Chapter 6
Neurotransmitters
The pioneering studies of Santiago Ramon y Cajal revealed that each cell (neuron) of the brain is an independent structure in close proximity but not physical contact with another neuron (see Shepherd, 1991). Although a half century elapsed before the introduction of the electron microscope led to the final confirmation of this hypothesis, a general acceptance of neurons as the independent building blocks of the brain emerged much more quickly. In turn, this resulted in a new debate: How do these neurons communicate? The answer to this question has evolved continuously over the past century and has become increasingly complex, but the major mode of communication is by chemical messengers.
Neurons share certain structural characteristics (see Chapter 3). They have a cell body (soma) from which processes emerge. One of these types of processes, the axon, is often long but can also be short and thus exert local effects or distant ones. Moreover, axons can branch extensively or have a very confined arbor. In contrast, dendrites are exclusively local structures. The identification of long (axon) and short (dendrite) processes led to the idea that the long axonal projections served to transmit information between neurons over (relatively) long distances, and dendrites were the receptive elements of neurons, responding to the information conveyed by long axons. The gap over which information between neurons must be relayed was termed the synapse by Charles Sherrington and came to identify pre- and postsynaptic cells. This general conceptual framework remains in place today, although there are many exceptions, including dendrites that release neuroactive substances and axons that receive inputs from other neurons. One other characteristic proposed by Sherrington that is central to the concept of chemical communication between neurons is that synaptic transmission does not follow all-or-none rules, but is graded in strength and is flexible.
Several Modes of Neuronal Communication Exist
Over the first half of the twentieth century, there was a vigorous debate on the nature of neuronal communication: as one overview of this debate described, it was a war of sparks (electrical communication) and soups (chemical messengers) (see Valenstein, 2005). The debate raged despite the fact that some evidence was marshaled in support of chemical transmission in the mid-nineteenth century. In 1849, Claude Bernard noted that curare, the active constituent of a poison that was applied to arrows in South America, blocked nerve-to-muscle signaling. This effect subsequently was shown to be due to the binding of curare to postsynaptic (muscle) receptors for acetylcholine (ACh), thus blocking neuro-muscular transmission. About 50 years later, Thomas Elliott found that epinephrine caused the contraction of smooth muscle that had been deprived of its nerve inputs, suggesting that muscle contraction depended on the action of chemical molecules liberated from nerves. In a key series of studies using isolated frog hearts, Otto Loewi provided firm evidence in support of chemical neurotransmission by showing that ACh was released upon nerve stimulation and activated a target muscle.
These and other data suggested that the major means of interneuronal communication is chemical in nature, but that neurons also use other processes for intercellular communication. Among these are electrical synaptic transmission, ephaptic interactions, and autocrine, paracrine, and long-range signaling, to which molecules produced by both neural and nonneural cells contribute. The nonsynaptic mode of intercellular communication with the longest range (distance) is hormonal signaling. For example, some hormones made outside of the brain can enter the central nervous system (CNS) to exert effects on neurons that express receptors for the hormones. These hormone actions may occur over the short term (changes in neuronal activity) or, more often, over a longer period (long-lasting changes in gene expression).
Molecules released from neurons can also be used in intercellular signaling that does not require synaptic specializations. A variety of molecules are secreted by neurons or diffuse passively from cells, ranging from conventional neurotransmitters to gases such as nitric oxide. These factors may act through autocrine mechanisms (activating receptors on the same cell that releases them) or paracrine pathways to influence nearby cells. Some of these compounds can be retrograde signaling molecules, providing chemically coded feedback to the presynaptic neuron and reversing the more common forward (anterograde) direction of information flow. The role of such molecules is thought to be primarily in modulating neural activity, although they may also provide guidance cues for neurons that are growing toward their final targets in the brain, as well as for the establishment and maintenance of synaptic connections (see Chapters 16 and 18).
Chemical Transmission
Chemically mediated transmission is the major mode of neuronal communication. The general acceptance of chemical neurotransmission as the means of conveying information between neurons resulted in specific criteria being required for designation of a compound as a neurotransmitter. Based on these “classical” criteria, a relatively small number of compounds have been designated as neurotransmitters. However, over the past generation it has become apparent that a large number of chemical messengers broadly qualify as intercellular transmitters, although these compounds often do not meet—and even seem to sharply differ from—the classical criteria.
Criteria for Designation as a Neurotransmitter
Neurotransmitters are usually considered to be endogenous substances that are released from neurons, act on receptor sites that are typically present on membranes of postsynaptic cells, and produce a functional change in the properties of the target cell. Over the years general agreement evolved that several criteria should be met for a substance to be designated a neurotransmitter:
1. A neurotransmitter must be synthesized by and released from neurons. Thus, the presynaptic neuron should contain both the transmitter and the appropriate enzymes needed to synthesize that neurotransmitter.
2. The substance should be released from nerve terminals in a chemically or pharmacologically identifiable form. It should therefore be possible to isolate the transmitter and characterize its structure.
