the cytoplasm.
8.2.6.6 Cytoskeleton
The cytoskeleton gives the cell its mobility and support. It serves to organize the cytoplasm, to provide the means for generating force within the cell, and to determine and maintain the shape of the cell and its structural integrity. The shape and movement of the cell is controlled by three main components. The microfilaments and the microtubules give the cell support and movement, while the intermediate filaments are built of several different proteins and play a supportive role. Actin filaments in the cytoskeleton are constantly being built and then broken down due to the dynamic nature of the cytoskeleton.
8.3 LIFE CYCLE AND GENETICS
8.3.1 Vegetative Reproduction
Most brewing strains are diploid, polyploid, or aneuploid, whereas most laboratory strains are haploid. The yeast cell cycle refers to the repeated pattern of events that occur between the formation of a cell by division of its mother cell and the time when the cell itself divides. Individual yeast cells are mortal. Yeast aging is a function of the number of divisions undertaken by the individual cell, and it is not a function of the cell’s chronological age. All yeast cells have a set lifespan determined both by genetics and environment. The maximum division capacity of a cell is called the “Hayflick limit.” Once a cell reaches this limit, it cannot replicate further and enters a stage of senescence and then death. A yeast cell can divide to produce 10 to 33 daughter cells. Research with industrial ale strains showed a high of 21.7 + 7.5 divisions to a low of 10.3 + 4.7 divisions.20 When yeast is examined under the microscope, the following are signs of aging: an increase in the number of bud scars, an increase in the cell size, surface wrinkles, granularity in the cytoplasm, and the retention of daughter cells. Older yeasts cell have a reduced capacity to adapt to stress and to ferment due to the progressive impairment of cellular mechanisms due to aging.
As the mother cell ages, there is protein damage within the cell. Before the yeast cell bud breaks off, the damaged and aged proteins in the newly formed yeast cell bud are transported back into the mother cell. Thus, the mother cell retains the problems and sends the daughter cell (bud) on its way in a healthy condition. This guarantees that the new daughter cell will be healthier than the parent cell.21
As the cell reproduces by budding, a birth scar is formed on the new daughter cell and a bud scar results on the mother cell. As budding continues, an individual cell ages and bud scars accumulate (usually 10 to 30) (Figure 8.4).
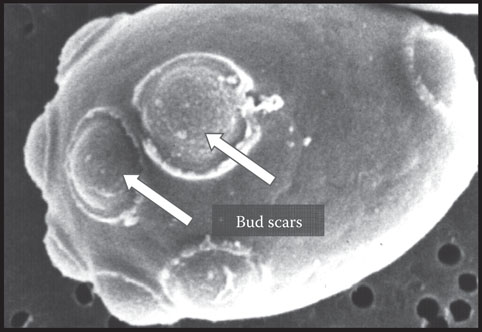
Figure 8.4 Electron micrograph of a Saccharomyces cerevisiae yeast cell showing numerous bud scars on the cell surface.
By counting the number of bud scars on a cell, it is possible to estimate a particular cell’s age. The important point to remember is that although individual cells age and die, the total ensemble of cells does not age, and, in theory, a well-cared-for yeast culture could be used indefinitely. A haploid yeast cell, in a rich medium such as wort and at its optimum temperature, has a doubling time of approximately 90 min.
The cell cycle can be divided into a number of different phases, with the two major phases being the S phase (synthesis phase where DNA is duplicated) and the M phase (mitosis phase where cell division occurs). Mitosis and nuclear division occupy a relatively short time in the cell cycle. The remainder is divided into a G1 period, (telophase to the beginning of DNA synthesis), an S period (time of DNA synthesis), and a G2 period (end of DNA synthesis to prophase). (Figure 8.5)

Figure 8.5 Phases of the yeast cell cycle.
The G1, S, and G2 phases together constitute the interphase, and this occupies more than 95% of the cell’s time. The G1 phase is where the cells synthesize the enzymes needed for genome duplication, and the G2 phase is where the duplicated centrosomes separate to organize the mitotic spindle. Salazar-Roa and Malumbres22 have recently reviewed the complex process of the cell cycle and the significant advances in our understanding of its regulation.
8.3.2 The Genome
Brewing yeasts are polyploid and, in particular, triploid, tetraploid, or aneuploid. Polyploidy brings benefits to the strain in that extra copies of genes for sugar utilization can improve fermentation performance. Polyploid yeasts are also more genetically stable as it takes multiple mutational events to change them.23, 24 As previously mentioned, the lager yeast S. carlsbergensis has been reclassified as S. pastorianus. Gibson and Liti25 have reviewed the evolutionary history of this lager yeast and the ancestral hybridization events between S. cerevisiae and S. eubayanus that led to this yeast’s strong fermentative ability and traits such as cold tolerance.
The chromosomes are located in the cell nucleus, and the total haploid yeast genome consists of more than 12 million base pairs. A widely studied laboratory haploid yeast,26 strain S228C, was chosen for the yeast genome project, and its DNA sequence has now been fully characterized. The Saccharomyces genome database (SGD) provides a wealth of information at http://www.yeastgenome.org.
8.3.3 Strain Improvement
There are many strategies to obtain superior industrial yeast,27 and some of these have recently been reviewed by Pretorius et al.28 and Steensels et al.29 They include natural and artificial diversity, mutagenesis, sexual hybridization, spheroplast fusion, cytoduction, and evolutionary engineering. In terms of natural genetic diversity, there is a huge unexplored pool of organisms that have not yet been examined in detail or even categorized as to genera and species.
In contrast to exploring natural genetic diversity, there is a whole new field of science emerging called “synthetic genome engineering.” Pretorius30 has recently reviewed this topic in terms of wine yeast and discusses the seismic shifts in this field. The convergence of the many different areas: bimolecular, chemical, physical, mathematical and computational sciences, engineering, and the rapid advances in high-throughput DNA sequencing and synthesis techniques. The use of CRISPR/Cas9 for genetic modifications in Saccharomyces has been referred to as “using a molecular Swiss army knife.”31
The work in synthetic genomes will continue to increase our understanding of yeast biochemical pathways and the global “Synthetic Yeast Genome (Sc2.0) Project” has the goal to synthesize all 16 chromosomes of S. cerevisiae by 2018. The first S. cerevisiae cell with a fully functional synthetic chromosome was accomplished by Annaluru et al.32 in 2014. Work at this intersection of biology and engineering is only beginning, and yeast is the ideal research organism for this task.33, 34 There is little doubt that, as this research progresses, new discoveries and opportunities for brewing yeast will also be seen with the implementation of so many new technologies. Will consumers react to the concept of a yeast for beer production containing synthetic DNA in a similar negative manner as they have expressed for recombinant yeast produced with the insertion of DNA from other yeast species or bacteria to enhance brewing qualities is a question with no answer as of yet.
8.3.4 Mutation and Selection
Classical approaches to strain improvement include mutation and selection, screening and selection, and crossbreeding. Mutation is any change that alters the sequence of bases along the DNA molecule, thus modifying the genetic material. The average spontaneous mutation frequency in S. cerevisiae at any particular locus is approximately 10−6 per generation.
Chemical mutagens and physical treatments such as ultraviolet light are used to induce mutation frequencies to detectable levels. Problems are often encountered with the use of mutagens for brewing strains as mutagenesis is a destructive process and can cause gross rearrangement of the genome. The mutagenized strains often no longer exhibit many of the desirable properties of the parent strain, and, in addition, may exhibit a slow growth rate and produce a number of undesirable taste and aroma compounds during fermentation. Moreover, mutagenesis is seldom employed with industrial strains; their polyploid nature obscures mutations because of the nonmutated genes. Casey34 cautioned on the use of mutagens as these hidden undesirable mutations can become expressed after a time lag by such events as chromosome loss, mitotic recombination, or mutation of other wild type alleles. He suggested an analogy to computer viruses in computer software that can surface months later. In a similar way, hidden mutations can later spoil the efforts of a protracted strain improvement program.
Screening of cultures to obtain spontaneous mutants or variants has proved to be a more successful technique as this avoids the use of destructive mutagens. Some early examples follow. To select for brewery yeast with improved maltose utilization rates, 2-deoxyglucose, a glucose analog, was employed and spontaneous mutants selected that were resistant to 2-deoxyglucose. These isolates were also found to be derepressed for glucose repression of maltose uptake, thus allowing the cells to take up maltose without first requiring a 50% to 60% drop in wort glucose levels. This resulted in faster fermentation rates and no alternation in the final flavor of the product.35, 36 Later research by Piña et al.37 reported similar results but this time using a lager yeast strain and high gravity (25°Plato) wort. However, the increased wort fermentation rate was not sufficient to introduce the 2-DOG mutants into commercial brewing. (Further details in Chapter 14 and in Figure 14.4.)
Galva’n et al.38 reported the isolation of brewing yeast strains with a dominant mutation and resistance to the herbicide sulfometuron methyl (SM). Yeast mutants resistant to SM are dominant and showed a decreased level of acetohydroxy acid synthase. This enzyme is the first enzyme in the isoleucine–valine biosynthetic pathway and produces acetolactate, the precursor of diacetyl in brewing fermentations. Workers at the Carlsberg Research Institute isolated low diacetyl-producing strains and conducted successful plant trials at the 4500 hL scale.39
There are three characteristics routinely encountered resulting from yeast mutation that can be harmful to a brewing fermentation. These are: the tendency of yeast strains to mutate from flocculence to nonflocculence; the loss of ability to ferment maltotriose; and the presence of respiratory deficient mutants. This last group usually consists of cytoplasmic mutants.
The most frequently identified spontaneous mutant found in brewing yeast strains is the respiratory deficient (RD) or “petite” mutation. The RD mutant arises spontaneously through a rearrangement of the mitochondrial genome. It normally occurs at frequencies of between 0.5% and 5% of the yeast population but, in some strains, figures as high as 50% have been reported. The mutant is characterized by deficiencies in mitochondrial function, resulting in a diminished ability to function aerobically and, as a result, these yeasts are unable to metabolize nonfermentable carbon sources, such as lactate, glycerol, or ethanol. Respiratory deficient mutants can range from point mutations (mit−) to deletion mutations (rho−) to the complete elimination of mitochondrial DNA (rho0). Many phenotypic effects occur due to this mutation, and these include alterations in sugar uptake, metabolic by-product formation, and tolerance to stress factors, such as ethanol and temperature. Flocculation, cell wall and plasma membrane structure, and cellular morphology are affected by this mutation.13
It should be remembered that beer produced with a yeast RD strain or with a strain that produces a high number of RD mutants is more likely to have flavor defects and fermentation problems. Ernandes et al.40 have reported that beer produced from these mutants contained elevated levels of diacetyl and some higher alcohols. Wort fermentation rates were slower, higher dead cell counts were observed, and biomass production and flocculation ability were reduced.
To test for the presence of respiratory deficient mutants, five-day-old colonies of yeast grown on peptone-yeast (PY) extract medium are overlaid with triphenyltetrazolium agar.41 Respiratory sufficient colonies stain red and RD mutants remain white. Respiratory deficient colonies growing on an agar plate are often smaller in size and hence the name “petites” (Figure 8.6). Because RD mutants lack respiratory chain enzymes, but can still grow by employing glycolysis as a source of ATP, a confirmative test involves streaking a suspect colony onto a PY glucose plate and a PY plate containing a nonfermentable carbon source, such as lactate or glycerol. An RD colony typically grows on the glucose plate but not on the lactate plate.42
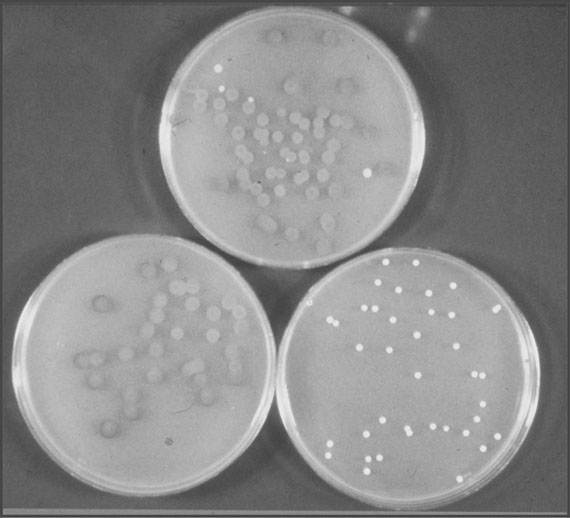
Figure 8.6 Triphenyltetrazolium overlay plates illustrating the presence of respiratory deficient (RD) mutants and respiratory sufficient (RS) colonies. Top plate—mix of RS and RD colonies. Bottom left plate—only RS colonies. Bottom right plate—only RD colonies.
8.3.5 Hybridization
The study of yeast genetics was pioneered by Winge43 in 1935 and his coworkers at the Carlsberg laboratory in Denmark; they established the haploid–diploid life cycle (Figure 8.7). Saccharomyces can alternate between the haploid (a single set of chromosomes) and diploid (two sets of chromosomes) states. Yeast can display two mating types, designated a and α, which are manifested by the extracellular production of an a- or an α-mating pheromone. When a haploids are mixed with α haploids, mating takes place and diploid zygotes are formed. Under conditions of nutritional deprivation, diploids undergo reduction division by meiosis and differentiate into asci, containing four uninucleate haploid ascospores, two of which are a mating type and two of which are α mating type. Ascus walls can be removed by the use of preparations such as zymolyase. The four spores from each ascus can be isolated by use of a micromanipulator, then induced to germinate, tested for their fermentation ability, and subsequently employed for further hybridization work. Both haploid and diploid organisms can exist stably and undergo cell division via mitosis and budding.
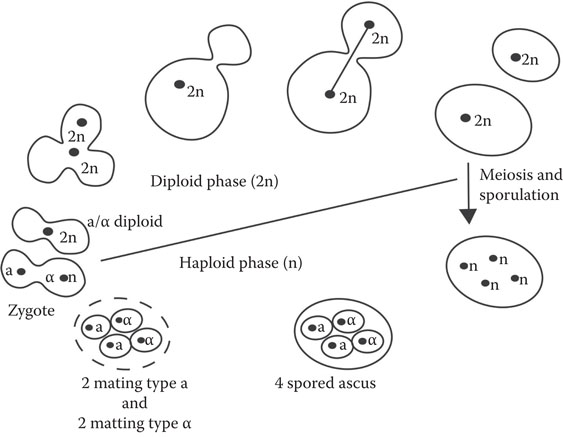
Figure 8.7 Haploid/diploid life cycle of Saccharomyces spp.
Although the technique of hybridization fell into disfavor for a number of years, when recombinant DNA was thought to be the solution to all future gene manipulation requirements, it has gradually come to be accepted again as one of the valuable techniques in the toolbox for strain improvement.44 There are three prerequisites for the production of a hybrid yeast by this technique. First, it must be possible to induce sporulation in the parent strains; second, single spore isolates must be viable; and, third, hybridization (mating) must take place when the spores of single-spore cultures are placed in contact with one another. The great stability of industrial strains has been attributed to the following characteristics: little or no mating ability, poor sporulation, and low spore viability. Nevertheless, it is possible to increase sporulation ability in many of these industrial strains, thus making them much more amenable to hybridization.45
Employing the classical techniques of spore dissection and cell mating, diploid strains were produced with multiple genes for carbohydrate utilization: for example, a diploid that is homozygous for all three known starch-hydrolyzing genes DEX1/DEX1, DEX2/DEX2, STA3/STA3. This starch hydrolyzing enzyme (glucoamylase) was heat stable and had no debranching ability. These strains could then be employed for specific purposes or further improved by fusing them to industrial polyploids with additional desirable characteristics.46
Gjermansen and Sigsgaard47 carried out extensive crossbreeding studies with a S. pastorianus strain and produced hybrids that were tested at the 575 hL scale and that produced beer of acceptable quality. Similarly, crosses between ale and lager meiotic segregants produced hybrids with faster attenuation rates and produced beers of good palate, which lacked the sulfury character of the lager but retained the estery aroma of the ale.48 The aforementioned examples demonstrate that although hybridization is an old and “classical” technique, it is still very useful. Delneri et al.49 reported on reconfiguring the S. cerevisiae genome so that it was collinear with that of S. mikatae. Researchers demonstrated that this imposed genomic collinearity allowed the generation of interspecific hybrids, which produced a large proportion of spores that were viable but extensively aneuploid. They obtained similar results in crosses between wild-type S. cerevisiae and the naturally collinear species S. paradoxus. One of the major advantages to crossbreeding is that this technique carries none of the burden of ethical questions and fears that can sometimes accompany the use of recombinant DNA technology.
8.3.6 Rare Mating
Rare mating, also called forced mating, is a technique that disregards ploidy and mating type and, thus, is ideal for the manipulation of polyploid/aneuploid strains where normal hybridization procedures cannot be utilized. When nonmating strains are mixed at a high cell density, a few hybrids that have fused nuclei form, and these can usually be isolated using appropriate selection markers. A possible disadvantage to this method is that while incorporating the nuclear genes from the brewing strain, the rare mating product can also inherit undesirable properties from the other partner, which is often a nonbrewing strain. An example of this was the construction of dextrin-fermenting brewing strains, where the POF gene (phenolic-off-flavor), which imparted the ability to decarboxylate wort ferulic acid to 4-vinyl guaiacol was also introduced50 and gave the resulting beer a phenolic clove-like off-flavor. This made the hybrid product unsuitable for commercial use from a taste perspective but acceptable from a dextrin utilization standpoint.
