Diabetes Insipidus
David T. Breault, Joseph A. Majzoub
Diabetes insipidus (DI) manifests clinically with polyuria and polydipsia and can result from either vasopressin deficiency (central DI) or vasopressin insensitivity at the level of the kidney (nephrogenic DI [NDI]). Both central DI and NDI can arise from inherited defects of congenital or neonatal onset or can be secondary to a variety of causes (Table 574.1 ).
Table 574.1
DI , Diabetes insipidus.
From Verbalis JG: Disorders of water balance. In Skorecki K, Chertow GM, Marsden PA, et al, editors: Brenner & Rector's the kidney , ed 10, Philadelphia, 2016, Elsevier, 2016. Table 16.2.
Physiology of Water Balance
The control of extracellular tonicity (osmolality) and volume within a narrow range is critical for normal cellular structure and function (see Chapter 68.2 ). Extracellular fluid tonicity is regulated almost exclusively by water intake and excretion, whereas extracellular volume is regulated by sodium intake and excretion. The control of plasma tonicity and intravascular volume involves a complex integration of endocrine, neural, behavioral, and paracrine systems (Fig. 574.1 ). Vasopressin, secreted from the posterior pituitary, is the principal regulator of tonicity, with its release largely stimulated by increases in plasma tonicity. Volume homeostasis is largely regulated by the renin-angiotensin-aldosterone system, with contributions from both vasopressin and the natriuretic peptide family.
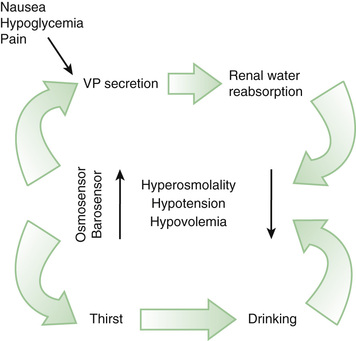
Vasopressin, a 9-amino-acid peptide, has both antidiuretic and vascular pressor activity and is synthesized in the paraventricular and supraoptic nuclei of the hypothalamus. It is transported to the posterior pituitary via axonal projections, where it is stored awaiting release into the systemic circulation. The half-life of vasopressin in the circulation is 5 min. In addition to responding to osmotic stimuli, vasopressin is secreted in response to significant decreases in intravascular volume and pressure (minimum of 8% decrement) via afferent baroreceptor pathways arising from the aortic arch (carotid sinus) and volume receptor pathways in the cardiac atria and pulmonary veins. Osmotic and hemodynamic stimuli interact synergistically.
The sensation of thirst and the release of vasopressin are regulated by cortical and hypothalamic neurons. The thirst threshold is approximately 10 mOsm/kg higher (i.e., 293 mOsm/kg) than the osmotic threshold for vasopressin release. Consequently, under conditions of hyperosmolality, vasopressin is released before thirst is initiated, allowing ingested water to be retained. Subsequently, anticipation of water ingestion by cortical and vasopressin-secreting neurons leads to a decrease in vasopressin release immediately before water ingestion, presumably to prevent subsequent hyponatremia. Chemoreceptors present in the oropharynx also downregulate vasopressin release following water ingestion. In addition, thirst drive decreases even before the ingested fluid lowers blood osmolality, presumably to prevent overdrinking leading to hyponatremia.
Vasopressin exerts its principal effect on the kidney via V2 receptors located primarily in the collecting tubule, the thick ascending limb of the loop of Henle, and the periglomerular tubules. The human V2 receptor gene is located on the long arm of the X chromosome (Xq28) at the locus associated with congenital, X-linked, vasopressin-resistant DI. Activation of the V2 receptor results in increases in intracellular cyclic adenosine monophosphate, which leads to the insertion of the aquaporin-2 water channel into the apical (luminal) membrane. This allows water movement along its osmotic gradient into the hypertonic inner medullary interstitium from the tubule lumen and excretion of concentrated urine. In contrast to aquaporin-2, aquaporin-3 and aquaporin-4 are expressed on the basolateral membrane of the collecting duct cells and aquaporin-1 is expressed in the proximal tubule. These channels may also contribute to urinary concentrating ability.
