Chapter 14. Making a Fully Reproducible Paper
Throughout this book, you’ve been learning how to use an array of individual tools and components to perform specific tasks. You now know everything you need to get work done—in small pieces. In this final chapter, we walk you through a case study that demonstrates how to bring all of the pieces together into an end-to-end analysis, with the added bonus of showcasing methods for ensuring full computational reproducibility.
The challenge posed in the case study is to reproduce a published analysis in which researchers identified the contribution of a particular gene to the risk for a form of congenital heart disease. The original study was performed on controlled-access data, so the first part of the challenge is to generate a synthetic dataset that can be substituted for the original. Then, we must re-create the data processing and analysis, which include variant discovery, effect prediction, prioritization, and clustering based on the information provided in the paper. Finally, we must apply these methods to the synthetic dataset and evaluate whether we can successfully reproduce the original result. In the course of the chapter, we derive lessons from the challenges we face that should guide you in your efforts to make your own work computationally reproducible.
Overview of the Case Study
We originally conceived this case study as a basis for a couple of workshops that we had proposed to deliver at conferences, starting with the general meeting of the American Society for Human Genetics (ASHG) in October 2018. The basic premise of our workshop proposal was that we would evaluate the barriers to computational reproducibility of genomic methods as they are commonly applied to human medical genetics and teach the audience to overcome those barriers.
We started out with certain expectations about some of the key challenges that might affect both authors and readers of genomic studies, and we were aware of existing solutions for most of those challenges. Our primary goal, therefore, was to highlight real occurrences of those known challenges and demonstrate how to overcome them in practice, based on approaches and principles recommended by experts in the open-science movement. We would then develop educational materials with the ultimate goal of popularizing a set of good practices for researchers to apply when publishing their own work.
Through a series of circumstances that we describe shortly, we selected a study about genetic risk factors for a type of congenital heart disease and worked with one of its lead authors, Dr. Matthieu J. Miossec, to reproduce the computational analysis at the heart of the paper (so to speak). In the course of the project, we verified some of our assumptions, but we also encountered obstacles that we had not foreseen. As a result, we learned quite a bit more than we had expected to, which is not the worst outcome one could imagine.
In this first section of the chapter, we set the stage by discussing the principles that guided our decision making, and then we start populating that stage by describing the research study that we set out to reproduce. We discuss the challenges that we initially identified and describe the logic that we applied to tackle them. Finally, we give you an overview of our implementation plans, as a prelude to the deep-dive sections in which we’ll examine the nitty-gritty details of each phase of the project.1
Computational Reproducibility and the FAIR Framework
Before we get into the specifics of the analysis that we sought to reproduce, it’s worth restating what we mean by reproducibility and making sure we distinguish it from replication. We’ve seen these terms used differently, sometimes even swapped for each other, and it’s not clear that any consensus exists on the ultimate correct usage. So let’s define them within the scope of this book. If you encounter these terms in a different context, we encourage you to verify the author’s intent.
We define reproducibility with a focus on the repeatability of the analysis process and results. When we say we’re reproducing an analysis, we’re trying to verify that if we put the same inputs through the same processing, we’ll get the same results as we (or someone else) did the first time. This is something we need to be able to do in order to build on someone else’s technical work, because we typically need to make sure that we’re running their analysis correctly before we can begin extending it for our own purposes. As such, it’s an absolutely vital accelerator of scientific progress. It’s also essential for training purposes, because when we give a learner an exercise, we usually need to ensure that they can arrive at the expected result if they follow instructions to the letter—unless the point is to demonstrate nondeterministic processes. Hopefully, you’ve found the exercises in this book to be reproducible!
Replication, on the other hand, is all about confirming the insights derived from experimental results (with apologies to Karl Popper, legendary contrarian). To replicate the findings of a study, we typically want to apply different approaches that are not likely to be subject to the same weaknesses or artifacts, to avoid simply falling in the same traps. Ideally, we’ll also want to examine data that was independently collected, to avoid confirming any biases originating at that stage. If the results still lead us to draw the same conclusions, we can say we’ve replicated the findings of the original work.
This difference, illustrated in Figure 14-1, comes down to plumbing versus truth. In one case, we are trying to verify that “the thing runs as expected,” and in the other, “yes, this is how this small part of nature works.”
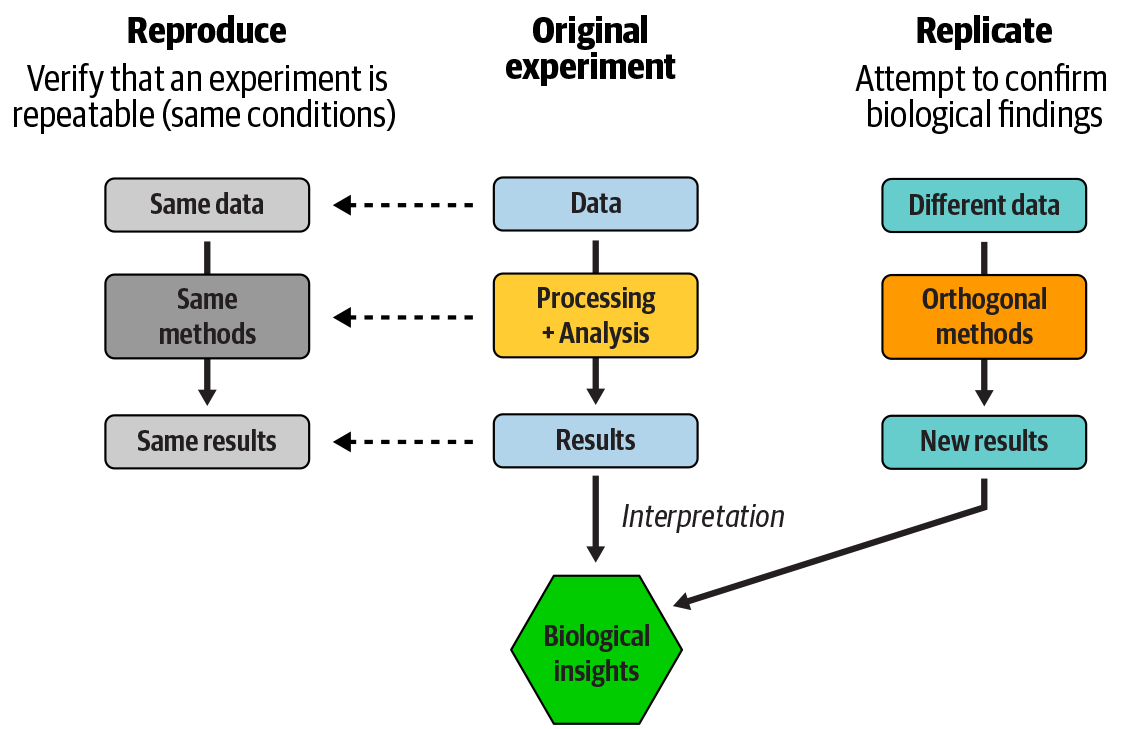
Figure 14-1. Reproducibility of an analysis versus replicability of study findings.
Those definitions shouldn’t sound too outlandish given that we’ve already mentioned this core concept of reproducibility in earlier chapters. For example, in Chapter 8, when we introduced workflows, we noted their great value as a way to encode the instructions for performing a complex analysis in a systematic, automatable way. We also touched on similar themes when we explored Jupyter Notebook as a more flexible, interactive approach to packaging analysis code. We’re confident that you won’t be surprised when both workflows and notebooks make an appearance later in this chapter.
However, reproducibility is only one facet of the kaleidoscope of open science. As we set out to tackle this case study, we made a deliberate decision to look at it through the lens of the FAIR framework. As we mentioned briefly in Chapter 1, FAIR is an acronym that stands for findability, accessibility, interoperability, and reusability. It refers to a framework for evaluating the openness of research assets and services based on those four characteristics. The underlying idea is simple enough: all four pillars are requirements that must be satisfied for an asset to be considered open (as in open science). For example, to consider that an analysis is open, it is not enough for the code to be technically reusable and interoperable with other computing tools; other researchers should also be able to find the code and obtain a complete working copy. The original publication of the FAIR principles highlighted the necessity of applying them to scientific data management purposes, but it also made clear that they could be applied to a variety of other types of assets or digital research objects such as code and tools.
Although the bulk of the case study is focused on reproducibility, which we take to be mostly synonymous with reusability as spelled out in the FAIR framework, the three other FAIR characteristics will be reflected in our various implementation decisions. We’ll circle back to this when we go over the methodology that we chose to follow, then again at the conclusion of the chapter when we discuss the final outcomes of this work. For now, it’s time to take a look at the original research study that we chose to showcase in this project.
Original Research Study and History of the Case Study
The original study was authored by Drs. Donna J. Page, Matthieu J. Miossec, et al., who set out to identify genetic components associated with a form of congenital heart disease called nonsyndromic tetralogy of Fallot by analyzing exome sequencing data from 829 cases and 1,252 controls collected from multiple research centers.2 We go over the analysis in more detail further down, but to summarize for now, they first applied variant discovery methods based on the GATK Best Practices to call variants across the full set of samples (including both cases and controls), and then they used functional effect prediction to identify probable deleterious variants. Finally, they ran a variant load analysis to identify genes more frequently affected by deleterious variants in case samples compared to the controls.
