Hypopituitarism
Briana C. Patterson, Eric I. Felner
Hypopituitarism denotes underproduction of 1 or multiple pituitary hormones. Affected children have postnatal growth impairment and other endocrine deficiencies that are specifically corrected by hormone replacement. The incidence of congenital hypopituitarism is thought to be between 1 in 4,000 and 1 in 10,000 live births. There is an epidemiologic association between hypopituitarism and breech delivery, but the causal relationship is not understood. With expanding knowledge of the genes that direct pituitary development or hormone production, an increasing proportion of cases can be attributed to specific genetic disorders. Mutations in 7 candidate genes account for 13% of isolated growth hormone deficiency (IGHD) and 20% of multiple pituitary hormone deficiency (MPHD) cases. The likelihood of finding mutations is increased by consanguinity and occurrence in siblings or across generations; however, in most cases of IGHD and MPHD, no specific genetic cause can be identified. The genes, hormonal phenotypes, associated abnormalities, and modes of transmission for such established genetic disorders are shown in Tables 573.1 and 573.3 through 573.5 . Acquired hypopituitarism usually has a later onset and different causes (Table 573.2 ).
Multiple Pituitary Hormone Deficiency
Genetic Forms (Table 573.1 )
Sequentially expressed transcriptional activation factors direct the differentiation and proliferation of anterior pituitary cell types. These proteins are members of a large family of DNA-binding proteins resembling homeobox genes. Mutations produce different forms of MPHD. PROP1 and POU1F1 genes are expressed fairly late in pituitary development only in cells of the anterior pituitary and result in hypopituitarism without anomalies of other organ systems. The HESX1, LHX3, LHX4, OTX2, SOX3, and PITX2 genes are expressed at earlier stages and are not restricted to the pituitary. Mutations in these genes tend to produce phenotypes that extend beyond hypopituitarism to include abnormalities in other organs, and the degree of hypopituitarism is typically variable.
Table 573.1
Etiologic Classification of Congenital and Genetic Forms of Multiple Pituitary Hormone Deficiency
| GENE OR LOCATION | PHENOTYPE | INHERITANCE |
|---|---|---|
| GENETIC FORMS | ||
| POU1F1 (PIT1) | GH, PRL deficiencies, variable TSH deficiency | R, D |
| PROP1 | GH, TSH, PRL, LH, FSH deficiencies, variable ACTH deficiency, variable AP | R |
| LHX3 | GH, TSH, PRL, LH, FSH deficiencies, variable AP, ±short neck, limited neck rotation, sensorineural deafness | R |
| LHX4 | GH, TSH, ACTH deficiencies, small AP, EPP, variable Arnold-Chiari, cerebellar abnormalities | D |
| TPIT | ACTH, severe neonatal form | R |
| HESX1 | GH deficiency, variable for others, small AP, EPP, optic nerve hypoplasia; septo-optic dysplasia | R, D |
| SOX2 | LH, FSH, variable GH, anophthalmia, microphthalmia, esophageal atresia, sensorineural hearing loss | D |
| SOX3 | Variable deficiencies, ±MR, EPP, small AP and stalk, developmental delay | XL |
| PITX2 | Axenfeld-Rieger syndrome | D |
| GLI2 | Hypopituitarism, holoprosencephaly, midline defects, polydactyly | D |
| GLI3 | Hall-Pallister syndrome, hypopituitarism | D |
| SHH (Sonic Hedgehog) | GH deficiency with single central incisor | D |
| OTX12 |
GH or combined deficiencies Anophthalmia or microphthalmia, coloboma, developmental delay |
D |
| TBX19 | ACTH, neonatal hypoglycemia or cholestatic jaundice | R |
| TGIF, SHH, CDON, GPR161, PROKR2 | Pituitary stalk interruption syndrome : thin or absent pituitary stalk, hypoplasia of adenohypophysis, ectopic neurohypophysis, neonatal hypoglycemia, cholestasis, micropenis | Holoprosencephaly related gene group |
| UNCERTAIN ETIOLOGY | ||
| Congenital absence of pituitary | ||
| Optic nerve hypoplasia syndrome/septo-optic dysplasia | Optic nerve hypoplasia, nystagmus, absent septum pellucidum, pituitary hypoplasia | |
ACTH, Adrenocorticotropic hormone; AP, anterior pituitary; D, dominant; EPP, ectopic posterior pituitary; FSH, follicle-stimulating hormone; GH, growth hormone; LH, luteinizing hormone; MR, mental retardation; PRL, prolactin; R, recessive; TSH, thyroid-stimulating hormone; XL, X-linked.