3. A neurotransmitter should reproduce at the postsynaptic cell the specific events (such as changes in membrane properties) that are seen after stimulation of the presynaptic neuron.
4. The effects of a putative neurotransmitter should be blocked by competitive antagonists of the receptor for that transmitter in a dose-dependent manner. In addition, treatments that inhibit synthesis of the transmitter should block the effects of presynaptic stimulation.
5. There should be active mechanisms to terminate the action of the neurotransmitter. Among such mechanisms are uptake of the transmitter by the presynaptic neurons or glial cells through specific transporter molecules and enzymatic inactivation of the chemical messenger.
The Five Steps of Chemical Neurotransmission
Synaptic transmission consists of a number of steps. The general mechanisms of chemical synaptic transmission are depicted in Figure 6.1.
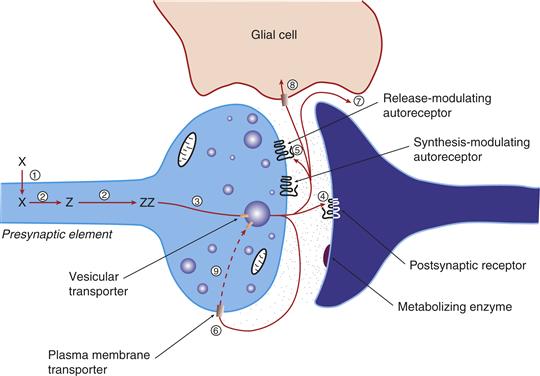
Figure 6.1 Schematic representation of the life cycle of a classical neurotransmitter. After accumulation of a precursor amino acid into the neuron (1), the amino acid precursor is metabolized sequentially (2) to yield the mature transmitter. The transmitter is then accumulated into vesicles by the vesicular transporter (3), where it is poised for release and protected from degradation. Once released, the transmitter can interact with postsynaptic receptors (4) or autoreceptors (5) that regulate transmitter release, synthesis, or firing rate. Transmitter actions are terminated by means of a high-affinity membrane transporter (6) that usually is associated with the neuron that released the transmitter. Alternatively, transmitter actions may be terminated by diffusion from the active sites (7) or accumulation into glia through a membrane transporter (8). When the transmitter is taken up by the neuron, it is subject to metabolic inactivation (9).
1. Synthesis of the neurotransmitter in the presynaptic neuron. In order for the transmitter to be synthesized, precursors should be present in appropriate places within neurons. Enzymes that convert the precursor(s) into the transmitter should be present in an active form and localized to the appropriate compartment in the neuron. For example, one would not expect synthetic enzymes for the transmitter to be found in the nucleus or the mitochondrion, but these enzymes should be found in the cytosol or in intracellular structures that store the transmitter. In addition, any cofactors that are required for enzyme activity should be present. Drugs that affect the synthesis of neurotransmitters have long been of value in medicine. An example is “α-methyl-p-tyrosine, a drug used to treat an adrenal gland tumor that sharply increases blood pressure by releasing very large amounts of norepinephrine (NE); α-methyl-p-tyrosine prevents the synthesis of NE and thereby lowers blood pressure.
2. Storage of the neurotransmitter and/or its precursor in the presynaptic nerve terminal. Classical and peptide transmitters are stored in synaptic vesicles, where they are sequestered and protected from enzymatic degradation and are ready for quick release. In the case of classical neurotransmitters such as acetylcholine, the synaptic vesicles are small (~50 nm in diameter), in contrast to the large dense-core vesicles (~100 nm in diameter) in which neuropeptide transmitters are stored. Vesicles are often found adjacent to the presynaptic membrane, where they are poised to be released in response to stimulation of the neuron. Because most neurotransmitters are synthesized in the cytosol of neurons, there must be some mechanism through which the transmitter enters the vesicle. This occurs through a vesicular transporter protein that accumulates the transmitter through an energy-dependent process.
3. Release of the neurotransmitter into the synaptic cleft. The vesicle in which the transmitter is stored fuses with the cell membrane and releases the transmitter. Neurons use two pathways to secrete proteins. The release of most neurotransmitters occurs by a regulated pathway that is controlled by extracellular signals. The neurotransmitter release process is discussed more fully in Chapter 7. A second (constitutive) pathway of release is not triggered by extracellular stimulation and is used to continuously secrete membrane components, viral proteins, and extracellular matrix molecules. Some unconventional transmitters (such as growth factors) may be synthesized and released by both constitutive and regulated pathways.
