Electrolyte and Acid-Base Disorders
Composition of Body Fluids
Larry A. Greenbaum
Total Body Water
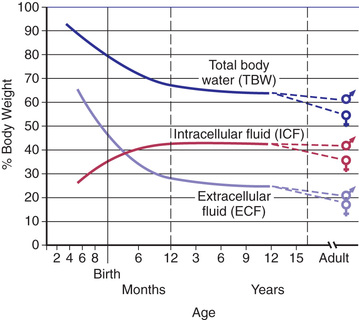
Total body water (TBW) as a percentage of body weight varies with age (Fig. 68.1 ). The fetus has very high TBW, which gradually decreases to approximately 75% of birthweight for a term infant. Premature infants have higher TBW than term infants. During the 1st yr of life, TBW decreases to approximately 60% of body weight and remains at this level until puberty. At puberty, the fat content of females increases more than that in males, who acquire more muscle mass than females. Because fat has very low water content and muscle has high water content, by the end of puberty, TBW in males remains at 60%, but TBW in females decreases to approximately 50% of body weight. The high fat content in overweight children causes a decrease in TBW as a percentage of body weight. During dehydration, TBW decreases and thus is a smaller percentage of body weight.
Fluid Compartments
TBW is divided between 2 main compartments: intracellular fluid (ICF) and extracellular fluid (ECF) . In the fetus and newborn, the ECF volume is larger than the ICF volume (Fig. 68.1 ). The normal postnatal diuresis causes an immediate decrease in the ECF volume. This is followed by continued expansion of the ICF volume, which results from cellular growth. By 1 yr of age, the ratio of ICF volume to ECF volume approaches adult levels. The ECF volume is approximately 20–25% of body weight, and the ICF volume is approximately 30–40% of body weight, close to twice the ECF volume (Fig. 68.2 ). With puberty, the increased muscle mass of males causes them to have a higher ICF volume than females. There is no significant difference in the ECF volume between postpubertal females and males.
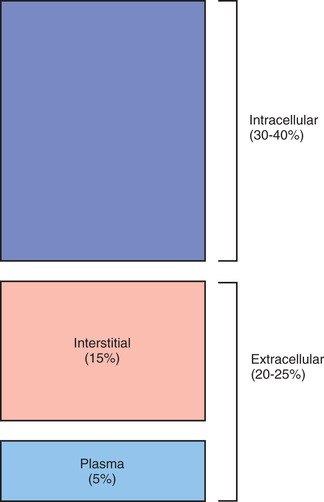
The ECF is further divided into the plasma water and the interstitial fluid (see Fig. 68.2 ). The plasma water is 5% of body weight. The blood volume, given a hematocrit of 40%, is usually 8% of body weight, although it is higher in newborns and young infants; in premature newborns it is approximately 10% of body weight. The volume of plasma water can be altered by pathologic conditions, including dehydration, anemia, polycythemia, heart failure, abnormal plasma osmolality, and hypoalbuminemia. The interstitial fluid, normally 15% of body weight, can increase dramatically in diseases associated with edema, such as heart failure, protein-losing enteropathy, liver failure, nephrotic syndrome, and sepsis. An increase in interstitial fluid also occurs in patients with ascites or pleural effusions.
There is a delicate equilibrium between the intravascular fluid and the interstitial fluid. The balance between hydrostatic and oncotic forces regulates the intravascular volume, which is critical for proper tissue perfusion. The intravascular fluid has a higher concentration of albumin than the interstitial fluid, and the consequent oncotic force draws water into the intravascular space. The maintenance of this gradient depends on the limited permeability of albumin across the capillaries. The hydrostatic pressure of the intravascular space, which is caused by the pumping action of the heart, drives fluid out of the intravascular space. These forces favor movement into the interstitial space at the arterial ends of the capillaries. The decreased hydrostatic forces and increased oncotic forces, which result from the dilutional increase in albumin concentration, cause movement of fluid into the venous ends of the capillaries. Overall, there is usually a net movement of fluid out of the intravascular space to the interstitial space, but this fluid is returned to the circulation via the lymphatics .
An imbalance in these forces may cause expansion of the interstitial volume at the expense of the intravascular volume. In children with hypoalbuminemia, the decreased oncotic pressure of the intravascular fluid contributes to the development of edema . Loss of fluid from the intravascular space may compromise the intravascular volume, placing the child at risk for inadequate blood flow to vital organs. This is especially likely in diseases in which capillary leak occurs because the loss of albumin from the intravascular space is associated with an increase in the albumin concentration in the interstitial space, further compromising the oncotic forces that normally maintain intravascular volume. In contrast, with heart failure , there is an increase in venous hydrostatic pressure from expansion of the intravascular volume, which is caused by impaired pumping by the heart, and the increase in venous pressure causes fluid to move from the intravascular space to the interstitial space. Expansion of the intravascular volume and increased intravascular pressure also cause the edema that occurs with acute glomerulonephritis.
Electrolyte Composition
The composition of the solutes in the ICF and ECF are very different (Fig. 68.3 ). Sodium (Na+ ) and chloride (Cl− ) are the dominant cation and anion, respectively, in the ECF. The sodium and chloride concentrations ([Na+ ], [Cl− ]) in the ICF are much lower. Potassium (K+ ) is the most abundant cation in the ICF, and its concentration ([K+ ]) within the cells is approximately 30 times higher than in the ECF. Proteins, organic anions, and phosphate are the most plentiful anions in the ICF. The dissimilarity between the anions in the ICF and the ECF is largely determined by the presence of intracellular molecules that do not cross the cell membrane, the barrier separating the ECF and the ICF. In contrast, the difference in the distribution of cations—Na+ and K+ —relies on activity of the Na+ ,K+ -adenosine triphosphatase (ATPase) pump and membrane ion channels.

The difference in the electrolyte compositions of the ECF and the ICF has important ramifications in the evaluation and treatment of electrolyte disorders. Serum concentrations of electrolytes—[Na+ ], [K+ ], and [Cl− ]—do not always reflect total body content. Intracellular [K+ ] is much higher than the serum concentration. A shift of K+ from the intracellular space (ICS) can maintain a normal or even an elevated serum [K+ ] despite massive losses of K+ from the ICS. This effect is seen in diabetic ketoacidosis, in which significant K+ depletion is masked by transmembrane shift of K+ from the ICF to the ECF. Therefore, for K+ and phosphorus, electrolytes with a high intracellular concentration, serum level may not reflect total body content. Similarly, the serum calcium concentration ([Ca2+ ]) does not reflect total body content of Ca2+ , which is largely contained in bone and teeth (see Chapter 64 ).
Osmolality
The ICF and the ECF are in osmotic equilibrium because the cell membrane is permeable to water. If the osmolality in 1 compartment changes, then water movement leads to a rapid equalization of osmolality, with a shift of water between the ICS and extracellular space (ECS) . Clinically, the primary process is usually a change in the osmolality of the ECF, with resultant shift of water into the ICF if ECF osmolality decreases, or vice versa if ECF osmolality increases. The ECF osmolality can be determined and usually equals ICF osmolality . Plasma osmolality , normally 285-295 mOsm/kg, is measured by the degree of freezing-point depression. The plasma osmolality can also be estimated by a calculation based on the following formula:
Osmolality=2×[Na]+[glucose]/18+[BUN]/2.8
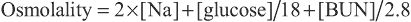
Glucose and blood urea nitrogen (BUN) are reported in mg/dL. Division of these values by 18 and 2.8, respectively, converts the units into mmol/L. Multiplication of the [Na+ ] value by 2 accounts for its accompanying anions, principally Cl− and bicarbonate. The calculated osmolality is usually slightly lower than measured osmolality.
Urea is not only confined to the ECS because it readily crosses the cell membrane, and its intracellular concentration approximately equals its extracellular concentration. Whereas an elevated [Na+ ] causes a shift of water from the ICS, with uremia there is no osmolar gradient between the two compartments and consequently no movement of water. The only exception is during hemodialysis , when the decrease in extracellular urea is so rapid that intracellular urea does not have time to equilibrate. This disequilibrium syndrome may result in shift of water into brain cells and leads to severe symptoms. Ethanol, because it freely crosses cell membranes, is another ineffective osmole. In the case of glucose, the effective osmolality can be calculated as follows:
Effective osmolality=2×[Na]+[glucose]/18
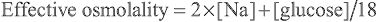
The effective osmolality (also called the tonicity ) determines the osmotic force that is mediating the shift of water between the ECF and the ICF.
Hyperglycemia causes an increase in the plasma osmolality because it is not in equilibrium with the ICS. During hyperglycemia, there is shift of water from the ICS to the ECS. This shift causes dilution of the Na+ in the ECS, causing hyponatremia despite elevated plasma osmolality. The magnitude of this effect can be calculated as follows:
[Na]corrected=[Na]measured+1.6×([glucose]−100 mg/dL)/100

where [Na]measured = Na+ concentration measured by the clinical laboratory and [Na]corrected = corrected Na+ concentration (the Na+ concentration if the glucose concentration were normal and its accompanying water moved back into the cells). The [Na]corrected is the more reliable indicator of the ratio of total body Na+ to TBW, the usual determinant of the [Na+ ].
Normally, measured osmolality and calculated osmolality are within 10 mOsm/kg. However, there are some clinical situations in which this difference does not occur. The presence of unmeasured osmoles causes measured osmolality to be significantly elevated in comparison with the calculated osmolality. An osmolal gap is present when the difference between measured osmolality exceeds calculated osmolality by >10 mOsm/kg. Examples of unmeasured osmoles include ethanol, ethylene glycol, methanol, sucrose, sorbitol, and mannitol. These substances increase measured osmolality but are not part of the equation for calculating osmolality. The presence of an osmolal gap is a clinical clue to the presence of unmeasured osmoles and may be diagnostically useful when there is clinical suspicion of poisoning with methanol or ethylene glycol.
Pseudohyponatremia is a second situation in which there is discordance between measured osmolality and calculated osmolality. Lipids and proteins are the solids of the serum. In patients with elevated serum lipids or proteins, the water content of the serum decreases because water is displaced by the larger amounts of solids. Some instruments measure [Na+ ] by determining the amount of Na+ per liter of serum, including the solid component. When the solid component increases, there is a decrease in [Na+ ] per liter of serum, despite a normal concentration when based on the amount of Na+ per liter of serum water. It is the concentration of Na+ in serum water that is physiologically relevant. A similar problem occurs when using instruments that require dilution of the sample prior to measurement of Na+ (indirect potentiometry). In both situations, the plasma osmolality is normal despite the presence of pseudohyponatremia, because the method for measuring osmolality is not appreciably influenced by the percentage of serum that is composed of lipids and proteins. Pseudohyponatremia is diagnosed by the finding of a normal measured plasma osmolality despite hyponatremia. This laboratory artifact does not occur if the [Na+ ] in water is measured directly with an ion-specific electrode, as with arterial blood gas (ABG) analyzers. Pseudohypernatremia may occur in patients with very low levels of serum proteins by a similar mechanism.
When there are no unmeasured osmoles and pseudohyponatremia is not a concern, the calculated osmolality provides an accurate estimate of the plasma osmolality. Measurement of plasma osmolality is useful for detecting or monitoring unmeasured osmoles and confirming the presence of true hyponatremia. Whereas many children with high plasma osmolality are dehydrated—as seen with hypernatremic dehydration or diabetic ketoacidosis—high osmolality does not always equate with dehydration. A child with salt poisoning or uremia has an elevated plasma osmolality but may be volume-overloaded.
Point-of-Care Testing
Point-of-care (POC) testing offers a number of advantages, including rapid turnaround and usually smaller blood sample volume required. POC devices may provide more accurate results in certain situations, such as pseudohyponatremia (see earlier) and pseudohyperkalemia (see Chapter 68.4 ). However, the agreement between POC and the laboratory is variable, and thus caution is needed when interpreting results. Because of bias, POC and laboratory results should not be used on an alternating basis when following critical trends (e.g., during correction of hypernatremia or hyponatremia; see Chapter 68.3 ).
Bibliography
Composition of Body Fluids
Allardet-Servent J, Lebsir M, Dubroca C, et al. Point-of-care versus central laboratory measurements of hemoglobin, hematocrit, glucose, bicarbonate and electrolytes: a prospective observational study in critically ill patients. PLoS ONE . 2017;12:e0169593.
Gavala A, Myrianthefs P. Comparison of point-of-care versus central laboratory measurement of hematocrit, hemoglobin, and electrolyte concentrations. Heart Lung . 2017;46:246–250.
Goldwasser P, Ayoub I, Barth RH. Pseudohypernatremia and pseudohyponatremia: a linear correction. Nephrol Dial Transplant . 2015;30:252–257.
Jain A. Body fluid composition. Pediatr Rev . 2015;36:141–150 [quiz 51–52].
Kraut JA. Diagnosis of toxic alcohols: limitations of present methods. Clin Toxicol (Phila) . 2015;53:589–595.
Liamis G, Filippatos TD, Liontos A, et al. Serum osmolal gap in clinical practice: usefulness and limitations. Postgrad Med . 2017;129:456–459.
Uysal E, Acar YA, Kutur A, et al. How reliable are electrolyte and metabolite results measured by a blood gas analyzer in the ED? Am J Emerg Med . 2016;34:419–424.
Regulation of Osmolality and Volume
Larry A. Greenbaum
The regulation of plasma osmolality and the intravascular volume is controlled by independent systems for water balance, which determines osmolality, and sodium balance, which determines volume status. Maintenance of normal osmolality depends on control of water balance. Control of volume status depends on regulation of sodium balance. When present, volume depletion takes precedence over regulation of osmolality, and retention of water contributes to the maintenance of intravascular volume.
Regulation of Osmolality
The plasma osmolality is tightly regulated and maintained at 285-295 mOsm/kg. Modification of water intake and excretion maintains normal plasma osmolality. In the steady state the combination of water intake and water produced by the body from oxidation balances water losses from the skin, lungs, urine, and gastrointestinal (GI) tract. Only water intake and urinary losses can be regulated.
Osmoreceptors in the hypothalamus sense plasma osmolality (see Chapter 572 ). An elevated effective osmolality leads to secretion of antidiuretic hormone (ADH) by neurons in the supraoptic and paraventricular nuclei in the hypothalamus. The axons of these neurons terminate in the posterior pituitary. Circulating ADH binds to its V2 receptors in the collecting duct cells of the kidney, and causes insertion of water channels (aquaporin-2) into the renal collecting duct cells. This produces increased permeability to water, permitting resorption of water into the hypertonic renal medulla. Urine concentration increases and water excretion decreases. Urinary water losses cannot be eliminated because there is obligatory excretion of urinary solutes, such as urea and sodium. The regulation of ADH secretion is tightly linked to plasma osmolality, responses being detectable with a 1% change in osmolality. ADH secretion virtually disappears when plasma osmolality is low, allowing excretion of maximally dilute urine. The resulting loss of free water (i.e., water without Na+ ) corrects plasma osmolality. ADH secretion is not an all-or-nothing response; there is a graded adjustment as the osmolality changes.