PROP1
PROP1 (prophet of PIT1 ) is found in the nuclei of somatotropes, lactotropes, and thyrotropes. Its roles include turning on POU1F1 expression and downregulating HESX1 expression. Although no genetic mutation can be identified in most patients with MPHD, mutations of PROP1 are the most common explanation for recessive MPHD and are 10 times as common as the combined total of mutations in other pituitary transcription factor genes. Deletions of 1 or 2 base pairs in exon 2 are most common, followed by missense, nonsense, and splice-site mutations. Anterior pituitary hormone deficiencies are seldom evident in the neonatal period. The median age at diagnosis of growth hormone (GH) deficiency is around 6 yr. Recognition of thyroid-stimulating hormone (TSH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and adrenocorticotropic hormone (ACTH) deficiencies is delayed relative to recognition of GH deficiency. Anterior pituitary size is small in most patients, but in others there is progressive enlargement of the pituitary.
POU1F1 (PIT1)
POU1F1 (formerly PIT1 ) was identified as a nuclear protein that binds to the GH and prolactin promoters. It is necessary for emergence and mature function of somatotropes, lactotropes, and thyrotropes. Dominant and recessive mutations in POU1F1 are responsible for complete deficiencies of GH and prolactin and variable TSH deficiency. Affected patients exhibit nearly normal fetal growth but experience severe growth failure in the 1st yr of life. With normal production of LH and FSH, puberty develops spontaneously, although at a later than normal age. These patients are not at risk for development of ACTH deficiency. Anterior pituitary size is normal to small.
HESX1
The HESX1 gene is expressed in precursors of all 5 cell types of the anterior pituitary early in embryologic development. Mutations result in heterogeneous phenotypes with defects in development of the optic nerve and pituitary. The anterior pituitary may be hypoplastic or aplastic, and the posterior pituitary may be orthotopic or ectopic. Patients may have IGHD or MPHDs, with or without the optic nerve hypoplasia syndrome, which is also called septo-optic dysplasia (incomplete development of the septum pellucidum with optic nerve hypoplasia and pituitary insufficiency). However, the great majority of patients with optic nerve hypoplasia syndrome do not have HESX1 mutations.
LHX3 and LHX4
The phenotype produced by recessive loss-of-function mutations of the LHX3 gene resembles that produced by PROP1 mutations. There are deficiencies of GH, prolactin, TSH, LH, and FSH but not ACTH. Some affected persons show enlargement of the anterior pituitary. The first patients to be described had the unusual findings of a short neck and a rigid cervical spine. Dominantly inherited mutations in the structurally similar LHX4 gene consistently produce GH deficiency, with the variable presence of TSH and ACTH deficiencies. Additional findings can include a very small V -shaped pituitary fossa, Chiari I malformation, and an ectopic posterior pituitary.
Other Congenital Forms
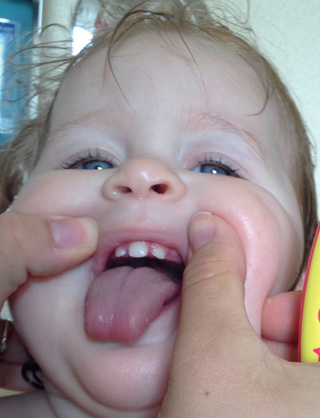
Pituitary hypoplasia can occur as an isolated phenomenon or in association with more extensive developmental abnormalities such as anencephaly or holoprosencephaly. Midline facial anomalies (cleft lip, palate; see Chapter 336 ) or the finding of a solitary maxillary central incisor indicate a high likelihood of GH or other anterior or posterior hormone deficiency (Fig. 573.1 ). At least 12 genes have been implicated in the complex genetic etiology of holoprosencephaly . The Hall-Pallister syndrome is caused by dominant loss of function mutations in the GLI3 gene. Absence of the pituitary gland is accompanied by hypothalamic hamartoma, polydactyly, nail dysplasia, bifid epiglottis, imperforate anus, and anomalies of the heart, lungs, and kidneys. The combination of anophthalmia and hypopituitarism has been associated with mutations in the SIX6, SOX2, and OTX2 genes.
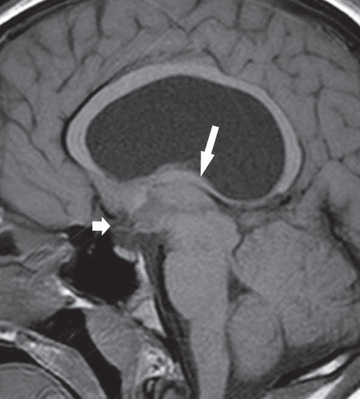
The optic nerve hypoplasia syndrome or septo-optic dysplasia may be detected due to clinical observation of nystagmus and visual impairment in infancy. Neuroimaging demonstrates optic nerve and brain abnormalities and is associated with anterior and/or posterior pituitary hormone deficiencies in up to 75% of the cases (Fig. 573.2 ). Although these patients often show the triad of a small anterior pituitary gland, an attenuated pituitary stalk, and an ectopic posterior pituitary bright spot, the primary etiology of the hypopituitarism in this condition is thought to be hypothalamic dysfunction. GH deficiency is the most commonly observed hormone deficiency, and other anterior pituitary hormone deficiencies are less common. Diabetes insipidus is reported in only about 5% of cases. The etiology is likely multifactorial and may involve interaction between genetic and environmental factors. In the vast majority of cases, no single gene defect can be identified.