As a result of this analysis, the authors identified 49 deleterious variants within the NOTCH1 gene that appeared to associate with the tetralogy of Fallot congenital heart disease. Others had previously identified NOTCH1 variants in families with congenital heart defects, including tetralogy of Fallot, so this was not a wholly unexpected result. However, this work was the first to scale variant analysis of tetralogy of Fallot to a cohort of nearly a thousand case samples, and to show that NOTCH1 is a significant contributor to tetralogy of Fallot risk. As of this writing, it is still the largest exome study of non-syndromic tetralogy of Fallot that we are aware of.
We connected with Dr. Miossec about a workshop on genomic analysis that we were developing for a joint conference of several societies in Latin America, ISCB-LA SOIBIO EMBnet, to be held in Chile in November in 2018, while the manuscript was still at the preprint stage. At time, our team had also committed to developing a case study on computational reproducibility for the ASHG 2018 workshop, mentioned earlier, which was scheduled for October. Serendipitously, Dr. Miossec’s preprint was a great fit for both purposes because it was typical of what our intended audience would find relatable: (1) a classic use case for variant discovery and association methods, (2) performed in a cohort of participants that was large enough to pose some scaling challenges but small enough to be within the means of most research groups, and (3) using exome sequencing data as was most common at the time. Given the Goldilocks-like “just right” character of the study itself and Dr. Miossec’s collaborative attitude during our initial discussions of the ISCB-LA SOIBIO EMBnet workshop development project, we approached him about building a case study from his preprint as basis for both workshops. We then worked together over the following months to develop the case study that we eventually delivered at the ASHG 2018 meeting and at the ISCB-LA SOIBIO EMBnet 2018 conference, respectively.
Note
To be clear, the selection of this study is not intended to demonstrate GATK Best Practices; technically, the authors’ original implementation shows deviations from the canonical Best Practices. In addition, this does not constitute an endorsement of the biological validity of the study. In this case study, we are focused solely on the question of computational reproducibility of the analysis.
Assessing the Available Information and Key Challenges
Before we even had the Tetralogy of Fallot study selected, we had been reviewing the key challenges that we expected to face based on our experience with scientific publishing. As illustrated in Figure 14-2, a fundamental asymmetry exists between the information and means available to the author of a scientific paper compared to what is available to the reader. As an author, you generally have full access to an original dataset, which in the case of human genomic data is almost always locked down by data access and data-use restrictions. You have your computing environment, which might be customized with a variety of hardware and software components, and you have tools or code that you apply to the data within that environment to generate results.
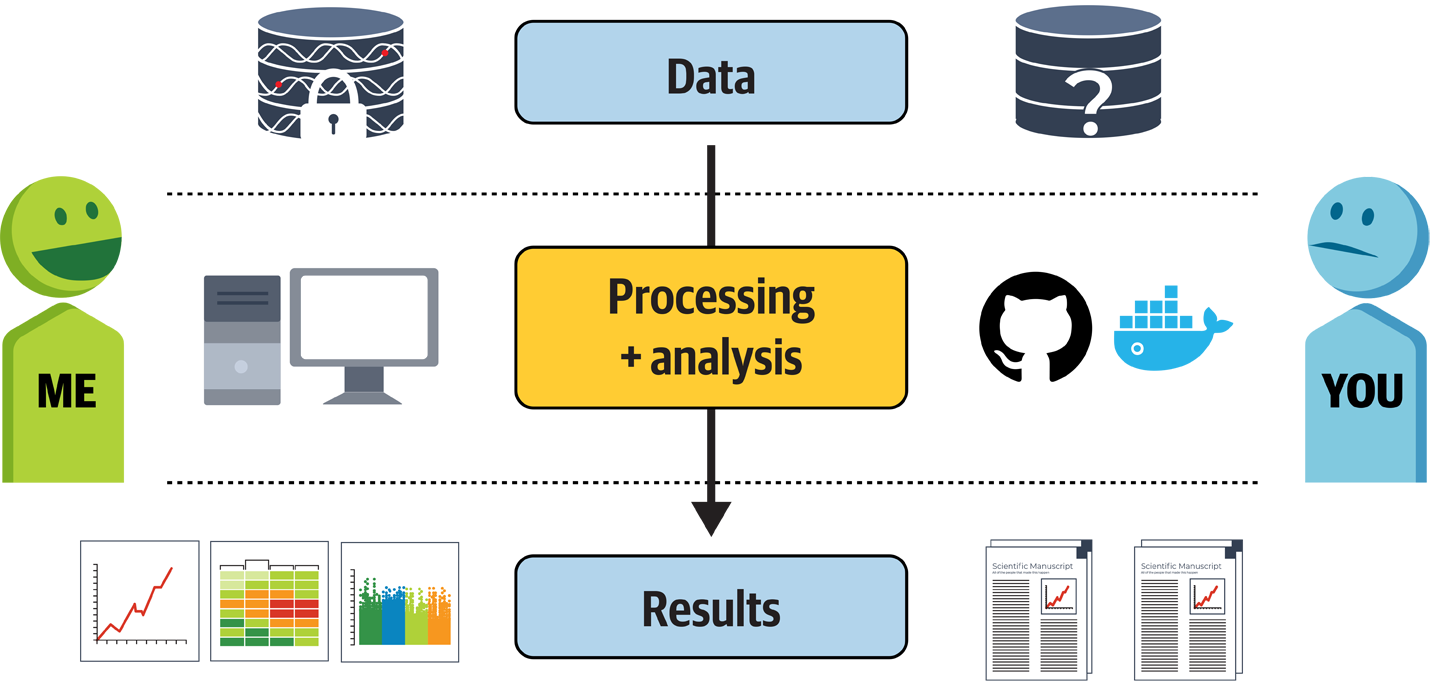
Figure 14-2. Typical asymmetry in the availability of information between author and reader.
On the other side, however, when you’re a reader, you usually first see the preprint or published paper, which is in itself a highly processed, curated, and, to some extent, censored view of the author’s original results. From the paper, you must walk backward up the stream to find a description of the methods that were applied, which are often incomplete, as in the dreaded “We called variants with GATK Best Practices as described in the online documentation.” (No indication of version, specific tools, nada.) If you’re lucky, an unformatted PDF buried in the supplemental materials includes a link to code or scripts in a GitHub repository, which might or might not be documented. If you’re extremely lucky, you might even find a reference to a Docker container image that encapsulates the software environment requirements, or at least a Dockerfile to generate one. Finally, you might find that the data is subject to access restrictions that could prove unsurmountable. You might be able to cobble together the analysis based on the available information, code, and software resources in the paper—which, to be fair, can sometimes be quite well presented—but without the original data, you have no way to know whether it’s running as you expect. So when you go to apply it to your own data, it can be difficult to know how much you can trust the results it produces.
Now that we’ve painted such a grim picture, how did our situation measure up? Well, the preprint that was available at the time for the Tetralogy of Fallot paper fell squarely within the norm. Figure 14-3 summarizes the methodological information it contained.
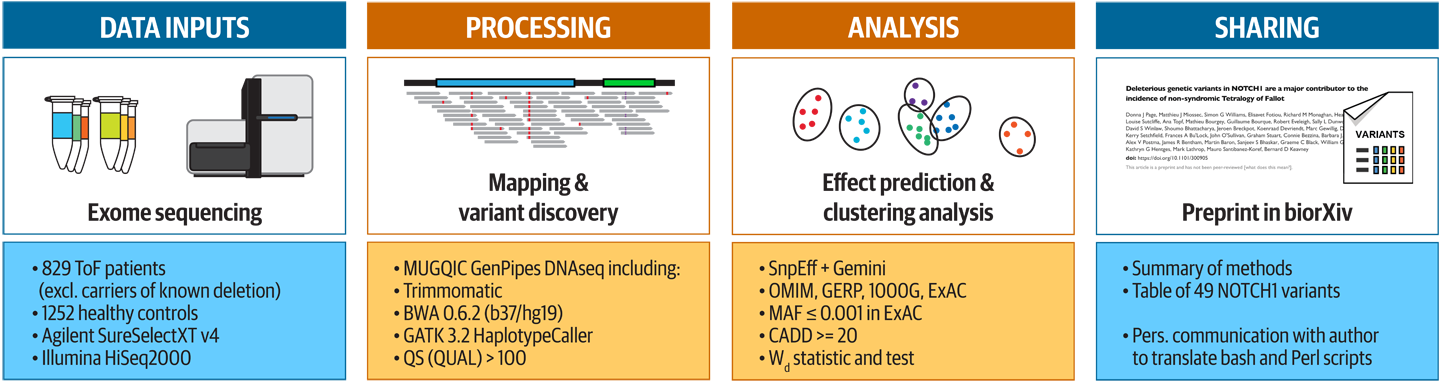
Figure 14-3. Summary of the information provided in the original preprint of the Tetralogy of Fallot paper.
The description of the methods used in the early stages of data processing (alignment up to variant calling) referenced a third-party pipeline operated at another institution, which was definitely too vague to be sufficient to reproduce the work exactly. On the bright side, it did reference key tools and their versions, such as HaplotypeCaller from GATK 3.2, and we knew enough about those tools to figure out their requirements by ourselves. For later parts of the analysis, the method descriptions were more detailed and pointed to scripts in Bash and Perl code, but these were still not entirely sufficient by themselves.
Note
Spoiler alert: we benefited immensely from having a lead author in our corner with detailed knowledge of how the analysis was done; we would not have been successful without direct help from Dr. Miossec.