Atrial natriuretic peptide , initially isolated from cardiac atrial muscle, has a number of important effects on salt and water balance, including stimulation of natriuresis, inhibition of sodium resorption, and inhibition of vasopressin secretion. Atrial natriuretic peptide is expressed in endothelial cells and vascular smooth muscle, where it appears to regulate relaxation of arterial smooth muscle. Atrial natriuretic peptide is also expressed in the brain, along with other natriuretic family members; the physiologic role of these factors has yet to be defined.
Approach to the Patient With Polyuria, Polydipsia, and Hypernatremia
The cause of pathologic polyuria or polydipsia (exceeding 2 L/m2 /24 hr) may be difficult to establish in children. Infants can present with irritability, failure to thrive, and intermittent fever. Patients with suspected DI should have a careful history taken, which should quantify the child's daily fluid intake and output and establish the voiding pattern, nocturia, and primary or secondary enuresis. A complete physical examination should establish the patient's hydration status, and the physician should search for evidence of visual and central nervous system dysfunction, as well as for other pituitary hormone deficiencies.
If pathologic polyuria or polydipsia is present, the following should be obtained: serum for osmolality, sodium, potassium, blood urea nitrogen, creatinine, glucose, and calcium; urine for osmolality, specific gravity, and glucose determination. The diagnosis of DI is established if the serum osmolality is >300 mOsm/kg, and the urine osmolality is <300 mOsm/kg. DI is unlikely if the serum osmolality is <270 mOsm/kg or the urine osmolality is >600 mOsm/kg. If the patient's serum osmolality is <300 mOsm/kg (but >270 mOsm/kg) and pathologic polyuria and polydipsia are present, a water deprivation test is indicated to establish the diagnosis of DI and to differentiate central from nephrogenic causes.
In the inpatient postneurosurgical setting, central DI is likely if hyperosmolality (serum osmolality >300 mOsm/kg) is associated with urine osmolality less than serum osmolality. It is important to distinguish between polyuria resulting from postsurgical central DI and polyuria resulting from the normal diuresis of fluids received intraoperatively. Both cases may be associated with a large volume (>200 mL/m2 /hr) of dilute urine, although in patients with DI, the serum osmolality is high in comparison with patients undergoing postoperative diuresis.
Causes of Hypernatremia
Hypernatremia is discussed in Chapter 68.3 .
Central Diabetes Insipidus
Central DI can result from multiple etiologies, including genetic mutations in the vasopressin gene; trauma (accidental or surgical) to vasopressin neurons; congenital malformations of the hypothalamus or pituitary; neoplasms; infiltrative, autoimmune, and infectious diseases affecting vasopressin neurons or fiber tracts; and increased metabolism of vasopressin. In approximately 10% of children with central DI, the etiology is idiopathic. Other pituitary hormone deficiencies may be present (see Chapter 573 ). Over time, up to 35% of those with idiopathic central DI will develop other hormone deficiencies or have an underlying etiology identified.
Autosomal dominant central DI usually occurs within the first 5 yr of life and results from mutations in the vasopressin gene, AVP . A number of mutations can cause gene-processing defects in a subset of vasopressin-expressing neurons, which have been postulated to result in endoplasmic reticulum stress and cell death. Wolfram syndrome, which includes DI, diabetes mellitus, optic atrophy, and deafness, also results in vasopressin deficiency. Mutations in 2 genes, which give rise to endoplasmic reticulum proteins, are associated with this condition. Congenital brain abnormalities (see Chapter 609 ) such as optic nerve hypoplasia syndrome with agenesis of the corpus callosum, the Niikawa-Kuroki syndrome, holoprosencephaly, and familial pituitary hypoplasia with absent stalk may be associated with central DI and defects in thirst perception (adipsia). Empty sella syndrome, possibly resulting from unrecognized pituitary infarction, can be associated with DI in children.