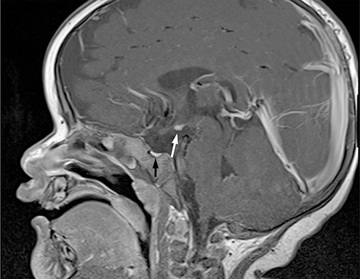
Severe, early-onset MPHD including deficiency of ACTH is often associated with the triad of anterior pituitary hypoplasia, absence or attenuation of the pituitary stalk, and an ectopic posterior pituitary bright spot on MRI. Most cases are sporadic, and there is a male predominance. Some are from abnormalities of the SOX3 gene, located on the X chromosome.
Acquired Forms
Any lesion that damages the hypothalamus, pituitary stalk, or anterior pituitary can cause pituitary hormone deficiency (Table 573.2 ). Because such lesions are not selective, multiple hormonal deficiencies are usually observed. Diabetes insipidus is more frequent in acquired than in congenital hypopituitarism. The most common lesion is the craniopharyngioma (see Chapter 524 ). Central nervous system germinoma, eosinophilic granuloma (histiocytosis), tuberculosis, sarcoidosis, toxoplasmosis, meningitis, pituitary abscess, and aneurysms can also cause hypothalamic-hypophyseal destruction. Children treated with radiation therapy for central nervous system or nasopharyngeal tumors are at increased risk for GH deficiency and other pituitary hormone deficiencies to the extent that the radiation field includes the hypothalamus and/or pituitary, even if the tumor itself is remote from the pituitary and hypothalamus. The magnitude of the risk and the timing of the emergence of pituitary hormone deficiencies depend on the dose of radiation to the hypothalamic-pituitary axis and the duration of elapsed time after radiotherapy is complete. High doses of radiation (>50 Gy) are likely to produce GH deficiency sooner than 1 yr after irradiation, whereas other anterior pituitary hormone deficiencies may not appear until later. GH production appears to be particularly vulnerable to the effects of irradiation, even at lower doses, whereas deficiencies of ACTH, gonadotropins, and TRH/TSH occur with declining frequency and typically occur at higher doses of radiation. Irradiation alone does not typically result in diabetes insipidus. Traumatic brain injury, including abusive head trauma, motor vehicle accidents, and chronic repetitive head injury, is an increasingly recognized cause of pituitary dysfunction due to damage to the pituitary, its stalk, or the hypothalamus.
Table 573.2
Causes of Acquired Pituitary Insufficiency
| TRAUMATIC |
| INFILTRATIVE/INFLAMMATORY |
| INFECTIONS |
| VASCULAR |
| NEOPLASTIC |
| FUNCTIONAL |
|
|
Modified from Kaiser U, Ho KKY: Pituitary physiology and diagnostic evaluation. In Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, editors: Williams textbook of endocrinology , ed 13, Philadelphia, 2016, Elsevier. Table 8.5, p. 193.
Isolated Growth Hormone Deficiency and Insensitivity
Genetic Forms of Growth Hormone Deficiency
IGHD is caused by abnormalities of the GH-releasing hormone receptor, GH genes, and genes located on the X chromosome (Table 573.3 ).
Table 573.3
Established Genetic Defects of the GH-IGF Axis Resulting in Isolated GH Deficiency, GH Insensitivity, or IGF-1 Deficiency
| MUTANT GENE | INHERITANCE | PHENOTYPE |
|---|---|---|
| ISOLATED GROWTH HORMONE DEFICIENCY | ||
| GHRHR | AR | Type IB form of IGHD; low levels of GH production, but less severe than type 1A IGHD; may also be caused by mutations in GH1 |
| GHS-R | AD | GHD and ISS |
| GH1 | AR | Type IA form of IGHD, in utero growth retardation; absent GH production due to gene deletion, antibodies to GH develop over time during treatment |
| GH1 | AR | Type IB form of IGHD; low levels of GH production, but less severe than Type 1A IGHD; may also be caused by mutations in GHRHR |
| GH1 | AD | Type II form of IGHD; dominant negative mutations in GH1 which decrease GH secretion |
| BTK | XL | Type III form of IGHD; hypogammaglobulinemia |
| GH1 | AD | Bioinactive GH molecule; rare, dominant negative mutation in GH1 that interferes with GHR signaling |
| GROWTH HORMONE INSENSITIVITY | ||
| GHR | AR, AD | IGF-I deficiency; high GH level; normal, decreased or increased GHBP (depending on which domain of the receptor is affected); unresponsive to GH treatment |
| IGF-1 DEFICIENCY | ||
| IGF1 | AR | IGF-1 deficiency; IUGR and postnatal growth failure, sensorineural deafness, insulin resistance, microcephaly |
| STAT5b | AR | IGF-1 deficiency, variable immune defect, hyperprolactinemia, chronic pulmonary infections, eczema |
| ALS | AR | IGF-1 deficiency; variable postnatal growth failure, delayed puberty |
AD, Autosomal dominant; ALS, acid labile subunit; AR, autosomal recessive; GH, growth hormone; GHBP, GH-binding protein; GHRHR, GH-releasing hormone receptor; IGF, insulin-like growth factor; IGHD, isolated GHD; ISS, idiopathic short stature; IUGR, intrauterine growth retardation; XL, X-linked.
