Adrenocortical Insufficiency
Perrin C. White
In primary adrenal insufficiency, congenital or acquired lesions of the adrenal cortex prevent production of cortisol and often aldosterone (Table 593.1 ). Acquired primary adrenal insufficiency is termed Addison disease . Dysfunction of the anterior pituitary gland or hypothalamus can cause a deficiency of corticotropin (adrenocorticotropic hormone [ACTH]) and lead to hypofunction of the adrenal cortex, termed secondary adrenal insufficiency; the term tertiary adrenal insufficiency is sometimes used to denote cases arising from hypothalamic dysfunction (Table 593.2 ).
Table 593.1
Causes of Primary Adrenal Insufficiency
| PATHOGENESIS OR GENETICS | CLINICAL FEATURES IN ADDITION TO ADRENAL INSUFFICIENCY | |
|---|---|---|
| CONGENITAL ADRENAL HYPERPLASIA | ||
| 21-Hydroxylase deficiency | CYP21A2 mutations | Hyperandrogenism |
| 11β-Hydroxylase deficiency | CYP11B1 mutations | Hyperandrogenism, hypertension |
| 3β-Hydroxysteroid dehydrogenase type 2 deficiency | HSD3B2 mutations | Ambiguous genitalia in males, postnatal virilization in females |
| 17α-Hydroxylase deficiency | CYP17A1 mutations | XY sex reversal, pubertal delay in both sexes, hypertension |
| P450 oxidoreductase deficiency | POR mutations | Skeletal malformation (Antley-Bixler syndrome), abnormal genitalia |
| P450 side-chain cleavage deficiency | CYP11A1 mutations | XY sex reversal |
| Congenital lipoid adrenal hyperplasia | STAR mutations | XY sex reversal |
| OTHER GENETIC DISORDERS | ||
| Adrenoleukodystrophy or adrenomyeloneuropathy | ABCD1 mutations | Weakness, spasticity, dementia, blindness, quadriparesis. Adrenomyeloneuropathy is a milder variant of adrenoleukodystrophy with slower progression |
| Triple A syndrome (Allgrove syndrome) | AAAS mutations | Achalasia, alacrima, cognitive deficits, neuromuscular deficits, hyperkeratosis |
| Smith-Lemli-Opitz syndrome | DHCR7 mutations | Craniofacial malformations, developmental delay growth failure, cholesterol deficiency |
| Wolman disease | LIPA mutations | Bilateral adrenal calcification, hepatosplenomegaly |
| Kearns-Sayre syndrome | Mitochondrial DNA deletions | External ophthalmoplegia, retinal degeneration, cardiac conduction defects, other endocrine disorders |
| Pallister-Hall syndrome | GLI3 mutations | hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly |
| IMAGe syndrome | CDKN1C or POLE mutations | Intrauterine growth retardation, metaphyseal dysplasia, genital abnormalities |
| Adrenal Hypoplasia Congenita | ||
| X-linked | NR0B1 mutations | Hypogonadotropic hypogonadism in males |
| Xp21 contiguous gene syndrome | Deletion of genes for Duchenne muscular dystrophy, glycerol kinase, and NR0B1 | Duchenne muscular dystrophy, glycerol kinase deficiency, psychomotor retardation |
| SF-1 linked | NR5A1 mutations | XY sex reversal |
| Familial Glucocorticoid Deficiency or Corticotropin Insensitivity Syndromes | ||
| Type 1 | MC2R mutations | Tall stature, characteristic facial features, such as hypertelorism and frontal bossing |
| Type 2 | MRAP mutations | |
| Variant of familial glucocorticoid deficiency | MCM4 mutations | Growth failure, increased chromosomal breakage, natural killer cell deficiency |
| Variant of familial glucocorticoid deficiency | NNT mutations | |
| AUTOIMMUNE | ||
| Isolated | Sporadic; associations with HLA-DR3-DQ2, HLA-DR4-DQ8, MICA, CTLA4, PTPN22, CIITA, CLEC16A | None |
| APS type 1 (APECED) | AIRE mutations | Chronic mucocutaneous candidosis, hypoparathyroidism, other autoimmune diseases |
| APS type 2 | Sporadic; associations with HLA-DR3, HLA-DR4, CTLA4 | Thyroid autoimmune disease, type 1 diabetes, other autoimmune diseases |
| APS type 4 | Sporadic; associations with HLA-DR3, CTLA4 | Other autoimmune diseases (autoimmune gastritis, vitiligo, coeliac disease, alopecia), excluding thyroid disease and type 1 diabetes |
| INFECTIOUS | ||
| Tuberculous adrenalitis | Tuberculosis | Tuberculosis-associated manifestations in other organs |
| AIDS | HIV-1 | Other AIDS-associated diseases |
| Fungal adrenalitis | Histoplasmosis, cryptococcosis, coccidioidomycosis | Opportunistic infections |
| Meningococcal sepsis (Waterhouse-Friderichsen syndrome) | Neisseria meningitidis | |
| African trypanosomiasis | Trypanosoma brucei | Other trypanosomiasis-associated organ involvement |
| OTHER ACQUIRED CAUSES | ||
| Bilateral adrenal hemorrhage | Meningococcal sepsis (Waterhouse-Friderichsen syndrome), primary antiphospholipid syndrome, traumatic birth, anticoagulation | Symptoms and signs of underlying disease |
| Bilateral adrenal metastases | Mainly cancers of the lung, stomach, breast, and colon | Symptoms and signs of underlying disease |
| Bilateral adrenal infiltration | Primary adrenal lymphoma, amyloidosis, hemochromatosis, sarcoidosis (rare) | Symptoms and signs of underlying disease |
| Bilateral adrenalectomy | Symptoms and signs of underlying disease | |
| DRUG INDUCED | ||
| Mitotane (o,p-DDD) | Cytotoxicity | None, unless related to drug |
| Aminoglutethimide | Inhibition of cholesterol side chain cleavage enzyme (CYP11A1) | None, unless related to drug |
| Trilostane | Inhibition of 3β-hydroxysteroid dehydrogenase type 2 | None, unless related to drug |
| Etomidate | Inhibition of 11β-hydroxylase (CYP11B1) | None, unless related to drug |
| Ketoconazole, fluconazole | Inhibition of mitochondrial cytochrome P450 enzymes (e.g., CYP11A1, CYP11B1) | None, unless related to drug |
AAAS , Achalasia, adrenocortical insufficiency, alacrima syndrome; ABCD , ATP-binding cassette, subfamily D; ABCG5 , ATP-binding cassette, subfamily G, member 5; ABCG8 , ATP-binding cassette, subfamily G, member 8; APECED , autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; APS , autoimmune polyendocrinopathy syndrome; CIITA , class II transactivator; CTLA-4 , cytotoxic T-lymphocyte antigen 4; DHCR7 , 7-dehydrocholesterol reductase; HLA , human leukocyte antigen; IMAGe , i ntrauterine growth restriction (IUGR), m etaphyseal dysplasia, a drenal hypoplasia congenita (AHC), and ge nitourinary abnormalities; LIPA , lipase A; MC2R , melanocortin 2 receptor; MCM4 , minichromosome maintenance complex component 4; MICA , major histocompatibility complex class I chain-related gene A; MRAP , melanocortin 2 receptor accessory protein; PTPN22 , protein tyrosine phosphatase, nonreceptor type 22; StAR , steroidogenic acute regulatory protein.
Adapted from Charmandari E, Nicolaides NC, Chrousos GP: Adrenal insufficiency, Lancet 383:2152–2164, 2014, Table 1, pp. 2153–2154.
Table 593.2
Causes of Secondary Adrenal Insufficiency
| ETIOLOGIES | CLINICAL MANIFESTATIONS IN ADDITION TO ADRENAL INSUFFICIENCY | |
|---|---|---|
| DRUG INDUCED | ||
| Abrupt cessation of glucocorticoid therapy (systemic or topical) | Suppression of CRH and ACTH secretion leading to atrophy of the adrenal cortex | Primary disease-associated symptoms |
| OTHER ACQUIRED CAUSES | ||
| Hypothalamic or pituitary tumors | Adenomas, cysts, craniopharyngiomas, ependymomas, meningiomas, rarely carcinomas, metastasis | Panhypopituitarism* ; primary disease-associated symptoms |
| Traumatic brain injury | Panhypopituitarism* ; primary disease-associated symptoms | |
| Hypothalamic or pituitary surgery or irradiation | Panhypopituitarism* ; primary disease-associated symptoms | |
| Infections or infiltrative processes | Lymphocytic hypophysitis, hemochromatosis, tuberculosis, meningitis, sarcoidosis, actinomycosis, histiocytosis X, Wegener granulomatosis | Panhypopituitarism* ; primary disease-associated symptoms |
| Pituitary apoplexy (when occurring in a peripartum mother, termed Sheehan syndrome) | High blood loss or hypotension | Abrupt onset of severe headache, visual disturbance, nausea, vomiting; panhypopituitarism* ; primary disease-associated symptoms |
| CONGENITAL OR GENETIC CAUSES | ||
| Abnormal Central Nervous System Development | ||
| Anencephaly | Multiple | Primary disease-associated symptoms |
| Holoprosencephaly | Multiple | Primary disease-associated symptoms |
| Combined Pituitary Hormone Deficiency (CPHD) † | ||
| CPHD2 | Mutations in PROP1 (paired-like homeobox 1) | Panhypopituitarism; corticotropin deficiency occurs in adolescence |
| CPHD3 | Mutations in LHX3 (LIM homeobox 3) | Panhypopituitarism; deafness, short neck |
| CPHD4 | Mutations in LHX4 (LIM homeobox 4) | Panhypopituitarism; small sella, cerebellar defects |
| Septo-optic dysplasia, CPHD5 | Mutations in HESX1 (HESX homeobox 1) | Panhypopituitarism; septo-optic dysplasia (blindness owing to hypoplastic optic nerves, absence of the septum pellucidum); developmental delay |
| CPHD6 | Mutations in OTX2 (orthodenticle homeobox 2) | Panhypopituitarism; ectopic posterior pituitary gland |
| X-linked panhypopituitarism | Mutations in SOX3 (SRY [sex-determining region Y] box 3) | Panhypopituitarism; infundibular hypoplasia, developmental delay |
| Other Genetic Syndromes Affecting Corticotropin Secretion | ||
| Congenital proopiomelanocortin deficiency | Mutations in POMC (proopiomelanocortin) | Early-onset severe obesity, hyperphagia, red hair |
Prohormone convertase  deficiency
deficiency |
Mutations in PC1
(prohormone convertase  )
) |
Obesity, malabsorption or diarrhea, hypogonadotropic hypogonadism |
| Isolated ACTH (corticotropin) deficiency | Mutations in TBX19 (T-box 19) | |
| Prader-Willi syndrome | Deletion or silencing of genes on the parental copy of genes within the imprinted chromosome region 15q11-q13 including SNRPN (small nuclear ribonucleoprotein polypeptide N) and NDN (necdin, melanoma antigen [MAGE] family member) | Dysmorphic features, hypotonia, developmental delay, obesity, growth hormone deficiency, hypogonadotropic hypogonadism |
* The associated anterior and/or posterior hormone deficiencies may vary.
