Viral Genetics of Chikungunya Virus and Zika Virus and Its Influence in Their Emergence and Application for Public Health Control Strategies
Yan-Jang S. Huang; Dana L. Vanlandingham; Stephen Higgs Kansas State University, Manhattan, KS, United States
Abstract
Emergence and dispersal of chikungunya virus (CHIKV) and Zika virus (ZIKV) clearly have been the most important human public health emergencies caused by arboviruses. While studies prior to the recent epidemics demonstrated that evolution of the two viruses led to establishment of different genetic lineages in different areas of endemic regions, the importance of viral genetics in the emergence and re-emergence of arboviruses was exemplified during these outbreaks, especially for CHIKV. The acquisition of an E1-A226V mutation by the East/Central/South African genotype of CHIKV allowed its rapid geographic expansion through viral adaptation to Aedes albopictus. Published findings suggest genetic mutations may have been involved in the emergence of ZIKV into the New World. In addition to the role genetic studies play as a determining factor in disease transmission and pathogenesis, genetics further provides the promise for developing novel antiviral therapeutics and efficacious vaccines. Together, these have the potential to significantly reduce the disease burden in endemic regions.
Keywords
Chikungunya virus; Zika virus; Arboviruses; Aedes albopictus; Aedes aegypti
Introduction
Chikungunya virus (CHIKV) and Zika virus (ZIKV) are arthropod-borne viruses (arboviruses) transmitted by mosquitoes. Based on the most current taxonomy of viruses, CHIKV and ZIKV belong to the Togaviridae family and the Flaviviridae family, respectively. As described in Chapter 4, both viruses were discovered in Africa. CHIKV was first isolated from a febrile patient during a dengue-like fever epidemic in Tanganyika during 1952 and 1953 (Mason and Haddow, 1957). Since its first isolation, CHIKV has been linked to several epidemics of febrile diseases in Africa and Southeast Asia and frequently vectored by Aedes aegypti, a vector for several medically important arboviruses (Hammon et al., 1960). In Africa, CHIKV’s isolation from Ae. africanus and its capability to replicate in nonhuman primates indicated that CHIKV is also maintained in a sylvatic cycle (Weinbren et al., 1958). Distinct from the human disease cases that led to the discovery of CHIKV, the first isolate of ZIKV was derived from a sentinel monkey exposed on a platform as part of yellow fever studies (Dick et al., 1952). Subsequently, the ZIKV maintenance cycle between nonhuman primates and arboreal mosquitoes was further delineated by isolates from Ae. africanus (Haddow et al., 1964). While human diseases caused by CHIKV and ZIKV have been continuously reported, the public health importance of both viruses was largely neglected prior to their emergence in the 21st century in various parts of the world. Within the last few years, our understanding of various aspects of CHIKV and ZIKV has grown exponentially, including the genetics and evolution of both viruses.
Even before the contemporary molecular biological techniques became available, characterization of both viruses and other related viruses used purified virions and identified that ribonucleic acid (RNA) was the form of genetic material and was infectious to vertebrate hosts (Cheng, 1958; Ada and Anderson, 1959; Igarashi et al., 1967). The single-stranded positive- sense structure of the genomes was determined through biochemical analysis of related viruses. Replication of CHIKV and ZIKV depends on similar strategies shared with other single-stranded positive-sense RNA viruses (Arif and Faulkner, 1972; Chu and Westaway, 1985). The synthesis of viral RNA genomes relies on a distinct mechanism from the replication of host genomes. The replication of CHIKV and ZIKV is achieved through incorporation of ribonucleoside triphosphates by the enzymatic action of virally encoded RNA-dependent RNA polymerases (RdRp). To the best of our knowledge, quantitative analysis of fidelity of viral RNA polymerases of CHIKV and ZIKV has never been published. However, the error-prone nature of viral RdRp that was first discovered in poliovirus and conserved in other RNA viruses is expected in the RdRp of CHIKV and ZIKV (Ward et al., 1988). This prediction is further supported by observed rapid adaptation of CHIKV and ZIKV when mice or cell lines were used for the passage of both viruses, and subsequent detection of genetic mutations (Dick, 1952; Levitt et al., 1986; Haddow et al., 2012).
In the first two decades of the 21st century, determining the mechanisms responsible for the emergence of CHIKV and ZIKV became one of the most urgent issues for the arbovirology community. There have been at least three major outbreaks associated with the emergence or introduction of specific genotypes or lineages of CHIKV and ZIKV. The observations clearly highlight the importance in understanding how viral genetics can significantly influence the epidemic potential and disease burden associated with both viruses. Both forward and reverse genetic approaches were used by scientists to investigate the cause of the emergence and dispersal to new regions. As transmission cycles became established and the case numbers of CHIKV and ZIKV infection continued to rise, viral genetics became an important approach toward developing control strategies. As described in Chapter 11, the development of live-attenuated vaccines was regarded as particularly important to protecting susceptible people and curtailing the spread of both viruses. This chapter reviews how viral genetics of CHIKV and ZIKV play a role in pathogenesis and transmission. Strategies to develop live-attenuated vaccine candidates are also discussed.
Genetics and Evolution of Arboviruses and the Importance to Public Health
The lack of proof-reading mechanisms associated with RdRp among RNA viruses leads to low fidelity associated with the replication of viral genomes. The accumulation of mutations is an inherently critical process for RNA viruses to evolve and adapt to new environments. This is particularly true for the evolution of arboviruses, which alternate between vertebrate hosts and arthropod vectors. With very few exceptions, such as in African swine fever virus, genetic information of almost all arboviruses is encoded in different forms of RNA genomes. Although mutations created by the error-prone nature of viral replication may often be lethal for viruses in the presence of selective pressure, this mechanism sometimes allows the emergence of mutants with a selective advantage at various steps of their life cycle. Several well-known mechanisms that provide the selective advantage for specific mutants include those that escape from immune responses, adapt to different intracellular environment of infected hosts, and increase efficiency for transmission.
For most arboviruses, the selection of mutants is a more complicated process than for RNA viruses that only infect a limited number of vertebrate host species. Because the transmission of arboviruses requires infection and replication in both vertebrate hosts and arthropod vectors, mutants of arboviruses must be able to adapt in very different physiological environments. For arboviruses like CHIKV, dengue viruses (DENV), yellow fever virus (YFV), and ZIKV, mutants must be able to propagate in both Aedes species mosquitoes and primate hosts. For zoonotic arboviruses like Japanese encephalitis virus (JEV), West Nile virus (WNV), and Venezuelan equine encephalitis virus (VEEV), mutations must allow viruses to survive in humans, zoonotic vertebrate hosts, and arthropod vectors. Importantly, infection of mutants must lead to sufficiently high viremic titers in amplification hosts in order to allow the infection of arthropod vectors to occur and for the transmission cycles to be sustained.
As the evolution of arboviruses and accumulation of mutations continues in nature, viral genetics has significantly impacted disease pathogenesis in vertebrate hosts, outcomes of infection in arthropod vectors, and ultimately the epidemic potential of given arboviruses. Although it is a relatively rare case scenario, single-point mutations can significantly alter the virulence or transmission efficiency of arboviruses. This can result in the emergence of specific viral populations that have higher selective advantage in nature or displacement of naturally occurring attenuating mutants by other viral populations that are more adapted to the transmission cycles. The most notable recent example of this was the single-point mutation resulting in the switch of competent vector species for CHIKV. As described in Chapters 2 and 4, during the outbreaks in the Indian Ocean between 2004 and 2006, the microevolution of CHIKV led to the emergence of the strain, which carries the A226V mutation in the E1 glycoprotein and shows the selective advantage in nature because of its higher infectivity to Ae. albopictus mosquitoes (Schuffenecker et al., 2006; Tsetsarkin et al., 2007).
The accumulation of point mutations can also lead to naturally occurring attenuated strains, which can cause silent or near-silent transmission of arboviruses. As an example, the DENV-2 isolates that appeared in Tonga in 1973 and 1974 only caused mild diseases in humans (Gubler et al., 1978). These isolates were later demonstrated to be genetically distinct from other strains that belong to the American genotype of DENV-2. This unique monophyletic clade was associated with three distinct point mutations, I46M in NS4A, H54R in prM, and S83G in NS2A (Steel et al., 2010). In addition to naturally occurring mutants, the deliberate attenuation of virulent strains under laboratory conditions has often been achieved by the introduction of genetic mutations with either the empirical passage or contemporary molecular virological techniques. Although the original intention was to develop vaccine candidates for disease control, these serially passaged strains of arboviruses later became models to study the links between viral genetics and phenotypes of arboviruses. The most successful example in the history of human public health is the development of live-attenuated YFV 17D vaccine strains from the parental virulent Asibi strain (Theiler and Smith, 1937a,b). A total of 67 nucleotide substitutions in the genome of the 17D-204 strain led to 32 amino acid mutations and several attenuated phenotypes including loss of viscerotropism in infected humans and nonhuman primates and disseminating phenotype from the midgut of an infected mosquito (Theiler and Smith, 1937b; Whitman, 1939; Hahn et al., 1987; Barban et al., 2007). The vaccine strains consist of a homogenous viral population with great genetic stability (Barban et al., 2007; Beck et al., 2014). This is consistent with the finding that NS5 RdRp of the 17D vaccine strains has significantly higher fidelity than the RdRp of other related flaviviruses (Pugachev et al., 2004; Jin et al., 2011). Yellow fever virus 17D vaccine has also become the pioneering model used to investigate the relationship between genetic diversity and virulence of arboviruses. Because of the high efficacy and safety profile, more than 450 million doses of 17D vaccines have been used in vaccination programs worldwide and this vaccine continues to be the most effective strategy for YF disease control. Similarly, the live-attenuated JEV SA14-14-2 vaccine strain was also generated by the empirical passage of the virulent SA14 strain in vitro. Because of the high immunogenicity and safety profile, this vaccine has become one of the most critical tools to prevent pediatric encephalitis caused by JEV in East Asia. In contrast to the homogeneous and stable genetic composition of the YFV 17D vaccine strains, significant genetic variations among the existing SA14-14-2 strains have been well documented in multiple studies (Nitayaphan et al., 1990; Aihara et al., 1991; Song et al., 2012). The accumulation of additional mutations and the change of phenotypes, especially the potential loss of immunogenicity, represent significant technical challenges for the production and international licensure of live-attenuated vaccines. However, successful stories of both live-attenuated YFV and JEV vaccines have demonstrated how live-attenuated vaccines can significantly reduce disease burden. Manipulation of viral genetics has constantly been a popular approach to produce attenuated vaccine candidates with the ultimate goal of achieving high immunogenicity and long-term protection among vaccinated individuals. In addition to the insertion of point mutations that are expected to disrupt normal functions of viral proteins, several significant advancements have been made by deletion of untranslated regions, chimerization of immunogenetic components with existing attenuated strains, and deliberate reduction in the genetic diversity of viral populations (Monath et al., 2000; Durbin et al., 2001; Van Slyke et al., 2012). Chapter 11 describes various approaches being used to develop vaccines that will protect people from infections with CHIKV and ZIKV.
