
KEY CONCEPTS
In recent years, evidence has accumulated in support of a role for neurodevelopmental insults in the etiopathogenesis of schizophrenia (Brown, 2011). The dramatic changes in brain structure and function from conception to birth underscore the particular vulnerability to insults during this stage of development with regard to both short- and long-term disease outcomes (Tau & Peterson, 2010). Hence, the determinants of fetal brain development—both genes and environmental factors—deserve consideration as potential risk factors for schizophrenia. Among many putative environmental risk factors that have been investigated in studies of schizophrenia, infection is generally deemed as one of the most plausible, because microbial pathogens are well-documented causes of congenital brain anomalies and behavioral disorders. For example, prenatal exposure to rubella, toxoplasmosis, herpes simplex virus type 2, and other infections are known causes of developmental disorders that include mental retardation, learning disabilities, sensorineural dysfunction, and several structural brain anomalies (Remington et al., 2006).
Studies of Prenatal Infection and Schizophrenia Based on Ecologic Data
The first clues that fetal infection plays a role in the origins of schizophrenia emerged from studies that utilized ecologic data. The term ecologic is used to refer to an event or events that are documented at the level of a population. In the case of a study of in utero infection and a disease outcome, the exposure in an ecologic-based study can be defined as the timing of an epidemic in a population. Among many potential infections, most ecologic studies of schizophrenia examined influenza because of its relatively high prevalence and excellent documentation in many populations, due to extensive surveillance efforts. These investigations assessed whether schizophrenia was more common if an influenza epidemic or epidemics coincided with particular periods of pregnancy in cases and noncases drawn from the population in which the epidemic or epidemics were documented. Two research designs were used in such studies.
The first design focused on a single epidemic, most commonly the 1957 type A2 influenza pandemic. The high morbidity of this epidemic, the circumscribed period of its peak incidence, and the availability of registry-based data on schizophrenia diagnoses among the offspring greatly expedited the ability of investigators to examine this question. The second design consisted of relating the longitudinal variations in influenza epidemics over intervals of many years to births of patients who later developed schizophrenia in the population. Although studies that employed the latter design generally made use of more sophisticated analytic approaches, the basic study aims of the two designs were the same, and both approaches allowed for the assessment of relationships between schizophrenia and the occurrences of epidemics at different periods of gestation.
These studies, conducted in different regions of the world—including Europe, Australia, Japan, and the United States—have been reviewed in a recent publication (Brown & Derkits, 2010). Initial studies yielded significant associations between influenza epidemics during all or part of the second trimester and schizophrenia (Mednick et al., 1988; O’Callaghan et al., 1991). Yet several attempts to replicate these findings in larger studies with more complete case ascertainment yielded negative findings (Erlenmeyer-Kimling et al., 1994; Susser et al., 1994). Ecologic studies of infections other than influenza have also been conducted. Associations between schizophrenia and epidemics of maternal respiratory viral infections (Watson et al., 1984; O’Callaghan et al., 1994), measles, varicella-zoster (Torrey, 1988), mumps (O’Callaghan et al., 1994), and polio (Suvisaari et al., 1999a; Cahill et al., 2002) have been reported.
Although these studies were novel, and the investigations offered approaches that were state-of-the-art at the time, there were a number of methodologic shortcomings that render interpretations difficult. First, studies based on epidemics in populations would undoubtedly misclassify individuals as being exposed who were in fact unexposed. Even during seasons with the highest incidence of influenza, such as the 1957 type A2 epidemic, the prevalence of this infection rarely rises above 30% of the population. Hence, studies that defined individuals as exposed to influenza based on whether they were in utero during the 1957 epidemic would have misclassified approximately 70% of the unexposed population. Influenza is not restricted to epidemic periods or even to the fall and winter months, and some epidemics recur within the same year, as evidenced by the H1N1 epidemic of 2009, which peaked both during the spring and the autumn and winter months. These would result in exposed subjects being incorrectly classified as unexposed. Of note is that none of the ecologic studies of infection and schizophrenia attempted to confirm infection in individual pregnancies.
A second limitation of these studies is that they assumed a full-term pregnancy. The significant number of pregnancies that do not extend to the expected date of birth and the increased rate of preterm and possibly also postterm birth in schizophrenia render estimates of the timing of exposure inaccurate. Each of these two limitations leads to misclassification of exposure. The occurrence of nondifferential misclassification of exposure, defined as no difference in misclassification between the cases and noncases, tends to bias effect sizes toward the null. This may explain some of the failures to replicate these findings.
Birth Cohort Studies of Prenatal Infection and Schizophrenia
To address these limitations, our group, as well as others, initiated birth cohort studies of prenatal infection and schizophrenia (Brown & Derkits, 2010). A birth cohort is a group of individuals born during a specified period and in a specified geographic region. In birth cohort studies, exposures are documented at birth or earlier, the subjects are followed up for an outcome of interest, and relationships between the exposures and the outcomes are examined. Birth cohorts of infection offer numerous advantages over ecologically based studies. First, the birth cohort studies allow for prospective documentation of prenatal infection in individual pregnancies. In some of these studies, this was achieved through obstetric and other medical records that were documented during pregnancy. Second, some existing birth cohorts provide a very valuable resource: archived biological maternal or infant specimens drawn during pregnancy or soon after delivery, which allow for documentation of infection during fetal or infant life using biomarkers such as antibodies to various infectious agents. Third, the longitudinal follow-up afforded by some birth cohort studies offers the potential for survival analytic methods aimed at addressing the effects of loss to follow-up. Fourth, birth cohort studies generally include access to information on a wide array of potential confounding or interacting influences, such as demographic variables, maternal medical conditions and health habits, other prenatal factors, and characteristics of the infant at birth.
In the case of birth cohort studies of infection and schizophrenia, our group and others documented infections by ascertainment of maternal antibody levels from archived serum specimens acquired in individual pregnancies (Brown et al., 2004a, 2005, 2006; Buka et al., 2001a, 2008) or in blood spots obtained from infants (Mortensen et al., 2007) and related these exposures to risk of schizophrenia following systematic assessment of the offspring later in life. Other studies relied on clinical diagnoses of infection documented during pregnancy (Brown & Derkits, 2010). These studies are reviewed in the sections that follow and the findings are summarized in tables 1.1 and 1.2.
Rubella
One of the first studies of prenatal infection and risk of schizophrenia documented through a birth cohort was based on the Rubella Birth Defects Evaluation Project (RBDEP), which was conducted in New York City following the 1964 rubella pandemic (Brown et al., 2000a, 2001). All mothers were documented as having been exposed to rubella during pregnancy, and offspring were serologically confirmed with infection. Our group demonstrated that exposure to rubella in utero was associated with a greater than fivefold increased risk of nonaffective psychosis (Brown et al., 2000a). In a follow-up study of the cohort in midadulthood, we demonstrated that more than 20% of subjects who were exposed in utero to rubella were diagnosed with schizophrenia or a schizophrenia spectrum disorder based on a structured research interview (Brown et al., 2001). Rubella-exposed subjects who developed schizophrenia had a substantially greater IQ decline than rubella-exposed controls, as well as greater premorbid neuromotor and behavioral anomalies.
