Congenital Adrenal Hyperplasia and Related Disorders
Perrin C. White
Congenital adrenal hyperplasia (CAH) is a family of autosomal recessive disorders of cortisol biosynthesis (normal adrenal steroidogenesis is discussed in Chapter 592 ). Cortisol deficiency increases secretion of corticotropin (adrenocorticotropic hormone [ACTH]), which, in turn, leads to adrenocortical hyperplasia and overproduction of intermediate metabolites. Depending on the enzymatic step that is deficient, there may be signs, symptoms, and laboratory findings of mineralocorticoid deficiency or excess; incomplete virilization or precocious puberty in affected males; and virilization or sexual infantilism in affected females (Figs. 594.1 and 594.2 , Table 594.1 ).
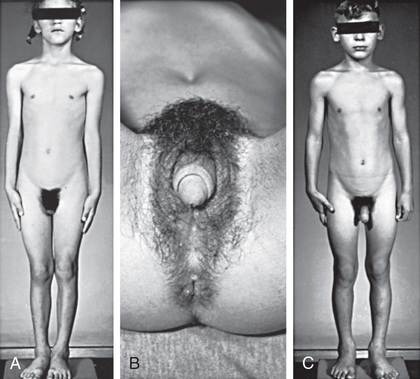

Table 594.1
Diagnosis and Treatment of Congenital Adrenal Hyperplasia
| DISORDER | AFFECTED GENE AND CHROMOSOME | SIGNS AND SYMPTOMS | LABORATORY FINDINGS | THERAPEUTIC MEASURES |
|---|---|---|---|---|
| 21-Hydroxylase deficiency, classic form |
CYP21 6p21.3 |
Glucocorticoid deficiency |
↓ Cortisol, ↑ACTH ↑↑ Baseline and ACTH-stimulated 17-hydroxy-progesterone |
Glucocorticoid (hydrocortisone) replacement |
| Mineralocorticoid deficiency (salt-wasting crisis) |
Hyponatremia, hyperkalemia ↑ Plasma renin |
Mineralocorticoid (fludrocortisone) replacement; sodium chloride supplementation | ||
| Ambiguous genitalia in females | ↑ Serum androgens | Vaginoplasty and clitoral recession | ||
| Postnatal virilization in males and females | ↑ Serum androgens | Suppression with glucocorticoids | ||
| 21-Hydroxylase deficiency, nonclassic form |
CYP21 6p21.3 |
May be asymptomatic; precocious adrenarche, hirsutism, acne, menstrual irregularity, infertility |
↑ Baseline and ACTH-stimulated 17-hydroxyprogesterone ↑ Serum androgens |
Suppression with glucocorticoids |
| 11β-Hydroxylase deficiency |
CYP11B1 8q24.3 |
Glucocorticoid deficiency | ↓ Cortisol, ↑ ACTH | Glucocorticoid (hydrocortisone) replacement |
| ↑↑ Baseline and ACTH-stimulated 11-deoxycortisol and deoxycorticosterone | ||||
| Ambiguous genitalia in females | ↑ Serum androgens | Vaginoplasty and clitoral recession | ||
| Postnatal virilization in males and females | ↑ Serum androgens | Suppression with glucocorticoids | ||
| Hypertension | ↓ Plasma renin, hypokalemia | Suppression with glucocorticoids | ||
| 3β-Hydroxysteroid dehydrogenase deficiency, classic form |
HSD3B2 1p13.1 |
Glucocorticoid deficiency |
↓ Cortisol, ↑ ACTH ↑↑ Baseline and ACTH-stimulated Δ5 steroids (pregnenolone, 17-hydroxy-pregnenolone, DHEA) |
Glucocorticoid (hydrocortisone) replacement |
| Mineralocorticoid deficiency (salt-wasting crisis) |
Hyponatremia, hyperkalemia ↑ Plasma renin |
Mineralocorticoid (fludrocortisone) replacement; sodium chloride supplementation | ||
| Ambiguous genitalia in females and males | ↑ DHEA, ↓ androstenedione, testosterone, and estradiol | Surgical correction of genitals and sex hormone replacement as necessary, consonant with sex of rearing | ||
| Precocious adrenarche, disordered puberty | ↑ DHEA, ↓ androstenedione, testosterone, and estradiol | Suppression with glucocorticoids | ||
| 17α-Hydroxylase/17,20-lyase deficiency |
CYP17 10q24.3 |
Cortisol deficiency (corticosterone is an adequate glucocorticoid) |
↓ Cortisol, ↑ ACTH ↑ DOC, corticosterone Low 17α-hydroxylated steroids; poor response to ACTH |
Glucocorticoid (hydrocortisone) administration |
| Ambiguous genitalia in males | ↓ Serum androgens; poor response to hCG | Orchidopexy or removal of intraabdominal testes; sex hormone replacement consonant with sex of rearing | ||
| Sexual infantilism | ↓ Serum androgens or estrogens | Sex hormone replacement consonant with sex of rearing | ||
| Hypertension | ↓ Plasma renin; hypokalemia | Suppression with glucocorticoids | ||
| Congenital lipoid adrenal hyperplasia |
STAR 8p11.2 |
Glucocorticoid deficiency |
↑ ACTH Low levels of all steroid hormones, with decreased or absent response to ACTH |
Glucocorticoid (hydrocortisone) replacement |
| Mineralocorticoid deficiency (salt-wasting crisis) |
Hyponatremia, hyperkalemia ↓ Aldosterone, ↑ plasma renin |
Mineralocorticoid (fludrocortisone) replacement; sodium chloride supplementation | ||
| Ambiguous genitalia in males | Decreased or absent response to hCG in males | Orchidopexy or removal of intraabdominal testes; sex hormone replacement consonant with sex of rearing | ||
| Poor pubertal development or premature ovarian failure in females | ↑ FSH, ↑ LH, ↓ estradiol (after puberty) | Estrogen replacement | ||
| P450 oxidoreductase deficiency |
POR 7q11.3 |
Glucocorticoid deficiency |
↓ Cortisol, ↑ ACTH ↑ Pregnenolone, ↑ progesterone |
Glucocorticoid (hydrocortisone) replacement |
| Ambiguous genitalia in males and females | ↑ Serum androgens prenatally, ↓ androgens and estrogens at puberty | Surgical correction of genitals and sex hormone replacement as necessary, consonant with sex of rearing | ||
|
Maternal virilization Antley-Bixler syndrome |
Decreased ratio of estrogens to androgens |
↓, Decreased; ↑, increased; ↑↑, markedly increased; ACTH , Adrenocorticotropic hormone; DHEA , dehydroepiandrosterone; DOC , 11-deoxycorticosterone; FSH , follicle-stimulating hormone; hCG , human chorionic gonadotropin; LH , luteinizing hormone.