While single-point mutations that cause significant changes in virulence or epidemic potential of arboviruses have been repeatedly observed, genetic mutations accumulated during the process of natural evolution do not all cause demonstrable differences in the phenotypes of arboviruses. Therefore, viral genetics of arboviruses are often studied by classifying available virus isolates into distinct genotypes. Historically, the definitions of genotypes have been established by constructing distinct clusters that reflect the divergence of genetic sequences and different epidemiological patterns (Chen et al., 1990; Chungue et al., 1995; Powers et al., 2000). Because the difference in epidemiological patterns of various arbovirus populations is often considered along with the actual genetic diversity, classification of arboviruses by categorically listing different populations into distinct genotypes becomes the most systematic and accepted approach when studying mosquito-borne arboviruses. Emergence of new genotypes and displacement of previously endemic genotypes have been repeatedly observed for different arboviruses as the throughput of sequencing techniques continue to be improved. Although the change of dominant viral genotypes may not result in a change of infectivity for host and vector species, genetic changes can often lead to viruses with new phenotypes that have great public health importance. For example, the continuous evolution of WNV in the United States has led to the emergence of at least four different genotypes since its introduction in 1999 (Mann et al., 2013). This process exemplifies how changes in viral genetics can not only lead to the emergence of specific populations of arboviruses through the increase in the transmission efficiency, but also result in the displacement or disappearance of attenuated genotypes. While the original NY99 genotype successfully spread through a majority of the North American continent within 4 years, the subsequent emergence of a new North America/WN 2002 (NA/WN02) genotype was associated with an increase of disease incidence between 2002 and 2003. Ultimately, this resulted in displacement of previously dominant NY99 genotype in certain regions (Ebel et al., 2004; Davis et al., 2005). In contrast to the observation made from the emergence of the NA/WN02 genotype, a distinct outcome of viral evolution was also observed in the evolution of WNV in the United States. A transient presence of the Coastal Texas Genotype in 2002 was documented (Davis et al., 2004). The genotype was shown to be attenuated and has not been detected since 2002. Because of its endemic status and the establishment of enzootic transmission cycles, we can expect the evolution of WNV will continue to have significant impact on human and veterinary public health in the United States.
As the determination of genetic sequences of novel variants remains an important tool to study the genetics of arboviruses and functional protein products, another significant advancement was made by development of reverse genetics systems. With the use of contemporary molecular biological techniques, reverse genetics systems enable the mutagenesis analyses of viral proteins to be performed at specific genetic loci. Reverse genetics has become the most powerful method to determine the functionally important residues of individual viral proteins. To study alphaviruses and flaviviruses that store genetic information on a single-stranded positive- sense RNA genome, reverse genetics systems often utilize positive- sense RNA transcripts derived from sequences of complementary DNA (cDNA). Positive-sense RNA transcripts transfected into permissive cell lines immediately become templates for the translation of viral proteins, which drive the subsequent replication to generate recombinant viruses. The use of cDNA sequences allows manipulation of genetic sequences through individual mutations or chimerization. Functions of viral proteins can therefore be studied by disrupting sequences of individual genes and rescuing mutant viruses. The reverse genetics system of CHIKV was first established based on the one-plasmid cDNA infectious clone of O’nyong-nyong virus (ONNV), a related alphavirus in the Semliki Forest virus (SFV) antigenic complex (Vanlandingham et al., 2005a). Similar to the technical challenges associated with the development of reverse genetics systems for flaviviruses, the use of low-copy-number plasmids or the separation of genomic fragments into multiple plasmids was applied to establish the reverse genetics systems of ZIKV. This was necessary in order to reduce the toxicity to the host Escherichia coli cells created by its genome (Shan et al., 2016; Weger-Lucarelli et al., 2017).
Molecular Biology and Intracellular Life Cycle of CHIKV
CHIKV is a single-stranded positive-sense RNA virus that belongs to the genus of alphavirus in the family of Togaviridae. The 11.8-kb genome is modified by cellular and viral biochemical machineries with the addition of a 7-methylguanosine cap and a polyadenylation tail on its 5′ and 3′ ends, respectively (Vasiljeva et al., 2000; Khan et al., 2002). Nonstructural proteins and structural proteins are encoded in two separate open reading frames (ORF) in the viral genome (Fig. 1). The first ORF from the 5′ terminus of the genome accounts for approximately two-thirds of the genome and consists of a nonstructural domain containing four nonstructural genes (nsP1, nsP2, nsP3, and nsP4). A recently established molecular interaction model suggests the physical association of the four nonstructural proteins in the replication complex (Rana et al., 2014). A Rossman-like methyltransferase (MT) motif in the nsP1 protein contains the capping mechanism required for the modification of the 5′ end of the genome (Ahola and Kaariainen, 1995). In the late replicase complex for synthesis of viral genomes, the MT domain of the nsP1 protein is in close association with the helicase domain of the nsP2 protein and the RdRp domain of the nsP4 protein (Lemm et al., 1998; Gomez de Cedron et al., 1999). RNA synthesis is achieved by the enzymatic activity of the GDD motif in the RdRp domain of the nsP4 protein (Koonin, 1991). The structural domain encodes the structural genes (capsid (C), E2, 6K, and E1) and forms the one-third portion of the genome from its 3′ terminus. Between the two ORFs, a subgenomic promoter drives the synthesis of a 26S subgenomic mRNA, which is subsequently translated into a polyprotein containing individual structural proteins (Clegg and Kennedy, 1974).

The surface of a mature viral particle of CHIKV shares extensive homology with other alphaviruses. There are 80 copies of the spike structure consisting of three copies of the E1-E2 heterodimers anchored through two independent transmembrane domains of E1 and E2 glycoproteins as summarized in Fig. 2 (Voss et al., 2010; Sun et al., 2013). The E1 glycoprotein consists of three discontinuous domains and contains a fusion loop structure that is inserted into cell membranes of susceptible cells and triggers viral membrane fusion in the acidic environment of late endosomes (Gibbons and Kielian, 2002). The E2 glycoprotein contains three distinct domains; A, B, and C. A putative receptor-binding region, based on its homology to the domain III of flavivirus envelope protein, is located in domain A of the E2 glycoprotein (Pierro et al., 2008). Interactions between the E1 and E2 glycoproteins protect the fusion loop from premature membrane fusion and are controlled by pH through distortion of the acid-sensitive region located in the β-ribbon structure between the E1 and E2 glycoproteins (Fields and Kielian, 2013). In the mature virion structure, the relative orientation between domains A and B of the E2 glycoprotein is stabilized by the presence of the E3 protein (Voss et al., 2010). The stabilization of domains A and B in the structure allows formation of the E1 fusion loop-binding groove, which subsequently becomes accessible in response to the acidic environment in the late endosome. During the endocytosis process, the acid environment in endosomes triggers the conformational changes of the E1-E2 heterodimer to expose the fusion loop structure in the E1 glycoprotein and mediate the fusion between viral and host membranes (Li et al., 2010).

Shortly after its entry through clathrin-mediated endocytosis and the release of viral genome, the general strategies of transcription and translation utilized by alphaviruses including CHIKV are summarized in Fig. 3. Translation of the positive-sense genome produces two distinct types of polyproteins, P123 and P1234, through the regulation of a “leaky” opal termination codon (Strauss et al., 1983). The translation termination mechanism controlled by the opal termination codon provides a mechanism to regulate the synthesis of negative-sense RNA replication intermediates and positive-sense viral genomic RNA through the stoichiometry of the P123 polyprotein and the nsP4 protein in infected cells. Nonstructural proteins functioning as replicases are subsequently produced from the polyproteins by the proteolytic processing of the nsP2 protein (Hardy and Strauss, 1989). After formation of the replication complex in the early phase of infection, negative-sense copies of viral RNA are produced as the template for synthesis of positive-sense copies of viral RNA by the RdRp domain of the nsP4 protein in the presence of uncleaved P123 polyprotein (Bruton and Kennedy, 1975). As proteolytic processing of the nsP2 protein continues, the level of uncleaved P123 polyprotein declines and leads to the switch of RNA synthesis by the nsP4 protein to production of positive-sense RNA as nascent viral genomes (Shirako and Strauss, 1994; Vasiljeva et al., 2003).
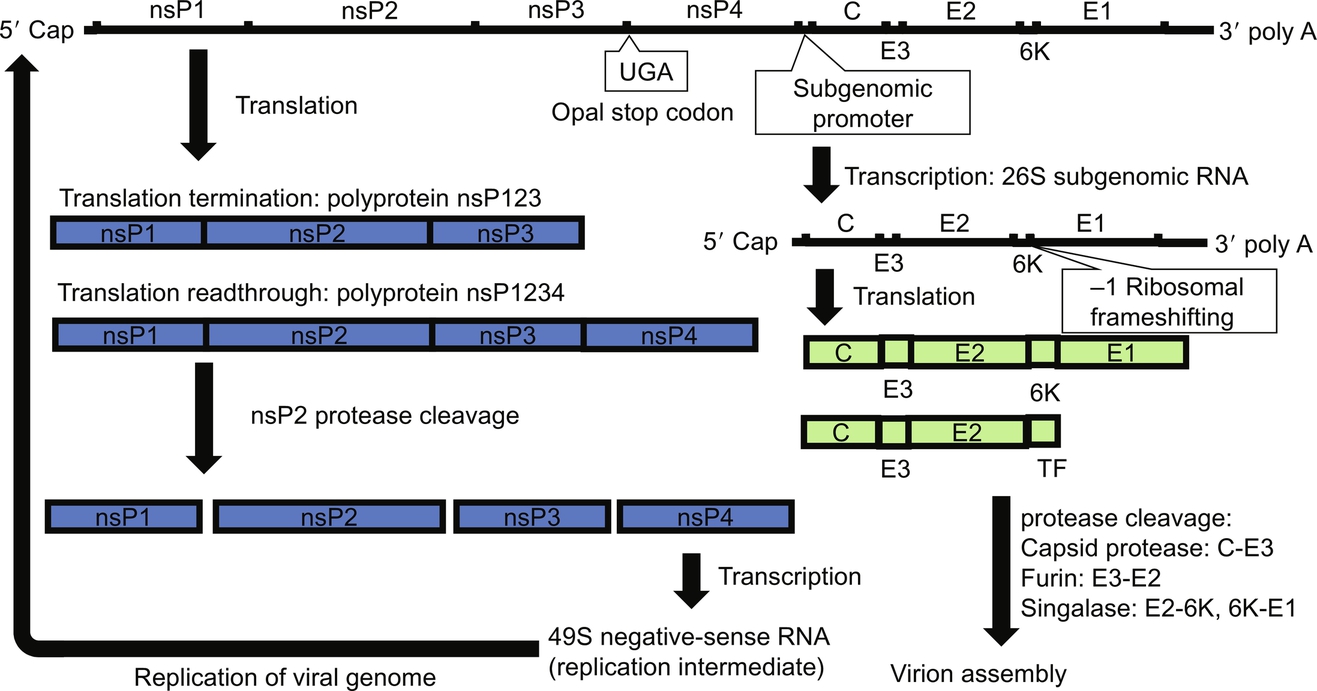
Transcription of the 26S subgenomic RNA from the negative-strand RNA template begins with recognition of the consensus subgenomic promoter sequence (Ou et al., 1982). Production of the polyprotein containing structural proteins for progeny virions is initiated by translation of the 26S subgenomic RNA (Cancedda et al., 1975). A recently discovered − 1 ribosomal frameshifting mechanism during translation of the 6k region in the 26S subgenomic RNA has identified an additional species of polyprotein that only produces C, E3, and E2 and transframe (TF) proteins (Firth et al., 2008). The result of ribosomal frameshifting, TF protein, has been shown to be critical for release of progeny virions and pathogenesis of alphaviruses in mice (Snyder et al., 2013). Cotranslational folding of the C protein in the N-terminus triggers the release from the polyprotein by its autoprotease activity (Melancon and Garoff, 1987; Nicola et al., 1999). The pE2 portion of the polyprotein is guided by the signaling sequence in the N-terminus for its insertion into endoplasmic reticulum (ER) and paused by the stop-transfer sequence in its C-terminus (Garoff et al., 1990). Similarly, a signaling sequence present in the 6K protein mediates the insertion of the E1 protein into the E1 and the stop-transfer sequence of the E1 glycoprotein controls its retention to ER (Melancon and Garoff, 1986; Migliaccio et al., 1989). Formation of the heterodimeric structure between the pE2 protein and the E1 protein takes place in ER and is essential for export of glycoproteins to the cell surface (Johnson et al., 1981). The two proteins must be folded simultaneously from the same translation unit. The folding efficiency of the E1 glycoprotein is significantly impaired when a functional E2 glycoprotein is absent (Andersson et al., 1997). Three copies of the E1-pE2 heterodimers are arranged into the spike structure protruding from the virions. As the assembly of virions begins, the critical interactions between the E1-pE2 heterodimer and the C protein occur between the hydrophobic pocket of the C protein and the cytoplasmic domain of the pE2 protein and allows formation and budding of virions (Metsikko and Garoff, 1990; Suomalainen et al., 1992). Incorporation of viral genomic RNA is mediated by interactions between positive charges in the N-terminus of the C protein and the packaging sequence in the nsP2 gene (Choi et al., 1997; Kim et al., 2011). During the process of evolution, CHIKV and other members of alphaviruses within the SFV complex have developed a different mechanism for RNA encapsidation from those alphaviruses in the VEEV, Eastern equine encephalitis virus, and Western equine encephalitis virus complexes (Kim et al., 2011). After formation of the virion structure, an additional processing step by the host furin protease releases the E3 protein, which remains physically associated with the E2 protein and protects premature fusion, and grants the infectivity to progeny virions (Simizu et al., 1984; Sjoberg et al., 2011).