Table 1.1 Serologic Studies of Prenatal Infection and Schizophrenia


a P ≤ 0.05 unless otherwise noted.
b Measures of effect (relative risks [RR], rate ratios, or odds ratios [OR]) are reported here, if reported in the original paper. CI = confidence intervals.
c “Nth month” indicates gestational month of exposure. Rather than estimating gestational age, some studies reported the number of months after the peak of an epidemic during which the number of babies born with schizophrenia increased. This distinction is noted in the table.
d All the studies in this table share the following strengths: direct biomarkers of infection obtained from archived maternal sera, population-based birth cohorts, diagnoses based on psychiatric interviews and medical chart reviews, and follow-up for psychosis or schizophrenia using specified protocols. The Child Health and Development Study featured two additional strengths: continuous follow-up and controls who were representative of the source population giving rise to cases.
e Child Health and Development Study (CHDS): population-based birth cohort (N = 12,094), Alameda County, California, born between 1959 and 1967, followed up for schizophrenia and other schizophrenia spectrum disorders (schizophrenia, schizoaffective disorder, schizotypal personality disorder, delusional disorder, and “other schizophrenia spectrum psychoses") in adulthood. Diagnoses were established using both psychiatric interview (Diagnostic Interview for Genetic Studies) and medical chart review. Two types of study designs were used. The first are cohort analytic designs in which classes of infections were defined on the basis of abstracted obstetric records; the second are nested case-control studies of archived maternal serum analyzed for antibodies to infectious exposures.
f Collaborative Perinatal Project (CPP): multisite study of population-based birth cohorts throughout the United States. The table includes three CPP population-based birth cohorts, from Boston, Massachusetts; Providence, Rhode Island; and Philadelphia, Pennsylvania, born between 1959 and 1967 (N = 25,025 from 19,471 pregnant women for combined cohorts) and followed up for psychotic disorders (schizophrenia, schizophreniform disorder, bipolar disorder with psychotic features, brief cohorts, from Boston, MA, Providence, RI, and Philadelphia, PA, born between 1959 and 1967 (N = 25,025 from 19,471 pregnant women for combined cohorts) and followed up for psychotic disorders (schizophrenia, schizophreniform disorder, bipolar disorder with psychotic features, brief psychosis, and psychosis not otherwise specified). Diagnoses were established using psychiatric interview (Structured Clinical Interview for DSM-IV), medical chart review, or both. All studies included herein are nested case-control investigations of archived maternal serum analyzed for antibodies to infectious exposures.
SOURCE: Reprinted from Brown and Derkits (2010) with permission from the American Journal of Psychiatry (copyright 2010). American Psychiatric Association.
Table 1.2 Other Birth Cohort Studies of Prenatal Infection and Schizophrenia
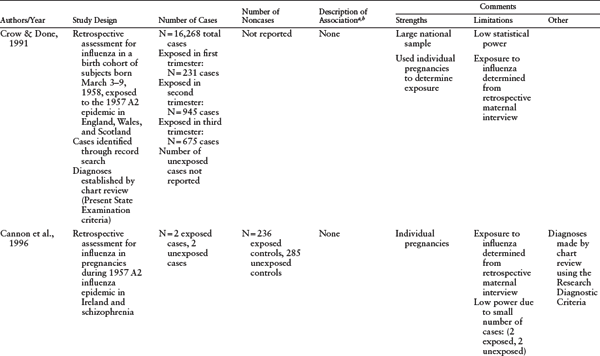
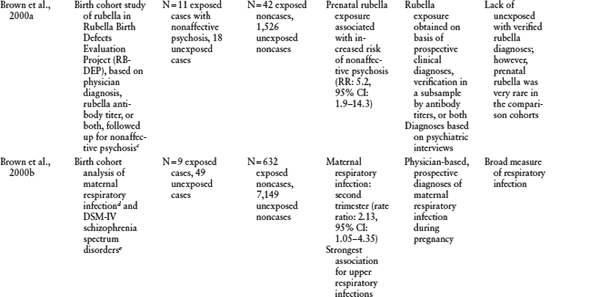




a P ≤ 0.05 unless otherwise noted.
b Measures of effect (relative risks [RR], rate ratios, or odds ratios [OR]) are reported here, if reported in the original paper. CI = confidence intervals.
c Nonaffective psychosis defined as (1) at least one psychotic symptom (delusions, hallucinations, or both) for a minimum of six months; (2) no evidence of a major affective disorder (bipolar or unipolar) by DSM-III-R criteria concurrent with the psychosis; and (3) no evidence that a medical condition or substance use initiated or maintained the psychosis. Diagnoses established using psychiatric interview (Diagnostic Interview Schedule for Children) and DSM-III-R criteria.
d Maternal respiratory infections included tuberculosis, influenza, influenza with pneumonia, bronchopneumonia, atypical pneumonia, pleurisy, emphysema/viral respiratory infections, acute bronchitis, and upper respiratory infections.
e Schizophrenia spectrum disorders included schizophrenia, schizoaffective disorder, schizotypal personality disorder, delusional disorder, and “other schizophrenia spectrum psychoses.” Diagnoses were established using both psychiatric interview (Diagnostic Interview for Genetic Studies) and medical chart review.
f Maternal genital-reproductive infection, including endometritis, cervicitis, pelvic inflammatory disease, vaginitis, syphilis, condylomata, “venereal disease,” or gonorrhea.
g Maternal bacterial infections include sinusitis, tonsillitis, pneumonia, cystitis, pyelonephritis, bacterial venereal infection, and any other bacterial infection.
SOURCE: Reprinted from Brown and Derkits (2010) with permission from the American Journal of Psychiatry (Copyright 2010). American Psychiatric Association
Influenza
In a second birth cohort, our group examined the relationship between serologically documented prenatal influenza infection and schizophrenia in the offspring (Brown et al., 2004a). This study was a follow-up of the Child Health and Development Study (CHDS), a population-based birth cohort born in Alameda County, California, between 1959 and 1967. Maternal sera were drawn prospectively during pregnancy, often at more than one time point, and stored frozen in a central repository. All of the mothers and offspring in this birth cohort were members of the Kaiser Permanente Medical Care Plan (KPMCP), a health maintenance organization that maintained computerized databases on all health visits by members, facilitating identification of offspring with schizophrenia, and diagnoses were confirmed by structured research interviews. In this study, the follow-up took place from 1996 to 1999, a minimum of nearly 30 years from the time of birth; hence, most of the cohort was followed up through the large majority of the period of risk for schizophrenia. Because the dates of membership were known throughout each subject’s time in KPMCP, it was possible to adjust for nondifferential loss to follow-up. An additional design advantage is that the membership dates of all the subjects made it possible to identify controls who represented the source population from which the cases were derived. This diminished the potential for bias from loss to follow-up.
For the serologic studies, a nested case-control design was used. In such a study, the cases and matched controls are “nested” within a defined birth cohort. Such a design is especially appropriate for large birth cohorts in which biospecimens are assayed and the costs for assays on the entire cohort are prohibitive.
In this study, our group demonstrated that exposure to influenza during the first half of pregnancy was associated with a threefold increase in risk of schizophrenia (Brown et al., 2004a). The risk of schizophrenia was increased sevenfold for influenza exposure during the first trimester. Influenza exposure during the second half of pregnancy was not related to schizophrenia. Although these findings warrant independent replication, they indicate that early to middle pregnancy may be a period of vulnerability for the development of schizophrenia. Unlike some of the other infections discussed, it is very unlikely that maternal influenza directly infects the fetus. This suggests that fetal infection is not required for an increase in schizophrenia risk.