8.3.7 Cytoduction
Cytoduction is a specialized form of rare mating in which only the cytoplasmic components of the donor strain are transferred into the brewing strain; that is, the cell receives the cytoplasm from both parents but retains the nucleus of only one parent. The process of cytoduction requires the presence of a specific nuclear gene mutation designated Kar, for karyogamy defective. This mutation impairs nuclear fusion.51 Cytoduction can be used in three ways: substitution of the mitochondrial genome; introduction of DNA plasmids; or transfer of dsRNA species. When used in the substitution of the mitochondrial genome, it is possible to study the effects of these genetic elements on various cell functions. Mitochondrial substitution has been demonstrated to bring about variations in respiratory functions, cell surface activities, and various other strain characteristics.52 In addition, rare mating, used to introduce DNA plasmids, has been successful in the introduction of specific genetic elements constructed by gene-cloning experiments and to transfer the “zymocidal” or “killer” factor from laboratory haploid strains to brewing yeast strains, without altering the primary fermentation characteristics of the brewing yeast strain.53, 54
8.3.8 Mass Mating and Genome Shuffling
Mass mating is a technique where large numbers of cells, using different parental strains, are mixed and allowed to randomly mate. It can be used to create interspecific hybrids if there are strong markers that can be used for selection.55 Related to this technique is genome shuffling where repeated rounds of genetic recombination (using protoplast fusion or mass mating) are carried out to combine many different beneficial mutations in the same cell.29
8.3.9 Killer Yeast
In 1963, Bevan and Makower discovered the killer phenomenon in a S. cerevisiae strain that was isolated as a brewery contaminant.56 During the past five decades, intensive investigations of the various killer systems in yeast have resulted in substantial progress in many different fields of biology, providing important insights into basic and more general aspects of eukaryotic cell biology, virus–host cell interactions, and yeast virology. The topic has been recently reviewed by Liu et al.57
In Saccharomyces, killer strains secrete a protein toxin that is lethal to sensitive strains of the same genus and, less frequently, strains of different genera. Among the yeasts, killer, sensitive, and neutral strains have been described. The killer toxin of S. cerevisiae kills sensitive cells of the same species by disturbing the ion gradient across the plasma membrane after binding to the receptor at cell wall β 1,6-glucan.58 The “killer” character of Saccharomyces spp. is determined by the presence of two species of cytoplasmically located dsRNA plasmids. The M-dsRNA (1.0 to 1.8 kb) “killer” plasmid is killer-strain specific and codes for killer toxin and also for the immunity factor, which is a protein or proteins that make the host immune to the toxin (i.e., prevents self-killing). The L-dsRNA, which is also present in many “nonkiller” yeast strains, codes for the production of a protein that encapsulates both forms of dsRNA, thereby yielding virus-like particles. These virus-like particles are not naturally transmitted from cell to cell by any infection process. The killer plasmid behaves as a true cytoplasmic element, showing dominant non-Mendelian segregation. It depends, however, on a number of chromosomal genes for its maintenance in the cell and for expression. Cells of killer strains normally contain about 12 copies of the M-dsRNA and 100 copies of the L-dsRNA. The yeast can be cured of the M-dsRNA by growth at elevated temperature or by treatment with cycloheximide.
Based on the lack of cross-immunity, their molecular mode of action, and their killing profiles, toxin-producing S. cerevisiae killer strains have been classified into three major groups (K1, K2, and K28). Each secretes a unique killer toxin as well as a specific but as yet unidentified immunity component that renders the killer cells immune to their own toxins. The production of killer toxin K1, K2, or K28 is associated with the presence of a cytoplasmically inherited M-dsRNA satellite virus (designated ScV-M1, ScV-M2, or ScV-M28 for S. cerevisiae virus). This virus depends on the coexistence of a helper virus (L-A) to be stably maintained and replicated within the cytoplasm of the infected host cell.57
Brewing strains can be modified such that they are both resistant to killing by a zymocidal yeast and so that they themselves have zymocidal activity, thereby eliminating contaminating yeasts. Rare mating has been successfully employed to produce brewing killer yeast by crossing a brewing lager yeast with a Kar killer strain.53 Beer produced with this strain was acceptable but contained an ester note that was not present in the control. This suggested that the cytoplasm of the killer strain appeared to exert more influence on the brewing strain than originally predicted. A question often asked is whether the toxin is still active in the finished beer. The toxin is extremely heat sensitive, and a brewery pasteurization cycle of eight pasteurization units was shown to completely inactivate it.54
To determine the effect of the zymocidal lager strain on a typical brewery fermentation, “killer” lager yeast was mixed at a concentration of 10% with an ale brewing strain. The control was the ale strain mixed with 10% “nonkiller” lager. Within 10 h the killer lager strain had almost totally eliminated the ale strain. When the concentration of the killer yeast was reduced to 1%, within 24 h the ale yeast was again eliminated59 (Figure 8.8).
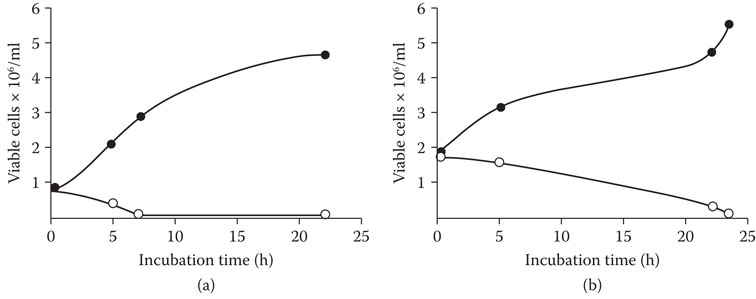
Figure 8.8 Effect of a ‘killer ‘ lager yeast on growth of a production ale yeast (12° P wort fermentation) at levels of 10% and 1%, assayed over a 24 hour period. (a) Legend control •--• : 90% ale yeast and 10% nonkiller lager yeast. Test ο—ο: 90% ale yeast and 10% killer lager yeast. (B) Legend control •--• : 90% ale yeast and 1% nonkiller lager yeast. Test ο—ο: 90% ale yeast and 1% killer lager yeast.
The speed at which this occurs may well make a brewer apprehensive about using such a yeast in the fermentation cellar, particularly where several yeasts are employed for the production of different beers. An error on an operator’s part in keeping lines and yeast tanks separate could have serious consequences. In a brewery with only one yeast strain, this would not be a cause for concern.
An alternative to the killer strain would be to produce a yeast strain that does not kill but is “killer resistant.” That is to say, it has received that genetic complement that makes it immune to zymocidal activity. The construction of such a yeast would perhaps be a good compromise because it would not itself kill; it would allay the brewer’s fear that this yeast might kill all other production strains in the plant; and, at the same time, it would not itself be killed by a contaminating yeast with “killer” ability.
8.3.10 Spheroplast Fusion
Spheroplast (protoplast) fusion, first described by van Solingen and van der Plaat,60 is a technique that can be employed in the genetic manipulation of industrial strains and circumvents the mating/sporulation barrier. The method does not depend on ploidy and mating type and, consequently, has great applicability to brewing yeast strains because of their polyploid nature and absence of mating type characteristic. Examples of fusions with commercial brewing strains include the construction of a brewing yeast with amylolytic activity by the fusion of S. cerevisiae with S. diastaticus,59 a polyploid capable of high ethanol production by fusion of a flocculent strain with saké yeasts,61 and the construction of industrial strains with improved osmotolerance by fusion of S. diastaticus with S. rouxii.62, 63
Two yeasts, one a S. diastaticus strain capable of dextrin fermentation (DEX) and the other a flocculent brewing lager strain (FLO), were converted to spheroplasts by the enzymic removal of the cell wall (Figure 8.9). This resulted in osmotically fragile spheroplasts (protoplasts), which were maintained in an osmotically stabilized medium of 1 M sorbitol. The spheroplasting enzyme was removed by thorough washing, and the spheroplasts were mixed and suspended in a fusing agent consisting of polyethylene glycol (PEG) and calcium ions in buffer. Subsequently, the fused spheroplasts were induced to regenerate their cell walls and recommence division. This was achieved in solid media containing 3% agar and sorbitol. The action of PEG as a fusing agent is not fully understood, but it is believed to act as a polycation, inducing the formation of small aggregates of spheroplasts. The PEG can be replaced by an electrical current or by short electric field pulses of high intensity.64, 65
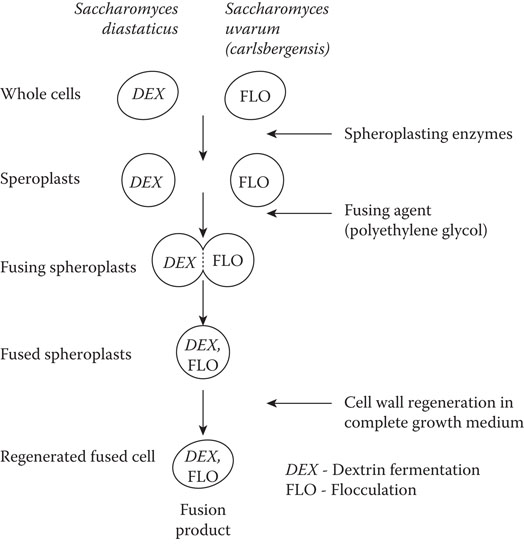
Figure 8.9 Spheroplast fusion to create novel industrial strains.
Although spheroplast fusion is an extremely efficient technique, it relies mainly on trial and error and does not modify strains in a predictable manner. The fusion product is nearly always different from both original fusion partners because the genome of both donors becomes integrated. Consequently, it is difficult to selectively introduce a single trait such as flocculation into a strain using this technique. For example, hybrid strains created by fusing lager yeast with S. diastaticus had unsatisfactory beer flavor/taste profiles, but could survive higher osmotic pressure and temperature and produced higher ethanol yields, and thus were of use to the fuel alcohol industry.66, 67 Similarly, a hybrid from a saké yeast with a brewing strain fermented high-gravity wort effectively, but the beers contained more ethanol and esters than the brew produced with the parental brewing strain.68
8.3.11 Recombinant DNA
Although the techniques of hybridization, rare mating, and spheroplast fusion have met with success, they have their limitations, the principal one being the lack of specificity in genetic exchange. Since 1978, when a transformation system for yeast became available,69 great strides have been made in yeast transformation. It is now possible to modify the genetic composition of a brewing yeast strain without disrupting the many other desirable traits of the strain, and it is possible to introduce genes from many other sources.
The applications of recombinant DNA methods to brewing and wine yeast have been reviewed in detail.28–30, 70 Examples include the production of brewing strains with glucoamylase, α-amylase, or pullulanase genes inserted from various sources. Work has been carried out cloning genes coding for β-glucanase into brewing strains. Research has been conducted on improving fermentation efficiency by increasing the gene dosage of the MAL genes for maltose utilization. The flocculation properties of brewing strains have been modified using cloning techniques. There is much interest in the use of transformation in terms of producing strains that have a reduced capacity to produce compounds such as diacetyl, hydrogen sulfide (H2S), sulfur dioxide (SO2), and dimethyl sulfide (DMS) as well as the increased ability to produce higher levels of esters such as isoamyl acetate. Genes for this cloning work come from a number of sources including Aspergillus niger, Bacillus subtilis, and Trichoderma reesii.
Strains have been constructed with the desirable traits expressed, but these strains are not used commercially at this time as there is still concern over consumer acceptance of beer made using a recombinant DNA yeast. The availability of alternative, inexpensive, traditional solutions for many of the problems it was hoped a genetically modified (GM) yeast could solve, such as inexpensive sources of β-glucanase and gluco- and α-amylase, has also retarded the implementation of these new strains.
There has been reluctance to use recombinant DNA technology in the brewing industry. The first genetically modified organism (GMO) for brewing to receive official government blessing was reported in the United Kingdom in 1994.70 However, this has not been used commercially to date. A gene coding for a glucoamylase was transferred from a diastatic strain of Saccharomyces to a brewing strain of Saccharomyces. The construct was considered “self-cloning” and was thus not treated as a recombinant organism, but this is not currently the legal designation in all countries.
The first commercially sold GMO-labeled beer in the world was launched in Sweden in 2004. Rather than using a recombinant DNA yeast, however, the beer was promoting the GM corn used in its manufacture. The company claimed to have launched the first beer to use genetic modification as a marketing tool. The beer, brewed in the southern Swedish town of Ystad, was made from corn—supplied by U.S. biotech giant Monsanto—genetically modified to resist attacks from pests.71
Researchers have produced strains using recombinant DNA that showed a reduction in diacetyl production by ILV2 gene disruption and BDH1 expression72 in laboratory fermentations. Strains have also been engineered to display α-acetolactate decarboxylase from Acetobacter aceti ssp. xylinum, and use of these strains resulted in a lower final diacetyl concentrations in the beers produced in the laboratory.73
Although it is now relatively easy with the strides made in the DNA technologies to manipulate yeast in the laboratory, and therefore to construct a GM yeast that has many additional useful brewing properties, the brewing industry today is still very hesitant to implement the use of a GM yeast due to the negative consumer perceptions regarding the use of such a yeast for beer production. The improvement of yeast for commercial beer production still relies on the more conventional techniques of breeding and directed evolution, although the newer gene editing techniques such as CRISPR will no doubt renew the discussion on, “What is a GM yeast?” and “What is the public willing to accept?”74
8.4 YEAST NUTRITION
8.4.1 Oxygen Requirements
Molecular oxygen has a multifaceted role in yeast physiology. Brewer’s yeast is a facultative aerobe. Wort fermentation in beer production, on the other hand, is largely anaerobic. However, this is not the case when the yeast is pitched into the wort, and at this time, some oxygen must be made available to the yeast. There is a need for oxygen because brewing yeasts require molecular oxygen to synthesize sterols and unsaturated fatty acids, which are present in wort but at suboptimal concentrations. Thus, yeast is capable of growth under strictly anaerobic conditions only when there is an exogenous supply of these compounds. Under aerobic conditions, the yeast can synthesize sterols and unsaturated fatty acids de novo from carbohydrates. Sterols and unsaturated fatty acids are abundant in malt, but normal manufacturing procedures prevent them from passing into the wort. When ergosterol and an unsaturated fatty acid, such as oleic acid, are added to wort, the requirement for oxygen disappears.
Cells prepared aerobically can grow to some extent anaerobically. Growth of yeast during the anaerobic phase of fermentation dilutes the preformed sterol pool between the mother cell and progeny. Cells can continue to divide until sterol depletion limits growth. Optimization of the dissolved oxygen (DO) supply for any brewing yeast strain is important to achieve good fermentation and a high-quality end product.75
The quantity of oxygen required for fermentation is yeast strain dependent. Ale yeast have been classified into four groups based on the oxygen concentration required to produce satisfactory fermentation performance.76 Similar groupings for lager yeast were reported by Jacobsen and Thorne.77 The amount of oxygen required for sterol synthesis and satisfactory fermentation varies widely, not only with the particular yeast employed but also with time of addition, whether in increments, and so on. With higher Plato worts, it is more difficult to ensure that there is enough oxygen available for the yeast, and pure sterile oxygen rather than air may be necessary.
Ensuring the correct yeast oxygenation at pitching ensures good yeast growth, full attenuation, and that the desired flavor compounds are produced. A general guideline on wort oxygenation is 1 ppm of oxygen for each degree Plato. Under-oxygenation is a bigger concern than a “slight” over-oxygenation because the yeast at pitching will very quickly remove all of the oxygen present in the wort. Excessive oxygen however will result in too much yeast growth and accompanying flavor issues. When air is used to oxygenate, 8 ppm is the maximum amount that will dissolve in a 10°Plato wort78 at 15°C. When higher wort values of oxygen are needed, pure oxygen must be used, and it should be remembered that, as the temperature and gravity of the wort increases, oxygen solubility decreases. (Further details in Chapters 9 and 14.)
8.4.2 Lipid Metabolism
Lipids are sparingly soluble in water but readily soluble in organic solvents. They are an integral part of the plasma membrane, where they are involved in the regulation of movement of compounds in and out of the cell, regulation of the activities of membrane-bound enzymes, and enhancement of the yeast’s ability to resist high ethanol concentrations.79 Saccharomyces yeast are able to take up fatty acids at low concentrations via facilitated diffusion and at high concentrations via simple diffusion.
Free fatty acids are powerful detergents and once taken up by the cell are quickly esterified to coenzyme A to reduce their potential for nonspecific enzyme inactivation. The addition of lipids, especially ergosterol and unsaturated long-chain fatty acids, has a pronounced effect on the growth and metabolism of yeast. The addition of unsaturated fatty acids (oleic, linoleic, and linolenic) has been reported to be a mechanism for the regulation of the concentration of flavor-active compounds in beer.80
The oxygen content of the wort at pitching is important with regard to lipid metabolism, yeast performance, and beer flavor. Under-aeration leads to suboptimal synthesis of essential membrane lipids and is reflected in limited yeast growth, a low fermentation rate, and concomitant flavor problems. Over-aeration results in overextending nutrients for the production of unnecessary yeast biomass, thus lowering fermentation efficiency because of the excess biomass and lower ethanol production. Negative effects on beer flavor and stability can result from over-aeration.
Studies have shown that the key factor in the successful application of wort or yeast aeration techniques is the physiological state of the yeast harvested at the end of the wort fermentation cycle.81 It has been suggested that the current regimes of yeast management produce variations in the physiological condition of the yeast, and that fluctuations in the ratio of glycogen to sterol may result in fermentation inconsistency, unless appropriate pitching rates and wort oxygen concentrations are selected. Devuyst et al.81 describe a procedure where the cropped yeast is suspended in water and aerated until maximum oxygen uptake rate is reached. The authors suggest that the formation of yeast sterols during the oxygenation process is fueled by glycogen dissimilation and that when the yeast is subjected to vigorous oxygenation pre-pitching, it provides a pitching yeast of standardized physiology. They believe that the best approach to improve fermentation control is to eliminate variability and that the oxygenation process they recommend produces pitching yeast of consistent and stable physiology, with no requirement for subsequent wort oxygenation to achieve satisfactory fermentation performance.
High metabolic activity of the cropped yeast is critical for efficient mobilization of reserve materials.82 Boulton et al.83 describe experiments to produce pitching yeast, replete in sterol, and thereby ostensibly remove the requirement for subsequent wort aeration. Yeast suspended in beer, cropped from the fermenter, was forcibly exposed to oxygen and was allowed to accumulate sterol. They found that it was necessary to increase pitching rates by 30% in order to achieve the same vessel residence times as control fermentations. Oxygenated yeast withstood the rigors of storage at elevated temperature less well than the untreated control.
Sterols are taken up by the yeast only while the yeast is growing under aerobic conditions. They are not taken up by stationary phase cells under anaerobic conditions. The poor physiological quality of cropped yeast is due to the depletion of sterols and unsaturated fatty acids. To restore the physiological activity of the plasma membrane, the yeast must take up its requirements from the wort, or synthesize the required lipids de novo, or convert them from the available pool of precursors. Callaerts et al.84 oxygenated an anaerobic brewing yeast to improve its physiological condition and measured the glycogen, sterol, and trehalose content of the yeast cell over time. In addition to the degradation of glycogen, and the expected synthesis of sterols, they observed an unexpected accumulation of trehalose and found that the trehalose and sterol concentrations correlated. The authors speculated that perhaps excessive contact with oxygen provoked a certain stress for the yeast and that the trehalose acted as a general stress protectant.85, 86
Fujiwara and Tamai87 showed that adequate aeration accelerates lager yeast fermentation and inhibits acetate ester formation. They demonstrated that complete depletion of trehalose adversely influences fermentation properties and ester synthesis and warned that aeration or agitation should be avoided during storage as high levels of dissolved oxygen enhanced the consumption of trehalose. Excess aeration resulted in trehalose exhaustion and did not stimulate anaerobic growth or decrease the synthesis of volatile esters. They suggest that intracellular trehalose concentration could be used as a marker that can reflect the appropriate level of aeration. Bleoanca et al.88 examined glycogen and trehalose protection mechanisms in brewing yeast as a response to oxidative stress and observed differences in the patterns of response based on the particular strain of yeast. (Further details in Chapter 14.)
8.4.3 Uptake and Metabolism of Wort Carbohydrates
When yeast is pitched into wort, it is introduced into an extremely complex environment consisting of simple sugars, dextrins, amino acids, peptides, proteins, vitamins, ions, nucleic acids, and other constituents too numerous to mention. One of the major advances in brewing science during the past 35 years has been the elucidation of the mechanisms by which the yeast cell, under normal circumstances, utilizes, in a very orderly manner, the plethora of wort nutrients.