Genetic Lineages of CHIKV
Even before the genetic sequences of alphaviruses became available, antigenic characterization has designated CHIKV as an Old World alphavirus placed in the SFV antigenic complex (Karabatsos, 1975; Tesh et al., 1975). The phylogenetic relationships among members within the complex were later found consistent with antigenic relationships based on sequences of the nsP1 gene and full-length genomes in two independent studies (Pfeffer et al., 1997; Forrester et al., 2012). While several viruses are included in the SFV complex, it contains the highest genetic heterogeneity among all known antigenic complexes within the genus of Alphavirus, largely reflecting the broad geographic distribution encompassing both the Old World and the New World (Pfeffer et al., 1997).
While the endemic status of CHIKV in Africa and Southeast Asia has been demonstrated by virus isolation and serological survey (Mason and Haddow, 1957; Hammon et al., 1960), determination of genetic sequences among different strains of CHIKV was not performed until the early 1990s. This apparent lack of interest was due to its public health importance (Strauss and Strauss, 1994). As described in Chapter 3, because of the difficulty in differential diagnosis of CHIKV from other febrile illness caused by arboviruses such as DENV, and the highly overlapped endemic regions among these viruses (Halstead, 2015), documentation of its disease burden prior to its emergence in the Indian Ocean in 2005 was relatively unclear and insufficient. However, results from the RNA-RNA hybridization experiments and two-dimensional gel electrophoresis led to the first interesting discovery in its genetics showing that CHIKV and ONNV share an unusually high degree of homology in their nucleotide sequences (Wengler et al., 1977). Because of the homology in the sequences of CHIKV and ONNV, it was perhaps not surprising that antisera prepared by immunization of CHIKV showed one-way crossreactive neutralization capacity against ONNV (Chanas et al., 1979). Therefore, ONNV was mistakenly considered as a subtype of CHIKV for nearly two decades until the analysis based on the full-length genomes was performed after the O’nyong-nyong fever outbreak in Southwest Uganda (Lanciotti et al., 1998).
The systematic examination on the phylogenetics of CHIKV and ONNV also subsequently began due to the status of CHIKV and ONNV as re-emerging pathogens (Rwaguma et al., 1997). The classification of three distinct genotypes based on the locations of virus isolation was first established by the partial nucleotide sequences of the E1 gene (Powers et al., 2000). CHIKV strains isolated from Africa form two distinct lineages, the West African genotype and the Central/East/South African genotype, which was later commonly described as East/Central/South African (ECSA) genotype in several published studies (Volk et al., 2010; Cui et al., 2011). Although location of virus isolation is used to differentiate between the West African genotype and the ECSA genotype, cocirculation of the two genotypes has at least been well documented in Senegal (Powers et al., 2000; Volk et al., 2010).
Infection with the West African genotype has been observed among susceptible populations in Senegal, Nigeria, and Cote d’Ivoire (Boorman and Draper, 1968; Diallo et al., 1999; Volk et al., 2010). Although isolates belonging to the West African genotype have been continuously made from infected humans, mosquitoes, and bats since 1964, and suggested as a criterion to categorize the genetic variants into two separate sublineages, disease burden caused by the West African genotype remains poorly understood (Diagne et al., 2014). Because of the relatively high percentage of available isolates derived from sylvatic Aedes species mosquitoes, sylvatic transmission cycles are likely to be important for the maintenance of the West African genotype.
Prior to its re-emergence in Kenya in 2004, presence of the ECSA genotype was documented in several African countries including Tanzania, South Africa, Senegal, the Central Africa Republic, and the Democratic Republic of Congo. Similar to the West African genotype, sylvatic transmission between nonhuman primates and arboreal mosquitoes is likely to be important for the maintenance of the ECSA genotype (Diallo et al., 1999). Historically, urban transmission of the ECSA genotype between humans and Ae. aegypti has also been reported in several urban areas in Africa (Filipe and Pinto, 1973). Although the actual incidence of disease caused by the ECSA genotype of CHIKV prior to its re-emergence in 2004 remains unknown due to difficulties in differentiation from other arboviral infections, it is well accepted that a majority of the disease burden of CHIKV in humans is caused by its urban transmission.
The intercontinental spread of CHIKV from Africa to Asia, estimated to have occurred between 1879 and 1956, resulted in establishment of the Asian genotype (Volk et al., 2010). As described in Chapter 5, since the first outbreak in Thailand documented in 1958 (Hammon et al., 1960), the Asian genotype has become endemic in several South and Southeast Asian countries including India, Thailand, Malaysia, Indonesia, and Philippines. Its maintenance is likely to rely on urban transmissions among humans vectored by Ae. aegypti and Ae. albopictus (Hammon et al., 1960; Rodrigues et al., 1972; Marchette et al., 1980).
Re-emergence of CHIKV ECSA Genotype in Kenya and the Epidemics in the Indian Ocean
The historical distribution of the ECSA genotype began to change at the end of 2004, when transmission on islands in the Indian Ocean was first observed. The origin of its re-emergence was traced back to a drought-associated outbreak of CHIKV in Kenya in 2004 (Chretien et al., 2007). The drought was likely the result in the transient increase of Ae. aegypti populations as previously observed in Kenya and subsequently increased the transmission of CHIKV (Subra, 1983). Although there were only 1400 human cases of CHIKV infection identified in Kenya in 2004, a retrospective serological survey indicated that the prevalence of the disease was significantly higher. For example, it was estimated that up to 75% of residents in Lamu, Kenya, was infected by CHIKV in 2004 (Sergon et al., 2008). The high attack rate and movement of viremic individuals between Kenya and islands in the Indian Ocean are likely the cause of subsequent dispersal of the ECSA genotype.
The eastward dispersal of CHIKV from Kenya to islands in the Indian Ocean was first indicated by the identification of cases reported from the Union of Coromos, a country on the shore of East Africa consisting of four individual islands. A serological survey conducted during the epidemic on the island of Grande Comore, the largest island under the Union of Coromos, indicated that up to 63% of the local residents were positive for anti-CHIKV IgM. Symptoms of CHIKV infection, including joint pains and myalgia, were reported among the majority of seropositive individuals (Sergon et al., 2007). An entomological survey indicated that Ae. aegypti was the principal vector responsible for the transmission cycle (Sang et al., 2008). There was no clear evidence that adaptation to Ae. albopictus had occurred at this phase of the epidemic because of the relatively low abundance of Ae. albopictus on the islands and the lack of the isolates containing the E1 A226V mutation (Sang et al., 2008; Wasonga et al., 2015).
Prior to the end of the epidemic in the Union of Coromos, CHIKV cases were reported in Mauritius and Réunion. The introduction of CHIKV to Mauritius resulted in two separate epidemics. The initial epidemic, leading to approximately 3500 reported cases, occurred between April and June 2005. The second epidemic, occurring between February and May 2006, led to 11,134 cases, approximately 1% of the 1.2 million population on the island (Beesoon et al., 2008). Similar to the epidemics in Mauritius, there were also two separate waves of epidemics observed on Réunion Island. However, the attack rate of CHIKV was significantly higher on Réunion Island, reportedly 34% at the end of the epidemic. The first peak of the epidemic occurred in May 2005 followed by initiation of the second peak starting in December 2006 (Renault et al., 2012). Between the two phases of the epidemic on Réunion Island, emergence of the E1 A226V genetic variant began approximately in September 2005. The mutation was later demonstrated to significantly enhance infectivity of the ECSA genotype in Ae. albopictus and increase its epidemic potential (Tsetsarkin et al., 2007). At the end of the epidemic, isolates made on Réunion Island and Mauritius all carried the E1 A226V mutation (Schuffenecker et al., 2006). This microevolution took place during the epidemic in the Indian Ocean and undoubtedly had one of the most profound effects in the history of human public health.
The introduction of CHIKV was detected in Mayotte between February and June 2005. The peak of reported cases in February and March 2006 lasted for several weeks. Similar to the isolates from the Réunion Island and Mauritius, all isolates obtained in Mayotte after February 2006 also carried the E1 A226V mutation (Schuffenecker et al., 2006). At the end of the epidemic, approximately 26% of the residents in Mayotte were estimated to be exposed to CHIKV (Sissoko et al., 2008). In addition to the infection of CHIKV reported in Coromos, Réunion Island, Mauritius, and Mayotte, the Indian Ocean lineage of CHIKV was also responsible for a concurrent outbreak of CHIKV and DENV-1 in Madagascar in January 2006. Although the precise number of infected individuals has never been available, detection of viral genome and anti-CHIKV IgG clearly confirmed the circulation of CHIKV (Ratsitorahina et al., 2008; Pistone et al., 2009). Interestingly, CHIKV isolated in Madagascar between February and March 2006 lacks the E1 A226V mutation (Schuffenecker et al., 2006).
At the end of the outbreaks, over one million individuals had been exposed to CHIKV. Except for the early phase of re-emergence in Kenya and the epidemics in Comoros and Madagascar, the large number of infected individuals on islands in the Indian Ocean was attributed to the adaptation of CHIKV to Ae. albopictus caused by the E1 A226V mutation. Before discussing molecular mechanisms in the latter part of the chapter, it is noteworthy to mention the likelihood that Ae. albopictus acting as a domestic anthropophilic vector species and large numbers of viremic individuals on these islands promoted the adaptation to occur. Historically, the presence of both Ae. aegypti and Ae. albopictus has been recorded in several islands in the Indian Ocean. Similar to other tropical regions, large numbers of Ae. aegypti were present in urban areas; whereas, Ae. albopictus, an invasive species from Asia, had mainly colonized in rural habitats (Paupy et al., 2001). Shortly after the Second World War, the population of Ae. aegypti in the urban environment experienced a significant decline because of dichlorodiphenyltrichloroethane (DDT) used in the malaria eradication program (Dowling, 1953). After use of DDT was terminated, reinfestation of areas by mosquitoes began and subsequently resulted in a shift in dynamics between the two vector species. Ae. albopictus was mainly found in urban areas; whereas the recovery of Ae. aegypti in the cities was not observed. Its presence has been limited to rural and natural settings on these islands (Salvan and Mouchet, 1994). The unique distribution of Ae. albopictus and Ae. aegypti led to a distinct use of vector species for DENV from other endemic regions of DENV, where Ae. aegypti has frequently been the primary vector. Since the DENV-2 outbreak in Seychelles in 1976, there have been repeated reports of Ae. albopictus as the primary vector species for arboviruses in the Indian Ocean (Metselaar et al., 1980; Zeller, 1998). Based on an entomological survey conducted in 2009, Ae. albopictus remains the predominant species in urban and suburban areas in Mayotte (Bagny et al., 2009). Therefore, it is reasonable to assume that engorgement of CHIKV from viremic individuals by Ae. albopictus frequently took place when large numbers of human infections were present during CHIKV outbreaks. Adaptation to Ae. albopictus caused by the E1 A226V mutation may be a result of virus evolution influenced by virus-vector interactions during the outbreak.