Congenital Adrenal Hyperplasia Caused by 21-Hydroxylase Deficiency
Perrin C. White
Keywords
- salt-wasting
- simple virilizing disease
- classic 21-hydroxylase deficiency
- nonclassic
- Ehlers-Danlos syndrome
- newborn screening
- rapid somatic growth
- accelerated skeletal maturation
- testicular adrenal rest tumors
- nonclassic 21-hydroxylase deficiency
- Hirsutism
- acne
- menstrual disorders
- infertility
- 17-hydroxyprogesterone
Etiology
More than 90% of CAH cases are caused by 21-hydroxylase deficiency. This P450 enzyme (CYP21, P450c21) hydroxylates progesterone and 17-hydroxyprogesterone to yield 11-deoxycorticosterone and 11-deoxycortisol, respectively (see Fig. 592.1 in Chapter 592 ). These conversions are required for synthesis of aldosterone and cortisol, respectively. Both hormones are deficient in the most-severe, salt-wasting form of the disease. Slightly less-severely affected patients are able to synthesize adequate amounts of aldosterone but have elevated levels of androgens of adrenal origin; this is termed simple virilizing disease . These 2 forms are collectively termed classic 21-hydroxylase deficiency . Patients with nonclassic disease have relatively mildly elevated levels of androgens and may be asymptomatic or have signs of androgen excess at any time after birth. Clinical presentation is dependent, in part, on the genotype (see later, Genetics) (Table 594.2 ).
Table 594.2
Genotype-Phenotype Correlations in Congenital Adrenal Hyperplasia Owing to 21-Hydroxylase Deficiency
| MUTATION GROUP | A | B | C |
|---|---|---|---|
| Enzymatic activity, % normal | Nil | 1–2% | 20–50% |
| CYP21 mutations (phenotype generally corresponds to the least affected allele) |
Gene deletion Exon 3 del 8 bp Exon 6 cluster Q318X R356W |
I172N |
P30L V281L P453S |
| Intron 2 splice* | |||
| Severity | Salt wasting | Simple virilizing | Nonclassic |
| Aldosterone synthesis | Low | Normal | Normal |
| Age at diagnosis (without newborn screening) | Infancy |
Infancy (females) Childhood (males) |
Childhood to adulthood, or asymptomatic |
| Virilization | Severe | Moderate to severe | None to mild |
| Incidence | 1/20,000 | 1/50,000 | 1/500 |
* This mutation is associated with both salt wasting and simple virilizing disease.
Epidemiology
Classic 21-hydroxylase deficiency occurs in approximately 1 in 15,000-20,000 births in most populations. Approximately 70% of affected infants have the salt-losing form, whereas 30% have the simple virilizing form of the disorder. In the United States, CAH is less common in African Americans compared with white children (1 : 42,000 vs 1 : 15,500). Nonclassic disease has a prevalence of approximately 1 in 1,000 in the general population but occurs more frequently in specific ethnic groups such as Ashkenazi Jews and Hispanics.
Genetics
There are 2 steroid 21-hydroxylase genes—CYP21P (CYP21A1P, CYP21A) and CYP21 (CYP21A2, CYP21B) —which alternate in tandem with 2 genes for the 4th component of complement (C4A and C4B) in the human leukocyte antigen (HLA) major histocompatibility complex on chromosome 6p21.3 between the HLA-B and HLA-DR loci. Many other genes are located in this cluster. CYP21 is the active gene; CYP21P is 98% identical in DNA sequence to CYP21 but is a pseudogene because of 9 different mutations. Although almost 300 mutations have been reported, more than 90% of mutant alleles causing 21-hydroxylase deficiency are the result of recombinations between CYP21 and CYP21P. Approximately 20% are deletions generated by unequal meiotic crossing-over between CYP21 and CYP21P, whereas the remainder are nonreciprocal transfers of deleterious mutations from CYP21P to CYP21, a phenomenon termed gene conversion.
The deleterious mutations in CYP21P have different effects on enzymatic activity when transferred to CYP21. Several mutations completely prevent synthesis of a functional protein, whereas others are missense mutations (they result in amino acid substitutions) that yield enzymes with 1–50% of normal activity. Disease severity correlates well with the mutations carried by an affected individual; for example, patients with salt-wasting disease usually carry mutations on both alleles that completely destroy enzymatic activity. Patients are frequently compound heterozygotes for different types of mutations (i.e., 1 allele is less-severely affected than the other), in which case the severity of disease expression is largely determined by the activity of the less-severely affected of the 2 alleles.
Closely adjacent to, but on the opposite DNA strand from, CYP21 is the tenascin-X (TNX) gene, which encodes a connective tissue protein. Rarely, deletions of CYP21 extend into TNX . Such patients may have a contiguous gene syndrome (see Chapter 98.1 ) consisting of CAH and Ehlers-Danlos syndrome (see Chapters 511 and 678 ).
Pathogenesis and Clinical Manifestations
Aldosterone and Cortisol Deficiency
Because both cortisol and aldosterone require 21-hydroxylation for their synthesis, both hormones are deficient in the most-severe, salt-wasting form of the disease. This form constitutes approximately 70% of cases of classic 21-hydroxylase deficiency. The signs and symptoms of cortisol and aldosterone deficiency, and the pathophysiology underlying them, are essentially those described in Chapter 593 . These include progressive weight loss, anorexia, vomiting, dehydration, weakness, hypotension, hypoglycemia, hyponatremia, and hyperkalemia. These problems typically first develop in affected infants at approximately 10-14 days of age. Without treatment, shock, cardiac arrhythmias, and death may occur within days or weeks.
CAH differs from other causes of primary adrenal insufficiency in that precursor steroids accumulate proximal to the blocked enzymatic conversion. Because cortisol is not synthesized efficiently, ACTH levels are high, leading to hyperplasia of the adrenal cortex and levels of precursor steroids that may be hundreds of times normal. In the case of 21-hydroxylase deficiency, these precursors include 17-hydroxyprogesterone and progesterone. Progesterone and perhaps other metabolites act as antagonists of the mineralocorticoid receptor and thus may exacerbate the effects of aldosterone deficiency in untreated patients.
It is not unusual for children with classic CAH to require hospitalization for intercurrent illnesses during childhood. This is most likely to occur in the first 2 yr of life and to be precipitated by gastroenteritis, because such illnesses may cause fluid and electrolyte losses and vomiting may interfere with medication dosing. Children requiring high fludrocortisone doses are most likely to be hospitalized, presumably because those patients have the greatest propensity to salt wasting.
Prenatal Androgen Excess
The most important problem caused by accumulation of steroid precursors is that 17-hydroxyprogesterone is shunted into the pathway for androgen biosynthesis, leading to high levels of androstenedione that are converted outside the adrenal gland to testosterone. This problem begins in affected fetuses by 8-10 wk of gestation and leads to abnormal genital development in females (see Figs. 594.1 and 594.2 ).
The external genitalia of males and females normally appear identical early in gestation (see Chapter 600 ). Affected females who are exposed in utero to high levels of androgens of adrenal origin have masculinized external genitalia (see Figs. 594.1 and 594.2 ). This is manifested by enlargement of the clitoris and by partial or complete labial fusion. The vagina usually has a common opening with the urethra (urogenital sinus). The clitoris may be so enlarged that it resembles a penis; because the urethra opens below this organ, some affected females may be mistakenly presumed to be males with hypospadias and cryptorchidism. The severity of virilization is usually greatest in females with the salt-losing form of 21-hydroxylase deficiency (see Table 594.2 ). The internal genital organs are normal, because affected females have normal ovaries and not testes and thus do not secrete antimüllerian hormone.