Modified from Sperling MA: Pediatric Endocrinology, ed 4, Philadelphia, 2014, Elsevier. Table 10.3, p. 333.
Growth Hormone–Releasing Hormone Receptor
Recessive loss-of-function mutations in the receptor for GH-releasing hormone interfere with proliferation of somatotropes during pituitary development and disrupt the most important signals for release of GH. The anterior pituitary is small, in keeping with the observation that somatotropes normally account for >50% of pituitary volume. There is some compromise of fetal growth followed by severe compromise of postnatal growth.
GH1
The GH1 gene is one of a cluster of 5 genes on chromosome 17q22-24. This cluster arose through successive duplications of an ancestral GH gene. Unequal crossing over at meiosis has produced a variety of gene deletions. Small deletions (<10 kb) remove only the GH1 gene, whereas large deletions (45 kb) remove 1 or more of the adjacent genes (CSL, CS1, GH2, and CS2 ). The growth phenotype is identical with deletion of GH1 alone or GH1 together with 1 or more of the adjacent genes. Loss of the CS1, GH2, and CS2 genes without loss of GH1 causes deficiency of chorionic somatomammotropin and placental GH in the maternal circulation, but it does not result in fetal or postnatal growth retardation. Most children with GH1 gene deletions respond very well to recombinant GH treatment, but some develop antibodies to GH and cease growing.
Recessively transmitted mutations in the GH1 gene produce a similar phenotype. Missense, nonsense, and frameshift mutations have been described. Autosomal dominant IGHD is also caused by mutations in GH1. The mutations usually involve splice-site errors in intron 3 and result in a variant protein that lacks the amino acids normally encoded by exon 3. Accumulation of this protein interferes with the processing, storage, and secretion of the normal GH protein and may result in additional deficiencies of TSH and/or ACTH. There are several reports of mutations in GH1 that lead to variant proteins with reduced biological activity.
X-Linked Isolated Growth Hormone Deficiency
Two loci on the X chromosome have been associated with hypopituitarism. The first lies at Xq21.3-q22 in the region of the Bruton thymidine kinase (BTK) gene. Mutations in this region produce hypogammaglobulinemia and IGHD. The second locus maps farther out on the long arm, at Xq24-q27.1, a region containing the SOX2 transcription factor gene. Abnormalities in this locus have been linked to IGHD with intellectual disability, as well as to MPHD with the triad of pituitary hypoplasia, missing pituitary stalk, and ectopic posterior pituitary gland.
Acquired Forms
The GH axis is more susceptible to disruption by acquired conditions than are other hypothalamic-pituitary axes. Recognized causes of acquired GH deficiency include the use of radiotherapy for malignancy, meningitis, histiocytosis, and trauma.
Children who receive radiotherapy for central nervous system tumors, leukemia, or total body irradiation prior to hematopoietic stem cell transplant are at risk for developing GH deficiency. Spinal irradiation results in disproportionately poor growth of the axial skeleton relative to the appendicular skeleton; this defect is not remediable with GH treatment. Growth typically slows during radiation therapy or chemotherapy, may improve for 1-2 yr after cancer treatment, and then declines with the development of GH deficiency. The dose and frequency of radiotherapy are important determinants of hypopituitarism. GH deficiency is almost universal 5 yr after therapy with a total dose ≥35 Gy. More subtle defects are seen with doses around 20 Gy. Deficiency of GH is the most common defect, but deficiencies of TSH and ACTH can also occur. Cranial irradiation may result in precocious puberty in conjunction with GH deficiency. The clinician is likely to encounter children in the 8- to 10-yr age range who are growing at rates that are normal for chronological age but subnormal for stage of pubertal development.