Where we hit the wall, however, was entirely predictable: we knew early on that the data was restricted-access, and as it turned out, we were not able to gain access to any of it at any point, even for development purposes. But hang on to your hat; that doesn’t mean the project was dead. We get to solutions in a few minutes.
Designing a Reproducible Implementation
Given these initial conditions, we began thinking about how we were going to tackle the project in practice. We wanted to use a methodology that would allow us not only to reproduce the analysis ourselves, but also to package the resulting implementation for others to reproduce. To that end, we followed the guidance provided by Justin Kitzes et al. in The Practice of Reproducible Research (University of California Press, 2018). You can learn a lot from their book that we won’t cover here, so we recommend that you check it out. One of the takeaways we quickly implemented was to break down the overall work based on the following questions:
-
What is the input data, and how are we going to make that accessible to others?
-
What (a) processing and (b) analysis methods need to be applied to the data, and how can we make them portable?
-
How can we bundle all the assets we produce and share them for others to use?
This was a simple but effective framework that reminded us of our priorities and guided our decisions. The distinction between processing and analysis methods was particularly useful after we agreed what those terms should mean. Processing would refer to all the up-front data transformations, cleaning up, format changes, and so on that are always the same and are typically automated to be run in bulk. This corresponds roughly to what is often described as secondary analysis. Meanwhile, analysis proper would refer to the more changing, context-dependent activities like modeling and clustering that are typically done interactively, on a case-by-case basis. This corresponds to tertiary analysis. There was some gray area regarding variant calling and effect prediction, but we ultimately decided that variant calling should fall into the processing bucket, whereas variant effect prediction would go into the analysis bucket.
Although those distinctions were somewhat arbitrary in nature, they were important to us because we had decided that they would guide our efforts regarding how closely we would seek to adhere to the original study. Obviously, we wanted to be as close to the original as possible, but we knew by then that some of the information was incomplete. It was clear that we would need to make compromises to strike a balance between achieving perfection and the amount of effort that we could afford to devote to the project, which was sadly not infinite. So we made a few assumptions. We posited that (1) basic data processing like alignment and variant calling should be fairly robust to adaptations as long as it is applied the same way to all of the data, avoiding batch effects, and that (2) the effect prediction and variant load analysis at the core of the study were more critical to reproduce exactly.
With these guiding principles in mind, we outlined the following implementation strategy:
-
Data input: To deal with the locked-down data, we decided to generate synthetic exome data that would emulate the properties of the original dataset, down to the variants of interest. The synthetic data would be completely open and therefore satisfy the Accessibility and Reusability requirements of the FAIR framework.
-
Data processing and analysis
-
Processing: Because the synthetic data would be fully aligned already, as we discuss shortly, we would need to implement only variant calling in this phase. Based on what we knew of the original pipeline, we decided to use a combination of existing and new workflows written in WDL. The open and portable nature of WDL would make the processing accessible, interoperable, and reusable from the perspective of FAIR.
-
Analysis: With Dr. Miossec’s help, we decided to reimplement his original scripts in two parts: the prediction of variant effects as a workflow in WDL, and the variant load analysis as R code in a Jupyter notebook. As with WDL workflows, using Jupyter notebooks would make the analysis accessible, interoperable, and reusable from the perspective of FAIR.
-
-
Sharing: We planned to do all the work in a Terra workspace, which we could then share with the community to provide full access to data, WDL workflows, and Jupyter notebooks. This easy sharing in Terra would help us round out the FAIR sweep by making our case study findable and accessible from the perspective of FAIR.
That last point was a nice bonus of doing the original work for this case study in Terra: we knew that after we were done, we could easily clone the development workspace to make a public workspace that would contain all of the workflows, notebook, and references to data. As a result, we would have only minimal work to do to turn our private development environment into a fully reproducible methods supplement.
In the next two sections, we explain how we implemented all of this in practice. At each stage, we point you to the specific elements of code and data that we developed in the workspace so that you can see exactly what we’re talking about. We’re not going to have you run anything as part of this chapter, but feel free to clone the workspace and try out the various workflows and notebook anyway.
Now, are you ready to dive into the details? Saddle up and fasten your seatbelt, because we’re going to start with what turned out to be the most challenging part: creating the synthetic dataset.
Generating a Synthetic Dataset as a Stand-In for the Private Data
The original analysis involved 829 cases and 1,252 controls, all produced using exome sequencing, and, unfortunately for us, all subject to access restrictions that we could not readily overcome. Indeed, even if we had been able to access the data, we would definitely not have been able to distribute it with the educational resources that we planned to develop. It would still have been useful to have the data available for testing, but in any case, we were going to need to resort to synthetic data to completely achieve our goals.
But wait...what does that mean exactly, you ask? In general, that can mean a lot of things, so let’s clarify that here, we’re specifically referring to synthetic sequence data: a dataset made entirely of read sequences generated by a computer program. The idea was to create such a synthetic dataset that would mimic the original data, as illustrated in Figure 14-4, including the presence of the variants of interest in specific proportions, so that we could swap it in and still achieve our goal of reproducing the analysis despite lacking access to the original data.
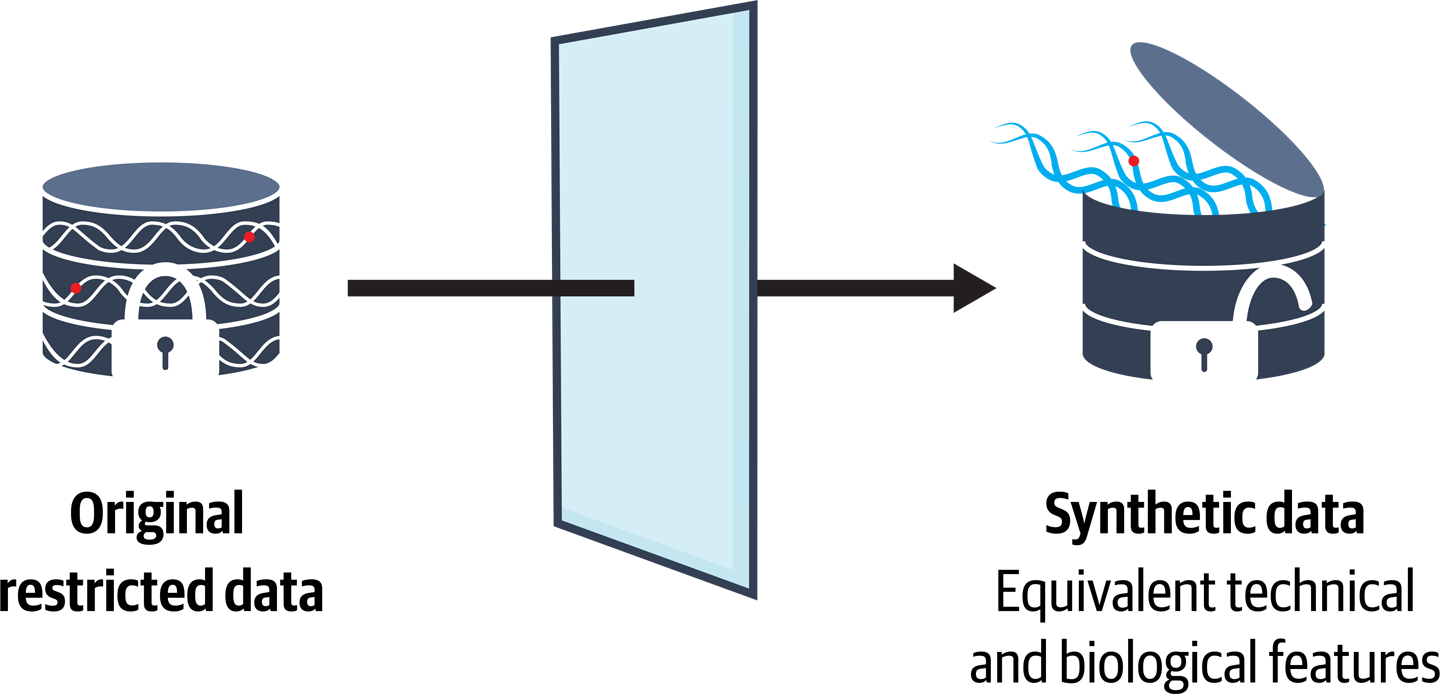
Figure 14-4. Replacing a real dataset that cannot be distributed with a synthetic dataset that mimics the original data’s characteristics.
Let’s talk about how that works in practice.
Overall Methodology
The idea of using synthetic genomic data is not new, far from it; people have been using synthetic data for some time. For example, the ICGC-TCGA DREAM Mutation challenges were a series of recurring competitions in which organizers provided synthetic data that had been engineered to carry specific known variants, and participants were challenged to develop analysis methods that could identify those mutations with a high degree of accuracy and specificity.
Multiple programs can generate synthetic sequence data for purposes like this; in fact, several were developed in part to enable those challenges. The basic mechanism is to simulate reads based on the sequence of the reference genome and output these as standard FASTQ or BAM files. The tools typically accept a VCF of variant calls as secondary input, which modifies the data simulation algorithm in such a way that the resulting sequence data supports the variants contained in the input VCF. There are also programs that can introduce (or spike in) variants into sequence data that already exists. You provide a set of variants to the tool, which then modifies a subset of reads to make them support the desired variant calls.
