Chapter 5
The Spark of Life
Sometime in the third century BCE, the Greek philosopher Aristotle (382–322 BCE) enunciated the theory of spontaneous generation. Synthesizing the work of earlier scholars, Aristotle argued that life could arise spontaneously from nonliving matter provided the latter was suffused with pneuma, or “vital heat.” After all, was it not obvious that maggots arose spontaneously in spoiled meat? This idea, as with so many of Aristotle’s claims, held fast through the Middle Ages, when it was still widely held that old rags mixed with wheat could give rise to fully formed adult mice.
One of the first to question spontaneous generation was the Tuscan (this being before Italy was “a thing”) naturalist Francesco Redi (1626–1697). Often called “the father of experimental biology,” Redi showed that maggots only appear in meat that has been exposed to adult flies; cover the opening of a meat-containing jar with fine gauze, and maggots no longer “form.” A hundred years later, in 1765, Redi’s studies were extended by the Italian Lazzaro Spallanzani (1729–1799), who used a microscope, unavailable in Redi’s time, to show that even microscopic organisms do not spontaneously form in broth that has been sterilized by boiling and left sealed and out of contact with air.
Redi’s observations, however, were not the final word on spontaneous generation. In his experiments, for example, he kept his flasks sealed—what if some component in the air was necessary for spontaneous generation to occur? Interest in this question grew over the next century until, in 1859, the French Academy of Sciences founded a prize of 2,500 francs for the scientist who could finally lay the question to rest by conclusively proving or disproving spontaneous generation. The prize was quickly won by Louis Pasteur (1822–1895), who would later go on to invent immunization, pasteurization, and, more or less, the germ theory of disease.
To clinch the prize, Pasteur placed sugar solutions, urine, and beet juice in swan-shaped flasks whose long, curved necks allowed communication with the air but would prevent, he postulated, any small, living particles from dropping into the liquids with the settling dust. In flasks that he sterilized by boiling, nothing happened. In contrast, the liquid in the unboiled flasks rapidly spoiled. Of course, that could simply have meant that boiling had somehow made the liquid unpalatable for the hypothesized organisms. Decisive proof, though, was provided when Pasteur snapped off the swan-shaped necks, allowing dust to settle into the previously boiled organic brews. The liquid then spoiled, demonstrating that the organisms responsible had settled out of the air and proving that the liquid’s prior sterility reflected the absence of spontaneous generation rather than ruined media that could no longer support life. Pasteur proclaimed, “Never will the doctrine of spontaneous generation recover from the mortal blow of this simple experiment.”*
Pasteur’s conclusions, though, were in conflict with a theory published just three years earlier by a scientist on the other side of the English Channel: Darwin’s theory on the origins of species. Charles Darwin (1809–1882) had obviated the need for God in the creation of species by showing how one species could slowly transmute into another. But Darwin’s theory left open the question of how the very first species arose. At the very least, the first species could not have arisen by the transmutation of some earlier species and thus could not have arisen without some form of spontaneous generation. Darwin seems to have been troubled by the subject, noting in 1866 that “as for myself I cannot believe in spontaneous generation, and though I expect that at some future time the principle of life will be rendered intelligible, at present it seems to me beyond the confines of science.”** Five years later, however, in a letter to his close friend and fellow naturalist Joseph Hooker (1817–1911), Darwin went on to speculate how life might have first arisen: “But if (and oh what a big if) we could conceive in some warm little pond, with all sorts of ammonia and phosphoric salts, light, heat, electricity etcetera present, that a protein compound was chemically formed, ready to undergo still more complex changes.”* More than a century and a half later, though, the details of life’s origins remain one of science’s greatest mysteries.
Panspermia
Within a decade of Pasteur’s publication of his results, several prominent European scientists—most notably the English physicist Lord Kelvin (1824–1907), after whom the absolute temperature scale is named, and the Prussian physicist Hermann von Helmholtz (1821–1894) of, for example, the Gibbs-Helmholtz equation in chemical thermodynamics—suggested a possible work-around. Could we not avoid the difficulty of spontaneous generation if life had originated elsewhere and been transported to the Earth through space? Their early speculations on this, the panspermia hypothesis, from the Greek pan sperma, meaning “all seed,” were fleshed out in great detail in a widely discussed body of work by the Swedish chemist and 1903 Nobel laureate (another example of how winning the prize provides a lot of leeway to pursue wacky ideas) Svante Arrhenius (1859–1927), who was already famous for relating the temperature dependence of chemical reaction rates. Originally in a 1903 journal article, then expanded in a popular book some five years later, Arrhenius argued that life, in the form of hardy, dormant spores, could survive in the cold dark vacuum of space for long enough to be transferred between the stars.
In Arrhenius’s version of events, bacterial spores could escape from the upper atmosphere of their home planet and be launched into interstellar space by the pressure of light; photons have momentum and thus can accelerate something as small as a bacterial spore to quite high speeds. Eventually, some of the spores would fall upon another planet (Arrhenius estimated that sunlight could push a bacterial spore from the Earth to Mars in 20 days, and from the Sun to the nearest star in as little as 9,000 years) and inoculate the virgin world.
In the century after he first published his work, others have explored Arrhenius’s hypothesis in quantitative detail. The well-known astronomer Carl Sagan, for example, calculated that, to leave the Solar System, a spaceborne spore must be less than 0.5 µm (a micrometer is a millionth of a meter) in diameter—any larger and light pressure, which depends on cross section (and thus goes with the square of radius), will lose out against the Sun’s gravitational pull, which depends on mass (and thus goes with the cube of radius). Only the very smallest of Earth’s bacteria, however, are this small. Sagan also described the various types of stars that spores could be blown from and to—from bright stars, which endow the spores with significant speed, to the solar systems of dimmer stars. He could not, however, come up with a solution to the vexing problem of radiation. Most Terrestrial organisms are rather quickly destroyed if exposed directly to the hazards of space; while the lichen species Rhizocarpon geographicum and Xanthoria elegans were once revived after a couple of weeks above the Earth’s atmosphere (in an experimental holder strapped to the outside of a Russian spacecraft), most of the organisms they shared their vessel with were quickly killed by the Sun’s raw UV light. That said, if protected from this by a layer of clay, spores of the bacterium Bacillus subtilis have been revived even after several years in space.
Putting aside our concerns regarding the viability of spores after millennia-long interstellar trips sans spacecraft, there is a deeper problem with the panspermia hypothesis: while it might explain the origins of life on Earth, it does not answer the more fundamental question of how life arose from inanimate matter in the first place. And thus, even if life could survive a trip between the stars, we astrobiologists can’t sweep the mystery of life’s origins under the rug simply by saying it took place elsewhere. Arrhenius himself sidestepped the issue of how life arose in the first place by suggesting that it might be eternal; after all, at the time, it was assumed that the Universe was immortal, so why couldn’t life have always existed as well? This argument was raised again half a century later by Fred Hoyle, the disparager of the big bang and champion of his alternative cosmology of continuous creation, which postulated that the Universe had no beginning (or end). But alas, continuous creation has since been thoroughly debunked. It is now well established that the Universe is “only” 13.8 billion years old. At least once during the past 13.8 billion years, life must have arisen spontaneously; if not here, then somewhere else. And, so, the question remains: how did it do so?
Theories of the Origins of Life
Modern, scientific consideration of the origins of life began, as mentioned in chapter 4, with the Soviet and Scottish scientists Aleksandr Oparin and J. B. S. Haldane, the latter of whom seems to have coined the phrase primordial soup. Oparin proposed a “cells-first” theory of origins. Impressed by the cell-like appearance that oily organic materials can adopt in water, he proposed that the physical structure of the cell came first in the form of a suspension of oily droplets and hollow, water-filled, oily “vesicles,” together called coacervates, from the Latin coacervātus, meaning “heaped together.” Oparin noted that, in particular, the water-filled vesicles could serve as a vessel, providing a sequestered environment isolated from potentially disruptive influences. Moreover, he noted, under some laboratory conditions such vesicles grow and divide in a manner reminiscent of cell division.
Oparin’s speculations aside, life is more than just swelling, water-filled vesicles of lipids.* Life, at least as we have defined it, requires genes and metabolism. The dispute over which of the two came first has divided researchers in the field, fueling a debate that many observers have likened to the classic chicken-or-egg argument, with both the metabolism-first and genes-first camps simultaneously claiming they have the upper hand.
Metabolism First
The metabolism-first camp argues that the first life was formed from a network of self-sustaining chemical reactions of monomeric organic molecules catalyzed by either organic or inorganic catalysts. This can be a bit hard to visualize, but we’ve seen examples of it. In chapter 1, we discussed fire as a self-replicating, albeit uncatalyzed, network of free radical reactions (see fig. 1.1). And in chapter 4, we discussed the formose reaction, a calcium hydroxide–catalyzed reaction network that consumes formaldehyde to produce sugars (see fig. 4.7). In both cases, if provided with sufficient input materials (fuel and oxygen for the former, formaldehyde for the latter), these reaction networks replicate. This replication is obvious in the case of fire, but it happens in the formose reaction too; the reaction starts off very slowly as the spontaneous formation of a molecule of glycolaldehyde is rare. But as the reaction progresses, one glycolaldehyde is converted into two with each turn of the cycle, exponentially accelerating (replicating) the reaction over time.