Water intake is regulated by hypothalamic osmoreceptors, which stimulate thirst when the serum osmolality increases. Thirst occurs with a small increase in the serum osmolality. Control of osmolality is subordinate to maintenance of an adequate intravascular volume. When volume depletion is present, both ADH secretion and thirst are stimulated, regardless of the plasma osmolality. The sensation of thirst requires moderate volume depletion but only a 1–2% change in the plasma osmolality.
A number of conditions can limit the kidney's ability to excrete adequate water to correct low plasma osmolality. In the syndrome of inappropriate antidiuretic hormone (SIADH) , ADH continues to be produced despite a low plasma osmolality (see Chapters 68.3 and 575 ).
The glomerular filtration rate (GFR) affects the kidney's ability to eliminate water. With a decrease in the GFR, less water is delivered to the collecting duct, limiting the amount of water that can be excreted. The impairment in the GFR must be quite significant to limit the kidney's ability to respond to an excess of water.
The minimum urine osmolality is approximately 30-50 mOsm/kg. This places an upper limit on the kidney's ability to excrete water; sufficient solute must be present to permit water loss. Massive water intoxication may exceed this limit, whereas a lesser amount of water is necessary in the child with a diet that has very little solute. This can produce severe hyponatremia in children who receive little salt and have minimal urea production as a result of inadequate protein intake. Volume depletion is an extremely important cause of decreased water loss by the kidney despite a low plasma osmolality. This “appropriate” secretion of ADH occurs because volume depletion takes precedence over the osmolality in the regulation of ADH.
The maximum urine osmolality is approximately 1,200 mOsm/kg. The obligatory solute losses dictate the minimum volume of urine that must be produced, even when maximally concentrated. Obligatory water losses increase in patients with high salt intake or high urea losses, as may occur after relief of a urinary obstruction or during recovery from acute kidney injury. An increase in urinary solute and thus water losses occurs with an osmotic diuresis , which occurs classically from glycosuria in diabetes mellitus as well as iatrogenically after mannitol administration. There are developmental changes in the kidney's ability to concentrate the urine. The maximum urine osmolality in a newborn, especially a premature newborn, is less than that in an older infant or child. This limits the ability to conserve water and makes such a patient more vulnerable to hypernatremic dehydration. Very high fluid intake, as seen with psychogenic polydipsia , can dilute the high osmolality in the renal medulla, which is necessary for maximal urinary concentration. If fluid intake is restricted in patients with this condition, the kidney's ability to concentrate the urine may be somewhat impaired, although this defect corrects after a few days without polydipsia. This may also occur during the initial treatment of central diabetes insipidus with desmopressin acetate; the renal medulla takes time to achieve its normal maximum osmolality.
Regulation of Volume
An appropriate intravascular volume is critical for survival; both volume depletion and volume overload may cause significant morbidity and mortality. Because sodium is the principal extracellular cation and is restricted to the ECF, adequate body sodium is necessary for maintenance of intravascular volume. The principal extracellular anion, Cl− , is also necessary, but for simplicity, Na+ balance is considered the main regulator of volume status because body content of sodium and that of chloride usually change proportionally, given the need for equal numbers of cations and anions. In some situations, Cl− depletion is considered the dominant derangement causing volume depletion (metabolic alkalosis with volume depletion).
The kidney determines sodium balance because there is little homeostatic control of sodium intake, even though salt craving does occasionally occur, typically in children with chronic renal salt loss. The kidney regulates Na+ balance by altering the percentage of filtered Na+ that is resorbed along the nephron. Normally, the kidney excretes <1% of the Na+ filtered at the glomerulus. In the absence of disease, extrarenal losses and urinary output match intake, with the kidney having the capacity to adapt to large variations in sodium intake. When necessary, urinary sodium excretion can be reduced to virtually undetectable levels or increased dramatically.
The most important determinant of renal Na+ excretion is the volume status of the child; it is the effective intravascular volume that influences urinary Na+ excretion. The effective intravascular volume is the volume status that is sensed by the body's regulatory mechanisms. Heart failure is a state of volume overload, but the effective intravascular volume is low because poor cardiac function prevents adequate perfusion of the kidneys and other organs. This explains the avid renal Na+ retention often present in patients with heart failure.
The renin-angiotensin system is an important regulator of renal Na+ excretion. The juxtaglomerular apparatus produces renin in response to decreased effective intravascular volume. Specific stimuli for renin release are decreased perfusion pressure in the afferent arteriole of the glomerulus, decreased delivery of sodium to the distal nephron, and β1 -adrenergic agonists, which increase in response to intravascular volume depletion. Renin, a proteolytic enzyme, cleaves angiotensinogen, producing angiotensin I. Angiotensin-converting enzyme (ACE) converts angiotensin I into angiotensin II. The actions of angiotensin II include direct stimulation of the proximal tubule to increase sodium resorption and stimulation of the adrenal gland to increase aldosterone secretion. Through its actions in the distal nephron—specifically, the late distal convoluted tubule and the collecting duct—aldosterone increases sodium resorption. Aldosterone also stimulates potassium excretion, increasing urinary losses. Along with decreasing urinary loss of sodium, angiotensin II acts as a vasoconstrictor, which helps maintain adequate blood pressure in the presence of volume depletion.
Volume expansion stimulates the synthesis of atrial natriuretic peptide (ANP) , which is produced by the atria in response to atrial wall distention. Along with increasing the GFR, ANP inhibits Na+ resorption in the medullary portion of the collecting duct, facilitating an increase in urinary Na+ excretion.
Volume overload occurs when Na+ intake exceeds output. Children with kidney failure have impaired ability to excrete Na+ . The GFR is low at birth, limiting a newborn's ability to excrete an Na+ load. In other situations, there is a loss of the appropriate regulation of renal Na+ excretion. This loss occurs in patients with excessive aldosterone, as seen in primary hyperaldosteronism or renal artery stenosis, where excess renin production leads to high aldosterone levels. In acute glomerulonephritis, even without significantly reduced GFR, the normal intrarenal mechanisms that regulate Na+ excretion malfunction, causing excessive renal retention of Na+ and volume overload.
Renal retention of Na+ occurs during volume depletion, but this appropriate response causes the severe excess in total body Na+ that is present in heart failure, liver failure, nephrotic syndrome, and other causes of hypoalbuminemia. In these diseases the effective intravascular volume is decreased, causing the kidney and the various regulatory systems to respond, leading to renal Na+ retention and edema formation.
Volume depletion usually occurs when Na+ losses exceed intake. The most common etiology in children is gastroenteritis. Excessive losses of sodium may also occur from the skin in children with burns, in sweat from patients with cystic fibrosis, or after vigorous exercise. Inadequate intake of Na+ is uncommon except in neglect, in famine, or with an inappropriate choice of liquid diet in a child who cannot take solids. Urinary Na+ wasting may occur in a range of renal diseases, from renal dysplasia to tubular disorders, such as Bartter syndrome. The neonate, especially if premature, has a mild impairment in the ability to conserve Na+ . Iatrogenic renal Na+ wasting takes place during diuretic therapy. Renal Na+ loss occurs as a result of derangement in the normal regulatory systems. An absence of aldosterone, seen most frequently in children with congenital adrenal hyperplasia caused by 21-hydroxylase deficiency, causes sodium wasting (see Chapter 594 ).
Isolated disorders of water balance can affect volume status and Na+ balance. Because the cell membrane is permeable to water, changes in TBW influence both the extracellular volume and the intracellular volume. In isolated water loss, as occurs in diabetes insipidus, the impact is greater on the ICS because it has a greater volume than the ECS. Thus, compared with other types of dehydration, hypernatremic dehydration has less impact on plasma volume; most of the fluid loss comes from the ICS. Yet, significant water loss eventually affects intravascular volume and will stimulate renal Na+ retention, even if total body Na+ content is normal. Similarly, with acute water intoxication or SIADH, there is an excess of TBW, but most is in the ICS. However, there is some effect on the intravascular volume, which causes renal excretion of Na+ . Children with SIADH or water intoxication have high urine Na+ concentration, despite hyponatremia. This finding reinforces the concept of independent control systems for water and Na+ , but the 2 systems interact when pathophysiologic processes dictate, and control of effective intravascular volume always takes precedence over control of osmolality.
Bibliography
Regulation of Osmolality and Volume
Knepper MA, Kwon TH, Nielsen S. Molecular physiology of water balance. N Engl J Med . 2015;372:1349–1358.
Kortenoeven ML, Pedersen NB, Rosenbaek LL, et al. Vasopressin regulation of sodium transport in the distal nephron and collecting duct. Am J Physiol Renal Physiol . 2015;309:F280–F299.
Mecawi Ade S, Ruginsk SG, Elias LL, et al. Neuroendocrine regulation of hydromineral homeostasis. Compr Physiol . 2015;5:1465–1516.
Schweda F. Salt feedback on the renin-angiotensin-aldosterone system. Pflugers Arch . 2015;467:565–576.
Sodium
Larry A. Greenbaum
Sodium Metabolism
Body Content and Physiologic Function
Sodium is the dominant cation of the ECF (see Fig. 68.3 ), and it is the principal determinant of extracellular osmolality. Na+ is therefore necessary for the maintenance of intravascular volume. Less than 3% of Na+ is intracellular. More than 40% of total body Na+ is in bone; the remainder is in the interstitial and intravascular spaces. The low intracellular [Na+ ], approximately 10 mEq/L, is maintained by Na+ ,K+ -ATPase, which exchanges intracellular Na+ for extracellular K+ .
Sodium Intake
A child's diet determines the amount of Na+ ingested—a predominantly cultural determination in older children. An occasional child has salt craving because of an underlying salt-wasting renal disease or adrenal insufficiency. Children in the United States tend to have very high salt intakes because their diets include a large amount of “junk” food or fast food. Infants receive sodium from breast milk (approximately 7 mEq/L) and formula (7-13 mEq/L, for 20 calorie/oz formula).
Sodium is readily absorbed throughout the GI tract. Mineralocorticoids increase sodium transport into the body, although this effect has limited clinical significance. The presence of glucose enhances sodium absorption owing to the presence of a co-transport system. This is the rationale for including sodium and glucose in oral rehydration solutions (see Chapter 366 ).
Sodium Excretion
Sodium excretion occurs in stool and sweat, but the kidney regulates Na+ balance and is the principal site of Na+ excretion. There is some Na+ loss in stool, but it is minimal unless diarrhea is present. Normally, sweat has 5-40 mEq/L of sodium. Sweat Na+ concentration is increased in children with cystic fibrosis, aldosterone deficiency, or pseudohypoaldosteronism. The higher sweat losses in these conditions may cause or contribute to Na+ depletion.
Sodium is unique among electrolytes because water balance, not Na+ balance, usually determines its concentration. When the [Na+ ] increases, the resultant higher plasma osmolality causes increased thirst and increased secretion of ADH, which leads to renal conservation of water. Both these mechanisms increase the water content of the body, and the [Na+ ] returns to normal. During hyponatremia, the decrease in plasma osmolality stops ADH secretion, and consequent renal water excretion leads to an increase in the [Na+ ]. Even though water balance is usually regulated by osmolality, volume depletion does stimulate thirst, ADH secretion, and renal conservation of water. Volume depletion takes precedence over osmolality; volume depletion stimulates ADH secretion, even if a patient has hyponatremia.
The excretion of Na+ by the kidney is not regulated by the plasma osmolality. The patient's effective plasma volume determines the amount of sodium in the urine. This is mediated by a variety of regulatory systems, including the renin-angiotensin-aldosterone system and intrarenal mechanisms. In hyponatremia or hypernatremia, the underlying pathophysiology determines the amount of urinary Na+ , not the serum [Na+ ].
Hypernatremia
Hypernatremia is a [Na+ ] >145 mEq/L, although it is sometimes defined as >150 mEq/L. Mild hypernatremia is fairly common in children, especially among infants with gastroenteritis. Hypernatremia in hospitalized patients may be iatrogenic—caused by inadequate water administration or, less often, by excessive Na+ administration. Moderate or severe hypernatremia has significant morbidity because of the underlying disease, the effects of hypernatremia on the brain, and the risks of overly rapid correction.
Etiology and Pathophysiology
There are 3 basic mechanisms of hypernatremia (Table 68.1 ). Sodium intoxication is frequently iatrogenic in a hospital setting as a result of correction of metabolic acidosis with sodium bicarbonate. Baking soda, a putative home remedy for upset stomach, is another source of sodium bicarbonate; the hypernatremia is accompanied by a profound metabolic alkalosis. In hyperaldosteronism, there is renal retention of sodium and resultant hypertension; hypernatremia may not be present or is usually mild.
The classic causes of hypernatremia from a water deficit are nephrogenic and central diabetes insipidus (see Chapters 548 and 574 ). Hypernatremia develops in diabetes insipidus only if the patient does not have access to water or cannot drink adequately because of immaturity, neurologic impairment, emesis, or anorexia. Infants are at high risk because of their inability to control their own water intake. Central diabetes insipidus and the genetic forms of nephrogenic diabetes insipidus typically cause massive urinary water losses and very dilute urine. The water losses are less dramatic, and the urine often has the same osmolality as plasma when nephrogenic diabetes insipidus is secondary to intrinsic renal disease (obstructive uropathy, renal dysplasia, sickle cell disease).
The other causes of a water deficit are also secondary to an imbalance between losses and intake. Newborns, especially if premature, have high insensible water losses. Losses are further increased if the infant is placed under a radiant warmer or with the use of phototherapy for hyperbilirubinemia. The renal concentrating mechanisms are not optimal at birth, providing an additional source of water loss. Ineffective breastfeeding, often in a primiparous mother, can cause severe hypernatremic dehydration. Adipsia , the absence of thirst, is usually secondary to damage to the hypothalamus, such as from trauma, tumor, hydrocephalus, or histiocytosis. Primary adipsia is rare.