† CPHD1 (mutations in POUF1 ) is not associated with corticotropin deficiency.
Primary Adrenal Insufficiency
Perrin C. White
Keywords
- adrenoleukodystrophy
- Cryptorchidism
- contiguous gene deletion
- Pallister-Hall syndrome
- very-long-chain fatty acids
- peroxisomes
- Zellweger (cerebrohepatorenal) syndrome
- Familial glucocorticoid deficiency
- melanocyte receptor accessory protein
- achalasia
- alacrima (triple A or Allgrove syndrome)
- Type I autoimmune polyendocrinopathy (APS-1)
- Chronic mucocutaneous candidiasis
- hypoparathyroidism
- familial hypercholesterolemia
- Smith-Lemli-Opitz syndrome
- Wolman disease
- antiadrenal cytoplasmic antibodies
- Type II autoimmune polyendocrinopathy (APS-2)
- Waterhouse-Friderichsen syndrome
- Ketoconazole
- Etomidate
- adrenal crisis
- Hyperpigmentation
- Hyponatremia
- Hyperkalemia
- cosyntropin
Primary adrenal insufficiency in children is most frequently caused by genetic conditions that are often but not always manifested in infancy and less often by acquired problems such as autoimmune conditions (Table 593.3 ). Susceptibility to autoimmune conditions often has a genetic basis, and so these distinctions are not absolute.
Table 593.3
Data from Perry R, Kecha O, Paquette J, et al: Primary adrenal insufficiency in children: twenty years' experience at the Sainte-Justine Hospital, Montreal, J Clin Endocrinol Metab 90:3243–3250, 2005; Hsieh S, White PC: Presentation of primary adrenal insufficiency in childhood, J Clin Endocrinol Metab 96:E925–E928, 2011.
Inherited Etiologies
Inborn Defects of Steroidogenesis
The most common causes of adrenocortical insufficiency in infancy are the salt-losing forms of congenital adrenal hyperplasia (see Chapter 594 ). Approximately 75% of infants with 21-hydroxylase deficiency, almost all infants with lipoid adrenal hyperplasia, and most infants with a deficiency of 3β-hydroxysteroid dehydrogenase manifest salt-losing symptoms in the newborn period because they are unable to synthesize either cortisol or aldosterone.
Adrenal Hypoplasia Congenita
Adrenal hypoplasia congenita (AHC) is a relatively frequent cause of adrenal failure in males, along with congenital adrenal hyperplasia, autoimmune disease, and adrenoleukodystrophy (ALD). The name of the disorder notwithstanding, AHC is predominantly a failure of development of the definitive zone of the adrenal cortex; the fetal zone may be relatively normal. Consequently, adrenal insufficiency generally becomes evident as the fetal zone involutes postnatally (see Chapter 592 ), with onset in infancy or in the first 2 yr of life but occasionally in later childhood or even adulthood. In some cases, aldosterone deficiency becomes evident before cortisol deficiency.
The disorder is caused by mutation of the DAX1 (NR0B1) gene, a member of the nuclear hormone receptor family, located on Xp21. Males with AHC often do not undergo puberty, owing to hypogonadotropic hypogonadism caused by the same mutated DAX1 gene. Cryptorchidism, sometimes noted in these males, is probably an early manifestation of hypogonadotropic hypogonadism, but often testicular function in infants is normal, with a typical or even an unusually prolonged testosterone surge in the 1st mo of life.
AHC occasionally occurs as part of a contiguous gene deletion syndrome together with Duchenne muscular dystrophy, glycerol kinase deficiency, cognitive impairment, or a combination of these conditions.
Other Genetic Causes of Adrenal Hypoplasia
The transcription factor SF-1 is required for adrenal and gonadal development (see Chapter 592 ). Males with a heterozygous mutation in SF-1 (NR5A1) have impaired development of the testes despite the presence of a normal copy of the gene on the other chromosome and can appear to be female, similar to patients with lipoid adrenal hyperplasia (see Chapter 594 ). Rarely, such patients also have adrenal insufficiency.
Adrenal hypoplasia is also occasionally seen in patients with Pallister-Hall syndrome caused by mutations in the GLI3 oncogene.
Adrenoleukodystrophy
In ALD, adrenocortical deficiency is associated with demyelination in the central nervous system (see Chapters 104.2 and 617.3 ). High levels of very-long-chain fatty acids are found in tissues and body fluids, resulting from their impaired β-oxidation in the peroxisomes.
The most common form of ALD is an X-linked disorder with various presentations. The most common clinical picture is of a degenerative neurologic disorder appearing in childhood or adolescence and progressing to severe dementia and deterioration of vision, hearing, speech, and gait, with death occurring within a few years. Neurologic symptoms may be subtle at onset, sometimes consisting only of behavioral changes or deteriorating academic performance. Generalized but incomplete alopecia, resembling that of chemotherapy, is a characteristic but inconsistent finding. A milder form of X-linked ALD is adrenomyeloneuropathy, which begins in later adolescence or early adulthood. Patients may have evidence of adrenal insufficiency before, at the time of, or after neurologic symptoms develop, often with years separating their presentation. X-linked ALD is caused by mutations in the ABCD1 gene located on Xq28. The gene encodes a transmembrane transporter involved in the importation of very-long-chain fatty acids into peroxisomes . More than 400 mutations have been described in patients with X-linked ALD. Clinical phenotypes can vary even within families, perhaps owing to modifier genes or other unknown factors. There is no correlation between the degree of neurologic impairment and severity of adrenal insufficiency. Prenatal diagnosis by DNA analysis and family screening by very-long-chain fatty acid assays and mutation analysis are available. Women who are heterozygous carriers of the X-linked ALD gene can develop symptoms in midlife or later; adrenal insufficiency is rare.
Neonatal ALD is a rare autosomal recessive disorder. Infants have neurologic deterioration and have or acquire evidence of adrenocortical dysfunction. Most patients have severe, progressive cognitive impairment and die before 5 yr of age. This disorder is a subset of Zellweger (cerebrohepatorenal) syndrome , in which peroxisomes do not develop at all owing to mutations in any of several genes (PEX5, PEX1, PEX10, PEX13, and PEX26 ) controlling the development of this organelle.
Familial Glucocorticoid Deficiency
Familial glucocorticoid deficiency is a form of chronic adrenal insufficiency characterized by isolated deficiency of glucocorticoids, elevated levels of ACTH, and generally normal aldosterone production, although salt-losing manifestations as are present in most other forms of adrenal insufficiency occasionally occur. Patients mainly have hypoglycemia, seizures, and increased pigmentation during the 1st decade of life. The disorder affects both sexes equally and is inherited in an autosomal recessive manner. There is marked adrenocortical atrophy with relative sparing of the zona glomerulosa. Mutations in the gene for the ACTH receptor (MCR2) have been described in approximately 25% of these patients, most of which affect trafficking of receptor molecules from the endoplasmic reticulum to the cell surface. Another 20% of cases are caused by mutations in MRAP, which encodes a melanocyte receptor accessory protein required for this trafficking. Mutations at new genetic loci have been identified, including the minichromosome maintenance-deficient 4 homolog (MCM4) and nicotinamide nucleotide transhydrogenase (NNT). These genes are involved in DNA replication and antioxidant defense, respectively. Patients with MCM4 mutations also have growth failure, increased chromosomal breakage, and natural killer cell deficiency.
Another syndrome of ACTH resistance occurs in association with achalasia of the gastric cardia and alacrima (triple A or Allgrove syndrome). These patients often have a progressive neurologic disorder that includes autonomic dysfunction, intellectual disability, motor neuropathy, and occasional deafness. This syndrome is also inherited in an autosomal recessive fashion, and the AAAS gene has been mapped to chromosome 12q13. The encoded protein, aladin, might help to regulate nucleocytoplasmic transport of other proteins.