Dispersal of CHIKV to Europe and Southeast Asia
Expansion of CHIKV’s geographic range continued as virus isolates were reported in Italy, India, Thailand, Cambodia, Malaysia, Bhutan, China, Singapore Myanmar, Philippines, Yemen, and Laos (Yergolkar et al., 2006; Pongsiri et al., 2010; Cui et al., 2011; Duong et al., 2012; Sam et al., 2012; Wangchuk et al., 2013; Tun et al., 2014; Phommanivong et al., 2016; Sy et al., 2016). Analysis of viral genetics, especially the E1 A226V mutation, became the fundamental approach to monitor the dispersal of CHIKV and disease burden caused by outbreaks. With very few exceptions, such as the outbreak in Bhutan in 2012 (Wangchuk et al., 2013), the majority of these epidemics was caused by the E1 A226V mutant of the ECSA genotype of CHIKV. As summarized in Table 1, its increased epidemic potential led to the first known significant dispersal of CHIKV in human public health history.
Table 1
| Location | Case number | A226V emergence | References (disease burden) | References (viral genetics) |
|---|---|---|---|---|
| Kenya | 1400 (reported) | Undetected | Sergon et al. (2008) | Kariuki Njenga et al. (2008) |
| Comoros | > 5000 | Undetected | Consigny et al. (2006) | Kariuki Njenga et al. (2008) |
| Seychelles | > 10,000 (reported) | Undetected | Renault et al. (2012) | Schuffenecker et al. (2006) |
| Réunion | 266,000 (estimated) | September 2005 | Renault et al. (2012) | Schuffenecker et al. (2006) |
| Mauritius | > 14,600 (reported) | February 2006 | Renault et al. (2012) | Schuffenecker et al. (2006) |
| Mayotte | 6443 (reported) | February 2006 | Renault et al. (2012) | Schuffenecker et al. (2006) |
| Madagascar | 55 (suspected, filed survey) | Undetected | Ratsitorahina et al. (2008) | Schuffenecker et al. (2006) |
| Italy | 205 (confirmed) | September 2007 | Rezza et al. (2007) | Bordi et al. (2008) |
| India | > 25,000 (reported) | April 2007 | Santhosh et al. (2008) | Santhosh et al. (2008) |
| Malaysia | 8320 (reported) | April 2008 | Chua (2010) | Sam et al. (2009) |
| Singapore | 123 (reported local) | May 2008 | Ng et al. (2009) | Hapuarachchi et al. (2010) |
| Sri Lanka | 40,000 (estimated in publication) | 2008 | Hapuarachchi et al. (2010) | Hapuarachchi et al. (2010) |
| Thailand | 46,000 (estimated in publication) | September 2008 | Auksornkitti et al. (2010) | Rianthavorn et al. (2010) |
| Maldives | 11,879 (suspected) | September 2009 | Yoosuf et al. (2009) | Hapuarachchi et al. (2010) and Pfeffer et al. (2010) |
| Myanmar | 107 (suspected) | July 2010 | Tun et al. (2014) | Tun et al. (2014) |
| China | 173 (reported) | October 2010 | Wu et al. (2012) | Li et al. (2012) |
| Gabon | 773 (suspected) | 2011 | Leroy et al. (2009) | Moyen et al. (2014) |
| Cambodia | 24 (laboratory confirmed) | May 2011 | Duong et al. (2012) | Duong et al. (2012) |
| Republic of Congo | 11,083 (reported) | July 2011 | Moyen et al. (2014) | Moyen et al. (2014) |
| Indonesia | 109 (suspected, field survey) | October 2011 | Maha et al. (2015) | Maha et al. (2015) |
| Philippines | Unclear (3 isolates) | 2012 | Tan et al. (2015) | Sy et al. (2016) |
| Papua New Guinea | 1590 (suspected) | September 2012 | Horwood et al. (2013) | Horwood et al. (2013) |
| Laos | 4638 (reported) | 2013 | Phommanivong et al. (2016) | Phommanivong et al. (2016) |
| France | 12 (reported)/11 (laboratory confirmed) | September 2014 | Delisle et al. (2015) | Delisle et al. (2015) |

Imported cases of CHIKV resulting from travel by people who were infected in an endemic region have been previously reported in Europe (Schwarz et al., 1996). However, the evidence derived from serological assays has sometimes failed to identify the viremic individuals, which can potentially initiate autochthonous transmissions and cannot rule out the potential of serological crossreactivity caused by the infection of related arboviruses (Filipe, 1974; Eisenhut et al., 1999). Imported cases caused by re-emergence of the CHIKV ECSA genotype in Africa and the Indian Ocean created an unprecedented concern by public health authorities because of the large numbers of cases and the likelihood of initiating autochthonous transmission in Europe. Although emergence of arboviruses that were previously exotic to the European continent had previously occurred, introduction of CHIKV accompanied by aggressive invasion of Ae. albopictus in Europe created a significant challenge to disease control. As described in Chapter 6, while imported cases with travel history to endemic regions were reported in multiple countries including Czech Republic, Denmark, France, Germany, Italy, Spain, the Netherlands, and United Kingdom, a more imminent threat to human public health in Europe was the autochthonous transmission recorded in Italy and France (Cordel et al., 2006; Fusco et al., 2006; Pfeffer and Loscher, 2006; Zelena et al., 2006; Amador Prous et al., 2007; Angelini et al., 2007; Edwards et al., 2007; Veber et al., 2007; Hassing et al., 2008; Delisle et al., 2015). Phylogenetic analyses demonstrated that all isolates from autochthonous transmission of the ECSA genotype in Europe carry the E1 A226V mutation and suggest that local Ae. albopictus can be competent vectors for establishment of transmission cycles (Bordi et al., 2008; Delisle et al., 2015). Since competent vectors for CHIKV are present in Europe, autochthonous transmission of CHIKV remains likely to occur in the near future (Vega-Rua et al., 2015).
In contrast to the two separate outbreaks in Europe, the eastward dispersal of the ECSA genotype continued and resulted in significant disease burden in India and Southeast Asia. As described in Chapter 5, as outbreaks in the Indian Ocean continued, the dispersal of the ECSA genotype in Asia was first confirmed in southern and western portions of the Indian subcontinent in 2006. Isolates in India were shown to be related to strains under the Indian Ocean lineage and subsequently became the origin of further dispersal throughout Southeast Asia (Yergolkar et al., 2006). However, arrival of the ECSA genotype in India can potentially be earlier than 2006. An isolate from a mosquito pool collected in Maharashtra, a state in west-central India, was made in 2000 and found to be closely related to the Ag41855 strain isolated from Uganda in 1982 (Arankalle et al., 2007; Tsetsarkin et al., 2009).
At the beginning of the epidemic, the presence of the E1 A226V mutation had not been reported and Ae. aegypti was the primary vector species for CHIKV in India (Yergolkar et al., 2006; Arankalle et al., 2007; Edwards et al., 2007). However, continuous transmission of CHIKV in India between 2006 and 2007 allowed the divergence of genetics to occur and resulted in creation of the Indian sublineage under the Indian Ocean lineage of the ECSA genotype (Cherian et al., 2009). Interestingly, the E1 A226V mutation began to be found among 2007 Indian isolates from Andhra Pradesh and Kerala, indicating the incidence of evolutionary convergence, resulted in emergence of the E1 A226V mutation independently in two distinct lineages (de Lamballerie et al., 2008; Kumar et al., 2008; Santhosh et al., 2008). An additional E2 L210Q mutation was subsequently detected in strains isolated from Kerala. This mutation promoted further adaptation of CHIKV to Ae. albopictus by increasing the incidence of disseminated infection (Tsetsarkin and Weaver, 2011). The adaptation to Ae. albopictus was also observed as multiple isolates of CHIKV was made from Ae. albopictus (Niyas et al., 2010; Kumar et al., 2012). Shortly after its emergence, the spread of the E1 A226V mutant also led to the outbreaks in Karnataka in 2008 and Orissa in 2010 (Santhosh et al., 2009; Das et al., 2012). However, emergence of the E1 A226V mutant did not lead to complete displacement of the strains without the E1 A226V mutations in the ECSA genotype. For example, the sequences of CHIKV isolates from Delhi and West Bengal in 2010 and Jharkhand in 2011 do not contain the A226V mutation in the E1 protein (Gurav et al., 2012; Shrinet et al., 2012; Taraphdar and Chatterjee, 2015).
It is no surprise that the epidemics caused by CHIKV in India became the source of multiple introductions of the ECSA genotype into various countries in Southeast Asia. In 2006, at least three countries, Sri Lanka, Malaysia, and Maldives, were affected by CHIKV outbreaks. Similar to the patterns observed in India, most of the viruses responsible for epidemics in 2006 did not contain the E1 A226V mutation. In Sri Lanka, total reported case numbers of CHIKV infection exceeded 40,000 between 2006 and 2008. However, the emergence of the E1 A226V mutation was not detected until 2008 (Lim et al., 2009; Hapuarachchi et al., 2010; Reller et al., 2013). While over 12,000 cases have been reported in Maldives, the E1 A226V mutant was not detected until 2009 (Yoosuf et al., 2009; Pfeffer et al., 2010). At the end of November 2006, the first autochthonous outbreak of CHIKV caused by the ECSA genotype in Malaysia was initiated by an imported case returning from Tamil Nadu, India (Noridah et al., 2007). The introduction of the ECSA genotype further complicated the epidemiology of CHIKV in Malaysia, where epidemics of the Asian genotype were also reported in 2006 (AbuBakar et al., 2007). In 2008, the first nationwide outbreak caused by the E1 A226V mutant of the ECSA genotype in Malaysia led to 8000 reported cases and became one of the largest outbreaks in recent history (Sam et al., 2009). An entomological survey also provided complementary evidence that Ae. albopictus was the major vector species in the outbreak (Rozilawati et al., 2011). While the case numbers slowly declined, this outbreak continued at least until the end of 2009 (Chua, 2010). In addition to disease burden among local populations, epidemics in India and Malaysia further became a source of imported cases leading to dispersal of CHIKV into other Southeast Asian countries. For example, analysis of genetic sequences among CHIKV isolated in Singapore suggests that the Indian sublineage of the ECSA genotype was continuously imported and responsible for initiation of different local transmissions. Although the first notable outbreak of CHIKV in Singapore took place during the first week of 2008 (Leo et al., 2009), screening of patients with acute febrile illness had repeatedly identified imported CHIKV cases since 2006 (Ho et al., 2011). Interestingly, the pattern of dispersal and the emergence process of the E1 A226V mutant resembled observations in both India and Malaysia. The first detection of the ECSA genotype without the E1 A226V mutation preceded emergence of the mutant virus. Initial local transmissions were mainly mediated by Ae. aegypti in urban areas prior to the switch of competent vector species to Ae. albopictus in suburban environments. Three individual epidemics were documented in Singapore in 2008. Based on sequences of the E1 gene, evidence suggested that each epidemic was likely to be associated with an independent event of dispersal from India, Malaysia, or Sri Lanka (Ng et al., 2009). The first epidemic took place in the southern part of the city, where infestation of Ae. aegypti had been reported. Isolates from this period were placed in the same clade with viruses isolated from India in 2006 and Maldives in 2007. The second epidemic was documented in central and northern suburbs of the city and coincided with multiple imported cases from Malaysia. The phylogenetic evidence revealed the presence of the E1 A226V mutation and the same trend was observed in epidemiological investigations of imported cases. During the second epidemic, virus isolates all carried a unique C300T synonymous nucleotide substitution in the E1 gene, which was also observed among the majority of isolates from Malaysia. Similarly, the third epidemic was also caused by transmission of the E1 A226V mutant. However, the virus isolates also contained a unique combination of genetic mutations that could be linked to the epidemics in Sri Lanka. Epidemics in Singapore exemplified how the E1 A226V mutation and the adaptation of CHIKV could have a profound effect on the epidemiology of CHIKV. Prior to emergence of the E1 A226V mutant, serological evidence indicated that the prevalence of CHIKV in the suburban environment in Singapore was relatively low, i.e., the local transmission has been nearly absent in the area (Ng et al., 2009). The abrupt onset of transmission can be quickly initiated in areas where competent vectors are present and herd immunity is low.