Growth Hormone Insensitivity
Abnormalities of the Growth Hormone Receptor
GH insensitivity is caused by disruption of pathways distal to production of GH (Table 573.4 ). Laron syndrome involves mutations of the GH receptor. Children with this condition clinically resemble those with severe IGHD. Birth length tends to be about 1 SD below the mean, and severe short stature with lengths >4 SD below the mean is present by 1 yr of age. Resting and stimulated GH levels tend to be high and insulin-like growth factor (IGF) 1 levels are low. The GH receptor has an extracellular GH-binding domain, a transmembrane domain, and an intracellular signaling domain. Mutations in the extracellular domain interfere with binding of GH. Serum GH-binding protein activity, representing the circulating form of the membrane receptor for GH, is generally low. Mutations in the transmembrane domain can interfere with anchoring of the receptor to the plasma membrane. In these cases, circulating GH-binding protein activity is normal or high. Mutations in the intracellular domain interfere with JAK/STAT signaling.
Table 573.4
Proposed Classification of Growth Hormone Insensitivity
| PRIMARY GH INSENSITIVITY (HEREDITARY DEFECTS) |
| SECONDARY GH INSENSITIVITY (ACQUIRED DEFECTS) |
ALS, Acid-labile subunit; GH, growth hormone; IGF, insulin-like growth factor.
From Sperling MA: Pediatric Endocrinology, ed 4, Philadelphia, 2014, Elsevier. Box 10.4.
Postreceptor Forms of Growth Hormone Insensitivity
Some children with severe growth failure, high GH and low IGF-1 levels, and normal GH-binding protein levels have abnormalities distal to the GH binding and activation of the GH receptor. Several have been found to have mutations in the gene encoding signal transducer and activator of transcription 5b (STAT5b). Disruption of this key intermediate connecting receptor activation to gene transcription produces growth failure similar to that seen in Laron syndrome. These patients also suffer from immunodeficiency and chronic pulmonary infections, consistent with important roles for STAT5b in interleukin cytokine signaling.
IGF-1 Gene Abnormalities
Abnormalities of the IGF-1 gene produce severe prenatal and postnatal growth impairment. Microcephaly, intellectual disability, and deafness are present in patients with exon deletion and a missense mutation. These patients can be expected to respond to recombinant IGF-1 treatment.
Insulin-Like Growth Factor–Binding Protein Abnormalities
Mutation of the gene encoding the acid-labile subunit of the circulating 165-kDa IGF-1, IGF-BP3, acid-labile subunit complex has been associated with short stature. Total IGF-1 levels were very low. The index case, with homozygosity for an acid-labile subunit mutation, did not show an increase in IGF-1 levels or an increase in growth rate during GH treatment.
IGF-1 Receptor Gene Abnormalities
Mutations of the IGF-1 receptor also compromise prenatal and postnatal growth. The phenotype does not appear to be as severe as that seen with absence of IGF-1. Adult heights are closer to the normal range, and affected patients do not have intellectual disability or deafness.
Clinical Manifestations
Congenital Hypopituitarism
The child with hypopituitarism is usually of normal size and weight at birth, although those with MPHD and genetic defects of the GH1 or GHR gene have birth lengths that average 1 SD below the mean. Children with severe defects in GH production or action typically fall more than 4 SD below the mean for length by 1 yr of age. Those with less-severe deficiencies grow at rates below the 25th percentile for age and gradually diverge from normal height percentiles. Delayed closure of the epiphyses permits growth beyond the normal age when growth should be complete. Features of GH insensitivity, including Laron syndrome, are noted in Table 573.5 .
Table 573.5
Clinical Features of Growth Hormone Insensitivity, Including Classic Laron Syndrome
| GROWTH AND DEVELOPMENT |
| OTHER PHYSICAL CHARACTERISTICS |
| LATE FINDINGS/OTHER COMPLICATIONS |
From Sperling MA: Pediatric endocrinology , ed 4, Philadelphia, 2014, Elsevier. Box 10.5.
Infants with congenital defects of the pituitary or hypothalamus may present with neonatal emergencies such as apnea, cyanosis, or severe hypoglycemia with or without seizures. Prolonged neonatal cholestatic jaundice is common. It involves elevation of conjugated and unconjugated bilirubin and may be associated with giant cell neonatal hepatitis. Nystagmus can suggest septo-optic dysplasia (see Chapter 591 ). Micropenis in boys provides an additional diagnostic clue. Deficiency of GH may be accompanied by hypoadrenalism (see Chapter 593 ) and hypothyroidism (see Chapter 581 ), as well as gonadotropin deficiency (see Chapters 601.2 and 604.2 ).