Type I Autoimmune Polyendocrinopathy Syndrome
Although autoimmune Addison disease most often occurs sporadically (see section on “Autoimmune Addison Disease ” in this chapter), it can occur as a component of 2 syndromes, each consisting of a constellation of autoimmune disorders (see Chapter 582 ). Type I autoimmune polyendocrinopathy syndrome (APS-1), also known as autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED) syndrome, is inherited in a mendelian autosomal recessive manner, whereas APS-2 (see section on “Autoimmune Addison Disease ” in this chapter) has complex inheritance. Chronic mucocutaneous candidiasis is most often the first manifestation of APS-1, followed by hypoparathyroidism and then by Addison disease, which typically develops in early adolescence. Other closely associated autoimmune disorders include gonadal failure, alopecia, vitiligo, keratopathy, enamel hypoplasia, nail dystrophy, intestinal malabsorption, and chronic active hepatitis. Hypothyroidism and type 1 diabetes mellitus occur in less than 10% of affected patients. Some components of the syndrome continue to develop as late as the 5th decade. Patients with APS-1 may have autoantibodies to the adrenal cytochrome P450 enzymes CYP21, CYP17, and CYP11A1. The presence of such antibodies indicates a high likelihood of the development of Addison disease or, in female patients, ovarian failure. Adrenal failure can evolve rapidly in APS-1; death in patients with a previous diagnosis and unexplained deaths in siblings of patients with APS-1 have been reported, indicating the need to closely monitor patients with APS-1 (or any child with hypoparathyroidism of unknown etiology) and to thoroughly evaluate apparently unaffected siblings of patients with this disorder.
The gene affected in APS-1 is designated autoimmune regulator-1 (AIRE1); it has been mapped to chromosome 21q22.3. The AIRE1 gene encodes a transcription factor that controls the expression of many proteins within the thymus, thus playing a critical role in the generation of immune tolerance. Many different mutations in the AIRE1 gene have been described in patients with APS-1, with 2 mutations (R257X and a 3-bp deletion) being most common. There has been autosomal dominant transmission in 1 kindred owing to a specific missense mutation (G228W).
Disorders of Cholesterol Synthesis and Metabolism
Patients with disorders of cholesterol synthesis or metabolism, including abetalipoproteinemia with deficient lipoprotein B–containing lipoproteins (such as low-density lipoprotein), and homozygous familial hypercholesterolemia , with impaired or absent low-density lipoprotein receptors, have mildly impaired adrenocortical function. Heterozygous familial hypercholesterolemia patients have normal adrenocortical function which is unaffected by treatment with statin (HMG-CoA reductase inhibitor) drugs. Adrenal insufficiency has been reported in patients with Smith-Lemli-Opitz syndrome , an autosomal recessive disorder manifesting with facial anomalies, microcephaly, limb anomalies, and developmental delay (see Chapter 104.3 ). Mutations in the gene coding for sterol Δ7 -reductase, mapped to 11q12-q13, resulting in impairment of the final step in cholesterol synthesis with marked elevation of 7-dehydrocholesterol, abnormally low cholesterol, and adrenal insufficiency, have been identified in Smith-Lemli-Opitz syndrome. Wolman disease is a rare autosomal recessive disorder caused by mutations in the gene encoding human lysosomal acid lipase on chromosome 10q23.2-23.3. Cholesteryl esters accumulate in lysosomes in most organ systems, leading to organ failure. Infants during the 1st or 2nd mo of life have hepatosplenomegaly, steatorrhea, abdominal distention, and failure to thrive. Adrenal insufficiency and bilateral adrenal calcification are present, and death usually occurs in the 1st yr of life.
Corticosteroid-Binding Globulin Deficiency and Decreased Cortisol-Binding Affinity
Corticosteroid-binding globulin deficiency and decreased cortisol-binding affinity result in low levels of plasma cortisol but normal urinary free cortisol and normal plasma ACTH levels. A high prevalence of hypotension and fatigue has been reported in some adults with abnormalities of corticosteroid-binding globulin deficiency.
Acquired Etiologies
Autoimmune Addison Disease
The most common cause of Addison disease is autoimmune destruction of the glands. The glands may be so small that they are not visible at autopsy, and only remnants of tissue are found in microscopic sections. Usually, the medulla is not destroyed, and there is marked lymphocytic infiltration in the area of the former cortex. In advanced disease, all adrenocortical function is lost, but early in the clinical course, isolated cortisol deficiency can occur. Most patients have antiadrenal cytoplasmic antibodies in their plasma; 21-hydroxylase (CYP21) is the most commonly occurring biochemically defined autoantigen.
Addison disease can occur as a component of 2 autoimmune polyendocrinopathy syndromes. Type I (APS-1) was discussed previously. Type II autoimmune polyendocrinopathy (APS-2) consists of Addison disease associated with autoimmune thyroid disease (Schmidt syndrome) or type 1 diabetes (Carpenter syndrome). Gonadal failure, vitiligo, alopecia, and chronic atrophic gastritis, with or without pernicious anemia, can occur. Frequencies of the human leukocyte antigen (HLA)-D3 and HLA-D4 alleles are increased in these patients and appear to confer an increased risk for development of this disease; particular alleles at the major histocompatibility complex class I chain–related genes A and B (MICA and MICB) also are associated with this disorder. Polymorphisms in genes involved in other autoimmune disorders have been inconsistently associated with primary adrenal insufficiency, and their contribution to its pathogenesis must be regarded as uncertain. These include the class II, major histocompatibility complex, transactivator (CIITA), C-type lectin domain family 16, member A (CLEC16A ), and protein tyrosine phosphatase, nonreceptor type 22 (PTPN22). The disorder is most common in middle-aged women and can occur in many generations of the same family. Antiadrenal antibodies, specifically antibodies to the CYP21, CYP17, and CYP11A1 enzymes, are also found in these patients. Autoimmune adrenal insufficiency may also be seen in patients with celiac disease and mitochondrial gene mutations.
Infection
Tuberculosis was a common cause of adrenal destruction in the past but is currently much less prevalent. The most common infectious etiology for adrenal insufficiency is meningococcemia (see Chapter 218 ); adrenal crisis from this cause is referred to as the Waterhouse-Friderichsen syndrome. Patients with AIDS can have a variety of subclinical abnormalities in the hypothalamic-pituitary-adrenal (HPA) axis, but frank adrenal insufficiency is rare. However, drugs used in the treatment of AIDS can affect adrenal hormone homeostasis.
Drugs
Ketoconazole , an antifungal drug, can cause adrenal insufficiency by inhibiting adrenal enzymes. Mitotane (o,p′-DDD), used in the treatment of adrenocortical carcinoma and refractory Cushing syndrome (see Chapters 595 and 597 ), is cytotoxic to the adrenal cortex and can also alter extraadrenal cortisol metabolism. Signs of adrenal insufficiency occur in a substantial percentage of patients treated with mitotane. Etomidate , used in the induction and maintenance of general anesthesia, inhibits 11β-hydroxylase (CYP11B1), and a single induction dose can block cortisol synthesis for 4-8 hr or longer. This may be problematic in severely stressed patients, particularly if repeated doses are used in a critical care setting. Abiraterone acetate, an androgen biosynthesis inhibitor which is used to treat metastatic prostate carcinoma, inhibits cortisol biosynthesis but leaves corticosterone biosynthesis unimpaired. This drug is not currently encountered in pediatric practice. Although not themselves a cause of adrenal insufficiency, rifampicin and anticonvulsive drugs such as phenytoin and phenobarbital reduce the effectiveness and bioavailability of corticosteroid replacement therapy by inducing steroid metabolizing enzymes in the liver.
Hemorrhage Into Adrenal Glands
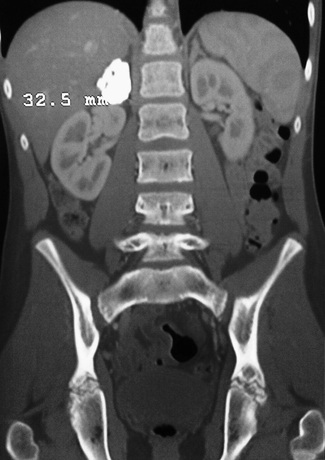
Hemorrhage into adrenal glands can occur in the neonatal period as a consequence of a difficult labor (especially breech presentation), or its etiology might not be apparent (Fig. 593.1 ). An incidence rate of 3 in 100,000 live births has been suggested. The hemorrhage may be sufficiently extensive to result in death from exsanguination or hypoadrenalism. An abdominal mass, anemia, unexplained jaundice, or scrotal hematoma may be the presenting sign. Often, the hemorrhage is asymptomatic initially and is identified later by calcification of the adrenal gland. Fetal adrenal hemorrhage has also been reported. Postnatally, adrenal hemorrhage most often occurs in patients being treated with anticoagulants. It can also occur as a result of child abuse.
Clinical Manifestations
Primary adrenal insufficiency leads to cortisol and often aldosterone deficiency. The signs and symptoms of adrenal insufficiency are most easily understood in the context of the normal actions of these hormones (see Chapter 592 ; Table 593.4 ).
Table 593.4
Clinical Manifestations and Biochemical Findings in Adrenal Insufficiency
| PATHOPHYSIOLOGIC MECHANISM | PREVALENCE (%)* | |
|---|---|---|
| SYMPTOMS | ||
| Fatigue | Glucocorticoid deficiency | 90 |
| Anorexia, weight loss | Glucocorticoid deficiency | 90 |
| Nausea, vomiting | Glucocorticoid deficiency, mineralocorticoid deficiency | 90 |
| Salt craving (primary adrenal insufficiency only) | Mineralocorticoid deficiency | 20 |
| Myalgia or joint pain | Glucocorticoid deficiency | |
| SIGNS | ||
| Low blood pressure, orthostatic hypotension | Mineralocorticoid deficiency, glucocorticoid deficiency | 70–100 |
| Skin or mucosal hyperpigmentation (primary adrenal insufficiency only) | Excess of proopiomelanocortin-derived peptides | 70 |
| LABORATORY FINDINGS | ||
| Hyponatremia | Mineralocorticoid deficiency, glucocorticoid deficiency (leading to decreased free water excretion) | 90 |
| Hyperkalemia (primary adrenal insufficiency only) | Mineralocorticoid deficiency | 50 |
| Hypoglycemia | Glucocorticoid deficiency | 30 |
| Ketosis | Glucocorticoid deficiency | 30 |
| Low random cortisol level | Glucocorticoid deficiency | 80 |
| Eosinophilia, lymphocytosis | Glucocorticoid deficiency | |
| High ACTH level (primary adrenal insufficiency only) | Glucocorticoid deficiency | 100 |
| High plasma renin activity (primary adrenal insufficiency only) | Mineralocorticoid deficiency | 100 |
* Prevalence data are for primary insufficiency only. Blanks indicate that no pediatric prevalence data are available.