Wort contains the sugars sucrose, fructose, glucose, maltose, and maltotriose, together with dextrin material. In the normal situation, brewing yeast species (i.e., S. cerevisiae and S. pastorianus) are capable of utilizing sucrose, glucose, fructose, maltose, and maltotriose in this approximate sequence, although some degree of overlap does occur. The majority of brewing strains leave the maltotetraose and other dextrins unfermented, but S. diastaticus is able to utilize some of the dextrin material due to the excretion of glucoamylase, which can hydrolyze smaller dextrins to metabolizable glucose.
The initial step in the utilization of any sugar by yeast is usually either its passage intact across the cell membrane or its hydrolysis outside the cell membrane, followed by entry into the cell by some or all of the hydrolysis products. Maltose and maltotriose are examples of sugars that pass intact across the cell membrane, whereas sucrose is hydrolyzed by an enzyme (invertase) located in the periplasmic space between the yeast cell wall and yeast cell membrane, and its hydrolysis products (glucose and fructose) are taken across the membrane into the cell (Figure 8.10). Dextrin material from wort is hydrolyzed outside of the yeast cell and simple sugars such as glucose can then enter the cell by facilitated diffusion.

Figure 8.10 Carbohydrate uptake by Saccharomyces spp.
Brewing yeast has several mechanisms for sensing the nutritional status of the environment, allowing it to adapt its uptake and metabolism to the surrounding conditions.89 Glucose and sucrose are always consumed first. Sucrose is hydrolyzed into glucose and fructose, and the monosaccharides are taken up by facilitated diffusion involving common membrane carriers.90 The yeast prefers glucose over fructose. Glucose slows down the uptake of fructose as the sugars have the same carriers, and these have a greater affinity for glucose. The presence of glucose and fructose in a fermentation causes the repression of gluconeogenesis, the glyoxylate cycle, respiration, and the uptake of less preferred carbohydrates, such as maltose and maltotriose. In addition, these two sugars activate cellular growth, mobilize storage compounds, and lower cellular stress resistance.91 Because yeast consumes fructose at a different rate from glucose, when large amounts of sucrose are used as adjunct, it can result in some of the very sweet fructose remaining in the beer and affecting the flavor profile. (Further details of adjuncts are in Chapter 6.)
Maltose and maltotriose are the major sugars in brewer’s wort and, therefore, the brewing strain’s ability to use these two sugars is vital and depends upon the correct genetic complement to transport the two sugars across the cell membrane into the cell. Once inside the cell, both sugars are hydrolyzed to glucose units by the α-glucosidase system.
8.4.4 Maltose and Maltotriose Uptake
Maltose fermentation in Saccharomyces requires at least one of five unlinked MAL loci. These five nearly identical regions are located at telomere-associated sites on different chromosomes92: MAL1, chromosome VII; MAL2, III; MAL3, II; MAL4, XI; and MAL6, VIII. Different maltose-fermenting strains carry at least one of these fully functional alleles, but often two or more loci are present in a strain. A typical MAL locus is a cluster of three genes, all of which are required for maltose fermentation. Gene 1, a member of the 12-transmembrane domain family of sugar transporters, encodes a maltose permease transporter; gene 2 encodes maltase (α-glucosidase); and gene 3 encodes the regulator/activator of transcription of the other two genes.
The expression of maltose permease and α-glucosidase are induced by maltose and repressed by glucose.93 The constitutive expression of the maltose transporter gene is critical, and the maltose-fermentation ability of brewing yeasts depends mostly on maltose permease activity.94 The maltose uptake system is an active process requiring cellular energy. Uptake involves a proton symport system, and potassium is exported to maintain electrochemical neutrality.
Brewing strains carry multiple copies of the maltose transporter gene, and it is speculated that the high number of MAL loci in brewing strains may have evolved as a mechanism to adapt to the high maltose environment of wort, giving the yeast strain a selective advantage.95
There are three different transporters known to carry maltose (Mal, Agt1, and Mtt1).96 Maltose and maltotriose can share the same transporter, the gene for which is closely linked to the maltase gene, and it is believed that brewing strains probably contain several copies of each of the two genes scattered as pairs around several different chromosomes. Yeast strains constructed with multiple MAL genes show increased rates of maltose uptake up from wort.97
The AGT1 (MAL1) permease is capable of transporting maltotriose as well as maltose, but although it is 57% identical to the MAL 6 permease gene, the MAL 6 permease gene cannot transport maltotriose—only maltose.98 More recent studies examining maltose and maltotriose uptake in brewing strains have shown that S. pastorianus strains utilize maltose more efficiently than S. cerevisiae strains99 and that there appear to be two subgroups of lager strains, which differ in sugar utilization due to the presence or absence of specific transmembrane transporters.100
During the brewing process, the rate and extent of wort sugar uptake are controlled by numerous factors including the yeast strains employed; the concentration and category of assimilable nitrogen; the concentration of ions; the fermentation temperature; the pitching rate; the tolerance of yeast cells to ethanol; the wort gravity; the wort oxygen level at yeast pitching and the wort sugar spectrum. These factors influence yeast performance either individually or in combination with others. The kinetics of maltose transport by an ale and a lager strain are affected by the presence of other sugars, ethanol, high gravity wort, and temperature, but maltose uptake is the dominant factor controlling the rate of wort maltose utilization.101
The uptake and hydrolysis of maltose and maltotriose from the wort is also dependent on the glucose concentration. When the glucose concentration is high (greater than 1% [w/v]), the MAL genes are repressed, and only when 40% to 50% of the glucose has been taken up from the wort will the uptake of maltose and maltotriose commence. Thus, the presence of glucose in fermenting wort exerts a major repressing influence on the wort fermentation rate. Mutants of brewing strains have been selected in which the maltose uptake was not repressed by glucose and, as a consequence, these strains exhibited increased fermentation rates.102
The presence of residual maltotriose in beer is not only due to the inability of yeast to utilize the sugar but also to the lower affinity for maltotriose uptake in conjunction with deteriorating conditions present at the later stages of fermentation. Indeed, transport, rather than hydrolysis of maltose, is the rate-limiting step determining fermentation performance.103 Meneses et al.104 used a model fermentation system to define the abilities of 25 industrial S. cerevisiae strains to utilize maltose and sucrose in the presence of glucose and fructose. Their survey exposed a number of novel phenotypes that could be harnessed as a means of producing strains with rapid and efficient utilization of fermentable carbohydrates. MTT1 (MTY1) is a transporter gene with a higher affinity for maltotriose than maltose.105 Magalhães et al.100 commented on how MTT1 plays an integral role in lager strains in terms of their ability to ferment effectively at lower temperatures.
8.4.5 Uptake and Metabolism of Wort Nitrogen
The nitrogen content of a yeast cell varies between 6% and 9% (w/w), and active yeast growth requires nitrogen, mainly in the form of amino acids, for the synthesis of new cell proteins and other nitrogenous components. Nitrogen uptake slows or ceases later in the fermentation as yeast multiplication stops. Wort nitrogen levels affect yeast growth and, at levels below 100 mg/L free amino nitrogen, growth is nitrogen-dependent. In wort, the main source of nitrogen for the synthesis of proteins, nucleic acids, and other nitrogenous cell components is the variety of amino acids formed from the proteolysis of barley protein. Lager wort nitrogen has been reported as approximately 30% to 40% amino acids, 30% to 40% polypeptides, 20% protein, and 10% nucleotides.106 Assimilable yeast nitrogen is the aggregation of the individual wort amino acids and small peptides (di-, tripeptides).107 These compounds are essential for the formation of new amino acids, synthesis of new structural and enzymic proteins, cell viability and vitality, fermentation rate, ethanol tolerance, and carbohydrate uptake. With all malt worts, there is usually enough nitrogen present, but with the use of adjuncts and high gravity brewing, the nitrogen levels must be monitored to ensure that the levels are adequate for yeast growth.
Wort contains 19 amino acids and, under brewery fermentation conditions, brewing yeast takes them up in an orderly manner, with different amino acids being removed at various points in the fermentation cycle and first described by Jones and Pierce108 in 1967. There are at least 16 different amino acid transport systems in yeast. In addition to permeases specific for individual amino acids, there is a general amino acid permease (GAP) with broad substrate specificity. Short chains of amino acids in the form of di- or tripeptides can also be taken up by the yeast cell. Uptake patterns are very complex, with a number of regulatory mechanisms interacting109 (Figure 8.11).
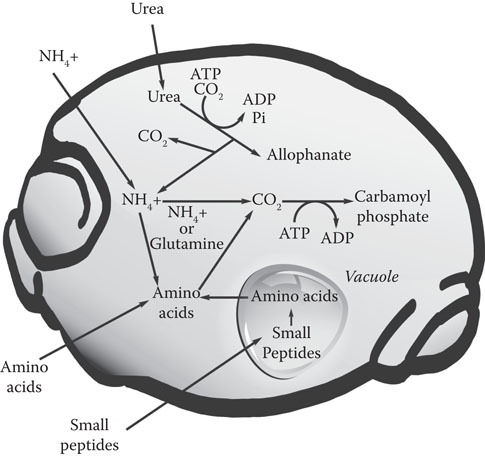
Figure 8.11 Nitrogen uptake by yeast.
Amino acids, like a number of sugars, do not permeate freely into the cell by simple diffusion; instead, there is a regulated uptake facilitated by a number of transport enzymes. At the start of fermentation, arginine, aspartic acid, asparagine, glutamic acid, glutamine, lysine, serine, methionine, and threonine are absorbed rapidly.109 The other amino acids are absorbed only slowly—or not until later in the fermentation. Under strictly anaerobic conditions, such as those encountered late in a brewery fermentation, proline, the most plentiful amino acid in wort, has scarcely been assimilated by the end of the fermentation, whereas over 95% of the other amino acids have disappeared. Proline is still present in the finished product at 200 to 300 mg/ml; however, under aerobic laboratory conditions, proline is assimilated after exhaustion of the other amino acids.
The inability of Saccharomyces spp. to assimilate proline under brewery conditions is the result of several phenomena. When other amino acids or ammonium ions are still present in the wort, the activity of proline permease, the enzyme that catalyzes the transport of proline across the cell membrane, is repressed. Once inside the cell, the first catabolic reaction of proline involves proline oxidase, which requires the participation of cytochrome c and molecular oxygen. By the time the other amino acids have been assimilated, thus removing the repression of the proline permease system, conditions are strongly anaerobic. As a result, the activity of proline oxidase is inhibited, and proline uptake does not occur.
Yeast strains exhibit different amino acid absorption rates and preferences. Amino acid uptake by yeast depends on a number of factors, including percentage of total assimilable nitrogen, individual amino acid concentrations, quality and absorption rate, amino acid competitive inhibition, yeast strain and generation, and yeast growth phase.
Free amino nitrogen (FAN) refers to free α-amino nitrogen and is expressed as milligrams N per liter assimilable nitrogen. FAN includes all of the amino acids minus proline. (Proline is not an α-amino acid and is not utilizable by Saccharomyces under anaerobic conditions.) FAN affects a great range of other fermentation factors, such as cell growth, biomass, viability, pH, and attenuation rate. The FAN target is usually stated as 160 to 190 mg/l wort (based on a 1040 wort). Optimization of the nitrogen content of wort is a complex issue as it is not only the initial FAN content in the wort but also the equilibrium of amino acids and ammonium ions, as well as other still undefined fermentation parameters, all of which will influence the final beer flavor.
S. cerevisiae is able to use different nitrogen sources for growth but not all nitrogen sources support growth equally well. Yeast prefers to use ammonium salts,110 but these are present in wort in only small amounts, and yeast cannot use inorganic nitrogen. S. cerevisiae selects nitrogen sources that enable the best growth by a mechanism called nitrogen catabolite repression. This mechanism is designed to prevent or reduce unnecessary divergence of the cell’s synthetic capacity to form enzymes and permeases for nonpreferred nitrogen sources.111, 112 The utilization of amino acids and the formation of fermentation by-products such as higher alcohols, esters, diketones, and organic acids, and their importance to beer flavor is discussed in later sections.
8.4.6 Uptake of Wort Minerals and Vitamins by Yeast
Yeast requires a number of vitamins and minerals for optimum growth and fermentation, and the levels needed for healthy yeast are adequate in most worts. Zinc is an essential element for yeast as it acts as a cofactor for many yeast enzymes, including the zinc-metalloenzyme alcohol dehydrogenase, the terminal step in yeast alcoholic fermentation. A lack of zinc can prevent a yeast from budding. The amount of zinc that a strain requires is determined to some extent by the particular strain’s genetic makeup. Some strains are very sensitive to low levels of zinc in the wort,113 and levels required for optimal fermentation range from 0.48 to 1.07 ppm. Problem fermentations can often be resolved by the addition of a source of bioavailable zinc, such as zinc chloride or sulfate, or yeast foods. (Further details in Chapter 10.) If magnesium is not present in sufficient amounts, the yeast cannot grow. Magnesium is important for key metabolic processes and is directly involved in ATP synthesis. Like zinc, magnesium also protects the cell from stress.114 Sufficient manganese in the wort is important when the wort is deficient in FAN. The control of flavor for beer consistency depends on a number of the metallic ions in the wort, where either not enough or too much can pose problems.115 Most of the yeast’s vitamin requirements can be met by the wort. Hucker et al.116 review and provide in-depth information on the roles of thiamine and riboflavin in wort.
8.5 YEAST EXCRETION PRODUCTS
One of the major excretion products produced during the fermentation of wort by yeast is ethanol. This primary alcohol impacts the final beer mainly by intensification of the alcoholic taste and aroma and by imparting a warming character. However, it is the types and concentrations of the other yeast excretion products that primarily determine the flavor of the product. The formation of these excretion products depends on the overall metabolic balance of the yeast culture, and there are many factors that can alter this balance and consequently the flavor of the product. Yeast strain, incubation temperature, adjunct level, wort pH, buffering capacity, wort gravity, oxygen, pressure, and so on, are all influencing factors.
Some volatiles are of great importance and contribute significantly to beer flavor, while others are of importance merely in building the background flavor of the product. The composition and concentration of beer volatiles depend upon the raw materials used, brewery procedures in mashing, fermentation parameters, and the yeast strain employed. The following groups of substances are to be found in beer: alcohols, esters, carbonyls, organic acids, sulfur compounds, amines, phenols, and a number of miscellaneous compounds.
8.5.1 Alcohols
In addition to ethanol, there several other alcohols found in beer, and these higher alcohols or fusel oils contribute significantly to flavor.117, 118 Their formation is linked to yeast protein synthesis. Higher alcohols can be synthesized via two routes: de novo from wort carbohydrates (the anabolic route) or as by-products of amino acid assimilation (the catabolic route). The contribution of the two routes is influenced by a number of factors, but generally, when low levels of amino acids are available, the anabolic route predominates and when high concentrations of amino acids are present the catabolic pathway is favored. For n-propanol, there is only the anabolic route as there is no corresponding amino acid for the catabolic pathway. The transport of branched-chain amino acids is important in brewing as the metabolites of these compounds are converted to higher alcohols. The composition of the wort, in particular the amino nitrogen content, influences the formation of these compounds. The yeast strain chosen for fermentation is of great significance and the levels of alcohols are dependent upon the fermentation temperature, with an increase in temperature resulting in increased concentrations of higher alcohols in the beer.
8.5.2 Glycerol
After ethanol and CO2, glycerol is the next highest compound produced by the yeast and present in beer. A 5% (v/v) ethanol beer will contain approximately 0.2% (v/v) glycerol. Glycerol has a sweet taste and thus contributes some sweetness to a beer’s flavor profile and is a palate contributor. Cells produce glycerol and accumulate it intracellularly, where it provides protection against various stress factors. It acts as an osmoprotectant for the cell, and it also protects against temperature stress.
The main pathway of glycerol formation is by the reduction of dihydroxyacetone phosphate (DHAP) to glycerol. It is also acts as a redox sink for the regeneration of cytosolic NAD+ from NADH. Yeast can be induced to overproduce glycerol in a number of ways, such as the addition of sulfite and salts to the growth media or by a pH change to above pH 7. Yeast can also catabolize glycerol, using it as a sole carbon source for growth.119, 120
8.5.3 Esters
Of the flavor-active substances produced by yeast, esters represent the largest and most important group. Esters are responsible for the highly desired fruity/floral character of beer. Esters are formed intracellularly by an enzyme-catalyzed condensation reaction between two co-substrates, a higher alcohol and an activated acyl-coenzyme A (acyl-CoA) molecule. The regulation of ester synthesis is complex (Figure 8.12). See reviews on this topic by Verstrepen et al.121 and Pires et al.122

Figure 8.12 Formation of esters and fatty acids as byproducts of yeast growth.
Beer contains more than 100 different esters, with the key ones being ethyl acetate (fruity/solvent), isoamyl acetate (banana/apple/fruity), isobutyl acetate (banana/fruity), and 2-phenylethyl acetate (honey/rose) aroma. The C6 to C10 medium-chain fatty acid ethyl esters, such as ethyl hexanoate (ethyl caproate) and ethyl octanoate (ethyl caprylate), have sour apple/aniseed aromas. Ethyl esters are present in the highest quantity presumably because ethanol is present in large amounts.
A number of factors have been found to influence the amount of esters formed during fermentation. The yeast strain is very important as are the fermentation parameters of temperature, pitching rate, and top pressure. Wort composition affects ester production; assimilable nitrogen compounds, the concentration of carbon sources, dissolved oxygen, and fatty acids all have an effect. Wort components that promote yeast growth tend to decrease ester levels.123 High-gravity worts can lead to disproportionate amounts of ethyl acetate and isoamyl acetate,124, 125 and the use of large-scale cylindro-conical fermentation vessels causes a dramatic drop in ester production, with a resultant imbalance in the ester profile.126 In order to obtain better control over ester synthesis, much research is being focused on the elucidation of the biochemical mechanisms of ester synthesis and on the factors influencing ester synthesis rates (further details in Chapters 9 and 14).
8.5.4 Sulfur Compounds
Sulfur compounds are of importance in brewing because traces of volatile sulfur compounds such as hydrogen sulfide, DMS, sulfur dioxide, and thiols significantly contribute to the flavor of the beer. Sulfur compounds are also one of the more difficult variables to control. Although small amounts of sulfur compounds can be acceptable or even desirable in beer, in excess they give rise to unpleasant off-flavors, and special measures such as purging with CO2 or prolonged maturation times are necessary to remove them. Although volatile organic sulfur compounds are contributed to the wort and beer by hops, adjuncts, and malt, a significant proportion of those present in finished beer are formed during or after fermentation. During fermentation, yeasts usually excrete significant amounts of hydrogen sulfide and sulfur dioxide. Sulfur dioxide can give indirect flavor effects by binding to compounds associated with beer flavor staling. In addition, sulfite acts as a natural antioxidant. Sulfur dioxide is usually present at concentrations below taste threshold. Excessive concentrations of hydrogen sulfide arise from deficiencies in wort composition, poorly controlled fermentations, and stressed yeast. Normal beers contain low levels of free hydrogen sulfide.