In fact, in an early round of brainstorming, we had considered simply editing real samples from the 1000 Genomes Project to carry the variants of interest, and have the rest of the dataset stand in as controls. This would allow us to avoid having to generate actual synthetic data. However, our preliminary testing showed that the exome data from the Low Coverage dataset was not of good-enough quality to serve our purposes. At the time, the new High Coverage dataset introduced in Chapter 13 was not yet available, and because that data was generated using WGS, not exome sequencing, it would not have been appropriate for us to use anyway.
We therefore decided to create a set of synthetic exomes, but base them on the 1000 Genomes Phase 3 variant calls (one per project participant) so that they would have realistic variant profiles. This would essentially amount to reconstructing sequencing data for real people based on their previously identified variants. We would then mutate a subset of the synthetic exomes, assigned as cases, with the variants that Dr. Miossec et al. discovered in the Tetralogy of Fallot study, which were listed in the paper. Because there were more participants in the 1000 Genomes dataset than in the Tetralogy of Fallot cohort, that would give us enough elbow room to generate as many samples as we needed. Figure 14-5 presents the key steps in this process.
In theory, this approach seemed fairly straightforward but, as we rapidly learned, the reality of implementing this strategy (especially at the scale that we required) was not trivial. The packages we ended up using were very effective and for the most part quite well documented, but they were not very easy to use out of the box. In our observation, these tools tend to be used mostly by savvy tool developers for small-scale testing and benchmarking purposes, which led us to wonder how much of the high threshold of effort was a consequence of having an expert target audience versus the cause of their limited spread to less savvy users. In any case, we have not seen biomedical researchers use them individually for providing the kind of reproducible research supplements that we were envisioning, and in retrospect we are not surprised given the difficulties we had to overcome in our project. As we discuss later, this further motivated us to consider how we could capitalize on the results that we achieved to make it easier for others to adopt the model of providing synthetic data as a companion to a research study.
In the next section, we expose the gory details of how we implemented this part of the work, occasionally pausing to provide additional color around specific challenges that we feel can provide either valuable insight for others or, failing that, a touch of comic relief to keep things light.
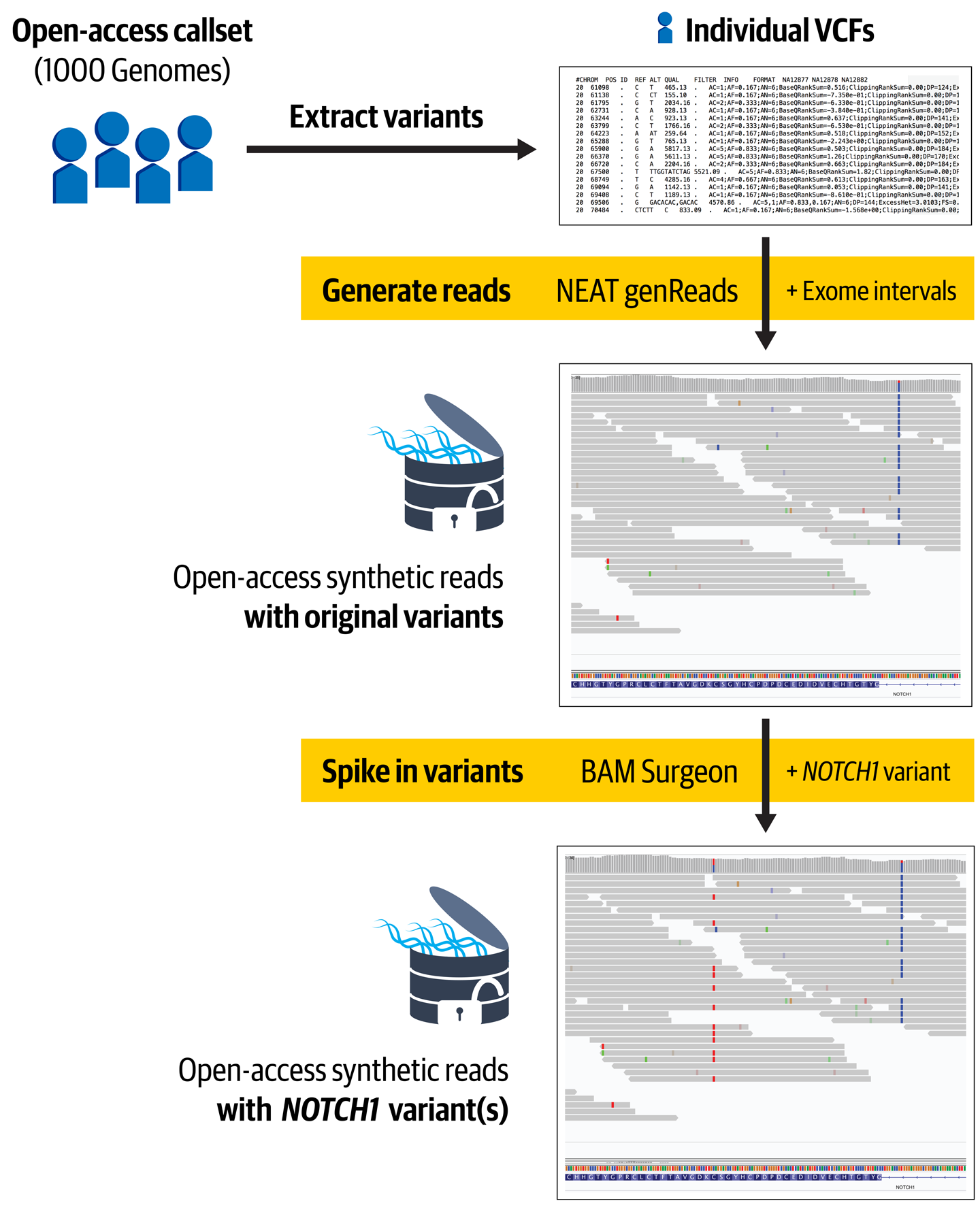
Figure 14-5. Overview of our implementation for generating appropriate synthetic data.
Retrieving the Variant Data from 1000 Genomes Participants
As we mentioned earlier, we decided to base the synthetic data simulation step on VCFs from participants from the 1000 Genomes Project. We chose that dataset because it was the largest available genomics dataset that was fully public, and a copy was freely available in GCS. The convenience stopped there, however. We had to jump through hoops from the get-go, first because the 1000 Genomes variant calls were provided at the time in the form of multisample VCFs containing variant calls from all project participants, split up by chromosome. For our purposes, we needed the opposite: a single-sample VCF containing all chromosomes’ worth of data for each project participant.
So the first thing we implemented was a WDL workflow that, given a participant’s identifier, would use GATK SelectVariants to extract the variant calls for the participant from each of the per-chromosome files, and then Picard MergeGvcfs to merge the data into a single VCF for that participant. This sounds simple enough, right? Well, yes, but this is bioinformatics, so of course things weren’t that simple. We ran into weird errors due to malformed file headers when trying to process the original multisample VCFs, so we had to add in a call to Picard FixVcfHeader to patch them up. Then, we ran into sorting errors after the SelectVariants step, so we had to add a call to Picard SortVcf to address those issues. We crammed all three commands into a single WDL step to avoid having to do a lot of back-and-forth file copying. It wasn’t pretty, but it worked. That became workflow 1_Collect-1000G-participants, which you can see in action in the case study workspace. For all tasks in this workflow, we used the standard GATK4 container image, which also contains Picard tools.
To run this in our workspace, we set up the participant table with minimal metadata from the 1000 Genomes Project, including participant identifiers and the cohort they originally belonged to, in case the latter came in handy down the line (it hasn’t). Then, we configured the collector workflow to run per row in the table of participants and output the resulting VCF to the same row under a sampleVcf column.
Note
In this workspace, we didn’t need the distinction between participant and sample, so we decided to use a very simple and flat data structure that would use only one of the two. At the time, the participant table was mandatory, so we had no choice but to use it. If we were to rebuild this today, we might switch to using samples instead to make our tables more directly compatible with what we have in other workspaces.
The workflow suffers from a few major inefficiencies, so it took a while to run (4.5 hours on average, 12 hours for a set of 100 files run in parallel, which includes some preemptions) but it did produce the results we were looking for: a complete individual VCF for each participant. With that in hand, we could now move on to the next step: generating the synthetic data!
Creating Fake Exomes Based on Real People
We wanted to create synthetic exome data that would stand in for the original access-controlled dataset, so our first task here was to choose a toolkit with the appropriate features because, as we mentioned earlier, there are quite a few and they display a fairly wide range of capabilities. After surveying available options, we selected the NEAT-genReads toolkit, an open source package developed by Zachary Stephens et al. It had a lot of features that we didn’t really need, but it seemed to be well regarded and had the core capability we were looking for: the ability to generate either whole genome or exome data based on an input VCF of existing variant calls (in our case, the 1000 Genome participant VCFs).
How does it work? In a nutshell, the program simulates synthetic reads based on the reference genome sequence and then modifies the bases in a subset of reads at the positions where variants are present in the input VCF, in a proportion that matches the genotype in the VCF.
Figure 14-6 illustrates this process. In addition, the program is capable of simulating sequencing errors and other artifacts by modifying other bases in reads, either randomly or based on an error model. By default, the program outputs FASTQ files containing unaligned synthetic sequence data, a BAM file containing the same data aligned to the reference genome, and a VCF file that serves as a truth set containing the variants that the program introduced into the data. For more information on how this works and other options, see the GitHub repository referenced earlier as well as the original publication describing the toolkit in PLoS ONE.