Data from Hsieh S, White PC: Presentation of primary adrenal insufficiency in childhood, J Clin Endocrinol Metab 96:E925–E928, 2011.
Cortisol deficiency decreases cardiac output and vascular tone; moreover, catecholamines such as epinephrine have decreased inotropic and pressor effects in the absence of cortisol. These problems are initially manifested as orthostatic hypotension in older children and can progress to frank shock in patients of any age. They are exacerbated by aldosterone deficiency, which results in hypovolemia owing to decreased resorption of sodium in the distal nephron.
Hypotension and decreased cardiac output decrease glomerular filtration and thus decrease the ability of the kidney to excrete free water. Vasopressin (AVP) is secreted by the posterior pituitary in response to hypotension and also as a direct consequence of lack of inhibition by cortisol. These factors decrease plasma osmolality and lead in particular to hyponatremia. Hyponatremia is also caused by aldosterone deficiency and may be much worse when both cortisol and aldosterone are deficient.
In addition to hypovolemia and hyponatremia, aldosterone deficiency causes hyperkalemia by decreasing potassium excretion in the distal nephron. Cortisol deficiency alone does not cause hyperkalemia.
Cortisol deficiency decreases negative feedback on the hypothalamus and pituitary, leading to increased secretion of ACTH. Hyperpigmentation is caused by ACTH and other peptide hormones (γ-melanocyte–stimulating hormone) arising from the ACTH precursor proopiomelanocortin. In patients with a fair complexion, the skin can have a bronze cast. Pigmentation may be more prominent in skin creases, mucosa, and scars. In dark-skinned patients, it may be most readily appreciated in the gingival and buccal mucosa.
Hypoglycemia is a feature of adrenal insufficiency. It is often accompanied by ketosis as the body attempts to use fatty acids as an alternative energy source. Ketosis is aggravated by anorexia, nausea, and vomiting, all of which occur frequently.
The clinical presentation of adrenal insufficiency depends on the age of the patient, whether both cortisol and aldosterone secretion are affected, and to some extent on the underlying etiology. The most common causes in early infancy are inborn errors of steroid biosynthesis, sepsis, AHC, and adrenal hemorrhage. Infants have a relatively greater requirement for aldosterone than do older children, possibly owing to immaturity of the kidney and also to the low sodium content of human breast milk and infant formula. Hyperkalemia, hyponatremia, and hypoglycemia are prominent presenting signs of adrenal insufficiency in infants. Ketosis is not consistently present because infants generate ketones less well than do older children. Hyperpigmentation is not usually seen because this takes weeks or months to develop, and orthostatic hypotension is obviously difficult to demonstrate in infants.
Infants can become ill very quickly. There may be only a few days of decreased activity, anorexia, and vomiting before critical electrolyte abnormalities develop.
In older children with Addison disease, symptoms include muscle weakness, malaise, anorexia, vomiting, weight loss, and orthostatic hypotension. These may be of insidious onset. It is not unusual to elicit, in retrospect, an episodic history spanning years with symptoms being noticeable only during intercurrent illnesses. Such patients can present with acute decompensation (adrenal crisis) during relatively minor infectious illnesses. Some of these patients have been initially misdiagnosed with chronic fatigue syndrome, postmononucleosis syndrome, chronic Lyme disease, or psychiatric disorders (depression or anorexia nervosa).
Hyperpigmentation is often, but not necessarily, present. Hyponatremia is present at diagnosis in almost 90% of patients. Hyperkalemia tends to occur later in the course of the disease in older children than in infants and is present in only half of patients at diagnosis. Normal potassium levels must never be presumed to rule out primary adrenal insufficiency.
Hypoglycemia and ketosis are common. Thus the clinical presentation can be easily confused with gastroenteritis or other acute infections. Chronicity of symptoms can alert the clinician to the possibility of Addison disease, but this diagnosis should be considered in any child with orthostatic hypotension, hyponatremia, hypoglycemia, and ketosis. Salt craving is seen in primary adrenal insufficiency with mineralocorticoid deficiency. Fatigue, myalgias, fever, eosinophilia, lymphocytosis, hypercalcemia, and anemia may be noted with glucocorticoid deficiency.
Laboratory Findings
Hypoglycemia, ketosis, hyponatremia, and hyperkalemia have been discussed. An electrocardiogram is useful for quickly detecting hyperkalemia in a critically ill child. Acidosis is often present, and the blood urea nitrogen level is elevated if the patient is dehydrated.
Cortisol levels are sometimes at the low end of the normal range but are invariably low when the patient's degree of illness is considered. ACTH levels are high in primary adrenal insufficiency but can take time to be reported by the laboratory. Similarly, aldosterone levels may be within the normal range but inappropriately low considering the patient's hyponatremia, hyperkalemia, and hypovolemia. Plasma renin activity is elevated. Blood eosinophils may be increased in number, but this is rarely useful diagnostically.
Urinary excretion of sodium and chloride are increased and urinary potassium is decreased, but these are difficult to assess in random urine samples. Accurate interpretation of urinary electrolytes requires more-prolonged (24 hr) urine collections and knowledge of the patient's sodium and potassium intake.
The most definitive test for adrenal insufficiency is measurement of serum levels of cortisol before and after administration of ACTH; resting levels are low and do not increase normally after administration of ACTH (Table 593.5 ). Occasionally, normal resting levels that do not increase after administration of ACTH indicate an absence of adrenocortical reserve. A low initial level followed by a significant response to ACTH can indicate secondary adrenal insufficiency. Traditionally, this test has been performed by measuring cortisol levels before and 30 or 60 min after giving 0.250 mg of cosyntropin (ACTH 1-24) by rapid intravenous infusion. Aldosterone will transiently increase in response to this dose of ACTH and may also be measured. A low-dose test (1 µg ACTH 1-24/1.73 m2 ) is a more sensitive test of pituitary-adrenal reserve but has somewhat lower specificity (more false-positive tests).
Table 593.5
Proposed Diagnostic Criteria for Autoimmune Addison Disease
Definitive diagnosis: 1, 2, 3, and 4.
Probable diagnosis: 1, 2, 4, and 5.
From Neto RAB, Freire de Carvalho J: Diagnosis and classification of Addison's disease (autoimmune adrenalitis), Autoimmune Rev 13:408–411, 2014, Table 3.
Differential Diagnosis
Upon presentation, Addison disease often needs to be distinguished from more acute illnesses such as gastroenteritis with dehydration or sepsis. Additional testing is directed at identifying the specific cause for adrenal insufficiency. When congenital adrenal hyperplasia is suspected, serum levels of cortisol precursors (17-hydroxyprogesterone) should be measured along with cortisol in an ACTH stimulation test (see Chapter 594 ) (Fig. 593.2 ). Elevated levels of very-long-chain fatty acids are diagnostic of ALD (see Chapter 617.3 ). Many genetic etiologies for primary adrenal insufficiency may be identified by direct genetic testing, but it can take many weeks for results to become available. The presence of antiadrenal antibodies suggests an autoimmune pathogenesis. Patients with autoimmune Addison disease must be closely observed for the development of other autoimmune disorders. In children, hypoparathyroidism is the most commonly associated disorder, and it is suspected if hypocalcemia and elevated phosphate levels are present.
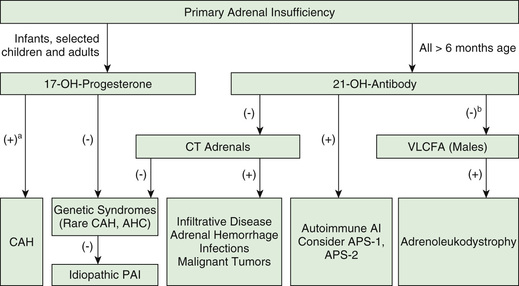
Ultrasonography (which requires an experienced operator), CT, or MRI can help to define the size of the adrenal glands.
Treatment
Treatment of acute adrenal insufficiency must be immediate and vigorous. If the diagnosis of adrenal insufficiency has not been established, a blood sample should be obtained before therapy to determine electrolytes, glucose, ACTH, cortisol, aldosterone, and plasma renin activity. If the patient's condition permits, an ACTH stimulation test can be performed while initial fluid resuscitation is underway. An intravenous solution of 5% glucose in 0.9% saline should be administered to correct hypoglycemia, hypovolemia, and hyponatremia. Hypotonic fluids (e.g., 5% glucose in water or 0.2% saline) must be avoided because they can precipitate or exacerbate hyponatremia. If hyperkalemia is severe, it can require treatment with intravenous calcium and/or bicarbonate, intrarectal potassium-binding resin (sodium polystyrene sulfonate, Kayexalate), or intravenous infusion of glucose and insulin. A water-soluble form of hydrocortisone, such as hydrocortisone sodium succinate, should be given intravenously. As much as 10 mg for infants, 25 mg for toddlers, 50 mg for older children, and 100 mg for adolescents should be administered as a bolus and a similar total amount given in divided doses at 6-hr intervals for the first 24 hr. These doses may be reduced during the next 24 hr if progress is satisfactory. Adequate fluid and sodium repletion is achieved by intravenous saline administration, aided by the mineralocorticoid effect of high doses of hydrocortisone.