Prenatal exposure of the brain to high levels of androgens may influence subsequent sexually dimorphic behaviors in affected females. Females may demonstrate aggressive play behavior, tend to be interested in masculine toys such as cars and trucks, and often show decreased interest in playing with dolls. Women may have decreased interest in maternal roles. There is an increased frequency of homosexuality in affected females. Nonetheless, most function heterosexually and do not have gender identity confusion or dysphoria. It is unusual for affected females to assign themselves a male role except in some with the severest degree of virilization.
Male infants appear normal at birth. Thus the diagnosis may not be made in males until signs of adrenal insufficiency develop. Because patients with this condition can deteriorate quickly, infant males are more likely to die than infant females. For this reason, all 50 American states and many countries have instituted newborn screening for this condition (see section on “Newborn Screening” in Chapter 594.2 ).
Postnatal Androgen Excess
Untreated or inadequately treated children of both sexes develop additional signs of androgen excess after birth. Males with the simple virilizing form of 21-hydroxylase deficiency often have a delayed diagnosis because they appear normal and rarely develop adrenal insufficiency.
Signs of androgen excess include rapid somatic growth and accelerated skeletal maturation . Thus affected patients are tall in childhood, but premature closure of the epiphyses causes growth to stop relatively early, and adult stature is stunted (see Fig. 594.1 ). Muscular development may be excessive. Pubic and axillary hair may appear, and acne and a deep voice may develop. The penis, scrotum, and prostate may become enlarged in affected males; however, the testes are usually prepubertal in size so that they appear small relative to the enlarged penis. Occasionally, ectopic adrenocortical cells in the testes of patients become hyperplastic similarly to the adrenal glands, producing testicular adrenal rest tumors (see Chapter 602 ). The clitoris may become further enlarged in affected females (see Fig. 594.1 ). Although the internal genital structures are female, breast development and menstruation may not occur unless the excessive production of androgens is suppressed by adequate treatment.
Similar but usually milder signs of androgen excess may occur in nonclassic 21-hydroxylase deficiency (see Table 594.2 ). In this attenuated form, cortisol and aldosterone levels are normal and affected females have normal genitals at birth. Males and females may present with precocious pubarche and early development of pubic and axillary hair. Hirsutism, acne, menstrual disorders, and infertility may develop later in life, but many females and males are completely asymptomatic.
Adrenomedullary Dysfunction
Development of the adrenal medulla requires exposure to the extremely high cortisol levels normally present within the adrenal gland. Thus patients with classic CAH have abnormal adrenomedullary function, as evidenced by blunted epinephrine responses, decreased blood glucose, and lower heart rates with exercise. Ability to exercise is unimpaired, and the clinical significance of these findings is uncertain. Adrenomedullary dysfunction may exacerbate the cardiovascular effects of cortisol deficiency in untreated or undertreated patients.
Laboratory Findings
See Table 594.1 .
Patients with salt-losing disease have typical laboratory findings associated with cortisol and aldosterone deficiency, including hyponatremia, hyperkalemia, metabolic acidosis, and, often, hypoglycemia, but these abnormalities can take 10-14 days or longer to develop after birth. Blood levels of 17-hydroxyprogesterone are markedly elevated. However, levels of this hormone are high during the first 2-3 days of life even in unaffected infants and especially if they are sick or premature. After infancy, once the circadian rhythm of cortisol is established, 17-hydroxyprogesterone levels vary in the same circadian pattern, being highest in the morning and lowest at night. Blood levels of cortisol are usually low in patients with the salt-losing type of disease. They are often normal in patients with simple virilizing disease but inappropriately low in relation to the ACTH and 17-hydroxyprogesterone levels. In addition to 17-hydroxyprogesterone, levels of androstenedione and testosterone are elevated in affected females; testosterone is not elevated in affected males, because normal infant males have high testosterone levels compared with those seen later in childhood. Levels of urinary 17-ketosteroids and pregnanetriol are elevated but are now rarely used clinically because blood samples are easier to obtain than 24-hr urine collections. ACTH levels are elevated but have no diagnostic utility over 17-hydroxyprogesterone levels. Plasma levels of renin are elevated, and serum aldosterone is inappropriately low for the renin level. However, renin levels are high in normal infants in the 1st few wk of life.
Diagnosis of 21-hydroxylase deficiency is most reliably established by measuring 17-hydroxyprogesterone before and 30 or 60 min after an intravenous bolus of 0.125-0.25 mg of cosyntropin (ACTH 1-24). Nomograms exist that readily distinguish normals and patients with nonclassic and classic 21-hydroxylase deficiency. Heterozygous carriers of this autosomal recessive disorder tend to have higher ACTH-stimulated 17-hydroxyprogesterone levels than genetically unaffected individuals, but there is significant overlap between subjects in these 2 categories. However, in infants with frank electrolyte abnormalities or circulatory instability, it may not be possible or necessary to delay treatment to perform this test, as levels of precursors will be sufficiently elevated on a random blood sample to make the diagnosis.
Genotyping is clinically available and may help to confirm the diagnosis, but it is expensive and may take weeks. Because the gene conversions that generate most mutant alleles may transfer more than one mutation, at least 1 parent should also be genotyped to determine which mutations lie on each allele.
Differential Diagnosis
Disorders of sexual development are discussed more generally in Chapter 606 . The initial step in evaluating an infant with ambiguous genitalia is a thorough physical examination to define the anatomy of the genitals, locate the urethral meatus, palpate the scrotum or labia and the inguinal regions for testes (palpable gonads almost always indicate the presence of testicular tissue and thus that the infant is a genetic male), and look for any other anatomic abnormalities. Ultrasonography is helpful in demonstrating the presence or absence of a uterus and can often locate the gonads. A rapid karyotype (such as fluorescence in situ hybridization of interphase nuclei for X and Y chromosomes) can quickly determine the genetic sex of the infant. These results are all likely to be available before the results of hormonal testing and together allow the clinical team to advise the parents as to the genetic sex of the infant and the anatomy of internal reproductive structures. Injection of contrast medium into the urogenital sinus of a virilized female demonstrates a vagina and uterus, and many surgeons use this information to formulate a plan for surgical management.
Prenatal Diagnosis
Prenatal diagnosis of 21-hydroxylase is possible late in the 1st trimester by analysis of DNA obtained by chorionic villus sampling or during the 2nd trimester by amniocentesis. This is usually done because the parents already have an affected child. Most often, the CYP21 gene is analyzed for frequently occurring mutations; more rare mutations may be detected by DNA sequencing.
Newborn Screening
Because 21-hydroxylase deficiency is often undiagnosed in affected males until they have severe adrenal insufficiency, all states in the United States and many other countries have instituted newborn screening programs. These programs analyze 17-hydroxyprogesterone levels in dried blood obtained by heelstick and absorbed on filter paper cards; the same cards are screened in parallel for other congenital conditions, such as hypothyroidism and phenylketonuria. Potentially affected infants are typically quickly recalled for additional testing (electrolytes and repeat 17-hydroxyprogesterone determination) at approximately 2 wk of age. Infants with salt-wasting disease often have abnormal electrolytes by this age but are usually not severely ill. Thus screening programs are effective in preventing many cases of adrenal crisis in affected males. The nonclassic form of the disease is not reliably detected by newborn screening, but this is of little clinical significance because adrenal insufficiency does not occur in this type of 21-hydroxylase deficiency.