When hypernatremia occurs in conditions with deficits of sodium and water, the water deficit exceeds the sodium deficit. This occurs only if the patient is unable to ingest adequate water. Diarrhea results in depletion of both Na+ and water. Because diarrhea is hypotonic—typical Na+ concentration of 35-65 mEq/L—water losses exceed Na+ losses, potentially leading to hypernatremia. Most children with gastroenteritis do not have hypernatremia because they drink enough hypotonic fluid to compensate for stool water losses (see Chapter 366 ). Fluids such as water, juice, and formula are more hypotonic than the stool losses, allowing correction of the water deficit and potentially even causing hyponatremia. Hypernatremia is most likely to occur in the child with diarrhea who has inadequate intake because of emesis, lack of access to water, or anorexia.
Osmotic agents, including mannitol, and glucose in diabetes mellitus , cause excessive renal losses of water and Na+ . Because the urine is hypotonic (Na+ concentration of approximately 50 mEq/L) during an osmotic diuresis, water loss exceeds Na+ loss, and hypernatremia may occur if water intake is inadequate. Certain chronic kidney diseases, such as renal dysplasia and obstructive uropathy, are associated with tubular dysfunction, leading to excessive losses of water and Na+ . Many children with such diseases have disproportionate water loss and are at risk for hypernatremic dehydration, especially if gastroenteritis supervenes. Similar mechanisms occur during the polyuric phase of acute kidney injury and after relief of urinary obstruction (postobstructive diuresis). Patients with either condition may have an osmotic diuresis from urinary losses of urea and an inability to conserve water because of tubular dysfunction.
Essential hypernatremia is rare in children and is thought to occur with injury to the hypothalamic-posterior pituitary axis. It is euvolemic, nonhypertensive, and associated with hypodipsia, possibly related to a reset osmol sensor.
Clinical Manifestations
Most children with hypernatremia are dehydrated and show the typical clinical signs and symptoms (see Chapter 70 ). Children with hypernatremic dehydration tend to have better preservation of intravascular volume because of the shift of water from the ICS to the ECS. This shift maintains blood pressure and urine output and allows hypernatremic infants to be less symptomatic initially and potentially to become more dehydrated before medical attention is sought. Breastfed infants with hypernatremia are often profoundly dehydrated, with failure to thrive (malnutrition). Probably because of intracellular water loss, the pinched abdominal skin of a dehydrated, hypernatremic infant has a “doughy” feel.
Hypernatremia, even without dehydration, causes central nervous system (CNS) symptoms that tend to parallel the degree of Na+ elevation and the acuity of the increase. Patients are irritable, restless, weak, and lethargic. Some infants have a high-pitched cry and hyperpnea. Alert patients are very thirsty, even though nausea may be present. Hypernatremia may cause fever, although many patients have an underlying process that contributes to the fever. Hypernatremia is associated with hyperglycemia and mild hypocalcemia; the mechanisms are unknown. Beyond the sequelae of dehydration, there is no clear direct effect of hypernatremia on other organs or tissues, except the brain.
Brain hemorrhage is the most devastating consequence of untreated hypernatremia. As the extracellular osmolality increases, water moves out of brain cells, leading to a decrease in brain volume. This decrease can result in tearing of intracerebral veins and bridging blood vessels as the brain moves away from the skull and the meninges. Patients may have subarachnoid, subdural, and parenchymal hemorrhages. Seizures and coma are possible sequelae of the hemorrhage, although seizures are more common during correction of hypernatremia. The cerebrospinal fluid protein is often elevated in infants with significant hypernatremia, probably because of leakage from damaged blood vessels. Neonates, especially if premature, seem especially vulnerable to hypernatremia and excessive sodium intake. There is an association between rapid or hyperosmolar sodium bicarbonate administration and the development of intraventricular hemorrhages in neonates. Even though central pontine myelinolysis is classically associated with overly rapid correction of hyponatremia, both central pontine and extrapontine myelinolysis can occur in children with hypernatremia (see Treatment ). Thrombotic complications occur in severe hypernatremic dehydration, including stroke, dural sinus thrombosis, peripheral thrombosis, and renal vein thrombosis. This is secondary to dehydration and possibly hypercoagulability associated with hypernatremia.
Diagnosis
The etiology of hypernatremia is usually apparent from the history. Hypernatremia resulting from water loss occurs only if the patient does not have access to water or is unable to drink. In the absence of dehydration, it is important to ask about sodium intake. Children with excess salt intake do not have signs of dehydration, unless another process is present. Severe Na+ intoxication causes signs of volume overload, such as pulmonary edema and weight gain. Salt poisoning is associated with an elevated fractional excretion of Na+ , whereas hypernatremic dehydration causes a low fractional excretion of Na+ . Gastric sodium concentrations are often elevated in salt poisoning. In hyperaldosteronism, hypernatremia is usually mild or absent and is associated with edema, hypertension, hypokalemia, and metabolic alkalosis.
When there is isolated water loss, the signs of volume depletion are usually less severe initially because much of the loss is from the ICS. When pure water loss causes signs of dehydration, the hypernatremia and water deficit are usually severe. In the child with renal water loss, either central or nephrogenic diabetes insipidus, the urine is inappropriately dilute and urine volume is not low. The urine is maximally concentrated and urine volume is low if the losses are extrarenal or caused by inadequate intake. With extrarenal causes of loss of water, the urine osmolality should be >1,000 mOsm/kg. When diabetes insipidus is suspected, the evaluation may include measurement of ADH and a water deprivation test, including a trial of desmopressin acetate (synthetic ADH analog) to differentiate between nephrogenic diabetes insipidus and central diabetes insipidus (see Chapters 548 and 574 ). A water-deprivation test is unnecessary if the patient has simultaneous documentation of hypernatremia and poorly concentrated urine (osmolality lower than that of plasma). In children with central diabetes insipidus, administration of desmopressin acetate increases the urine osmolality above the plasma osmolality, although maximum osmolality does not occur immediately because of the decreased osmolality of the renal medulla as a result of the chronic lack of ADH. In children with nephrogenic diabetes insipidus, there is no response to desmopressin acetate. Hypercalcemia or hypokalemia may produce a nephrogenic diabetes insipidus–like syndrome.
With combined Na+ and water deficits, analysis of the urine differentiates between renal and nonrenal etiologies. When the losses are extrarenal, the kidney responds to volume depletion with low urine volume, concentrated urine, and Na+ retention (urine [Na+ ] <20 mEq/L, fractional excretion of Na+ <1%). With renal causes, the urine volume is not appropriately low, the urine is not maximally concentrated, and the urine [Na+ ] may be inappropriately elevated.
Treatment
As hypernatremia develops, the brain generates idiogenic osmoles to increase the intracellular osmolality and prevent the loss of brain water. This mechanism is not instantaneous and is most prominent when hypernatremia has developed gradually. If the serum [Na+ ] is lowered rapidly, there is movement of water from the serum into the brain cells to equalize the osmolality in the 2 compartments. The resultant brain swelling manifests as seizures or coma.
Because of the associated dangers, chronic hypernatremia should not be corrected rapidly. The goal is to decrease the serum [Na+ ] by <10 mEq/L every 24 hr. The most important component of correcting moderate or severe hypernatremia is frequent monitoring of the serum [Na+ ] value so that fluid therapy can be adjusted to provide adequate correction, neither too slow nor too fast. If a child has seizures as a result of brain edema secondary to rapid correction, administration of hypotonic fluid should be stopped. An infusion of 3% saline can acutely increase the serum [Na+ ], reversing the cerebral edema.
Chapter 70 outlines a detailed approach to the child with hypernatremic dehydration. Acute, severe hypernatremia, usually secondary to sodium administration, can be corrected more rapidly with 5% dextrose in water (D5W) because idiogenic osmoles have not had time to accumulate. This fact balances the high morbidity and mortality rates associated with hypernatremia with the dangers of overly rapid correction. When hypernatremia is severe and is caused by sodium intoxication, it may be impossible to administer enough water to correct the hypernatremia rapidly without worsening the volume overload. In this situation, dialysis allows for removal of the excess Na+ , with the precise strategy dependent on the mode of dialysis. In less severe cases, the addition of a loop diuretic increases the removal of excess Na+ and water, decreasing the risk of volume overload. With Na+ overload, hypernatremia is corrected with Na+ -free intravenous (IV) fluid (D5W).
Hyperglycemia from hypernatremia is not usually a problem and is not treated with insulin because the acute decrease in glucose may precipitate cerebral edema by lowering plasma osmolality. Rarely, the glucose concentration of IV fluids must be reduced (from 5% to 2.5% dextrose in water). The secondary hypocalcemia is treated as needed.
It is important to address the underlying cause of the hypernatremia, if possible. The child with central diabetes insipidus should receive desmopressin acetate. Because this treatment reduces renal excretion of water, excessive intake of water must be avoided to prevent both overly rapid correction of the hypernatremia and the development of hyponatremia. Over the long term, reduced sodium intake and the use of medications can somewhat ameliorate the water losses in nephrogenic diabetes insipidus (see Chapter 548 ). The daily water intake of a child receiving tube feeding may need to be increased to compensate for high losses. The patient with significant ongoing losses, such as through diarrhea, may need supplemental water and electrolytes (see Chapter 69 ). Sodium intake is reduced if it contributed to the hypernatremia.
Hyponatremia
Hyponatremia, a very common electrolyte abnormality in hospitalized patients, is a serum sodium level <135 mEq/L. Both total body sodium and TBW determine the serum sodium concentration. Hyponatremia exists when the ratio of water to Na+ is increased. This condition can occur with low, normal, or high levels of body Na+ . Similarly, body water can be low, normal, or high.
Etiology and Pathophysiology
Table 68.2 lists the causes of hyponatremia. Pseudohyponatremia is a laboratory artifact present when the plasma contains very high concentrations of protein (multiple myeloma, IVIG infusion) or lipid (hypertriglyceridemia, hypercholesterolemia). It does not occur when a direct ion-selective electrode determines the [Na+ ] in undiluted plasma, a technique that is used by ABG analyzers or POC instruments (see Chapter 68.1 ). In true hyponatremia, the measured osmolality is low, whereas it is normal in pseudohyponatremia. Hyperosmolality, as may occur with hyperglycemia, causes a low [Na+ ] because water moves down its osmotic gradient from the ICS into the ECS, diluting the [Na+ ]. However, because the manifestations of hyponatremia are a result of the low plasma osmolality, patients with hyponatremia resulting from hyperosmolality do not have symptoms of hyponatremia. When the etiology of the hyperosmolality resolves, such as hyperglycemia in diabetes mellitus, water moves back into the cells, and the [Na+ ] rises to its “true” value. Mannitol or sucrose, a component of intravenous immune globulin (IVIG) preparations, may cause hyponatremia because of hyperosmolality.
Classification of hyponatremia is based on the patient's volume status. In hypovolemic hyponatremia the child has lost Na+ from the body. The water balance may be positive or negative, but Na+ loss has been higher than water loss. The pathogenesis of the hyponatremia is usually a combination of Na+ loss and water retention to compensate for the volume depletion. The patient has a pathologic increase in fluid loss, and this fluid contains Na+ . Most fluid that is lost has a lower [Na+ ] than that of plasma. Viral diarrhea fluid has an average [Na+ ] of 50 mEq/L. Replacing diarrhea fluid, which has [Na+ ] of 50 mEq/L, with formula, which has only approximately 10 mEq/L of Na+ , reduces [Na+ ]. Intravascular volume depletion interferes with renal water excretion, the body's usual mechanism for preventing hyponatremia. The volume depletion stimulates ADH synthesis, resulting in renal water retention. Volume depletion also decreases the GFR and enhances water resorption in the proximal tubule, thereby reducing water delivery to the collecting duct.
Diarrhea as a result of gastroenteritis is the most common cause of hypovolemic hyponatremia in children. Emesis causes hyponatremia if the patient takes in hypotonic fluid, either intravenously or enterally, despite the emesis. Most patients with emesis have either a normal [Na+ ] or hypernatremia. Burns may cause massive losses of isotonic fluid and resultant volume depletion. Hyponatremia develops if the patient receives hypotonic fluid. Losses of sodium from sweat are especially high in children with cystic fibrosis, aldosterone deficiency, or pseudohypoaldosteronism, although high losses can also occur in a hot climate. Third space losses are isotonic and can cause significant volume depletion, leading to ADH production and water retention, which can cause hyponatremia if the patient receives hypotonic fluid. In diseases that cause volume depletion through extrarenal Na+ loss, the urine Na+ level should be low (<10 mEq/L) as part of the renal response to maintain the intravascular volume. The only exceptions are diseases that cause both extrarenal and renal Na+ losses: adrenal insufficiency and pseudohypoaldosteronism.
Renal Na+ loss may occur in a variety of situations. In some situations the urine [Na+ ] is >140 mEq/L; thus hyponatremia may occur without any fluid intake. In many cases the urine Na+ level is less than the serum [Na+ ]; thus the intake of hypotonic fluid is necessary for hyponatremia to develop. In diseases associated with urinary Na+ loss, the urine Na+ level is >20 mEq/L despite volume depletion. This may not be true if the urinary Na+ loss is no longer occurring, as is frequently the case if diuretics are discontinued. Because loop diuretics prevent generation of a maximally hypertonic renal medulla, the patient can neither maximally dilute nor concentrate the urine. The inability to maximally retain water provides some protection against severe hyponatremia. The patient receiving thiazide diuretics can concentrate the urine and is at higher risk for severe hyponatremia. Osmotic agents, such as glucose during diabetic ketoacidosis, cause loss of both water and Na+ . Urea accumulates during renal failure and then acts as an osmotic diuretic after relief of urinary tract obstruction and during the polyuric phase of acute tubular necrosis. Transient tubular damage in these conditions further impairs Na+ conservation. The serum [Na+ ] in these conditions depends on [Na+ ] of the fluid used to replace the losses. Hyponatremia develops when the fluid is hypotonic relative to the urinary losses.
Renal salt wasting occurs in hereditary kidney diseases, such as juvenile nephronophthisis and autosomal recessive polycystic kidney disease. Obstructive uropathy, most often a result of posterior urethral valves, produces salt wasting, but patients with the disease may also have hypernatremia as a result of impaired ability to concentrate urine and high-water loss. Acquired tubulointerstitial nephritis, usually secondary to either medications or infections, may cause salt wasting, along with other evidence of tubular dysfunction. CNS injury may produce cerebral salt wasting, which is theoretically caused by the production of a natriuretic peptide that causes renal salt wasting. In type II renal tubular acidosis (RTA) , usually associated with Fanconi syndrome (see Chapter 547.1 ), there is increased excretion of Na+ and bicarbonate in the urine. Patients with Fanconi syndrome also have glycosuria, aminoaciduria, and hypophosphatemia because of renal phosphate wasting.