Particular caution should be exercised in the rare patient with concomitant adrenal insufficiency and hypothyroidism, because thyroxine can increase cortisol clearance. Thus an adrenal crisis may be precipitated if hypothyroidism is treated without first ensuring adequate glucocorticoid replacement.
After the acute manifestations are under control, most patients require chronic replacement therapy for their cortisol and aldosterone deficiencies. Hydrocortisone (cortisol) may be given orally in daily doses of 10 mg/m2 /24 hr in 3 divided doses; some patients require 15 mg/m2 /24 hr to minimize fatigue, especially in the morning. Timed-release preparations of hydrocortisone are undergoing clinical trials but are not yet generally available. Subcutaneous infusion of hydrocortisone with an insulin pump has also been examined in clinical trials; although this has the advantage that it can very closely mimic normal diurnal variation in cortisol secretion, it is very expensive and has not yet entered routine clinical practice. Equivalent doses (20–25% of the hydrocortisone dose) of prednisone or prednisolone may be divided and given twice daily. ACTH levels may be used to monitor adequacy of glucocorticoid replacement in primary adrenal insufficiency; in congenital adrenal hyperplasia, levels of precursor hormones are used instead. Blood samples for monitoring should be obtained at a consistent time of day and in a consistent relation to (i.e., before or after) the hydrocortisone dose. Normalizing ACTH levels is unnecessary and can require excessive doses of hydrocortisone; in general, morning ACTH levels high in the normal range to 3-4 times normal are satisfactory. Because untreated or severely undertreated patients can acutely decompensate during relatively minor illnesses, assessment of symptoms (or lack thereof) must not be used as a substitute for biochemical monitoring. During situations of stress, such as periods of infection or minor operative procedures, the dose of hydrocortisone should be increased 2-3–fold. Major surgery under general inhalation anesthesia requires high intravenous doses of hydrocortisone similar to those used for acute adrenal insufficiency.
If aldosterone deficiency is present, fludrocortisone, a synthetic mineralocorticoid, is given orally in doses of 0.05-0.2 mg daily. Measurements of plasma renin activity are useful in monitoring the adequacy of mineralocorticoid replacement. Chronic overdosage with glucocorticoids leads to obesity, short stature, and osteoporosis, whereas overdosage with fludrocortisone results in hypertension and occasionally hypokalemia.
Replacement of dehydroepiandrosterone (DHEA) in adults remains controversial; prepubertal children do not normally secrete large amounts of DHEA. Many adults with Addison disease complain of having decreased energy, and replacing DHEA can improve this problem, particularly in women in whom adrenal androgens represent approximately 50% of total androgen secretion.
Additional therapy might need to be directed at the underlying cause of the adrenal insufficiency in regard to infections and certain metabolic defects. Therapeutic approaches to ALD include administration of glycerol trioleate and glycerol trierucate (Lorenzo's oil), bone marrow transplantation, and lovastatin (see Chapter 617.3 ).
Bibliography
Afroze B, Amjad N, Ibrahim SH, et al. Adrenal insufficiency in a child with MELAS syndrome. Brain Dev . 2014;36:924–927.
Akirav EM, Ruddle NH, Herold KC. The role of AIRE in human autoimmune disease. Nat Rev Endocrinol . 2011;7:25–33.
Bornstein SR. Predisposing factors for adrenal insufficiency. N Engl J Med . 2009;360:2328–2339.
Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab . 2016;101:364–389.
Calderwood L, Holm IA, Teot LA, Anselm I. Adrenal insufficiency in mitochondrial disease: a rare case of GFER-related mitochondrial encephalomyopathy and review of the literature. J Child Neurol . 2016;31(2):190–194.
Charmandari E, Nicolaides NC, Chrousos GP. Adrenal insufficiency. Lancet . 2014;383:2152–2164.
Chung TT, Chan LF, Metherell LA, et al. Phenotypic characteristics of familial glucocorticoid deficiency (FGD) type 1 and 2. Clin Endocrinol (Oxf) . 2010;72:589–594.
Debono M, Ghobadi C, Rostami-Hodjegan A, et al. Modified-release hydrocortisone to provide circadian cortisol profiles. J Clin Endocrinol Metab . 2009;94:1548–1554.
Ferraz-de-Souza B, Achermann JC. Disorders of adrenal development. Endocr Dev . 2008;13:19–32.
Hannah-Shmouni F, Demidowich A, Stratakis CA. Cortisol in the evaluation of adrenal insufficiency. JAMA . 2016;316(5):535–536.
Hsieh S, White PC. Presentation of primary adrenal insufficiency in childhood. J Clin Endocrinol Metab . 2011;96:E925–E928.
Hughes CR, Guasti L, Meimaridou E, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest . 2012;122:814–820.
Llano JP, Beaufils E, Nicolino M. Uncommon cause of large paravertebral calcification in a child. J Pediatr . 2013;162:881.
Logan CV, Murray JE, Parry DA, et al. DNA polymerase epsilon deficiency causes IMAGe syndrome with variable immunodeficiency. Am J Hum Genet . 2018;103:1038–1044.
Meimaridou E, Kowalczyk J, Guasti L, et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet . 2012;44:740–742.
Michels A, Michels N. Addison disease: early detection and treatment principles. Am Fam Physician . 2014;89:563–568.
Mitchell AL, Pearce SH. Autoimmune Addison disease: pathophysiology and genetic complexity. Nat Rev Endocrinol . 2012;8:306–316.
Neto RAB, Freire de Carvalho J. Diagnosis and classification of Addison's disease (autoimmune adrenalitis). Autoimmune Rev . 2014;13:408–411.
Perlman SJ, Mar S. Leukodystrophies. Adv Exp Med Biol . 2012;724:154–171.
Schimmer BP, White PC. Minireview: steroidogenic factor 1: its roles in differentiation, development, and disease. Mol Endocrinol . 2010;24:1322–1337.
Shulman DI, Palmert MR, Kemp SF. Adrenal insufficiency: still a cause of morbidity and death in childhood. Pediatrics . 2007;119:e484–e494.
Wass JAH, Arlt W. How to avoid precipitating an acute adrenal crisis. BMJ . 2012;345:9.
Secondary and Tertiary Adrenal Insufficiency
Perrin C. White
Etiology
Abrupt Cessation of Administration of Corticosteroids
Secondary adrenal insufficiency most commonly occurs when the HPA axis is suppressed by prolonged administration of high doses of a potent glucocorticoid and that agent is suddenly withdrawn or the dose is tapered too quickly. Patients at risk for this problem include those with leukemia, asthma (particularly when patients are transitioned from oral to inhaled corticosteroids), and collagen vascular disease or other autoimmune conditions and those who have undergone tissue transplants or neurosurgical procedures. The maximal duration and dosage of glucocorticoid that can be administered before encountering this problem is not known, but it is assumed that high-dose glucocorticoids (the equivalent of >10 times physiologic cortisol secretion) can be administered for at least 1 wk without requiring a subsequent taper of dose. On the other hand, when high doses of dexamethasone are given to children with leukemia, it can take 6 mo or longer after therapy is stopped before tests of adrenal function return to normal. Signs and symptoms of adrenal insufficiency are most likely in patients who are subsequently subjected to stresses such as severe infections or additional surgical procedures.
Corticotropin (Adrenocorticotropic Hormone) Deficiency
Pituitary or hypothalamic dysfunction can cause corticotropin deficiency (see Chapter 573 ), usually associated with deficiencies of other pituitary hormones such as growth hormone and thyrotropin. Destructive lesions in the area of the pituitary, such as craniopharyngioma and germinoma, are the most common causes of corticotropin deficiency. In many cases the pituitary or hypothalamus is further damaged during surgical removal or radiotherapy of tumors in the midline of the brain. Traumatic brain injury (see Chapter 728 ) frequently causes pituitary dysfunction, especially in the 1st days after the injury. However, corticotropin deficiency is difficult to detect then owing to frequent use of high doses of dexamethasone to minimize brain swelling, and permanent corticotropin deficiency is unusual after traumatic brain injury. In rare instances, autoimmune hypophysitis is the cause of corticotropin deficiency.
Congenital lesions of the pituitary also occur. The pituitary alone may be affected, or additional midline structures may be involved, such as the optic nerves or septum pellucidum. The latter type of abnormality is termed septo-optic dysplasia, or de Morsier syndrome (see Chapter 609.9 ). More severe developmental anomalies of the brain, such as anencephaly and holoprosencephaly, can also affect the pituitary. These disorders are usually sporadic, although a few cases of autosomal recessive inheritance have occurred. Isolated deficiency of corticotropin has been reported, including in several sets of siblings. Patients with multiple pituitary hormone deficiencies caused by mutations in the PROP1 gene have been described with progressive ACTH/cortisol deficiency. Isolated deficiency of corticotropin-releasing hormone has been documented in an Arab kindred as an autosomal recessive trait.
Up to 60% of children with Prader-Willi syndrome (see Chapter 98.8 ) have some degree of secondary adrenal insufficiency as assessed by provocative testing with metyrapone, although diurnal cortisol levels are normal. The clinical significance of this finding is uncertain, but it might contribute to the relatively high incidence of sudden death with infectious illness that occurs in this population. Although it is not yet a standard of care, some endocrinologists advocate treating patients who have Prader-Willi syndrome with hydrocortisone during febrile illness.
Clinical Presentation
Aldosterone secretion is unaffected in secondary adrenal insufficiency because the adrenal gland is, by definition, intact and the renin-angiotensin system is not involved. Thus signs and symptoms are those of cortisol deficiency. Newborns often have hypoglycemia. Older children can have orthostatic hypotension or weakness. Hyponatremia may be present.