On physical examination, the head is round and the face is short and broad. The frontal bone is prominent, and the bridge of the nose is depressed and saddle shaped. The nose is small, and the nasolabial folds are well developed. The mandible and the chin are underdeveloped, and the teeth, which erupt late, are often crowded. The neck is short and the larynx is small. The voice is high-pitched and remains high after puberty. The extremities are well proportioned, with small hands and feet. Weight for height is usually normal, but an excess of body fat and a deficiency of muscle mass contribute to a pudgy appearance. The genitals are usually small for age, and sexual maturation may be delayed or absent. Facial, axillary, and pubic hair usually is lacking, and the scalp hair is fine. Intelligence is usually normal for age, unless there are other structural brain abnormalities, and the children may seem precocious compared with children of a similar size.
Acquired Hypopituitarism
The child is normal initially, and manifestations similar to those seen in idiopathic pituitary growth failure gradually appear and progress. When complete or almost complete destruction of the pituitary gland occurs, signs of pituitary insufficiency are present. Atrophy of the adrenal cortex, thyroid, and gonads results in loss of weight, asthenia, sensitivity to cold, mental torpor, and absence of sweating. Sexual maturation fails to take place or regresses if already present. There may be atrophy of the gonads and genital tract with amenorrhea and loss of pubic and axillary hair. There is a tendency to hypoglycemia. Growth slows dramatically. Diabetes insipidus (see Chapter 574 ) may be present but can by obscured by the development of central adrenal insufficiency.
If the lesion is an expanding tumor, symptoms such as headache, vomiting, visual disturbances, pathologic sleep patterns, decreased school performance, seizures, polyuria, and growth failure can occur. Slowing of growth can antedate neurologic signs and symptoms, especially with craniopharyngioma. In other cases, evidence of pituitary insufficiency may first appear after surgical intervention. In children with craniopharyngioma, visual field defects, optic atrophy, papilledema, obesity, and cranial nerve palsy are common.
Laboratory Findings
GH deficiency should be suspected in children with severe postnatal growth failure (Table 573.6 ). Criteria for short stature include height below the 1st percentile for age and sex or height >2 SD below sex-adjusted mid-parent height. Acquired GH deficiency can occur at any age, and when it is of acute onset, height may be within the normal range. In both congenital and acquired GH deficiency, height velocity will be low relative to sex- and bone age–matched peers. A strong clinical suspicion is important in establishing the diagnosis because laboratory measures of GH sufficiency lack specificity. Observation of low serum levels of IGF-1 and the GH-dependent IGF-BP3 can be helpful, but IGF-1 and IGF-BP3 levels should be matched to normal values for skeletal age rather than chronological age. Values in the upper part of the normal range for age effectively exclude GH deficiency. IGF-1 values in the lower part of the normal range may occur in normally growing children, children with impaired nutrition, or in those with hypopituitarism. The expected range for IGF-1 in normal and GH-deficient children overlaps somewhat during infancy and early childhood. IGF-1 levels in isolation should not be used to diagnose GH deficiency.
Table 573.6
Evaluation of Suspected Growth Hormone Deficiency
| History | |
| Physical exam | |
| Imaging | |
| Laboratory evaluation | |
| Treatment Considerations |
GH, Growth hormone; IGF, insulin-like growth factor; rhGH, human recombinant growth hormone.
Definitive diagnosis of GH deficiency traditionally requires demonstration of absent or low levels of GH in response to stimulation, but provocative testing may be omitted if the patient has the expected auxologic findings, a documented hypothalamic or pituitary defect, and at least one other pituitary hormone deficiency. A variety of provocative tests have been devised that rapidly increase the level of GH in normal children. These include administration of insulin, arginine, clonidine, levodopa, or glucagon. Because thyroid hormone is a prerequisite for normal GH synthesis, it must always be assessed before provocative GH testing. In chronic GH deficiency, the demonstration of subnormal linear growth, a delayed skeletal age, and low peak levels of GH (<10 ng/mL) in each of 2 provocative tests are compatible with GH deficiency. In acute GH deficiency, a high clinical suspicion of GH deficiency and low peak levels of GH (<10 ng/mL) in each of 2 provocative tests are compatible with GH deficiency. This rather arbitrary cutoff point is higher than the criteria used for diagnosis of adult GH deficiency. There is no consensus regarding adoption of criteria that take into account age, sex, and GH assay characteristics. Some studies indicate that many GH-sufficient prepubertal children fail to achieve GH values >10 ng/mL with 2 pharmacologic tests; pre-test, short-term sex steroid priming has been proposed to increase the diagnostic specificity of this testing.
In addition to establishing the diagnosis of GH deficiency, it is necessary to examine other pituitary functions. Levels of TSH, free thyroxine or total thyroxine with T3 resin uptake, ACTH, cortisol, gonadotropins, and gonadal steroids might provide evidence of other pituitary hormonal deficiencies. Antidiuretic hormone deficiency may be established by appropriate studies.