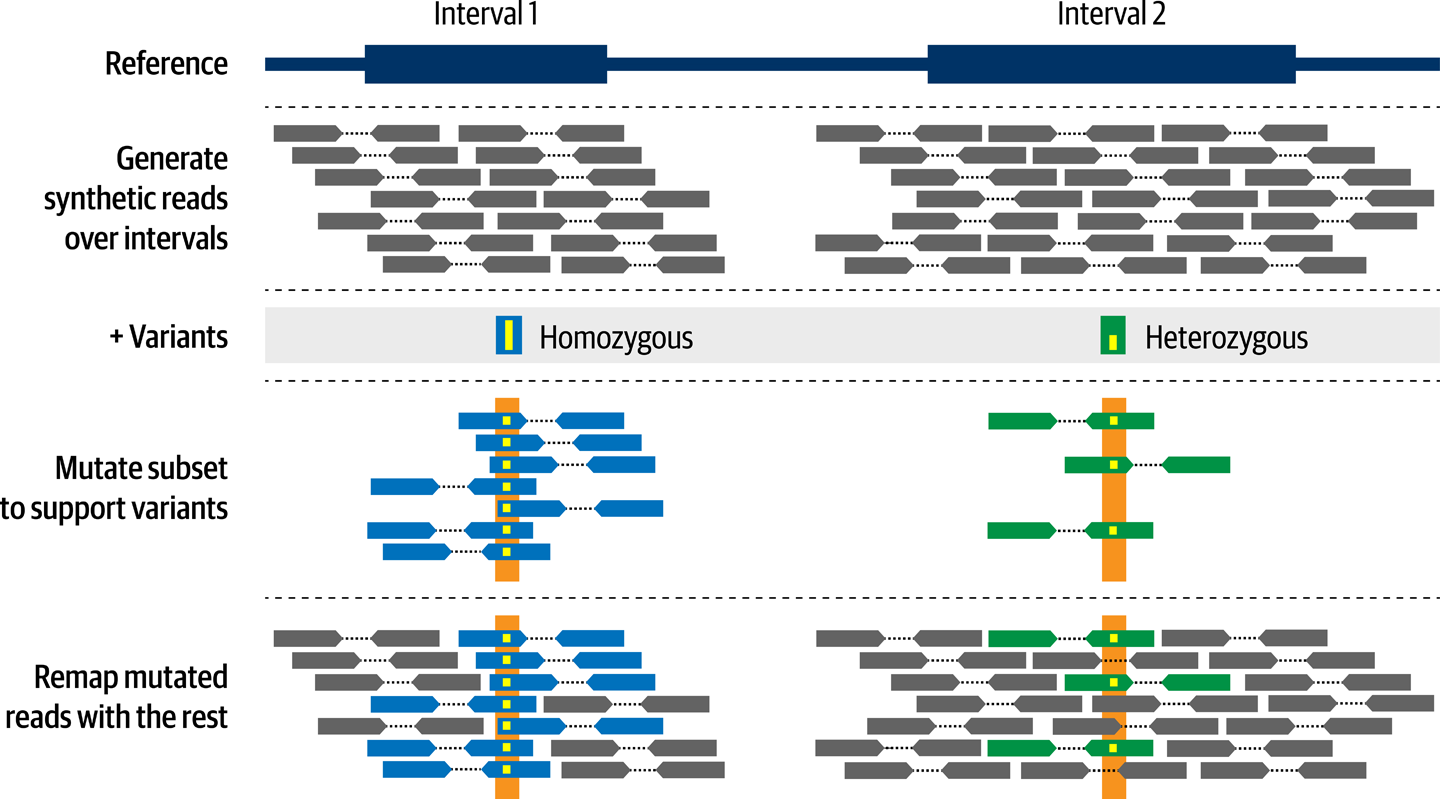
Figure 14-6. NEAT-genReads creates simulated read data based on a reference genome and list of variants.
In addition to this core functionality, NEAT-genReads accepts various parameters that control different aspects of the simulated data. For example, you can specify which regions should be covered or not using an intervals file, and at what target coverage they should be covered. You can also control read length, fragment length, and so on, as well as the mutation rate to use for errors and spontaneous mutations. We took advantage of all of these to tailor the resulting synthetic data to resemble the original study cohort as closely as possible based on the information available in the preprint.
To implement this, we developed the WDL workflow 2_Generate-synthetic-reads, which takes a reference genome and a VCF of variants to use in generating the sequence read, and produces a BAM file as well as the truth VCF file. Again, you can see it in action in the workspace.
We’d love to say that this one went more smoothly than the first workflow, but no. We ran into even more problems that appeared to be due to either malformed data or the tools choking on elements that are allowed by the VCF format specification. For example, NEAT-genReads did not tolerate variant records that had multiple identifiers in the rsID field, so we had to apply an awk command to sanitize the VCFs by removing the contents of the rsID field from every record. Another fun one was that NEAT-genReads emitted incomplete VCF headers in the truth VCFs, so again we trotted out Picard FixVcfHeader. It also occasionally emitted malformed read names, so to fix that we deployed an unholy combination of awk and samtools. Finally, we also had to add readgroup information to the BAM files produced by NEAT-genReads, which clearly didn’t consider that metadata important (GATK responds: blasphemy!).
Is your head spinning yet? Ours certainly was. Don’t worry if you’re not familiar with the more nitty-gritty details, though. Ultimately, the point is that after you get into a certain level of rolling your own analysis, this is the kind of thing you’re likely to encounter, even with a great toolkit like NEAT-genReads (yes, we really like it, warts and all). What’s worse is that this is the kind of thing that gets left out of methodology descriptions. Imagine that we had simply written, “We used NEAT-genReads with parameter x, y, and z to generate the synthetic data” and withheld the workflow code. If you tried to reproduce this work without our workflow (which includes comments about the issues), you would have to rediscover and troubleshoot all of that yourself.
Meanwhile, the other slight complication here was that we couldn’t find a Docker container image of NEAT-genReads anywhere, so we had to create one ourselves. And this is where we realize with horror that we’re 13-plus chapters into this book and we haven’t even shown you how to create your own Docker image. Oops? Well, keep in mind that these days most commonly used tools are available on Docker images through projects like BioContainers, which provide a great service to the community—they even take requests. Depending on what you do for a living, there’s a good chance that you can get most, if not all, of your work done on the cloud without ever worrying about creating a Docker image. Yet we can’t really let you finish this book without at least showing you the basics, so if you ever do need to package your own tool, you’ll know what to expect. Let’s take advantage of this NEAT-genReads exposé to do that in the accompanying sidebar.
We used the Dockerfile shown in the sidebar to create the container image and then we published it to a public repository on Docker Hub.
To run the workflow in our Terra workspace, we configured it to run on individual rows of the participant table, just like the first workflow. We wired up the sampleVcf column, populated by the output of the previous workflow, to serve as input for this second one. Finally, we configured the two resulting outputs, the BAM file and the VCF file, to be written to new columns in the table, synthExomeBam and originalCallsVcf, along with their index files.
Just like our first workflow, this one too suffers from a few inefficiencies due in part to the cleanup tasks that we had to add. It took about the same time as the first one to run, though within that amount of time it did quite a bit more work: it yielded a complete exome BAM file for each of the participants that we ran it on, with coverage and related metrics modeled after the original Tetralogy of Fallot cohort exomes reported in the preprint. We opened a few of the output BAM files in IGV, and were impressed to see that we could not readily distinguish a synthetic exome from a real exome with similar coverage properties.
This brings us to the final step in the process of generating the synthetic dataset: adding in the variants of interest.
Mutating the Fake Exomes
Walking into this, we were already familiar with an open source tool developed by Adam Ewing called BAMSurgeon, which some of our colleagues had previously used successfully to create test cases for variant-calling development work. In a nutshell, BAMSurgeon is designed to modify read records in an existing BAM file to introduce variant alleles, as illustrated in Figure 14-7. The tool takes a BED file (a tab-delimited file format) listing the position, allele, and allele fraction of variants of interest, and will modify the corresponding fraction of reads accordingly. Because we had a list of the variants of interest identified in the Tetralogy of Fallot cohort, including their alleles and positions, we figured that we should be able to introduce them into a subset of our synthetic exomes to re-create the relevant characteristics of the case samples.