DMS is an important beer flavor compound derived from the wort production process and via yeast metabolism. The flavor of DMS is described as cooked sweet corn or cooked vegetable. At low levels, it is considered an essential flavor component contributing to the distinctive flavor and aroma of lager beer, but at high concentrations it is objectionable. Precursors of DMS are S-methylmethionine (SMM) and dimethylsulfoxide (DMSO), both of which originate from malted barley. DMS in beer may be derived through thermal degradation of SMM to DMS during kilning and wort preparation.127 Further, yeast during fermentation can reduce DMSO to DMS.128
8.5.5 Carbonyl Compounds
Carbonyl compounds are important because they have a high flavor potential and a significant influence on the flavor stability of beer. More than 200 carbonyl compounds have been detected in beer. Excessive concentrations of carbonyl compounds are known to cause a stale flavor in beer. The carbonyl found in highest concentration in beer is acetaldehyde.
The effects of aldehydes on finished beer flavor are a grassy aroma (propanol, 2-methyl butanol, pentanal) and a papery taste (trans-2-nonenal, furfural). Acetaldehyde is formed by yeast during fermentation in the final step of alcoholic fermentation. Acetaldehyde is reduced to ethanol by an enzymic reaction. The concentration of acetaldehyde varies during fermentation and aging/conditioning, reaching a maximum during the main fermentation and then decreasing. Removal of acetaldehyde is favored by a vigorous secondary fermentation, warmer maturation, sufficient wort aeration, and increased yeast content during maturation. High levels of acetaldehyde can also be caused by high air levels during fermentation. Acetaldehyde levels in bottled beer have been observed to increase during pasteurization and storage, especially if there is a high air content in the bottle headspace. Excessive quantities of acetaldehyde in beer can also be the result of bacterial spoilage, especially by strains of Zymomonas mobilis.
8.5.6 Diacetyl and 2,3-Pentanedione
The vicinal diketones, diacetyl and 2,3-pentanedione, are normal products of brewery fermentations that impart to beer a characteristic aroma and taste described as butterscotch, honey, or toffee. The taste threshold concentration for diacetyl in lagers is 0.1 to 0.14 mg/L, and the levels are somewhat higher in ales. Over the years, there has been a great deal of interest in the factors that influence the concentration of diacetyl in beer and in the methodology for its control129 and measurement. Excessive levels of diacetyl in beer can also be the result of beer spoilage by certain strains of bacteria such as Pediococcus and Lactobacillus.
Diacetyl and 2,3-pentanedione are formed outside the yeast cell by the oxidative decarboxylation of α-acetolactate and α-acetohydroxybutyrate, respectively. These α-acetohydroxy acids are intermediates in the biosynthesis of leucine and valine (acetolactate), and isoleucine (acetobutyrate), and are leaked into the wort by yeast during fermentation. In the wort, they are chemically decarboxylated into diacetyl and 2,3-pentanedione, respectively, and subsequently converted to acetoin or 2,3-pentanediol by the yeast (Figure 8.13). Thus, the final concentration of diacetyl in beer is the net result of three separate steps: (i) synthesis and excretion of α-acetohydroxy acids by yeast; (ii) oxidative decarboxylation of α-acetohydroxy acids to their respective diketones; and (iii) reduction of diacetyl and 2,3-pentanedione by yeast. (Further details in Chapter 15.)
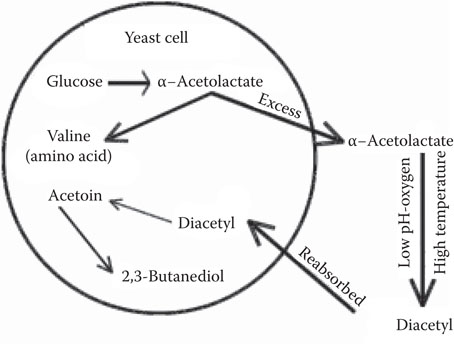
Figure 8.13 Diacetyl pathways.
The presence of diacetyl in beer at above-threshold levels occurs when α-acetolactate is decarboxylated to give diacetyl, at a time when the yeast cells are either absent or have lost their ability to reduce diacetyl to acetoin. Commonly, the fault arises because α-acetolactate breakdown has been curtailed by the use of temperatures conducive to yeast settling when the potential to produce diacetyl remains. When the beer becomes warm, which is usually when it is packaged and pasteurized—but may not be until the beer is disposed at the point of sale, diacetyl is produced and, in the absence of yeast, this diacetyl is not converted to acetoin and, therefore, accumulates. Diacetyl levels can thus be controlled by ensuring that there is sufficient active yeast in contact with the beer at the end of fermentation to reduce diacetyl to acetoin. Diacetyl formation from α-acetolactate has been shown to be dependent upon pH, the concentration of α-acetolactate, temperature, the presence of oxygen, the vigor of the fermentation, and certain metal ions. Vigorous fermentations produce more acetohydroxy acids, but the decomposition of acetohydroxy acids to vicinal diketones is also more rapid. In addition, since diacetyl is formed earlier in the fermentation, there is more time for diacetyl removal by the yeast. Gibson et al.130 studied the genetics of diacetyl production in lager yeast and stated that although much is known about the pathways, the “biological significance of diacetyl assimilation and reduction by yeast remains obscure and requires elucidation.” (further details of diacetyl metabolism are in chapter 15).
8.6 FLOCCULATION
Flocculation, a property of the yeast cell wall, is also the characteristic of a brewing yeast that allows the separation of yeast from beer, and is strongly correlated to the physical surface properties of the cell. Flocculation can be defined as: “The phenomenon wherein yeast cells adhere in clumps and either sediment rapidly from the medium in which they are suspended or rise to the surface.”131
This definition excludes other forms of aggregation, such as chain formation, which is different from flocculation and occurs when the younger buds fail to separate from the mother cells, resulting in aggregates of 30 to 50 cells. These aggregates are not able to re-aggregate after mechanical disruption. In addition to yeast separation from the wort by sedimentation, there is also flotation, where aggregates of cells are entrapped in the bubbles of CO2, as seen with some hydrophobic top-cropping ale brewing yeast strains. Other aggregate phenomena in Saccharomyces spp. include biofilms and pseudohyphae.132 Figure 8.14 shows the appearance of flocculent and nonflocculent yeast cells (wet mount using a light microscope).
Figure 8.14 Saccharomyces cerevisiae cells: (a) nonflocculent cells and (b) flocculent cells.
The correct flocculation properties are needed so that the yeast can be cropped at the end of primary fermentation and reused in subsequent fermentations. However, flocculation changes in a yeast culture can occur for many reasons. Mutations can often occur in the yeast’s flocculation ability, but this characteristic is very strain-dependent, with some strains being more stable than others. The method of harvesting can also select for specific populations.133 Individual strains of yeast differ considerably in their flocculating power. At one extreme are the very flocculent strains referred to as “gravelly,” and at the other extreme are totally nonflocculent strains sometimes referred to as “powdery.” The strongly flocculating yeasts can sediment out of the fermentation broth prematurely, giving rise to sweeter and less-fermented beers, whereas the weakly flocculating strains can remain in the beer during aging and cause yeasty flavor and filtration difficulties.
To produce a high-quality beer, it is axiomatic that not only must the yeast culture be effective in removing the required nutrients from the wort, be able to tolerate the prevailing environmental conditions (e.g., high ethanol levels), and impart the desired flavor to the beer, but the yeasts themselves must be effectively removed from the wort by flocculation, centrifugation, and filtration after they have fulfilled their metabolic role. Consequently, the flocculation properties—or conversely, lack of flocculation—of a particular brewing yeast culture is very important when considering important factors affecting wort fermentation.134
The measurement of yeast flocculation is a controversial topic. There is little standardization of the wide variety of tests available. The Helm’s flocculation test135 has been adopted as a recommended method by various brewing societies, and suggestions for improvements continue to be published by researchers. A concern with flocculation tests is that laboratory test results often do not mimic what happens at the plant scale. For reviews on this topic see Speers et al.136, 137
In addition to flocculation, there is the phenomenon of coflocculation.138 Coflocculation is defined as the phenomenon where two strains are nonflocculent alone but flocculent when mixed together. To date, coflocculation has only been observed with ale strains, and there are no reports of coflocculation between two lager strains of yeast. There is a third flocculation reaction that has been described, where the yeast strain has the ability to aggregate and co-sediment with contaminating bacteria in the culture.139, 140 Again, this phenomena appears to be confined to ale yeast and co-sedimentation of lager yeast with bacteria has not been reported. The exact mechanism of yeast flocculation onset and flocculent bond formation are far from understood.141, 142 However, newer technologies, such as “atomic force microscopy,” where actions of a single cell can be observed, continue to shed light on this topic.143
Flocculation is determined by the genotype of the cell. Dominant and recessive flocculation genes and flocculation suppresser genes have been described, but much remains to be discovered regarding the regulation of flocculation. It is generally agreed that there are at least two dominant flocculation genes present in brewing yeast. The existence of multiple gene copies and the possibility of there being more than one flocculation mechanism emphasize the difficulties researchers encounter when studying the genetics of brewing yeast flocculation.
The “lectin theory,” that yeast flocculation is mediated by specific interactions between cell wall proteins and carbohydrates from neighboring cells, is generally accepted to explain flocculation.144 It is believed that flocculation is due in part to mannose-specific lectin-like adhesion, but that it is also modulated by electrostatic and hydrophobic interactions. The yeast cell wall is a matrix, with an enriched outer layer of o-mannosylated proteins over an enriched layer of glucan, to which hyperglycosylated proteins are covalently attached. Stratford and Carter145 describe a specific lectin–receptor interaction, and van de Aar et al.146 describe a correlation between cell hydrophobicity and flocculence. Kock et al.147 observed the accumulation of hydrophobic carboxylic acids (3-hydroxy oxylipins) on the cell surfaces of a S. cerevisiae strain during the initiation of flocculation. Current studies on flocculation by El-Kirat-Chatel143 confirm that Flo 1 binds mannose via Ca2+ dependent interactions (the lectin model) and that the strength and duration of the cell–cell interactions depend not only on the weak lectin-sugar interactions but also on the hydrophobic interactions that result from the unfolding of proteins.
Yeast flocculation types can be classified into two groups distinguished by sugar inhibition: the NewFlo phenotype, which is inhibited by mannose, glucose, maltose, and sucrose but not by galactose, and the Flo1 type, which is inhibited by mannose but not by glucose, maltose, sucrose, or galactose.148 These two phenotypes are thought to be caused by two different lectin-like proteins. In some NewFlo-type yeasts, flocculation develops only toward the early stationary growth phase through the binding of zymolectins with the cell wall sugar receptors of neighboring cell surfaces. These receptors are available throughout the growth of the yeasts. The presence of glucose inhibits the flocculation of the yeast by binding to specific lectins. The onset of flocculation coincides with the termination of budding and glucose limitation, with a concomitant increase in cell surface hydrophobicity. In other yeasts, flocculation of the FLO 1 type occurs throughout the growth curve in all types of media.149 The FLO 1 gene150 comprises a 4.6-kb open reading frame, which includes repetitive sequences. The FLO 1 gene product is believed to be a hydrophobic cell wall protein, located on fimbriae-like structures, which are absent in nonflocculent cells.151 Straver et al.149 have suggested that cell surface hydrophobicity is a major determinant for yeast cells to become flocculent during growth in wort. Increased hydrophobicity of the cells may facilitate cell contact leading to calcium-dependent lectin–sugar binding. They also suggested that oxygen might be an indirect growth-limiting factor and that shortage of sterols and unsaturated fatty acids preceded flocculence under brewing conditions.
Verstrepen and Klis141 have discussed the origins of variations in the fungal cell surface and how, in S. cerevisiae, the FLO genes confer adhesion to agar and plastic as well as to other yeast cells. The genome sequence shows that there are at least five unlinked FLO genes in the adhesin family (FLO 1, FLO 5, FLO 9, FLO 10, and FLO 11). With the diversity in strains in regard to the flocculation genes, the differential regulation of the FLO genes, and the complexity of the interactions, this suggests that there is still much more research required before the interaction of all of the factors is understood. Because yeast is a living evolving organism, the flocculation characteristic can change and result in fermentation problems when the yeast falls out of suspension too early, when fermentation is not yet complete, or alternatively in other cases, if the natural flocculating ability has mutated to nonflocculent, and it cannot be induced to fall out of suspension. It is not only an issue of genetics but also of how the genes interact with the local environment. How quickly the flocculation ability changes due to mutations is very strain dependent, with some strains being more stable than other strains. Regular use of fresh yeast cultures after a number of repitchings avoids using a yeast that has accumulated too many mutations over time. Flocculation in brewing yeast has been reviewed in detail by Vidgren and Londesborough.152
Premature yeast flocculation (PYF) is a unique phenomenon thought to arise from fungal infections of the barley or malt (rather than the genetics of the yeast strain), and this type of flocculation can cause significant problems during the brewing process, resulting in under-attenuated worts and the resultant problems are consistent with an incomplete fermentation.153–155
8.7 YEAST MANAGEMENT IN THE BREWERY
8.7.1 Glycogen and Trehalose—The Yeast’s Carbohydrate Storage Polymers
Yeast cells produce two storage polymers, glycogen and trehalose. Both are accumulated by the yeast toward the end of the fermentation.
8.7.1.1 Glycogen
Glycogen, the yeast’s major reserve carbohydrate, is a multiple-branched molecule consisting of numerous chains of α-1,4-linked glucose residues. Chains containing 10 to 14 glucose residues form a tree-like structure, the branching being formed by α-1,6-linkages.156 Typically, under appropriate conditions, 20% to 30% of the yeast dry weight can consist of glycogen. The glycogen serves as a store of biochemical energy for use during the lag phase of fermentation, when energy demand is high for the synthesis of compounds such as sterols and fatty acids. This intracellular source of glucose fuels lipid synthesis at the same time that oxygen is available to the cell. The dissimilation of glycogen and lipid are both rapid. Hydrolysis of glycogen from approximately 25% to 5% and the corresponding production of lipid from approximately 5% to 11.5% of cell dry weight occurs in the first few hours after pitching. Toward the later stages of fermentation, the yeast restores its reserve of glycogen. (Further details in Chapter 14, see Figure 14.11 for the structure of glycogen.)
Once wort constituents are taken up by the yeast cell, the glycogen level increases to a maximum and then it decreases slightly toward the end of the fermentation. Glycogen accumulation occurs where growth is limited by a nutrient other than carbon. Yeast glycogen can be determined by a number of methods, but a simple indication of glycogen concentration can be obtained by staining the yeast with tincture of iodine (Lugol’s stain). A yeast cell rich in glycogen stains deep brown, whereas a yeast cell depleted in glycogen stains a pale yellow.
Ideally, the glycogen content of the pitching yeast should be high.157 Because glycogen reserves are depleted rapidly in storage, yeast that is repitched 24 to 48 h after collection is preferred. The rate of glycogen depletion is dependent on a number of factors, including yeast strain and brewing conditions. (Further details in Chapter 14.) Low glycogen levels in the pitching yeast result from unsatisfactory yeast handling practices (such as high storage temperature and extended storage time), and this correlates with low yeast in suspension and poor cell viability, extended fermentation times, and high end-of-fermentation diacetyl, acetaldehyde, and sulfur dioxide levels.
8.7.1.2 Trehalose
Trehalose is a nonreducing disaccharide, consisting of two glucose residues linked by an α-1,1-glycosidic bond. It is present in high concentrations in resting and stressed yeast cells and serves as a natural osmoprotectant. It stabilizes the structure of the lipid bilayer in the cell membrane and protects membrane proteins. It is much more than just a storage compound.158 It has the ability to increase the thermotolerance of proteins. It gives the cells an increased tolerance to desiccation, osmotic shock, freeze-thaw stress, oxidative stress and high temperatures, and high sugar and ethanol concentrations. (Further details in Chapter 14, see Figure 14.14 for structure of trehalose.)
When cells of Saccharomyces spp. encounter starvation conditions, ATP is produced by the catabolism of trehalose, which involves the action of the enzyme trehalase to yield glucose.159 Trehalose is utilized only after prolonged starvation conditions and when the glycogen pool is relatively depleted. Disappearance of trehalose correlates with a rapid loss in viability. Under certain conditions, yeast cells can store up to 10% to 20% of their dry weight as trehalose, but in the standard brewing process, levels of less than 5% are more typical.160 Very high gravity brewing leads to higher levels of trehalose. Trehalose accumulation occurs in late stationary phase and in response to environmental changes.
Trehalose’s stabilizing effect on the yeast membrane ensures yeast viability during sporulation, starvation, and dehydration by protecting the plasma membrane against autolysis. It plays a protective role in nutrient depletion, against toxic compounds, and improves cell resistance to temperature changes.161 A recent study by Petitjean et al.162 has suggested that yeast tolerance to various stresses does not rely on trehalose directly but rather on the trehalose-6P synthase (Tps1) protein, the first protein in the trehalose biosynthetic pathway.
When dried yeast, which requires rehydrating, is purchased for brewing and distilling, the levels of trehalose in the yeast are very important to maintain yeast viability during storage. When this yeast is rehydrated, it is important to pitch it into wort promptly (following the particular manufacturer’s instructions for rehydration) as on reactivation it will quickly lose its store of trehalose and weaken if there is no fresh carbohydrate source available for immediate use.
8.7.2 Pure Yeast Cultures
8.7.2.1 History
Yeasts belonging to the genus Saccharomyces are often referred to as the oldest plants cultivated by humans. The practice of using a pure yeast culture for brewing was started by Emil C. Hansen in the Carlsberg Laboratories more than 133 years ago. Hansen, using dilution techniques, was able to isolate single cells of the brewing yeast. This allowed him to test and then select the specific yeast strains that gave the desired brewing properties. The first pure yeast culture, a bottom fermenting lager, was introduced into a Carlsberg brewery on a production scale in 1883, and the benefits of pure culture quickly became clear. The Saccharomyces carlsbergensis (now Saccharomyces pastorianus) yeast of Hansen had its genome sequenced163 in 2014 and was identified as belonging to the group of I/Saaz-type lager yeast strains, which are better adapted to cold growth conditions than the group II/Frohberg-type (characterized by Weihenstephan strain WS34/70), although they have a joint evolutionary ancestry.