Radiologic Findings
Neurologic imaging should be obtained when the cause of hypopituitarism is not known. CT is appropriate for recognizing suprasellar calcification associated with craniopharyngiomas and bony changes accompanying histiocytosis. MRI provides a much more detailed view of hypothalamic and pituitary anatomy. Many cases of severe early-onset MPHD show the triad of a small anterior pituitary gland, a missing or attenuated pituitary stalk, and an ectopic posterior pituitary bright spot at the base of the hypothalamus (Fig. 573.3 ). Subnormal anterior pituitary height, implying a small anterior pituitary, is common in genetic and idiopathic causes of IGHD. Craniopharyngiomas are common, and pituitary adenomas are rare in children as causes of acquired hypopituitarism. Both hypoplastic and markedly enlarged anterior pituitary glands are seen in patients with PROP1 or LHX3 mutations.
Skeletal maturation may be assessed with a plain film of the hand (bone age) and is delayed in patients with IGHD and may be even more delayed when there is combined GH and TSH deficiency. Dual-photon x-ray absorptiometry shows deficient bone mineralization, deficiencies in lean body mass, and a corresponding increase in adiposity, but it is not routinely recommended in the evaluation of pediatric GH deficiency.
Differential Diagnosis
The causes of growth disorders are numerous. Differential diagnosis can be summarized broadly as follows: hormonal disorders, chronic illness, undernutrition, genetic conditions, nonsyndromic family trait, and constitutional delay of growth and development. Hormonal disorders include primary hypothyroidism and Cushing disease. Systemic conditions, such as inflammatory bowel disease, celiac disease, occult renal disease, and anemia must be considered. Patients with systemic conditions often have a greater deficit of weight than length. Severe psychosocial deprivation may result in growth failure that mimics GH deficiency. Many syndromic genetic conditions include short stature as a manifestation. Some genetic conditions, such as Turner syndrome and SHOX gene defects have variable phenotypes, and isolated short stature may be the clinical presentation.
Some otherwise normal children are short (i.e., >2.25 SD below the mean for age) and grow 5 cm/yr or less but have normal levels of GH in response to provocative tests and normal spontaneous episodic secretion; this is often termed idiopathic short stature . Most of these children show increased rates of growth when treated with GH in doses comparable with those used to treat children with hypopituitarism. Plasma levels of IGF-1 in these patients may be normal or low. Several groups of treated children have achieved final or near-final adult heights. Different studies have found changes in adult height that range from −2.5 to +7.5 cm compared with pretreatment predictions. There are no methods that can reliably predict which of these children will become taller in adulthood as a result of GH treatment and which will have compromised adult height.
Diagnostic strategies for distinguishing between permanent GH deficiency and other causes of impaired growth are imperfect. Children with a combination of genetic short stature and constitutional delay of growth have short stature, below-average growth rates, and delayed bone ages. Many of these children exhibit minimal GH secretory responses to provocative stimuli. When children in whom idiopathic or acquired GH deficiency is diagnosed are treated with human GH (hGH) and retested as adults, the majority have peak GH levels within the normal range.
Constitutional Growth Delay
Constitutional growth delay is one of the variants of normal growth commonly encountered by the pediatrician. Length and weight measurements of affected children are normal at birth, and growth is normal for the first 4-12 mo of life. Height is sustained at a lower percentile during childhood. The pubertal growth spurt is delayed, so their growth rates continue to decline after their classmates have begun to accelerate. Detailed questioning often reveals other family members (often 1 or both parents) with histories of short stature in childhood, delayed puberty, and eventual normal stature. IGF-1 levels tend to be low for chronological age but within the normal range for bone age. GH responses to provocative testing tend to be lower than in children with a more typical timing of puberty. The prognosis for these children to achieve normal adult height is guarded. Predictions based on height and bone age tend to overestimate eventual height to a greater extent in boys than in girls. Boys with >2 yr of pubertal delay can benefit from a short course of testosterone therapy to hasten puberty after 14 yr of age. The cause of this variant of normal growth is thought to be persistence of the relatively hypogonadotropic state of childhood.
Treatment
Recombinant hGH (rhGH) has been available by prescription since the 1980s. Multiple brands are marketed in the United States. They are therapeutically equivalent, with the major differences consisting of proprietary devices for subcutaneous injection and availability of solubilized liquid forms versus powders needing reconstitution before injection. At present, none of the products are available in long-acting forms; clinical trials to develop such products are underway.