The main difficulty with current newborn screening programs is that to reliably detect all affected infants, the cutoff 17-hydroxyprogesterone levels for recalls are set so low that there is a very high frequency of false-positive results (i.e., the test has a low positive predictive value of as little as 1%). This problem is worst in premature infants. Positive predictive value can be improved by using cutoff levels based on gestational age and by using more specific 2nd-tier screening methods such as liquid chromatography followed by tandem mass spectrometry.
Treatment
Glucocorticoid Replacement
Cortisol deficiency is treated with glucocorticoids. Treatment also suppresses excessive production of androgens by the adrenal cortex and thus minimizes problems such as excessive growth and skeletal maturation and virilization. This often requires larger glucocorticoid doses than are needed in other forms of adrenal insufficiency, typically 15-20 mg/m2 /24 hr of hydrocortisone daily administered orally in 3 divided doses. Affected infants usually require dosing at the high end of this range. Double or triple doses are indicated during periods of stress, such as infection or surgery. Glucocorticoid treatment must be continued indefinitely in all patients with classic 21-hydroxylase deficiency but may not be necessary in patients with nonclassic disease unless signs of androgen excess are present. Therapy must be individualized. It is desirable to maintain linear growth along percentile lines; crossing to higher height percentiles may suggest undertreatment, whereas loss of height percentiles often indicates overtreatment with glucocorticoids. Overtreatment is also suggested by excessive weight gain. Pubertal development should be monitored by periodic examination, and skeletal maturation is evaluated by serial radiographs of the hand and wrist for bone age. Hormone levels, particularly 17-hydroxyprogesterone and androstenedione, should be measured early in the morning, before taking the morning medications, or at a consistent time in relation to medication dosing. In general, desirable 17-hydroxyprogesterone levels are in the high-normal range or several times normal; low-normal levels can usually be achieved only with excessive glucocorticoid doses. Alternative delivery modalities have undergone small clinical trials, including delayed release hydrocortisone tablets and the use of a continuous subcutaneous insulin infusion device (insulin pump) to deliver hydrocortisone in a pattern more closely approximating the normal diurnal variation in cortisol secretion. At present, these approaches have not entered clinical practice.
Menarche occurs at the appropriate age in most females in whom good control has been achieved; it may be delayed in females with suboptimal control. Children with simple virilizing disease, particularly males, are frequently not diagnosed until 3-7 yr of age, at which time skeletal maturation may be 5 yr or more in advance of chronological age. In some children, especially if the bone age is 12 yr or more, spontaneous central (i.e., gonadotropin-dependent) puberty may occur when treatment is instituted, because therapy with hydrocortisone suppresses production of adrenal androgens and thus stimulates release of pituitary gonadotropins if the appropriate level of hypothalamic maturation is present. This form of superimposed true precocious puberty may be treated with a gonadotropin hormone–releasing hormone analog such as leuprolide (see Chapter 578.1 ).
Males with 21-hydroxylase deficiency who have had inadequate corticosteroid therapy may develop testicular adrenal rest tumors , which usually regress with increased steroid dosage. Testicular MRI, ultrasonography, and color flow Doppler examination help to define the character and extent of disease. Testis-sparing surgery for steroid-unresponsive tumors has been reported.
Mineralocorticoid Replacement
Patients with salt-wasting disease (i.e., aldosterone deficiency) require mineralocorticoid replacement with fludrocortisone. Infants may have very high mineralocorticoid requirements in the 1st few mo of life, usually 0.1-0.3 mg daily in 2 divided doses but occasionally up to 0.4 mg daily, and often require sodium supplementation (sodium chloride, 8 mmol/kg) in addition to the mineralocorticoid. Older infants and children are usually maintained with 0.05-0.1 mg daily of fludrocortisone. In some patients, simple virilizing disease may be easier to control with a low dose of fludrocortisone in addition to hydrocortisone even when these patients have normal aldosterone levels in the absence of mineralocorticoid replacement. Therapy is evaluated by monitoring of vital signs; tachycardia and hypertension are signs of overtreatment with mineralocorticoids. Serum electrolytes should be measured frequently in early infancy as therapy is adjusted. Plasma renin activity is a useful way to determine adequacy of therapy; it should be maintained in or near the normal range but not suppressed.
Additional approaches to improve outcome have been proposed but have not yet become the standard of care. These include an antiandrogen such as flutamide to block the effects of excessive androgen levels, and/or an aromatase inhibitor such as anastrozole, which blocks conversion of androgens to estrogen and thus retards skeletal maturation, a process that is sensitive to estrogens in both males and females. Aromatase inhibitors generally should not be used in pubertal females because they will retard normal puberty and may expose the ovaries to excessive levels of gonadotropins. Growth hormone, with or without luteinizing hormone–releasing hormone agonists to retard skeletal maturation, has been suggested to improve adult height.
Surgical Management of Ambiguous Genitals
Significantly virilized females usually undergo surgery between 2 and 6 mo of age. If there is severe clitoromegaly, the clitoris is reduced in size, with partial excision of the corporal bodies and preservation of the neurovascular bundle; however, moderate clitoromegaly may become much less noticeable even without surgery as the patient grows. Vaginoplasty and correction of the urogenital sinus usually are performed at the time of clitoral surgery; revision in adolescence is often necessary.
Risks and benefits of surgery should be fully discussed with parents of affected females. There is limited long-term follow-up of functional outcomes in patients who have undergone modern surgical procedures. It appears that female sexual dysfunction increases in frequency and severity in those with the most significant degrees of genital virilization and with the degree of enzymatic impairment (prenatal androgen exposure) caused by each patient's mutations (see Table 594.2 ). Sex assignment of infants with disorders of sexual differentiation (including CAH) is usually based on expected sexual functioning and fertility in adulthood with early surgical correction of the external genitalia to conform with the sex assignment. Gender dysphoria is not common with CAH; it occurs mostly in females with the salt-wasting form of the disease and the greatest degree of virilization.
Lay and medical opponents of genital surgery for other disorders of sexual differentiation bring up the concern that it ignores any prenatally biased gender role predisposition from androgen exposure and precludes the patient from having any decision as to the patient's own preferred sexual identity and what surgical correction of the genitals should be performed. They advocate that treatment should be aimed primarily at educating the patient, family, and others about the medical condition, its treatment, and how to deal with the intersex condition. They propose that surgery should be delayed until the patient decides on what, if any, correction should be performed. Not all lay groups support delayed surgery and many agree with appropriate surgery during infancy. Severely virilized genotypic (XX) females raised as males have generally functioned well in the male gender as adults.
In adolescent and adult females with poorly controlled 21-hydroxylase deficiency (hirsutism, obesity, amenorrhea), bilateral laparoscopic adrenalectomy (with hormone replacement) may be an alternative to standard medical hormone replacement therapy, but because the adrenal glands have been removed, patients treated in this way may be more susceptible to acute adrenal insufficiency if treatment is interrupted. Moreover, they may exhibit signs of elevated ACTH levels such as abnormal pigmentation.