By 1892, 23 countries had installed Hansen’s pure culture plant, and in North America, Pabst, Schlitz, Anheuser Busch, and 50 smaller breweries were using pure lager yeast cultures. For a detailed history of research on yeasts from 1789 to 1900, see articles by Barnett164, 165 and Barnett and Lichtenthaler.166
8.7.2.2 Multiple Cultures in a Plant Environment
When a brewery is using multiple yeast strains for various products in a plant, it is important to have quality tests in place to ensure that pure cultures remain pure over time. To ensure that lager yeasts have not been contaminated with ale yeasts (or with wild yeasts that typically can grow at the higher temperature), there is a relatively easy test. Lager strains do not have the ability to grow above 37°C; therefore, to test if a lager yeast strain is contaminated with an ale yeast, plate growth on nutrient agar plates (PYN or MYGP) at 37°C can be used. Immediately after inoculation (not longer than 10 min), the plate is incubated at 37°C to 39°C. The only growth of colonies on the plate will be from any contaminating brewery ale strains or wild yeast.167
To determine if an ale culture is contaminated with a lager strain is more difficult. An x-α-gal test plate media can be utilized. Lager yeast possess the enzyme melibiase (which ale yeast do not) and, by using an x-α-gal media, any lager yeast colonies present will appear blue-green in coloration on the plate as only lager yeast can break down the indicator compound.167
A culture’s giant colony morphology can be useful when looking at ale yeast purity. Colonies of the yeast are grown on very large plates using a gelatin medium for three to four weeks.168 This results in very distinctive colony morphologies. Because the plates have to be grown over a longer time period, this is not a rapid test but still useful for examining the purity of a culture, especially for ale yeasts, which are much more distinctive than lagers in their appearance. Distinguishing contaminating yeast in a brewery often requires a mix of methods and media (e.g., WLN media)169 depending on the concern. Media for testing for the presence of wild yeast and newer genetic technologies for yeast characterization are discussed in Chapter 17.
Rapid molecular biology methods,170, 171 which differentiate and identify yeast strains, continue to become simpler to use. Molecular biology techniques, once only feasible in research environments, will continue to become available in kits for routine testing in breweries as more and more methods become adapted to be used with brewing yeast strains. (Further details in Chapter 17 on microbiological methods.)
8.7.2.3 Brettanomyces and Specialty Yeasts
Pure cultures of Brettanomyces and other unique yeasts are now being used for various specialty beers in the craft brewing industry172 and not just for traditional beers such as Belgian ale styles (Lambic, Gueuze). Contamination of regular beers in a plant usually is not an issue with Brettanomyces because it is a slow grower. It can utilize carbohydrates such as starch and it produces lactic and acetic acid, as well as a number of traditional aroma compounds such as ethyl acetate. It also produces some nontraditional beer aroma compounds. Some of these aromas are deemed pleasant (sour cherry pie) and some not so pleasant (sweaty horse, mousey). For a review of Brettanomyces yeast, see Schifferdecker et al.173
8.7.2.4 Strain Selection
In a laboratory or on a small pilot plant scale, it is not difficult to keep a culture pure and healthy. However, once the culture enters the brewery environment, large-scale management of the yeast is a more difficult task. It should include the following: a regular and high-quality pure yeast culture source, meticulous maintenance and diligent monitoring of the brewery yeast supply, and extensive microbiological quality checks.
In many breweries, fresh yeast is propagated every 8 to 10 generations (fermentation cycles), or earlier if contamination or a fermentation problem is identified. The systematic use of clean, pure, and highly viable yeast cells ensures that bacteria, wild yeast, or yeast mutants, such as RD yeast (also called petites due to their smaller colony size), do not lead to inconsistent fermentations or the development of off-flavors. RD mutants were discussed earlier in this chapter.
Lager yeast is normally a pure culture, whereas ale yeasts have often been a mixture of strains. The pure culture practice is invaluable in ensuring that any wild yeast is quickly detected and not allowed to proliferate into a significant problem for the brewer. Moreover, undesirable mutations of the parent strain, which often occur over long usage, are kept to a minimum. Various procedures can be used to isolate pure cultures from pitching yeast such as culturing from a single colony isolate from an agar or gelatin plate, or the use of a micromanipulator.
8.8 STORAGE OF CULTURES
The most important consideration in the maintenance of a culture collection of brewing yeasts is that the stored cultures and their subsequent progeny continue to accurately represent the strains originally deposited. The yeast preservation method should confer maximum survival and stability and be appropriate to the laboratory facilities available. There are many methods available to store yeast in culture collections.174 Boundy-Mills et al.175 discuss the challenges of maintaining culture collections, including intellectual property issues, and they list global culture collections and their areas of specific focus in terms of the organisms held therein. The most common preservation methods currently in use for storing microorganisms are subculture, drying or desiccation, freeze-drying, and freezing or cryopreservation.
8.8.1 Subculture
Subculture, a traditional and popular method, involves the use of two vials—one for transfer and one for laboratory use, that is, for inoculation to scale up the culture for plant use. The cultures are maintained on a medium suitable for yeast growth, such as malt yeast extract glucose peptone (MYGP) media or peptone yeast extract nutrient media (PYN),176 incubated between 20°C and 30°C to stationary phase (approximately 72 h), and then stored for up to six months at 1°C to 4°C. At six months, the culture is transferred to two fresh slopes from the vial reserved exclusively for transfer. Few cultures are lost using this method, but the cultures do change over time. Studies have shown that in 600 yeast strains studied, after 10 to 25 years of storage, 46% of the ascosporogenous strains had lost their ability to sporulate and 50% of the strains that carried amino acid markers had lost some of their nutritional markers. In addition, of the 300 brewery strains studied, 25% of these strains lost their ability to utilize maltotriose, and 10% showed a change in flocculation ability.177 In summary, this method is inexpensive and versatile, and the slopes are convenient for distribution purposes, but the method can lead to unacceptable levels of strain degeneration and is not recommended for long-term storage. Another concern is the danger of poor technique and cross-contamination, compromising the strain identity or purity.
8.8.2 Drying
There are a number of methods that use drying or desiccation. For example, silica gel can be used as a desiccant, but this method is generally reported to be more successful for genetically marked research strains rather than for industrial strains. The damaging effects appear to be very strain-specific, and substantial changes in fermentation patterns have been observed. Another popular drying method uses squares of filter paper and tinned milk as the suspending medium. Again, this method is favored for use by culture collection curators because of the ease of mailing cultures and is used primarily for genetically marked strains.
8.8.3 Freeze-Drying
Freeze-drying, or lyophilization, is a popular technique used for storing bacteria. It differs from desiccation in that water is removed by sublimation from the frozen material using a centrifugal dryer. The organism is sealed under a vacuum in a glass ampoule. However, for yeast, the survival levels tend to be low using this method, and when 580 strains of Saccharomyces were examined, the mean percentage of survival was only 5%. There is also the important question as to whether the surviving cells represent the original population. Studies have shown little change in morphological, physiological, or industrial characteristics, one exception being the increased level of RD mutants in some strains of Saccharomyces.176, 177 The advantages of this method include longevity of the freeze-dried culture and easy storage and distribution of ampoules. The major disadvantage is the initial diminished activity of the culture. In addition, the technique is labor intensive and requires special equipment.
8.8.4 Cryopreservation
Cryopreservation is the method of choice as little molecular activity takes place at the lower temperatures. For long-term storage, with maximum genetic stability, storage at −196°C in liquid nitrogen is ideal. Storage at −20°C to −90°C is also acceptable but for shorter storage periods. At very low temperatures, there are few reports of genetic instability, phenotypic and industrial characteristics are reported to be unchanged, yeast plasmids are retained, and the petite mutation (RD) is not a problem. Of 75 Saccharomyces strains studied,177 the mean survival rate was 66%. This method clearly yields the highest viability and superior stability, but this must be balanced against the disadvantages of using liquid nitrogen (cost, handling, delivery) and the inconvenience of culture distribution.
Mechanical freezers that operate at −70°C to −130°C are now available, and this eliminates many of the disadvantages associated with the use of liquid nitrogen. Cryogenic storage is clearly the method of choice178 as it holds the yeast in suspended animation and there are few genetic changes (if any) during storage. There is minimal cell damage on resuscitation from storage.176 When this method is employed, it is wise, as a safeguard, to keep a duplicate set of the most critical cultures on solid medium at 4°C in case of mechanical failure or a prolonged interruption of the electrical supply.
In addition, for convenient short-term storage (up to 6 months), a number of agar slopes can be prepared from a resuscitated culture and the slopes stored at 4°C in a refrigerator—using one slope each time to start a new propagation. It is important to remember that this slope should not be subcultured repeatedly as it is produced for one time use.179, 180 Again, it should be emphasized that freeze-drying brewer’s yeast for starter culture storage should be avoided and that storage under liquid nitrogen, or below −70°C, is the superior method to maintain the integrity of the culture.
8.9 PROPAGATION AND SCALE UP
The first yeast propagation plant, as already discussed, was developed by Hansen and Kuhle and consisted of a steam-sterilizable wort receiver and propagation vessel equipped with a supply of sterile air and impeller. The basic principles of propagation devised by Hansen in 1890 have changed little. The propagation can be batch or semicontinuous. There are usually three stainless steel vessels of increasing size equipped with attemperation control, sight glasses, and noncontaminating venting systems. They are equipped with a clean-in-place (CIP) system and often have in-place heat sterilizing and cooling systems for both the equipment and the wort. The yeast propagation system is ideally located in a separate room from the fermenting area with positive air pressure as well as humidity control and air sterilizing systems, disinfectant mats in doorways, and limited access by brewing staff.
During yeast propagation, the brewer wishes to obtain a maximum yield of yeast but also wishes to keep the flavor of the beer similar to a normal fermentation so that it can be blended into the production stream. As a result, the propagation is often carried out at only a slightly increased temperature and with intermittent aeration to stimulate yeast growth. The propagation of the master culture to the plant fermentation scale is a progression of fermentations of increasing size (typically 4 to 10 times), until enough yeast is grown to pitch a half-size or full commercial size brew.
Wort sterility is normally achieved by boiling for 30 min, or the wort can be pasteurized using a plate heat exchanger and passed into a sterile vessel and then cooled. Wort gravities range from 10°Plato to 16°Plato. Depending on the yeast, zinc or a commercial yeast food can be added. Aeration is important for yeast growth; the wort is aerated using oxygen or sterile air, and antifoam is added if necessary. Agitation is not normally necessary as the aeration process and CO2 evolved during active fermentation are sufficient to keep the yeast in suspension.
The exact details of the yeast propagation will vary whether it is a small brewery181 or a larger brewery utilizing high-gravity fermentation182 and depending on the propagation equipment available.183 Propagating yeast from a slope to obtain sufficient yeast for pitching a propagation tank in a brewery requires extra care. Sterile equipment should be used and the manipulation for scale up should be performed under aseptic conditions, preferably in a laminar flow hood. The media should be autoclaved to ensure no contamination enters in these very early laboratory stages. Orbital shakers are ideal for the first stage as stirring on a magnetic plate can cause the temperature to rise. Cells are then transferred to the next scale while they are still in exponential growth phase. The dilution during scale up is very important as too large a jump in dilution will result in poor growth.
The following is a typical procedure. The initial inoculum from the slope/plate of fresh yeast goes into 10 ml of sterile hopped wort for 24 h at 25°C. This is then scaled up to approximately 100 ml in a 200-ml shake flask, 1000 ml in a 2000-ml shake flask, and 5 l in a 10-l Van Laer flask or equivalent using 24- to 48-h increments. The steps can be larger and the temperature varied from 12°C to 25°C with resultant longer propagation times at the lower temperature. Scale-up steps are kept small at the early stages to ensure good growth. In the yeast propagation plant, use can be made of a three-vessel procedure (i.e., 10 hL at 16°C → 30 hL at 14°C → 300 hL at 12°C to 14°C for 4 to 5 days), or two vessels of 10 and 100 hL are also commonly used with the yeast inoculum being transferred from an 18-l Cornelius Spartan vessel. The European Brewing Convention (EBC) Analytica 3.4.4 provides a detailed method for scale up.184 There are many variations on the scale-up methods depending on the particular equipment available at the brewery location, but the key is careful attention to sterility and incremental stages of volume scale up. Yields can vary from 8 to 25 g yeast/L, depending on growth conditions. Kurz et al.185 describe a model for yeast propagation in breweries and present the basis for a control strategy aimed at the provision of optimal inoculum at the starting time of subsequent beer fermentations.
8.9.1 Contamination of Cultures
Various bacteria can contaminate the pure culture pitching yeast. These organisms originate from a number of sources: the wort, the yeast inoculum, or unclean equipment.186–188 Great care must be taken to ensure that there is no contamination during yeast propagation. There are a number of excellent textbooks that discuss the bacteria encountered during propagation and beer fermentation and the tests required for their detection and isolation,189–191 and further details can be found in Chapter 17.
Wild yeasts can originate from very diverse sources192 and, in addition to various Saccharomyces strains, include species of the genera Brettanomyces, Candida, Debaromyces, Hansenula, Kloeckera, Pichia, Rhodotorula, Torulaspora, and Zygosaccharomyces. The potential of the wild yeast to cause adverse effects varies with the specific contaminant. If the contaminant wild yeast is another culture yeast, the primary concern is with rate of fermentation, final attenuation, flocculation, and taste implications. If the contaminating yeast is a nonbrewing strain and can compete with the culture yeast for the wort constituents, inevitably problems will arise as these yeasts can produce a variety of off-flavors and aromas often similar to those produced by contaminating bacteria. Some wild yeasts can utilize wort dextrins, resulting in an over-attenuated beer that lacks body. These yeasts are found as both contaminants of fermentation and as postfermentation contaminants. In addition, wild yeasts often produce a phenolic off-flavor due to the presence of the POF gene. However, under controlled conditions, such as in the production of a German wheat beer or “weiss beer,” this phenolic clove-like aroma, produced when the yeast decarboxylates wort ferulic acid to 4-vinylguaiacol, can be a positive attribute of the beer.
8.9.2 Yeast Cropping from a Vertical Fermenter
When yeast is cropped from a vertical fermenter, it should be selected from the middle of the cone rather than from the very top or bottom. If cropped from the bottom, the brewer is selecting yeast that has fallen out of suspension early, and when cropping from the top, the brewer is selecting yeast that has stayed in suspension too long. Cropping from the middle of the cone yields the highest chance of selecting a good viable pitching yeast with the desired flocculation profile. When cropping from an ale fermentation vessel, where yeast rises to the surface, it is the middle skim that the brewer should select for re-pitching. Good practice is to avoid harvesting yeast from atypical fermentations. Further details on vertical fermenters can be found in Chapter 14.
8.10 YEAST WASHING
If there is evidence of bacterial contamination, the yeast can be washed to decontaminate the culture. Washing will not remove all of the bacteria, but it will reduce the bacterial load present. It is important to remember that washing will not remove wild yeast—only bacteria are removed because they are more susceptible to a lower pH than yeast. Some breweries incorporate a yeast wash into their process as a routine part of the operation, especially if there are concerns over eliminating bacteria responsible for the production of apparent total N-nitroso compounds (ATNC). There has been much controversy over the use of yeast washing and the effects on subsequent fermentations, but these problems (reduced cell viability, vitality, reduced rate of fermentation, changes in flocculation, fining, yeast crop size, and excretion of cell components) generally only surface if yeast washing is carried out incorrectly.193
Historically, there are three commonly used procedures for washing yeast:
Many brewers have a strong preference for a certain regime of yeast washing, and a number of factors must be taken into account when choosing the method, such as food grade quality of the acid, hazards involved in using the acid, and cost. Phosphoric and citric acid offer the advantage of being weak acids and yeast pH is more easily controlled, whereas with strong acids, such as sulfuric acid, there are special handling procedures required for the operators and a slight overdose will yield excessively low pH values.
A very popular method is that described by Simpson and Hammond193 in 1989, where cold phosphoric acid is used. The cooled yeast slurry (in water or beer at 2°C to 5°C) is acidified with cold food-grade phosphoric acid (citric, tartaric, and sulfuric are also options) to a pH of 2.0/2.2. (Note that it is important to check the pH to make sure it is correct.) Agitation should be continuous through the acid addition stage and during the wash. Poor viability after washing has been reported if one does not carefully follow the criteria of Simpson and Hammond. Proper washing has been shown to have no negative effects on the yeast if performed with care, but many things can go wrong with improper washing techniques, resulting in problems with viability in the subsequent fermentation when using an incorrectly washed yeast. Washing an unhealthy yeast is not recommended, and if it is yeast from a high alcohol fermentation, extra care must be taken to reduce the alcohol concentration to 5% (v/v) or less before washing.196 (Further details in Chapter 14.)
Some alternatives to acid washing have been used and one of these is the use of chlorine dioxide, which is available for this purpose from various suppliers. It is very easy to kill the yeast with this washing process if there is poor mixing or the chlorine dioxide concentration is not adjusted carefully for the amount of yeast present. Care must be taken especially in terms of time, temperature, and pH in order not to damage the yeast cells with this particular treatment.197, 198
8.11 CELL VIABILITY/VITALITY AND YEAST PITCHING
8.11.1 Microscopic Examination
Microscopic examination of brewery pitching yeast is as important today as it was when first described by Pasteur. It is a rapid way to ensure that there is not a major contaminant or viability problem with the yeast. When a sample of pitching yeast in water, wort, or beer is examined under the microscope, it can be difficult to distinguish a small number of bacteria from the trub and other extraneous nonliving material. The trub material, however, is irregular in size and outline, and it dissolves readily in dilute alkali. A trained microbiologist becomes very familiar with the typical appearance of the production yeast: the appearance of the cytoplasm, the shape of the yeast cells, whether the cells are chain formers, and so on, and thus, one can sometimes identify the presence of a wild yeast due to the presence of cells with unusual shapes or differences in budding or flocculating behavior.
8.11.2 Viability of Yeast and the Methylene Blue Test
The use of a viability stain such as methylene blue199 or methylene violet200 gives a good indication of the health of the cells.201 The most common stain is methylene blue, where in theory only dead cells stain blue. Methylene violet is reported to provide less ambiguity in color variation, while other researchers prefer the stains fluorescein diacetate (FDA), acridine orange, or alcian blue.
Methylene blue (and methylene violet) staining, due to its simplicity, remains very popular, but care must be taken when using it. As the viability of the cells decreases, the stain becomes less and less accurate and, at more than 15% dead cells (85% viable count) in a sample, the true dead cell numbers are much higher, so viability will be overestimated. The methylene blue stain can report viabilities of 30% to 40% when the true viability is almost zero. With cultures that are in very poor shape, this stain does not give accurate values.202 Also important to remember is that cell counts should be performed within 10 min of mixing the stain with the yeast as the stain is toxic to the cell. Methylene blue powder is shelf stable, but the solution once made up is only stable for three months and should be replaced on a regular basis.
Although there are a number of other stains and techniques available,203–207 in experienced hands, methylene blue can still quickly identify a viability problem before the yeast is pitched. New technologies continue to emerge to address better ways of determining yeast cell viability. A recent report by Feizi et al.208 discussed the possible application of “lense free chip microscopy” where machine learning replaced the issues with operator variance and fatigue in using the classical methylene blue viability stain.
8.11.3 Vitality of Yeast
When yeast is pitched, a high number of viable cells with high vitality is key. It is important to define in the brewing context what we mean when we talk about a yeast cell that is viable and a yeast cell that exhibits good vitality. A viable yeast cell has the ability to perform certain functions such as fermentation but it may not be able to reproduce (i.e., bud) because, as discussed earlier, yeast has a maximum number of times that it can bud. Vitality is about the health of the yeast, how metabolically active the cell is, whereas viability is merely about “Is the cell dead or alive?” There is not yet an ideal accepted method for total cell count, total viable cell count, and vitality measurement—all in one easy and inexpensive test. However, there are a number of very good methods that allow the brewer to set a baseline for the particular yeast.