Aldosterone is necessary for renal Na+ retention and for the excretion of K+ and acid. In congenital adrenal hyperplasia caused by 21-hydroxylase deficiency, the block of aldosterone production results in hyponatremia, hyperkalemia, and metabolic acidosis. Decreased aldosterone secretion may be seen in Addison disease (adrenal insufficiency). In pseudohypoaldosteronism, aldosterone levels are elevated, but there is no response because of either a defective Na+ channel or a deficiency of aldosterone receptors. A lack of tubular response to aldosterone may occur in children with urinary tract obstruction, especially during an acute urinary tract infection.
In hypervolemic hyponatremia there is an excess of TBW and Na+ , although the increase in water is greater than the increase in Na+ . In most conditions that cause hypervolemic hyponatremia, there is a decrease in the effective blood volume , resulting from third space fluid loss, vasodilation, or poor cardiac output. The regulatory systems sense a decrease in effective blood volume and attempt to retain water and Na+ to correct the problem. ADH causes renal water retention, and the kidney, under the influence of aldosterone and other intrarenal mechanisms, retains sodium. The patient's sodium concentration decreases because water intake exceeds sodium intake and ADH prevents the normal loss of excess water.
In these disorders, there is low urine [Na+ ] (<10 mEq/L) and an excess of both TBW and Na+ . The only exception is in patients with renal failure and hyponatremia. These patients have an expanded intravascular volume, and hyponatremia can therefore appropriately suppress ADH production. Water cannot be excreted because very little urine is being made. Serum Na+ is diluted through ingestion of water. Because of renal dysfunction, the urine [Na+ ] may be elevated, but urine volume is so low that urine Na+ excretion has not kept up with Na+ intake, leading to sodium overload. The urine [Na+ ] in renal failure varies. In patients with acute glomerulonephritis, because it does not affect the tubules, the urine Na+ level is usually low, whereas in patients with acute tubular necrosis, it is elevated because of tubular dysfunction.
Patients with hyponatremia and no evidence of volume overload or volume depletion have euvolemic hyponatremia. These patients typically have an excess of TBW and a slight decrease in total body Na+ . Some of these patients have an increase in weight, implying that they are volume-overloaded. Nevertheless, from a clinical standpoint, they usually appear normal or have subtle signs of fluid overload. In SIADH the secretion of ADH is not inhibited by either low serum osmolality or expanded intravascular volume (see Chapter 575 ). The result is that the child with SIADH is unable to excrete water. This results in dilution of the serum Na+ and hyponatremia. The expansion of the extracellular volume because of the retained water causes a mild increase in intravascular volume. The kidney increases Na+ excretion to decrease intravascular volume to normal; thus the patient has a mild decrease in body Na+ . SIADH typically occurs with disorders of the CNS (infection, hemorrhage, trauma, tumor, thrombosis, Guillain-Barré syndrome), but lung disease (infection, asthma, positive pressure ventilation) and malignant tumors (producing ADH) are other potential causes. A variety of medications may cause SIADH, including recreational use of 3,4-methylenedioxymethylamphetamine (MDMA, or “Ecstasy”), opiates, antiepileptic drugs (carbamazepine, oxcarbazepine, valproate), tricyclic antidepressants, vincristine, cyclophosphamide, and selective serotonin reuptake inhibitors (SSRIs). The diagnosis of SIADH is one of exclusion, because other causes of hyponatremia must be eliminated (Table 68.3 ). Because SIADH is a state of intravascular volume expansion, low serum uric acid and BUN levels are supportive of the diagnosis. A rare gain-of-function mutation in the renal ADH receptor causes nephrogenic syndrome of inappropriate antidiuresis . Patients with this X-linked disorder appear to have SIADH but have undetectable levels of ADH.
Hyponatremia in hospitalized patients is frequently caused by inappropriate production of ADH and administration of hypotonic IV fluids (see Chapter 69 ). Causes of inappropriate ADH production include stress, medications such as narcotics or anesthetics, nausea, and respiratory illness. The synthetic analog of ADH, desmopressin acetate, causes water retention and may cause hyponatremia if fluid intake is not appropriately limited. The main uses of desmopressin acetate in children are for the management of central diabetes insipidus and nocturnal enuresis.
Excess water ingestion can produce hyponatremia. In these cases, [Na+ ] decreases as a result of dilution. This decrease suppresses ADH secretion, and there is a marked water diuresis by the kidney. Hyponatremia develops only because the intake of water exceeds the kidney's ability to eliminate water. This condition is more likely to occur in infants because their lower GFR limits their ability to excrete water.
Hyponatremia may develop in infants <6 mo of age when caregivers offer water to their infant as a supplement, during hot weather, or when they run out of formula. Hyponatremia may result in transient seizures, hypothermia, and poor tone. With cessation of water intake, the hyponatremia rapidly corrects. Infants <6 mo of age should not be given water to drink; infants 6-12 mo of age should not receive >1-2 ounces. If the infant appears thirsty, the parent should offer formula or breastfeed the child.
In some situations the water intoxication causes acute hyponatremia and is caused by a massive acute water load . Causes include infant swimming lessons, inappropriate use of hypotonic IV fluids, water enemas, and forced water intake as a form of child abuse. Chronic hyponatremia occurs in children who receive water but limited sodium and protein. The minimum urine osmolality is approximately 50 mOsm/kg, the kidney can excrete 1 L of water only if there is enough solute ingested to produce 50 mOsm for urinary excretion. Because Na+ and urea (a breakdown product of protein) are the principal urinary solutes, a lack of intake of Na+ and protein prevents adequate water excretion. This occurs with the use of diluted formula or other inappropriate diets. Subsistence on beer, a poor source of Na+ and protein, causes hyponatremia because of the inability to excrete the high water load (“beer potomania”). Exercise-induced hyponatremia , reported frequently during marathons, is caused by excessive water intake, salt losses from sweat, and secretion of ADH.
The pathogenesis of the hyponatremia in glucocorticoid deficiency (adrenal insufficiency) is multifactorial and includes increased ADH secretion. In hypothyroidism there is an inappropriate retention of water by the kidney, but the precise mechanisms are not clearly elucidated.
Cerebral salt wasting , an uncommon disorder in children, may be confused with SIADH and is often associated with CNS injury or lesions. Cerebral salt wasting produces renal salt losses and hypovolemia (orthostatic hypotension and elevated hematocrit, BUN, or creatinine).
Clinical Manifestations
Hyponatremia causes a decrease in the osmolality of the ECS. Because the ICS then has a higher osmolality, water moves from the ECS to the ICS to maintain osmotic equilibrium. The increase in intracellular water causes cells to swell. Although cell swelling is not problematic in most tissues, it is dangerous for the brain, which is confined by the skull. As brain cells swell, there is an increase in intracranial pressure, which impairs cerebral blood flow. Acute, severe hyponatremia can cause brainstem herniation and apnea; respiratory support is often necessary. Brain cell swelling is responsible for most of the symptoms of hyponatremia. Neurologic symptoms of hyponatremia include anorexia, nausea, emesis, malaise, lethargy, confusion, agitation, headache, seizures, coma, and decreased reflexes. Patients may have hypothermia and Cheyne-Stokes respirations. Hyponatremia can cause muscle cramps and weakness; rhabdomyolysis can occur with water intoxication.
The symptoms of hyponatremia are mostly a result of the decrease in extracellular osmolality and the resulting movement of water down its osmotic gradient into the ICS. Brain swelling can be significantly obviated if the hyponatremia develops gradually, because brain cells adapt to the decreased extracellular osmolality by reducing intracellular osmolality. This reduction is achieved by extrusion of the main intracellular ions (K+ , Cl− ) and a variety of small organic molecules. This process explains why the range of symptoms in hyponatremia is related to both the serum [Na+ ] and its rate of decrease. A patient with chronic hyponatremia may have only subtle neurologic abnormalities with a serum [Na+ ] of 110 mEq/L, but another patient may have seizures because of an acute decline in serum [Na+ ] from 140 to 125 mEq/L.
Diagnosis
The history usually points to a likely etiology of the hyponatremia. Most patients with hyponatremia have a history of volume depletion. Diarrhea and diuretic use are common causes of hyponatremia in children. A history of polyuria, perhaps with enuresis, and/or salt craving is present in children with primary kidney diseases or absence of aldosterone effect. Children may have signs or symptoms suggesting a diagnosis of hypothyroidism or adrenal insufficiency (see Chapters 581 and 593 ). Brain injury raises the possibility of SIADH or cerebral salt wasting, with the caveat that SIADH is much more likely. Liver disease, nephrotic syndrome, renal failure, or congestive heart failure may be acute or chronic. The history should include a review of the patient's intake, both intravenous and enteral, with careful attention to the amounts of water, Na+ , and protein.
The traditional first step in the diagnostic process is determination of the plasma osmolality. This is done because some patients with a low serum [Na+ ] do not have low osmolality. The clinical effects of hyponatremia are secondary to the associated low osmolality. Without a low osmolality, there is no movement of water into the intracellular space.
A patient with hyponatremia can have a low, normal, or high osmolality. A normal osmolality in combination with hyponatremia occurs in pseudohyponatremia. Children with elevation of serum glucose concentration or of another effective osmole (mannitol) have a high plasma osmolality and hyponatremia. The presence of a low osmolality indicates “true” hyponatremia. Patients with low osmolality are at risk for neurologic symptoms and require further evaluation to determine the etiology of the hyponatremia.
In some situations, true hyponatremia is present despite a normal or elevated plasma osmolality. The presence of an ineffective osmole, usually urea, increases the plasma osmolality, but because the osmole has the same concentration in the ICS, it does not cause fluid to move into the ECS. There is no dilution of the serum Na+ by water, and the [Na+ ] remains unchanged if the ineffective osmole is eliminated. Most importantly, the ineffective osmole does not protect the brain from edema caused by hyponatremia. Therefore, a patient may have symptoms of hyponatremia despite having a normal or increased osmolality because of uremia.
In patients with true hyponatremia, the next step in the diagnostic process is to clinically evaluate the volume status. Patients with hyponatremia can be hypovolemic, hypervolemic, or euvolemic. The diagnosis of volume depletion relies on the usual findings with dehydration (see Chapter 70 ), although subtle volume depletion may not be clinically apparent. Children with hypervolemia are edematous on physical examination. They may have ascites, pulmonary edema, pleural effusion, or hypertension.
Hypovolemic hyponatremia can have renal or nonrenal causes. The urine [Na+ ] is very useful in differentiating between renal and nonrenal causes. When the losses are nonrenal and the kidney is working properly, there is renal retention of Na+ , a normal homeostatic response to volume depletion. Thus the urinary [Na+ ] is low, typically <10 mEq/L, although Na+ conservation in neonates is less avid. When the kidney is the cause of the Na+ loss, the urine [Na+ ] is >20 mEq/L, reflecting the defect in renal Na+ retention. The interpretation of the urine Na+ level is challenging with diuretic therapy because it is high when diuretics are being used but low after the diuretic effect is gone. This becomes an issue only when diuretic use is surreptitious. The urine [Na+ ] is not useful if a metabolic alkalosis is present; the urine [Cl− ] must be used instead (see Chapter 68.7 ).
Differentiating among the nonrenal causes of hypovolemic hyponatremia is usually facilitated by the history. Although the renal causes are more challenging to distinguish, a high serum [K+ ] is associated with disorders in which the Na+ wasting is caused by absence of or ineffectiveness of aldosterone.
In the patient with hypervolemic hyponatremia, the urine [Na+ ] is a helpful parameter. It is usually <10 mEq/L, except in the patient with renal failure.
Treatment
The management of hyponatremia is based on the pathophysiology of the specific etiology. The management of all causes requires judicious monitoring and avoidance of an overly quick normalization of the serum [Na+ ]. A patient with severe symptoms (seizures), no matter the etiology, should be given a bolus of hypertonic saline to produce a small, rapid increase in serum sodium. Hypoxia worsens cerebral edema, and hyponatremia may exacerbate hypoxic cell swelling. Therefore, pulse oximetry should be monitored and hypoxia aggressively corrected.
With all causes of hyponatremia, it is important to avoid overly rapid correction, which may cause central pontine myelinolysis (CPM) . This syndrome, which occurs within several days of rapid correction of hyponatremia, produces neurologic symptoms, including confusion, agitation, flaccid or spastic quadriparesis, and death. There are usually characteristic pathologic and radiologic changes in the brain, especially in the pons, but extrapontine lesions are quite common and may cause additional symptoms. Despite severe symptoms, full recovery does occur in some patients.
CPM is more common in patients who are treated for chronic hyponatremia than for acute hyponatremia. Presumably, this difference is based on the adaptation of brain cells to the hyponatremia. The reduced intracellular osmolality, an adaptive mechanism for chronic hyponatremia, makes brain cells susceptible to dehydration during rapid correction of the hyponatremia, which may be the mechanism of CPM. Even though CPM is rare in pediatric patients, it is advisable to avoid correcting the serum [Na+ ] by >10 mEq/L/24 hr or >18 mEq/L/48 hr. Desmopressin is a potential option if the serum [Na+ ] is increasing too rapidly. This guideline does not apply to acute hyponatremia, as may occur with water intoxication, because the hyponatremia is more often symptomatic, and the adaptive decrease in brain osmolality has not had time to occur. The consequences of brain edema in acute hyponatremia exceed the small risk of CPM.
Patients with hyponatremia can have severe neurologic symptoms, such as seizures and coma. The seizures associated with hyponatremia generally are poorly responsive to anticonvulsants. The child with hyponatremia and severe symptoms needs treatment that will quickly reduce cerebral edema. This goal is best accomplished by increasing the extracellular osmolality so that water moves down its osmolar gradient from the ICS to the ECS.
Intravenous hypertonic saline rapidly increases serum [Na+ ], and the effect on serum osmolality leads to a decrease in brain edema. Each mL/kg of 3% NaCl increases the serum [Na+ ] by approximately 1 mEq/L. A child with active symptoms often improves after receiving 4-6 mL/kg of 3% NaCl.
The child with hypovolemic hyponatremia has a deficiency in Na+ and may have a deficiency in water. The cornerstone of therapy is to replace the Na+ deficit and any water deficit present. The first step in treating any dehydrated patient is to restore the intravascular volume with isotonic saline. Ultimately, complete restoration of intravascular volume suppresses ADH production, thereby permitting excretion of the excess water. Chapter 70 discusses the management of hyponatremic dehydration.
The management of hypervolemic hyponatremia is difficult; patients have an excess of both water and Na+ . Administration of Na+ leads to worsening volume overload and edema. In addition, patients are retaining water and Na+ because of their ineffective intravascular volume or renal insufficiency. The cornerstone of therapy is water and Na+ restriction, because patients have volume overload. Diuretics may help by causing excretion of both Na+ and water. Vasopressin antagonists (vaptans ), by blocking the action of ADH and causing a water diuresis, are effective in correcting the hypervolemic hyponatremia caused by heart failure. Vaptans are contraindicated if there are moderate to severe CNS symptoms.