TOXOPLASMA GONDII (T. GONDII). T. gondii is an intracellular parasite. Numerous studies have documented that this infectious exposure is a cause of central nervous system (CNS) congenital anomalies, and have led to recommendations to minimize exposure among pregnant mothers. Moreover, subtle, delayed neurologic sequelae of T. gondii have been observed (Dukes et al., 1997; Remington et al., 2006). In the CHDS cohort, prenatal levels of elevated maternal T. gondii IgG antibody (titers ≥ 1: 128) were associated with a greater than twofold increased risk of schizophrenia (Brown et al., 2005). In a subsequent study, from a sample in Denmark in which T. gondii IgG was assayed in filter paper blood spots from the infant taken within the first week of birth, elevated T. gondii IgG antibody was also associated with risk of schizophrenia (Mortensen et al., 2007). This finding is essentially a replication of the findings from the study in the CHDS cohort because T. gondii IgG antibody in maternal blood crosses the placenta and enters the fetal circulation, and it is very unlikely that infant exposure through a source other than the mother could have occurred in the first week of the infant’s life.
HERPES SIMPLEX VIRUS TYPE 2 (HSV-2). HSV-2 is a sexually transmitted disease, and transmission from mother to fetus usually occurs at the time of birth, when the fetus comes into contact with maternal cervicovaginal secretions of the virus, which is shed during primary exposure or reactivation. Like T. gondii, neonatal HSV-2 also causes congenital CNS anomalies, including adverse neuropsychiatric outcomes in childhood, such as mental retardation, low-normal IQ, language deficits, and motor disability (Kropp et al., 2006; Engman et al., 2008).
Previous findings from epidemiologic studies of prenatal HSV-2 and schizophrenia are conflicting. Two of these studies were based on selected sites of the Collaborative Perinatal Project (CPP), a multisite population-based study of a birth cohort born between 1959 and1967 (as in the CHDS). Similar to the CHDS, the cohort featured archived maternal serum samples that were drawn prospectively during pregnancy. An additional strength of the CPP study is that diagnoses were also confirmed by psychiatric interviews and medical record reviews, as in the CHDS follow-up study of schizophrenia.
The first of these studies revealed an association between maternal anti-HSV-2 IgG antibody levels and risk of psychotic disorders among offspring (Buka et al., 2001a). The risk of schizophrenia was increased threefold to fourfold, depending on the antibody cutoff definition for exposure. The investigators followed up this study with a larger sample made up of 200 subjects with a psychotic disorder and more than 500 matched control subjects from the CPP cohort. In that study, a 1.6-fold increase in risk of psychosis was observed among subjects seropositive for HSV-2, based on assays for HSV-2 antibody in the maternal sera. When schizophrenic psychoses were analyzed separately, HSV-2 was related to a 1.8-fold increase in risk (Buka et al., 2008). In the latter study, the finding was isolated to subjects who did not report regular use of contraception and who had frequent intercourse. These findings were not replicated, however, in the CHDS birth cohort study of 60 schizophrenia cases and 120 controls (Brown et al., 2006), with odds ratios close to 1 for all analyses.
Several methodologic explanations might be invoked to explain these discrepant findings. The greater proportion of African Americans in the CPP cohort may have resulted in increased statistical power, given that HSV-2 is more prevalent among African Americans (Buka et al., 2008), and statistical power may have been further enhanced by the larger number of cases with schizophrenic psychoses in the latter CPP study than in the CHDS study. It is therefore conceivable that the lack of association between prenatal HSV-2 and schizophrenia in the latter study may have resulted from a lack of statistical power.
A second issue to be considered regards variations in the way in which exposure status was defined. Different criteria for exposure were applied to the two CPP studies. In the first, exposure was defined on the basis of HSV-2 IgG antibody levels, whereas the primary analysis of the second study was based on HSV-2 seropositivity. The study from the CHDS cohort, which classified HSV-2 with regard to seropositivity and IgG antibody, did not detect an association by either definition of exposure.
A third consideration relates to quantification of loss to follow-up. In both CPP studies, there were no population denominators over the period of follow-up. Consequently, survival analytic methods aimed at mitigating bias from loss to follow-up could not be used in both CPP studies, in contrast to the CHDS study, which offered continuous follow-up of the cohort. If subjects with HSV-2 were more likely to remain in the CPP cohort than controls without HSV-2, a spurious finding may have resulted because of bias.
Other Prenatal Infections Identified from Birth Cohort Studies
Birth cohort studies have also indicated associations between maternal respiratory infection (Brown et al., 2000b), maternal genital-reproductive infection (Babulas et al., 2006), and bacterial infection (Sorensen et al., 2009) and schizophrenia. These studies extend the classes of infections that may increase schizophrenia risk further, suggesting that the type of infection may be less important than common consequences of infectious insults.
In summary, birth cohort studies have provided several key methodologic advantages that have allowed for more rigorous testing of relationships between maternal infection and schizophrenia. These advantages include prospective assessment of infection, maternal biomarkers of infection in individual pregnancies, longitudinal follow-up, and control for confounding influences. Birth cohort studies conducted to date have provided further support for the hypothesis that maternal viral, protozoal, and bacterial infections are related to schizophrenia in adult offspring.
Common Pathways that Mediate Relationships Between Infection and Risk of Schizophrenia
Cytokines
A key question posed by these findings concerns the mechanisms by which maternal infection might alter development of the brain and lead to schizophrenia. As discussed in chapter 10, animal models of maternal immune activation are shedding new light on these pathogenic mechanisms. In this parsimonious model, a wide array of infections is postulated to act through stimulation of the cytokine response (Gilmore & Jarskog, 1997) to alter brain development, resulting in behavioral, neurochemical, neurophysiologic, and neuroanatomic abnormalities similar to those observed in schizophrenia (Patterson, 2009). Cytokines are small proteins that act as the systemic mediators of the host response to infection. These immune factors become markedly elevated both systemically and within the target organs following primary infections, and they are also increased in a number of noninfectious inflammatory conditions (Weizman et al., 1999).
Evidence from clinical studies of neurodevelopmental disorders provides further support for this model (Dammann & Leviton, 1997). Maternal or fetal infection-induced cytokine elevations have been related to an increased occurrence of chorioamnionitis, periventricular leukomalacia, and cerebral palsy. Although the mechanisms are not clearly defined, one potential scenario includes cytokine stimulation of microglia and astroglia in the fetal brain to produce nitric oxide and excitatory amino acids, which are toxic to neurons. A second mechanism could involve disrupted maturation of oligodendrocytes contributing to white matter abnormalities, which have been previously associated with schizophrenia (Davis et al., 2003). A third mechanism involves direct activation of fetal neurons, as cytokines are used for signaling during normal fetal brain development and infection-induced increases can alter developmental processes (Meyer, Feldon, & Yee, 2008; Deverman & Patterson, 2009)). A fourth mechanism could involve activation of cytokine-responsive pathways in the placenta, with consequences for the fetus.
Consistent with a role for cytokines in infection as a risk factor for schizophrenia, the CHDS birth cohort follow-up study revealed that elevated levels of the proinflammatory chemokine interleukin-8 (IL-8) during the second or early third trimester were associated with an increased risk for schizophrenia by a factor of nearly two, in comparison to the risk for matched controls (Brown et al., 2004b). Elevated maternal serum IL-8 levels are also related to histologic chorioamnionitis in term infants (Shimoya et al., 1997). Moreover, maternal and fetal levels of IL-8 have been correlated with one another significantly. IL-8 may be particularly important for neutrophil attraction and for discharge of lysosomal enzymes from neutrophils, the latter of which leads to oxygen-free radicals. In the CPP follow-up study, maternal perinatal levels of tumor necrosis factor-α (TNF-α), another proinflammatory cytokine, were increased in subjects with psychotic disorders (Buka et al., 2001b). Like IL-8, TNF-α has also been associated with chorioamnionitis (Saji et al., 2000; Buka et al., 2001b), and elevated amniotic fluid levels of this cytokine have been related to maternal and fetal infection (Baud et al., 1999; Buka et al., 2001b). Intriguingly, polymorphisms in TNF-α genes have also been associated with schizophrenia (Boin et al., 2001; Buka et al., 2001b).