4. Binding and recognition of the neurotransmitter by target receptors. Once released, neurotransmitters interact with receptors located on the target cell. Most transmitter receptors fall into two broad classes. The first are membrane proteins called metabotropic receptors, which are coupled to intracellular G proteins as effectors (see Chapter 8). Ionotropic receptors form channels through which ions such as Na+ and Ca2+ can enter the neuron. Receptors are also found on the neuron that releases the transmitter. This presynapatic receptor can respond to the transmitter released from the same cell as part of a feedback process to the presynaptic neuron. Such receptors are called autoreceptors, and regulate transmitter release, synthesis, or impulse flow. Alternatively, presynaptic receptors can respond to a chemical signal elaborated from the postsynaptic neuron, another type of feedback mechanism. Finally, receptors on axon terminals can respond to transmitter released from the axon of a different cell, in which case the receptor is called a heteroceptor.
5. Termination of the action of the released transmitter. If a cell cannot stop the continued actions of a neurotransmitter, chaotic information flow and sometimes overt cell damage results. Sustained activation of postsynaptic targets can result in tetanus (sustained muscle contraction) or seizures. If one thinks of chemical transmission as being the flow of information, continuous unregulated transmitter release is not conveying to the target cell temporally specific data, such as the rate of firing of the presynaptic neuron or the pattern of firing of the neuron. Neurotransmitter actions may be terminated actively or passively. Among the active termination processes is reuptake of the neurotransmitter through specific transporter proteins on the presynaptic neuron or on glial cells. Another common means of terminating the action of a transmitter is by enzymatic degradation to an inactive substance. Finally, inactivation can happen by simple diffusion of the transmitter from the synaptic region. All three processes often work cooperatively to terminate the action of a neurotransmitter.
These five steps form a logical scaffold for understanding classical and peptide chemical neurotransmission. There are, however, differences across the various transmitters. We discuss in detail one class of neurotransmitters, the catecholamines, to illustrate the process of chemical neurotransmission, and then examine more briefly other classical neurotransmitters. We also discuss differences between classical and nonclassical transmitters or chemical messengers, including peptide transmitters and unconventional transmitters (such as nitric oxide, growth factors, and endocannabinoids).
Classical Neurotransmitters
The term classical is used to differentiate acetylcholine, the biogenic amines, and the amino acid transmitters from other transmitters. The designation is somewhat arbitrary, although these compounds were all accepted as neurotransmitters by the late 1950s. Several factors differentiate classical and other transmitters. First, storage vesicles for classical transmitters are smaller. Second, an energy-dependent reuptake process accumulates classical transmitters that have been released by the presynaptic neuron, allowing subsequent reuse of the transmitter. In contrast, there is no energy-dependent, high-affinity reuptake process for nonclassical transmitters. Third, most classical transmitters are synthesized in the nerve terminal by the actions of one or more enzymes, while peptides are synthesized in the soma from a precursor protein and are then transported to the nerve terminal. The remainder of this chapter deals with the biochemistry of several different transmitter groups, discussing their synthesis, storage, and release, and concludes with new developments that have recently been discovered and expand significantly our understanding of neuronal communication. Other aspects of chemical transmission, including detailed discussions of transmitter release and the receptors for various transmitters is presented in Chapters 7 and 8.
Catecholamine Neurotransmitters
Catecholamines are organic compounds that contain a catechol nucleus (a benzene ring with two adjacent hydroxyl substitutions) and an amine group. The term catecholamine is normally used to refer to the three transmitters dopamine (DA), norepinephrine (NE), and epinephrine (Epi). These three neurotransmitters derived from a common precursor are formed by successive enzymatic steps requiring separate enzymes (Fig. 6.2). The presence of these specific synthesizing enzymes in different cells results in different groups of neurons in the brain that synthesize and use as a transmitter either DA, NE, or Epi.
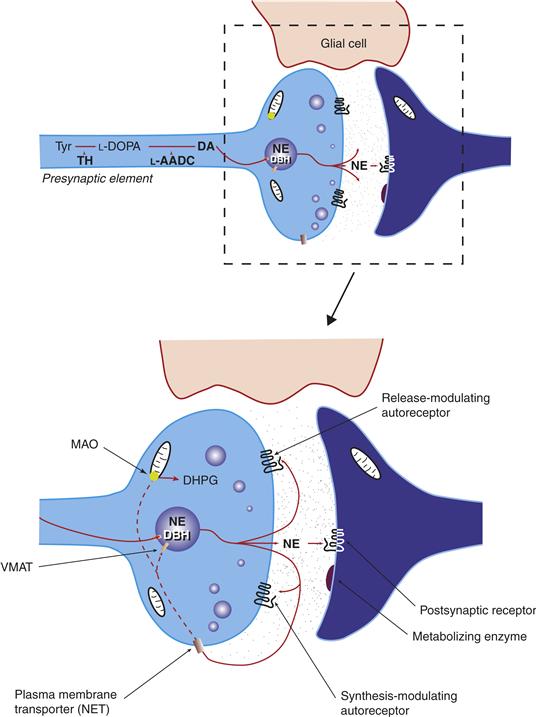
Figure 6.2 Characteristics of a norepinephrine (NE)-containing catecholamine neuron. Tyrosine (Tyr) is accumulated by the neuron and then is metabolized sequentially by tyrosine hydroxylase (TH) and L-aromatic amino acid decarboxylase (L-AADC) to dopamine (DA). The DA is then taken up through the vesicular monoamine transporter into vesicles. In DA neurons, this is the final step. However, in this NE-containing cell, DA is metabolized to NE by dopamine-b-hydroxylase (DBH), which is found in the vesicle. Once NE is released, it can interact with postsynaptic noradrenergic receptors or presynaptic noradrenergic autoreceptors. The accumulation of NE by the high-affinity membrane NE transporter (NET) terminates the actions of NE. Once taken back up by the neuron, NE can be metabolized to inactive compounds (DHPG) by degradative enzymes such as monoamine oxidase (MAO) or taken back up by the vesicle.