Trauma to the base of the brain and neurosurgical intervention in the region of the hypothalamus or pituitary are common causes of central DI. The triphasic response following surgery refers to an initial phase of transient DI, lasting 12-48 hr, followed by a 2nd phase of syndrome of inappropriate antidiuretic hormone secretion (SIADH), lasting up to 10 days, which may be followed by permanent DI. The initial phase may be the result of local edema interfering with normal vasopressin secretion; the 2nd phase results from unregulated vasopressin release from dying neurons, whereas in the 3rd phase, permanent DI, results if more than 90% of the neurons have been destroyed.
Given the anatomic distribution of vasopressin neurons over a large area within the hypothalamus, tumors causing DI must either be very large and infiltrative or be strategically located near the base of the hypothalamus, where vasopressin axons converge before their entry into the posterior pituitary. Germinomas and pinealomas typically arise in this region and are among the most common primary brain tumors associated with DI. Germinomas can be very small and undetectable by MRI for several years following the onset of polyuria. Quantitative measurement of α-fetoprotein and β-human chorionic gonadotropin, often secreted by germinomas, should be performed in children with idiopathic or unexplained DI, in addition to serial MRI scans. Craniopharyngiomas and optic gliomas can also cause central DI when they are very large, although this is more often a postoperative complication of the treatment for these tumors (see Chapter 524 ). Hematologic malignancies, such as acute myelocytic leukemia, can cause DI via infiltration of the pituitary stalk and sella.
Langerhans cell histiocytosis (see Chapter 534.1 ) and lymphocytic hypophysitis are common types of infiltrative disorders causing central DI, with hypophysitis as the cause in 50% of cases of “idiopathic” central DI. Infections involving the base of the brain (see Chapter 621 ), including meningitis (meningococcal, cryptococcal, listerial, toxoplasmal), congenital cytomegalovirus infection, and nonspecific inflammatory diseases of the brain may give rise to central DI that is often transient. Drugs associated with the inhibition of vasopressin release include ethanol, phenytoin, opiate antagonists, halothane, and α-adrenergic agents.
Nephrogenic Diabetes Insipidus
NDI can result from genetic or acquired causes. Genetic causes are less common but more severe than acquired forms of NDI. The polyuria and polydipsia associated with genetic NDI usually occur within the 1st several weeks of life but may become apparent only after weaning or with longer periods of nighttime sleep. Many infants initially present with fever, vomiting, and dehydration. Failure to thrive may be secondary to the ingestion of large amounts of water, resulting in caloric malnutrition. Long-standing ingestion and excretion of large volumes of water can lead to nonobstructive hydronephrosis, hydroureter, and megabladder.
Congenital X-linked NDI results from inactivating mutations of the vasopressin V2 receptor, AVPR2 . Congenital autosomal recessive NDI results from defects in the aquaporin-2 gene, AQP2 . An autosomal dominant form of NDI is associated with processing mutations of the aquaporin-2 gene.
Acquired NDI can result from hypercalcemia or hypokalemia and is associated with lithium, demeclocycline, foscarnet, clozapine, amphotericin, methicillin, and rifampin. Impaired renal concentrating ability can also be seen with ureteral obstruction, chronic renal failure, polycystic kidney disease, medullary cystic disease, Sjögren syndrome, and sickle cell disease. Decreased protein or sodium intake or excessive water intake, as in primary polydipsia, can lead to diminished tonicity of the renal medullary interstitium and NDI.