When secondary adrenal insufficiency is the consequence of an inborn or acquired anatomic defect involving the pituitary, there may be signs of associated deficiencies of other pituitary hormones. The penis may be small in male infants if gonadotropins are also deficient. Infants with secondary hypothyroidism are often jaundiced. Children with associated growth hormone deficiency grow poorly after the 1st yr of life.
Some children with pituitary abnormalities have hypoplasia of the midface. Children with optic nerve hypoplasia can have obvious visual impairment. They usually have a characteristic wandering nystagmus, but this is often not apparent until several months of age.
Laboratory Findings
Because the adrenal glands themselves are not directly affected, the diagnosis of secondary adrenal insufficiency is sometimes challenging. Historical gold standard dynamic tests include insulin-induced hypoglycemia, which provides a potent stress to the entire HPA axis. This test requires constant attendance by a physician and is considered by many endocrinologists to be too dangerous for routine use. A second gold standard test uses metyrapone, a specific inhibitor of steroid 11β-hydroxylase (CYP11B1) to block cortisol synthesis, thus removing the normal negative feedback of cortisol on ACTH secretion. There are several protocols for this test; one version administers 30 mg/kg of metyrapone orally at midnight, with a blood sample obtained for cortisol and 11-deoxycortisol (the substrate for 11β-hydroxylase) at 8 AM . A low cortisol level (<5 µg/dL) demonstrates adequate suppression of cortisol synthesis, and an 11-deoxycortisol level >7 µg/dL indicates that ACTH has responded normally to the cortisol deficiency by stimulating the adrenal cortex. This test should be used with caution outside the research setting because it can precipitate adrenal crises in patients with marginal adrenal function; the drug is not available in all locales.
Currently, the most commonly used test to diagnose secondary adrenal insufficiency is low-dose ACTH stimulation testing (1 µg/1.73 m2 of cosyntropin given intravenously), the rationale being that there will be some degree of atrophy of the adrenal cortex if normal physiologic ACTH stimulation is lacking. Thus this test may be falsely negative in cases of acute compromise of the pituitary (e.g., injury or surgery). Such circumstances rarely pose a diagnostic dilemma; in general, this test provides excellent sensitivity and specificity. Although assays vary somewhat, a threshold cortisol level of 18-20 µg/dL 30 min after cosyntropin administration may be used to dichotomize normal and abnormal responses.
Currently, there seems to be little reason to use stimulation with corticotropin-releasing hormone instead of ACTH; although the corticotropin-releasing hormone test has the theoretical advantage of testing the ability of the anterior pituitary to respond to this stimulus by secreting ACTH (thus distinguishing secondary and tertiary adrenal insufficiency), in practice it does not provide improved sensitivity and specificity, and the agent is not as widely available.
Treatment
Iatrogenic secondary adrenal insufficiency (caused by chronic glucocorticoid administration) is best avoided by use of the smallest effective doses of systemic glucocorticoids for the shortest period of time. When a patient is thought to be at risk, tapering the dose rapidly to a level equivalent to or slightly less than the physiologic replacement (~10 mg/m2 /24 hr of hydrocortisone) and further tapering over several weeks can allow the adrenal cortex to recover without the patient developing signs of adrenal insufficiency. Patients with anatomic lesions of the pituitary should be treated indefinitely with glucocorticoids. Mineralocorticoid replacement is not required. In patients with panhypopituitarism, treating cortisol deficiency can increase free water excretion, thus unmasking central diabetes insipidus. Electrolytes must be monitored carefully when initiating cortisol therapy in panhypopituitary patients.
Bibliography
Ahmad T, Borchert M, Geffner M. Optic nerve hypoplasia and hypopituitarism. Pediatr Endocrinol Rev . 2008;5:772–777.
Ahmet A, Brienza V, Tran A, et al. Frequency and duration of adrenal suppression following glucocorticoid theryapy in children with rheumatic diseases. Arthritis Care Res . 2017;69(8):1224–1230.
Broersen LHA, Pereira AM, Jorgensen OL, Dekkers OM. Adrenal insufficiency in corticosteroids use: systematic review and meta-analysis. J Clin Endocrinol Metab . 2015;100(6):2171–2180.
de Lind van Wijngaarden RF, Joosten KF, van den Berg S, et al. The relationship between central adrenal insufficiency and sleep-related breathing disorders in children with Prader-Willi syndrome. J Clin Endocrinol Metab . 2009;94:2387–2393.
Gordijn MS, Rensen N, Gemke RJ, et al. Hypothalamic-pituitary-adrenal (HPA) axis suppression after treatment with glucocorticoid therapy for childhood acute lymphoblastic leukaemia. Cochrane Database Syst Rev . 2015;(8) [CD008727].
Grugni G, Beccaria L, Corrias A, et al. Central adrenal insufficiency in young adults with Prader-Willi Syndrome. Clin Endocrinol (Oxf) . 2013;79(3):371–378.
Hsu AA, von Elten K, Chan D, et al. Characterization of the cortisol stress response to sedation and anesthesia in children. J Clin Endocrinol Metab . 2012;97:E1830–E1835.
Kazlauskaite R, Evans AT, Villabona CV, et al. Corticotropin tests for hypothalamic-pituitary-adrenal insufficiency: a metaanalysis. J Clin Endocrinol Metab . 2008;93:4245–4253.
Kelberman D, Dattani MT. Role of transcription factors in midline central nervous system and pituitary defects. Endocr Dev . 2009;14:67–82.
Zollner EW. Hypothalamic-pituitary-adrenal axis suppression in asthmatic children on inhaled corticosteroids (Part 2)—the risk as determined by gold standard adrenal function tests: a systematic review. Pediatr Allergy Immunol . 2007;18:469–474.
Adrenal Insufficiency in the Critical Care Setting
Perrin C. White
Etiology
Adrenal insufficiency in the context of critical illness is encountered in up to 20–50% of pediatric patients, often as a transient condition. In many cases, it is considered to be functional or relative in nature, meaning that cortisol levels are within normal limits but cannot increase sufficiently to meet the demands of critical illness. The causes are heterogeneous, and some were discussed in Chapter 593.1 . They include adrenal hypoperfusion from shock, particularly septic shock, as is often seen in meningococcemia. Inflammatory mediators during septic shock, particularly interleukin-6, can suppress ACTH secretion, directly suppress cortisol secretion, or both. Etomidate, used as sedation for intubation, inhibits steroid 11β-hydroxylase and thus blocks cortisol biosynthesis. Neurosurgical patients with closed head trauma or with tumors that involve the hypothalamus or pituitary might have ACTH deficiency in the context of panhypopituitarism. Some children have been previously treated with systemic corticosteroids (e.g., children with leukemia) and have suppression of the HPA axis for that reason. In the intensive care nursery, premature infants have not yet developed normal cortisol biosynthetic capacity and thus may not be able to secrete adequate amounts of this hormone when ill.
Clinical Manifestations
Cortisol is required for catecholamines to have their normal pressor effects on the cardiovascular system (see Chapters 592.4 and 592.5 ). Accordingly, adrenal insufficiency is often suspected in hypotensive patients who do not respond to intravenous pressor agents. Patients may be at increased risk for hypoglycemia or a presentation resembling the syndrome of inappropriate antidiuretic hormone secretion, but these conditions commonly occur in the context of sepsis, and the contribution of adrenal insufficiency may be difficult to distinguish.
Laboratory Findings
Although low random cortisol levels in severely stressed patients are certainly abnormal, very high levels are also associated with a poor outcome in such patients; the latter situation presumably reflects a maximally stimulated adrenal cortex with diminished reserve. ACTH (cosyntropin) stimulation testing is generally considered the best way to diagnose adrenal insufficiency in this setting (see Chapter 593.1 ); evidence suggests that the low-dose (1 µg/1.73 m2 ) test may be superior to the 250 µg standard dose test, although this remains controversial. In general, a peak cortisol level <18 µg/dL or an increment of <9 µg/dL from baseline is considered suggestive for adrenal insufficiency in this context. In evaluating cortisol levels, it should be remembered that cortisol in the circulation is normally mostly bound to cortisol-binding globulin; in hypoproteinemic states, total cortisol levels may be decreased, whereas free cortisol levels might be normal. It may be prudent to measure free cortisol before initiating treatment when total cortisol is low and albumin is <2.5 g/dL, but such measurements are not readily available in all institutions.
Treatment
There are limited data regarding treatment efficacy in critically ill children. Based on studies of both children and adults, it is likely that moderate stress doses of hydrocortisone (e.g., 50 mg/m2 /day) improve responses to pressor agents in patients with shock and documented adrenal insufficiency. It is uncertain if there is a beneficial effect on overall survival. There seems to be no benefit in using pharmacologic doses of potent synthetic glucocorticoids such as dexamethasone.
Bibliography
Aneja R, Carcillo JA. What is the rationale for hydrocortisone treatment in children with infection-related adrenal insufficiency and septic shock? Arch Dis Child . 2007;92:165–169.
Brierley J, Carcillo JA, Choong K, et al. Clinical practice parameters for hemodynamic support of pediatric and neonatal septic shock: 2007 update from the American College of Critical Care Medicine. Crit Care Med . 2009;37:666–688.
Hebbar KB, Stockwell JA, Leong T, et al. Incidence of adrenal insufficiency and impact of corticosteroid supplementation in critically ill children with systemic inflammatory syndrome and vasopressor-dependent shock. Crit Care Med . 2011;39:1145–1150.
Lundy JB, Slane ML, Frizzi JD. Acute adrenal insufficiency after a single dose of etomidate. J Intensive Care Med . 2007;22:111–117.