Individual Effects of Infections
In an alternative scenario, individual prenatal infections act to influence risk of schizophrenia by a variety of effects, rather than by a single common mediator. Infections that have been associated previously with schizophrenia differ significantly from one another with regard to several characteristics, such as duration of symptoms, antigenicity and antibody response, and gestational specificity to known congenital outcomes. In the paragraphs that follow, we consider putative unique mechanisms by which three of the maternal infections already discussed here—influenza, toxoplasmosis, and genital-reproductive infection—may act to increase schizophrenia risk.
INFLUENZA. Influenza is believed to cross the placenta only rarely and largely remains confined to the respiratory tract. Hence, induction of developmental brain changes is more likely to result from the response of the host organism to the infection, which in turn adversely influences fetal brain development, rather than through direct fetal infection. One previously proposed mechanism includes the elicitation of IgG antibodies to the infection and their cross-reaction with fetal brain antigens by molecular mimicry (Wright et al., 1999). Another potential mechanism by which influenza could increase risk of schizophrenia is via nonspecific effects, including hyperthermia, fetal hypoxia, and the use of over-the-counter remedies, such as aspirin.
As reviewed in chapter 10, preclinical studies have suggested that maternal influenza infection leads to neuropathologic alterations of particular relevance to schizophrenia.
TOXOPLASMA GONDII. T. gondii is a microbial pathogen that may exist in an active as well as a latent form. Following primary infection, the parasite may become sequestered as inactive oocysts within the brain and a number of other tissues and become reactivated over the life of the host. Hence, there may be different mechanisms by which T. gondii can elevate risk for schizophrenia. Although recommendations to protect against T. gondii are aimed at preventing acute infection, it is unlikely that active primary infection by this parasite is by itself sufficient to account for its association with schizophrenia, since the presence of maternal or neonatal anti-T. gondii IgM antibodies, a robust marker of recently acquired infection, was not observed in the two studies cited here (Brown et al., 2005; Mortensen et al., 2007). A second possible mechanism consists of reactivation of a previous T. gondii infection, leading to an inflammatory response in the developing fetal brain. This inflammatory response is expected to be accompanied by an increase in maternal anti-T. gondii IgG antibodies, although to our knowledge these assays have not been conducted in reactivated T. gondii infection. In a third scenario, elevated anti-T. gondii IgG antibodies—which exist for many years after infection, including the dormant phase—could cause neuropathology, because the IgG antibody is small enough to cross the placenta. It is worth noting in this regard that elevated total IgG antibody is teratogenic in some autoimmune disorders (Nadler et al., 1995).
GENITAL-REPRODUCTIVE INFECTION. Maternal genital-reproductive infections have the potential to disrupt fetal neurodevelopment by direct fetal invasion. These infections are known to infect the fetus by ascending the perineum, vagina, or cervix, and several have been associated with known congenital brain anomalies (Remington et al., 2006).
Implications
Preventive Approaches
One of the most important implications of research on prenatal infections in the etiology of schizophrenia is the potential for preventing this disorder. In this regard, the concept of population-attributable proportion (PAP) offers the potential to estimate the relative importance of preventive efforts. The PAP represents the proportion of cases in a population that would have been prevented if the risk factor was completely removed. This statistic is based on the magnitude of the effect on risk of the outcome and the prevalence of the risk factor in the population studied (Rothman & Greenland, 1998). A number of infections, including influenza, T. gondii, and genital-reproductive infections, are highly prevalent in the U.S. and global populations. Moreover, pregnant women are believed to be more vulnerable to these infections than nonpregnant women. We demonstrated that the PAP corresponding to these three maternal infections alone is approximately 30% (Brown & Derkits, 2010), suggesting that if preventive efforts removed these three infections from the pregnant population in our sample, then nearly one-third of schizophrenia cases in the population that we studied would have been eliminated. On the one hand, this may be an overestimate, because the true effect size could be smaller, and the prevalence of the exposure may vary in other populations. On the other hand, this figure does not take into account the documented risk associated with bacterial infection or the possible risks associated with other microbial infections. This figure nonetheless indicates that greater attention to efforts to prevent infection during pregnancy could play a significant role in reducing the risk of schizophrenia. Further, a more complete estimate of the PAP will need to await studies of the risk of schizophrenia conferred by additional infections, such as other viral and bacterial microbes. Confirming these associations would have the effect of increasing the PAP.
Infections are among the most preventable of all risk factors. Vaccination is one of the oldest, safest, and most effective approaches for preventing many infections, including some of those associated with schizophrenia. Influenza vaccination has prevented millions of cases and saved tens of thousands of lives. Because of a process known as antigenic drift, in which mutations occur in the strains of influenza, it is necessary to develop new vaccinations each year, requiring ongoing surveillance and vigilance on the part of public health administrators and experts (Kilbourne, 1987). Occasionally, the subtypes of influenza mutate as well, in a process known as antigenic shift, which resulted in the H1N1 “swine flu” influenza virus. However, technologic advances to identify the genomic and antigenic sequences of these viruses are hastening the development of new influenza vaccines.
Given the increased vulnerability of pregnant women to influenza, as well as the association between influenza during pregnancy and fetal morbidity and mortality (Zaman et al., 2008), pregnant women are considered to be a high-risk group that should be targeted for influenza vaccination. Clearly, this approach will reduce the adverse effects of influenza during pregnancy to both the mothers and offspring in a significant proportion of the pregnant population. It must also be kept in mind, however, that influenza vaccination will also trigger a cytokine response, which could be detrimental to fetal brain development (Dammann & Leviton, 1997). Although more research into the effects of influenza vaccination during pregnancy is necessary, one strategy that might address this issue is to target for vaccination nonpregnant women of reproductive age. Administering influenza vaccination to this subgroup will provide protection in the event of a pregnancy in the ensuing months, while minimizing the risk of fetal exposure to the release of cytokines during pregnancy, given that the cytokine response ends approximately one week following vaccination.
With regard to the prevention of genital-reproductive infections, there are two approaches that have long been in use: (1) antibiotics, which are highly effective against many bacterial genital-reproductive pathogens, including gonococcus and syphilis, and (2) barrier contraceptives to reduce the transmission of sexually transmitted diseases.
Approaches aimed at the prevention of T. gondii represent standard obstetric practices. Because T. gondii can be found in cat litter boxes, soil, and uncooked meats and fish, current recommendations during pregnancy are to avoid exposure to these sources of pathogens, to use proper hygienic measures, and to thoroughly cook meats, poultry, and fish to kill T. gondii oocysts. The fact that in the cited studies schizophrenia was related to elevated anti-T. gondii IgG antibodies (Brown et al., 2005; Mortensen et al., 2007) rather than to the parasite itself suggests that most infections were acquired before pregnancy. Hence, measures to reduce exposure to this infection may need to be initiated long before reproductive age.