Hyponatremic patients with low albumin from nephrotic syndrome have a better response to diuretics after an infusion of 25% albumin; the [Na+ ] often normalizes as a result of expansion of the intravascular volume. A child with heart failure may have an increase in renal water and Na+ excretion if there is an improvement in cardiac output. This improvement will “turn off” the regulatory hormones causing renal water (ADH) and Na+ (aldosterone) retention. The patient with renal failure cannot respond to any of these therapies except fluid restriction. Insensible fluid losses eventually result in an increase in the [Na+ ] as long as insensible and urinary losses are greater than intake. A more definitive approach in children with renal failure is to perform dialysis, which removes water and Na+ .
In isovolumic hyponatremia there is usually an excess of water and a mild Na+ deficit. Therapy is directed at eliminating the excess water. The child with acute excessive water intake loses water in the urine because ADH production is turned off as a result of the low plasma osmolality. Children may correct their hyponatremia spontaneously over 3-6 hr. For acute, symptomatic hyponatremia as a result of water intoxication, hypertonic saline may be needed to reverse cerebral edema. For chronic hyponatremia from poor solute intake, the child needs an appropriate formula, and excess water intake should be eliminated.
Children with iatrogenic hyponatremia caused by the administration of hypotonic IV fluids should receive 3% saline if symptomatic. Subsequent management is dictated by the patient's volume status. The hypovolemic child should receive isotonic IV fluids. The child with nonphysiologic stimuli for ADH production should undergo fluid restriction. Prevention of this iatrogenic complication requires judicious use of IV fluids (see Chapter 69 ).
Specific hormone replacement is the cornerstone of therapy for the hyponatremia of hypothyroidism or cortisol deficiency. Correction of the underlying defect permits appropriate elimination of the excess water.
SIADH is a condition of excess water, with limited ability of the kidney to excrete water. The mainstay of its therapy is fluid restriction with normal sodium intake. Furosemide and NaCl supplementation are effective in the patient with SIADH and severe hyponatremia. Even in a patient with SIADH, furosemide causes an increase in water and Na+ excretion. The loss of Na+ is somewhat counterproductive, but this Na+ can be replaced with hypertonic saline. Because the patient has a net loss of water and the urinary losses of Na+ have been replaced, there is an increase in the [Na+ ], but no significant increase in blood pressure. Vaptans, which block the action of ADH and cause a water diuresis, are effective at correcting euvolemic hyponatremia , but overly rapid correction is a potential complication. Vaptans are not appropriate for treating symptomatic hyponatremia because it can take a few hours before the water diuresis occurs.
Treatment of chronic SIADH is challenging. Fluid restriction in children is difficult for nutritional and behavioral reasons. Other options are long-term furosemide therapy with Na+ supplementation, an oral vaptan (tolvaptan), or oral urea.
Bibliography
Sodium
Berl T. Vasopressin antagonists. N Engl J Med . 2015;372:2207–2216.
Cuesta M, Garrahy A, Thompson CJ. Siad: Practical recommendations for diagnosis and management. J Endocrinol Invest . 2016;39:991–1001.
Cuesta M, Thompson CJ. The syndrome of inappropriate antidiuresis (SIAD). Best Pract Res Clin Endocrinol Metab . 2016;30:175–187.
Henry DA. Hyponatremia. Ann Intern Med . 2015;163(3):ITC1–ITC19.
Hew-Butler T, Rosner MH, Fowkes-Godek S, et al. Statement of the Third International Exercise-Associated Hyponatremia Consensus Development Conference, Carlsbad, California, 2015. Clin J Sport Med . 2015;25:303–320.
Leth Møller K, Hansen A, Torstensson M, et al. Antidepressants and the risk of hyponatremia: a Danish register-based population study. BMJ Open . 2016;6:e011200.
Manish S, Nader M, Kuppachi S. Essential hypernatremia: evidence of reset osmostat in the absence of demonstrable hypothalamic lesions. Am J Med Sci . 2014;347(4):341–342.
Oude Lansink-Hartgring A, Hessels L, Weigel J, et al. Long-term changes in dysnatremia incidence in the ICU: a shift from hyponatremia to hypernatremia. Ann Intensive Care . 2016;6:22.
Powell CVE. Not enough salt in maintenance fluids. Arch Dis Child . 2015;100(11):1013–1015.
Powers KS. Dehydration: isonatremic, hyponatremic, and hypernatremic recognition and management. Pediatr Rev . 2015;36:274–283 [quiz 84–85].
Sinha VK, Ko B. Hyponatremia in cirrhosis: pathogenesis, treatment, and prognostic significance. Adv Chronic Kidney Dis . 2015;22:361–367.
Sterns RH. Disorders of plasma sodium: causes, consequences, and correction. N Engl J Med . 2015;372:55–65.
Suarez-Rivera M, Bonilla-Felix M. Fluid and electrolyte disorders in the newborn: Sodium and potassium. Curr Pediatr Rev . 2014;10:115–122.
Wallace D, Lichtarowicz-Krynska E, Bockenhauer D. Non-accidental salt poisoning. Arch Dis Child . 2017;102(2):119–122.
Potassium
Larry A. Greenbaum
Potassium Metabolism
Body Content and Physiologic Function
The intracellular [K+ ], approximately 150 mEq/L, is much higher than the plasma [K+ ] (see Fig. 68.3 ). The majority of body K+ is contained in muscle. As muscle mass increases, there is an increase in body K+ . Thus an increase in body K+ occurs during puberty, and it is more significant in males. The majority of extracellular K+ is in bone; <1% of total body K+ is in plasma.
Because most K+ is intracellular, the plasma concentration does not always reflect the total body K+ content. A variety of conditions alter the distribution of K+ between the intracellular and extracellular compartments. Na+ ,K+ -ATPase maintains the high intracellular [K+ ] by pumping Na+ out of the cell and K+ into the cell. This activity balances the normal leak of K+ out of cells via potassium channels that is driven by the favorable chemical gradient. Insulin increases K+ movement into cells by activating Na+ ,K+ -ATPase. Hyperkalemia stimulates insulin secretion, which helps mitigate the hyperkalemia. Acid-base status affects K+ distribution, probably via K+ channels and the Na+ ,K+ -ATPase. A decrease in pH drives potassium extracellularly; an increase in pH has the opposite effect. β-Adrenergic agonists stimulate the Na+ ,K+ -ATPase, increasing cellular uptake of K+ . This increase is protective, in that hyperkalemia stimulates adrenal release of catecholamines. α-Adrenergic agonists and exercise cause a net movement of K+ out of the ICS. An increase in plasma osmolality, as with mannitol infusion, leads to water movement out of the cells, and K+ follows as a result of solvent drag. The serum [K+ ] increases by approximately 0.6 mEq/L with each 10 mOsm rise in plasma osmolality.
The high intracellular concentration of K+ , the principal intracellular cation, is maintained through Na+ ,K+ -ATPase. The resulting chemical gradient is used to produce the resting membrane potential of cells. K+ is necessary for the electrical responsiveness of nerve and muscle cells and for the contractility of cardiac, skeletal, and smooth muscle. The changes in membrane polarization that occur during muscle contraction or nerve conduction make these cells susceptible to changes in serum [K+ ]. The ratio of intracellular to extracellular K+ determines the threshold for a cell to generate an action potential and the rate of cellular repolarization. The intracellular [K+ ] affects cellular enzymes. K+ is necessary for maintaining cell volume because of its important contribution to intracellular osmolality.
Potassium Intake
Potassium is plentiful in food. Dietary consumption varies considerably, even though 1-2 mEq/kg is the recommended intake. The intestines normally absorb approximately 90% of ingested K+ . Most absorption occurs in the small intestine, whereas the colon exchanges body K+ for luminal Na+ . Regulation of intestinal losses normally has a minimal role in maintaining potassium homeostasis, although renal failure, aldosterone, and glucocorticoids increase colonic secretion of K+ . The increase in intestinal losses in the setting of renal failure and hyperkalemia, which stimulates aldosterone production, is clinically significant, helping to protect against hyperkalemia.
Potassium Excretion
Some loss of K+ occurs in sweat but is normally minimal. The colon has the ability to eliminate some K+ . In addition, after an acute K+ load, much of the K+ , >40%, moves intracellularly, through the actions of epinephrine and insulin, which are produced in response to hyperkalemia. This process provides transient protection from hyperkalemia, but most ingested K+ is eventually excreted in the urine. The kidneys principally regulate long-term K+ balance, and they alter excretion in response to a variety of signals. K+ is freely filtered at the glomerulus, but 90% is resorbed before reaching the distal tubule and collecting duct, the principal sites of K+ regulation that have the ability to absorb and secrete K+ . The amount of tubular secretion regulates the amount of K+ that appears in the urine. The plasma [K+ ] directly influences secretion in the distal nephron. As the [K+ ] increases, secretion increases.
The principal hormone regulating potassium secretion is aldosterone , which is released by the adrenal cortex in response to increased plasma K+ . Its main site of action is the cortical collecting duct, where aldosterone stimulates Na+ movement from the tubule into the cells. This movement creates a negative charge in the tubular lumen, facilitating K+ excretion. In addition, the increased intracellular Na+ stimulates the basolateral Na+ ,K+ -ATPase, causing more K+ to move into the cells lining the cortical collecting duct. Glucocorticoids, ADH, a high urinary flow rate, and high Na+ delivery to the distal nephron also increase urinary K+ excretion. Insulin, catecholamines, and urinary ammonia decrease K+ excretion. Whereas ADH increases K+ secretion, it also causes water resorption, decreasing urinary flow. The net effect is that ADH has little overall impact on K+ balance. Alkalosis causes potassium to move into cells, including the cells lining the collecting duct. This movement increases K+ secretion, and because acidosis has the opposite effect, it decreases K+ secretion.
The kidney can dramatically vary K+ excretion in response to changes in intake. Normally, approximately 10–15% of the filtered load is excreted. In an adult, excretion of K+ can vary from 5-1,000 mEq/day.
Hyperkalemia
Hyperkalemia—because of the potential for lethal arrhythmias—is one of the most alarming electrolyte abnormalities.
Etiology and Pathophysiology
Three basic mechanisms cause hyperkalemia (Table 68.4 ). In the individual patient, the etiology is sometimes multifactorial.
Spurious hyperkalemia or pseudohyperkalemia is very common in children because of the difficulties in obtaining blood specimens. This laboratory result is usually caused by hemolysis during a heelstick or phlebotomy, but it can be the result of prolonged tourniquet application or fist clenching, either of which causes local potassium release from muscle.
The serum [K+ ] is normally 0.4 mEq/L higher than the plasma value, secondary to K+ release from cells during clot formation. This phenomenon is exaggerated with thrombocytosis because of K+ release from platelets. For every 100,000/m3 increase in the platelet count, the serum [K+ ] rises by approximately 0.15 mEq/L. This phenomenon also occurs with the marked white blood cell (WBC) count elevations sometimes seen with leukemia. Elevated WBC counts, typically >200,000/m3 , can cause a dramatic elevation in the serum [K+ ]. Analysis of a plasma sample usually provides an accurate result. It is important to analyze the sample promptly to avoid K+ release from cells, which occurs if the sample is stored in the cold, or cellular uptake of K+ and spurious hypokalemia, which occurs with storage at high temperatures. Pneumatic tube transport can cause pseudohyperkalemia if cell membranes are fragile (leukemia). Occasionally, heparin causes lysis of leukemic cells and a false elevation of the plasma sample; a blood gas syringe has less heparin and may provide a more accurate reading than a standard tube. There are rare genetic disorders causing in vitro leakage of K+ from red blood cells (RBCs) that may causes familial pseudohyperkalemia.
Because of the kidney's ability to excrete K+ , it is unusual for excessive intake, by itself, to cause hyperkalemia. This condition can occur in a patient who is receiving large quantities of IV or oral K+ for excessive losses that are no longer present. Frequent or rapid blood transfusions can acutely increase the [K+ ] because of the K+ content of blood, which is variably elevated. Increased intake may precipitate hyperkalemia if there is an underlying defect in K+ excretion.
The ICS has a very high [K+ ], so a shift of K+ from the ICS to the ECS can have a significant effect on the plasma [K+ ]. This shift occurs with metabolic acidosis, but the effect is minimal with an organic acid (lactic acidosis, ketoacidosis). A respiratory acidosis has less impact than a metabolic acidosis. Cell destruction, as seen with rhabdomyolysis, tumor lysis syndrome, tissue necrosis, or hemolysis, releases K+ into the extracellular milieu. The K+ released from RBCs in internal bleeding, such as hematomas, is resorbed and enters the ECS.
Normal doses of succinylcholine or β-blockers and fluoride or digitalis intoxication all cause a shift of K+ out of the intracellular compartment. Succinylcholine should not be used during anesthesia in patients at risk for hyperkalemia. β-Blockers prevent the normal cellular uptake of K+ mediated by binding of β-agonists to the β2 -adrenergic receptors. K+ release from muscle cells occurs during exercise, and levels can increase by 1-2 mEq/L with high activity. With an increased plasma osmolality, water moves from the ICS, and K+ follows. This process occurs with hyperglycemia, although in nondiabetic patients the resultant increase in insulin causes K+ to move intracellularly. In diabetic ketoacidosis (DKA) , the absence of insulin causes potassium to leave the ICS, and the problem is compounded by the hyperosmolality. The effect of hyperosmolality causes a transcellular shift of K+ into the ECS after mannitol or hypertonic saline infusions. Malignant hyperthermia , which is triggered by some inhaled anesthetics, causes muscle release of potassium (see Chapter 629.2 ). Hyperkalemic periodic paralysis is an autosomal dominant disorder caused by a mutated Na+ channel. It results in episodic cellular release of K+ and attacks of paralysis (see Chapter 629.1 ).
The kidneys excrete most of the daily K+ intake, so a decrease in kidney function can cause hyperkalemia. Newborn infants in general, and especially premature infants, have decreased kidney function at birth and thus are at increased risk for hyperkalemia despite an absence of intrinsic renal disease. Neonates also have decreased expression of K+ channels, further limiting K+ excretion.