Pizarro CF, Troster EJ, Damiani D, et al. Absolute and relative adrenal insufficiency in children with septic shock. Crit Care Med . 2005;33:855–859.
Altered End-Organ Sensitivity to Corticosteroids
Perrin C. White
Diseases can result from altered actions of hormones at their physiologic targets. These may be caused by abnormal metabolism of hormones, mutations in hormone receptors, or defects in cellular effectors (such as ion channels) that are targets of hormone action.
Generalized Glucocorticoid Resistance
Etiology
Patients with generalized glucocorticoid resistance have target-tissue insensitivity to glucocorticoids. The condition is usually inherited in an autosomal dominant manner, but sporadic cases occur. Impairment of normal negative feedback of cortisol at the levels of the hypothalamus and pituitary activates the HPA axis, with consequent increases in ACTH and cortisol concentrations. Generalized glucocorticoid resistance is caused by mutations in the glucocorticoid receptor (encoded by the NR3C1 gene) that impair its action by interfering with ligand binding, DNA binding, transcriptional activation, or some combination of these. Most mutations are heterozygous; glucocorticoid receptors usually bind DNA as dimers, and 3 out of every 4 dimers will contain at least 1 abnormal receptor molecule when a heterozygous mutation is present.
Clinical Manifestations
The excess ACTH secretion causes adrenal hyperplasia with increased production of adrenal steroids with mineralocorticoid activity, including cortisol, deoxycorticosterone, and corticosterone, and also androgens and precursors, including androstenedione, DHEA, and DHEA sulfate. The high cortisol concentrations do not cause Cushing syndrome (see Chapter 597 ) because of the insensitivity to glucocorticoids; conversely, most signs and symptoms of adrenal insufficiency are absent except for the frequent occurrence of chronic fatigue and occasional anxiety (neonatal hypoglycemia was reported in one very unusual patient with a homozygous null mutation). On the other hand, the mineralocorticoid and androgen receptors are normally sensitive to their ligands. Signs of mineralocorticoid excess, such as hypertension and hypokalemic alkalosis, are frequently noted. The increased concentrations of adrenal androgens may cause ambiguous genitalia in females and gonadotropin-independent precocious puberty in children of either gender; acne; hirsutism, and infertility in both sexes; menstrual irregularities in females; and oligospermia in males. Testicular adrenal rest tumors and ACTH-secreting pituitary adenomas occasionally occur.
Laboratory Findings
The diagnosis of generalized glucocorticoid resistance is suggested by elevated serum cortisol concentrations and increased 24-hr urinary free cortisol excretion in the absence of Cushing syndrome. Levels of other adrenal steroids are also increased. Plasma concentrations of ACTH may be normal or high. The circadian pattern of ACTH and cortisol secretion is preserved, although at higher-than-normal concentrations, and there is resistance of the HPA axis to dexamethasone suppression. Sequencing of the NR3C1 gene can confirm the diagnosis but is not routinely available.
Differential Diagnosis
Generalized glucocorticoid resistance should be distinguished from relatively mild cases of Cushing syndrome (whether caused by a pituitary adenoma or adrenal tumor, see Chapter 595 ); the latter is more likely to be associated with excessive weight gain or poor linear growth. Adrenocortical tumors may secrete mineralocorticoids such as deoxycorticosterone and also androgens, but ACTH levels are often suppressed and of course the tumor can usually be visualized with appropriate imaging techniques. Congenital adrenal hyperplasia (see Chapter 594 ), particularly 11β-hydroxylase deficiency, may present with hypertension and signs of androgen excess, but in that condition cortisol levels are low and levels of cortisol precursors (17-hydroxyprogesterone, 11-deoxycortisol) are elevated. Obese patients may be hypertensive and have hyperandrogenism, but cortisol secretion should be readily suppressed by dexamethasone.
Treatment
The goal of treatment is to suppress the excess secretion of ACTH, thereby suppressing the increased production of adrenal steroids with mineralocorticoid and androgenic activity. This requires administration of high doses of a pure glucocorticoid agonist such as dexamethasone (typically ~20-40 µg/kg/day) with careful titration to suppress endogenous corticosteroid secretion without causing signs of glucocorticoid excess such as excessive weight gain or suppression of linear growth.
Cortisone Reductase Deficiency
Etiology
Levels of active glucocorticoids in target tissues are modulated by 2 isozymes of 11β-hydroxysteroid dehydrogenase. The HSD11B2 isozyme converts cortisol to an inactive metabolite, cortisone; the 2 steroids differ in the presence of an 11β-hydroxyl versus an 11-oxo group, respectively. Mutations in this enzyme cause the syndrome of apparent mineralocorticoid excess (discussed later in this chapter). Conversely, the HSD11B1 isozyme converts cortisone to cortisol, and so it is sometimes referred to as cortisone reductase. This isozyme is expressed at high levels in glucocorticoid target tissues, particularly the liver, where it ensures adequate levels of active glucocorticoids (cortisol and corticosterone) to meet metabolic demands without requiring excessive adrenal cortisol secretion.
The HSD11B1 isozyme is located in the endoplasmic reticulum (i.e., it is a microsomal enzyme) and functions as a dimer. It accepts electrons from reduced nicotine–adenine dinucleotide phosphate, which is generated within the endoplasmic reticulum by hexose-6-phosphate dehydrogenase, an enzyme distinct from cytoplasmic glucose-6-phosphate dehydrogenase.
Apparent cortisone reductase deficiency is caused by homozygous mutations in hexose-6-phosphate dehydrogenase that prevent generation of reduced nicotine–adenine dinucleotide phosphate within the endoplasmic reticulum and thus starve HSD11B1 of its essential cofactor for the reductase reaction. Very rare patients have been reported to have heterozygous mutations in the HSD11B1 gene itself and thus have “true” cortisone reductase deficiency; because the enzyme functions as a homodimer, heterozygous mutations are able to impair three fourths of all dimers.
Clinical Manifestations
Because circulating cortisone is not converted to cortisol, the circulating half-life of cortisol is decreased and the adrenal cortex must secrete additional cortisol to compensate. This leads to adrenocortical overactivity analogous to, but generally much milder than, that seen in generalized glucocorticoid resistance. This is usually not severe enough to cause hypertension, presenting instead with mild to moderate signs of androgen excess such as hirsutism, oligomenorrhea or amenorrhea, and infertility in females and precocious pseudopuberty (axillary and pubic hair, and penile enlargement, but not testicular enlargement) in males.
Laboratory Findings
The ratio of cortisol to cortisone in blood is lower than usual. The same is true of urinary metabolites, typically measured as a ratio of the sum of the tetrahydrocortisol and allotetrahydrocortisol excretion to that of tetrahydrocortisone. These determinations are best accomplished by gas chromatography followed by mass spectrometry and are available in specialized reference laboratories. Absolute levels of cortisol and ACTH are within normal limits.
Differential Diagnosis
Cortisone reductase deficiency has to be distinguished from, and is much less common than, other causes of androgen excess such as polycystic ovarian syndrome and nonclassical congenital adrenal hyperplasia as a result of 21-hydroxylase deficiency.
Treatment
Treatment is aimed at decreasing adrenal overactivity and thus reducing secretion of androgens. This can be accomplished by administration of hydrocortisone.
Altered End-Organ Sensitivity to Mineralocorticoids
Pseudohypoaldosteronism
Etiology
Pseudohypoaldosteronism type 1 (PHA1) is a monogenic disease in which aldosterone action is deficient and patients are thus unable to resorb urinary sodium or excrete potassium properly. There are 2 forms. A relatively mild autosomal dominant form is caused by mutations in the NR3C2 gene encoding the human mineralocorticoid receptor. A heterozygous mutation is sufficient to cause disease because the mineralocorticoid receptor interacts with DNA as a dimer, and three fourths of the dimers are defective in individuals carrying heterozygous mutations (assuming mutant protein is synthesized). A more-severe autosomal recessive form is usually the result of homozygous mutations in the α (SCNN1A), β (SCNN1B), or γ (SCNN1G) subunits of the epithelial Na+ channel, but 1 reported case of severe autosomal recessive disease was caused by homozygous mutations in NR3C2 .
PHA1 should not be confused with pseudohypoaldosteronism type 2 , a rare Mendelian syndrome characterized by hyperkalemia and, in contrast to PHA1, by hypertension from excessive renal salt reabsorption. This disorder is caused by mutations in the renal regulatory kinases WNK1 and WNK4 or components of an E3 ubiquitin ligase complex Kelch-like 3 (KLHL3) and Cullin 3 (CUL3).
Clinical Manifestations
Infants with PHA1 present with hyperkalemia, hyponatremia, hypovolemia, hypotension, and failure to thrive. In more-severe (usually autosomal recessive) cases, salt loss is not confined to the kidney but instead occurs from most epithelia. Mothers may report that the skin of their affected infants tastes salty. Some infants suffer from cystic fibrosis–like pulmonary symptoms. It is often difficult to control electrolyte abnormalities in patients with the autosomal recessive form, leading to frequent hospitalizations and a need for close clinical monitoring.
It is noteworthy that signs and symptoms of aldosterone deficiency tend to remit as the patients get older, particularly in the autosomal dominant form. This is similar to what is seen in actual aldosterone deficiency as occurs in the salt-losing forms of congenital adrenal hyperplasia or aldosterone synthase deficiency. The kidney matures after early infancy to become more efficient at excreting potassium, and although breast milk and infant formula are low in sodium, the normal adult Western diet is relatively high in sodium, thus compensating for the renal salt wasting.