In summary, the findings of associations between maternal infectious exposures and schizophrenia have important implications for the prevention of schizophrenia, given that maternal infection occurs commonly during pregnancy, and practical, effective, and relatively inexpensive approaches aimed at reducing or eliminating exposure to these pathogens are already available. Therefore, if the findings reviewed in the previous paragraphs can be replicated in independent cohorts in concert with efforts to identify new, relevant microbial pathogens, these studies may have significant public health implications for reducing the risk of schizophrenia in the global population. In fact, it has been argued previously that the overall decline in bacterial illnesses and initiation of immunization programs may be responsible for the reduction in the incidence of schizophrenia beginning in the 1950s (Suvisaari et al., 1999b).
Relationship Between Infection and Genetic Vulnerability
As discussed in chapters 7 and 8, family, twin, and adoption studies have established a role for genetic vulnerability in the etiology of schizophrenia, and association studies have revealed evidence for individual susceptibility genes and a greater occurrence of copy number variants in this disorder. Given the relatively small effect sizes of individual susceptibility alleles on schizophrenia risk, and the approximately 40–50% discordance rate of schizophrenia in monozygotic twins, it appears likely that a substantial portion of the variance in risk will not be explained solely by genetic effects. Rather, it is now increasingly recognized that most complex disorders, including psychiatric ones, result from an interplay between genetic mutations and environmental exposures (Caspi et al., 2010). This interplay may take several forms, including gene-environment interaction and epigenetic effects, which may include environmental effects on gene expression (see chapter 9, this book). We focus here on gene-environment interaction.
Gene-Environment Interaction
There is increasing evidence that gene-environment interaction may play a significant role in schizophrenia and other neuropsychiatric disorders. Gene-environment interaction is defined as an increase in the effect of an environmental exposure when a susceptibility gene variant is present.
In a recent investigation of a Finnish cohort, prenatal exposure to pyelonephritis had a fivefold greater effect on risk of schizophrenia among subjects with a family history of psychosis than in those with no family history (Clarke et al., 2009). However, there has yet to be a study of interactions between individual susceptibility gene variants and prenatal infections in schizophrenia. In the paragraphs that follow, I discuss strategies that may lead toward the detection of such genes.
GENES THAT PLAY A ROLE IN NEURODEVELOPMENT. Genes involved in neurodevelopmental processes, including those that occur in utero, represent logical candidates to investigate in studies of interactions with prenatal infections. As discussed in more detail in chapters 14 and 15, some of the most important effects of Disrupted-in-Schizophrenia 1 (DISC1) and neuregulin, two leading susceptibility gene candidates, occur during the prenatal period. Intriguingly, there are a number of compelling similarities between the effects of mutations in these two genes and those of maternal immune activation (Brown & Derkits, 2010).
GENES AND THE IMMUNE RESPONSE. Genes that regulate immune function represent a second class of logical candidates to investigate in studies of gene-infection interactions. The Major Histocompatibility Complex (MHC) Class I proteins, which present antigens to T lymphocytes at the cell surface, are of particular interest in this regard (Boulanger, 2009). In addition to their role in immune regulation, these proteins are critical for brain development and function. These molecules are enriched at the synapse, are essential for normal synaptic function and for synaptic remodeling, and play an especially important role in graded fine-tuning of plasticity. It is also relevant that the expression of MHC Class I proteins is regulated by cytokines (Boulanger & Shatz, 2004). In three of the largest genomewide association studies of schizophrenia, one of the few results to meet statistical significance was that of genetic variants in the extended MHC (Purcell et al., 2009; Shi et al., 2009; Stefansson et al., 2009) (see chapter 7, this book). This finding is particularly striking given that this approach was not hypothesis driven.
GENE IDENTIFICATION. Recent findings from molecular genetic studies of schizophrenia (chapter 7, this book) and from animal models based on mutations of putative susceptibility genes (chapters 14 and 15, this book) offer the promise of discovering the true origins of schizophrenia. Yet, if gene-environment interaction plays a significant role in schizophrenia, strategies that are focused only on the search for genes may be limited in their ability to identify definitive etiologies in the majority of cases. For example, if certain genes produce their effects on the risk of schizophrenia only in the presence of in utero infection, it may be difficult to detect the effects of these genes on risk of the disorder if infection is not measured in these cases. Hence, one strategy that may facilitate gene identification in this scenario is to scan the genome in a subsample of cases whose subjects have been exposed to maternal infection.
This concept also has implications for prevention. If maternal infection and a gene are each necessary but not individually sufficient causes of a hypothetical subset of schizophrenia cases, then it may not be necessary to modify the genetic defect in order to prevent the disorder in this subset of cases. Instead, a much more practical and feasible approach would be the elimination of the environmental factor, such as maternal infection, for which well-known strategies for treatment and prevention exist (see “Implications").
Research on epigenetics and schizophrenia is covered in detail in chapter 9. In this context, an epigenetic approach might involve examining the effects of maternal infection on gene expression. This can occur through different processes, including DNA methylation and histone modifications. These changes represent a mechanism that may lead to lifelong changes in gene expression. In a seminal work on this question, Smith et al. (2007) demonstrated that maternal immune activation in mice leads to dysregulation of gene expression, with overexpression and underexpression of particular subsets of genes that persist in the adult offspring, and virtually all of these changes are mediated by the cytokine interleukin-6 (see chapter 10, this book). These findings suggest that effects of maternal infection might be mediated in part by epigenetic changes in the brain, and that cytokines may play an essential role in infection-induced epigenesis. Although this field is still in its infancy, with no known human studies of infection and epigenetically induced modifications, future birth cohort studies may, at least in theory, have the capacity to test these associations by relating well-documented prenatal infections to epigenetic alterations in offspring with schizophrenia.
Recommendations for Future Epidemiologic Studies of Prenatal Infection and Schizophrenia
Challenges of Previous Research Efforts
In spite of the intriguing findings generated to date from seroepidemiologic studies of prenatal infection and schizophrenia, there have been few attempts to replicate most of these results, and there are numerous additional infectious and immune candidates that have not yet been investigated. This is not surprising given the dearth of opportunities to address these hypotheses. The aforementioned birth cohort studies represent major methodologic advances, including archived prenatal biospecimens, rich data sets to address potential confounders, and follow-up into the age of risk for schizophrenia. Yet, there were also certain limitations to these studies.
First, the sample sizes were small to modest. This may have had several consequences. Statistical power to detect effects of certain exposure variables was somewhat compromised. This may have accounted for the lack of association between some infectious and immune measures and risk of schizophrenia in these studies. A second consequence is that the precision of the observed effects was also less than ideal, as manifested by relatively wide confidence intervals. Third, acute and less common infections were not investigated. It is conceivable that there may be a substantial number of such exposures that in total make an important contribution to the risk of schizophrenia. Fourth, the sample sizes from these studies generally were too small to allow for adequate statistical power to test for gene-environment interaction, which generally requires large sample sizes. Fifth, sample sizes in previous studies also were insufficient to test for interactive and mediating effects of other exposures; for example, power was not adequate to detect maternal cytokines as mediators of the effects of maternal infection on risk of schizophrenia. There are also numerous potential factors, such as sex and race, that may interact with maternal infection to increase schizophrenia risk. Larger sample sizes will be necessary to test for these and other interactive effects. In recent years, new birth cohorts with much larger samples have been established. These include the Finnish Prenatal Studies (FiPS), the Danish National Longitudinal Study (DNLS) (Olsen et al., 2001), and the Mother and Baby Study (MoBA) in Norway (Magnus et al., 2006). The number of births in these cohorts ranges from approximately 100,000 (DNLS, MoBA) to 1.5 million (FiPS).