Günter Wächtershäuser, a German chemist turned patent lawyer who started dabbling in origins-of-life chemistry in his spare time, proposed just such a metabolic network in the late 1980s. Wächtershäuser postulated that an assembly-line network of chemical reactions, which he dryly described as “two-dimensional chemi-autotrophic surface metabolism in an iron-sulfur world,” was set up on the surface of catalytic minerals, such as iron sulfide, in hydrothermal vents deep beneath the sea. The hydrothermal vent environment can drive the reactions by using the chemical disequilibria set up when the hot, briny water emerges into the colder ocean.
Wächtershäuser’s hypothetical “pioneer organism” was composed of a mineral substrate on which transition metal atoms, such as iron or nickel, catalyzed the reduction of carbon dioxide to other small, organic molecules using the reducing power of hydrogen sulfide. These molecules remained on the mineral surface and served as ligands—small molecules that bind to and thus modify the chemical activity of the transition metals. By improving a metal’s catalytic activity, some of these new ligands formed an autocatalytic “surface metabolism” in which the formation of new reactive centers was accelerated by the very same small molecules that they produced. Specifically, Wächtershäuser proposed that the key set of reactions (fig. 5.1) in his “metabolic life” formed a backward, reducing version of the Krebs (citric acid) cycle—the central oxidative biochemical pathway in aerobic organisms. As the mineral substrate for this activity, Wächtershäuser proposed troilite (FeS), a now rare form of iron sulfide that was probably more plentiful on the early Earth (under current conditions, the more oxidized iron-sulfur mineral pyrite, FeS2, is far more common). The troilite, in turn, can catalytically reduce various organic molecules, converting itself back into pyrite. Wächtershäuser also suggested that the surface of iron sulfide would constrain the distribution and orientation of the products of each reduction in such a way as to support a complex, self-sustaining sequence of metabolic reactions that, ultimately, would lead to the formation of new catalysts and new metabolic pathways. Thus, argued Wächtershäuser, once an autocatalytic metabolism is established, it will go on to produce increasingly complex catalysts, increasingly complex product molecules, and increasingly numerous and diverse metabolic pathways.
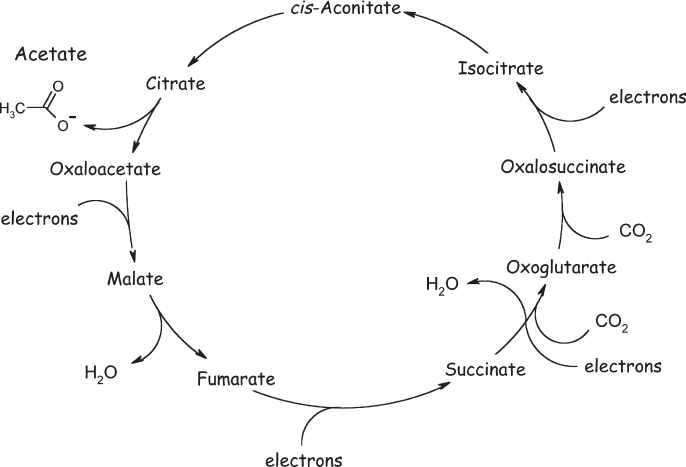
Figure 5.1 Günter Wächtershäuser argues that life could start as purely metabolic networks spontaneously arising on the surface of iron sulfide minerals. For example, he postulates that a reverse (reductive) Krebs (or citric acid) cycle can be catalyzed on the mineral troilite (FeS), which (in net) takes the reducing potential of hydrogen sulfide and uses it to reduce carbon dioxide to larger organic molecules. To date, however, few if any of the steps in this postulated reaction network have been demonstrated in the laboratory.
In support of his hypothesis, Wächtershäuser pointed to at least a few potentially relevant reduction reactions that can take place on iron sulfides. For example, in collaboration with Claudia Huber at the Technical University of Munich, Wächtershäuser showed that, in the presence of iron and nickel sulfides, hydrogen sulfide reduces carbon monoxide to acetic acid—carbon monoxide, though, not the carbon dioxide that is essential to his postulated reductive citric acid cycle. The two researchers have similarly shown that, in the presence of ammonia and hydrogen sulfide, iron and nickel sulfides can reduce a specific class of molecules called α-ketoacids to the α-amino acids used in proteins. So, it seems that biochemistry-like reductive reactions can take place in the presence of mineral sulfides. It remains very much an open question, though, whether a complete metabolic network could spontaneously self-organize (and operate autonomously) on an iron sulfide surface under plausible prebiotic conditions. What, after all, would drive a collection of seemingly dissimilar chemical reactions to spontaneously self-organize?
Moving forward from Wächtershäuser, William Martin of the University of Düsseldorf and Michael Russell of NASA’s Jet Propulsion Laboratory in California have argued that similar metabolic pathways formed within small, iron sulfide–lined cavities in the rocks of hydrothermal vents are a better candidate for the first life. In part, this is because the cavities would tend to retain any newly formed metabolic products at higher concentrations than would occur on a mineral surface in the open ocean. Moreover, steep temperature gradients inside the walls of the vent would increase the range of reactions that are feasible. Martin and Russell argue, for example, that the synthesis of small, monomeric molecules such as amino acids, which requires the input of substantial energy, would occur in the hotter regions and their polymerization, which is reversed at high temperatures, would progress more readily in cooler regions.
Critical to the work of Wächtershäuser and of Martin and Russell is the argument that the self-organization inherent in their putative metabolic pathways stems from the constraint of molecules being adsorbed onto (and synthesized on) the surface of a mineral. The organization imposed by the mineral surface, they argue, would foster the formation of reaction networks and increase their specificity. To date, however, no such mineral-driven guidance has been observed for any set of chemical reactions even remotely approaching the complexity that would be required to form a self-sustaining network that fed on simple precursors and synthesized larger molecules. Perhaps this is to be expected: any one mineral is unlikely to specifically catalyze more than one or two of the many distinctly different chemical reactions required for a metabolic network as complex as the Krebs cycle, much less catalyze only those reactions that are productive and not competing side reactions that would disrupt the network. Moreover, even if such multicatalytic minerals did exist, there is no laboratory evidence that complex catalytic networks could spontaneously self-organize on their surfaces. Thus, while the iron-sulfur theory has stimulated discussion and research, it is, at least currently, not based on firm laboratory verification. Nor are there any laboratory-based hints that something like this might be possible under some as yet to be defined set of conditions.
Of course, we might still feel confident about the possibility of iron-sulfur life, even in the absence of laboratory examples, if we could come up with an argument in favor of evolution in the iron-sulfur world and, likewise, could compellingly explain how we could have evolved from such a start. That is, even though our interest is in defining the range of possible forms that life can adopt anywhere, not just on Earth, if we could argue that we had evolved from iron-sulfur life, we’d feel more confident about the possibility of iron-sulfur-based metabolism as a precursor to life on both the early Earth and elsewhere in the cosmos. And while there are small “iron-sulfur” clusters (consisting of up to four irons and four sulfurs) in some of the proteins that catalyze oxidative biochemistry, no clear route from “there” to “here” has yet been described. For example, why would nucleic acids, so central to biology on Earth, provide a selective advantage for these systems? For that matter, given that the FeS itself is not replicating, what, if anything, would provide a selective advantage—which is first and foremost about improving the ability to reproduce—for such a system? In the absence of compelling answers to these questions, we consider it unlikely that our very first ancestor was a gene-free metabolic network quietly chugging away on the surface of some mineral, and in the absence of a compelling demonstration of such chemistry in the laboratory, we must question whether any life anywhere could arise from such a start.
An alternative twist on the metabolism-first idea is that the first living organisms were not made up of the sort of complex, multistep networks that we typically envision when we say “metabolism” but instead were built from metabolic-like pathways involving lipid aggregates. Lipids are “water-hating” molecules (termed “hydrophobic” in biochemistry) and thus, in solution, lipids tend to spontaneously organize into compact spheres called micelles or into hollow, water-filled, membrane-bound balls called vesicles (soap is a lipid that spontaneously forms micelles around grease, solubilizing it and allowing it to wash down the drain). Even today, most biological membranes are not created from scratch but rather through a growth-and-division process that is at least somewhat analogous to life. And the coacervates Oparin studied in the 1920s could sometimes be coaxed to adsorb smaller precursors, and even to divide. But replication alone does not life make. Is there a plausible chemical scenario in which a blob of membranes could be said to evolve? Doron Lancet of the Weizmann Institute in Israel and Dave Deamer at the University of California, Santa Cruz, have argued that there might be (fig. 5.2).