Catecholamines also have transmitter roles in the peripheral (autonomic) nervous system or have certain hormonal functions. For example, catecholamine-containing cells are present in the lining of the intestines where they regulate gut motility. NE is the transmitter of the sympathetic nervous system in mammals, whereas Epi is the sympathetic transmitter in frogs. Despite this species difference, biochemical aspects of neurotransmission are remarkably constant across different vertebrate species and even invertebrates.
Biosynthesis of Catecholamines
The amino acids phenylalanine and tyrosine are precursors for catecholamines. Both amino acids are found in high concentrations in the plasma and brain. In mammals, tyrosine can be formed from dietary phenylalanine by the enzyme phenylalanine hydroxylase, which is found in large amounts in the liver. Insufficient amounts of phenylalanine hydroxylase result in phenylketonuria, a metabolic disorder that leads to intellectual deficits unless treated by dietary manipulation.
Catecholamine synthesis in the brain is usually considered to begin with tyrosine. The enzyme tyrosine hydroxylase (TH) converts the amino acid L-tyrosine into 3,4-dihydroxyphenylalanine (L-DOPA). In turn, L-DOPA is metabolized by L-aromatic amino acid decarboxylase (AADC; see Iversen, Iversen, Bloom, & Roth, 2008) to the transmitter dopamine. This latter step occurs so rapidly that it is almost impossible to measure L-DOPA in the brain without first inhibiting AADC. In neurons that use DA as a transmitter, the decarboxylation of L-DOPA to DA is the final step in transmitter synthesis. However, in neurons that use norepinephrine (also known as noradrenaline) or epinephrine (adrenaline) as transmitters, the enzyme dopamine-β-hydroxylase (DBH), which converts DA to yield NE, is also present. In still other neurons in which epinephrine is the transmitter, a third enzyme (phenylethanolamine N-methyltransferase, PNMT) converts NE into Epi. Thus, a cell that uses Epi as its transmitter contains four key enzymes (TH, AADC, DBH, and PNMT), whereas NE neurons contain three enzymes (lacking PNMT), while DA cells have but two transmitter biosynthetic enzymes (TH and AADC).
Tyrosine Hydroxylase
In human beings, a single TH gene is alternatively spliced to yield four TH mRNAs, leading to four TH protein isoforms. In most other primates, only two TH isoforms are present, and rodents have but a single form of TH. It has been suggested that the human TH isoforms are differentially regulated and have different activities.
TH is a mixed-function oxidase with moderate substrate specificity, hydroxylating phenylalanine, as well as tyrosine. The actions of TH require the cofactor tetrahydrobiopterin (BH4) and iron (Fe2+). BH4 is also an essential cofactor for tryptophan hydroxylase, an enzyme that synthesizes another transmitter, serotonin. Because BH4 is not present in saturating concentrations under basal conditions, it is crucial in regulating TH activity. Intracellular levels of BH4 are determined by its own synthesizing enzyme, GTP cyclohydrolase. Mutations in the gene encoding GTP cyclohydrolase cause DOPA-responsive dystonia, a movement disorder in which DA synthesis is markedly decreased; treatment consists of bypassing the disruption of TH synthesis by administering L-DOPA, the immediate precursor of dopamine.
The two major processes that determine the function of enzymes are mechanisms that control enzyme activity (the rate at which the enzyme converts a precursor into its product) and the amount of newly synthesized enzyme protein. The major factor that determines the activity of TH is phosphorylation of the enzyme at four key serine residues. A second means of regulating enzyme activity is through end-product inhibition: catecholamines can inhibit the activity of TH by competing for a required cofactor for the enzyme (Iversen et al., 2008).
When neurons require additional catecholamine transmitter to keep demand with release, this can be accomplished by synthesizing new TH protein or increasing the activity of the TH activity. The degree to which increases in catecholamine synthesis depend on de novo synthesis of new enzyme protein or changes in the activity of existing enzymes differs across brain regions. For example, chronic increases in the activity of most norepinephrine neurons are accompanied by increases in TH gene expression and subsequent translation of the enzyme. But midbrain dopamine neurons do not appear to meet increased neurotransmitter demand by making more of the enzyme, instead primarily relying on increasing activity of the enzyme through phosphorylation.