Outbreaks caused by the ECSA genotype in Malaysia and Singapore further resulted in its dispersal into the Indochinese peninsula, where the Asian genotype of CHIKV has been known to be endemic in Thailand since at least 1958. The first detection of the ECSA genotype was first reported in the Narathiwat province close to the Malaysian-Thailand border. Genetic sequences of virus isolates obtained during this outbreak resemble those isolated in India in 2007 and Singapore in 2008 (Theamboonlers et al., 2009). Different from previous epidemics in Southeast Asia, the E1 A226V mutant was involved in the initial emergence of the ECSA genotype in Thailand (Rianthavorn et al., 2010). Virological surveillance further demonstrated that the E1 A226V mutant spread to other provinces in central and northern parts of the country in 2010 and 2013, respectively (Sasayama et al., 2014; Wanlapakorn et al., 2014). Additionally, epidemics were reported in China in 2010, Myanmar in 2010, Cambodia in 2011, and Laos in 2013 (Duong et al., 2012; Wu et al., 2012; Tun et al., 2014; Phommanivong et al., 2016; Sy et al., 2016). Similar to the northward spread to the Indochinese peninsula, outbreaks in Malaysia also became the source of eastward dispersal to Indonesia in 2011, Papua New Guinea in 2012, and Philippines in 2013 (Horwood et al., 2013; Maha et al., 2015; Sy et al., 2016).
While the E1 A226V mutant resulted in a significant fitness gain of infectivity for Ae. albopictus and was quickly dispersed throughout the Indian Ocean and Southeast Asia, it is important to highlight that not all emergences of the ECSA genotype were associated with the mutation. One of the regions affected by the ECSA genotype but not experiencing the dispersal of the E1 A226V mutant was Yemen, where Ae. aegypti remains the principal vector for CHIKV since 2011 (Ciccozzi et al., 2014). The lack of Ae. albopictus populations in the region is a clear limitation on establishment of the E1 A226V mutant (Kraemer et al., 2015). Another interesting example is the report of the 2012 CHIKV epidemic in southern Bhutan, where Ae. aegypti and Ae. albopictus are presumably present and act as competent vectors. Surprisingly, in spite of the high degree of genetic homologies to the E1 A226V mutants isolated in Thailand in 2009 and in India in 2010, the virus responsible for the epidemic in Bhutan contains an alanine residue in the 226 residue of the E1 glycoprotein (Wangchuk et al., 2013). It remains unclear if the lack of reports on the E1 A226V mutant in Bhutan was due to the actual absence of the E1 A226V mutant in the epidemic or due to the limitation of surveillance systems that failed to detect the virus.
While the E1 A226V variant continues to cause outbreaks in the Old World, another significant expansion of the CHIKV endemic region took place in the Western Hemisphere after introduction of the Asian genotype. However, the initiation of the Asian genotype dispersal happened much earlier than the epidemics in the New World. Emergence of the Asian genotype, which was historically found in its endemic region of Southeast Asia and through the Pacific Islands in the 21st century, could be traced back to the autochthonous transmission reported in New Caledonia in 2011 (Roth et al., 2014). Molecular epidemiological analyses indicated that the introduction of CHIKV was a consequence of imported cases from Indonesia and that Ae. aegypti remained as the competent vector during the outbreak (Dupont-Rouzeyrol et al., 2012). Interestingly, during this period, the same region was also experiencing outbreaks caused by the ECSA genotype that carries the A226V mutation. Similar to the outbreak in 2011, another epidemic caused by the Asian genotype was reported in New Caledonia and Yap Island, where Ae. hensilli also may have been involved in transmission (Savage et al., 2015). The observation of Ae. hensilli as a vector species was likely caused by the ubiquitous distribution of the species on the island as no specific mutations were found from isolates. The spread of the Asian genotype further resulted in observations of the disease in multiple countries including Tonga, American Samoa, Independent State of Samoa, and Tokelau prior to its arrival in French Polynesia, where approximately 25% of local residents were infected (Nhan et al., 2014; Nhan and Musso, 2015).
Although historical evidence suggests that CHIKV was introduced into the Americas during the 19th century, the virus subsequently failed to establish itself in the New World (Halstead, 2015). The first clearly documented autochthonous transmission of CHIKV in the Americas was reported from Saint Martin Island, French West Indies, in December of 2013 (Leparc-Goffart et al., 2014). Distinct from the ECSA genotype causing expansion in the Old World, outbreaks in the New World are associated with the Asian genotype. Results from genetic characterization demonstrated that strains isolated in the New World possess a unique deletion of four amino acids in the nsP3 protein, only known to be present among the strains within the Asian lineage that are closely related to strains responsible for epidemics in Malaysia and Indonesia (Huang et al., 2009). The Asian genotype has been shown to have a slower rate of evolution than the ECSA genotype (Chen et al., 2016). Between December 2013 and March 2014, continuous spread of the virus was observed in multiple Caribbean Islands and French Guiana on the Northern Atlantic Coast of South America (Van Bortel et al., 2014). With the large numbers of travelers between the Caribbean Islands and the United States, the first local transmission in the United States occurred in Florida in June 2014 (Kendrick et al., 2014). By October of 2014, multiple countries in Central and South America reported local transmission including Brazil, Colombia, and Venezuela (Laiton-Donato et al., 2015; Nunes et al., 2015). By the end of this outbreak, it was estimated that by 2015 the number of infections in the Americas exceeded 1.7 million. CHIKV strains collected during the epidemics in the Americas share the same origin under clade II of the Asian genotype, which had previously caused epidemics in Indonesia, China, Philippines, Micronesia, and American Samoa (Chen et al., 2016). Interestingly, the spread of CHIKV after its introduction to the New World was likely to be bidirectional (Sahadeo et al., 2017). In a published deep sequencing analysis based on clinical samples obtained during the outbreaks in Martinique and Guadeloupe, it was found that genetic mutations were detected in a significantly higher frequency in the nonstructural proteins and 3′ untranslated region (UTR) than the structural proteins (Stapleford et al., 2016). A unique but functionally undetermined genetic characteristic is a 177-nucleotide duplication present in the 3′ UTR among available isolates in the Americas. Since there is no consistent selective advantage caused by the duplication in arthropod vectors and vertebrate hosts, it remains unclear why such a genetic signature is conserved in the Asian genotype (Chen et al., 2013; Stapleford et al., 2016).
After two major outbreaks of CHIKV in the 21st century, the geographic distribution of at least two genotypes of CHIKV has significantly changed. The acquisition of the E1 A226V mutation led to the selective advantage in nature by utilizing Ae. albopictus as its vector and resulted in the expansion of the ECSA genotype to islands in the Indian Ocean and Southeast Asia. After the introduction of the Asian genotype into the Western Hemisphere, the introduction of the ECSA genotype has also been observed in Brazil in 2014. Interestingly, the strains isolated in the autochthonous transmissions in the New World do not carry the E1 A226V mutation indicating that dispersal is possibly an independent event from the previously observed outbreaks (Souza et al., 2017). Collection of infected mosquitoes in the field further suggests that Ae. aegypti remains the primary vector species for the strains of CHIKV ECSA genotype in the Americas (Costa-da-Silva et al., 2017). After 2015, as case numbers of CHIKV decline in the New World, whether both genotypes of CHIKV can be established through autochthonous transmission remains an important but unanswered question.
Viral Genetics of CHIKV and Molecular Mechanisms Determining Vector Competence
Genetic studies on CHIKV and its relationship with mosquito vectors have provided several excellent models for our understanding in the range of competent vector species utilized by alphaviruses. Prior to Ae. albopictus becoming a known competent vector species for CHIKV, the first scientific question to be addressed was the difference in the choice of competent vector species in nature in spite of the high degree of sequence homology between the genomes of CHIKV and ONNV. Historically, CHIKV is vectored by several arboreal Aedes species mosquitoes and Ae. aegypti in its sylvatic and urban transmission cycles, respectively. Although Ae. aegypti can be experimentally infected with ONNV under laboratory conditions, epidemiological evidence suggests that in nature, Anopheles species mosquitoes are competent vectors for the transmission of ONNV (Vanlandingham et al., 2005b). During two major epidemics of ONNV outbreaks in Africa in the 20th century, transmission of ONNV was likely to be mediated by Anopheles gambiae and An. funestus since large number of isolates were derived from these species (Williams et al., 1965; Lutwama et al., 1999).
After the reverse genetics system of both viruses became available (Keene et al., 2004; Vanlandingham et al., 2005a), a common approach taken by multiple groups of arbovirologists to investigate the mechanisms responsible for the vector choice of CHIKV and ONNV was to characterize the infection process of chimeric viruses consisting of the genetic fragments from CHIKV and ONNV in Ae. aegypti and An. gambiae. The evidence derived from oral infection of chimeric viruses suggests that multiple genetic loci in both structural and nonstructural genes are involved in the choice of competent vector species. While Ae. aegypti can be experimentally infected by ONNV and chimeric CHIKV-ONNV viruses, the persistent infection of An. gambiae up to 14 days postinfection (d.p.i.) could only be efficiently established by chimeric CHIKV-ONNV viruses that contained the structural genes of ONNV (Vanlandingham et al., 2006). The importance of the nonstructural genes of ONNV in its establishment of infection was subsequently demonstrated as insertion of the nsP3 gene significantly increases the chimeric CHIKV-ONNV in An. gambiae at 8 d.p.i. (Saxton-Shaw et al., 2013). Together, the available evidence highlights the complexity of the infection process of CHIKV and ONNV in two different mosquito species as multiple viral proteins have been shown to control infection and dissemination processes.
After its emergence in the Indian Ocean, the focus of research on CHIKV quickly shifted to identification of mechanisms responsible for the increased incidence of diseases and transmission. The characterization of genetic variants of CHIKV further expanded our knowledge in arbovirology by showing how changes in viral genetics can change the phenotypes of viruses in mosquitoes and ultimately increase public health consequences associated with the virus. As the case numbers continued to increase and severe and neurological forms of the diseases were reported, the first systematic examination of genetic sequences involved in the Indian Ocean outbreaks was performed on specimens collected from 127 patients (Schuffenecker et al., 2006). Characterization of viral genetics identified several unique mutations in both the structural and nonstructural genes. Among all the mutations present in the genome, purification of variants containing the A226V mutation in the E1 glycoprotein was observed during multiple outbreaks. Although it was initially absent in the early phase of outbreaks in 2005, frequency of the E1 A226V mutation increased over time and was found in over 90% of isolates obtained after 2006, indicating a selective advantage associated with the point mutation (Schuffenecker et al., 2006). Although severe neurological and hemorrhagic symptoms, which were rarely associated with CHIKV in humans (Borgherini et al., 2007), were observed in hospitalized cases, the characterization of isolates obtained during the outbreak did not show demonstrable differences in virulence in mammalian hosts (Sourisseau et al., 2007; Chen et al., 2010).