The “lipid-world” argument starts with the observation that lipids are an extremely diverse set of compounds. For example, modern eukaryotic cells contain three broad classes of lipids in their cell membranes: phospholipids, sphingolipids, and sterols. The first two differ in terms of the polar, water-loving “head group” attached to the water-hating fatty acid tail of the lipid. Sterols, however, represent a different design, the most common example of which is cholesterol. Each of these three classes, in turn, can be subdivided by chemical details, such as the chemistry of the water-hating tail, chemistry of the water-loving head group, and so on. There are dozens of distinct types of lipids in the membranes of our cells, dozens and dozens of others in the membranes of prokaryotes, and countless more that can be synthesized in the laboratory and may have arisen prebiotically. With such tremendous diversity, who can say what range of chemical and physical properties is within reach? More specifically, notes Lancet, is it not possible that among the myriad of possible lipid compositions in a simple vesicle there might be some particular mixture that encourages the adsorption of other lipids in exactly the right ratios to grow and produce offspring with almost exactly the same composition as, and thus very similar physical and chemical properties to, the parent vesicle? That is, might there exist some “special” composition of lipids that has the ability to “breed true” and thus accurately pass down its traits to the generations that follow, such that lipid “A” attracts lipid “B” into the blob, and then lipid “B” attracts lipid “C,” and so on until, finally, lipid “Z” completes the cycle (network) by drawing in lipid “A”? And, if this is so, is it much more of a stretch to postulate that these lipid aggregates could sometimes accidentally take on some new lipid component that would improve the efficiency with which they reproduced or improve their ability to survive and grow in some new environment and that also would allow them to breed true and pass these properties to their offspring? Were such lipid aggregates possible, they could be said to be both reproducing and evolving. They could be said to be living things.
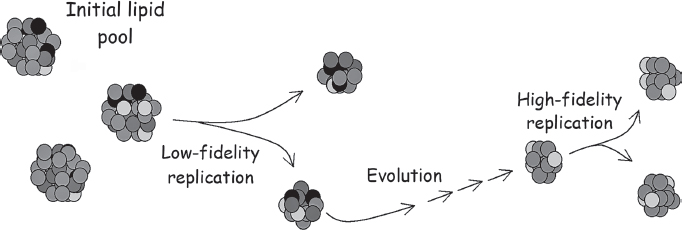
Figure 5.2 The lipid-world hypothesis is predicated on the thought that there are so many different types of lipid that some rare collections of them might exhibit the property of adsorbing new lipid components in the same ratio as the existing ratio in the aggregate, thus swelling and eventually splitting to produce offspring similar to themselves. If this “breeding” were not quite perfectly “true,” any mistakes—“mutations”—that improved the aggregate’s rate of growth and division would cause the system to evolve.
The lipid-world hypothesis nicely explains how a presumably very dilute primordial soup could spontaneously organize into complex structures; to minimize the extent to which their hydrophobic elements are exposed to water, lipids readily self-assemble into micelles or vesicles, even when at extremely low concentrations. That said, the lipid-world hypothesis is still just that, a hypothesis. For example, to date, the micelle and vesicle chemistry we have observed in the laboratory just isn’t selective; no one has yet come up with a mixture of lipids that even comes close to breeding true, much less one that starts there and slowly evolves. One problem is that the aggregation of lipids to form micelles and vesicles is generally nonspecific; only relatively poor differentiation is observed. And, thus, there isn’t much in the way of laboratory evidence supporting the ability of lipid vesicles to breed true, a key element of the lipid-world hypothesis. A second problem is shared with the iron-sulfur hypothesis: how would this evolve into us? Even though our cells are enclosed within membranes (and, indeed, membranes may have played an absolutely critical role in the evolution of life—just not a solo role!), lipid-world chemistry is hardly reminiscent of our most fundamental metabolic and genetic chemistry. To quote its proponents, “A complex chain of evolutionary events, yet to be deciphered, could then have led to the common ancestors of today’s free-living cells, and to the appearance of DNA, RNA and protein enzymes.”* And thus, the lipid-world hypothesis remains an interesting speculation, backed perhaps by theoretical models but lacking much in the way of supporting empirical observations.
Metabolism-first theories, such as the iron-sulfur world or the network of interactions inherent in the lipid-world model, face yet another, perhaps even more fundamental, difficulty: gene-free networks are generally resistant to evolutionary change because such change would require that multiple mutations occur simultaneously.** Let’s look at a network in which product A catalyzes the formation of product B, and vice versa (fig. 5.3). It’s always possible that there is some “mutant” version of A, say A′, that is a better catalyst of the formation of B. Thus, the mutant A′ would provide a selective advantage for the putative metabolism-only organism. But while A′ is a better catalyst, it is still likely to catalyze the formation of B, and B catalyzes the formation of A, not A′. To provide an “inheritable” selective advantage, the mutant A′ must instead catalyze the formation of a mutant B′ that, in turn, catalyzes the formation of A′. That is, for networks to evolve, they require multiple, simultaneous mutations.* And mutation probability does not change algebraically with the number of mutations but changes geometrically: the formation of two simultaneous mutations is not twice as improbable as the formation of one mutation but is, instead, the square of the improbability of forming one. Given that even single mutations must be relatively rare events—if mutations occur too readily, too few of the offspring will be viable—if we square the probability (or cube it, if it is a three-part network, and so on), the ease with which mutations produce a selective advantage diminishes very rapidly. It is thus difficult for complex networks to evolve via the slow accumulation of stepwise mutations; at a very fundamental level, it seems unlikely that networks could give rise to life before the emergence of genes because, in contrast to networks, genes allow for the stepwise, additive accumulation of favorable mutations.
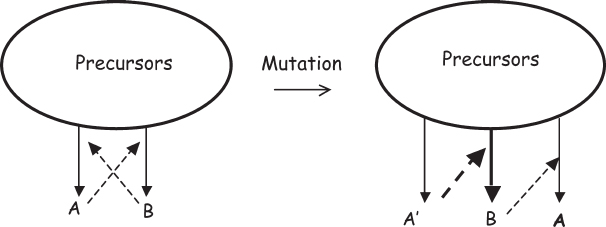
Figure 5.3 A generic problem with metabolism-first theories of the origins of life is that gene-free networks are generally resistant to change. Let’s consider a network in which product A catalyzes the formation of product B, which catalyzes the formation of more product A. This hypothetical network would be a self-replicating chemical system, but it does not meet our definition of life as it is exceedingly difficult for such a system to evolve. Even though there may be an A-like molecule, A′, that is a better catalyst of the formation of B, the B so produced catalyzes the formation of A, not A′. Only in the extremely unlikely event that A′ catalyzes the formation of B′ (which catalyzes the formation not of A but A′) will this system have evolved to a more fit state. For more complex networks (e.g., A catalyzes the formation of B, which catalyzes the formation of C, which catalyzes the formation of more A), the problem compounds exponentially with the number of nodes in the network.
Genes First
The genes-first camp argues that the first living organisms were likely to be genes, that is, information-containing single molecules that could catalyze their own replication. These simple, self-replicating molecules would then, under the influence of selective pressures, evolve into increasingly complex organisms and, on Earth at least, could eventually have evolved into organisms with complex biochemistry such as ours. The genes-first hypothesis has a significant advantage over the metabolism-first camp. Namely, while networks are fundamentally resistant to evolutionary change, a single molecule that catalyzes its own formation can evolve more readily if modifications of the catalyst breed true and can be passed down through the generations (fig. 5.4). The question, then, is, Can we think up chemically plausible catalysts for which this holds? Excitingly, several possibilities have been suggested.
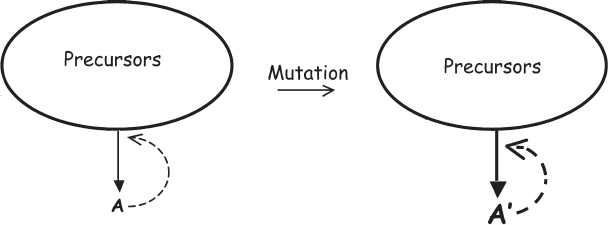
Figure 5.4 A solution to the network problem is possible if there is a single molecule, A, that can catalyze the template-directed formation of copies of itself. In this case, a mutation producing a more fit molecule, A′, say with better catalytic activity, would be a heritable change.
Even the simplest self-replicating chemical systems, crystals, have been seen to exhibit the property of modifications that breed true. For example, irregularities in the surface of a crystal, such as the so-called screw dislocations that cause the surface of the growing crystal to spiral upward rather than form discrete layers, can continue to propagate as a crystal grows and, in some circumstances, can breed true if the crystal shatters and nucleates the formation of more crystals. Once again, though, we must remember that even with mutations, replication alone is not sufficient to meet our definition of life. Even self-propagating modifications do not constitute evolution unless they provide a selective advantage for the “organism.”