A wide range of primary adrenal disorders , both hereditary and acquired, can cause decreased production of aldosterone, with secondary hyperkalemia (see Chapters 593 and 594 ). Patients with these disorders typically have metabolic acidosis and salt wasting with hyponatremia. Children with subtle adrenal insufficiency may have electrolyte problems only during acute illnesses. The most common form of congenital adrenal hyperplasia , 21-hydroxylase deficiency, typically manifests in male infants as hyperkalemia, metabolic acidosis, hyponatremia, and volume depletion. Females with this disorder usually are diagnosed as newborns because of their ambiguous genitalia; treatment prevents the development of electrolyte problems.
Renin, via angiotensin II, stimulates aldosterone production. A deficiency in renin, a result of kidney damage, can lead to decreased aldosterone production. Hyporeninemia occurs in many kidney diseases, with some of the more common pediatric causes listed in Table 68.4 . These patients typically have hyperkalemia and a metabolic acidosis, without hyponatremia. Some of these patients have impaired renal function, partially accounting for the hyperkalemia, but the impairment in K+ excretion is more extreme than expected for the degree of renal insufficiency.
A variety of renal tubular disorders impair renal excretion of K+ . Children with pseudohypoaldosteronism type 1 have hyperkalemia, metabolic acidosis, and salt wasting (kidney, colon, sweat) leading to hyponatremia and volume depletion; aldosterone values are elevated. In the autosomal recessive variant, there is a defect in the renal Na+ channel that is normally activated by aldosterone. Patients with this variant have severe symptoms (failure to thrive, diarrhea, recurrent respiratory infections, miliaria-rubra like rash), beginning in infancy. Patients with the autosomal dominant form have a defect in the aldosterone receptor, and the disease is milder, often remitting in adulthood. Pseudohypoaldosteronism type 2 (familial hyperkalemic hypertension), also called Gordon syndrome, is an autosomal dominant disorder characterized by hypertension caused by salt retention and impaired excretion of K+ and acid, leading to hyperkalemia and hyperchloremic metabolic acidosis. Activating mutations in either WNK1 or WNK4 , both serine-threonine kinases located in the distal nephron, cause Gordon syndrome. Patients may respond well to thiazide diuretics. In Bartter syndrome , caused by mutations in the potassium channel ROMK (type 2 Bartter syndrome), there can be transient hyperkalemia in neonates, but hypokalemia subsequently develops (see Chapter 549.1 ).
Acquired renal tubular dysfunction, with an impaired ability to excrete K+ , occurs in a number of conditions. These disorders, all characterized by tubulointerstitial disease, are often associated with impaired acid secretion and a secondary metabolic acidosis. In some affected children, the metabolic acidosis is the dominant feature, although a high K+ intake may unmask the defect in K+ handling. The tubular dysfunction can cause renal salt wasting, potentially leading to hyponatremia. Because of the tubulointerstitial damage, these conditions may also cause hyperkalemia as a result of hyporeninemic hypoaldosteronism.
The risk of hyperkalemia resulting from medications is greatest in patients with underlying renal insufficiency. The predominant mechanism of medication-induced hyperkalemia is impaired renal excretion, although ACE inhibitors may worsen hyperkalemia in anuric patients, probably by inhibiting GI potassium loss, which is normally upregulated in renal insufficiency. The hyperkalemia caused by trimethoprim generally occurs only at the very high doses used to treat Pneumocystis jiroveci pneumonia. Potassium-sparing diuretics may easily cause hyperkalemia, especially because they are often used in patients receiving oral K+ supplements. Oral contraceptives containing drospirenone, which blocks the action of aldosterone, may cause hyperkalemia and should not be used in patients with decreased renal function.
Clinical Manifestations
The most important effects of hyperkalemia result from the role of K+ in membrane polarization. The cardiac conduction system is usually the dominant concern. Changes in the electrocardiogram (ECG) begin with peaking of the T waves. This is followed, as K+ level increases, by ST-segment depression, an increased PR interval, flattening of the P wave, and widening of the QRS complex. However, the correlation between K+ level and ECG changes is poor. This process can eventually progress to ventricular fibrillation. Asystole may also occur. Some patients have paresthesias, fasciculations, weakness, and even an ascending paralysis, but cardiac toxicity usually precedes these clinical symptoms, emphasizing the danger of assuming that an absence of symptoms implies an absence of danger. Chronic hyperkalemia is generally better tolerated than acute hyperkalemia.
Diagnosis
The etiology of hyperkalemia is often readily apparent. Spurious hyperkalemia is very common in children, so obtaining a 2nd potassium measurement is often appropriate. If there is a significant elevation of WBC or platelet count, the 2nd measurement should be performed on a plasma sample that is evaluated promptly. The history should initially focus on potassium intake, risk factors for transcellular shifts of K+ , medications that cause hyperkalemia, and signs of renal insufficiency, such as oliguria and edema. Initial laboratory evaluation should include creatinine, BUN, and assessment of the acid-base status. Many etiologies of hyperkalemia cause metabolic acidosis , which worsens hyperkalemia through the transcellular shift of K+ out of cells. Renal insufficiency is a common cause of the combination of metabolic acidosis and hyperkalemia, also seen in diseases associated with aldosterone insufficiency or aldosterone resistance. Children with absent or ineffective aldosterone often have hyponatremia and volume depletion because of salt wasting. Genetic diseases, such as congenital adrenal hyperplasia and pseudohypoaldosteronism, usually manifest in infancy and should be strongly considered in the infant with hyperkalemia and metabolic acidosis, especially if hyponatremia is present.
It is important to consider the various etiologies of a transcellular K+ shift. In some of these disorders, the K+ level continues to increase, despite the elimination of all K+ intake, especially with concurrent renal insufficiency. This increase is potentially seen in tumor lysis syndrome, hemolysis, rhabdomyolysis, and other causes of cell death. All these entities can cause concomitant hyperphosphatemia and hyperuricemia. Rhabdomyolysis produces an elevated creatinine phosphokinase (CPK) value and hypocalcemia, whereas children with hemolysis have hemoglobinuria and a decreasing hematocrit. For the child with diabetes, elevated blood glucose suggests a transcellular shift of K+ .
Treatment
The plasma K+ level, the ECG, and the risk of the problem worsening determine the aggressiveness of the therapeutic approach. High serum [K+ ] and the presence of ECG changes require vigorous treatment. An additional source of concern is the patient in whom plasma K+ levels are rising despite minimal intake. This situation can happen if there is cellular release of K+ (tumor lysis syndrome), especially in the setting of diminished excretion (renal failure).
The first action in a child with a concerning elevation of plasma [K+ ] is to stop all sources of additional K+ (oral, intravenous). Washed RBCs can be used for patients who require blood transfusions. If the [K+ ] is >6.5 mEq/L, an ECG should be obtained to help assess the urgency of the situation. Peak T waves are the first sign of hyperkalemia, followed by a prolonged PR interval, and when most severe, prolonged QRS complex. Life-threatening ventricular arrhythmias may also develop. The treatment of hyperkalemia has 2 basic goals: (1) to stabilize the heart to prevent life-threatening arrhythmias and (2) to remove K+ from the body. The treatments that acutely prevent arrhythmias all have the advantage of working quickly (within minutes) but do not remove K+ from the body. Calcium stabilizes the cell membrane of heart cells, preventing arrhythmias; it is given intravenously over a few minutes, and its action is almost immediate. Calcium should be given over 30 min in a patient receiving digitalis; otherwise the calcium may cause arrhythmias. Bicarbonate causes potassium to move intracellularly, lowering the plasma [K+ ]; it is most efficacious in a patient with a metabolic acidosis. Insulin causes K+ to move intracellularly but must be given with glucose to avoid hypoglycemia. The combination of insulin and glucose works within 30 min. Nebulized albuterol , by stimulation of β1 -adrenergic receptors, leads to rapid intracellular movement of K+ . This has the advantage of not requiring an IV route of administration, allowing it to be given concurrently with the other measures.
It is critical to begin measures that remove K+ from the body. In patients who are not anuric, a loop diuretic increases renal excretion of K+ . A high dose may be required in a patient with significant renal insufficiency. Sodium polystyrene sulfonate (SPS ; Kayexalate) is an exchange resin that is given either rectally or orally. Patiromer is an oral exchange resin for treating hyperkalemia. Some patients require dialysis for acute K+ removal. Dialysis is often necessary if the patient has either severe renal failure or an especially high rate of endogenous K+ release, as is sometimes present with tumor lysis syndrome or rhabdomyolysis. Hemodialysis rapidly lowers plasma [K+ ]. Peritoneal dialysis is not nearly as quick or reliable, but it is usually adequate as long as the acute problem can be managed with medications and the endogenous release of K+ is not high.
Long-term management of hyperkalemia includes reducing intake through dietary changes and eliminating or reducing medications that cause hyperkalemia (see Chapter 550 ). Some patients require medications to increase potassium excretion, such as SPS, patiromer and loop or thiazide diuretics. Some infants with chronic renal failure may need to start dialysis to allow adequate caloric intake without hyperkalemia. It is unusual for an older child to require dialysis principally to control chronic hyperkalemia. The disorders caused by aldosterone deficiency respond to replacement therapy with fludrocortisone.
Hypokalemia
Hypokalemia is common in children, with most cases related to gastroenteritis.
Etiology and Pathophysiology
There are 4 basic mechanisms of hypokalemia (Table 68.5 ). Spurious hypokalemia occurs in patients with leukemia and very elevated WBC counts if plasma for analysis is left at room temperature, permitting the WBCs to take up K+ from the plasma. With a transcellular shift, there is no change in total body K+ , although there may be concomitant potassium depletion resulting from other factors. Decreased intake, extrarenal losses, and renal losses are all associated with total body K+ depletion.
Because the intracellular [K+ ] is much higher than the plasma level, a significant amount of K+ can move into cells without greatly changing the intracellular [K+ ]. Alkalemia is one of the more common causes of a transcellular shift. The effect is much greater with a metabolic alkalosis than with a respiratory alkalosis. The impact of exogenous insulin on K+ movement into the cells is substantial in patients with DKA. Endogenous insulin may be the cause when a patient is given a bolus of glucose. Both endogenous (epinephrine in stress) and exogenous (albuterol) β-adrenergic agonists stimulate cellular uptake of K+ . Theophylline overdose, barium intoxication, administration of cesium chloride (a homeopathic cancer remedy), and toluene intoxication from paint or glue sniffing can cause a transcellular shift hypokalemia, often with severe clinical manifestations. Children with hypokalemic periodic paralysis, a rare autosomal dominant disorder, have acute cellular uptake of K+ (see Chapter 629 ). Thyrotoxic periodic paralysis, which is more common in Asians, is an unusual initial manifestation of hyperthyroidism. Affected patients have dramatic hypokalemia as a result of a transcellular shift of potassium. Hypokalemia can occur during refeeding syndrome (see Chapters 58 and 364.8 ).
Inadequate K+ intake occurs in anorexia nervosa ; accompanying bulimia and laxative or diuretic abuse exacerbates the K+ deficiency. Sweat losses of K+ can be significant during vigorous exercise in a hot climate. Associated volume depletion and hyperaldosteronism increase renal losses of K+ (discussed later). Diarrheal fluid has a high concentration of K+ , and hypokalemia as a result of diarrhea is usually associated with metabolic acidosis resulting from stool losses of bicarbonate. In contrast, normal acid-base balance or mild metabolic alkalosis is seen with laxative abuse. Intake of SPS or ingestion of clay because of pica increases stool losses of potassium.
Urinary potassium wasting may be accompanied by a metabolic acidosis (proximal or distal RTA). In DKA, although it is often associated with normal plasma [K+ ] from transcellular shifts, there is significant total body K+ depletion from urinary losses because of the osmotic diuresis, and the K+ level may decrease dramatically with insulin therapy (see Chapter 607 ). Both the polyuric phase of acute tubular necrosis and postobstructive diuresis cause transient, highly variable K+ wasting and may be associated with metabolic acidosis. Tubular damage, which occurs either directly from medications or secondary to interstitial nephritis, is often accompanied by other tubular losses, including magnesium, Na+ , and water. Such tubular damage may cause a secondary RTA with metabolic acidosis. Isolated magnesium deficiency causes renal K+ wasting. Penicillin is an anion excreted in the urine, resulting in increased K+ excretion because the penicillin anion must be accompanied by a cation. Hypokalemia from penicillin therapy occurs only with the sodium salt of penicillin, not with the potassium salt.
Urinary K+ wasting is often accompanied by a metabolic alkalosis. This condition is usually associated with increased aldosterone, which increases urinary K+ and acid losses, contributing to the hypokalemia and the metabolic alkalosis. Other mechanisms often contribute to both the K+ losses and the metabolic alkalosis. With emesis or nasogastric suction, there is gastric loss of K+ , but this is fairly minimal, given the low K+ content of gastric fluid, approximately 10 mEq/L. More important is the gastric loss of hydrochloric acid (HCl), leading to metabolic alkalosis and a state of volume depletion. The kidney compensates for metabolic alkalosis by excreting bicarbonate in the urine, but there is obligate loss of K+ and Na+ with the bicarbonate. The volume depletion raises aldosterone levels, further increasing urinary K+ losses and preventing correction of metabolic alkalosis and hypokalemia until the volume depletion is corrected.
Urinary chloride (Cl− ) is low as a response to the volume depletion. Because the volume depletion is secondary to Cl− loss, this is a state of Cl − deficiency . There were cases of Cl− deficiency resulting from infant formula deficient in Cl- , which caused a metabolic alkalosis with hypokalemia and low urine [Cl− ]. Current infant formula is not deficient in Cl− . A similar mechanism occurs in cystic fibrosis because of Cl− loss in sweat. In congenital chloride-losing diarrhea , an autosomal recessive disorder, there is high stool loss of Cl− , leading to metabolic alkalosis, an unusual sequela of diarrhea. Because of stool K+ losses, Cl− deficiency, and metabolic alkalosis, patients with this disorder have hypokalemia. During respiratory acidosis, there is renal compensation, with retention of bicarbonate and excretion of Cl− . After the respiratory acidosis is corrected, the patients have Cl− deficiency and post–hypercapnic alkalosis with secondary hypokalemia. Patients with Cl− deficiency, metabolic alkalosis, and hypokalemia have a urinary [Cl− ] of <10 mEq/L. Loop and thiazide diuretics lead to hypokalemia, metabolic alkalosis, and Cl− deficiency. During treatment, these patients have high urine chloride levels resulting from the effect of the diuretic. However, after the diuretics are discontinued, there is residual Cl− deficiency, the urinary [Cl− ] is appropriately low, and neither the hypokalemia nor the alkalosis resolves until the Cl− deficiency is corrected.