In contrast to the lack of phenotypic differences observed in mammalian hosts, the characterization of epidemic strains of CHIKV in mosquitoes provided critical evidence that the emergence of the ECSA genotype was attributed to the greater epidemic potential caused by the E1 A226V mutation. Historically, entomological surveys indicated a majority of islands in the Indian Ocean were not infested with the urban vector of CHIKV, namely, Ae. aegypti. The finding was supported by observations that transmissions of DENV during previous epidemics were mainly supported by Ae. albopictus (Coulanges et al., 1979; Metselaar et al., 1980). Therefore, it became evident that the anthropophilic populations of Ae. albopictus on the islands are likely to be the competent vector to support the transmission (World Health Organization, 2006; Higgs, 2006; Pialoux et al., 2007). This hypothesis was subsequently validated by the study demonstrating that virus isolates obtained during outbreaks are more adapted to Ae. albopictus (Vazeille et al., 2007). Available isolates were capable of developing disseminated infection at a higher incidence and replicating to higher titers in Ae. albopictus than in Ae. aegypti. Using the reverse genetics system based on the LR2006 OPY1 strain (Tsetsarkin et al., 2006), the mechanism for the selective advantage observed in epidemic strains in the Indian Ocean was determined. The presence of the A226V mutation in the E1 glycoprotein significantly reduced the viremic titers required for the establishment of infection and increased the incidence of disseminated infection in Ae. albopictus. The competition between two lineages of recombinant viruses further demonstrated that an engineered virus developed in the laboratory possessing the E1 A226V mutation consistently surpass the wild-type recombinant virus in replication kinetics and transmission efficiency in Ae. albopictus (Tsetsarkin et al., 2007). Since the outbreaks in the Indian Ocean in 2006, variants of the ECSA genotype carrying the E1 A226V mutation have been extensively found in the autochthonous transmissions in Africa, Southeast Asia, and Europe (de Lamballerie et al., 2008). Its rapid dispersal combined with the invasion of Ae. albopictus in various geographic locations continues to post a significant threat to human public health around the world.
Clearly, emergence of the Indian Ocean lineage within the ECSA genotype due to the acquisition of the E1 A226V mutation has been a significant turning point in the public health significance of CHIKV. Another critical knowledge gap that needed to be addressed was why the acquisition of the E1 A226V mutation specifically increased the epidemic potential of the Indian Ocean lineage under the ECSA genotype but not other genotypes. Ae. albopictus is widely distributed and can be abundantly found in Asia. The earliest report of its breeding populations in Africa could be dated back to 1991 (Savage et al., 1992). It seems reasonable, therefore, to assume that the endemic status of CHIKV in both regions could have already created an environment for adaptation in Ae. albopictus to occur. This assumption is supported by the observation of “evolutionary convergence” of CHIKV indicating that emergence of the E1 A226V mutation occurred independently multiple times in different genotypes (de Lamballerie et al., 2008). For example, after the outbreaks occurred in the Indian Ocean, the E1 A226V mutation was also detected in isolates of the Indian lineage of the ECSA genotype in India and Sri Lanka and the Central African genotype in Gabon (Pages et al., 2009; Hapuarachchi et al., 2010). Interestingly, it was subsequently found that the adaptation to Ae. albopictus caused by acquisition of the E1 A226V mutation was specific to the Indian Ocean lineage of the ECSA genotype. When the mutation was engineered into the Ag41855 strain, which belongs to the ECSA genotype, there was no significant increase in the selective advantage compared to the effects created by the G60D and I211T mutations in the E2 glycoprotein (Tsetsarkin et al., 2009). Additionally, the attempt to introduce the A226V mutations in the E1 glycoprotein of the Asian lineage demonstrated that the endemic genotype in Asia was unable to be adapted to Ae. albopictus by the acquisition of the single E1 A226V mutation due to the presence of the A98T substitution in the E1 protein (Tsetsarkin et al., 2011). This genetic signature has been conserved throughout the majority of strains within both clades of the Asian genotypes, including those currently present in the New World (Chen et al., 2016). These findings further indicate that the adaptation of CHIKV to Ae. albopictus observed during the outbreaks in the Indian Ocean might be a consequence of viral evolution influenced by available vector species and ecological conditions. Therefore, after introduction of the ECSA genotype into regions in the New World where Ae. albopictus is endemic, whether or not its adaptation to Ae. albopictus will occur has become another critical question in assessing the epidemic potential of CHIKV in the future.
Deliberate Attenuation of CHIKV by Genetic Mutations
Although the need for efficacious vaccines for CHIKV was not recognized until the 21st century, vaccine development has been driven by severe arthritis that immobilizes infected individuals. In addition to considering formalin-inactivated or killed vaccines, the call for vaccine development also included attempts to develop live-attenuated vaccine candidates by serially passage of the virulent AF15561 strain in vitro. The result was the attenuated 181/25 strain as a vaccine candidate for long-lasting immune responses (Levitt et al., 1986). Although the vaccine candidate induced neutralizing antibodies among vaccinees and was unable to be transmitted by mosquitoes (Turell and Malinoski, 1992; Edelman et al., 2000), the incidence of adverse events, especially arthritis, precluded its further advancement in clinical trials. By introducing the mutations identified in the genome of the 181/25 strain into the virulent LR2006 OPY1 strain, a potential mechanism for its attenuation was proposed based on an increased binding affinity between the E2 G82R mutation and glycosaminoglycans on the cell surface (Gorchakov et al., 2012). This mechanism was subsequently applied to attenuation of the LR2006 OPY1 strain by introducing positively charged amino acid mutations in the same region (Gardner et al., 2014). Although this mechanism has been demonstrated to cause attenuation of various alphaviruses and flaviviruses, it remains unclear if the phenotype of high-binding affinity to glycosaminoglycans will be sufficient for the production of safe vaccine candidates.
In addition to the conventional approach of generating attenuated vaccine candidates by the empirical passage of virulent strains in vitro, the other interesting approach taken to attenuate CHIKV was achieved by decreasing the genetic heterogeneity in the viral population. As demonstrated in several RNA viruses including polio, West Nile, and yellow fever (Vignuzzi et al., 2008; Van Slyke et al., 2012; Beck et al., 2014), the establishment of infection and disease pathogenesis in vivo is often promoted by the higher level of genetic heterogeneity. By deliberately introducing mutations into the RdRp, especially the structure of nucleotide-binding pocket, fidelity of viral replication can be significantly increased thereby suppressing the error-prone nature of viral replication among RNA viruses (Vignuzzi et al., 2008; Van Slyke et al., 2012). Because of the lack of available structures for the RdRp of CHIKV, an efficient alternative approach was to select RNA mutagen-resistant phenotypes among serially passage CHIKV. While the mechanisms that induce high replication fidelity remain unknown, the C483Y mutation in the nsP4 protein was shown to significantly reduce the mutation rate, subsequently resulting in attenuation in mosquitoes and vertebrate hosts (Coffey et al., 2011). The finding is consistent with others’ findings in other arboviruses and potentially provides an approach for the attenuation of CHIKV through genetic engineering.
As the emergence of CHIKV continues in multiple geographic locations, it has become apparent that the conventional definition of genetic lineages based on the locations of virus isolation has changed. Its establishment in the New World has brought significant changes to its epidemiology. As the numbers of urban transmission have significantly declined since 2016, it is still unclear if sylvatic transmission cycles can be established among the primate species in the Americas. More importantly, after the introduction of the ECSA genotype was reported in Brazil, whether or not the evolution will ultimately lead to the similar adaptation to Ae. albopictus, which is also present as an invasive species in the Americas, remains an unanswered question. It is clear that an effective surveillance system that detects transmission and monitors the evolution of CHIKV is still needed to understand how the continuous evolution of CHIKV will determine its epidemic potential and disease burden.
ZIKV and Its Molecular Biology
Shortly after the emergence of CHIKV in the Indian Ocean, an outbreak of ZIKV took place in Yap State, Federal States of Micronesia, and initiated the unprecedented emergence in the 21st century. As described in Chapters 4 and 5, while the virus has been known to be endemic in Africa and Asia, it has long been regarded a neglected tropical disease due to the lack of knowledge regarding its pathogenesis and recognition of severe forms of diseases. This significant lack of knowledge about the evolution and viral genetics of ZIKV had not begun to be addressed until outbreaks in the New World were observed.
The first isolate of ZIKV named as the prototypic Rhesus 766 strain was made from a sentinel monkey in Uganda in 1947 (Dick et al., 1952). As described in Chapter 2, additional isolates were subsequently made from Ae. africanus and Ae. luteocephalus and suggested that the virus is likely to be maintained in the sylvatic cycles between arboreal Aedes mosquitoes and primates in Africa (Weinbren and Williams, 1958; Lee and Moore, 1972). Evidence of its transmission in the Middle East, South Asia, and Southeast Asia was first established by detection of neutralizing antibodies in serological surveys (Smithburn, 1954; Smithburn et al., 1954a,b; Hammon et al., 1958). Isolation of ZIKV from Ae. aegypti and febrile patients indicated that continuous urban transmission among immunologically naive humans of ZIKV may be an important mechanism for viral maintenance in Asia (Marchette et al., 1969; Olson et al., 1981). While isolations of ZIKV continued to be made in the endemic region, our understanding of its molecular epidemiology was very limited compared to other medically important flaviviruses transmitted by Ae. aegypti such as DENV and YFV. Genetic characterization of ZIKV began based on the full-length genome sequence of the prototype MR-766 strain (Kuno and Chang, 2007). Based on the genomic sequences, ZIKV is placed in the Spondweni virus (SPONV) group under the family of Flaviviridae (Grard et al., 2010). The genetic homology between ZIKV and SPONV is consistent with findings in the resemblance of clinical symptoms and serological crossreactivity, which subsequently led to misidentification of etiological agents in several outbreaks in Africa (Macnamara, 1954; Simpson, 1964). While both viruses are genetically related, it remains unclear why ZIKV has a greater epidemic potential and a more aggressive dispersal pattern than SPONV, which remains confined to Subsaharan Africa. A likely explanation for the distinct epidemiological patterns between the two related viruses may be multifactorial as multiple urban and sylvatic vertebrate hosts and arthropod vectors are known to be involved in the maintenance and transmission of both ZIKV and SPONV.