A. G. Cairns-Smith (1931–2016), while at the University of Glasgow, suggested that some minerals have the right stuff to form “mineral genes” that can evolve and thus could have been the first living things. Clay is made up of sheets of charged alumina and silicates packed together in alternating layers (fig. 5.5); the slickness of wet clay arises when water intercalated between the layers lubricates them and allows them to slide past one another. One common clay, montmorillonite, has the formula (Na, Ca)⅓(Al, Mg)2(Si4O10)(OH)2•nH2O. Now, you may have noticed several odd things about this formula. For one, there’s the “nH2O,” which simply reflects the fact that different amounts of water can be inserted between the layers of clay. For another, have you ever seen a chemical formula that contains commas? This one does because positively charged sodium (Na) and calcium (Ca) ions can replace one another at some of the negatively charged “sites” on the surfaces of the alumina or silica layers and, thus, while the sum of the sodium plus calcium in the mineral is fixed, their ratio is not. Likewise, aluminum (Al) and magnesium (Mg) ions are similarly sized and thus can replace one another in the clay’s alumina layers, leading to an effectively infinite number of molecules each differing from the others in its ratios of sodium to calcium and magnesium to aluminum. These clays form two-dimensional, water-separated sheets in which one sheet might or might not look exactly like the sheet above or below it.
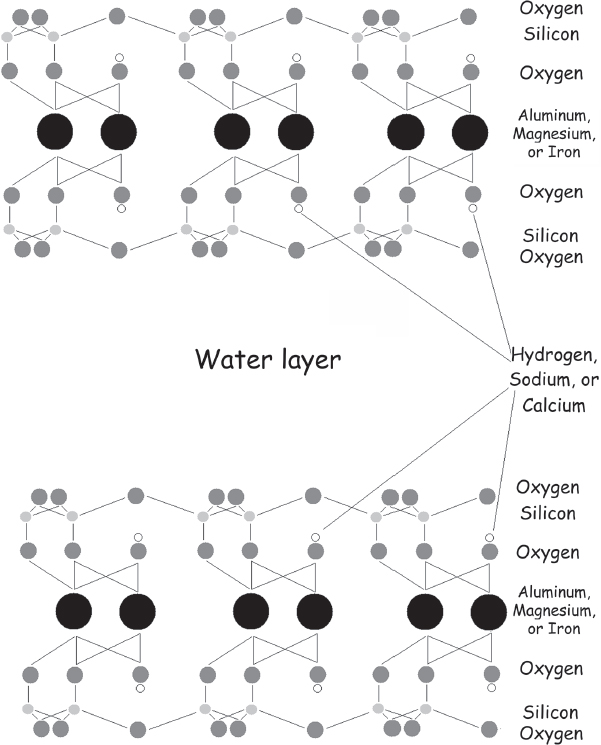
Figure 5.5 Clays are aluminosilicate minerals consisting of alternating sheets of alumina and silica. Oxygen atoms at the faces of these sheets take on a negative charge and bind positively charged ions, such as sodium or calcium. Other ions, such as iron or magnesium, sit within the mineral lattice itself. Shown here is the structure of the montmorillonite family of clays, in which an alumina layer is sandwiched between two silica layers, and this triple-layer structure is linked to others by weak, ionic interactions. Water disrupts these ionic interactions, allowing adjacent layers to slide against one another, which accounts for the slickness of wet clay.
Cairns-Smith theorized that the sheet-to-sheet variability in clays might allow them to act as “genes.” That is, the specific array of charged ions in one layer of clay could act as a template and catalyze the formation of a complementary new layer of clay—the “next-generation” layer. Any “mutations”—that is, mistakes in the packing of the alumina or silica, or changes in the positive ions that associate with them to neutralize their negative charge—that occur during copying would be inherited by all follow-on layers, and if these mistakes improved the efficiency of the replication process, they would provide a selective advantage, thus fulfilling our definition of life. These first inorganic organisms, Cairns-Smith suggested, provided the scaffold on which life as we know it—built of sugar and spice and everything nice—later evolved. Cairns-Smith also noted that ions in clay can act as catalysts to speed up organic chemical reactions, including, as we noted in chapter 4, the polymerization of RNA, and thus over the course of millions of years could have been a significant source of biologically relevant polymers in the primordial soup. Despite this potential strength, though, Cairns-Smith’s theory is not without serious weaknesses.
Perhaps the dominant weakness in this “clay-world” hypothesis is that no chemistry anything like it has ever been demonstrated in the laboratory. It thus remains very much an open question whether defects and irregularities in mineral sheets can be made to breed true, such that they propagate with reasonable fidelity to the next layer of clay. Likewise, it may be telling that no remnants of the clay-based metabolism exist in modern metabolism on Earth: there is nothing in current biochemistry that looks remotely like silicate minerals. Cairns-Smith argued that this simply means that the original clay genes and catalysts have been completely and utterly replaced, leaving not the slightest vestigial traces. This is, of course, quite possible: clays are not particularly good catalysts and thus we might expect them to be completely abandoned after evolution invented better catalysts based on organic materials. Still, were such vestiges present, they would be nice, tangible evidence supporting such origins. In their absence, we are left only with thought-provoking, but as yet experimentally unsupported, speculations.
What we really want is a “replicase.” That is, some relatively simple molecule that, unlike the hypothetical clay chemistry of Cairns-Smith, has been shown in the laboratory to be able, if not to make more copies of itself (which is our goal), at least to make molecules similar to itself. The first simple examples of such molecules were provided by two independent research groups at the Scripps Research Institute in La Jolla, California. In the first, Reza Ghadiri and his research team designed a polypeptide (a short protein) that autocatalytically directs the synthesis of copies of itself from two smaller, chemically activated polypeptide fragments, each consisting of half of the template sequence. Likewise, Gerald Joyce has demonstrated not only molecular replication but even rudimentary evolution in the test tube using RNA molecules that catalyze the fusion of smaller RNA molecules into new molecules that support the linkage reaction.
Nevertheless, systems such as these are probably poor analogs for the origins of life itself: as Ghadiri argues, these examples simply show how molecules that copy themselves from (slightly) simpler precursors are physically possible. But they are poor candidates for the origins of life because plausible prebiotic reactions are very unlikely to generate the short polymers from which these reactions proceed. A similar problem has arisen in efforts to invoke DNA as the original self-replicating molecule. In 1968, Leslie Orgel began to explore the idea that a single strand of DNA could serve as a template on which activated nucleotides could spontaneously polymerize to form the complementary sequence. After decades of work, the conclusions are mixed. Polymerization can proceed reasonably efficiently starting from phosphorimidazolide monomers (an activated nucleotide that we discussed in chapter 4), but only on templates that are rich in the nucleobase cytosine. For example, the sequence GGCGG is obtained in 18% yield from the template CCGCC. But cytosine-poor polymers make very poor templates, and thus the GGCGG product cannot be used to synthesize more of the CCGCC starting material; the reaction grinds to a halt after just one round and is therefore not self-replicating. Orgel’s idea also fails our second test—there is no evidence of such origins remaining in our biochemistry. That is, the high-energy nitrogen-phosphorous bonds that drive the polymerization of phosphorimidazolide-activated monomers are not seen anywhere in current biology, dealing this idea another blow with respect to its having played a role in the origins of life on Earth.
Orgel’s work with DNA-templated DNA polymerization, Ghadiri’s work with self-replicating peptides, and Joyce’s work with self-linking RNA raise another chicken-or-egg conundrum. Namely, in current Terrestrial biochemistry, DNA encodes the information necessary to make proteins, and proteins are required in order to copy DNA. It is thus not at all clear whether either of these two species can replicate without the other, and thus it is not at all clear that either of these two could have been the first, self-replicating molecule. A more promising candidate for the chemistry of the origins of life would be a molecule that has been shown to have the capacity, at least in a small way, to copy itself from simpler, monomeric precursors. Just such a molecule may be at hand in the form of RNA.
The RNA World
As we hinted above, by the late 1960s, Francis Crick (1916–2004; codiscoverer of the structure of DNA), Leslie Orgel (we’ve met him before), and Carl Woese (1928–2012; we’ll get to know him better in chapter 7) had each independently developed the hypothesis that nucleic acids could have formed the basis of the first living things, a hypothesis that motivated much of Orgel’s later work on DNA-templated DNA polymerization. But underlying that work was the assumption that some inorganic catalyst would be responsible for the polymerization reaction. While the ability of RNA molecules to fold up into the sort of intricate, three-dimensional structures associated with protein-based catalysts was well established by the early 1970s, the intellectual stance that proteins are the biological catalysts was so firmly entrenched—indeed, it was considered a major element of biology’s central dogma—that the idea of nucleic acids as catalysts seemed far-fetched. Molecular biologists had relegated RNA to secondary, or more precisely, tertiary importance. In cellular organisms like us, for example, RNA is neither the genetic material (a job carried out by DNA) nor the catalytic machinery by which most of our metabolism is conducted (a job carried out by proteins). Instead, RNA was long thought to serve such lowly (if essential) tasks as messenger RNA’s role in transporting genetic information from the DNA, where it resides, to the protein-synthesizing ribosomes. That said, RNA was known to serve as the genetic material in a few small viruses (HIV and the coronaviruses, for example, encode their genetic information in RNA). If only RNA could catalyze reactions as well! Then we’d have what we are looking for: a molecule that could form the basis of both genetics and metabolism. And in 1981, just such catalytic activity was discovered, like many truly revolutionary observations, essentially by accident.