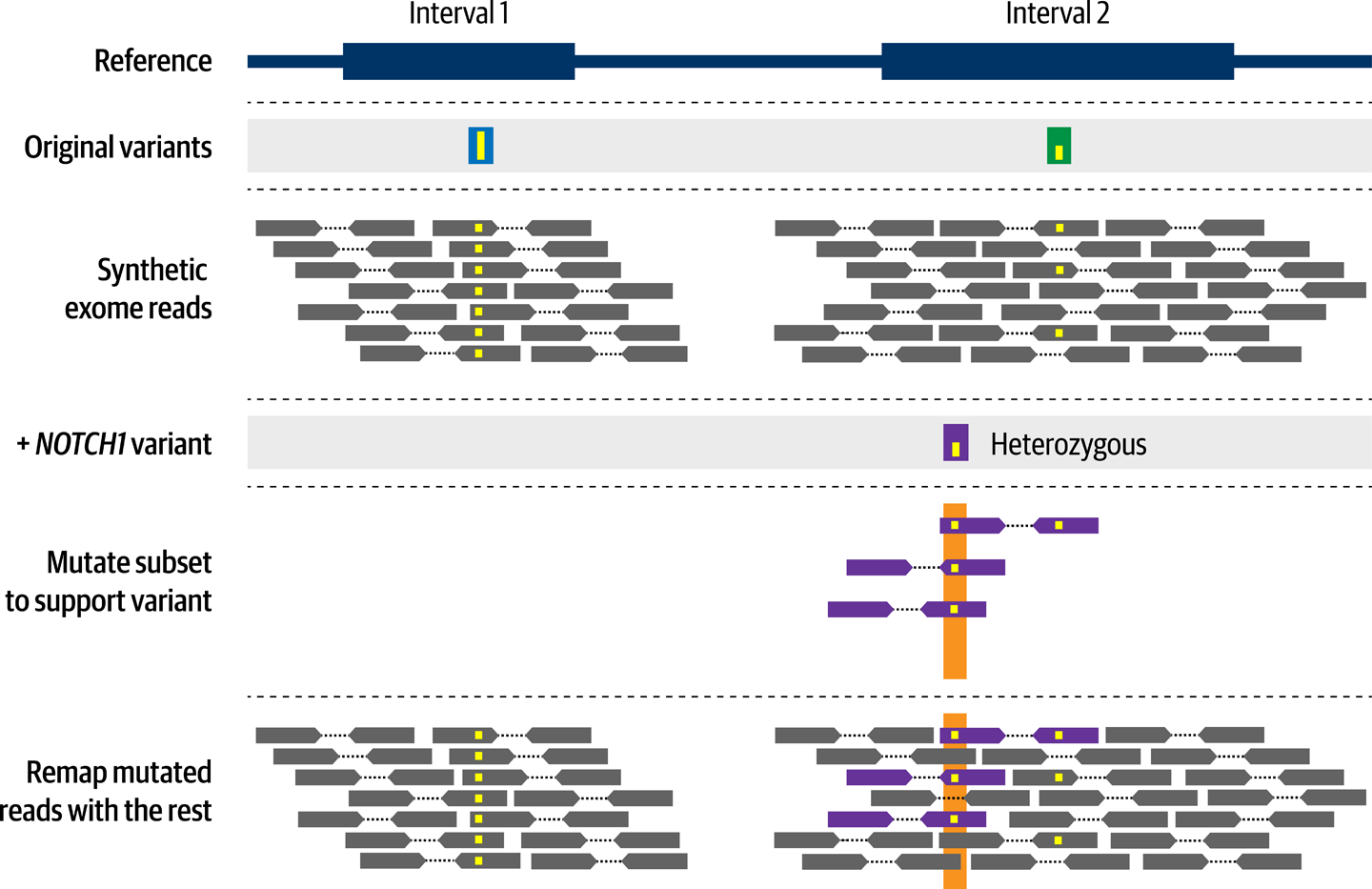
Figure 14-7. BAMSurgeon introduces mutations in read data.
We implemented this as the WDL workflow 3_Mutate-reads-with-BAMSurgeon, which takes the reference genome, a BAM file, and a BED file listing the variants to introduce, and produces the mutated BAM file. In the first iteration, we limited it to just adding SNPs, because BAMSurgeon uses different commands for different types of variants. In later work, contributors from the community added some logic to the workflow to also handle indels and copy-number variants using the relevant BAMSurgeon commands.
How much trouble did we have with this one? Hardly any trouble at all, if you can believe it. The only real problem we had was when we initially tested this on the 1000 Genomes Low Coverage data, because BAMSurgeon refused to add variants at positions where the sequence coverage was too low. However, when we switched to running on the lovely, plump data produced by NEAT-genReads (which we had given a target coverage of 50X), it worked like a charm.
As previously, in our Terra workspace, we configured the workflow to run on individual rows of the participant table. Then, we just needed to determine how we would label cases versus controls and how to set up which cases would be mutated with which variant.
That’s when we realized that we had oversimplified the experimental design for this part. Our original plan was simply to introduce the variants listed in the paper into a subset of exomes that would serve as case samples, whereas the rest would be left untouched as control samples. Unfortunately, this reasoning had two flaws.
First, not all case samples had yielded variants of interest in the original study, so there were fewer variants than case samples. In addition, a number of variants in that list fell in genes for which the variant load results were less convincing, so we had decided early on to focus on just the NOTCH1 variants for the case study. Because NOTCH1 was the main focus of the paper itself, we felt that reproducing the NOTCH1 result would be sufficient as a proof of concept. Yet we did believe that we should keep the proportion of cases and controls the same as in the paper. As a result, many of the synthetic exomes labeled as case samples would actually not receive a variant of interest.
Second, putting some data through different processing steps compared to the rest would expose us to batch effects. What if something in the differential processing caused an artifact that affected the analysis? We needed to eliminate this source of bias and ensure that all of the data would be processed the same way.
To address these flaws, we decided to introduce a neutral variant into any exome that would not receive a NOTCH1 variant, whether it was labeled as a case sample or as a control sample. To that end, we designed a synonymous mutation that we predicted should not have any effect.
In the Terra workspace, we added a role column to the participant table, so that we could label participants as neutral or case based on the mutation they would receive. We used the term neutral rather than control because we wanted to give ourselves the flexibility to use participants with the neutral mutation either as actual control samples or as case samples that do not have a NOTCH1 mutation. We put the list of participants through a random selection process to pick the ones that would become NOTCH1 case samples and assigned them the case role in the participant table, labeling all other participants as neutral.
Finally, we created BED files for each of the NOTCH1 variants reported in the paper as well as for the neutral variant we had designed. We randomly assigned individual NOTCH1 variants to the participants labeled case and assigned the neutral variant to all others. For each participant, we linked the corresponding BED file in a new column of the participant table named mutation. We configured the BAMSurgeon workflow to take that field (this.mutation) as input for the variant to be introduced in each participant’s exome BAM file.
Even though the preparation that we had to do to allocate variants appropriately was not trivial, the workflow was straightforward and ran quickly (0.5 hours on average, 2.5 hours for a set of 100 files run in parallel, again including some preemptions). As planned, it produced mutated exome BAMs for all the participants we ran it on. We checked a few of the output BAM files in IGV to verify that the desired variant had indeed been introduced, and were satisfied that the process had worked as expected.
Generating the Definitive Dataset
Having successfully harnessed existing data and tools into the aforementioned workflows, we were now in a position to generate a synthetic dataset that could adequately stand in for the original Tetralogy of Fallot cohort. However, we had to make one more compromise, which was to limit the number of exomes that we created to 500 instead of the full 2,081 that we had originally planned on to emulate the original cohort. The reason? It’s so pedestrian that it hurts: we simply ran out of time before the conference. Sometimes, you just have to move forward with what you have.
So we generated 500 mutated synthetic exomes, which we further organized into two participant sets in the workspace, imaginatively named A100 and B500. The A100 set was a subset of 100 participants that included eight NOTCH1 cases. The B500 set was the superset of all participants for which we created synthetic exomes, and included all 49 NOTCH1 cases reported in the paper. You might notice that these compositions don’t quite match the proportion of NOTCH1 cases in the overall Tetralogy of Fallot cohort, but the two sets have similar proportions to each other. Later, we discuss how this affects the interpretation of the final results.
At this point, we’re ready to move on to the next section, in which we attempt to reproduce the study methodology itself.
Re-Creating the Data Processing and Analysis Methodology
We wouldn’t blame you if you had forgotten all about the original study by now, so let’s go through a quick refresher. As illustrated in Figure 14-8, Dr. Miossec et al. started out with exome sequencing data from a cohort of 2,081 study participants, split almost evenly between cases; that is, patients suffering from the congenital heart disease tetralogy of Fallot (specifically, a type called nonsyndromic), and controls (people not affected by the disease). They applied fairly standard processing to the exome data using a pipeline based on the GATK Best Practices, consisting mainly of mapping to the reference genome and variant calling, which you are an expert in since Chapter 6. Then, they applied a custom analysis that involved predicting the effects of variants in order to focus on deleterious variants, before attempting to identify genes with a higher variant load than would be expected by chance.

Figure 14-8. Summary of the two phases of the study: Processing and Analysis.
We go into more detail about the meaning and purpose of these procedures in a few moments. For now, the key concept we’d like to focus on is the difference between these two phases of the study. In the Processing phase, which can be highly standardized and automated, we’re mainly trying to clean up the data and extract what we consider to be useful information out of the immense haystack of data we’re presented. This produces variant calls across the entire sequenced territory, a long list of differences between an individual’s genome, and a rather arbitrary reference. In itself, that list doesn’t provide any real insights; it’s still just data. That’s where the Analysis phase comes in: we’re going to start asking specific questions of the data and, if all goes well, produce insights about a particular aspect of the biological system that we’re investigating.
We highlight this distinction because it was a key factor in our decision making about how closely we believed we needed to get to reproducing the exact methods used in the original study. Let’s dive in, and you’ll see what we mean.