8.11.4 Slide Viability Test
To accurately determine the health of a culture yeast with a possible low viability level (the case where the stain technique is the most unreliable), the slide culture technique is the method of choice.209 A suitably diluted suspension of yeast is applied to a microscope slide covered with a thin layer of nutrient medium. A sterile cover slip is positioned over the yeast, and the slide is incubated for no longer than 18 h at room temperature. The slide is examined at a magnification of 200×. Cells that give rise to microcolonies are viable (and clearly vital). Single cells not giving rise to microcolonies are scored as dead. Using this technique, the budding percent capability can be ascertained and a high percentage of cells capable of budding indicates a very vital culture.
8.11.5 Metabolic Activity Tests
Metabolic activity tests in the literature that test different aspects of the cell also to try to capture the concept of vitality. They include some of the following tests: ATP content, vicinal diketone (VDK) reduction, intracellular pH (ICP), enzyme activity (alcohol dehydrogenase [ADH], pyruvate dehydrogenase [PDH], pyruvate decarboxylase [PDC], maltase, protease, and so on), oxygen reduction capacity, oxygen consumption rate (OCR), ethanol and CO2 generation rate (respirometer), potentiometric titration, acidification power test (APT), and magnesium release. Often a combination of the aforementioned methods is required to obtain a truly accurate picture of the yeast’s state of metabolic activity.210
8.11.6 Yeast Pitching
Yeast pitching is governed by a number of factors, such as wort gravity, wort constituents, temperature, degree of wort aeration, and previous history of the yeast. Ideally, one wants a minimum lag in order to obtain a rapid start to fermentation, which then results in a fast pH drop, and ultimately assists in the suppression of bacterial growth. Pitching rates employed vary from 5 to 20 million cells per ml, but 10 million cells per ml is considered an optimum level by many and results in a lager yeast reproducing four to five times. Increasing the pitching rate results in fewer doublings as yeast cells, under given conditions, multiply to a maximum number of cells/unit volume regardless of the original pitching rate. The pitching rate can be determined by various methods, such as dry weight, turbidimetric sensors, hemocytometer, and electronic cell counting. With flow cytometry and the use of fluorescent probes, the physiological state of the yeast (viability, vitality, and intracellular pH) can be determined.201, 202, 211–213 Commercially available in-line biomass sensors have been introduced that utilize the passive dielectric properties of microbial cells and can discriminate among viable and nonviable cells and trub.214 The importance of levels of glycogen and trehalose in the stored pitching yeast are discussed in Chapter 14.
8.11.7 Pitching Rate Calculations
First measuring the yeast viability and then calculating the correct pitching rate are key to having consistent fermentations, for both micro- and mega-breweries. Too little yeast can cause problems of under-attenuation, long lag times, and inconsistent flavor profiles in the final beer, with elevated esters and sulfur compounds. Too much yeast, can lead to yeast autolysis flavors, poor head retention, and low levels of esters.
Pitching by wet weight is carried out by some brewers using the following calculations. A yeast cell weighs approximately 8 × 10− 11 g. In a yeast slurry that has settled, the general rule is that it will contain approximately 1 to 3 billion cells per ml (depending on the yeast size). A slurry that is 40% to 60% yeast solids correlates to 1.2 billion cells/ml. With a dried yeast, the general rule is that it contains approximately 7 to 20 billion cells/g. Viability counts are very important in both cases to calculate what amount is required for the optimal “healthy” pitching yeast concentration.
For yeast being re-pitched from a previous fermentation, a general brewer’s guideline is to use 1 × 106 cells/° Plato (and, with oxygen, a similar guideline is 1 ppm of oxygen for each degree Plato—so a 12°P wort would be pitched to a level of 12 × 106 cells/ml and 12 ppm oxygen/ml, and an 18°P wort would require 18 × 106 cells/ml and 18 ppm oxygen, etc.). Ales tend to require a slightly lower pitching rate than lagers because the fermentation progresses at a warmer temperature. Pitching yeast harvested from a previous fermentation should be used as soon as possible. It should be stored cold (1°C to 2°C, with care to avoid accidental freezing), in an oxygen-free environment under a layer of water or beer. The goal is to avoid the presence of oxygen during the storage phase. A blanket of CO2 with low positive pressure and minimal trub levels help with the yeast storage process.
8.12 YEAST COLLECTION
Yeast collection techniques differ among traditional ale top-fermentation systems, traditional lager bottom-fermentation systems, and the cylindro-conical fermentation system (further details in Chapter 14, see Figure 14.5). With the traditional ale top-fermentation, although there are many variations on this system, a single-, dual-, or multistrain yeast system can be employed, and the timing of the skimming can be critical to maintain the flocculation characteristics of the strains. Traditionally, the first skim or “dirt skim” with the trub present is discarded as is the final skim in most cases. The middle skim is normally kept for repitching. With the traditional lager bottom-fermentation, the yeast is deposited on the floor of the vessel at the end of fermentation. Yeast cropping is nonselective, and the yeast contains entrained trub. With the cylindro-conical fermentation vessel, now widely adopted for both ale and lager fermentations, the angle at the bottom of the tank allows for effective yeast plug removal.
Today, the use of centrifuges for the removal of yeast and the collection of pitching yeast is commonplace. (Further details in Chapter 14, see Figure 14.9.) There are a number of advantages, such as shorter process time, cost reduction, increased productivity, and reduced shrinkage. Care must be taken to ensure high temperatures (i.e., greater than 20°C) are not generated during centrifugation and that the design ensures low dissolved oxygen pickup and a high throughput. This is usually accomplished by use of a self-desludging and low heat-induction unit. Timing control of the desludge cycle is important; it allows for a more frequent cycle for yeast for the pitching tank and resultant lower solids or a longer frequency for yeast being sent to waste with higher solids and resulting reduced product shrink. (Further details in Chapters 14 and 15.)
8.12.1 Yeast Storage
Ideally, the yeast is stored in a room that is designed to be easily sanitized and that contains a plentiful supply of sterile water, a separate filtered air supply with positive pressure to prevent the entry of contaminants, and a temperature controlled at 0°C. Alternatively, insulated tanks in a dehumidified room are employed. When open vessels were commonly used, great care had to be taken to ensure that sources of contamination were eliminated. Reduction of moisture levels to retard mold growth and elimination of difficult-to-clean surfaces and unnecessary equipment and tools from the room are useful.
Yeast is most commonly stored as a slurry at 2°C to 4°C under six inches of beer, or under a water or 2% potassium dihydrogen phosphate solution. With high gravity brewing, it is important to remember that the ethanol levels are significantly higher and that this can affect the viability of the stored yeast. As more sophisticated systems have become available, storage tanks with external cooling and equipped with low-shear stirring devices have become popular. Reduction of available oxygen is important during storage, and minimal yeast surface areas exposed to air is desirable. Low dead cell counts and minimum storage time are desirable with the yeast being cropped “just in time” for repitching.
8.12.2 Shipping of Yeast
The pure culture is often maintained and supplied from a commercial laboratory and sent through the mail growing on a slope,180 in a pressed form, or the brewery can supply a slope from its own central laboratory to branch plants. More often, if a brewery has a number of branch plants, a pure yeast culture system is situated in only one plant, and this plant supplies the other branch plants with the yeast needed to pitch a larger fermentation vessel. For breweries with multiple plants, interplant homogeneity of the culture yeast is always a concern when consistency of flavor among plants is critical. Sending yeast from a central culture plant in sufficient quantity ensures conformity among plants.
General principles were established in the 1980s for shipping yeast from a central brewery or culture plant to other breweries and these principles are still valid.215, 216
Shipping Rules
When a yeast culture has been shipped it is important that it is monitored for temperature, viability, and bacterial contamination before use. Disposable time/temperature monitors can be attached to shipping containers that alert the receiver that temperature extremes have occurred during the shipping process. Care must be taken so that the receiver is expecting the shipment and that it does not sit on a receiving dock in the heat or cold for any length of time.
8.13 SUMMARY
It is important to remember that yeast is a living, breathing organism and not just another raw material process input. It must be handled with care! Although it is a very resilient organism, yeast encounters many different stress factors in its environment during transport, fermentation and the storage process. Remember that stress factors are additive, and that it is the cumulative effect, when care is not taken that results in a yeast that can no longer function optimally in a brewery can no longer function optimally in a brewery.
REFERENCES
1. Barnett, J.A., The taxonomy of the genus Saccharomyces Meyen ex Reess: A short review for non-taxonomists, Yeast, 8:1–23, 1992.
2. Chen, P., Dong, J., Yin, H., Bao, X., Chen, L., He, Y., Chen, R., Wan, X., Zhao, Y., and Hou, X., Genome comparison and evolutionary analysis of different industrial lager yeasts (Saccharomyces pastorianus), J. Instit. Brew., 122:42–47, 2016.
3. Kopecká, J., Němec, M., and Matoulková, D., Comparison of DNA‐based techniques for differentiation of production strains of ale and lager brewing yeast, J. Appl. Microbiol., 120:1561–1573, 2016.
4. Pederson, M.B., Recent views and methods for the classification of yeasts, Cerevisia — Belg. J. Brew. Biotechnol., 20:28–33, 1995.
5. Libkind, D., Hittinger, C., Valerio, E., Gonçalves, C., Dover, J., Johnston, M., Gonçalves, P., and Sampaio, J., Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast, Proc. Natl. Acad. Sci. U S A, 108:14539–14544, 2011.
6. Hebly, M., Brickwedde, A., Bolat, I., Driessen, M.R., de Hulster, E.A., van den Broek, M., Pronk, J.T., Geertman, J.M., Daran, J.M., and Daran-Lapujade, P., S. cerevisiae × S. eubayanus interspecific hybrid, the best of both worlds and beyond, FEMS Yeast Res., 15(3), p.fov005, 2015.
7. Baker, E.C., Wang, B., Bellora, N., Peris, D., Hulfachor, A.B., Koshalek, J.A., Adams, M., Libkind, D., and Hittinger, C.T., The genome sequence of Saccharomyces eubayanus and the domestication of lager-brewing yeast, Molec. Biol. Evol., 32:2818–2831, 2015.
8. Gallone, B., Steensels, J., Prahl, T., Soriaga, L., Saels, V., Herrera-Malaver, B., Merlevede, A., Roncoroni, M., Voordeckers, K., Miraglia, L., Teiling, C., Steffy, B., Taylor, M., Schwartz, A., Richardson, T., White, C., Baele, G., Maere, S., and Verstrepen, K.J., Domestication and divergence of Saccharomyces cerevisiae beer yeasts, Cell, 166:1397–1410, 2016.
9. Krogerus, K., Magalhães, F., Vidgren, V., and Gibson, B., Novel brewing yeast hybrids: Creation and application, Appl. Microbiol. Biotechnol., 101:65–78, 2017.
10. Walker, G.M., Yeast cytology, in Yeast Physiology and Biotechnology, John Wiley & Sons Ltd, West Sussex, UK, pp. 11–42, 1998.
11. Klis, F.M., Boorsma, A., and De Groot, P.W., Cell wall construction in Saccharomyces cerevisiae, Yeast, 23:185–202, 2006.
12. Schweizer, M., Lipids and membranes, in The Metabolism and Molecular Physiology of Saccharomyces cerevisiae, Dickinson, J.R. and Schweizer, M., Eds., CRC Press, London, pp. 140–223, 2004.
13. Stewart, G.G., Yeast mitochondria—Their influence on brewer’s yeast fermentation and medical research, Tech. Q. Master Brew. Assoc. Am., 51:3–11, 2014.
14. Jensen, R.E., Hobbs, A.A., Cerveny, K.L., and Sesaki, H., Yeast mitochondrial dynamics: Fusion, division, segregation, and shape, Microscopy Res. Tech., 51:573–583, 2000.
15. Mishra, P. and Chan, D.C., Metabolic regulation of mitochondrial dynamics, J. Cell Biology, 212:379–387, 2016.
16. Hecht, K.A., O’Donnell, A.F., and Brodsky, J.L., The proteolytic landscape of the yeast vacuole, Cellular Logistics, 4: e28023, 2014.
17. Yuan, W., Veenhuis, M., and van der Klei, I.J., The birth of yeast peroxisomes, Biochim. Biophys. (BBA)-Mol. Cell Res., 1863:902–910, 2016.
18. English, A.R. and Voeltz, G.K., Endoplasmic reticulum structure and interconnections with other organelles, Cold Spring Harbor Perspect. Biol., 5:p.a013227, 2013.
19. Borgese, N., Getting membrane proteins on and off the shuttle bus between the endoplasmic reticulum and the Golgi complex, J. Cell Sci., 129:1537–1545, 2016.
20. Smart, K., The death of the yeast cell, in Brewing Yeast Fermentation Performance, Smart, K., Ed., 1st ed., London, Blackwell Science, pp. 105–113, 2000.
21. Liu, B., Larsson, L., Caballero, A., Hao, X., Öling, D., Grantham, J., and Nyström, T., The polarisome is required for segregation and retrograde transport of protein aggregates, Cell, 140:257–267, 2010.
22. Salazar-Roa, M. and Malumbres, M., Fueling the cell division cycle. Trends Cell Biol., 27:69–81, 2017.
23. Bond, U., Neal, C., Donnelly, D., and James, T.C., Aneuploidy and copy number breakpoints in the genome of lager yeasts mapped by microarray hybridization, Curr. Genet., 45:360–370, 2004.
24. Krogerus, K., Arvas, M., Chiara, M., Magalhães, F., Mattinen, L., Oja, M., Vidgren, V., Yue, J.X., Liti, G., and Gibson, B., Ploidy influences the functional attributes of de novo lager yeast hybrids, Appl. Microbiol. Biotechnol., 100:7203–7222, 2016.
25. Gibson, B. and Liti, G., Saccharomyces pastorianus: Genomic insights inspiring innovation for industry, Yeast, 28:17–27, 2015.
26. Mortimer, R.K. and Johnson, J., Genealogy of principal strains of the yeast genetic stock center, Genetics, 113:35–43, 1986.
27. Bellon, J.R., Yang, F., Day, M.P., Inglis, D.L., and Chambers, P.J., Designing and creating Saccharomyces interspecific hybrids for improved, industry relevant, phenotypes, Appl. Microbiol. Biotechnol., 99:8597–8609, 2015.
28. Pretorius, I.S., Curtin, C.D., and Chambers, P.J., The winemaker’s bug: From ancient wisdom to opening new vistas with frontier yeast science, Bioengineering Bugs , 3:147–156, 2012.
29. Steensels, J., Snoek, T., Meersman, E., Nicolino, M.P., Voordeckers, K., and Verstrepen, K.J., Improving industrial yeast strains: Exploiting natural and artificial diversity, FEMS Microbiol. Rev., 38:947–955, 2014.
30. Pretorius, I.S., Synthetic genome engineering forging new frontiers for wine yeast, Crit. Rev. Biotechnol., 2016, doi: 10.1080/07388551.2016.1214945.
31. Mans, R., van Rossum, H.M., Wijsman, M., Backx, A., Kuijpers, N.G., van den Broek, M., Daran-Lapujade, P., Pronk, J.T., van Maris, A.J., and Daran, J.M., CRISPR/Cas9: A molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae, FEMS Yeast Res. 15, 2015. doi: 10.1093/femsyr/fov004.
32. Annaluru, N., Muller, H., Mitchell, L.A., Ramalingam, S., Stracquadanio, G., Richardson, S.M., Dymond, J.S., et al., Total synthesis of a functional designer eukaryotic chromosome, Science, 344(6179):55–58, 2014.
33. Coudreuse, D., Insights from synthetic yeasts, Yeast, 33:483–492, 2016.
34. Casey, G.P., Yeast selection in brewing, in Yeast Strain Selection, Panchal C.J., Ed., Vol. 7, Marcel Dekker, New York, pp. 65–111, 1990.
35. Jones, R.M., Russell, I., and Stewart, G.G., The use of catabolite derepression as a means of improving the fermentation rate of brewing yeast strains, J. Am. Soc. Brew. Chem., 44:161–166, 1986.
36. Novak, S., D’Amore, T., Russell, I., and Stewart, G.G., Sugar uptake in a 2-deoxy-D-glucose resistant mutant of Saccharomyces cerevisiae, J. Ind. Microbiol., 7:35–40, 1991.
37. Piña, B., Casso, D., Orive, M., Fité, B., Torrent, J., and Vidal, J. F. A new source to obtain lager yeast strains adapted to very high gravity brewing: 2-deoxy-D-glucose resistant colonies with fermentation capacity up to 25°Plato, Proc. Eur. Brew. Conv. Congr., Venice, Fachverlag Hans Carl, Nürenberg, pp. 186–198, 2007.
38. Galván, L., Pérez, A., Delgado, M., and Conde, J., Diacetyl production by sulfometron resistant mutants of brewing yeast, Proc. Eur. Brew. Conv. Congr., Zurich, IRL Press, Oxford, pp. 385–392, 1987.
39. Kielland-Brandt, M.C., Gjermansen, G., Nilsson-Tillgren, T., Holmberg, S., and Petersen, J.G.L., Diacetyl and brewer’s yeast, Yeast, 4:470, 1988 (special issue).
40. Ernandes, J.R., Williams, J.W., Russell, I., and Stewart, G.G., Respiratory deficiency in brewing yeast strains—Effects on fermentation, flocculation, and beer flavor components, J. Am. Soc. Brew. Chem., 51:16–20, 1993.
41. American Society of Brewing Chemists, Yeast-3.C., Triphenyltetrazolium chloride for identification of respiratory deficient cells, in Methods of Analyses, 9th ed., American Society of Brewing Chemists, St. Paul, MN, 2004.
42. Ogur, M., St. John, R., and Nagai, S., Tetrazolium overlay technique for population studies of respiration deficiency in yeast, Science, 125:928–929, 1957.
43. Winge, O., On haplophase and diplophase in some Saccharomycetes, C. R. Trav. Lab. Carlsberg, Ser. Physiol., 21:77–111, 1935.
44. Bonciani, T., Solieri, L., De Vero, L., and Giudici, P., Improved wine yeasts by direct mating and selection under stressful fermentative conditions. Eur. Food Res. Technol., 242:899–910, 2016.
45. Bilinski, C.A., Russell, I., and Stewart, G.G., Analysis of sporulation in brewer’s yeast: Induction of tetrad formation, J. Inst. Brew., 92:594–598, 1986.
46. Jones, R.M., Russell, I., and Stewart, G.G., Classical genetic and protoplast fusion techniques in yeast, in Yeast Biotechnology, Berry, D.R., Russell, I., and Stewart, G.G., Eds., Allen and Unwin, London, pp. 55–79, 1987.
47. Gjermansen, C. and Sigsgaard, P., Construction of a hybrid brewing strain of Saccharomyces carlsbergensis by mating of meiotic segregants, Carlsberg Res. Commun., 46:1–11, 1981.