The combination of metabolic alkalosis, hypokalemia, high urine [Cl− ], and normal blood pressure is characteristic of Bartter syndrome, Gitelman syndrome, and current diuretic use. Patients with any of these conditions have high urinary losses of Cl− despite a state of relative volume depletion with secondary hyperaldosteronism (high plasma renin). Bartter and Gitelman syndromes are autosomal recessive disorders caused by defects in tubular transporters (see Chapter 549 ). Bartter syndrome is usually associated with hypercalciuria, and often with nephrocalcinosis, whereas children with Gitelman syndrome have low urinary calcium losses but hypomagnesemia because of urinary magnesium losses. Some patients with Bartter syndrome have hypomagnesemia. A transient antenatal form of Bartter syndrome is associated with severe polyhydramnios and mutations in MAGED2 .
Some patients with hypoparathyroidism and hypocalcemia caused by an activating mutation of the calcium-sensing receptor (autosomal dominant hypoparathyroidism ) have hypokalemia, hypomagnesemia, and metabolic alkalosis. The reason is that activation of the calcium-sensing receptor in the loop of Henle impairs tubular resorption of sodium and chloride, causing volume depletion and secondary hyperaldosteronism. EAST syndrome , an autosomal recessive disorder caused by mutations in the gene for a potassium channel in the kidney, inner ear, and brain, consists of e pilepsy, a taxia, s ensorineural hearing loss, and t ubulopathy (hypokalemia, metabolic alkalosis, hypomagnesemia, and hypocalciuria).
In the presence of high aldosterone levels, there is urinary loss of K+ , hypokalemia, metabolic alkalosis, and elevated urinary [Cl− ]. Also, renal retention of Na+ leads to hypertension. Primary hyperaldosteronism caused by adenoma or hyperplasia is much less common in children than in adults (see Chapters 597 and 598 ). Glucocorticoid-remediable aldosteronism , an autosomal dominant disorder that leads to high levels of aldosterone (but low renin levels), is often diagnosed in childhood, although hypokalemia is not always present.
Increased aldosterone levels may be secondary to increased renin production. Renal artery stenosis leads to hypertension from increased renin and secondary hyperaldosteronism. The increased aldosterone can cause hypokalemia and metabolic alkalosis, although most patients have normal electrolyte levels. Renin-producing tumors, which are extremely rare, can cause hypokalemia.
A variety of disorders cause hypertension and hypokalemia without increased aldosterone levels. Some are a result of increased levels of mineralocorticoids other than aldosterone. Such increases occur in two forms of congenital adrenal hyperplasia (see Chapter 594 ). In 11β-hydroxylase deficiency , which is associated with virilization, the value of 11-deoxycorticosterone is elevated, causing variable hypertension and hypokalemia. A similar mechanism, increased 11-deoxycorticosterone, occurs in 17α-hydroxylase deficiency , but patients with this disorder are more uniformly hypertensive and hypokalemic, and they have a defect in sex hormone production. Cushing syndrome , frequently associated with hypertension, less frequently causes metabolic alkalosis and hypokalemia, secondary to the mineralocorticoid activity of cortisol. In 11β-hydroxysteroid dehydrogenase deficiency , an autosomal recessive disorder, the enzymatic defect prevents the conversion of cortisol to cortisone in the kidney. Because cortisol binds to and activates the aldosterone receptor, children with this deficiency have all the features of excessive mineralocorticoids, including hypertension, hypokalemia, and metabolic alkalosis. Patients with this disorder, which is also called apparent mineralocorticoid excess , respond to spironolactone therapy, which blocks the mineralocorticoid receptor. An acquired form of 11β-hydroxysteroid dehydrogenase deficiency occurs from the ingestion of substances that inhibit this enzyme. A classic example is glycyrrhizic acid, which is found in natural licorice. Liddle syndrome is an autosomal dominant disorder that results from an activating mutation of the distal nephron sodium channel that is normally upregulated by aldosterone. Patients have the characteristics of hyperaldosteronism—hypertension, hypokalemia, and alkalosis—but low serum renin and aldosterone levels. These patients respond to the potassium-sparing diuretics (triamterene and amiloride) that inhibit this sodium channel (see Chapter 549.3 ). Hypertension exacerbated by pregnancy and syndrome of apparent mineralocortical excess are associated with hypokalemia and low renin levels.
Clinical Manifestations
The heart and skeletal muscle are especially vulnerable to hypokalemia. ECG changes include a flattened T wave, a depressed ST segment, and the appearance of a U wave, which is located between the T wave (if still visible) and the P wave. Ventricular fibrillation and torsades de pointes may occur, although usually only in the context of underlying heart disease. Hypokalemia makes the heart especially susceptible to digitalis-induced arrhythmias, such as supraventricular tachycardia, ventricular tachycardia, and heart block (see Chapter 462 ).
The clinical consequences of hypokalemia in skeletal muscle include muscle weakness and cramps. Paralysis is a possible complication, generally only at [K+ ] <2.5 mEq/L. It usually starts in the legs and moves to the arms. Respiratory paralysis may require mechanical ventilation. Some patients have rhabdomyolysis; the risk increases with exercise. Hypokalemia slows GI motility. This effect manifests as constipation; with K+ levels <2.5 mEq/L, an ileus may occur. Hypokalemia impairs bladder function, potentially leading to urinary retention.
Hypokalemia causes polyuria and polydipsia by impairing urinary concentrating ability, which produces nephrogenic diabetes insipidus. Hypokalemia stimulates renal ammonia production, an effect that is clinically significant if hepatic failure is present, because the liver cannot metabolize the ammonia. Consequently, hypokalemia may worsen hepatic encephalopathy. Chronic hypokalemia may cause kidney damage, including interstitial nephritis and renal cysts.
Diagnosis
Most causes of hypokalemia are readily apparent from the history. It is important to review the child's diet, GI losses, and medications. Both emesis and diuretic use can be surreptitious. The presence of hypertension suggests excess mineralocorticoid effects or levels. Concomitant electrolyte abnormalities are useful clues. The combination of hypokalemia and metabolic acidosis is characteristic of diarrhea and distal and proximal RTA. A concurrent metabolic alkalosis is characteristic of emesis or nasogastric losses, aldosterone excess, use of diuretics, and Bartter and Gitelman syndromes. Fig. 68.4 shows an approach to persistent hypokalemia.

If a clear etiology is not apparent, the measurement of urinary K+ distinguishes between renal and extrarenal losses. The kidneys should conserve K+ in the presence of extrarenal losses. Urinary K+ losses can be assessed with a 24 hr urine collection, spot K+ :creatinine ratio, fractional excretion of K+ , or calculation of the transtubular K+ gradient (TTKG), which is the most widely used approach in children:
TTKG=[K]urine/[K]plasma×(plasma osmolality/urine osmolality)
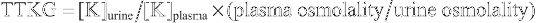
where [K]urine = urine potassium concentration and [K]plasma = plasma potassium concentration.
The urine osmolality must be greater than the serum osmolality for the result of this calculation to be valid. A TTKG >4 in the presence of hypokalemia suggests excessive urinary losses of K+ . The urinary K+ excretion value can be misleading if the stimulus for renal loss, such as a diuretic, is no longer present.
Treatment
Factors that influence the treatment of hypokalemia include the K+ level, clinical symptoms, renal function, the presence of transcellular shifts of K+ , ongoing losses, and the patient's ability to tolerate oral K+ . Severe, symptomatic hypokalemia requires aggressive treatment. Supplementation is more cautious if renal function is decreased because of the kidney's limited ability to excrete excessive K+ . The plasma potassium level does not always provide an accurate estimation of the total body K+ deficit because there may be shifts of K+ from the ICS to the plasma. Clinically, such shifts occur most often with metabolic acidosis and the insulin deficiency of DKA; the plasma [K+ ] measurement underestimates the degree of total body K+ depletion. When these problems are corrected, K+ moves into the ICS, so more K+ supplementation is required to correct the hypokalemia. Likewise, the presence of a transcellular shift of K+ into the cells indicates that the total body K+ depletion is less severe. In an isolated transcellular shift, as in hypokalemic periodic paralysis, K+ supplementation should be used cautiously, given the risk of hyperkalemia when the transcellular shift resolves. This caution is especially required in thyrotoxic periodic paralysis, which responds dramatically to propranolol, with correction of weakness and hypokalemia. Patients who have ongoing losses of K+ need correction of the deficit and replacement of the ongoing losses.
Because of the risk of hyperkalemia, intravenous K+ should be used very cautiously. Oral K+ is safer, but not as rapid in urgent situations. Liquid preparations are bitter tasting; microencapsulated or wax matrix formulations are less irritating than tablets to the gastric mucosa. Oral dosing is variable depending on the clinical situation. A typical starting dose is 1-2 mEq/kg/day, with a maximum of 60 mEq/day in divided doses. The dose of IV potassium is 0.5-1.0 mEq/kg, usually given over 1 hr. The adult maximum dose is 40 mEq. Conservative dosing is generally preferred. Potassium chloride is the usual choice for supplementation, although the presence of concurrent electrolyte abnormalities may dictate other options. Patients with acidosis and hypokalemia can receive potassium acetate or potassium citrate. If hypophosphatemia is present, some of the potassium deficit can be replaced with potassium phosphate. It is sometimes possible to decrease ongoing K+ losses. For patients with excessive urinary losses, potassium-sparing diuretics are effective, but they need to be used cautiously in patients with renal insufficiency. If hypokalemia, metabolic alkalosis, and volume depletion are present (with gastric losses), restoration of intravascular volume with adequate NaCl will decrease urinary K+ losses. Correction of concurrent hypomagnesemia is important because it may cause hypokalemia. Disease-specific therapy is effective in many of the genetic tubular disorders.
Bibliography
Potassium
Emma F, Pizzini C, Tessa A, et al. “Bartter-like” phenotype in Kearns-Sayre syndrome. Pediatr Nephrol . 2006;21:355–360.
Gumz ML, Rabinowitz L, Wingo CS. An integrated view of potassium homeostasis. N Engl J Med . 2015;373:60–72.
Jain G, Ong S, Warnock DG. Genetic disorders of potassium homeostasis. Semin Nephrol . 2013;33(3):300–309.
Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilcate on potassium lowering for 28 days among outpatients with hyperkalemia. JAMA . 2014;312(21):2223–2233.
Lee AC, Reduque LL, Luban NL, et al. Transfusion-associated hyperkalemic cardiac arrest in pediatric patients receiving massive transfusion. Transfusion (Paris) . 2014;54:244–254.
Matsunoshita N, Nozu K, Shono A, et al. Differential diagnosis of Bartter syndrome, Gitelman syndrome, and pseudo-Bartter/Gitelman syndrome based on clinical characteristics. Genet Med . 2016;18:180–188.
Meng QH, Wagar EA. Pseudohyperkalemia: A new twist on an old phenomenon. Crit Rev Clin Lab Sci . 2015;52:45–55.
Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med . 2015;372(23):222–231.
Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol . 2015;10:1050–1060.
Sterns RH, Grieff M, Bernstein PL. Treatment of hyperkalemia: something old, something new. Kidney Int . 2016;89:546–554.
Suarez-Rivera M, Bonilla-Felix M. Fluid and electrolyte disorders in the newborn: sodium and potassium. Curr Pediatr Rev . 2014;10:115–122.
Weir MR, Bakris GL, Bushinsky DA, et al. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med . 2015;372(3):211–220.
Zieg J, Gonsorcikova L, Landau D. Current views on the diagnosis and management of hypokalaemia in children. Acta Paediatr . 2016;105:762–772.
Magnesium
Larry A. Greenbaum
Magnesium Metabolism
Body Content and Physiologic Function
Magnesium is the 4th most common cation in the body and the 3rd most common intracellular cation (see Fig. 68.3 ). From 50–60% of body magnesium is in bone, where it serves as a reservoir because 30% is exchangeable, allowing movement to the ECS. Most intracellular magnesium is bound to proteins; only approximately 25% is exchangeable. Because cells with higher metabolic rates have higher magnesium concentrations, most intracellular magnesium is present in muscle and liver.
The normal plasma magnesium concentration is 1.5-2.3 mg/dL (1.2-1.9 mEq/L; 0.62-0.94 mmol/L), with some variation among clinical laboratories. Infants have slightly higher plasma magnesium concentrations than older children and adults. Only 1% of body magnesium is extracellular (60% ionized; 15% complexed; 25% protein bound). In the United States, serum magnesium is reported as mg/dL (Table 68.6 ). Values in the left-column unit are converted into the right-column unit by multiplying the conversion factor (e.g., calcium of 10 mg/dL × 0.25 = 2.5 mmol/L). Dividing the right-column unit by the conversion factor converts to the units of the left-column unit.
Table 68.6
Conversion Factors for Calcium, Magnesium, and Phosphorus
| UNIT | CONVERSION FACTOR | UNIT | |
|---|---|---|---|
| Calcium | mg/dL | 0.25 | mmol/L |
| mEq/L | 0.5 | mmol/L | |
| mg/dL | 0.5 | mEq/L | |
| Magnesium | mg/dL | 0.411 | mmol/L |
| mEq/L | 0.5 | mmol/L | |
| mg/dL | 0.822 | mEq/L | |
| Phosphorus | mg/dL | 0.32 | mmol/L |
Magnesium is a necessary cofactor for hundreds of enzymes. It is important for membrane stabilization and nerve conduction. Adenosine triphosphate (ATP) and guanosine triphosphate need associated magnesium when they are used by ATPases, cyclases, and kinases.
Magnesium Intake
Between 30% and 50% of dietary magnesium is absorbed. Good dietary sources include green vegetables, cereals, nuts, meats, and hard water, although many foods contain magnesium. Human milk contains approximately 35 mg/L of magnesium; formula contains 40-70 mg/L. The small intestine is the major site of magnesium absorption, but the regulation of magnesium absorption is poorly understood. There is passive absorption, which permits high absorption in the presence of excessive intake. It probably occurs by a paracellular mechanism. Absorption is diminished in the presence of substances that complex with magnesium (free fatty acids, fiber, phytate, phosphate, oxalate); increased intestinal motility and calcium also decrease magnesium absorption. Vitamin D and parathyroid hormone (PTH) may enhance absorption, although this effect is limited. Intestinal absorption does increase when intake is decreased, possibly by a saturable, active transport system. If there is no oral intake of magnesium, obligatory secretory losses prevent the complete elimination of intestinal losses.
Magnesium Excretion
Renal excretion is the principal regulator of magnesium balance. There is no defined hormonal regulatory system, although PTH may increase tubular resorption. Approximately 15% of resorption occurs in the proximal tubule, and 70% in the thick ascending limb (TAL) of the loop of Henle. Proximal resorption may be higher in neonates. High serum magnesium levels inhibit resorption in the TAL, suggesting that active transport is involved. Approximately 5–10% of filtered magnesium is resorbed in the distal tubule. Hypomagnesemia increases absorption in the TAL and the distal tubule.