A second challenge relates to the measurement of infection. As discussed previously, most prior studies assigned exposure status on the basis of whether subjects were in utero at the time of an infection in the population or on records indicating that maternal infection occurred. Studies that classified infection status on the basis of maternal or infant sera frequently used IgG antibody titers rather than direct markers of acute infection. As already noted here, IgG antibody provides evidence that infection occurred in the individual in whom it is measured, but it does not localize the infection to a specified period. It would be preferable to utilize antibody markers, such as IgM, which indicate that infection occurred soon before the time during which the blood sample was drawn; this would allow infection to be diagnosed during pregnancy or infancy. However, acute infection during pregnancy is more difficult to detect than chronic or previous infection, as the corresponding antibody markers are elevated only transiently in active infection. This issue can be surmounted by use of larger sample sizes, which will increase the likelihood of identifying a sufficient number of pregnancies in which the time window of the infection will coincide with the time during which the prenatal serum specimen was drawn.
A third challenge concerns potential biasing effects. First, incomplete ascertainment of cases was a frequent limitation of some cohort studies. In previous birth cohort studies of prenatal infection and schizophrenia, it is likely that a substantial number of cases were missed because of the fact that most ascertainment strategies are based on recruitment of cases from a selected number of facilities rather than from all facilities from which care is received. In this regard, national birth cohorts from countries with universal health insurance—coupled with comprehensive, centralized registries—offer the advantage of more complete case ascertainment. A second issue that can compromise both case and control ascertainment is loss to follow-up, which is more likely to occur when follow-up is conducted over long intervals, such as in disorders with long latency periods, including schizophrenia. Third, nonrepresentative control samples can also contribute to bias. This can occur, for example, when controls are selected from the original birth cohort rather than from the population that would have been identified as cases if they developed schizophrenia. A powerful approach toward addressing these issues is to utilize population registries with systematically collected longitudinal data, including migration.
A fourth limitation is the fact that few studies have adequately examined the specificity of maternal infection for schizophrenia. It is conceivable that this risk factor may be involved in the etiology of a wide range of disorders.
Finally, previous studies of maternal infection and schizophrenia did not consider phenotypes of schizophrenia beyond the presence or absence of diagnosis, which is based only on symptom criteria. In an initial attempt to address this question, we assessed relationships between maternal exposure to influenza and anti-toxoplasma IgG and neuropsychological function in offspring with schizophrenia in the Developmental Insult and Brain Anomaly in Schizophrenia Study (DIBS), which is based on schizophrenia cases in the CHDS birth cohort. We found that maternal exposure to these infections was significantly associated with anomalies in measures of executive function, including the Wisconsin Card Sort Test and the Trails B test, both of which are also specific to cognitive set-shifting (Brown et al., 2009a). These findings suggest that maternal exposure to infection may account for some portion of the neuropsychological disruption in schizophrenia, analogous to findings on genetic variants and neurocognitive anomalies in this disorder (Egan et al., 2001; Hennah et al., 2005). In a second DIBS study, we demonstrated that maternal infection was related to substantially increased volume of the cavum septum pellucidum (CSP), a neuroembryologic anomaly that arises during the prenatal or early postnatal period (Brown et al., 2009b). This provides biologically based evidence in a human study that indicates that prenatal infection disrupts the closure of the CSP, an anomaly that has been repeatedly observed in patients with schizophrenia (Brown et al., 2009a).
Infection and Molecular Genetic Studies
First, as implied from the preceding section, we suggest that future molecular genetic studies, including genomewide association studies, should incorporate prospectively documented, specific, and reliably measured maternal infectious exposures. As already noted here, such studies have the potential to facilitate the identification of susceptibility genes by enriching the sample for maternal infectious exposures.
Translational Studies
As discussed in chapter 10, translational approaches involving maternal immune activation models have helped validate the epidemiologic findings by demonstrating brain and behavioral phenotypes that are consistent with those observed in schizophrenia. These studies are also beginning to suggest potential causal mechanisms by which maternal infection alters brain development and function, most notably by the increase of specific cytokines. Increased interdisciplinary efforts between epidemiologists and molecular and cellular neuroscientists should enhance this work.
Critical Periods of Vulnerability and Developmental Trajectories
As illustrated by this review, the gestational timing of infection appears to be a critical factor that influences vulnerability to schizophrenia. Although initial emphasis was placed on the second trimester as a period of susceptibility, this notion was largely based on ecologic studies. Birth cohort studies, which featured more refined methodologic approaches, have since suggested that, at least for certain infections, exposure earlier in pregnancy, including periconception and the first trimester, may also represent important periods of susceptibility. Hence, it would be of benefit to devote more attention to “windows” of vulnerability to infectious insults. Our understanding of the developmental mechanisms by which these insults increase risk for schizophrenia could also be facilitated by concurrent documentation of the developmental trajectories that potentially mediate the effects of infection on risk of schizophrenia. For this purpose, longitudinal studies on early and later manifestations of schizophrenia in subjects with in utero exposure data who are followed into the age of risk for schizophrenia could begin to address these questions.
Conclusions
In the past two decades, epidemiologic studies of maternal infection and schizophrenia have evolved from work based on ecologic data to sophisticated, longitudinal birth cohort designs that have documented maternal infection by analysis of biomarkers. Most birth cohort studies that have utilized this resource have provided evidence consistent with maternal infection having a role in the etiology of schizophrenia. Although these findings are intriguing, considerable work remains to replicate them and to generate further evidence of a causal association. The study of maternal infection and of other environmental exposures may facilitate the identification of susceptibility genes that interact with these exposures.
These studies offer the potential for the prevention of schizophrenia, which for many infectious exposures can be accomplished through greater implementation of relatively inexpensive and standard interventions, including vaccination, antibiotics, and improved hygiene. Moreover, coupled with translational studies of animal models of infection, this work may elucidate pathogenic mechanisms by which prenatal infection might lead to schizophrenia.
KEY AREAS FOR FUTURE RESEARCH
Acknowledgments
The author wishes to acknowledge National Institute of Mental Health grant 1K02-MH65422 for support of this work, Paul Patterson for his valuable suggestions, and Patric Prado for technical assistance.
Selected Readings
Brown, A. S., Begg, M. D., Gravenstein, S., Schaefer, C. A., Wyatt, R. J., Bresnahan, M. A., Babulas, V., & Susser, E. (2004). Serologic evidence for prenatal influenza in the etiology of schizophrenia. Archives of General Psychiatry 61(8): 774–780.
Brown, A. S. & Derkits, E. J. (2010). Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. American Journal of Psychiatry 167(3): 261–280.
Buka, S. L., Cannon, T. D., Torrey, E. F., & Yolken, R. H. (2008). Maternal exposure to herpes simplex virus and risk of psychosis among adult offspring. Biological Psychiatry 63(8): 809–815.
Meyer, U., Feldon, J., & Yee, B. K. (2008). A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophrenia Bulletin 35(5): 959–972.
Patterson, P. H. (2009). Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behavioural Brain Research 204(2): 313–321.
Smith, S. E., Li, J., Garbett, K., Mirnics, K., & Patterson, P. H. (2007). Maternal immune activation alters fetal brain development through interleukin-6. Journal of Neuroscience 27(40): 10695–10702.
References
Babulas, V., Factor-Litvak, P., Goetz, R., Schaefer, C. A., & Brown, A. S. (2006). Prenatal exposure to maternal genital and reproductive infections and adult schizophrenia. American Journal of Psychiatry 163(5): 927–929.