The catalytic possibilities of RNA were discovered in the laboratory of Thomas Cech at the University of Colorado. Cech and his students were studying the mechanism by which ribosomal RNAs, key components of the ribosome, are “processed.” Two years earlier, it had been discovered that the information contained in many RNA molecules is not continuous; the coding regions of freshly synthesized RNAs are interrupted by apparently meaningless sequences called introns that must be spliced out of the immature RNA before it is used. Cech found that cellular extracts from his favorite study organism, the single-celled paramecium Tetrahymena, could carry out the splicing reaction in the test tube. When faced with such an observation, the natural thing for a biochemist to do is to try to “fractionate” the extract into its component parts in order to discover which protein in the system is responsible for the catalytic activity. The first fractionation experiment they attempted, using extracts from the cell nucleus, carried out the splicing reaction perfectly, suggesting that the investigators were on the right track. But there was a fly in the ointment. The test tube “next door” to the nuclear extract was a negative control that contained the RNA but lacked the nuclear extract, and yet splicing seemed to have occurred there as well. At first, they thought they had just mixed up the tubes, but repeating the experiment another five times produced the same puzzling result. Was the problem that the RNA was contaminated with a small amount of Tetrahymena proteins, including, perhaps, the splicing protein? After all, the RNA had also been purified from Tetrahymena extracts, so some of the relevant proteins could have come along for the ride. Treatment of the RNA with proteases, though, which should destroy any leftover proteins, did not stop the reaction, suggesting that contaminants were not responsible for the observed splicing. Finally, in desperation, the research group synthesized the RNA in Escherichia coli, which is quite unrelated to Tetrahymena and thus produces RNA samples entirely free of Tetrahymena proteins. And still the supposed control RNA spliced. With such compelling and undeniable evidence in hand showing that the reaction could occur even in the absence of Tetrahymena proteins, Cech finally went public with a positively revolutionary idea: an RNA molecule could splice itself without the help of proteins. And while, in the strictest sense, the RNA splicing reaction is not catalysis—a true catalyst remains above the fray and emerges unchanged from the chemical reaction it fosters—this result was the first hint that RNA could accelerate biochemical reactions in a manner previously thought solely the realm of proteins.
It quickly became apparent that Cech’s autocatalytic RNA was not a one-off example. While Cech’s group was conducting these experiments, Sidney Altman at Yale University had been studying another catalytic RNA-processing step—the maturation of transfer RNA (more about this type of small RNA in chapter 6). One of the last steps in the process is cleavage of the RNA to remove several nucleotides from one end. The purified cellular component that carries out this process is a two-molecule complex consisting of a protein tightly bound to an RNA. Altman had been assuming that, as the central dogma stated, the protein part of the complex is the catalytic agent and the RNA plays some structural role or serves to bind the enzyme’s target. But when Altman’s students attempted to run the reaction using the purified RNA component alone, they found that, in the presence of a large quantity of magnesium ions (presumably to take the place of the protein in its role of neutralizing the negative charges on the RNA and allowing it to fold), the RNA was the catalyst. Indeed, it was the protein that serves a merely structural role! For these discoveries, Cech and Altman shared the 1989 Nobel Prize in Chemistry.
In the years since the first ribozymes, short for ribonucleic acid (RNA) enzymes, were discovered, the list of catalytic roles that RNA can play has grown (fig. 5.6). For example, it has since been shown that the ribosome, the massive protein and RNA “factory” in which proteins are synthesized, is a ribozyme: proteins, it turns out, are synthesized by the catalytic activity of RNA, an important point to which we’ll be returning in the next chapter. Meanwhile, the catalytic activity of RNA is far more diverse than indicated by the few examples that natural selection provides; many more catalytic functions have been produced by artificial selection in the laboratory. These additional catalytic activities include the cleavage of DNA, the ligation (linking together) of two RNA molecules, the ligation of DNA, acyl transfer (the transfer of an organic group from a phosphate to another molecule), the cleavage of peptide bonds (the type of bond that links amino acids together in a protein), the formation of peptide bonds from activated, acylated compounds, and the insertion of a metal into a porphyrin (a large, nitrogen-containing organic ring structure) to form the catalytic organometallic compounds at the heart of such diverse metabolic processes as photosynthesis (in the form of chlorophyll) and oxygen transport (in hemoglobin).
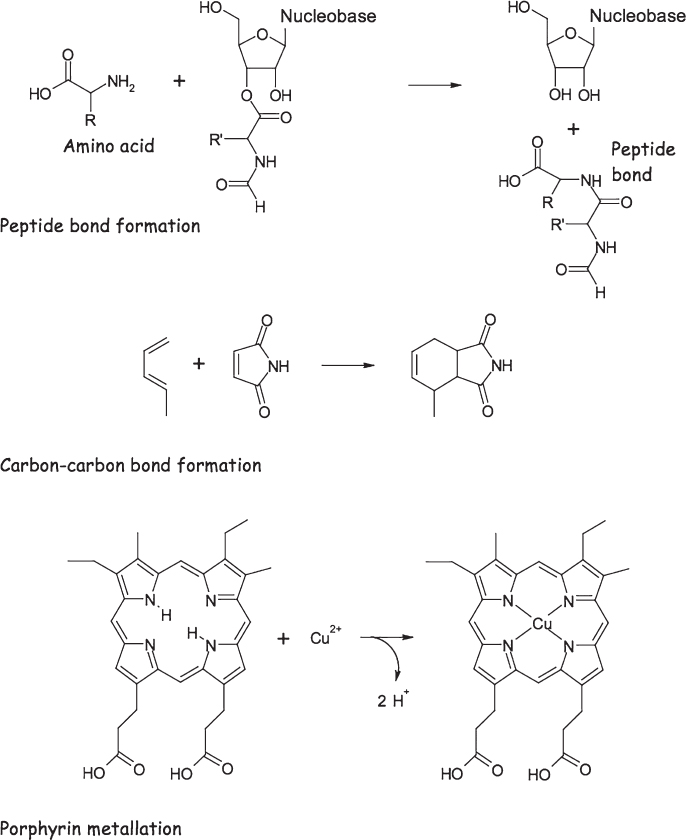
Figure 5.6 While only a handful of naturally occurring ribozymes have been identified, a number of artificial ribozymes have been generated in the laboratory. Shown here are three examples that illustrate the broad range of reactions within reach of ribozyme-based catalysis.
Did the discovery of ribozymes provide a solution to the origins-of-life question? As we’ve noted, it had long been known that RNA can serve as the repository of genetic information, and by the early 1980s it was established that RNA could serve as a catalyst as well. Could RNA, then, have been the original self-replicator? To serve this role requires a very special kind of catalysis: a self-replicating molecule must be able to catalyze the formation of copies of itself. RNA, like other polymers, forms from simpler precursors by polymerization. The formation of a specific polymer sequence based on the sequence of another, templating polymer is called template-directed polymerization. Was the first living thing a ribozyme that carried out template-directed RNA polymerization? Is such a ribozyme even physically plausible?
One of the first scientists to head down this road was Walter Gilbert, the Nobel Prize–winning molecular biologist from Harvard who, in 1986, first coined the phrase “RNA world.” In the spring of that same year, when on a tour of potential grad schools, I (Plaxco) was sitting in Professor Gilbert’s office when a student interrupted, popping her head in the door and simply saying, “nothing’s come up yet.” Professor Gilbert looked at me thoughtfully for a moment, said “You look like you can keep a secret,” and then proceeded to describe the experiment his student was conducting. She had synthesized a test tube full of random RNA sequences, fed them with activated nucleotides, and then, once a week, was checking to see whether any one sequence was replicating and thus increasing in number. Given that, in the many years that have since passed, the results of said experiment have not appeared in print, it’s a safe guess that none of the sequences ever started to dominate the mix (it likewise seems that the statute of limitations has run out on the promise not to tell this story). The experiment’s failure, though, might have been quantitative rather than fundamental. That is, while a test tube full of completely random, 70-base RNA polymers contains 1014 different sequences, this represents only a miniscule (one in 1028) fraction of the more than 1042 possible sequences that can be encoded by a 70-base RNA polymer. Perhaps sequences with the properties we want are less common than one in 1028?