The synthesis of catecholamines starts with the entry of tyrosine into the brain. This process is an energy-dependent one in which tyrosine competes with large neutral amino acids as a substrate for a transporter. Because brain tyrosine levels are high enough to saturate TH, catecholamine synthesis cannot usually be increased by administration of tyrosine. Exceptions to this rule are catecholamine synthesis in cells that have a very high activity and thus release the catecholamine transmitter at rapid rates, or in disease states in which entry of tyrosine into the brain is retarded.
L-Aromatic Amino Acid Decarboxylase
As noted above, the hydroxylation of tyrosine by TH generates L-DOPA, which is then decarboxylated to dopamine by the enzyme L-aromatic amino acid decarboxylase (AADC; also sometimes referred to as DOPA decarboxylase). AADC has low substrate specificity and decarboxylates tryptophan as well as tyrosine. Because AADC is found in serotonin as well as catecholamine neurons, it plays an important role in the synthesis of both serotonin and catecholamines. In dopaminergic neurons, AADC is the final enzyme of the synthetic pathway.
Administration of L-DOPA is the primary treatment for Parkinson’s disease, which is due to loss of dopamine. Dopamine itself cannot readily enter the brain, but the dopamine precursor L-DOPA freely enters the brain and, after being accumulated by dopamine neurons, is metabolized to dopamine. Although L-DOPA crosses the blood–brain barrier to enter the brain, systemically administered L-DOPA is so rapidly metabolized in the blood that little of the precursor actually reaches the brain. To circumvent this problem L-DOPA is administered together with a second drug that does not enter the brain but inhibits decarboxylases in the bloodstream.
Dopamine-β-Hydroxylase
Noradrenergic and adrenergic neurons contain the enzyme dopamine-β-hydroxylase (DBH), which converts DA into NE. In noradrenergic neurons, this is the final step in catecholamine synthesis. Humans have two different DBH mRNAs that are generated from a single gene. DBH has poor substrate specificity and in vitro oxidizes almost any phenylethylamine to its corresponding phenylethanolamine. For example, in addition to forming NE from DA, DBH converts tyramine into octopamine. Receptors that have a high affinity for the trace amines tyramine and octopamine have recently been discovered (see Zucchi, Chiellini, Scanlan, & Grandy, 2006) and may be involved in certain psychiatric disorders and in the side effects of certain drugs used to treat major depression.
Phenylethanolamine-N-Methyltransferase
Phenylethanolamine-N-methyltransferase is present at high levels in a small number of brainstem neurons that use epinephrine as a transmitter. PNMT is also found in the inner portion (medulla) of the adrenal gland, where it methylates NE to form Epi, the major adrenal catecholamine. There is a single PNMT gene. Brain PNMT regulation has not been extensively studied, but expression of the mRNA encoding PNMT is regulated by adrenal glucocorticoids and nerve growth factor.
Storage of Catecholamines and Their Enzymes
Catecholamines are stored in specialized subcellular organelles called vesicles. Accordingly, a specialized vesicular transporter protein is required in order for dopamine and other transmitters that are formed in the cytosol to enter the vesicle (El Mestikawy, Wallén-Mackenzie, Fortin, Descarries, & Trudeau, 2011).
Vesicular Storage
The vesicle serves as a depot for the transmitter until it is released by appropriate physiological stimuli (see Chapters 5 and 7). In addition, by storing transmitters in an organelle, vesicles protect the transmitter from metabolic inactivation by cytosolic enzymes.
The norepinephrine-synthesizing enzyme DBH differs from the other enzymes in catecholamine synthesis by being found in the vesicle rather than the cytosol. This means that only after DA is taken up by vesicles through a vesicular monoamine transporter (VMAT) is it metabolized to NE. This vesicular storage of DBH has an interesting consequence: DBH and dopamine can both be released into the synapse along with NE.
Vesicular Monoamine Transporters
The ability of vesicles to accumulate DA and other compounds depends on VMAT. The vesicular transporter differs from the neuronal membrane transporter that terminates the action of catecholamines in terms of substrate affinity and localization. Two VMAT genes have been cloned: VMAT1 is found in adrenal gland cells that synthesize and release catecholamines, and VMAT2 is found in the catecholamine and serotonin-containing neurons of the brain. VMAT2 is not very specific and accumulates catecholamines, indoleamines such as serotonin, and histamine into vesicles.
The VMATs are inhibited by reserpine. This drug has been used for centuries in India as a folk medicine to treat high blood pressure and psychoses. These uses of reserpine were reported in international journals in the early 1930s, but these therapeutic actions were not appreciated in Western medicine until a generation later; the unpleasant side effects of reserpine initially garnered more attention. In the 1950s Bernard Brodie and coworkers discovered that reserpine depleted brain serotonin. The contemporaneous discovery that the hallucinogen LSD is structurally similar to serotonin led to the proposal that the antipsychotic actions of reserpine were due to its ability to deplete serotonin in the brain. However, it was soon realized that reserpine depletes both serotonin and catecholamines in the brain; thus, antipsychotic effects of reserpine might be due to serotonin or catecholamine depletion, or both.