48. Bilinski, C.A., Russell, I., and Stewart, G.G., Crossbreeding of Saccharomyces cerevisiae and Saccharomyces uvarum (carlsbergensis) by mating of meiotic segregants. Isolation and characterization of species hybrids, Proc. 21st Eur. Brew. Conv. Congr., Madrid, IRL Press, Oxford, pp. 499–504, 1987.
49. Delneri, D., Colson, I., Grammenoudi, S., Roberts, I.N., Louis, E.J., and Oliver, S.G., Engineering evolution to study speciation in yeasts, Nature, 422:68–72, 2003.
50. Goodey, A.R. and Tubb, R.S., Genetic and biochemical analysis of the ability of Saccharomyces cerevisiae to decarboxylate cinnamic acids, J. Gen. Microbiol., 128:2615–2620, 1982.
51. Conde, J. and Fink, G.R., A mutant of Saccharomyces cerevisiae defective for nuclear fusion, Proc. Natl. Acad. Sci. U. S. A., 73:3651–3655, 1976.
52. Conde Zurita, J. and Mascort, S.J.L., Effect of the mitochondrial genome on the fermentation behavior of brewing yeast, Proc. Eur. Brew. Conv. Congr., Copenhagen, IRL Press, Oxford, pp. 177–186, 1981.
53. Russell, I. and Stewart, G.G., Valuable techniques in the genetic manipulation of industrial yeast strains, J. Am. Soc. Brew. Chem., 43:84–90, 1985.
54. Hammond, J.R.M. and Eckersley, K.W., Fermentation properties of brewing yeast with killer character, J. Inst. Brew., 90:167–177, 1984.
55. Kunicka‐Styczyńska, A. and Rajkowska, K., Physiological and genetic stability of hybrids of industrial wine yeasts Saccharomyces sensu stricto complex, J. Appl. Microbiol., 110:1538–1549, 2011.
56. Bevan, E.A. and Makower, M., The physiological basis of the killer character in yeast, in Genetics Today, XI International Congress on Genetics, Geerts, S.J., Ed., Vol. 1, Pergamon Press, Oxford, pp. 202–203, 1963.
57. Liu, G.L., Chi, Z., Wang, G.Y., Wang, Z.P., Li, Y., and Chi, Z.M., Yeast killer toxins, molecular mechanisms of their action and their applications, Crit. Rev. Biotechnol., 35:222–234, 2015.
58. American Society of Brewing Chemists, Yeast-8 killer yeast identification, in Methods of Analyses, 9th ed., American Society of Brewing Chemists, St. Paul, MN, 2004.
59. Stewart, G.G. and Russell, I., Centenary review—One hundred years of yeast research and development in the brewing industry, J. Inst. Brew., 92:537–558, 1986.
60. Van Solingen, D. and van der Plaat, J.B., Fusion of yeast spheroplasts, J. Bacteriol., 130:946–947, 1977.
61. Seki, T.S., Myoga, S., Limtong, S., Uedono, S., Kamnuanta, J., and Taguchi, J., Genetic construction of yeast strains for high ethanol production, Biotechnol. Lett., 5:351–356, 1983.
62. Spencer, J.F.T., Bizeau, C., Reynolds, N., and Spencer, D.M., The use of mitochondrial mutants in hybridization of industrial yeast strains. VI. Characterization of the hybrid, Saccharomyces diastaticus and Saccharomyces rouxii, obtained by protoplast fusion, and its behavior in simulated dough-raising tests, Curr. Genet., 9:649–652, 1985.
63. Crumplen, R.M., D’Amore, T., Russell, I., and Stewart, G.G., The use of spheroplast fusion to improve yeast osmotolerance, J. Am. Soc. Brew. Chem., 48:58–61, 1990.
64. Weber, H.W., Forster, W., Jacob, H.E., and Berg, H., Enhancement of yeast protoplast fusion by electric field effects, in Current Developments in Yeast Research, Stewart, G.G. and Russell, I., Eds., Pergamon Press, Toronto, pp. 219–224, 1981.
65. Halfmann, H.J., Emeis, C.C., and Zimmermann, U., Electro-fusion of haploid Saccharomyces yeast of identical mating type, Arch. Microbiol., 134:1–4, 1983.
66. Whitney, K., Murray, C.R., Russell, I., and Stewart, G.G., Potential cost savings for fuel ethanol production by employing a novel hybrid yeast strain, Biotechnol. Lett., 7:349–354, 1985.
67. Stewart, G.G., Russell, I., and Panchal, C.J., Genetically stable allopolyploid somatic fusion product useful for the production of fuel alcohols, Canadian Patent No. 1,199,593, 1986.
68. Mukai, N., Nishimori, C., Wilson-Fujishige, I., Mizuno, A., Takahashi, T., and Sato, K., Beer brewing using a fusant between a sake yeast and a brewer’s yeast, J. Biosci. Bioeng., 91:482–86, 2001.
69. Hinnen, A., Hicks, J.B., and Fink, G.R., Transformation of yeast, Proc. Natl. Acad. Sci. U. S. A., 75:1929–1933, 1978.
70. Hammond, J.R.M. and Bamforth, C.W., Practical use of gene technology in food production, Brewer, 90:65–69, 1994.
71. Anon., News in brief, Nat. Biotechnol., 22:259, 2004.
72. Shi, T.T., Guo, X.W., Li, P., Zhou, Z., and Xiao, D.G., Diacetyl content reduction in industrial brewer’s yeast through ILV2 disruption and BDH1 expression, Eur. Food Res. Technol., 242:919–926, 2016.
73. Cejnar, R., Hložková, K., Kotrba, P., and Dostálek, P., Surface-engineered Saccharomyces cerevisiae displaying α-acetolactate decarboxylase from Acetobacter aceti ssp xylinum, Biotechnol. Lett., 38:2145–2151, 2016.
74. Callaway, E., Tapping genetics for better beer, Nature/News Feature, 535(7613): 484–486, 2016.
75. Verbelen, P.J., Saerens, S.M.G., Van Mulders, S.E., Delvaux, F., and Delvaux, F.R., The role of oxygen in yeast metabolism during high cell density brewery fermentations, Appl. Microbiol. Biotechnol., 82:1143–1156, 2009.
76. Kirsop, B.H., Oxygen in brewery fermentations, J. Inst. Brew., 80:252–259, 1974.
77. Jacobsen, M. and Thorne, R.S.W., Oxygen requirements of brewing strains of Saccharomyces uvarum (carlsbergensis)—Bottom fermentation yeast, J. Inst. Brew., 86:284–289, 1980.
78. Baker, C. D. and Morton, S., Oxygen levels in air-saturated worts, J. Inst. Brew,. 83:348–349, 1977.
79. Henderson, C.M. and Block, D.E., Examining the role of membrane lipid composition in determining the ethanol tolerance of Saccharomyces cerevisiae, Appl. Environ. Microbiol., 80:2966–2972, 2014.
80. Rosi, I. and Bertuccioli, M., Influences of lipid addition on fatty acid composition of Saccharomyces cerevisiae and aroma characteristics of experimental wines, J. Inst. Brew., 98:305–314, 1992.
81. Devuyst, R., Dyon, D., Ramos-Jeunehomme, C., and Masschelein, C.A., Oxygen transfer efficiency with a view to the improvement of lager yeast performance and beer quality, Proc. Eur. Brew. Conv. Congr., Lisbon, IRL Press, Oxford, pp. 377–384, 1991.
82. Boulton, C.A., Jones, A.R., and Hinchliffe, E., Yeast physiological condition and fermentation performance, Proc. Eur. Brew. Conv. Congr., Lisbon, IRL Press, Oxford, pp. 385–392, 1991.
83. Boulton, C., Clutterbuck, V., and Durnin, S., Yeast oxygenation and storage, in Brewing Yeast Fermentation Performance, Smart, K., Ed., Blackwell Science, Oxford, pp. 140–151, 2000.
84. Callaerts, G., Iserentant, D., and Verachtert, H., Relationship between trehalose and sterol accumulation during oxygenation of cropped yeast, J. Am. Soc. Brew. Chem., 51:75–77, 1993.
85. van Laere, A., Trehalose, reserve and/or stress metabolite? FEMS Microbiol. Rev., 63:201–210, 1989.
86. Wiemken, A., Trehalose in yeast, stress protectant rather than reserve carbohydrate, Antonie van Leeuw., 58:209–217, 1990.
87. Fujiwara, D. and Tamai, Y., Aeration prior to pitching increases intracellular enzymatic and transcriptional responses under nonnutritional conditions, J. Am. Soc. Brew. Chem., 61:99–104, 2003.
88. Bleoanca, I., Silva, A.R.C., Pimentel, C., Rodrigues-Pousada, C., and de Andrade Menezes, R., Relationship between ethanol and oxidative stress in laboratory and brewing yeast strains, J. Bioscience Bioeng., 116:697–705, 2013.
89. Conrad, M., Schothorst, J., Kankipati, H.N., Van Zeebroeck, G., Rubio-Texeira, M., and Thevelein, J.M., Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae, FEMS Microbiol. Rev., 38:254–299, 2014.
90. Kruckeberg, A.L. and Dickinson, J.R., Carbon metabolism, in The Metabolism and Molecular Physiology of Saccharomyces cerevisiae, Dickinson, J.R. and Schweizer, M., Eds., CRC Press, London, pp. 42–105, 2004.
91. Verstrepen, K.J., Iserentant, D., Malcorps, P., Derdelinckx, G., Van Dijck, P., Winderickx, J., Pretorius, I.S., Thevelein, J.M., and Delvaux, F.R., Glucose and sucrose: Hazardous fast-food for industrial yeast? Trends Biotechnol., 22:531–537, 2004.
92. Charron, M.J., Read, E., Haut, S.R., and Michels, C.A., Molecular evolution of the telomere-associated MAL loci of Saccharomyces, Genetics, 122:307–316, 1989.
93. Wang, X., Bali, M., Medintz, I., and Michels, C.A., Intracellular maltose is sufficient to induce MAL gene expression in Saccharomyces cerevisiae, Eukaryot. Cell, 1:696–703, 2002.
94. Kodama, Y., Fukui, N., Ashikari, T., and Shibano, Y., Improvement of maltose fermentation efficiency: Constitutive expression of MAL genes in brewing yeasts, J. Am. Soc. Brew. Chem., 53:24–29, 1995.
95. Ernandes, J., Williams, J., Russell, I., and Stewart, G.G., Effect of yeast adaptation to maltose utilization on sugar uptake during the fermentation of brewer’s wort, J. Inst. Brew., 96:67–91, 1993.
96. Vidgren, V., Ruohonen, L., and Londesborough, J., Characterization and functional analysis of the MAL and MPH loci for maltose utilization in some ale and lager yeast strains, Appl. Environ. Microbiol., 71:7846–7857, 2005.
97. Stewart, G.G., Panchal, C.J., and Russell, I., Current developments in the genetic manipulation of brewing yeast strains—A review, J. Inst. Brew., 89:170–188, 1983.
98. Han, E.K., Cotty, F., Sottas, C., Jiang, H., and Michels, C.A., Characterization of AGT1 encoding a general alpha-glucoside transporter from Saccharomyces, Mol. Microbiol., 17:1093–1097, 1995.
99. Duval, E.H., Alves, S.L., Jr., Dunn, B., Sherlock, G., and Stambuk, B.U., Microarray karyotyping of maltose‐fermenting Saccharomyces yeasts with differing maltotriose utilization profiles reveals copy number variation in genes involved in maltose and maltotriose utilization, J. Appl. Microbiol., 109:248–259, 2010.
100. Magalhães, F., Vidgren, V., Ruohonen, L., and Gibson, B., Maltose and maltotriose utilisation by group I strains of the hybrid lager yeast Saccharomyces pastorianus, FEMS Yeast Res., 16(5):fow053, 2016.
101. Rautio, J. and Londesborough, J., Maltose transport by brewer’s yeasts in brewer’s wort, J. Inst. Brew., 109:251–261, 2003.
102. Russell, I., Jones, R.M., Berry, D.R., and Stewart, G.G., Isolation and characterization of derepressed mutants of Saccharomyces cerevisiae and Saccharomyces diastaticus, in CRC Biological Research on Industrial Yeasts II, Stewart, G.G., Russell, I., Klein, R.D., and Hiebsch, R.R., Eds., CRC Press, Boca Raton, FL, pp. 7–65, 1987.
103. Zheng, X., D’Amore, T., Russell, I., and Stewart, G.G., Factors influencing maltotriose utilization during brewery wort fermentations, J. Am. Soc. Brew. Chem., 52:41–47, 1994.
104. Meneses, F.J., Henschke, P.A., and Jiranek, V., A survey of industrial strains of Saccharomyces cerevisiae reveals numerous altered patterns of maltose and sucrose utilization, J. Instit. Brew., 108:310–321, 2002.
105. Dietvorst, J., Londesborough, J., and Steensma, H.Y., Maltotriose utilization by lager yeast strains: MTT1 encodes a maltotriose transporter, Yeast, 22:775–788, 2005.
106. Ingledew, W.M., Utilization of wort carbohydrates and nitrogen by Saccharomyces carlsbergensis, Tech. Q. Master Brew. Assoc. Am., 12:146–150, 1975.
107. Lekkas, C., Hill, A.E., Taidi, B., Hodgson, J., and Stewart, G.G., The role of small wort peptides in brewing fermentations, J. Inst. Brew., 115:134–139, 2009.
108. Jones, M. and Pierce, J.S., The role of proline in the amino acid metabolism of germinating barley, J. Instit. Brew., 73:577–583, 1967.
109. Lekkas, C., Stewart, G.G., Hill, A.E., Taidi, B., and Hodgson, J., Elucidation of the role of nitrogenous wort components in yeast fermentation, J. Inst. Brew., 113:3–8, 2007.
110. Cueto-Rojas, H.F., Seifar, R.M., Ten Pierick, A., van Helmond, W., Pieterse, M.M., Heijnen, J.J., and Wahl, S.A., In vivo analysis of NH4+ transport and central nitrogen metabolism in Saccharomyces cerevisiae during aerobic nitrogen-limited growth, Appl. Environ. Microbiol., 82:6831–6845, 2016.
111. Magasanik, B. and Kaiser C.A., Review—Nitrogen regulation in Saccharomyces cerevisiae, Gene, 290:1–18, 2002.
112. Dickinson, J.R., Nitrogen metabolism, in The Metabolism and Molecular Physiology of Saccharomyces cerevisiae, Dickinson, J.R. and Schweizer, M., Eds., CRC Press, London, pp. 104–116, 2004.
113. De Nicola, R. and Walker, G.M., Zinc interactions with brewing yeast: Impact on fermentation performance, J. Am. Soc. Brew. Chem., 69:214–219, 2011.
114. Walker, G.M., DeNicola, R., Anthony, S. and Learmonth, R., Yeast metal interactions: Impact of brewing and distilling fermentations, Proceedings of the Institute of Brewing and Distilling, Asia Pacific Section 20.6 Convention, Hobart, Tasmania, pp. 1–19, 2006. Available from: http://www.ibdasiapac.com.au/asia-pacific-activities/convention-proceedings/2006/program_wed.htm (accessed December 15, 2016).
115. Bourke, E. J. and Datsomor, J., Effect of manganese and some FAN supplements on fermentations. IBD Africa Conference 2013. Available from: http://www.ibdafrica.co.za/wp-content/uploads/2013/05/Bourke-Manganese.pdf (accessed December 15, 2016).
116. Hucker, B., Wakeling, L., and Vriesekoop, F., Vitamins in brewing: The impact of wort production on the thiamine and riboflavin vitamer content of boiled sweet wort, J. Inst. Brew., 120:164–173, 2014.
117. Hazelwood, L.A., Daran, J.M., van Maris, A.J., Pronk, J.T., and Dickinson, J.R., The Ehrlich pathway for fusel alcohol production: A century of research on Saccharomyces cerevisiae metabolism, Appl. Environ. Microbiol., 74:2259–2266.
118. He, Y., Dong, J., Yin, H., Zhao, Y., Chen, R., Wan, X., Chen, P., Hou, X., Liu, J., and Chen, L., Wort composition and its impact on the flavour‐active higher alcohol and ester formation of beer—A review, J. Inst. Brew., 120:157–163, 2014.
119. Goold, H.D., Kroukamp, H., Williams, T.C., Paulsen, I.T., Varela, C., and Pretorius, I.S., Yeast’s balancing act between ethanol and glycerol production in low‐alcohol wines, Microbial Biotechnol., 10(2):264–278, 2017. doi:10.1111/1751-7915.12488
120. Klein, M., Swinnen, S., Thevelein, J., and Nevoigt, E., Glycerol metabolism and transport in yeast and fungi: Established knowledge and ambiguities, Environ. Microbiol., 19(3):878–893, 2017. doi: 10.1111/1462-2920.13617
121. Verstrepen, K.J., Derdelinckx, G., Dufour, J.P., Winderickx, J., Thevelein, J.M., Pretorius, I.S., and Delvaux, F.R., Flavor-active esters: Adding fruitiness to beer, J. Biosci. Bioengin., 96:110–118, 2003.
122. Pires, E.J., Teixeira, J.A., Brányik, T., and Vicente, A.A., Yeast: The soul of beer’s aroma—A review of flavour-active esters and higher alcohols produced by the brewing yeast, Appl. Microbiol. Biotechnol., 98:1937–1949, 2014.
123. Boulton, C. and Quain, D., Formation of flavor compounds, in Brewing Yeast and Fermentation, Blackwell Science, Oxford, pp. 116–142, 2001.
124. Younis, O.S. and Stewart, G.G., Effect of malt wort, very-high-gravity malt wort, and very-high-gravity adjunct wort on volatile production in Saccharomyces cerevisiae, J. Am. Soc. Brew. Chem., 57:39–45, 1999.
125. Stewart, G.G., High gravity brewing and distilling—Past experiences and future prospects, J. Am. Soc. Brew. Chem., 68:1–9, 2010.
126. Meilgaard, M.C., Effects on flavor of innovations in brewery equipment and processing: A review, J. Inst. Brew., 107:271–286, 2001.
127. Scheuren, H., Baldus, M., Methner, F.J., and Dillenburger, M., Evaporation behaviour of DMS in an aqueous solution at infinite dilution—A review, J. Inst. Brew., 122:181–190, 2016.
128. Duan, W., Roddick, F.A., Higgins, V.J., and Rogers, P.J., A parallel analysis of H2S and SO2 formation by brewing yeast in response to sulfur-containing amino acids and ammonium ions, J. Am. Soc. Brew. Chem., 62:35–41, 2004.
129. Krogerus, K. and Gibson, B.R., 125th Anniversary review: Diacetyl and its control during brewery fermentation, J. Instit. Brew., 119:86–97, 2013.
130. Gibson, B., Krogerus, K., Ekberg, J., Monroux, A., Mattinen, L., Rautio, J., and Vidgren, V., Variation in α‐acetolactate production within the hybrid lager yeast group Saccharomyces pastorianus and affirmation of the central role of the ILV6 gene, Yeast, 32:, 301–316, 2015.