The question then becomes how to improve the odds. One approach would be to start with a larger test tube (more random sequences), but the most RNA that can reasonably be synthesized and handled is only a few hundred times the amount that Gilbert’s team used. Realizing this, David Bartel’s group at the Massachusetts Institute of Technology took a different approach. Specifically, they applied two new tricks. First, to avoid having to search through a huge morass of unfolded RNA polymers (no well-defined three-dimensional structure equals no complex chemical activities), they started with a folded, catalytically active ribozyme sequence. Specifically, they employed a sequence that Bartel had discovered when he worked in Jack Szostak’s lab and that already performed chemistry similar to the kind they wanted: the ribozyme was a ligase, a type of enzyme that catalyzes the splicing together of two longer RNA polymers and thus creates the kind of phosphodiester bonds that are also formed during polymerization. Indeed, the ligase ribozyme is already capable of polymerizing RNA, but only up to three nucleotides and not in the necessary template-directed fashion. Bartel’s group then mutagenized the ligase to create a “random” starting pool of some 1015 different RNA sequences, all of which had significant similarity to the parent ligase. Second, they employed a clever trick to ensure they could pick out sequences that had any template-directed polymerase activity, even if the activity was insufficient to actually support full-fledged self-replication. To do so, they added to every element in their mutated library a short “priming sequence” that acted as the initiation site for the polymerization reaction, along with a short RNA that would act as an internal template to guide the sequence-specific polymerization reaction.
To fish for the (presumably) rare sequences with the desired activity, Bartel’s group added some activated nucleoside triphosphates to the pool of molecules, including the modified, sulfur-containing nucleotide 4-thiouracil triphosphate (4-thioUTP). Any RNA molecule in the pool that could polymerize a short stretch of RNA on its template would incorporate this 4-thioUTP into its structure as 4-thiouracil. Bartel’s students then separated out any such 4-thiouracil-containing RNA molecules by forcing the RNA products of their reaction to migrate through a mercury-containing gel; mercury binds to the sulfur, impeding the motion of thio-containing RNAs. They then isolated the slowly migrating RNA molecules, converted these into their complementary DNA sequences (using an enzyme called reverse transcriptase), copied and amplified this pool of DNA sequences (using the enzyme DNA polymerase), and, finally, converted the amplified pool of DNA sequences back into RNA (using the protein RNA polymerase)—phew!—before starting the cycle over again. During the first few cycles, they uncovered mostly mutant RNA that had “evolved” the property of covalently binding 4-thioUTP without catalyzing template-directed polymerization. But with each additional round, the population of RNA molecules that exhibited polymerase activity (and thus incorporated more than one 4-thiouracil) increased. Ultimately, after 18 rounds of selection, copying, reengineering, reamplification, and reselection, Bartel group member Wendy Johnsen recovered a 189-nucleotide-long RNA, the “18.12 polymerase,” that could use a template to polymerize a specific, complementary sequence of RNA. That is, they’d found molecules that could catalyze the copying of molecules of their own kind.
But is the 18.12 polymerase ribozyme the self-replicating “replicase” we are looking for? It’s promisingly close, but it’s not there. Even this “best” RNA polymerase extends its primer by only 14 nucleotides before the ribozyme, which itself is relatively unstable, breaks down. Moreover, it only works if given an unstructured (i.e., unfolded) template; it is incapable of unwinding the sort of structure it, itself, adopts, and thus it cannot use something as complex as itself as a template.
Building on the Bartel group’s pioneering work, a number of other researchers have since “evolved” more efficient ribozyme polymerases. A promising example is the work of David Horning and Jerry Joyce, both then at the Scripps Institute in San Diego, California. Horning started with the 18.12 polymerase, mutagenized it, and then selected from among a pool of a hundred trillion variants those best able to copy two structurally complex RNA templates. Specifically, Horning selected for the ability to generate RNA sequences that fold into the precise three-dimensional shapes required to bind the small molecules guanosine triphosphate and vitamin B12 and then used a combination of binding to these molecules and size separation as a means of fishing out those sequences that had succeeded in copying these longer templates. Repeating this cycle a total of 24 times, each time increasing the selective pressure by increasing the length of the required polymerization and decreasing the time allowed to generate it, Horning identified a polymerase that not only exhibited a dramatically improved ability to copy complex templates, but that also polymerized RNA a hundred times faster than the best rate previously achieved. The catalytic efficiency of the new ribozyme is so great that it can synthesize a highly structured, 76-base yeast transfer RNA (more on this type of RNA in the next chapter), albeit at a yield of only 0.07%.
Moving forward, in 2020, Katrina Tjhung, also working with Joyce, reported the best ribozyme RNA polymerase described to date. To do so, she devised a new selection scheme in which successful polymerases would generate the hammerhead ribozyme, which is an RNA sequence that performs RNA cleavage at a specific RNA sequence. She then added the complement of the hammerhead ribozyme as a template to Horning’s best polymerase, mutagenized the polymerase, and attached the library of mutant sequences to a solid support. Any ribozyme that copied the hammerhead complement to form a hammerhead ribozyme would, by doing so, cleave itself off the support such that it could be collected, amplified, and mutagenized to set up the next round of selection. Tjhung performed this cycle 14 times (coupled with Horning’s work, this pushed her 38 generations past the 18.12 polymerase) while progressively reducing the time allowed for polymerization from two hours to five minutes in order to select for more rapid catalysis. Using this approach, the best ribozyme polymerase Tjhung derived was able to synthesize the above-described transfer RNA in 2.4% yield; while this is still low, it is more than 300 times better than the yield its immediate ancestor was able to achieve.
This 179-base ribozyme efficiently copies sequences more than a fifth of its length and can, with low yield, even copy something more than a third of its length. Thus, if the catalytic abilities of the ribozyme were increased just a few fold, it would be efficient enough to create copies of itself. Product length, however, is not the only metric we need to concern ourselves with: the fidelity of replication is also crucial. Specifically, self-replicating things must breed true enough that a fair fraction of their offspring are functional enough to reproduce. Since deleterious mutations are more common than neutral or helpful mutations, this means replication fidelity must be high. Conversely, too high a reproductive fidelity inhibits evolution; if all your offspring are perfect carbon copies of you, then none of them will have any selective advantage over any of the others. These two contravening effects together produce an optimal level of fidelity that is somewhere between “high enough that sufficient numbers of offspring survive and reproduce” and “low enough that evolution has at least the occasional mutation to work with.” This trade-off is seen in the fact that, even today, evolution optimizes, rather than maximizes, the fidelity of replication: the mutation rates in today’s organisms are tuned such that, irrespective of the size of the genome, there is, on average, one mutation per genome per generation.
The above arguments suggest that an optimal level of fidelity for the RNA polymerase ribozyme would be about one mutation per generation, which corresponds to an accuracy of about 99.4% for ribozymes of the length we’re discussing. In making three mistakes per hundred nucleotides polymerized, however, Bartel’s original ribozyme missed this mark by a factor of five. The fidelity of the catalytically improved polymerase of Horning and Joyce likewise clocks in at 97%, and the improved polymerase of Tjhung is just 96%. The poor fidelity of these ribozymes, however, likely reflects a technical hurdle rather than some fundamental limitation of their chemistry. The problem is that, while in vitro selections for improved catalytic efficiency are conceptually easy (if perhaps difficult in practice)—one can select for longer products by separating larger molecules from smaller molecules—how one would select for improved fidelity is not so clear. Or at least it is not clear how one would do this before one has a self-replicating system that can evolve itself.
The fidelity and catalytic efficiency problems aside, the best RNA polymerase ribozymes identified to date do have many of the characteristics that we are looking for in an RNA-based replicase. They are, for example, quite unselective in their choice of templates and thus, were they more efficient, they most likely could produce copies of themselves and any other genes that might be of use. And they do show the right type of activity—they synthesize RNA polymers with linkages between the same set of hydroxyl groups seen in naturally occurring RNAs. It thus appears likely that an appropriately catalytic, self-replicating ribozyme could exist, an observation that provides a major boost for the idea at the core of the RNA-world hypothesis: that the first living thing on Earth used RNA for both genetic material and catalytic activity, including the replication of the genetic material.
RNA World Redux
In the RNA-world scenario, the first living thing, your very, very, very first ancestor, consisted of three parts: a ribozyme with RNA polymerase activity, a template RNA to direct the polymerization, and a membrane or some other type of physical container. The need for two RNA molecules, and not just one, stems from how ribozymes catalyze reactions. Because ribozymes need to fold into specific, complex structures to perform their functions, it is exceedingly unlikely that any “self”-replicating molecule can actually serve as its own template. In order to serve as a template on which a new RNA molecule can be synthesized, a molecule must be unfolded and exposed to the monomers that will polymerize on it. And unfolded molecules are not catalytic; catalysis is intimately related to the precise, three-dimensional placement of atoms. Thus, in the RNA world, the spark that separates life from nonlife is the creation of not one ribozyme sequence, but two. The need for compartmentalization (e.g., a surrounding membrane) stems from the need that genes remain physically associated with the products they encode. Without such association, the metabolic products of this primitive earliest life-form would simply diffuse away and thus would not provide a selective advantage for the genes that created them. Putting this all together, the RNA world predicts that our very earliest ancestor was two RNA molecules floating together inside a membrane sac, taking up monomers from the surrounding environment and polymerizing them into more copies of the RNA until the RNA load grew so great that the membrane split. Grow, split, grow. The first life.