Prenatal Treatment
Besides genetic counseling, the main goal of prenatal diagnosis is to facilitate prenatal treatment of affected females. Mothers with pregnancies at risk may be given dexamethasone, a steroid that readily crosses the placenta, in an amount of 20 µg/kg prepregnancy maternal weight daily in 2 or 3 divided doses. This suppresses secretion of steroids by the fetal adrenal, including secretion of adrenal androgens. If started by 6 wk of gestation, it ameliorates virilization of the external genitals in affected females. Chorionic villus biopsy is then performed to determine the sex and genotype of the fetus; therapy is continued only if the fetus is an affected female. DNA analysis of fetal cells isolated from maternal plasma for sex determination and CYP21 gene analysis may permit earlier identification of the affected female fetus. Treatment should be considered only in affected female fetuses. Children exposed to this therapy have slightly lower birthweights. Effects on personality or cognition, such as increased shyness, have been suggested but not consistently observed. At present there is insufficient information to determine whether the long-term risks are acceptable, particularly in the males and unaffected females who derive no direct benefit from the treatment. Maternal side effects of prenatal treatment have included edema, excessive weight gain, hypertension, glucose intolerance, cushingoid facial features, and severe striae. Consensus statements from professional societies recommend that prenatal treatment be carried out only under institutional protocols, but it is sometimes offered as an option outside the research setting by high-risk obstetricians in some locales.
Bibliography
DiSandro M, Merke DP, Rink RC. Review of current surgical techniques and medical management considerations in the treatment of pediatric patients with disorders of sex development. Horm Metab Res . 2015;47:321–328.
El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet . 2017;390:2194–2208.
Finkielstain GP, Chen W, Mehta SP, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab . 2011;96:E161–E172.
Finkielstain GP, Kim MS, Sinaii N, et al. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab . 2012;97:4429–4438.
Gidlöf S, Falhammar H, Thilén A, et al. One hundred years of congenital adrenal hyperplasia in Sweden: a retrospective, population-based cohort study. Lancet . 2013;1:35–42.
Gidlöf S, Wedell A, Guthenberg C, et al. Nationwide neonatal screening for congenital adrenal hyperplasia in Sweden. JAMA Pediatrics . 2014;168:567.
Mallappa A, Sinaii N, Kumar P, et al. A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab . 2015;100:1137–1145.
Merce Fernandez-Balsells M, Muthusamy K, Smushkin G, et al. Prenatal dexamethasone use for the prevention of virilization in pregnancies at risk for classical congenital adrenal hyperplasia because of 21-hydroxylase (CYP21A2) deficiency: a systematic review and meta-analyses. Clin Endocrinol (Oxf) . 2010;73:436–444.
Meyer-Bahlburg HF, Dolezal C, Baker SW, et al. Sexual orientation in women with classical or non-classical congenital adrenal hyperplasia as a function of degree of prenatal androgen excess. Arch Sex Behav . 2008;37:85–99.
Minette MS, Hoyer AW, Pham PP, et al. Cardiac function in congenital adrenal hyperplasia: a pattern of reversible cardiomyopathy. J Pediatr . 2013;162:1193–1198.
Muthusamy K, Elamin MB, Smushkin G, et al. Clinical review: adult height in patients with congenital adrenal hyperplasia: a systematic review and metaanalysis. J Clin Endocrinol Metab . 2010;95:4161–4172.
Nella AA, Mallappa A, Perritt AF, et al. A phase 2 study of continuous subcutaneous hydrocortisone infusion in adults with congenital adrenal hyperplasia. J Clin Endocrinol Metab . 2016;101:4690–4698.
Nordenstrom A, Frisen L, Falhammar H, et al. Sexual function and surgical outcome in women with congenital adrenal hyperplasia due to CYP21A2 deficiency: clinical perspective and the patients’ perception. J Clin Endocrinol Metab . 2010;95:3633–3640.
Speiser PW, Arlt W, Auchus RJ, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab . 2018;103(11):4043–4088.
White P. Update on diagnosis and management of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Curr Opin Endocrinol Diabetes Obes . 2018;25:178–184.
White PC. Neonatal screening for congenital adrenal hyperplasia. Nat Rev Endocrinol . 2009;5:490–498.
Witchel SF. Nonclassic congenital adrenal hyperplasia. Curr Opin Endocrinol Diabetes Obes . 2012;19:151–158.
Yang M, White PC. Risk factors for hospitalization of children with congenital adrenal hyperplasia. Clin Endocrinol (Oxf) . 2017;86(5):669–673.
Congenital Adrenal Hyperplasia Caused by 11β-Hydroxylase Deficiency
Perrin C. White
Etiology
Deficiency of 11β-hydroxylase is caused by a mutation in the CYP11B1 gene located on chromosome 8q24. CYP11B1 mediates 11-hydroxylation of 11-deoxycortisol to cortisol. Because 11-deoxycortisol is not converted to cortisol, levels of corticotropin are high. In consequence, precursors—particularly 11-deoxycortisol and deoxycorticosterone—accumulate and are shunted into androgen biosynthesis in the same manner as occurs in 21-hydroxylase deficiency. The adjacent CYP11B2 gene encoding aldosterone synthase is generally unaffected in this disorder, so patients are able to synthesize aldosterone normally.
Epidemiology
11β-Hydroxylase deficiency accounts for approximately 5% of cases of adrenal hyperplasia; its incidence in the general population has been estimated as 1 in 250,000 to 1 in 100,000. The disorder occurs relatively frequently in Israeli Jews of North African origin (1 in 15,000-17,000 live births). In this ethnic group, almost all alleles carry an Arg448 to His (R448H) mutation in CYP11B1 , but many other mutations have been identified. This disorder presents in a classic, severe form and very rarely in a nonclassic, milder form.
Clinical Manifestations
Although cortisol is not synthesized efficiently, aldosterone synthetic capacity is normal, and some corticosterone is synthesized from progesterone by the intact aldosterone synthase enzyme. Thus it is unusual for patients to manifest signs of adrenal insufficiency such as hypotension, hypoglycemia, hyponatremia, and hyperkalemia. Approximately 65% of patients become hypertensive, although this can take several years to develop. Hypertension is probably a consequence of elevated levels of deoxycorticosterone, which has mineralocorticoid activity. Infants may transiently develop signs of mineralocorticoid deficiency after treatment with hydrocortisone is instituted. This is presumably from sudden suppression of deoxycorticosterone secretion in a patient with atrophy of the zona glomerulosa caused by chronic suppression of renin activity.
All signs and symptoms of androgen excess that are found in 21-hydroxylase deficiency may also occur in 11β-hydroxylase deficiency.
Laboratory Findings
Plasma levels of 11-deoxycortisol and deoxycorticosterone are elevated. Because deoxycorticosterone and some metabolites have mineralocorticoid activity, plasma renin activity is suppressed. Consequently, aldosterone levels are low even though the ability to synthesize aldosterone is intact. Hypokalemic alkalosis occasionally occurs.
Treatment
Patients are treated with hydrocortisone in doses similar to those used for 21-hydroxylase deficiency. Mineralocorticoid replacement is sometimes transiently required in infancy but is rarely necessary otherwise. Hypertension often resolves with glucocorticoid treatment but may require additional therapy if it is of long standing. Calcium channel blockers may be beneficial under these circumstances.