Baud, O., Emilie, D., Pelletier, E., Lacaze-Masmonteil, T., Zupan, V., Fernandez, H., Dehan, M., Frydman, R., & Ville, Y. (1999). Amniotic fluid concentrations of interleukin-1beta, interleukin-6 and TNF-alpha in chorioamnionitis before 32 weeks of gestation: Histological associations and neonatal outcome. British Journal of Obstetric Gynaecology 106(1): 72–77.
Boin, F., Zanardini, R., Pioli, R., Altamura, C. A., Maes, M., & Gennarelli, M. (2001). Association between -G308A tumor necrosis factor alpha gene polymorphism and schizophrenia. Molecular Psychiatry 6(1): 79–82.
Boulanger, L. M. (2009). Immune proteins in brain development and synaptic plasticity. Neuron, 64(1): 93–109.
Boulanger, L. M. & Shatz, C. J. (2004). Immune signalling in neural development, synaptic plasticity and disease. Nature Reviews Neuroscience 5(7): 521–531.
Brown, A. S. (2011). The environment and susceptibility to schizophrenia. Progress in Neurobiology 93(1): 23–58.
Brown, A. S., Begg, M. D., Gravenstein, S., Schaefer, C. A., Wyatt, R. J., Bresnahan, M. A., Babulas, V., & Susser, E. (2004a). Serologic evidence for prenatal influenza in the etiology of schizophrenia. Archives of General Psychiatry 61(8): 774–780.
Brown, A. S., Cohen, P., Greenwald, S., & Susser, E. (2000a). Nonaffective psychosis after prenatal exposure to rubella. American Journal of Psychiatry 157(3): 438–443.
Brown, A. S., Cohen, P., Harkavy-Friedman, J., Babulas, V., Malaspina, D., Gorman, J. M., & Susser, E. S. (2001). A. E. Bennett Research Award. Prenatal rubella, premorbid abnormalities, and adult schizophrenia. Biological Psychiatry 49(6): 473–486.
Brown, A. S., Deicken, R. F., Vinogradov, S., Kremen, W. S., Poole, J. H., Penner, J. D., Kochetkova, A., Kern, D., & Schaefer, C. A. (2009a). Prenatal infection and cavum septum pellucidum in adult schizophrenia. Schizophrenia Research 108(1–3): 285–287.
Brown, A. S. & Derkits, E. J. (2010). Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. American Journal of Psychiatry 167(3): 261–280.
Brown, A. S., Hooton, J., Schaefer, C. A., Zhang, H., Petkova, E., Babulas, V., Perrin, M., Gorman, J. M., & Susser, E. S. (2004b). Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. American Journal of Psychiatry 161(5): 889–895.
Brown, A. S., Schaefer, C. A., Quesenberry, C. P., Jr., Liu, L., Babulas, V. P., & Susser, E. S. (2005). Maternal exposure to toxoplasmosis and risk of schizophrenia in adult offspring. American Journal of Psychiatry 162(4): 767–773.
Brown, A. S., Schaefer, C. A., Quesenberry, C. P., Jr., Shen, L., & Susser, E. S. (2006). No evidence of relation between maternal exposure to herpes simplex virus type 2 and risk of schizophrenia. American Journal of Psychiatry 163(12): 2178–2180.
Brown, A. S., Schaefer, C. A., Wyatt, R. J., Goetz, R., Begg, M. D., Gorman, J. M., & Susser, E. S. (2000b). Maternal exposure to respiratory infections and adult schizophrenia spectrum disorders: A prospective birth cohort study. Schizophrenia Bulletin 26(2): 287–295.
Brown, A. S., Vinogradov, S., Kremen, W. S., Poole, J. H., Deicken, R. F., Penner, J. D., McKeague, I. W., Kochetkova, A., Kern, D., & Schaefer, C. A. (2009b). Prenatal exposure to maternal infection and executive dysfunction in adult schizophrenia. American Journal of Psychiatry 166(6): 683–690.
Buka, S. L., Cannon, T. D., Torrey, E. F., & Yolken, R. H. (2008). Maternal exposure to herpes simplex virus and risk of psychosis among adult offspring. Biological Psychiatry 63(8): 809–815.
Buka, S. L., Tsuang, M. T., Torrey, E. F., Klebanoff, M. A., Bernstein, D., & Yolken, R. H. (2001a). Maternal infections and subsequent psychosis among offspring. Archives of General Psychiatry 58(11): 1032–1037.
Buka, S. L., Tsuang, M. T., Torrey, E. F., Klebanoff, M. A., Wagner, R. L., & Yolken, R. H. (2001b). Maternal cytokine levels during pregnancy and adult psychosis. Brain, Behavior, and Immunology 15(4): 411–420.
Cahill, M., Chant, D., Welham, J., & McGrath, J. (2002). No significant association between prenatal exposure poliovirus epidemics and psychosis. Australian and New Zealand Journal of Psychiatry 36(3): 373–375.
Cannon, M., Cotter, D., Coffey, V. P., Sham, P. C., Takei, N., Larkin, C., Murray, R. M., & O’Callaghan, E. (1996). Prenatal exposure to the 1957 influenza epidemic and adult schizophrenia: A follow-up study. British Journal of Psychiatry 168(3): 368–371.
Caspi, A., Hariri, A. R., Holmes, A., Uher, R., & Moffitt, T. E. (2010). Genetic sensitivity to the environment: The case of the serotonin transporter gene and its implications for studying complex diseases and traits. American Journal of Psychiatry 167(5): 509–527.
Clarke, M. C., Tanskanen, A., Huttunen, M., Whittaker, J. C., & Cannon, M. (2009). Evidence for an interaction between familial liability and prenatal exposure to infection in the causation of schizophrenia. American Journal of Psychiatry 166(9): 1025–1030.
Crow, T. J. & Done, D. J. (1991). Schizophrenia and influenza. Lancet 338(8759): 116–117.
Dammann, O. & Leviton, A. (1997). Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatric Research 42(1): 1–8.
Davis, K. L., Stewart, D. G., Friedman, J. I., Buchsbaum, M., Harvey, P. D., Hof, P. R., Buxbaum, J., & Haroutunian, V. (2003). White matter changes in schizophrenia: Evidence for myelin-related dysfunction. Archives of General Psychiatry 60(5): 443–456.
Deverman, B. E. & Patterson, P. H. (2009). Cytokines and CNS development. Neuron 64(1): 61–78.
Dukes, C. S., Luft, B. J., Durack, D. T., Scheld, W. M., & Whitley, R. J. (1997). Toxoplasmosis Infections of the Central Nervous System (vol. 2, pp. 785–806). Philadelphia: Lippincott-Raven.
Egan, M. F., Goldberg, T. E., Kolachana, B. S., Callicott, J. H., Mazzanti, C. M., Straub, R. E., Goldman, D., & Weinberger, D. R. (2001). Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proceedings of the National Academy of Sciences U.S.A. 98(12): 6917–6922.
Engman, M. L., Adolfsson, I., Lewensohn-Fuchs, I., Forsgren, M., Mosskin, M., & Malm, G. (2008). Neuropsychologic outcomes in children with neonatal herpes encephalitis. Pediatric Neurology 38(6): 398–405.
Erlenmeyer-Kimling, L., Folnegovic, Z., Hrabak-Zerjavic, V., Borcic, B., Folnegovic-Smalc, V., & Susser, E. (1994). Schizophrenia and prenatal exposure to the 1957 A2 influenza epidemic in Croatia. American Journal of Psychiatry 151(10): 1496–1498.