Mapping and Variant Discovery
Right off the bat, we bump into an important deviation. The original study started from exome sequencing data in FASTQ format, which had to be mapped before anything else could happen. However, when we generated the synthetic exome dataset, we produced BAM files containing sequencing data that was already mapped and ready to go. We could have reverted the data to an unmapped state and started the pipeline from scratch, but we chose not to do that. Why? Time, cost, and convenience, not necessarily in that order. We made the deliberate decision to take a shortcut because we estimated that the effect on our results would be minimal. In our experience, as long as some key requirements are met and the data is of reasonably good quality, the exact implementation of the data processing part of the pipeline does not matter quite as much as whether it has been applied consistently in the same way to the entire dataset. We did, however, make sure that we were using the same reference genome, of course.
Note
Note the preconditions; we’re not saying that anything goes. Sadly, a full discussion on that topic is beyond the scope of this book, but hit us up on Twitter if you want to talk.
So with that, we fast-forward to the variant discovery part of the processing. The original study used a pipeline called GenPipes DNAseq developed and operated at the Canadian Centre for Computational Genomics in Montreal. Based on the information we found in the preprint and in the online documentation for GenPipes DNAseq, the pipeline followed the main tenets of the GATK Best Practices for germline short variant discovery. This included performing single-sample variant calling with GATK HaplotypeCaller to generate a GVCF for each sample and then joint-calling all samples together. We did flag one deviation, which was that according to the preprint, the variant callset filtering step was done by hard filtering instead of using VQSR.
Accordingly, we repurposed some GATK workflows that we had on hand and customized them to emulate the processing described in the materials available to us. This produced the two WDL workflows 4_Call-single-sample-GVCF-GATK (featuring your favorite HaplotypeCaller) and 5_Joint-call-and-hard-filter-GATK4 (featuring GenotypeGVCFs and friend), which you can find in the Terra workspace. We configured the first to run the synthetic exome BAM file of each participant in the table and output the GVCF to the corresponding row. In contrast, we configured the second to run at the sample set level, taking in the GVCFs from all participants in the set and producing a filtered multisample VCF for the set.
Here we allowed ourselves another notable deviation: instead of using version 3.2 of the GATK as the original study did, we used GATK 4.0.9.0 in order to take advantage of the substantial improvements in speed and scalability that GATK4 versions offer compared to older versions of GATK. We expected that given the high quality of our synthetic data, there would be few differences in the output of HaplotypeCaller between these versions. We would expect important differences to arise only on lower-quality data and in the low-confidence regions of the genome. Again, some caveats apply that we don’t get into here, and we were careful to apply the same methods consistently across our entire dataset.
All in all, we made quick work of the Processing phase, in part thanks to those two deviations from the original methods. Since then, we’ve considered going back and reprocessing the data from scratch with a more accurate reimplementation of the original methods, to gauge exactly how much difference it would make. It’s not exactly keeping us awake at night, but we’re curious to see whether the results would support our judgment calls. If you end up doing it as a take-home exercise, don’t hesitate to let us know how badly wrong we were.
For now, it’s time to move on to the really interesting part of the analysis, in which we discover whether we’re able to reproduce the main result of the original study.
Variant Effect Prediction, Prioritization, and Variant Load Analysis
Let’s take a few minutes to recap the problem statement and go over the experimental design for this part given that, so far, we’ve mostly waved off the details. First, what do we have in hand, and what are we trying to achieve? Well, we’re starting from a cohort-level VCF, which is a long inventory of everyone’s variants, and we’re hoping to identify genes that contribute to the risk of developing a form of congenital heart disease called tetralogy of Fallot. To be clear, we’re not trying to find a particular variant that is associated with the disease; we’re looking for a gene that is associated with the disease, with the understanding that different patients can carry different variants located within the same gene. What makes this difficult is that variants occur naturally in all genes, and if you look at enough variants across enough samples, you can easily find spurious associations that mean nothing.
The first step to tackling this problem is to narrow the list of variants to eliminate as many of them as you can based on how common they are, whether they’re present in the control samples, and what kind of biological effect they are likely to have, if any. Indeed, the overwhelming majority of the variants in the callset are common, boring, and/or unlikely to have any biological effect. We want to prioritize; for instance, focus on rare variants that are found only in the case samples and are likely to have a deleterious effect on gene function.
With that much-reduced list of variants in hand, we can then perform a variant load analysis, as illustrated in Figure 14-9. This involves looking for genes that appear to be more frequently mutated than you would expect to observe by chance given their size.
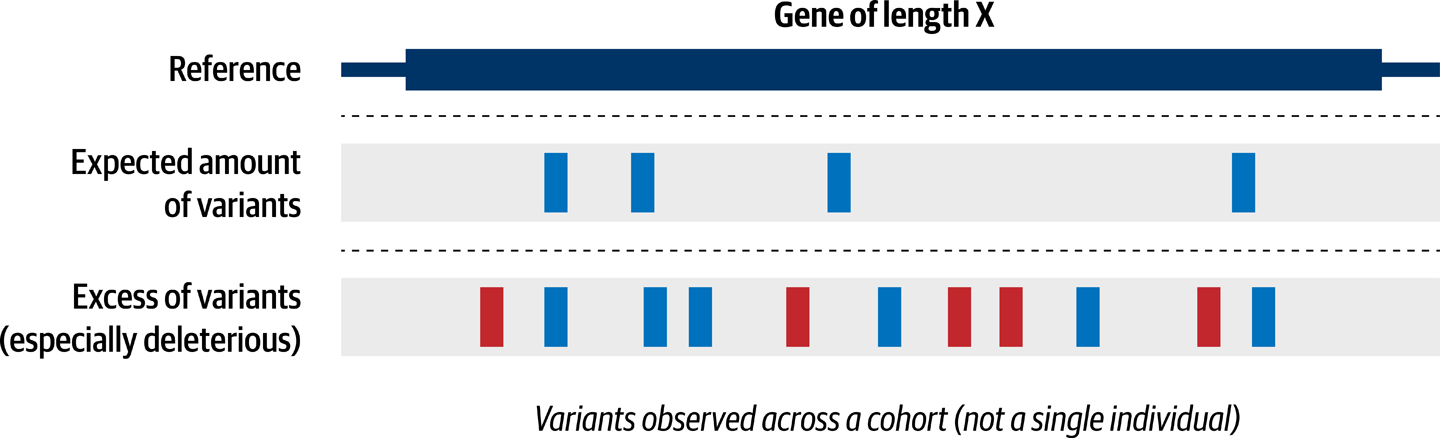
Figure 14-9. Comparing variant load in a gene across multiple samples.
The original study used a tool called SnpEff to predict variant effects, and then vt from GEMINI to prioritize variants based on their predicted effect, their frequency in population databases, and their presence in only the case samples. We implemented this as a two-step WDL workflow, 6_Predict-variant-effects-GEMINI, that used the same versions and commands for both tools, as well as the same population resources. In Terra, we configured the workflow to run at the participant set level and output the list of prioritized variants to the same row in the table. For this step, we had to add a pedigree file to each participant, specifying whether it should be counted as a case sample or as a control sample. For a given participant set, we selected one half of the participants, all carrying the neutral mutation, to be control samples. We then assigned the rest of the participants in the set, including all those carrying NOTCH1 mutations, to be case samples.
Finally, Dr. Miossec helped us rewrite the original Perl scripts for the clustering analysis into R in a Jupyter notebook, which you can also find in the workspace. The notebook is set up to pull in the output file produced by GEMINI. It then runs through a series of analysis steps, culminating in a clustering test that looks for an excess of rare, deleterious variants in the case samples. The analysis is documented with explanations for each step, so be sure to check it out if you’d like to learn more about this analysis.
In both the workflow and the notebook, we were careful to reproduce the original analysis to the letter, because we considered that these were the parts that would have the most influence on the final results.
Analytical Performance of the New Implementation
So the burning question is…did it work? Were we able to pull out NOTCH1 from the haystack? The short answer is, yes, mostly; the long answer is rather more interesting.
As we mentioned earlier, we ended up being able to generate only 500 synthetic exomes in the time we had, so we defined a 100-participant set with eight NOTCH1 cases and a 500-participant set with all 49 NOTCH1 cases. Although these proportions of NOTCH1 cases were higher than those in the original dataset, what mattered to us at that point was that our two participant sets were roughly proportional to each other. Because we could not test the method at full scale as originally intended, we would at least be able to gauge how the results changed proportionally to the dataset size, which was a point of interest from early on.
We ran the full set of processing and analysis workflows on both participant sets and then loaded the results of both into the notebook, where you can see the final clustering analysis repeated on both sets. In both cases, NOTCH1 came up as a candidate gene, but with rather different levels of confidence, as shown in Figure 14-10. In the 100-participant set, NOTCH1 was ranked only second in the table of candidate genes, failing to rise above the background noise. In contrast, in the 500-participant set, NOTCH1 emerged as the uncontested top candidate, clearing the rest of the pack by a wide margin.