To determine if serotonin or catecholamines were more important for the antipsychotic effect of reserpine, Arvid Carlsson and colleagues administered catecholamine and serotonin precursors to rats treated with reserpine in an attempt to replenish the levels of the transmitters. At the time of these studies dopamine was not thought to be a transmitter, but was considered to be present solely as a precursor of norepinephrine. Carlsson and collaborators examined locomotor activity, which is severely depressed by reserpine. Motor function was restored by the DA precursor L-DOPA but not by the serotonin precursor 5-hydroxytryptophan. This work led to the characterization of DA as a neurotransmitter. The demonstration that L-DOPA increases brain DA concentrations in animals treated with reserpine to deplete vesicular stores of DA also suggested that the primary mechanism through which reserpine exerts antipsychotic effects was through its ability to disrupt DA transmission. This idea led directly to the hypothesis that the dysfunction of central DA systems underlies schizophrenia.
VMATs have significant homology with bacterial antibiotic drug resistance transporters, suggesting a role of VMAT in detoxification. This is indeed the case: VMAT can sequester toxins (as it sequesters transmitters) in vesicles. This is perhaps best illustrated by studies of genetically modified mice with low VMAT2 expression. In these mice, the toxin MPTP (which disrupts mitochondrial function and elicits dopamine neuron degeneration) results in a greater loss of dopamine than seen in control mice. This is because less MPTP is sequestered in vesicles, with more MPTP in the cytosol where it may disrupt mitochondria.
Release of Catecholamines
Catecholamine release usually occurs through a calcium-dependent exocytotic process involving brief fusion of the transmitter-containing vesicle with the plasma membrane (see Chapter 8). However, catecholamine release can also occur through at least two other mechanisms. First, catecholamines can be released by a reversal of the dopamine and NE transporters to extrude the catecholamines; this mode of release is seen after administration of amphetamine. Second, dopamine can be released from dendrites through a process that does not appear to involve calcium.
Regulation of Catecholamine Synthesis and Release by Autoreceptors
Enzymes that control catecholamine synthesis can be regulated at both the transcriptional level and by posttranslational modifications (such as phosphorylation) that alter enzymatic activity. In addition, DA and NE synthetic enzymes can be regulated by autoreceptors on the nerve terminal.
Autoreceptors are found on most parts of the neuron and are defined functionally by the events that they regulate. Thus, synthesis-, release-, and impulse-modulating DA autoreceptors have been described (see Iversen et al., 2008). All three types of DA autoreceptors belong to the D2 family of DA receptors, which includes three different receptors (D2, D3, and D4). It is clear that D2 autoreceptors exist, but the presence of D3 DA autoreceptors remains controversial; there are no D4 autoreceptors. Because all three types of autoreceptors are D2 receptors, there must be different coupling to distinct intracellular signaling cascades to acount for the functional differences between release-, synthesis, and impulse-modulating autoreceptors.
The autoreceptor can be thought of as a key part of a feedback mechanism. Thus, DA that is released from a neuron stimulates an autoreceptor to inhibit further DA release. Release-modulating autoreceptors are a common regulatory feature of all neurons that use classical transmitters. Because intracellular DA levels regulate tyrosine hydroxylase activity by interfering with the binding of TH to its cofactor, changes in the release of DA may also alter transmitter synthesis. Synthesis-modulating autoreceptors directly regulate DA synthesis: DA release and subsequent binding to the D2 autoreceptor decreases DA synthesis, while DA antagonists that block the autoreceptor increase catecholamine synthesis. Interestingly, synthesis-modulating autoreceptors are not found on all DA neurons: some midbrain and hypothalamic DA neurons lack synthesis-modulating autoreceptors. Finally, impulse-modulating autoreceptors are found on the soma and dendrites of DA neurons and regulate the firing rates of these cells. Because the release of DA can alter synthesis of the transmitter by changing feedback inhibition on TH, it follows that impulse-modulating autoreceptors also change DA synthesis. Thus, all three types of DA autoreceptors may ultimately regulate synthesis. The interdependence of regulatory processes governing DA cells is characteristic of monoaminergic neurons.
Inactivation of Catecholamine Transmission
Constant levels of a neurotransmitter in the synapse will not accurately convey information about the dynamic state of the presynaptic neuron to its follower cell. In addition, continuous stimulation of certain receptors is pathological and can damage postsynaptic cells. For example, when certain receptors for the transmitter glutamate are activated continuously, the result is “excitotoxic” cell death. In order to prevent continuous presynaptic signaling, different mechanisms have evolved for terminating the actions of a transmitter. The simplest is diffusion of the transmitter and its subsequent dilution in extracellular fluid to subthreshold concentrations. More important, however, are active modes of halting transmitter action, including uptake of the transmitter by membrane-associated transporter proteins and enzymatic inactivation of the transmitter.