Congenital Adrenal Hyperplasia Caused by 3β-Hydroxysteroid Dehydrogenase Deficiency
Perrin C. White
Etiology
Deficiency of 3β-hydroxysteroid dehydrogenase (3β-HSD) occurs in fewer than 2% of patients with adrenal hyperplasia. This enzyme is required for conversion of Δ5 steroids (pregnenolone, 17-hydroxypregnenolone, dehydroepiandrosterone [DHEA]) to Δ4 steroids (progesterone, 17-hydroxyprogesterone, and androstenedione). Thus deficiency of the enzyme results in decreased synthesis of cortisol, aldosterone, and androstenedione but increased secretion of DHEA (see Fig. 592.1 in Chapter 592 ). The 3β-HSD isozyme expressed in the adrenal cortex and gonad is encoded by the HSD3B2 gene located on chromosome 1p13.1. More than 30 mutations in the HSD3B2 gene have been described in patients with 3β-HSD deficiency.
Clinical Manifestations
Because cortisol and aldosterone are not synthesized in patients with the classic form of the disease, infants are prone to salt-wasting crises. Because androstenedione and testosterone are not synthesized, males are incompletely virilized. Varying degrees of hypospadias may occur, with or without bifid scrotum or cryptorchidism. Because DHEA levels are elevated and this hormone is a weak androgen, females are mildly virilized, with slight to moderate clitoral enlargement. Postnatally, continued excessive DHEA secretion can cause precocious adrenarche. During adolescence and adulthood, hirsutism, irregular menses, and polycystic ovarian disease occur in females. Males manifest variable degrees of hypogonadism, although appropriate male secondary sexual development may occur. However, a persistent defect of testicular 3β-HSD is demonstrated by the high Δ5:Δ4 steroid ratio in testicular effluent.
Laboratory Findings
The hallmark of this disorder is the marked elevation of the Δ5 steroids (such as 17-hydroxypregnenolone and DHEA) preceding the enzymatic block. Patients may also have elevated levels of 17-hydroxyprogesterone because of the extraadrenal 3β-HSD activity that occurs in peripheral tissues; these patients may be mistaken for patients with 21-hydroxylase deficiency. The ratio of 17-hydroxypregnenolone:17-hydroxyprogesterone is markedly elevated in 3β-HSD deficiency, in contrast to the decreased ratio in 21-hydroxylase deficiency. Plasma renin activity is elevated in the salt-wasting form.
Differential Diagnosis
It is not unusual for children with premature adrenarche, or women with signs of androgen excess, to have mild to moderate elevations in DHEA levels. It has been suggested that such individuals have nonclassic 3 β-HSD deficiency . Mutations in the HSD3B2 gene are usually not found in such individuals, and a nonclassic form of this deficiency must actually be quite rare. The activity of 3β-HSD in the adrenal zonae fasciculata and reticularis, relative to CYP17 (17-hydroxylase/17,20-lyase) activity, normally decreases during adrenarche to facilitate DHEA synthesis, and so modest elevations in DHEA in preteenage children or women usually represent a normal variant.
Treatment
Patients require glucocorticoid and mineralocorticoid replacement with hydrocortisone and fludrocortisone, respectively, as in 21-hydroxylase deficiency. Incompletely virilized genetic males in whom a male sex of rearing is contemplated may benefit from several injections of 25 mg of a depot form of testosterone every 4 wk early in infancy to increase the size of the phallus. They may also require testosterone replacement at puberty.
Congenital Adrenal Hyperplasia Caused by 17-Hydroxylase Deficiency
Perrin C. White
Etiology
Less than 1% of CAH cases are caused by 17-hydroxylase deficiency, but the condition is apparently more common in Brazil and China. A single polypeptide, CYP17, catalyzes 2 distinct reactions: 17-hydroxylation of pregnenolone and progesterone to 17-hydroxypregnenolone and 17-hydroxyprogesterone, respectively, and the 17,20-lyase reaction mediating conversion of 17-hydroxypregnenolone to DHEA and, to a lesser extent, 17-hydroxyprogesterone to Δ4-androstenedione. DHEA and androstenedione are steroid precursors of testosterone and estrogen (see Fig. 592.1 in Chapter 592 ). The enzyme is expressed in both the adrenal cortex and the gonads and is encoded by a gene on chromosome 10q24.3. Most mutations affect both the hydroxylase and lyase activities, but rare mutations can affect either activity alone.
Mutations in genes other than CYP17 can have the same phenotype as 17,20-lyase deficiency (i.e., deficient androgen synthesis with normal cortisol synthesis). These include an accessory electron transfer protein, cytochrome-b5 , and mutations in 2 aldo-keto reductases, AKR1C2 and AKR1C4. These AKR1C isozymes normally catalyze 3α-HSD activity, which allows synthesis of the potent androgen dihydrotestosterone through an alternative backdoor biosynthetic pathway that does not include testosterone as an intermediate.
Clinical Manifestations and Laboratory Findings
Patients with 17-hydroxylase deficiency cannot synthesize cortisol, but their ability to synthesize corticosterone is intact. Because corticosterone is an active glucocorticoid, patients do not develop adrenal insufficiency. Deoxycorticosterone, the immediate precursor of corticosterone, is synthesized in excess. This can cause hypertension, hypokalemia, and suppression of renin and aldosterone secretion, as occurs in 11β-hydroxylase deficiency. In contrast to 11β-hydroxylase deficiency, patients with 17-hydroxylase deficiency are unable to synthesize sex hormones. Affected males are incompletely virilized and present as phenotypic females (but gonads are usually palpable in the inguinal region or the labia) or with sexual ambiguity. Affected females usually present with failure of sexual development at the expected time of puberty. 17-Hydroxylase deficiency in females must be considered in the differential diagnosis of primary hypogonadism (see Chapter 604 ). Levels of deoxycorticosterone are elevated, and renin and aldosterone are consequently suppressed. Cortisol and sex steroids are unresponsive to stimulation with ACTH and human chorionic gonadotropin, respectively.
Patients with isolated 17,20-lyase deficiency have deficient androgen synthesis with normal cortisol synthesis and therefore do not become hypertensive.
Treatment
Patients with 17-hydroxylase deficiency require glucocorticoid replacement with hydrocortisone to suppress secretion of deoxycorticosterone and thus control hypertension. Additional antihypertensive medication may be required. Females require estrogen replacement at puberty. Genetic males may require either estrogen or androgen supplementation depending on the sex of rearing. Because of the possibility of malignant transformation of abdominal testes, as is more commonly encountered with androgen insensitivity syndrome (see Chapter 606.2 ), genetic males with severe 17-hydroxylase deficiency being reared as females require gonadectomy at or before adolescence.
Bibliography
Auchus RJ. The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab . 2004;15:432–438.
Idkowiak J, Randell T, Dhir V, et al. A missense mutation in the human cytochrome b5 gene causes 46,XY disorder of sex development due to true isolated 17,20 lyase deficiency. J Clin Endocrinol Metab . 2012;97:E465–E475.
Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab . 2009;23:181–192.
Miller WL. The syndrome of 17,20 lyase deficiency. J Clin Endocrinol Metab . 2012;97:59–67.