Gilmore, J. H. & Jarskog, L. F. (1997). Exposure to infection and brain development: Cytokines in the pathogenesis of schizophrenia. Schizophrenia Research 24(3): 365–367.
Hennah, W., Tuulio-Henriksson, A., Paunio, T., Ekelund, J., Varilo, T., Partonen, T., Cannon, T. D., Lonnqvist, J., & Peltonen, L. (2005). A haplotype within the DISC1 gene is associated with visual memory functions in families with a high density of schizophrenia. Molecular Psychiatry 10(12): 1097–1103.
Kilbourne, E. D. (1987). Influenza. New York: Plenum Medical.
Kropp, R. Y., Wong, T., Cormier, L., Ringrose, A., Burton, S., Embree, J. E., & Steben, M. (2006). Neonatal herpes simplex virus infections in Canada: Results of a 3-year national prospective study. Pediatrics 117(6): 1955–1962.
Magnus, P., Irgens, L. M., Haug, K., Nystad, W., Skjaerven, R., & Stoltenberg, C. (2006). Cohort profile: The Norwegian Mother and Child Cohort Study (MoBa). International Journal of Epidemiology 35(5): 1146–1150.
Mednick, S. A., Machon, R. A., Huttunen, M. O., & Bonett, D. (1988). Adult schizophrenia following prenatal exposure to an influenza epidemic. Archives of General Psychiatry 45(2): 189–192.
Meyer, U., Feldon, J., & Yee, B. K. (2008). A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophrenia Bulletin 35(5): 959–972.
Mortensen, P. B., Norgaard-Pedersen, B., Waltoft, B. L., Sorensen, T. L., Hougaard, D., Torrey, E. F., & Yolken, R. H. (2007). Toxoplasma gondii as a risk factor for early-onset schizophrenia: Analysis of filter paper blood samples obtained at birth. Biological Psychiatry 61(5): 688–693.
Nadler, D. M., Klein, N. W., Aramli, L. A., Chambers, B. J., Mayes, M., & Wener, M. H. (1995). The direct embryotoxicity of immunoglobulin-G fractions from patients with systemic lupus-erythematosus. American Journal of Reproductive Immunology 34(6): 349–355.
O’Callaghan, E., Gibson, T., Colohan, H. A., Walshe, D., Buckley, P., Larkin, C., & Waddington, J. L. (1991). Season of birth in schizophrenia. Evidence for confinement of an excess of winter births to patients without a family history of mental disorder. British Journal of Psychiatry 158(6): 764–769.
O’Callaghan, E., Sham, P. C., Takei, N., Murray, G., Glover, G., Hare, E. H., & Murray, R. M. (1994). The relationship of schizophrenic births to 16 infectious diseases. British Journal of Psychiatry 165(3): 353–356.
Olsen, J., Melbye, M., Olsen, S. F., Sorensen, T. I., Aaby, P., Andersen, A. M., Taxbol, D., Hansen, K. D., Juhl, M., Schow, T. B., et al. (2001). The Danish National Birth Cohort—Its background, structure and aim. Scandavian Journal of Public Health 29(4): 300–307.
Patterson, P. H. (2009). Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behavioural Brain Research 204(2): 313–321.
Purcell, S. M., Wray, N. R., Stone, J. L., Visscher, P. M., O’Donovan, M. C., Sullivan, P. F., & Sklar, P. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460(7256): 748–752.
Remington, J. S., Klein, J. O., Wilson, C. B., & Baker, C. J. (2006). Infectious Diseases of the Fetus and Newborn Infant (6th ed.). Philadelphia: Elsevier Saunders.
Rothman, K. J. & Greenland, S. (1998). Modern Epidemiology. Philadelphia: Lippincott Williams & Wilkins.
Saji, F., Samejima, Y., Kamiura, S., Sawai, K., Shimoya, K., & Kimura, T. (2000). Cytokine production in chorioamnionitis. Journal of Reproductive Immunology 47(2): 185–196.
Shi, J., Levinson, D. F., Duan, J., Sanders, A. R., Zheng, Y., Pe’er, I., Dudbridge, F., Holmans, P. A., Whittemore, A. S., Mowry, B. J., et al. (2009). Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460(7256): 753–757.
Shimoya, K., Matsuzaki, N., Taniguchi, T., Okada, T., Saji, F., & Murata, Y. (1997). Interleukin-8 level in maternal serum as a marker for screening of histological chorioamnionitis at term. International Journal of Gynaecology and Obstetrics 57(2): 153–159.
Smith, S. E., Li, J., Garbett, K., Mirnics, K., & Patterson, P. H. (2007). Maternal immune activation alters fetal brain development through interleukin-6. Journal of Neuroscience 27(40): 10695–10702.
Sorensen, H. J., Mortensen, E. L., Reinisch, J. M., & Mednick, S. A. (2009). Association between prenatal exposure to bacterial infection and risk of schizophrenia. Schizophrenia Bulletin 35(3): 631–637.
Stefansson, H., Ophoff, R. A., Steinberg, S., Andreassen, O. A., Cichon, S., Rujescu, D., Werge, T., Pietilainen, O. P., Mors, O., Mortensen, P. B., et al. (2009). Common variants conferring risk of schizophrenia. Nature 460(7256): 744–747.
Susser, E., Lin, S. P., Brown, A. S., Lumey, L. H., & Erlenmeyer-Kimling, L. (1994). No relation between risk of schizophrenia and prenatal exposure to influenza in Holland. American Journal of Psychiatry 151(6): 922–924.
Suvisaari, J., Haukka, J., Tanskanen, A., Hovi, T., & Lonnqvist, J. (1999a). Association between prenatal exposure to poliovirus infection and adult schizophrenia. American Journal of Psychiatry 156(7): 1100–1102.
Suvisaari, J. M., Haukka, J. K., Tanskanen, A. J., & Lonnqvist, J. K. (1999b). Decline in the incidence of schizophrenia in Finnish cohorts born from 1954 to 1965. Archives of General Psychiatry 56(8): 733–740.
Tau, G. Z. & Peterson, B. S. (2010). Normal development of brain circuits. Neuropsychopharmacology 35(1): 147–168.
Torrey, E. F. (1988). Stalking the schizovirus. Schizophrenia Bulletin 14(2): 223–229.
Watson, C. G., Kucala, T., Tilleskjor, C., & Jacobs, L. (1984). Schizophrenic birth seasonality in relation to the incidence of infectious diseases and temperature extremes. Archives of General Psychiatry 41(1): 85–90.
Weizman, R., Bessler, H., Plotnikoff, N. P., Faith, R. E., Murgo, A. J., & Good, R. A. (1999). Cytokines: Stress and Immunity. Boca Raton, FL: CRC Press.
Wright, I. C., Sharma, T., Ellison, Z. R., McGuire, P. K., Friston, K. J., Brammer, M. J., Murray, R. M., & Bullmore, E. T. (1999). Supra-regional brain systems and the neuropathology of schizophrenia. Cerebral Cortex 9(4): 366–378.
Zaman, K., Roy, E., Arifeen, S. E., Rahman, M., Raqib, R., Wilson, E., Omer, S. B., Shahid, N. S., Breiman, R. F., & Steinhoff, M. C. (2008). Effectiveness of maternal influenza immunization in mothers and infants. New England Journal of Medicine 359(15): 1555–1564.