The virion of ZIKV resembles the structures of other related mosquito- borne flaviviruses (Sirohi et al., 2016). The single-stranded positive-sense RNA genome is encapsidated in the icosahedral protein shell and complexed with multiple copies of the C protein. The icosahedral shell contains 180 copies of the envelope (E) protein and membrane (M) protein inserted through the transmembrane domain of each protein. Each monomer of the E protein can be further divided into three distinct domains, domains I (EDI), II (EDII), and III (EDIII). Two neighboring copies of EDII create the dimerization interface and arrange two monomers of E protein into the antiparallel dimeric structure as shown in Fig. 4. Similar to other flaviviruses, the structure of ZIKV EDIII projects from the surface of the virion and promotes its binding with cellular receptors. Such a protruding structure of EDIII often contributes to the antigenic properties of flaviviruses including ZIKV. Although ZIKV and DENV share a sympatric endemic geographic distribution and utilize similar vector species, a distinction in the antigenic structures of both viruses can be found in the CD loop structure of EDIII. Similar to other flaviviruses in JEV and YFV serocomplexes, the CD loop of ZIKV EDIII contains the insertion of a single amino acid residue containing small hydrophobic sidechains (Xie et al., 2017). While the CD loop of ZIKV EDIII is different from that of DENV and its involvement in the formation of hydrogen bonds (Kostyuchenko et al., 2016), this residue is less likely to be a genetic determinant for virulence and tissue tropism because of its presence in the attenuated YFV 17D strains (Hahn et al., 1987; Volk et al., 2007). The envelope protein of ZIKV is further modified by glycosylation at the Asn154 residue. This glycosylation pattern resembles the envelope protein of YFV, which is only glycosylated at the Asn155 residue, and is distinct from four serotypes of DENV, which contains two glycosylation sites, Asn67 and Asn153, in the E protein (Post et al., 1992; Bryant et al., 2007). The distinct patterns of glycosylation among different flaviviruses possibly reflect the difference in the utilization of cellular receptors as the Asn67 and Asn155 residues of DENV-2 E protein are critical for binding with dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN) (Alen et al., 2012). It may also suggest the possibility that ZIKV may utilize a different set of receptor molecules than DENV (Sirohi et al., 2016). Interestingly, the glycosylation site among variants of the MR766 prototype strain is removed due to deletions spanning 4–6 amino acids in the neighboring region, which presumably occurred during the process of in vitro passage. A similar deletion that removes the glycosylation site was found in the envelope protein of the IbH 30656 strain, an isolate from an infected human in Nigeria in 1968. Because of the heterogeneity in sequences containing the glycosylation site among isolates with different passage history, modification of glycosylation in the envelope protein is not essential for the replication of ZIKV in vitro (Haddow et al., 2012). It also suggests that loss of the glycosylation site may be involved in its adaptation mechanisms to the environment in tissue culture as absence of the glycosylation site delays the peak of infectious titers in vitro and may likely prevent the early cytopathic effects (Widman et al., 2017).

Similar to other flaviviruses, one single ORF is encoded in the positive-sense genome of ZIKV and later translated into a polyprotein in infected cells. The overall genome organization of ZIKV also resembles other members within the genus of flavivirus under the family of Flaviviridae as shown in Fig. 5 (Kuno and Chang, 2007). Three structural genes, C, premembrane (prM), and E, are located in the 5′ end of the genome. Seven nonstructural genes, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5, are encoded in the 3′ end of the genome. The modification in the 5′ end of the genome with a methylated guanosine nucleoside forms the cap structure that is critical for stability of the viral genome in infected hosts (Coutard et al., 2017). The 3′ end of the genome lacks a polyadenylation structure but is presumably involved in circularization of the viral genome as observed with other flaviviruses (Villordo and Gamarnik, 2009).

Infectious viruses infect permissive tissues through receptor-mediated viral entry through the interactions between cellular receptors and viral EDIII. Recent publications suggest that ZIKV may utilize the tyrosine-protein kinase receptor UFO encoded by the AXL gene in humans as its unique receptor (Hamel et al., 2015; Liu et al., 2016b; Meertens et al., 2017). The acidic environment in late endosomes triggers the viral membrane fusion by inserting the fusion loop located in domain II of the E protein and allows release of the positive-sense viral genome, which serves as the messenger RNA for the polyprotein (Harrison, 2008; Nour et al., 2013). Production of the nonstructural proteins shortly after viral entry allows formation of a viral replication complex, which produces negative-sense RNA templates for replication and the nascent viral genomic RNA. Cotranslational and posttranslational modifications by viral and host proteases are made to the polyprotein inserted into the membrane structure of ER to release individual viral proteins. The complex structure between NS2B and NS3 functions as a serine protease that processes the polyprotein structure and gives rise to individual nonstructural proteins required for viral replication (Chambers et al., 1990). Prior to formation of the ribonucleic acid complex encapsidated in progeny virions, the C protein is released by cleavage of the NS2B-NS3 serine protease in the basic amino acid residues in its C-terminus (Stocks and Lobigs, 1998). The conserved amino acid residues in the polyprotein of ZIKV for posttranslational processing indicate release of the C protein of ZIKV is likely to share the same mechanism with other flaviviruses. Signalase cleavage in the C-prM, prM-E, and E-NS1 junctions releases prM and E proteins, the two major surface components of infectious virions, and allows the morphogenesis of progeny virions. Images derived from electron microscopy indicate that virion assembly through the interactions between the prM and E proteins occurs close to the ER (Mackenzie and Westaway, 2001). The prM protein functions as a molecular chaperone for the proper folding and secretion of the E protein (Konishi and Mason, 1993; Lorenz et al., 2002). The initial assembly of the heterodimeric structure of the prM and E proteins in the neutral environmental of ER leads to the formation of 60 copies of spike structure, each consisting of three copies of the heterodimer. The reduced pH in the secretory pathway promotes the maturation of the virion and ultimately leads to the smooth surface covered by 90 copies of the homodimers of E proteins arranged in an antiparallel fashion (Yu et al., 2008). During this process, the fusion loop structure in EDII is protected by the pr peptide in the prM protein to prevent premature fusion. Removal of the pr peptide is achieved by the host furin protease in the trans-Golgi network and subsequently grants infectivity to the virions secreted into the extracellular space.
Phylogenetics of ZIKV and Genetic Mutations Associated With Emerging Strains
Since the end of its outbreak in Micronesia in 2007, the phylogenetics and evolution of ZIKV began to be systematically examined in multiple studies based on available sequences of individual genes or entire open reading frames. The endemic strains found in Africa and Asia represent two major lineages (clades) in the phylogenetic trees (Lanciotti et al., 2008; Haddow et al., 2012). Retrospective analyses on the available African isolates further suggest that ZIKV strains found in Africa can be further divided into two distinct clusters: the MR766 prototype cluster and the Nigerian cluster (Faye et al., 2014). Viruses under these two clusters have been continuously detected in two West African countries, Senegal and Côte d’Ivoire, indicating that at least since 1990, simultaneous circulation of genetically distinct viruses is highly likely in this region. Except for an isolate from Ae. aegypti and two isolates from humans, the majority of African isolates used to demonstrate the cocirculation of two clusters of ZIKV in West Africa were obtained from sylvatic Aedes species mosquitoes and nonhuman primates. While a retrospective screening of ZIKV viral RNA in human sera and mosquitoes in Gabon suggests that strains under the Nigerian cluster may have been associated with the outbreaks in 2007 (Grard et al., 2014), a critical knowledge gap remains to be addressed concerning whether or not viruses within both clusters have also been maintained in urban transmission cycles and caused human diseases in West Africa.
In contrast to the genetic diversity observed in Africa, isolates of ZIKV in Asia belong to one single cluster (Haddow et al., 2012; Faye et al., 2014). This remains unchanged after large numbers of isolates were obtained after outbreaks in French Polynesia and Brazil. Genetically, isolates in Latin America all share the common ancestor found in French Polynesia (Faria et al., 2016). In addition to the observation that all isolates within the Asian lineage are placed in a common clade, a fundamental difference in its evolution from the African lineage was found to be a relatively higher mutation rate (Yokoyama and Starmer, 2017). The rapid evolution may result in a higher likelihood for accumulation of mutations that alter the tissue tropism and pathogenic outcomes and increase the epidemic potential. Therefore, the search for genetic mutations becomes an important approach to investigate pathogenic mechanisms of the emerging strains, especially for the causal link between genetic mutations and neurotropic diseases. However, there is still no definitive evidence indicating that emerging strains within the Asian lineage possess any fitness advantage when replicating in mammalian hosts or arthropod vectors (Weger-Lucarelli et al., 2016).
Several mutations in the genomes of ZIKV isolates from French Polynesia, where Guillian-Barré syndrome caused by ZIKV was first reported, and from Brazil, where microcephaly was reported to be associated with infection during pregnancy, have been identified (Faria et al., 2016; Pettersson et al., 2016). Notably, a D394E point mutation in the E protein was reported among the majority of isolates made since the outbreak in Micronesia (Pettersson et al., 2016). The mutation is located in the FG loop structure of EDIII. Based on evidence from available structures of flavivirus EDIIIs and mutagenesis studies, the FG loop region is located on the upper lateral region of EDIII and has been considered as a putative receptor-binding region of EDIII. Mutations in this region have previously been shown to alter the efficiency of systemic spread in mammalian hosts and also to modulate the infectivity of DENV-2 and YFV in Ae. aegypti (Lee and Lobigs, 2008; Erb et al., 2010; Huang et al., 2014). Since the substitution of an aspartate residue with a glutamate residue is likely to cause only minor changes in structural and biochemical properties, the impact of such a conservative mutation remains unclear in the functions of ZIKV EDIII. Another mutation observed in the structural proteins was the S17N mutation in the prM protein. This mutation emerged coincidentally with the report of neurotropic diseases as described in two independent studies (Faria et al., 2016; Pettersson et al., 2016). Because of its location in the pr peptide portion of the prM protein, which is removed by host furin cleavage in order to remove its steric hindrance to the fusion peptide in EDII (Li et al., 2008), this mutation is therefore absent from the structure of infectious virions. Based on the available structure of the prM-E heterodimer of DENV-2, the S17N mutation is likely to be located between the second and third ß-strands positioned away from the residues directly interacting with the fusion loop. While the mutation cannot have direct influence on the structure and function of infectious virions secreted from infected cells, mutations in the pr peptide region of flavivirus prM protein may still have significant functional importance in vitro and in vivo. For instance, the L36F mutation in the prM protein of YFV 17D vaccine strains significantly impaired the infection process of the virulent Asibi strain in Ae. aegypti (McElroy et al., 2006). The introduction of the same phenylalanine substitution into the same residue in the prM protein of JEV was also shown to reduce the efficiency in the assembly of progeny virions in vitro and the virulence in vivo (de Wispelaere et al., 2015). Further characterization of the S17N mutation with reverse genetics system and recombinant virus-like particle systems will be necessary to further advance our understanding on the potential phenotypic changes associated with the mutation.
In addition to the hypothesis suggesting that the increased epidemic potential and incidence of neurotropic diseases can be caused by genetic mutations in the structural proteins, the enhancement of pathological outcomes can also be due to mutations in the nonstructural proteins, which are involved in replication and immunomodulation of flaviviruses in infected hosts. Although the functions of flavivirus NS1 protein in vivo are not fully understood, as mentioned in Chapter 2, a recent report demonstrated that the secreted form of ZIKV NS1 protein in infected vertebrate hosts plays a significant role in the establishment of infection in Ae. aegypti. This finding is similar to those observed in DENV and JEV (Liu et al., 2016a). Additionally, a significant gain in the function of NS1 protein was reported to be associated with an A188V mutation, which was observed among endemic strains of ZIKV isolated in various outbreaks since 2012. The infection of ZIKV, promoted by the antigenemia of NS1, in mosquitoes was shown to be further enhanced by the A188V mutation (Liu et al., 2017). The functional importance of the NS1 A188V mutation is established by at least two lines of evidence. The NS1-A188V mutant was demonstrated to be significantly more infectious in Ae. aegypti. Additionally, blockade of the function of the NS1 protein by polyclonal antibodies further supports its functional importance in vivo. The causal link between the mutation and the increased epidemic potential of ZIKV can only be established when tested with infectious mutant viruses.