Lipoid Adrenal Hyperplasia
Perrin C. White
Etiology
Lipoid adrenal hyperplasia is a rare disorder, most frequently found in Japanese persons. Patients with this disorder exhibit marked accumulation of cholesterol and lipids in the adrenal cortex and gonads, associated with severe impairment of all steroidogenesis. Lipoid adrenal hyperplasia is usually caused by mutations in the gene for steroidogenic acute regulatory protein (StAR), a mitochondrial protein that promotes the movement of cholesterol from the outer to the inner mitochondrial membrane. However, mutations in the CYP11A1 gene (which encodes the cholesterol side chain cleavage enzyme) have been reported in several patients.
Some cholesterol is able to enter mitochondria even in the absence of StAR, so it might be supposed that this disorder would not completely impair steroid biosynthesis. However, the accumulation of cholesterol in the cytoplasm is cytotoxic, eventually leading to death of all steroidogenic cells in which StAR is normally expressed. This occurs prenatally in the adrenals and testes. The ovaries do not normally synthesize steroids until puberty, so cholesterol does not accumulate and the ovaries can retain the capacity to synthesize estrogens until adolescence.
Although estrogens synthesized by the placenta are required to maintain pregnancy, the placenta does not require StAR for steroid biosynthesis. Thus mutations of StAR are not prenatally lethal.
Clinical Manifestations
Patients with lipoid adrenal hyperplasia are usually unable to synthesize any adrenal steroids. Thus affected infants are likely to be confused with those with adrenal hypoplasia congenita. Salt-losing manifestations are typical, and many infants die in early infancy. Genetic males are unable to synthesize androgens and thus are phenotypically female but with gonads palpable in the labia majora or inguinal areas. Genetic females appear normal at birth and may undergo feminization at puberty with menstrual bleeding. They, too, progress to hypergonadotropic hypogonadism when accumulated cholesterol kills granulosa (i.e., steroid synthesizing) cells in the ovary.
Laboratory Findings
Adrenal and gonadal steroid hormone levels are low in lipoid adrenal hyperplasia, with a decreased or absent response to stimulation (ACTH, human chorionic gonadotropin). Plasma renin levels are increased.
Imaging studies of the adrenal gland demonstrating massive adrenal enlargement in the newborn help to establish the diagnosis of lipoid adrenal hyperplasia.
Treatment
Patients require glucocorticoid and mineralocorticoid replacement. Genetic males are usually assigned a female sex of rearing; thus both genetic males and females require estrogen replacement at the expected age of puberty.
Bibliography
Miller WL. StAR search—what we know about how the steroidogenic acute regulatory protein mediates mitochondrial cholesterol import. Mol Endocrinol . 2007;21:589–601.
Stocco DM. Clinical disorders associated with abnormal cholesterol transport: mutations in the steroidogenic acute regulatory protein. Mol Cell Endocrinol . 2002;191:19–25.
Tee MK, Abramsohn M, Loewenthal N, et al. Varied clinical presentations of seven patients with mutations in CYP11A1 encoding the cholesterol side-chain cleavage enzyme, P450scc. J Clin Endocrinol Metab . 2013;98:713–720.
Deficiency of P450 Oxidoreductase (Antley-Bixler Syndrome)
Perrin C. White
Etiology, Pathogenesis, and Clinical Manifestations
P450 oxidoreductase (POR; gene located on chromosome 7q11.3) is required for the activity of all microsomal cytochrome P450 enzymes (see Chapter 592 ), including the adrenal enzymes CYP17 and CYP21. Thus complete POR deficiency abolishes all microsomal P450 activity. This is embryonically lethal in mice and presumably also in humans. Patients with mutations that decrease but do not abolish POR activity have partial deficiencies of 17-hydroxylase and 21-hydroxylase activities in the adrenals. A single recurrent mutation A287P (alanine-287 to proline) is found on approximately 40% of alleles.
Deficiency of 17-hydroxylase leads to incomplete masculinization in males; 21-hydroxylase deficiency may lead to virilization in females. In addition, aromatase (CYP19) activity in the placenta is decreased, leading to unopposed action of androgens produced by the fetal adrenal. This exacerbates virilization of female fetuses and may virilize the mother of an affected fetus as well. Although it is puzzling that affected females could be virilized despite a partial deficiency in CYP17 (which is required for androgen biosynthesis), an alternative (backdoor) biosynthetic pathway is used in which 17-hydroxyprogesterone is converted to 5α-pregnane-3α,17α-diol-20-one, a metabolite that is a much better substrate for the 17,20-lyase activity of CYP17 than the usual substrate, 17-hydroxypregnenolone (see Chapter 592 ). The metabolite is then converted in several enzymatic steps to dihydrotestosterone, a potent androgen.
Because many other P450 enzymes are affected, patients often (but not invariably) have other congenital anomalies collectively referred to as Antley-Bixler syndrome. These include craniosynostosis; brachycephaly; frontal bossing; severe midface hypoplasia with proptosis and choanal stenosis or atresia; humeroradial synostosis; medial bowing of ulnas; long, slender fingers with camptodactyly; narrow iliac wings; anterior bowing of femurs; and malformations of the heart and kidneys. Studies of mutant mice suggest that the metabolic defects responsible for these anomalies include defective metabolism of retinoic acid, leading to elevated levels of this teratogenic compound, and deficient biosynthesis of cholesterol.
Epidemiology
The prevalence is not known with certainty. It must be rare compared with 21-hydroxylase deficiency but might occur at similar frequencies to the other forms of CAH.
Laboratory Findings
Serum steroids that are not 17- or 21-hydroxylated are most increased, including pregnenolone and progesterone. 17-Hydroxy, 21-deoxysteroids are also increased, including 17-hydroxypregnenolone, 17-hydroxyprogesterone, and 21-deoxycortisol. Urinary steroid metabolites may be determined by quantitative mass spectrometry. Metabolites excreted at increased levels include pregnanediol, pregnanetriol, pregnanetriolone, and corticosterone metabolites. Urinary cortisol metabolites are decreased. Genetic analysis demonstrates mutations in the POR gene.
Differential Diagnosis
This disorder must be distinguished from other forms of CAH, particularly 21-hydroxylase deficiency in females, which is far more common and has similar laboratory findings. Suspicion for POR deficiency may be raised if the mother is virilized or if the associated abnormalities of Antley-Bixler syndrome are present. Conversely, virilization of both the mother and her daughter can result from a luteoma of pregnancy , but in this case postnatal abnormalities of corticosteroid biosynthesis should not be observed. Antley-Bixler syndrome may also occur without abnormalities of steroid hormone biosynthesis, resulting from mutations in the fibroblast growth factor receptor FGFR2.
Bibliography
Krone N, Reisch N, Idkowiak J, et al. Genotype-phenotype analysis in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab . 2012;97:E257–E267.
Miller WL, Agrawal V, Sandee D, et al. Consequences of POR mutations and polymorphisms. Mol Cell Endocrinol . 2011;336:174–179.
Schmidt K, Hughes C, Chudek JA, et al. Cholesterol metabolism: the main pathway acting downstream of cytochrome P450 oxidoreductase in skeletal development of the limb. Mol Cell Biol . 2009;29:2716–2729.