Laboratory Findings
Infants have marked hyperkalemia and hyponatremia. Both plasma renin and aldosterone are markedly elevated. Levels of cortisol and ACTH are normal. If hypovolemia is severe, patients may develop prerenal azotemia. With severe hyperkalemia, the electrocardiogram may include tall peaked T or ventricular tachycardia.
Differential Diagnosis
PHA in infants should be distinguished from other causes of hyperkalemia and hyponatremia. These include renal failure of any cause, congenital adrenal hyperplasia, aldosterone synthase deficiency, and other causes of adrenocortical insufficiency such as AHC. Patients with renal failure will have elevated blood urea nitrogen and creatinine, but these may also be seen in severely dehydrated patients with PHA or adrenal insufficiency. Patients with any form of adrenal insufficiency in this clinical context will have low or low-normal aldosterone levels (with elevated plasma renin), in contrast to the elevated aldosterone levels seen in PHA. Patients with congenital adrenal hyperplasia have elevated levels of steroid precursors such as 17-hydroxyprogesterone (in patients with 21-hydroxylase deficiency), and patients with most forms of adrenal insufficiency have elevated ACTH levels.
Treatment
Infants must be given sodium supplementation (initially intravenous and then oral or enteral), typically approximately 8 mEq/kg/day. Potassium levels in the infant formula often need to be reduced, which may be accomplished by mixing the formula with polystyrene resin (Kayexalate) and then decanting the formula prior to feeding. Fludrocortisone, a synthetic mineralocorticoid, may be efficacious in milder autosomal dominant cases if administered in high doses (titrating up to ~0.5 mg daily). Significant electrolyte abnormalities require treatment with intravenous normal saline and rectal polystyrene resin. Severe hyperkalemia may require glucose and insulin infusions to control.
Apparent Mineralocorticoid Excess
Etiology
The syndrome of apparent mineralocorticoid excess is an autosomal recessive disorder caused by mutations in the HSD11B2 gene encoding the type 2 isozyme of 11β-hydroxysteroid dehydrogenase. The mineralocorticoid receptor actually has nearly identical affinities for aldosterone (the main mineralocorticoid hormone) and cortisol, yet cortisol is normally only a weak mineralocorticoid in vivo. This is because HSD11B2 is expressed along with the mineralocorticoid receptor in most target tissues such as the renal cortical collecting duct epithelium. It converts cortisol to cortisone, which is not an active steroid, thus preventing it from occupying the mineralocorticoid receptor. In contrast, aldosterone is not a substrate for the enzyme because its 11β-hydroxyl group forms a hemiketal with the 18-aldehyde group of the steroid and is thus not accessible to the enzyme. Thus, in the absence of HSD11B2, cortisol is able to efficiently occupy the mineralocorticoid receptor, and because cortisol concentrations are normally far higher than those of aldosterone, this results in signs and symptoms of mineralocorticoid excess.
A similar clinical picture occurs with excessive consumption of licorice or licorice-flavored chewing tobacco; licorice contains compounds including glycyrrhetinic and glycyrrhizic acids that inhibit HSD11B2. Carbenoxolone, an antihypertensive drug that is not marketed in the United States, has similar effects.
Clinical Manifestations
Affected infants often have some degree of intrauterine growth restriction, with birthweights of 2 kg typical for term infants. Infants and children often fail to thrive. Severe hypertension (to ~200/120 mm Hg) is almost always present. In some patients, the hypertension tends to be labile or paroxysmal with severe emotional stress as a precipitating factor. Complications of hypertension have included cerebrovascular accidents. Several patients have died during infancy or adolescence, either from electrolyte imbalances leading to cardiac arrhythmias or from vascular sequelae of hypertension. Hypokalemic alkalosis can eventually cause nephrocalcinosis (often visible on renal ultrasound) and nephrogenic diabetes insipidus leading to polyuria and polydipsia. Deleterious effects on muscle range from elevations in serum creatine phosphokinase to frank rhabdomyolysis . Electrocardiograms show left ventricular hypertrophy.
Laboratory Findings
Hypokalemia and alkalosis are common but not consistently present. Sodium levels are generally in the upper part of the reference range. Aldosterone and renin levels are very low because the hypertension and hypervolemia are independent of aldosterone concentrations. Serum cortisol and ACTH levels are generally within normal limits. The serum half-life of cortisol is increased, but the test for this requires a radioactive tracer and is not clinically available. Total urinary excretion of cortisol metabolites is markedly decreased. The urinary ratio of free cortisol to free cortisone is elevated, as is the ratio of urinary tetrahydrocortisol plus allotetrahydrocortisol to tetrahydrocortisone.
Differential Diagnosis
The differential diagnosis includes other forms of severe childhood hypertension such as renal artery anomalies, but relatively few conditions present with suppressed renin and aldosterone levels. Liddle syndrome (see below) has a similar presentation but no abnormalities in the steroid profile, typically has an autosomal dominant mode of inheritance, and does not respond to treatment with mineralocorticoid receptor antagonists. Hypertensive forms of congenital adrenal hyperplasia (see Chapter 594 ) also have suppressed renin and aldosterone levels, but they present with signs of androgen excess (11β-hydroxylase deficiency) or androgen deficiency (17α-hydroxylase deficiency); the latter can be difficult to appreciate in young children. The steroid profiles in congenital adrenal hyperplasia differ from those seen in apparent mineralocorticoid excess syndrome.
Patients with severe Cushing syndrome may have high enough cortisol levels to overwhelm renal HSD11B2, leading to severe hypertension with alterations in urinary cortisol-to-cortisone ratios. This occurs most often in patients with the ectopic ACTH syndrome. This generally does not present a diagnostic dilemma, because other signs of Cushing syndrome are present including high cortisol levels.
Treatment
Treatment includes a low-salt diet, potassium supplementation, and mineralocorticoid receptor blockade with spironolactone or eplerenone; a sodium channel blocker, such as amiloride or triamterene may work at least as well. In principle, suppression of cortisol secretion with dexamethasone (which does not bind the mineralocorticoid receptor) should work, but in practice it is much less effective than mineralocorticoid receptor blockade.
Liddle Syndrome
Etiology
Liddle syndrome is a form of hypertension and hypokalemia that is clinically similar to the syndrome of apparent mineralocorticoid excess, but it is inherited in an autosomal dominant manner. It is caused by activating mutations in the β (SCNN1B) or γ (SCNN1G) subunits of the epithelial sodium channel. Most of these mutations prevent the channel subunits from being ligated to ubiquitin and targeted to the proteasome for degradation, a process that is normally regulated indirectly by aldosterone. The net effect is to increase the number of open channels at the apical surface of epithelial cells of the renal collecting duct, thus facilitating sodium resorption and potassium excretion. This disorder is thus the exact opposite of the autosomal recessive form of pseudohypoaldosteronism discussed previously.
Clinical Manifestations, Laboratory Findings, and Differential Diagnosis
Liddle syndrome is characterized by severe early-onset hypertension and by hypokalemia, which may not be persistent. Aldosterone and renin levels are suppressed but all steroid hormone levels are normal.
The differential diagnosis is the same as that for apparent mineralocorticoid excess.
Treatment
The mainstays of treatment are a low-salt diet, potassium supplementation, and a sodium channel blocker such as amiloride or triamterene. Mineralocorticoid receptor antagonists such as spironolactone are ineffective.
Bibliography
Generalized Glucocorticoid Resistance
Charmandari E, Kino T, Chrousos GP. Primary generalized familial and sporadic glucocorticoid resistance (Chrousos syndrome) and hypersensitivity. Endocr Dev . 2013;24:67–85.
van Rossum EF, Lamberts SW. Glucocorticoid resistance syndrome: a diagnostic and therapeutic approach. Best Pract Res Clin Endocrinol Metab . 2006;20:611–626.
Cortisone Reductase Deficiency
Lavery GG, Walker EA, Tiganescu A, et al. Steroid biomarkers and genetic studies reveal inactivating mutations in hexose-6-phosphate dehydrogenase in patients with cortisone reductase deficiency. J Clin Endocrinol Metab . 2008;93:3827–3832.
Lawson AJ, Walker EA, Lavery GG, et al. Cortisone-reductase deficiency associated with heterozygous mutations in 11beta-hydroxysteroid dehydrogenase type 1. Proc Natl Acad Sci USA . 2011;108:4111–4116.
White PC, Rogoff D, McMillan DR. Physiological roles of 11 beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase. Curr Opin Pediatr . 2008;20:453–457.
Altered End-Organ Sensitivity to Mineralocorticoids
Boyden LM, Choi M, Choate KA, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature . 2012;482:98–102.
Fernandes-Rosa FL, Hubert EL, Fagart J, et al. Mineralocorticoid receptor mutations differentially affect individual gene expression profiles in pseudohypoaldosteronism type 1. J Clin Endocrinol Metab . 2011;96:E519–E527.
Hubert EL, Teissier R, Fernandes-Rosa FL, et al. Mineralocorticoid receptor mutations and a severe recessive pseudohypoaldosteronism type 1. J Am Soc Nephrol . 2011;22:1997–2003.
Zennaro MC, Hubert EL, Fernandes-Rosa FL. Aldosterone resistance: structural and functional considerations and new perspectives. Mol Cell Endocrinol . 2012;350:206–215.
Apparent Mineralocorticoid Excess
Chapman K, Holmes M, Seckl J. 11beta-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev . 2013;93:1139–1206.
Hassan-Smith Z, Stewart PM. Inherited forms of mineralocorticoid hypertension. Curr Opin Endocrinol Diabetes Obes . 2011;18:177–185.
Liddle Syndrome
Bubien JK. Epithelial Na+ channel (ENaC), hormones, and hypertension. J Biol Chem . 2010;285:23527–23531.
Ellison DH. Ubiquitylation and the pathogenesis of hypertension. J Clin Invest . 2013;123:546–548.