131. Stewart, G.G., Hill, A.E. and Russell, I., 125th Anniversary review: Developments in brewing and distilling yeast strains, J. Instit. Brew., 119:202–220, 2013.
132. Soares, E.V., Flocculation in Saccharomyces cerevisiae: A review, J. Appl. Microbiol., 110:1–18, 2011.
133. Stewart, G.G., The concept of nature-nurture applied to brewer’s yeast and wort fermentation, Tech. Q. Master Brew. Ass. Am., 51(3): 2014. doi: 10.1094/TQ-51-3-0905-01.
134. Speers, A., Brewing fundamentals, part 3: Yeast settling—Flocculation, Tech. Q. Master Brew. Assoc. Am., 53:17–22, 2016.
135. American Society of Brewing Chemists, Yeast-11 A yeast flocculation. Helm assay, in Methods of Analyses, 9th ed., American Society of Brewing Chemists, St. Paul, MN, 2004.
136. Speers, R.A., Tung, M.A., Durance, T.D., and Stewart, G.G., Biochemical aspects of yeast flocculation and its measurement: A review, J. Inst. Brew., 98:292–300, 1992.
137. Speers, R.A., Tung, M.A., Durance, T.D., and Stewart, G.G., Colloidal aspects of yeast flocculation: A review, J. Inst. Brew., 98:525–531, 1992.
138. Stewart, G.G., Russell, I., and Garrison, I., Further studies on flocculation and co-flocculation in brewer’s yeast strains, Proc. Am. Soc. Brew. Chem., 31:100–108, 1973.
139. Zarattini, R. de A., Williams, J.W., Ernandes, J.R., and Stewart, G.G., Bacterial- induced flocculation in selected brewing strains of Saccharomyces, Cerevisia Biotechnol., 18:65–70, 1993.
140. Peng, X., Sun, J., Iserentant, D., Michiels, C., and Verachtert, H., Flocculation and coflocculation of bacteria by yeasts, Appl. Microbiol. Biotechnol., 55:, 777–781, 2001.
141. Verstrepen, K.J. and Klis, F.M., Flocculation, adhesion and biofilm formation in yeasts, Molecular Microbiol., 60:5–15, 2006.
142. Van Mulders, S.E., Christianen, E., Saerens, S.M., Daenen, L., Verbelen, P.J., Willaert, R., Verstrepen, K.J., and Delvaux, F.R., Phenotypic diversity of Flo protein family-mediated adhesion in Saccharomyces cerevisiae, FEMS Yeast Res., 9:178–190, 2009.
143. El-Kirat-Chatel, S., Beaussart, A., Vincent, S.P., Flos, M.A., Hols, P., Lipke, P.N., and Dufrêne, Y.F., Forces in yeast flocculation, Nanoscale, 7:1760–1767, 2015.
144. Miki, B.L.A., Poon, N.H., James, A.P., and Seligy, V.L., Possible mechanism for flocculation interactions governed by the FLO 1 gene in Saccharomyces cerevisiae, J. Bacteriol., 150:878–889, 1982.
145. Stratford, M. and Carter, A.T., Yeast flocculation: Lectin synthesis and activity, Yeast, 9:371–378, 1993.
146. van der Aar, P.C., Straver, M.H., and Teunissen, A.W.R.H., Flocculation of brewers’ lager yeast, Proc. Eur. Brew. Conv. Congr., Oslo, IRL Press, Oxford, pp. 259–266, 1993.
147. Kock, J.L.F., Venter, P., Smith, D.P., Van Wyk, P.W.J., Botes, P.J., Coetzee, D.J., Pohl, C.H., Botha, A., Riedel, K.-H., and Nigam, S., A novel oxylipin-associated “ghosting” phenomenon, Antonie van Leeuw., 77:401–406, 2000.
148. Stratford, M. and Assinder, S., Yeast flocculation: Flo1 and NewFlo phenotypes and receptor structure, Yeast, 7:559–574, 1991.
149. Straver, M.H., van der Aar, P.C., Smit, G., and Kijne, J.W., Determinants of flocculence of brewers’ yeast during fermentation in wort, Yeast, 9:527–532, 1993.
150. Russell, I., Stewart, G.G., Reader, H.P., Johnston, J.R., and Martin, P.A., Revised nomenclature of genes that control yeast flocculation, J. Instit. Brew., 86:120–121, 1980.
151. Day, A.W., Poon, N.H., and Stewart, G.G., Fungal fimbriae III. The effect on flocculation in Saccharomyces cerevisiae, Can. J. Microbiol., 21:558–564, 1975.
152. Vidgren, V. and Londesborough, J., 125th Anniversary review: Yeast flocculation and sedimentation in brewing, J. Instit. Brew., 117:475–487, 2011.
153. Kaur, M., Bowman, J.P., Stewart, D.C., Sheehy, M., Janusz, A., Speers, R.A., Koutoulis, A., and Evans, D.E., TRFLP analysis reveals that fungi rather than bacteria are associated with premature yeast flocculation in brewing, J. Ind. Microbiol. Biotechnol., 39:1821–1832, 2012.
154. Panteloglou, A.G., Smart, K.A., and Cook, D.J., Malt-induced premature yeast flocculation: Current perspectives, J. Ind. Microbiol. Biotechnol., 39:813–822, 2012.
155. Shang, Y.L., Li, X.M., Cai, G.L., Lu, J., and Chen, J., Premature yeast flocculation factors from barley malt present in both malt husk and the non‐husk part, J. Instit. Brew., 120:220–224, 2014.
156. Wilson, W.A., Roach, P.J., Montero, M., Baroja-Fernández, E., Muñoz, F.J., Eydallin, G., Viale, A.M., and Pozueta-Romero, J., Regulation of glycogen metabolism in yeast and bacteria, FEMS Microbiol. Rev., 34:952–985, 2010.
157. Deshpande, P.S., Sankh, S.N., and Arvindekar, A.U., Study of two pools of glycogen in Saccharomyces cerevisiae and their role in fermentation performance, J. Instit. Brew., 117:113–119, 2011.
158. Eleutherio, E., Panek, A., De Mesquita, J.F., Trevisol, E., and Magalhães, R., Revisiting yeast trehalose metabolism, Curr. Genetics, 61:263–274, 2015.
159. Reinman, M. and Londesborough, J., Rapid mobilization of intracellular trehalose by fermentable sugars: A comparison of different strains, in Brewing Yeast Fermentation Performance, Smart, K., Ed., Blackwell Science, Oxford, pp. 20–26, 2000.
160. Boulton, C., Trehalose, glycogen and sterol, in Brewing Yeast Fermentation Performance, Smart, K., Ed., Blackwell Science, Oxford, pp. 10–19, 2000.
161. Trevisol, E.T., Panek, A.D., Mannarino, S.C., and Eleutherio, E.C., The effect of trehalose on the fermentation performance of aged cells of Saccharomyces cerevisiae, Appl. Microbiol. Biotechnol., 90:697–704, 2011.
162. Petitjean, M., Teste, M.A., François, J.M., and Parrou, J.L., Yeast tolerance to various stresses relies on the trehalose-6P synthase (Tps1) protein, not on trehalose, J.Biol. Chem., 290:16177–16190, 2015.
163. Walther, A., Hesselbart, A., and Wendland, J., Genome sequence of Saccharomyces carlsbergensis, the world’s first pure culture lager yeast, G3: Genes| Genomes| Genetics, 4:783–793, 2014.
164. Barnett, J.A., A history of research on yeasts 1: Work by chemists and biologists 1789–1850, Yeast, 14:1439–1451, 1998.
165. Barnett, J.A., A history of research on yeasts 2: Louis Pasteur and his contemporaries, 1850–1880, Yeast, 16:755–771, 2000.
166. Barnett, J.A. and Lichtenthaler, F.W., A history of research on yeasts 3: Emil Fischer, Eduard Buchner and their contemporaries, 1880–1900, Yeast, 18:363–388, 2001.
167. American Society of Brewing Chemists, Methods of analysis—Yeast 10. Differentiation of ale and lager yeast. (A) by X-α-Gal medium. (B) by growth at 37°C. (C) by growth on melibiose, in Methods of Analyses, 9th ed., The Society, St. Paul, MN, 2011.
168. American Society of Brewing Chemists, Yeast 9. Morphology of giant yeast colonies, in Methods of Analyses, 9th ed., The Society, St. Paul, MN, 2011.
169. EBC Analytica, Method 3.3.2.2 Morphology on WLN agar. Fachverlag Hans Carl, Nuremberg, 2011. Available from: http://www.analytica-ebc.com/ (December 15, 2016).
170. American Society of Brewing Chemists, Yeast 13. Differentiation of brewing yeast strains by PCR fingerprinting, in Methods of Analyses, 9th ed., ASBC, Ed., The Society, St. Paul, MN, 2011.
171. Pham, T., Wimalasena, T., Box, W.G., Koivuranta, K., Storgårds, E., Smart, K.A., and Gibson, B.R., Evaluation of ITS PCR and RFLP for differentiation and identification of brewing yeast and brewery ‘wild’ yeast contaminants, J. Instit. Brew., 117:556–568, 2011.
172. Michel, M., Meier‐Dörnberg, T., Jacob, F., Methner, F.J., Wagner, R.S., and Hutzler, M., Review: Pure non‐ Saccharomyces starter cultures for beer fermentation with a focus on secondary metabolites and practical applications, J. Instit. Brew., 122:569–587, 2016.
173. Schifferdecker, A.J., Dashko, S., Ishchuk, O.P., and Piškur, J., The wine and beer yeast Dekkera bruxellensis, Yeast, 31(9): 323–332, 2014. doi: 10.1002/yea.3023.
174. Kirsop, B.E., Maintenance of yeasts, in Maintenance of Microorganisms and Cultured Cells—A Manual of Laboratory Methods, 2nd ed., Kirsop, B.E. and Doyle, A., Eds., Academic Press, San Diego, CA, pp. 161–182, 1991.
175. Boundy‐Mills, K.L., Glantschnig, E., Roberts, I.N., Yurkov, A., Casaregola, S., Daniel, H.M., Groenewald, M., and Turchetti, B., Yeast culture collections in the twenty‐first century: New opportunities and challenges, Yeast, 33:243–260 , 2016.
176. Russell, I. and Stewart, G.G., Liquid nitrogen storage of yeast cultures compared to more traditional storage methods, J. Am. Soc. Brew. Chem., 39:19–24, 1981.
177. Kirsop, B., Yeast identification and maintenance, in Yeast Biotechnology, Berry, D.R., Russell, I., and Stewart, G.G., Eds., Unwin Hyman, London, pp. 1–32, 1987.
178. EBC Analytica, Method 3.4.1. Yeast Storage at Ultra–Low Temperatures, Fachverlag Hans Carl, Nuremberg, 2011. Available from: http://www.analytica-ebc.com/ (accessed December 15, 2016).
179. EBC Analytica, Method 3.4.2. Yeast Subculturing for Short-Term Storage, Fachverlag Hans Carl, Nuremberg, 2011. Available from: http://www.analytica-ebc.com/
180. EBC Analytica, Method 3.4.3. Yeast strain Transport. Fachverlag Hans Carl, Nuremberg, 2011. Available from: http://www.analytica-ebc.com/ (accessed December 15, 2016).
181. Edgerton, J., A primer on yeast propagation technique and procedures, Tech. Q. Master Brew. Assoc. Am., 38:167–175, 2001.
182. Cholerton, M., Yeast management under high-gravity brewing conditions, Tech. Q. Master Brew. Assoc. Am., 40:181–185, 2003.
183. Birle, S., Hussein, M.A. and Becker, T., On‐line yeast propagation process monitoring and control using an intelligent automatic control system. Eng. Life Sci., 15:83–95, 2015.
184. EBC Analytica, Method 3.4.4. Yeast Propagation (Laboratory Stages), Fachverlag Hans Carl, Nuremberg, 2011. Available from: http://www.analytica-ebc.com/ (accessed December 15, 2016).
185. Kurz, T., Mieleitner, J., Becker, T., and Delgado, A., A model based simulation of brewing yeast propagation, J. Inst. Brew., 108:248–255, 2002.
186. Suzuki, K., 125th Anniversary review: Microbiological instability of beer caused by spoilage bacteria, J. Instit. Brew., 117:131–155, 2011.
187. Bokulich, N.A., Bergsveinson, J., Ziola, B., and Mills, D.A., Mapping microbial ecosystems and spoilage-gene flow in breweries highlights patterns of contamination and resistance, Elife, 4:p.e04634, 2015. doi: 10.7554/eLife.04634.
188. Ashtavinayak, P. and Elizabeth, H.A., Review: Gram negative bacteria in brewing, Adv. Microbiol., 6:195 –209, 2016.
189. Priest, F.G. and Campbell, I., Eds., Brewing Microbiology, 3rd ed., Elsevier Applied Science, London, 399 p., 2003.
190. Hill, A., Ed., Brewing Microbiology: Managing Microbes, Ensuring Quality and Valorising Waste, Woodhead Publishing, Cambridge, 2015.
191. Bamforth, C.W., Ed., Beer: A Quality Perspective, Academic Press, Cambridge, MA, 304 p., 2011.
192. Back, W., Detection and identification of wild yeasts in the brewery, Brauwelt Int., II:174–177, 1987.
193. Simpson, W.J. and Hammond, J.R.M., The response of brewing yeasts to acid washing, J. Inst. Brew., 95:347–354, 1989.
194. Brenner, M.W., Disinfection of brewing yeast with acidified ammonium persulfate, J. Inst. Brew., 71:290, 1965.
195. Simpson, W.J., Kinetic studies of the decontamination of yeast slurries with phosphoric acid and acidified ammonium persulphate and a method for the detection of surviving bacteria involving solid medium repair in the presence of catalase, J. Inst. Brew., 93:313–318, 1987.
196. Cunningham, S. and Stewart, G., Acid washing and serial repitching a brewing ale strain of Saccharomyces cerevisiae in high gravity wort and the role of wort oxygenation conditions, J. Instit. Brew., 106:389–402, 2000.
197. Johnson, D., Coming clean: A new method of washing yeast with chlorine dioxide, New Brewer, September/October, 1998. Available from: http://www.birkocorp.com/brewery/white-papers/coming-clean-a-new-method-of-washing-yeast-with-chlorine-dioxide/ (accessed December 15, 2016).
198. White, C. and Zainasheff, J., Yeast growth, handling and storage, in Yeast- The Practical Guide to Beer Fermentation, Brewers Publications, Boulder, CO, p. 170, 2010.
199. American Society of Brewing Chemists, Yeast-3. Yeast stains, in Methods of Analyses, 9th ed., American Society of Brewing Chemists, St. Paul, MN, 2004.
200. EBC Analytica, Method 3.2.11. Methylene blue/violet stain, Fachverlag Hans Carl, Nuremberg, 2011. Available from: http://www.analytica-ebc.com/ (accessed December 15, 2016).
201. Layfield, J. B. and Sheppard, J.D., What brewers should know about viability, vitality, and overall brewing fitness: A mini-review, Tech. Q. Master Brew. Ass. Am., 52:132–140, 2015.
202. White, L.R., Richardson, K.E., Schiewe, A. J. and White, C.E., Comparison of yeast viability/vitality methods and their relationship to fermentation performance, in Brewing Yeast Fermentation Performance, 2nd ed., Smart, K., Ed., Blackwell Science, UK, pp. 38–147, 2008.
203. Heggart, H.M., Margaritis, A., Pilkington, H., Stewart, R.J., Dowhanick, T.M., and Russell, I., Factors affecting yeast viability and vitality characteristics, Tech. Q. Master Brew. Assoc. Am., 36:383–406, 1999.
204. Heggart, H., Margaritis, A., Stewart, R., Pilkington, H., Sobczak, J., and Russell, I., Measurement of yeast viability and vitality, Tech. Q. Master Brew. Assoc. Am., 37:409–430, 2000.
205. Schlee, C., Miedl, M., Leiper, K.A., and Stewart, G.G., The potential of confocal imaging for measuring physiological changes in brewer's yeast, J. Instit. Brew., 112:134–147, 2006.
206. Kwolek-Mirek, M. and Zadrag-Tecza, R. , Comparison of methods used for assessing the viability and vitality of yeast cells, FEMS Yeast Res., 14:1068–1079, 2014.
207. Cadena-Herrera, D., Esparza-De Lara, J.E., Ramírez-Ibañez, N.D., López-Morales, C.A., Pérez, N.O., Flores-Ortiz, L.F., and Medina-Rivero, E., Validation of three viable-cell counting methods: Manual, semi-automated, and automated, Biotechnol. Rep., 7:9–16, 2015.
208. Feizi, A., Zhang, Y., Greenbaum, A., Guziak, A., Luong, M., Chan, R.Y.L., Berg, B., Ozkan, H., Luo, W., Wu, M., and Wu, Y., Rapid, portable and cost-effective yeast cell viability and concentration analysis using lensfree on-chip microscopy and machine learning, Lab on a Chip, 16:4350–4358, 2016.
209. American Society of Brewing Chemists, Yeast-6. Yeast viability by slide culture, in Methods of Analyses, 9th ed., American Society of Brewing Chemists, ASBC, Ed., St. Paul, MN, 2004.
210. Russell, I., Yeast, in Brewing Materials and Processes: A Practical Approach to Beer Excellence, Bamforth, C.W., Ed., Elsevier, UK, pp. 77–96, 2016.
211. Sugihara, M., Imai, T., Muro, M., Yasuda, Y., Ogawa, Y., and Ohkochi, M., Application of flow cytometer to analysis of brewing yeast physiology, IBD Asia Pacific Conference, Hobart Tasmania, 2006. Available from: http://www.ibdasiapac.com.au/asia-pacific-activities/convention-proceedings/2006/program_tues.htm (accessed December 15, 2016).
212. Chlup, P.H., Wang, T., Lee, E.G., and Stewart, G.G., Assessment of the physiological states of yeast during high- and low-gravity wort fermentations determined by flow cytometry, Tech. Q. Master Brew. Assoc. Am., 44:286–295, 2007.
213. Weigert, C., Steffler, F., Kurz, T., Shellhammer, T.H., and Methner, F.J., Application of a short intracellular pH method to flow cytometry for determining Saccharomyces cerevisiae vitality, Appl. Environ. Microbiol., 75:5615–5620, 2009.
214. Carvell, J.P., Austin, G., Matthee, A., Van de Spiegle, K., Cunningham, S., and Harding, C., Developments in using off-line radio frequency impedance methods for measuring the viable cell concentration in the brewery, J. Am. Soc. Brew. Chem., 58:57–62, 2000.
215. Maule, D.R., Propagation and handling of pitching yeast, Brew. Dig., 55:36–40, 1980.
216. Casey, G.P., Hass, C.A., Hysert, D.W., and Ingledew, W.M., Effective transportation of brewer's yeast slurry, J. Instit. Brew., 89:393–396, 1983.