Enzymatic Inactivation of Catecholamines
Enzymatic inactivation originally was thought to be the major means of terminating catecholamine actions in the CNS. Two enzymes contribute to catecholamine catabolism: monoamine oxidase (MAO) and catechol-0-methyltransferase (COMT). These enzymes can act independently or can act on the products generated by the other enzyme, leading to catecholamine metabolites that are deaminated, O-methylated, or both. COMT is a relatively nonspecific enzyme that transfers methyl groups from the donor S-adenosylmethionine to the m-hydroxy group of catechols. COMT is found both in peripheral tissues and the central nervous system and is the major means of inactivating catecholamines that are released from the adrenal gland.
MAOs oxidatively deaminate catecholamines and their O-methylated derivatives to form inactive and unstable derivatives that can be further degraded by other enzymes. Two forms of MAO have been identified. MAOA has high affinities for NE and serotonin and is selectively inhibited by drugs such as clorgyline. In contrast, MAOB has a higher affinity for o-phenylethylamines and is selectively inhibited by different compounds, such as deprenyl. The MAOs are important targets of drugs used to treat several neuropsychiatric disorders (Box 6.1).
Box 6.1 Mao and Comt Inhibitors in the Treatment of Neuropsychiatric Disorders
One hypothesis of the pathophysiology of depression posits a decrease in noradrenergic levels in the brain. MAOA inhibitors, such as tranylcypromine, effectively increase NE levels (as well as DA and 5-HT concentrations) and were once a mainstay in the treatment of depression. However, the use of MAO inhibitors in depression is less common today, with the major pharmacological treatments being drugs that increase extracellular NE levels by blocking the NE transporter (tricyclic antidepressants) and other drugs that increase 5-HT or DA levels by blocking SERT or DAT (such as Prozac and Welbutrin). The treatment of depression with MAOA inhibitors, although still useful for certain patients who do not respond to other antidepressants, is marred by a large number of side effects. Among the most serious is hypertensive crisis. Patients treated with MAOA inhibitors cannot metabolize tyramine efficiently, which is present in large amounts in foods such as aged cheeses and red wines. Because tyramine releases catecholamines peripherally, small amounts of tyramine increase blood pressure significantly and increase the risk for a stroke.
Deprenyl, a specific inhibitor of MAOB, has been used as an initial treatment for Parkinson’s disease (PD; see Chapter 31). The use of deprenyl in the treatment of PD and the rationale for its use were based on data from studies of a neurotoxin, 1-methyl-4-pheny 1-1,2,3,6-tetrahydropyridine (MPTP). MPTP causes degeneration of midbrain DA neurons and a parkinsonian syndrome. MPTP-induced parkinsonism was first noted in a group of opiate addicts: in an attempt to synthesize a designer drug, the structurally related MPTP was inadvertently produced. Addicts who injected this drug developed a severe parkinsonian syndrome. Subsequent animal studies showed that MPTP itself is not toxic, but that its active metabolite, MPP+, is highly toxic. The formation of MPP+ from MPTP is catalyzed by MAOB, and thus treatment with MAO inhibitors such as deprenyl can prevent MPTP toxicity. The realization that MPTP administration reproduces the motor deficits on PD reawakened interest in environmental toxins as a cause of PD. The MPTP saga also led to the idea that deprenyl treatment might slow the progression of PD by preventing metabolism of some environmental compound to an active toxin. Although clinical studies initially were interpreted to suggest that deprenyl treatment slowed progression of Parkinson’s disease, later studies showed that deprenyl increases DA levels slightly and thus gives some symptomatic relief.
Catechol O-methyl transferase, which together with MAO degrades catecholamines, also plays a role in the treatment of PD. Two COMT inhibitors are used to prevent the enzymatic inactivation of DOPA. By inhibiting COMT, these drugs prolong the therapeutic action of DOPA and may smooth out fluctuations in the therapeutic response to DOPA.
Changes in catecholamine function also have been the object of intense scrutiny in schizophrenia, with recent attention focusing on possible changes in dopamine. One allelic variant of the COMT gene results in a much reduced activity of the enzyme. Data have examined COMT alleles for full vs. low COMT activity in normal subjects and schizophrenics. Individuals bearing the allele that confers lower COMT activity display improved performance on cognitive tasks that involve DA actions in the prefrontal cortex; the performance of schizophrenic persons on these tasks is impaired. It therefore has been proposed that high COMT activity may confer an increased risk to schizophrenia.
Ariel Y. Deutch and Robert H. Roth
Neuronal Catecholamine Transporters
The reuptake of a transmitter released by a neuron is the major mode of inactivation of classical neurotransmitters in the brain. Accumulation of the transmitter also allows intracellular enzymes that degrade the transmitter to act, thus bolstering the actions of extracellular enzymes.