Aldosterone Synthase Deficiency
Perrin C. White
Etiology
This is an autosomal recessive disorder in which conversion of corticosterone to aldosterone is impaired; a group of Iranian Jewish patients has been the most thoroughly studied. The majority of cases result from mutations in the CYP11B2 gene coding for aldosterone synthase; however, linkage to CYP11B2 has been excluded in other kindreds. When not caused by CYP11B2 mutations, the disorder has been termed familial hyperreninemic hypoaldosteronism type 2; the causative gene or genes have not yet been identified.
Aldosterone synthase mediates the 3 final steps in the synthesis of aldosterone from deoxycorticosterone (11β-hydroxylation, 18-hydroxylation, and 18-oxidation). Although 11β-hydroxylation is required to convert deoxycorticosterone to corticosterone, this conversion can also be catalyzed by the related enzyme, CYP11B1, located in the fasciculata, which is unaffected in this disorder. For the same reason, these patients have normal cortisol biosynthesis.
The disease has been classified into 2 types, termed corticosterone methyloxidase deficiency types I and II. They differ only in levels of the immediate precursor of aldosterone, 18-hydroxycorticosterone; levels are low in type I deficiency and elevated in type II deficiency. These differences do not correspond in a simple way to particular mutations and are of limited clinical importance.
Clinical Manifestations
Infants with aldosterone synthase deficiency may have severe electrolyte abnormalities with hyponatremia, hyperkalemia, and metabolic acidosis. Because cortisol synthesis is unaffected, infants rarely become as ill as untreated infants with salt-losing forms of CAH such as 21-hydroxylase deficiency. Thus some infants escape diagnosis. Later in infancy or in early childhood they may exhibit failure to thrive and poor growth. Adults often are asymptomatic, although they may develop electrolyte abnormalities when depleted of sodium through procedures such as bowel preparation for a barium enema.
Laboratory Findings
Infants have elevated plasma renin activity. Aldosterone levels are decreased; they may be at the lower end of the normal range but are always inappropriately low for the degree of hyperkalemia or hyperreninemia. Corticosterone levels are often elevated.
Some, but not all, patients have marked elevation of 18-hydroxycorticosterone; however, low levels of this steroid do not exclude the diagnosis. In those kindreds in which 18-hydroxycorticosterone levels are elevated in affected individuals, this biochemical abnormality persists in adults even when they have no electrolyte abnormalities.
Differential Diagnosis
It is important to distinguish aldosterone synthase deficiency from primary adrenal insufficiency in which both cortisol and aldosterone are affected (including salt-wasting forms of CAH), because the latter condition is usually associated with a much greater risk of shock and hyponatremia. This becomes apparent after the appropriate laboratory studies. Adrenal hypoplasia congenita may initially present with aldosterone deficiency; all male infants with apparently isolated aldosterone deficiency should be carefully monitored for subsequent development of cortisol deficiency. Pseudohypoaldosteronism (see Chapter 593.4 ) may have similar electrolyte abnormalities and hyperreninemia, but aldosterone levels are high, and this condition usually does not respond to fludrocortisone treatment.
Treatment
Treatment consists of giving enough fludrocortisone (0.05-0.3 mg daily) or sodium chloride, or both, to return plasma renin levels to normal. With increasing age, salt-losing signs usually improve and drug therapy can often be discontinued.
Bibliography
Leshinsky-Silver E, Landau Z, Unlubay S, et al. Congenital hyperreninemic hypoaldosteronism in Israel: sequence analysis of CYP11B2 gene. Horm Res . 2006;66:73–78.
Nguyen HH, Hannemann F, Hartmann MF, et al. Five novel mutations in CYP11B2 gene detected in patients with aldosterone synthase deficiency type I: functional characterization and structural analyses. Mol Genet Metab . 2010;100:357–364.
White PC. Aldosterone synthase deficiency and related disorders. Mol Cell Endocrinol . 2004;217:81–87.
Glucocorticoid-Suppressible Hyperaldosteronism
Perrin C. White
Etiology
Glucocorticoid-suppressible hyperaldosteronism (glucocorticoid-remediable aldosteronism, familial hyperaldosteronism type I) is an autosomal dominant form of low-renin hypertension in which hyperaldosteronism is rapidly suppressed by glucocorticoid administration. This unusual effect of glucocorticoids suggests that aldosterone secretion in this disorder is regulated by ACTH instead of by the renin-angiotensin system. In addition to abnormally regulated secretion of aldosterone, there is marked overproduction of 18-hydroxycortisol and 18-oxocortisol. The synthesis of these steroids requires both 17-hydroxylase (CYP17) activity, which is expressed only in the zona fasciculata, and aldosterone synthase (CYP11B2) activity, which is normally expressed only in the zona glomerulosa. Together, these features imply that aldosterone synthase is being expressed in a manner similar to the closely related enzyme steroid 11-hydroxylase (CYP11B1). The disorder is caused by unequal meiotic crossing-over events between the CYP11B1 and CYP11B2 genes, which are closely linked on chromosome 8q24. An additional “chimeric” gene is produced, having regulatory sequences of CYP11B1 juxtaposed with coding sequences of CYP11B2. This results in the inappropriate expression of a CYP11B2 -like enzyme with aldosterone synthase activity in the adrenal fasciculata.
Clinical Manifestations
Some affected children have no symptoms, the diagnosis being established after incidental discovery of moderate hypertension, typically approximately 30 mm Hg higher than unaffected family members of the same age. Others have more symptomatic hypertension with headache, dizziness, and visual disturbances. A strong family history of early-onset hypertension or early strokes may alert the clinician to the diagnosis. Some patients have chronic hypokalemia, but this is not a consistent finding and is usually mild.
Laboratory Findings
Patients have elevated plasma and urine levels of aldosterone and suppressed plasma renin activity. Hypokalemia is not consistently present. Urinary and plasma levels of 18-oxocortisol and 18-hydroxycortisol are markedly increased. The hybrid CYP11B1/CYP11B2 gene can be readily detected by molecular genetic methods.
Differential Diagnosis
This condition should be distinguished from primary aldosteronism based on bilateral hyperplasia or an aldosterone-producing adenoma (see Chapter 598 ). Most cases of primary aldosteronism are sporadic, although several affected kindreds have been reported. Patients with primary aldosteronism may also have elevated levels of 18-hydroxycortisol and 18-oxocortisol, and these biochemical tests should be used cautiously when attempting to distinguish primary and glucocorticoid-suppressible aldosteronism. By definition, a therapeutic trial of dexamethasone should suppress aldosterone secretion only in glucocorticoid-suppressible hyperaldosteronism, and genetic testing should identify the hybrid gene of glucocorticoid-suppressible hyperaldosteronism if it is present.
Treatment
Glucocorticoid-suppressible hyperaldosteronism is managed by daily administration of a glucocorticoid, usually dexamethasone, 25 µg/kg/day in divided doses. If necessary, effects of aldosterone can be blocked with a potassium-sparing diuretic such as spironolactone, eplerenone, or amiloride. Hypertension resolves in patients in whom the hypertension is not severe or of long standing. If hypertension is long standing, additional antihypertensive medication may be required, such as a calcium channel blocker.
Genetic Counseling
Because of the autosomal dominant mode of inheritance, at-risk family members should be investigated for this easily treated cause of hypertension.