There we have it: the RNA-world hypothesis in a nutshell. We wrap a leaky lipid membrane around a self-replicating ribozyme, allow nucleotide monomers to diffuse in, and the monomers are polymerized into new copies of the ribozyme. The high molecular weight of the ribozyme traps it within the lipid membrane, perhaps driving its growth and subsequent division. Neat idea, right? It’s easy to see, for example, how the lipid membrane could have evolved into today’s cell membranes, which have abolished the putative leakiness, presumably by employing longer, better “sealing” lipids and (now protein-based) pores that are highly selective and let in only those small molecules needed by the cell. Pursuing this idea in the mid-2000s, the research team of Jack Szostak (2009 Nobel laureate in Medicine and Physiology) at Harvard Medical School in Boston demonstrated, if not quite this exact system, at least some interesting analogs. Abandoning the long-chain lipids used in our cells, which produce virtually impenetrable membranes, the Szostak lab fabricated vesicles from simpler, less ideal lipids that allowed the transport of small molecules. In 2004, for example, Szostak team member Irene Chen showed that vesicles containing high concentrations of RNA “steal” lipids from vesicles lacking RNA, presumably to relieve some of the osmotic pressure created by their contents. And in 2008, Szostak team member Sheref Mansy trapped a synthetic, modified DNA that could act as a template for the polymerization of a DNA analog inside these leaky vesicles and supply the requisite phosphorimidazolide-activated nucleosides to the solution surrounding the vesicles to form “protocells.” Not surprisingly, if the vesicles had just the right leakiness—if they trapped the DNA polymer but allowed in the nucleotide monomers to flow in and out—the polymer grew within the vesicles. As the (so far) culmination of this work, in 2013, Szostak group member Katarzyna Adamala showed that, with a magnesium citrate complex as catalyst, activated RNA monomers could diffuse into template-containing fatty acid vesicles and drive templated-directed RNA polymerization in the fashion of Orgel’s work described above, albeit, as with Orgel’s work, this produced only rather short sequences.
Of course, the membranes that surround our cells are more than just passive sacks: they also have to expand and divide as the cell grows. Exploring this, in 2009, Szostak team member Ting Zhu showed that vesicle growth under some conditions produces long, thread-like tubes that easily rupture under mechanical agitation, producing multiple daughter “cells.” And, in 2011, Szostak and Itay Budin described ways of getting cycles of growth and division driven by increasing the concentration of lipids followed by their redilution under turbulent conditions, as might happen in a tide pool that evaporates at low tide only to be filled again at the next high tide.
All told, the RNA-world hypothesis remains the most chemically plausible and best experimentally supported theory of the origins of life. In RNA, we have a molecule whose credentials as both a genetic material and as a catalyst are well established. And not just any type of catalysis, but RNA-templated RNA polymerase activity, the very type of activity that would be fundamental to the simplest living thing, were that living thing built of RNA. And while, admittedly, the very best ribozyme RNA polymerase described so far can’t copy itself, or even copy a copy of itself, this is presumably a quantitative issue and does not reflect a fundamental inability of RNA to perform such action. Moreover, while it’s not clear how RNA polymers might have been synthesized under prebiotic conditions, at least we understand how most of the precursors of RNA were probably synthesized in the primordial soup. Finally, as we’ll explore in great depth in the next chapter, the RNA-world hypothesis is also the most biologically plausible theory we have of the origins of life. That is, as biologists we can spin a compelling narrative explaining how a single, self-replicating RNA polymer could have evolved, ultimately, into us through plausible, stepwise evolution in which each stepwise increase in complexity provided a selective advantage over the last. The RNA world has a great deal going for it; infinitely more, really, than any other theory as to the origins of Terrestrial life. But is the whole package now neatly wrapped up? In a word: no. (But what would be the fun of that, anyway?)
Several serious questions remain regarding the plausibility of the RNA world. One is the sheer improbability of spontaneously generating one of the rare sequences that could copy itself through the random, prebiotic polymerization of RNA monomers. This becomes even more of a hurdle when one considers that, as noted above, the first living thing had to consist of two such sequences together. If the first living chemical system did not arise until a ribozyme template-dependent RNA polymerase sequence was spontaneously generated in the presence of a second sequence that encoded the same catalytic function (such as a copy of itself, although any sequence that worked as a polymerase would be suitable) and could serve as a template, then it was a rare event indeed: the need for two sequences squares the improbability of the random synthesis event (see sidebar 5.1). Given this, Szostak and others continue to search for plausible, prebiotic routes to templated-directed polymerization that don’t require a ribozyme. But while our mechanistic understanding of these reactions has improved significantly in recent years, polymerization efficient enough and general enough to support self-replication remains, so far, out of reach.
Mirror Molecules
Beyond the debunking of spontaneous generation, Louis Pasteur’s career had many other highlights, including the invention of pasteurization. He was also the first person to note, some 15 years before his work on “corpuscles that exist in the atmosphere” (i.e., germs), that the component chemicals of life are chiral, from the Greek cheir, meaning “hand.” This coinage, which came from Lord Kelvin, stems from the fact that your hands are a convenient example of chirality; that is, they are mirror images of each other that are not superimposable. This is illustrated by the fact that, no matter how many ways you twist and turn it, your left glove will not fit on your right hand. Amino acids and sugars are similarly chiral, coming in left- and right-handed versions that, while similar in many aspects, are mirror images of one another that are not interchangeable.
On Earth, proteins are composed exclusively of l-amino acids (the l being taken from the Latin laevus, or “left”). The equivalent d-amino acids (from dexter, meaning “right”) do show up now and then, such as in the cell walls of some bacteria, but they are far from common. The nucleic acids DNA and RNA likewise contain only “right-handed” sugars, termed d-deoxyribose and d-ribose, respectively. (Note, however, that the historical designations “right” and “left” were assigned arbitrarily and do not imply that l-amino acids are in some way opposite in structure to d-sugars.) Because the chemistries of mirror-image molecular pairs (i.e., pairs of enantiomers) are indistinguishable, which chirality was selected does not appear to be at all important. For example, a protease (a protein that catalytically cleaves other proteins) synthesized in the laboratory using only d-amino acids folds fine, but as expected, it folds into the mirror image of the naturally occurring protein. This mirror-image protein, in turn, is just as catalytically active as its naturally occurring counterpart, but, consistent with its mirrored structure, it cleaves only protein chains composed of only d-amino acids. Just as your right-hand glove fits only your right hand, this right-handed protein bound to and cleaved only right-handed substrates. And yet, while polymer chains of either handedness appear to work equally well, polymers of mixed handedness do not. If the chirality of the monomers changes randomly from left to right along the length of a polymer, this tends to preclude folding into nice, regular structures. And without folding, there is little in the way of catalysis. Homochirality thus appears to be critical.
The requirement for homochirality represents another potentially serious problem for the RNA world. The problem is that biological processes, such as the prebiotic chemistry of the early Earth, produce equal amounts of left- and right-handed molecules, rendering it improbable in the extreme that random chemistry would produce a polymer containing only one. To be precise, the probability of Bartel’s 189-nucleotide ribozyme being polymerized from an equal-molar mixture of left- and right-handed monomers is one in 2189 (one in 8 × 1056)! This number of 189-nucleotide RNA sequences, taken together, would have the mass of 40,000 Suns, which nicely illuminates the extent to which the requirement for homochirality increases the improbability of a self-replicating RNA sequence arising spontaneously from an activated mixture of l- and d-ribonucleotides.
Like the poor efficiency with which RNA is synthesized under plausible prebiotic conditions, the requirement for homochirality reduces the probability that RNA-world-type chemistry is at the root of the origins of life. But does this render the RNA-world hypothesis invalid? It does not. For one, the need for homochirality does not push the probability of spontaneously generating a self-copying ribozyme to zero, and the anthropic principle says that even super-astronomically improbable mechanisms may lie at the heart of the origins of life—our existence only says that the probability of life arising in our Universe is not zero, but it could be infinitesimally close to zero. Disliking this explanation, though, a good number of researchers have spent much effort over the last few decades trying to come up with approaches that avoid or solve the problem of homochirality. Four broad classes of theories have been suggested in an attempt to address or circumvent this problem. The first is that there are abiological processes that preferentially degrade or create one enantiomer. Alternatively, there may be abiological processes that can amplify small, random deviations in the ratio of l and d monomers or chiral catalysts that polymerize only one monomer out of a mixture of two. Finally, it is possible that life arose starting with polymers lacking chirality and only later evolved to include chiral polymers like RNA and proteins.