Treatment of Central Diabetes Insipidus
Fluid Therapy
With an intact thirst mechanism and free access to oral fluids, a person with complete DI can maintain plasma osmolality and sodium in the high normal range, although at great inconvenience. Neonates and young infants are often best treated solely with fluid therapy, given their requirement for large volumes (~3 L/m2 /24 hr) of nutritive fluid. The use of vasopressin analogs in patients with obligate high fluid intake is difficult given the risk of life-threatening hyponatremia. Although not FDA approved, the use of diluted parenteral and lyophilized long-acting vasopressin analog DDAVP (desmopressin) has been successfully administered to infants with central DI both subcutaneously and orally without causing severe hyponatremia. Patients with both central and NDI should ingest a diet without excessive solute (e.g., sodium chloride) to help decrease urine output when vasopressin action wanes.
Vasopressin Analogs
Treatment of central DI in older children is best accomplished with the use of DDAVP. DDAVP is available in an intranasal preparation (onset 5-10 min) and as tablets (onset 15-30 min). The intranasal preparation of DDAVP (10 µg/0.1 mL) can be administered by rhinal tube (allowing dose titration) or by nasal spray (10 µg/puff). Use of DDAVP oral tablets requires at least a 10-fold increase in the dosage compared with the intranasal preparation. Oral dosages of 25-300 µg every 8-12 hr are safe and effective in children. The appropriate dosage and route of administration is determined empirically based on the desired length of antidiuresis and patient preference. The use of oral DDAVP for the treatment of enuresis in older children should be regarded as a temporizing measure, given it does not affect the underlying condition, and should be used with great caution given the risk of hyponatremia if water intake exceeds the capacity for renal clearance. To prevent water intoxication, patients should have at least 1 hr of urinary breakthrough between doses each day and be advised to drink only in response to thirst sensation, if present. The use of DDAVP nasal spray for childhood enuresis is no longer approved due to its risk of causing hyponatremia.
Aqueous Vasopressin
Central DI of acute onset following neurosurgery is best managed with continuous administration of synthetic aqueous vasopressin (Pitressin). Under most circumstances, total fluid intake must be limited to 1 L/m2 /24 hr during antidiuresis. A typical dosage for intravenous vasopressin therapy is 1.5 mU/kg/hr, which results in a blood vasopressin concentration of approximately 10 pg/mL. On occasion, following hypothalamic (but not transsphenoidal) surgery, higher initial concentrations of vasopressin may be required to treat acute DI, which has been attributed to the release of a vasopressin inhibitory substance. Vasopressin concentrations >1,000 pg/mL should be avoided because they can cause cutaneous necrosis, rhabdomyolysis, cardiac rhythm disturbances, and hypertension. Postneurosurgical patients treated with vasopressin infusion should be switched from intravenous to oral fluids as soon as possible to allow thirst sensation, if intact, to help regulate osmolality.
Treatment of Nephrogenic Diabetes Insipidus
The treatment of acquired NDI focuses on eliminating, if possible, the underlying disorder, such as offending drugs, hypercalcemia, hypokalemia, or ureteral obstruction. Congenital NDI is often difficult to treat. The main goals are to ensure the intake of adequate calories for growth and to avoid severe dehydration. Foods with the highest ratio of caloric content to osmotic load (Na <1 mmol/kg/24 hr) should be ingested to maximize growth and to minimize the urine volume required to excrete the solute load. However, even with the early institution of therapy, growth failure and developmental disabilities are common.
Pharmacologic approaches to the treatment of NDI include the use of thiazide diuretics and are intended to decrease the overall urine output. Thiazides appear to induce a state of mild volume depletion by enhancing sodium excretion at the expense of water and by causing a decrease in the glomerular filtration rate, which results in proximal tubular sodium and water reabsorption. Indomethacin and amiloride may be used in combination with thiazides to further reduce polyuria. High-dose DDAVP therapy, in combination with indomethacin, has been used in some subjects with NDI. This treatment could prove useful in patients with genetic defects in the V2 receptor associated with a reduced binding affinity for vasopressin.