Hypomagnesemia
Hypomagnesemia is relatively common in hospitalized patients, although most cases are asymptomatic. Detection requires a high index of suspicion because magnesium is not measured in most basic metabolic panels.
Etiology and Pathophysiology
Gastrointestinal and renal losses are the major causes of hypomagnesemia (Table 68.7 ). Diarrheal fluid contains up to 200 mg/L of magnesium; gastric contents have only approximately 15 mg/L, but high losses can cause depletion. Steatorrhea causes magnesium loss as a result of the formation of magnesium-lipid salts; restriction of dietary fat can decrease losses. The potassium-lowering agent patiromer binds magnesium and may cause hypomagnesemia.
Hypomagnesemia with secondary hypocalcemia , a rare autosomal recessive disorder, is caused by decreased intestinal absorption of magnesium and renal magnesium wasting. Patients with this disorder have mutations in a gene expressed in intestine and kidney; TRPM6 codes for a transient receptor potential cation channel. The patients have seizures, tetany, tremor, or restlessness at 2-8 wk of life as a result of severe hypomagnesemia (0.2-0.8 mg/dL) and secondary hypocalcemia.
Renal losses may occur because of medications that are direct tubular toxins. Amphotericin frequently causes significant magnesium wasting and is typically associated with other tubular defects (especially potassium wasting). Cisplatin produces dramatic renal magnesium losses. Diuretics affect tubular handling of magnesium. Loop diuretics cause a mild increase in magnesium excretion, and thiazide diuretics have even less effect. Chronic use of proton pump inhibitors may cause hypomagnesemia. Potassium-sparing diuretics reduce magnesium losses. Osmotic agents, such as mannitol, glucose in diabetes mellitus, and urea in the recovery phase of acute tubular necrosis, increase urinary magnesium losses. Epidermal growth factor (EGF) receptor inhibitors cause renal magnesium wasting. IV fluid, by expanding the intravascular volume, decreases renal resorption of sodium and water, thereby impairing magnesium resorption. Hypercalcemia inhibits magnesium resorption in the loop of Henle, although this inhibition does not occur in hypercalcemia caused by familial hypercalcemic hypocalciuria or lithium.
A number of rare genetic diseases cause renal magnesium loss. Gitelman and Bartter syndromes, both autosomal recessive disorders, are the most common entities (see Chapter 549 ). Gitelman syndrome , caused by a defect in the thiazide-sensitive Na+ -Cl− co-transporter in the distal tubule, is usually associated with hypomagnesemia. Hypomagnesemia occurs in a minority of patients with Bartter syndrome , which can be caused by mutations in multiple genes necessary for Na+ and Cl− reabsorption in the loop of Henle. In both disorders there is hypokalemic metabolic alkalosis. Typically, hypomagnesemia is not severe and is asymptomatic, although tetany as a result of hypomagnesemia occasionally occurs.
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (Michelis-Castrillo syndrome) , an autosomal recessive disorder, is caused by mutations in the gene for claudin 16 (paracellin-1), located in the tight junctions of the TAL of the loop of Henle. Patients with the disease have severe renal wasting of magnesium and calcium with secondary hypomagnesemia and nephrocalcinosis; serum calcium levels are normal. Chronic renal failure frequently occurs during childhood. Other features include kidney stones, urinary tract infections, hematuria, increased PTH levels, tetany, seizures, incomplete distal RTA, hyperuricemia, polyuria, and polydipsia. Patients with familial hypomagnesemia with hypercalciuria, nephrocalcinosis, and severe ocular involvement have mutations in the gene for claudin 19.
Autosomal recessive renal magnesium wasting with normocalciuria is caused by mutations in the EGF gene. Clinical manifestations include seizures, mild to moderate psychomotor retardation, and brisk tendon reflexes.
Autosomal dominant renal magnesium wasting is caused by mutations in a number of different genes. A dominant-negative mutation in the gene encoding the Na+ ,K+ -ATPase γ subunit is associated with hypomagnesemia, increased urinary magnesium losses, hypocalciuria, and normocalcemia. Patients may present with seizures; most are asymptomatic, despite serum magnesium levels of 0.8-1.5 mg/dL. Mutations in CNNM2 , which encodes a protein that mediates magnesium-sensitive sodium currents, cause isolated hypomagnesemia. A mutation in KCNA1 , a gene that encodes a K+ channel, also causes an autosomal dominant form of hypomagnesemia; symptoms may be severe.
Renal cysts and diabetes syndrome , which is caused by mutations in the gene for hepatocyte nuclear factor-1β, is associated with hypomagnesemia, despite the frequent presence of renal insufficiency. The hypomagnesemia is usually mild but may cause symptomatic hypocalcemia. EAST syndrome is caused by mutations in a potassium channel, and patients with this autosomal recessive disorder have hypokalemia, metabolic alkalosis, and hypomagnesemia. Autosomal dominant hypoparathyroidism is caused by an activating mutation in the calcium-sensing receptor, which also senses magnesium levels in the kidney (see Chapter 589 ). The mutated receptor inappropriately perceives that magnesium and calcium levels are elevated, leading to urinary wasting of both cations. Hypomagnesemia, if present, is usually mild. A mutation in a mitochondrially encoded transfer RNA is associated with hypomagnesemia, hypertension, and hypercholesterolemia. Hypomagnesemia is occasionally present in children with other mitochondrial disorders.
Poor intake is an unusual cause of hypomagnesemia, although it can be seen in children who are hospitalized and receive only IV fluids without magnesium. In hungry bone syndrome , which most frequently occurs after parathyroidectomy in patients with hyperparathyroidism, magnesium moves into bone as a result of accelerated bone formation. These patients usually have hypocalcemia and hypophosphatemia through the same mechanism. A similar mechanism can occur during the refeeding phase of protein-calorie malnutrition in children, with high magnesium use during cell growth depleting the patient's limited reserves. Insulin therapy stimulates uptake of magnesium by cells, and in DKA, in which total body magnesium is low because of osmotic losses, hypomagnesemia frequently occurs. In pancreatitis there is saponification of magnesium and calcium in necrotic fat, causing both hypomagnesemia and hypocalcemia.
Transient hypomagnesemia in newborns , which is sometimes idiopathic, is more common in infants of diabetic mothers, presumably as a result of maternal depletion from osmotic losses. Other maternal diseases that cause magnesium losses predispose infants to hypomagnesemia. Hypomagnesemia is more common in infants with intrauterine growth restriction. Hypomagnesemia may develop in newborn infants who require exchange transfusions because of magnesium removal by the citrate in banked blood.
Clinical Manifestations
Hypomagnesemia causes secondary hypocalcemia by impairing the release of PTH by the parathyroid gland and through blunting of the tissue response to PTH. Thus, hypomagnesemia is part of the differential diagnosis of hypocalcemia. It usually occurs only at magnesium levels <0.7 mg/dL. The dominant manifestations of hypomagnesemia are caused by hypocalcemia: tetany, presence of Chvostek and Trousseau signs, and seizures. However, with severe hypomagnesemia, these same signs and symptoms may be present despite normocalcemia. Persistent hypocalcemia caused by hypomagnesemia is a rare cause of rickets.
Many causes of hypomagnesemia also result in hypokalemia. Hypomagnesemia may produce renal potassium wasting and hypokalemia that corrects only with magnesium therapy. ECG changes with hypomagnesemia include flattening of the T wave and lengthening of the ST segment. Arrhythmias may occur, almost always in the setting of underlying heart disease.
Diagnosis
The etiology of hypomagnesemia is often readily apparent from the clinical situation. The child should be assessed for GI disease, adequate intake, and kidney disease, with close attention paid to medications that may cause renal magnesium wasting. When the diagnosis is uncertain, an evaluation of urinary magnesium losses distinguishes between renal and nonrenal causes. The fractional excretion of magnesium (FEMg ) is calculated via the following formula:
FEMg=(UMg×PCr)/([0.7×PMg]×UCr)×100
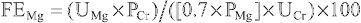
where UMg = urinary magnesium concentration, PCr = plasma creatinine concentration, PMg = plasma magnesium concentration, and UCr = urinary magnesium concentration. The plasma magnesium concentration is multiplied by 0.7 because approximately 30% is bound to albumin and not filtered at the glomerulus.
The FEMg does not vary with age, but it does change according to the serum magnesium concentration. The FEMg ranges from 1–8% in children with normal magnesium levels. In the patient with hypomagnesemia as a result of extrarenal causes, FEMg should be low because of renal conservation, typically <2%. The FEMg is inappropriately elevated in the setting of renal magnesium wasting; values are usually >4% and frequently >10%. The measurement should not be made during a magnesium infusion, because the acute increase in serum magnesium increases urinary magnesium. Other approaches for evaluating urinary magnesium losses include calculation of 24 hr urinary magnesium losses and the urine magnesium/creatinine ratio, both of which vary with age.
The genetic causes of renal magnesium loss are distinguished on the basis of the measurement of other serum and urinary electrolytes. Children with Gitelman or Bartter syndrome have hypokalemia and metabolic alkalosis.
Treatment
Severe hypomagnesemia is treated with parenteral magnesium. Magnesium sulfate is given at a dose of 25-50 mg/kg (0.05-0.1 mL/kg of a 50% solution; 2.5-5.0 mg/kg of elemental magnesium). It is administered as a slow IV infusion, although it may be given intramuscularly in neonates. The rate of IV infusion should be slowed if a patient experiences diaphoresis, flushing, or a warm sensation. The dose is often repeated every 6 hr (every 8-12 hr in neonates), for a total of 2-3 doses, before the plasma magnesium concentration is rechecked. Lower doses are used in children with renal insufficiency.
Long-term therapy is usually given orally. Preparations include magnesium gluconate (5.4 mg elemental magnesium/100 mg), magnesium oxide (60 mg elemental magnesium/100 mg), and magnesium sulfate (10 mg elemental magnesium/100 mg). Sustained-release preparations include Slow-Mag (60 mg elemental magnesium/tablet) and Mag-Tab SR (84 mg elemental magnesium/tablet). Oral magnesium dosing should be divided to decrease cathartic side effects. Alternatives to oral magnesium are intramuscular injections and nighttime nasogastric infusion, both designed to minimize diarrhea. Magnesium supplementation must be used cautiously in the context of renal insufficiency.
Hypermagnesemia
Clinically significant hypermagnesemia is almost always secondary to excessive intake. It is unusual, except in neonates born to mothers who are receiving IV magnesium for preeclampsia or eclampsia (see Chapter 119.5 ).
Etiology and Pathophysiology
There is no feedback mechanism to prevent magnesium absorption from the GI tract. Magnesium is present in high amounts in certain laxatives, enemas, cathartics used to treat drug overdoses, and antacids. It is also usually present in total parenteral nutrition (TPN), and neonates may receive high amounts transplacentally if maternal levels are elevated. Usually the kidneys excrete excessive magnesium, but this ability is diminished in patients with chronic renal failure. In addition, neonates and young infants are vulnerable to excessive magnesium ingestion because of their reduced GFR. Most pediatric cases not related to maternal hypermagnesemia occur in infants as a result of excessive use of antacids or laxatives. Mild hypermagnesemia may occur in chronic renal failure, familial hypocalciuric hypercalcemia, DKA, lithium ingestion, milk-alkali syndrome, and tumor lysis syndrome. The hypermagnesemia in DKA occurs despite significant intracellular magnesium depletion as a result of urinary losses; hypomagnesemia often occurs after insulin treatment.
Clinical Manifestations
Symptoms usually do not appear until the plasma magnesium level is >4.5 mg/dL. Hypermagnesemia inhibits acetylcholine release at the neuromuscular junction, producing hypotonia, hyporeflexia, and weakness; paralysis occurs at high concentrations. The neuromuscular effects may be exacerbated by aminoglycoside antibiotics. Direct CNS depression causes lethargy and sleepiness; infants have a poor suck. Elevated magnesium values are associated with hypotension because of vascular dilation, which also causes flushing. Hypotension can be profound at higher concentrations from a direct effect on cardiac function. ECG changes include prolonged PR interval, QRS complex, and QT interval. Severe hypermagnesemia (>15 mg/dL) causes complete heart block and cardiac arrest. Other manifestations of hypermagnesemia include nausea, vomiting, and hypocalcemia.
Diagnosis
Except for the case of the neonate with transplacental exposure, a high index of suspicion and a good history are necessary to make the diagnosis of hypermagnesemia. Prevention is essential; magnesium-containing compounds should be used judiciously in children with renal insufficiency.
Treatment
Most patients with normal renal function rapidly clear excess magnesium. Intravenous hydration and loop diuretics can accelerate this process. In severe cases, especially in patients with underlying renal insufficiency, dialysis may be necessary. Hemodialysis works faster than peritoneal dialysis. Exchange transfusion is another option in newborn infants. Supportive care includes monitoring of cardiorespiratory status, provision of fluids, monitoring of electrolyte levels, and the use of pressors for hypotension. In acute emergencies, especially in the context of severe neurologic or cardiac manifestations, 100 mg/kg of IV calcium gluconate is transiently effective.
Bibliography
Magnesium
Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol . 2015;10:1257–1272.
Costello R, Wallace TC, Rosanoff A. Magnesium. Adv Nutr . 2016;7:199–201.
Huang JW, Famure O, Li Y, et al. Hypomagnesemia and the risk of new-onset diabetes mellitus after kidney transplantation. J Am Soc Nephrol . 2016;27:1793–1800.
Janett S, Bianchetti MG, Milani GP, et al. Hypomagnesemia following prolonged use of proton-pump inhibitors. J Pediatr Gastroenterol Nutr . 2016;62:e39.
Jiang L, Chen C, Yuan T, et al. Clinical severity of Gitelman syndrome determined by serum magnesium. Am J Nephrol . 2014;39:357–366.
Konrad M, Schlingmann KP. Inherited disorders of renal hypomagnesaemia. Nephrol Dial Transplant . 2014;29(Suppl 4):iv63–iv71.
Tilman M, Wolf F. Inherited and acquired disorders of magnesium homeostasis. Curr Opin Pediatr . 2017;29:187–198.
Van der Made CI, Hoorn EJ, de la Faille R, et al. Hypomagnesemia as first clinical manifestation of ADTKD-HNF1B: A case series and literature review. Am J Nephrol . 2015;42:85–90.
William JH, Danziger J. Magnesium deficiency and proton-pump inhibitor use: a clinical review. J Clin Pharmacol . 2016;56:660–668.