The U.S. Food and Drug Administration (FDA) has approved 8 pediatric indications for rhGH treatment to promote linear growth. They are GH deficiency, Turner syndrome, chronic renal failure before transplantation, idiopathic short stature, small-for-gestational-age short stature, Prader-Willi syndrome, SHOX gene abnormality, and Noonan syndrome. FDA approval for a given indication does not ensure that a patient's insurance carrier will approve payment for the drug. Treatment should be started as soon as possible to narrow the gap in height between patients and their classmates during childhood and to have the greatest effect on mature height. The recommended initial dose of rhGH for treatment of GH deficiency is 0.16-0.24 mg/kg/wk (22 to 35 µg/kg/day). Higher doses have been used during puberty and for non-GH deficiency indications. RhGH is administered subcutaneously once daily. Maximal response to rhGH occurs in the 1st yr of treatment. Growth velocity during this 1st yr is typically above the 95th percentile for age. With each successive year of treatment, the growth rate tends to decrease. If growth rate drops below the 25th percentile, adherence should be evaluated before the dose is increased. IGF-1 may be measured as an objective assessment of adherence. GH therapy should be continued until near-final height is achieved. Criteria for stopping GH treatment include a decision by the patient that he or she is tall enough, a growth rate <1 inch/yr, and a bone age >14 yr in girls and >16 yr in boys.
Concurrent treatment with rhGH and a gonadotropin-releasing hormone agonist has been used in the hope that interruption of puberty will delay epiphyseal fusion and prolong growth. This strategy can increase adult height. It can also increase the discrepancy in physical maturity between GH-deficient children and their age peers and can impair bone mineralization. There have also been attempts to forestall epiphyseal fusion in boys by giving aromatase inhibitors, which inhibit the enzyme responsible for converting androgens to estrogens, and clinical trials to determine the efficacy of this approach are underway.
Some patients develop either primary or central hypothyroidism while under treatment with GH. Similarly, there is a risk of developing adrenal insufficiency as an associated component of hypopituitarism. If unrecognized, this can be fatal. Periodic evaluation of thyroid and adrenal function is indicated for all patients diagnosed with GH deficiency.
RhGH treatment may enhance the growth of non–GH-deficient children as well. Intensive investigation is in progress to determine the full spectrum of short children who may benefit from treatment with GH. The FDA approval for use of GH in idiopathic short stature specifies a height below the 1.2 percentile (−2.25 SD) for age and sex, a predicted height below the 5th percentile, and open epiphyses. Studies of the effect of GH treatment on adult height suggest a median gain of 2-3 inches, depending on dose and duration of treatment.
In children with MPHD, replacement should also be directed at other hormonal deficiencies. In TSH-deficient patients, thyroid hormone is given in full replacement doses. In ACTH-deficient patients, hydrocortisone should be prescribed in physiologic doses, about 8-12 mg/m2 /day. Individualized dose adjustment is needed to minimize the risk of side effects associated with excess glucocorticoid administration and prevent symptoms of adrenal insufficiency. Increased doses are required to provide stress coverage during illness, or during and after surgical procedures. In patients with a deficiency of gonadotropins, gonadal steroids are given when bone age reaches the age at which puberty usually takes place. For infants with micropenis, one or two 3-mo courses of monthly intramuscular injections of 25 mg of testosterone cypionate or testosterone enanthate can bring the penis to normal size without an inordinate effect on osseous maturation.
Recombinant IGF-1 (mecasermin) is approved for use in the United States for primary IGF-1 deficiency. It is given subcutaneously twice a day. Side effects are similar to rhGH, except that mecasermin can cause hypoglycemia. The risk of hypoglycemia is reduced by giving the injections concurrently with a meal or snack. In some situations, its use may be more efficacious than use of GH. These conditions include abnormalities of the GH receptor and STAT5b genes that alter GH signaling downstream. It may have utility for severe GH deficiency in the rare patients who have developed clinically significant antibodies to administered rhGH. However, mecasermin is not an indicated treatment for the majority of patients with GH deficiency.
Complications and Adverse Effects of Growth Hormone Treatment
GH treatment influences glucose homeostasis. Fasting and postprandial insulin levels are characteristically low before treatment, and they normalize during GH replacement. GH treatment is associated with an increase in the risk for type 2 diabetes and no significant increase in the risk for type 1 diabetes.
Concerns have been raised about the safety of GH treatment in children who become deficient after treatment of brain tumors, leukemia, and other neoplasms. Long-term studies show no increase in risk of recurrence of craniopharyngioma, other brain tumors, or leukemia. At least 3 studies indicate an increased risk of second neoplasms in cancer survivors treated with GH.
An unconfirmed study documents an increased risk of hemorrhagic stroke and a 30% increase in mortality among young adults who received GH in childhood, particularly if the GH dose exceeds 0.35 mg/kg/wk (50 µg/kg/day).
Other reported side effects include pseudotumor cerebri, slipped capital femoral epiphysis, gynecomastia, coarsening of features, and worsening of scoliosis. The risk of late development of Creutzfeldt-Jakob disease was limited to recipients of contaminated lots of extracted pituitary GH. No comparable risks have been seen with rhGH, which is the only pharmacologic form of hGH currently in clinical use.