At least four mutations have been identified in the NS4B and NS5 proteins of ZIKV isolates associated with neurotropic diseases through phylogenetic analysis (Pettersson et al., 2016). The acquisition of the A/G36S mutation in the NS4B protein was first observed among the French Polynesian isolates and conserved in the isolates in Latin America. It was also shown to be a genetic determinant distinguishing the recent isolates from the first isolate of the Asian lineage, Malaysia_HQ234499 strain in 1966. Although the structures of full-length flavivirus NS4B proteins have not yet become available, its potential importance in the disease pathogenesis of ZIKV has been suggested by previously published data on related flaviviruses. Recombinant expression studies have shown that NS4B is a small hydrophobic viral protein inserted into the intracellular membrane and shown to promote the viral RNA synthesis in infected cells (Puig-Basagoiti et al., 2007). One of its known functions is the impairment of type-I interferon (IFN) signaling pathways mainly through the functions mediated by its N-terminus to create an advantageous environment for viral infection (Munoz-Jordan et al., 2003, 2005). Mutations in the NS4B proteins of different flaviviruses have further been shown to contribute to disease pathogenesis in mammalian hosts in vivo (Wicker et al., 2006, 2012; Puig-Basagoiti et al., 2007; Grant et al., 2011). Therefore, the acquisition of the NS4B A/G36S mutation is a likely cause for the change of tissue tropism or the augmentation of immunomodulation and disease pathogenesis in vertebrate hosts. Based on the available structure and predicted membrane topology of the DENV-2 NS4B protein, the naturally occurring A/G36S mutation is located in the N-terminus of NS4B protein and exposed in the ER lumen (Li et al., 2015). This region has previously been demonstrated to contain the motif required for the suppression of type-I IFN immunity (Munoz-Jordan et al., 2005). Interestingly, a single P38G mutation in the homologous region of WNV NS4B protein led to the loss of neurovirulence and differential outcomes in the induction of immune responses. As previously demonstrated in the animal models, the NS4B P38G mutation in WNV led to different innate and T-cell immune responses in vivo indicating genetic compositions in this region may be critical for protective immunity and disease pathogenesis (Welte et al., 2011). It is highly likely that the NS4B A/G36S mutation of ZIKV may lead to different capabilities in immunomodulation and, ultimately, changes in the outcomes of infection. In addition to the potential enhanced capability in immunomodulation, an alternative hypothesis for severe diseases caused by ZIKV is the direct enhancement in viral replication as three other additional mutations, A/T526I, G/R587K, and R/S647N, are found in the NS5 protein. Because all three mutations are located in motif A of ZIKV NS5 protein, critical for the binding of divalent ions in order to promote nucleotide polymerization, the hypothesis is challenged by the reality that none of the three mutations are located in the regions that are involved in the nucleotide-binding or enzymatic functions of the NS5 RdRp domain and there is no demonstrable difference in the enzymatic activities between the MR766 prototype strain and the Brazil/PE243/205 strain (Zhao et al., 2017).
While mutations can be quickly identified by nucleotide sequencing, there have been several apparent knowledge and technical gaps that preclude our further understanding in how viral genetics can be a critical factor for the emergence of ZIKV. Because of its previous status as a highly neglected arbovirus, the first significant challenge in identifying the link between genetic mutations and phenotypic changes is to determine whether identification of genetic mutations is the result of a more intensive attempt in sequencing the available isolates, i.e., a relatively small number of previously available isolates failed to identify the polymorphism in the amino acid residues. In addition to the lack of available sequences from earlier isolates, the development of its reverse genetics systems did not begin until its emergence. Although several cDNA-based infectious clones have become available (Shan et al., 2016; Tsetsarkin et al., 2016; Weger-Lucarelli et al., 2017), the toxicity of flavivirus genomes requires the manipulation of genetic materials to take place in platforms based upon low-copy-number plasmids and further creates the technical difficulties in characterizing genetic mutations. However, the interdisciplinary research using reverse genetics to study changes in virulence, disease pathogenesis and vector competence caused by novel genetic mutations remains the fundamental approach to investigate the mechanisms responsible for the emergence of ZIKV.
Manipulation of Viral Genetics to Generate Vaccine Candidates for ZIKV
As the etiological agent for a previously neglected tropical disease, functional characterization of individual viral proteins and other genomic regions of ZIKV through mutagenesis analyses did not begin until its recent emergence. The lack of such analyses was a significant obstacle in identifying the virulence factors responsible for human diseases observed during its recent emergence but also precluded vaccine development, especially the development of live-attenuated vaccines. As described in Chapter 11, similar to other viral diseases, live-attenuated vaccines for ZIKV are expected to have several critical advantages including higher immunogenicity, long-lasting memory immune responses, and less booster immunizations required. While the knowledge directly related to ZIKV is not immediately available, strategies for manipulation of viral genetics to generate live-attenuated vaccine candidates have been quickly developed based on previous success in attenuating related flaviviruses.
One of the proposed approaches to generate live-attenuated vaccine candidates for ZIKV is the deletion of the 3′ UTR (Shan et al., 2017). Although the region in ZIKV 3′ UTR subjected to mutagenesis is different, the removal of noncoding elements in the 3′ UTR has been highly successfully in attenuating at least three different serotypes of DENV and further applied to development of DENV vaccines (Men et al., 1996; Durbin et al., 2001). ZIKV deletion mutants in this study were generated by removing 10–30 nucleotides in close proximity to the conserved sequence 2 (CS2) region in the 3′ UTR of ZIKV genome (Kuno and Chang, 2007). The CS2 region forms the highly conserved dumbbell 1 (DB1) RNA secondary structure present in the majority of mosquito-borne flaviviruses (Olsthoorn and Bol, 2001; Gritsun and Gould, 2006). Disruption of the DB1 structure in the genome of DENV-2 led to reduction of translation and replication of the viral genome (Manzano et al., 2011). The phenotype of reduced viral RNA synthesis in vitro was also observed in ZIKV deletion mutants in this study. The vaccine candidate ZIKV 10-del further showed a low viremic profile and high immunogenicity in the A129 mouse model. This live-attenuated vaccine candidate is also significantly less infectious to Ae. aegypti than its wild-type parental strain. While the initial observations on immunogenicity and safety provide the basis for further advancement in trials and evaluations, further evaluation in nonhuman primates and clinical trials remains to be completed.
The other promising approach to developing live-attenuated vaccine candidates for ZIKV is through the chimerization of immunogenic structural proteins with attenuated strains of related flaviviruses (Xie et al., 2017). Chimerization of two related flaviviruses allows the presentation of immunogenic components and often leads to the attenuation of wild-type virulent viruses in the presence of genetic mutations that contribute to the phenotype of attenuated strains. The strategy has been known for its success in generating vaccine candidates for DENV, JEV, and WNV by inserting structural genes of wild-type viruses into the backbone of attenuated YFV 17D vaccine strain (Guirakhoo et al., 2001). Similarly, the approach has also been used by chimerizing structural proteins derived from four different serotypes of DENV on the backbones of attenuated strains. This approach can be regarded as a general platform to generate live-attenuated vaccine candidates for medically important flaviviruses. In addition to protective humoral immune responses induced by structural proteins, cell-mediated immune responses can also partially be induced by conserved T-cell epitopes present in nonstructural proteins (Gao et al., 2008; Koo et al., 2009). Unlike the previous approach using an attenuated strain as its backbone, a vaccine candidate for ZIKV was generated by the chimerization between two wild-type flaviviruses, ZIKV strain FSS13025 and DENV-2 strain D2Y98P (Xie et al., 2017). The chimeric virus was constructed by replacing the prM and E genes of DENV-2 D2Y98P strain with the prM and E genes of ZIKV FS13025 strain. Interestingly, the choice of using the virulent DENV-2 D2Y98P strain as the backbone of the vaccine candidate did not result in virulent phenotypes in vivo even if an attenuated variant of DENV-2 D2Y98P is available (Grant et al., 2011). The initial evaluation of virulence showed reduced viremic titers caused by the chimeric virus in the immunocompromised A129 mouse model compared to both parental viruses. Presence of neutralizing antibodies and protection against the challenge of wild-type viruses further demonstrate its immunogenicity in vaccinated animals. A plausible explanation for attenuated phenotypes of the chimeric virus is likely to be attenuation caused by chimerization. Although detailed mechanisms remain unknown, the general attenuating effect as a result of genetic chimerization has also been reported in DENV and YFV in several independent studies (Whitehead et al., 2003; Blaney Jr. et al., 2004; McElroy et al., 2006).
Despite the initial success in the safety and immunogenicity of live-attenuated vaccine candidates generated by manipulation of viral genetics, several major knowledge gaps remain to be addressed. Although protection induced by vaccination has been observed in immunocompromised mouse models, it is evident that the evaluation of safety and immunogenicity among attenuated strains must be performed in nonhuman primates prior to advancement into clinical trials. The most significant concern is that long-lasting immunity induced by live-attenuated vaccines against ZIKV may result in an increase in disease outcomes of other flaviviruses through antibody-dependent enhancement (ADE). This becomes an apparent challenge if immunization programs targeting DENV and ZIKV are implemented in the same region. Potential solutions to ADE may be the continued research to identify crossreactive B-cell epitopes, which can be strategically removed in the process of rationale-based vaccine development.
Conclusions
While the scope of this chapter is confined to CHIKV and ZIKV, it is certain that emergence of arboviruses will continue to be a significant challenge to human public health worldwide. Genetic mutations will clearly act as a potential mechanism contributing to emergence of some obscure arboviruses. This was the case for CHIKV; however, only limited evidence indicating the recent emergence of ZIKV is a consequence of the accumulation of mutations. The continuous advancement of technologies will certainly promote growth of our knowledge in how genetic changes can shape phenotypes of arboviruses and ultimately change the epidemiology and disease burden. Laboratory tools will continue to be improved for higher sensitivity and throughput. In addition to the delineation of mechanisms responsible for the emergence, increase of virulence and attenuation by searching for specific mutations that carry functional importance, several knowledge gaps remain to be addressed. In contrast to dependence on specific host species involved in transmission cycles and maintenance of all other viruses, a shared distinction among all arboviruses is the involvement of arthropod vectors in nature. Therefore, in addition to the presence of susceptible amplification hosts, how the environmental factors driving expansion of vector species can be involved and shape the evolution of arboviruses remain unclear. When new susceptible host and competent vector species are present, much in the process of adaptation remains unknown. The clear involvement of genetic mutations in adaptation has been repeatedly demonstrated for multiple arboviruses during various outbreaks. In addition to the adaptation of CHIKV for Ae. albopictus, the dispersal of WNV through North America after its introduction in 1999 is one of the best examples. The success of WNV is attributed to its capability of infecting hundreds of avian species and greater than sixty mosquito species in the United States. As a result of the rapid dispersal, continuous evolution taking place in the past two decades leads to the emergence of new genotypes that can be efficiently transmitted and maintained in enzootic cycles (Ebel et al., 2004; Mann et al., 2013). Detailed mechanisms responsible for selection and dispersal of new genetic variants remain to be further investigated. As the information of viral genetics increases and becomes more readily available, a new focus of viral genetics will be translating knowledge at the molecular level into practical applications to improve human public health. Although determination of genetic sequences allows the descriptive analysis of variants and formulation of hypotheses for functional studies, incorporation of structural biological and biophysical knowledge remains limited. Ultimately, further investigations of these factors will lead to a comprehensive understanding of how genetic variants of arboviruses can adapt and be selected in vertebrate hosts or arthropod vectors.
Because transmission and maintenance of arboviruses often require multiple species in complicated ecological conditions, the ultimate goal of formulating predictive models that anticipate the outcomes of evolution and adaptation remains challenging. Optimistically, as more mutagenesis studies are conducted, one hopes that universal models for the attenuation of medically important arboviruses will become more realistic and be applied as a solution to public health threats created by the emergence of arboviruses.