Two of the abiological processes postulated to produce enantiomeric excesses are the weak nuclear force and the action of circularly polarized light. Neither idea, however, appears all that compelling. In the 1950s, it was discovered that β-particles, energetic electrons emitted from atomic nuclei during some forms of radioactive decay, are preferentially emitted with a specific handedness (in terms of their direction of travel and direction of spin). This implies that the weak nuclear force, the force that holds nuclei together, is itself asymmetric, an effect that could, potentially, have implications in the origins of homochirality. Despite intensive investigations, however, no one has demonstrated the selective degradation of one enantiomer over another through the effects of β-radiation, so its role in prebiotic chemistry must be seriously questioned. The theory suggesting that circularly polarized light produced enantiomeric excesses on the early Earth is on perhaps better footing, but even this one is only very weakly supported by laboratory experiments. As a charged object moving through a magnetic field, an electron traveling through the intense magnetic fields of a neutron star will spiral, causing it, per Maxwell’s equations, to emit light, typically, under these conditions, as UV light. Due to the spiral motion of the electrons, the light will be circularly polarized. That is, the electric field vector that describes it oscillates in a circular fashion, as opposed to the more well-known plane polarization caused by your sunglasses, in which the electric field vector of the polarized light is constrained within a plane. The two enantiomers of a chiral molecule absorb circularly polarized light to different extents. Given this, it has been postulated that if (1) there were a nearby neutron star producing copious amounts of circularly polarized UV light, and (2) the prebiotic precursors to life arose in the pre-solar nebula (where they would feel the full brunt of any UV light), then this might account for, for example, the selection of l-amino acids and d-ribose on Earth. Laboratory studies of this mechanism, however, indicate that it is a very weak effect. When amino acids were blasted with so much UV light that 99% of all the molecules (of both enantiomers) were destroyed, the remaining dregs of amino acids showed enantiomeric enrichment of at most only a few percent. If such an effect is a mandatory step in the formation of life, then the origins of life were a fortuitous event indeed.
Given the lack of plausible mechanisms for generating homochiral monomers without biology,* researchers are now focusing on mechanisms that might amplify small enantiomeric excesses (caused, for example, by small, random fluctuations) into larger excesses. These include autocatalytic reactions in which the product of a reaction catalyzes its own production or inhibits the formation of the other enantiomer. The “Soai reaction,” for example, produces enantiomerically enriched products even when starting from a seemingly completely racemic mixture of materials. This said, this reaction is of no specific relevance to prebiotic chemistry, and no plausibly prebiotic reaction has yet been reported that achieves similar autocatalytic production of near homochirality. Alternatively, Donna Blackmond and her team at the Scripps Institute in California have shown that, if two enantiomers that can interconvert in solution but preferentially form only single-enantiomer crystals, then a mixture of crystals of both enantiomers will spontaneously resolve into a single handedness given some energetic input (such as grinding) via a process called “Viedma ripening,” in which one crystal form, and thus one enantiomer, eventually wins over time.
Still another mechanism potentially supporting chiral amplification would be catalysts that could selectively polymerize only l or pure d monomers out of a mixture of the two. To this end, Kenso Soai of Tokyo University has shown that, when added to various non-polymerization reactions that normally produce 50:50 mixtures of l and d products, pure, powdered d- or pure, powdered l-quartz* can produce significant excesses of one handedness over the other. As yet, however, this has not been shown to hold for RNA polymerization on mineral surfaces.
Given the lack of any clear solution to the “chirality problem,” several researchers have proposed that there was a pre-RNA world in which a nonchiral, RNA-like polymer was the basis of the first life, and chiral RNA came into the picture only later, under the influence of selective pressures. Several potential pre-RNA polymers have been suggested. For example, Albert Eschenmoser, then at the Scripps Research Institute in La Jolla, California, demonstrated that nucleotides containing the sugar threose can be strung together into RNA-like polymers that form complementary duplexes, and he has argued that threose, being a four-carbon sugar, is likely to be synthesized in greater yield than the five-carbon ribose under prebiotic conditions. Threose, however, is also chiral, so Eschenmoser’s TNAs (threose nucleic acids) do not solve the homochirality problem. In contrast, Pernilla Wittung, while working in Peter Nielsen’s laboratory in Copenhagen, demonstrated in the early 1990s that a nucleic acid composed of a nonchiral, polypeptide-like backbone—peptide nucleic acid, or PNA, composed of nucleobases linked via the molecule N-(2-aminoethyl)glycine, which is produced in the Miller-Urey experiment—can couple with itself to form stable double helices. Other than RNA, however, no polymer has ever been demonstrated in the laboratory to support template-directed polymerization of like polymers in the absence of protein catalysts, and so speculation that there was a pre-RNA world remains just that: speculation.
Conclusions
In 1859, Louis Pasteur won the French Academy’s prize for showing us how life does not start. And now, more than a century and a half on, we still do not know how it does. Self-replicating metabolic pathways? Self-replicating clays? An RNA (or RNA-like) replicase that can copy a copy of itself? We simply do not know, because every one of these theories faces major, unsolved hurdles. But, at least in the RNA-world hypothesis, we have a chemically and biologically plausible—if perhaps quantitatively improbable—theory concerning our origins.
Further Reading
Metabolism-First Hypothesis
Orgel, Leslie E. “Self-Organizing Biochemical Cycles.” Proceedings of the National Academy of Sciences USA 97, no. 23 (2000): 12503–07.
The FeS World
Cody, George D. “Transition Metal Sulfides and the Origins of Metabolism.” Annual Review of Earth and Planetary Sciences 32 (2004): 569–99.
The Lipid World
Lancet, Doron, Daniel Segre, and Amit Kahana. “Twenty Years of ‘Lipid World’: A Fertile Partnership with David Deamer.” Life (Basel) 9, no 4 (2019): 77–88.
The RNA World
Atkins, John F., Raymond F. Gesteland, and Thomas R. Cech, eds. RNA Worlds: From Life’s Origins to Diversity in Gene Regulation. Woodbury, NY: Cold Spring Harbor Laboratory Press, 2011.
RNA Polymerase Ribozymes
Horning, David P., and Gerald F. Joyce. “Amplification of RNA by an RNA Polymerase Ribozyme.” Proceedings of the National Academy of Sciences USA 113, no. 35 (2016): 9786–91.
Johnston, Wendy K., Peter J. Unrau, Michael S. Lawrence, Margaret E. Glasner, and David P. Bartel. “RNA-Catalyzed RNA Polymerization: Accurate and General RNA-Templated Primer Extension.” Science 292, no. 5520 (2001): 1319–25.
Protocells and RNA Self-Replication
Joyce, Gerald F., and Jack W. Szostak. “Protocells and RNA self-Replication.” Cold Spring Harbor Perspectives in Biology 10, no. 9 (2018): a034801.
Szostak, Jack W. “The Narrow Road to the Deep Past: In Search of the Chemistry of the Origin of Life.” Angewandte Chemie 56, no. 37 (2017): 11037–43.
The Origins of Chirality
Blackmond, Donna G. “The Origin of Biological Homochirality.” Cold Spring Harbor Perspectives in Biology 2, no. 5 (2010): a002147.
- * In a lecture at the Sorbonne in Paris in 1864; quoted by his son-in-law, René Vallery-Radot, in his book Life of Pasteur (New York: Doubleday, Page and Co., 1919), 109.
- ** As quoted in Charles Darwin, Francis Darwin, and A. C. Seward, More Letters of Charles Darwin: Volume 1 (London: J. Murray, 1903), 273.
- * Quoted in F. Darwin, ed., The Life and Letters of Charles Darwin (New York: Basic Books, 1898), II: 202–3.
- * Though sometimes, midway through a dull work week, it may not seem so.
- * Daniel Segré and Doron Lancet, “Composing Life,” EMBO Reports 1, no. 3 (2000): 217–22.
- ** We should note that, over a beer or two, Lancet and Deamer have argued to us that their lipid-world hypothesis is a “genes-first” (discussed next) rather than “metabolism-first” hypothesis, but to our thinking, their model envisions a cooperative, complex network of interactions between lipids and not the one-to-one sort of templating we think of when we say gene.
- * The internet, with its complex network of servers, is similarly robust. If any single node is knocked out, others are there to take its place. And thus, changing one node does not change the network itself. In fact, the internet was originally built under funding from the US Department of Defense as a communications network that could survive nuclear war.
- * This said, a few percent excesses of one handedness have been identified among some of the amino acids found in the Murchison meteorite, including in some of the nonproteinogenic amino acids, suggesting that this asymmetry is not due to biological contamination that occurred on Earth. The origin of these small, if tantalizing, asymmetries remains completely unknown.
- * The silica (SiO2) units that make up quartz are not, themselves, chiral, but the crystal forms that quartz adopts are. When molten silica crystallizes, it forms a 50:50 mixture of left- and right-handed crystals. Next time you’re shopping in a New Age store, be sure to check out its collection of quartz crystals and see whether you can identify the two mirror-image crystal forms.