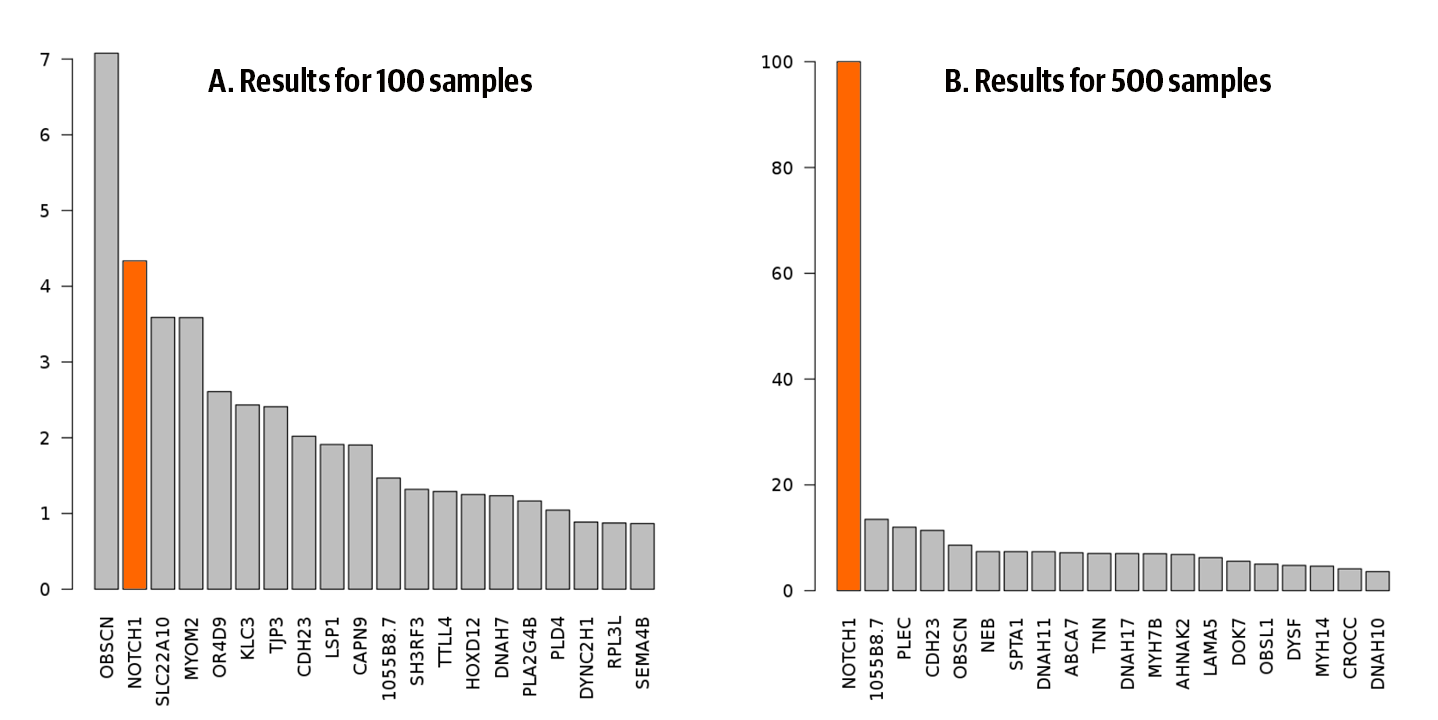
Figure 14-10. Ranking from the clustering test for A) 100-participant set, and B) 500-participant set.
We considered that this constituted a successful enough reproduction of the analysis because we were able to generate a result that matched our expectations, even though we had to make compromises in terms of fidelity.
We were particularly encouraged to see that this approach could be used to test the scaling of statistical power depending on the dataset size. We can easily imagine setting up additional experiments to test the scaling observed here to a finer granularity and a larger scale. We would also want to test how varying the proportion of NOTCH1 cases relative to the overall cohort size would affect the results, as well, starting with the proportion that was actually observed in the original study.
So many possibilities, so little time. And speaking of which, it’s getting late; we’re almost at the end of...
The Long, Winding Road to FAIRness
Given what we just described, you should now be able to reproduce the Tetralogy of Fallot analysis in Terra as well as reuse the data and/or the methods for your own work if they are applicable. You should also be able to do so outside of Terra for the most part because most of the components are directly usable on other platforms. You can download the data to use on different platforms, and the Docker images and WDL workflows can be run just about anywhere. The notebook might take more effort to reuse given that the environment is not as cleanly bundled as the Docker images used for workflows. You can access all necessary information regarding the computing environment, but you would still need to independently set up an equivalent environment. We’re hopeful that this is something that will improve further over time; for example, imagine if there were an option to emit a Dockerfile that could re-create the software environment of a particular notebook. We would also like to see a standardized way to obtain a digital object identifier (DOI) for a Terra workspace, to use in publications that include a companion workspace as supplemental material. This ties in with important work that others are doing to bundle research artifacts for easier archival, retrieval, and reuse. It’s a complicated topic, however, as tends to be the case when developing standards.
So what can you do to achieve computational reproducibility and FAIRness in your own work? In the course of this case study, we pointed out several key factors that are typically under your direct control. We discussed many of them in the context in which they arose, but we thought it might be helpful to provide a summary of what we consider to be the most important guidelines:
- Open source
- It’s hard to overstate the importance of using open source tools and workflows. Using open source tooling helps keep your work reproducible in two ways: it ensures accessibility to all, and it increases the transparency of the methodology. If someone isn’t able to run the exact same code for any reason, they still have a chance to read the code and reimplement the algorithm.
- Version control
- It’s imperative that you track the versions of tools, workflows, and notebooks you use as systematically as possible because changes from one version to the next can have a major effect on how tools work and what results they produce. If you are using other people’s tools, record the versions you use and include that information when you publish your analysis. If you develop your own tools and workflows, make sure to use a version-control system like GitHub. In Terra, workflows are systematically versioned, either through GitHub and Dockstore or through the internal Methods Repository. Notebooks are currently not versioned in Terra, so we recommend downloading your notebooks regularly and checking them into a version-control system like GitHub.
- Automation and portability
- When it comes to developing the part of your analysis that you will run many times on a lot of data, choose a method that will allow you to automate as much as possible and reduce your dependence on a specific environment. For example, choose a pipelining language like WDL or CWL, which can easily be run by others, rather than writing elaborate scripts in Bash, Python, or R that others will have a difficult time running even if you provide the code with your publications.
- Built-in documentation
- For analyses that involve a lot of interaction with the data and judgment calls in how to go from one step to the next, consider providing a Jupyter notebook that recapitulates your analysis with built-in commentary explaining what’s happening at each step. Even if you prefer working in an environment that gives you more flexibility in your day-to-day work, like RStudio, for example, packaging the finished product as a Jupyter notebook will greatly enhance both its reproducibility and its intelligibility. Think about the last time you helped a new lab member or classmate get up to speed on a new analysis method; imagine being able to give them a notebook that explained what to do step by step rather than a loose collection of scripts and a README document that might or might not be up-to-date.
- Open data
- Finally, the elephant in the room is often going to be the data. There will be many valid reasons you might not be able to share the original data you worked with, especially if you’re working with protected human data. However, you might be able to refer to open-access data that is sufficient to demonstrate how your analysis works. When that’s not an option, as in the case study we just described, consider whether it might be possible to use synthetic data instead.
Does that last point mean you should go through what we did to generate the synthetic dataset? Well, hopefully not. You’re most welcome to use the synthetic exomes we generated, which are freely accessible, or use the workflows we showed you to create your own. Our workflows are admittedly not very efficient in their present form and would benefit from some optimization to make them cheaper to run and more scalable, but they work. And, hey, they’re open source, so feel free to play with them and propose some improvements!
Looking at the bigger picture, however, we believe there is an opportunity here to develop a community resource to minimize the amount of duplicative work that anyone needs to do in this space. Considering how standardized the generation of sequencing data has become (at least for the short read technologies), it should be possible to identify the data types that are most commonly used and generate a collection of generic synthetic datasets. These could then be hosted in the cloud and used off the shelf in combination with tools like BAMSurgeon by researchers who want to reproduce someone else’s work, or to make their own work more readily reproducible, along the lines of what we described in this chapter.
Final Conclusions
Well, here we are at the end of the final chapter. How does it feel? You’ve covered a lot of ground in the cloud, so to speak. You started on a puny little Cloud Shell, and then moved quickly on to a beefier virtual machine, where you ran real genomic analyses with GATK; first manually, step by step, and then through simple WDL workflows with Cromwell. You applied your inner detective to deciphering mystery workflows, learning in the process how to approach new workflows methodically. Next, you graduated to using the Pipelines API, scaling up your workflow chops and tasting the freedom of launching a parallelized workflow into a wide-open space. From there, you jumped to Terra, where you peeled back layers of functionality—workflows, notebooks, data management—to finally find yourself on solid ground with our case study of a fully reproducible paper.
Based on what you learned in this book, you can now head down the path of using these tools in your own work. Take advantage of massive cloud-hosted datasets and a wealth of Dockerized tools that you don’t need to figure out how to install. Develop your own analyses, execute them at scale, and share them with the world—or just your labmates. Whether you continue to use the specific tools we used here for those purposes, or with other tools within the growing cloud-based life sciences ecosystem, you can rest assured that similar principles and paradigms will apply.
Be sure to check back for updates to the book’s GitHub repository, companion blog, and latest developments around GATK, WDL, Docker, and Terra.
Have fun, and keep it reproducible!
1 This case study was originally developed by the Support and Education Team in the Data Sciences Platform at the Broad Institute. Because that work was led at the time by one of us (Geraldine), we’ve kept the description in the first person plural, but we want to be clear that other team members and community members contributed directly to the material presented here, either toward the development and delivery of the original workshop or in later improvements of the materials.
2 Page, et al. originally shared their manuscript with the research community as a preprint in bioRxiv in April 2018, and ultimately published it in the peer-reviewed journal Circulation Research in November 2018: “Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot,” https://doi.org/10.1161/CIRCRESAHA.118.313250.