Several characteristics define the high-affinity reuptake process for transmitters. The process is energy dependent and saturable, depends on Na+ cotransport, and requires extracellular Cl–. Because reuptake depends on coupling to the Na+ gradient across the neuronal membrane, toxins that inhibit Na+,K+-ATPase inhibit reuptake. Under certain conditions, the coupling of transporter function to Na+ flow may cause local changes in membrane Na+ gradients and thereby paradoxically cause the transporter to operate in “reverse” to “release” the transmitter from the cell to the extracellular space.
Membrane catecholamine transporters are not Mg2+ dependent and are not inhibited by reserpine, which distinguishes neuronal and vesicular membrane transporters. Catecholamine transporters are localized to neurons. Although a reuptake process accumulates catecholamines into astrocytes, a type of glial cell, this is not a high affinity process and its functional significance is unclear.
Two distinct mammalian catecholamine transporters, one for dopamine (dopamine transporter, DAT) and one for norepinephrine (NET), have been identified. The two are closely related members of a class of transporter proteins (including serotonin and amino acid transmitter transporters) with 12 transmembrane domains. Neither transporter is specific, with each accumulating both DA and NE. In fact, NET has a higher affinity for dopamine than for NE.
The regional distribution of DAT and NET largely follows the expected localization to DA and NE neurons, respectively. However, DAT does not appear to be expressed in all DA cells. Certain hypothalamic cells that release DA into the blood system of the pituitary lack detectable DAT mRNA and protein. Because DA released from these neurons is carried away in the blood, there is no need for a transporter protein on these DA cells.
Immunohistochemical studies of the subcellular localization of DAT found that the transporter is not present at the synaptic junction, but is located just outside of this region. Thus, the transporter may be used to accumulate DA that has escaped from the synaptic cleft. This suggests that diffusion is the initial process by which DA is removed from the synapse. The extrasynaptic localization of the catecholamine transporters, coupled with a similar extrasynaptic localization of dopamine receptors, suggests that extrasynaptic (“volume”) neurotransmission may be of major importance in transmitter actions (see Agnati, Guidolin, Guescini, Genedani, & Fuxe, 2010).
Mice in which the gene encoding DAT has been deleted have been particularly useful in clarifying the function of transporters. Transgenic mice that lack DAT have a remarkable number of changes in DA function, ranging from increased extracellular DA levels and delayed clearance of released DA to a striking decrease in tissue concentrations of DA in the face of increased DA synthesis (Gainetdinov, Jones, Fumagalli, Wightman, & Caron, 1998). Some of the deficits in DAT knockout mice have been suggested to reflect a disinhibition of tyrosine hydroxylase due to a lack of intraneuronal DA (removing feedback inhibition of the enzyme), resulting in a marked increased in DA synthesis and release.
The catecholamine transporters are important targets of many drugs. Cocaine and amphetamine both increase extracellular levels of catecholamines by blocking transporters. In particular, cocaine shows a very high affinity for DAT; amphetamine is a less potent inhibitor but also “releases” catecholamine by reversing the normal direction of transporter function, extruding the transmitter from neurons. The tricyclic antidepressant drugs work by blocking the norepinephrine transporter, as do newer norepinephrine-selective uptake blockers.
Serotonin
Well over a century ago, scientists were aware of a substance in the blood that induced powerful contractions of smooth muscle organs. In the mid-twentieth century, Page and collaborators succeeded in isolating the compound, which they suggested to be a possible cause of high blood pressure, from platelets. At the same time, Italian researchers were studying a substance in intestinal mucosa that caused contractions of intestinal smooth muscle. The material isolated from platelets was called “serotonin,” and the substance isolated from the intestinal tract was named “enteramine.” Studies soon revealed that the two substances were the same compound, 5-hydroxytryptamine (5-HT), which now is commonly referred to as serotonin.
Serotonin is found in neurons and in several other types of cells in the body. In fact, the brain accounts for only about 1% of total body stores of serotonin. Although the isolation and identification of serotonin were made from peripheral tissues, much of the subsequent interest in serotonin was based on its potential involvement in psychiatric disorders. The finding that serotonin’s chemical structure is similar to that of LSD led to theories that associated abnormalities in serotonin function to schizophrenia and depression.
The basic process of serotonin synthesis is very similar to that of catecholamine transmitters: a peripheral amino acid (tryptophan) gains entry into the brain and is metabolized in serotonergic neurons via a series of enzymatic steps. Once tryptophan enters the serotonergic neuron, it is hydroxylated by tryptophan hydroxylase, the rate-limiting step in serotonin synthesis, giving rise to 5-hydroxytryptophan (5-HTP). In turn, 5-HTP is decarboxylated by L-aromatic amino acid decarboxylase to the transmitter serotonin. Thus, only two critical enzymes (tryptophan hydroxylase and AADC) are involved in the synthesis of 5-HT (Iversen et al., 2008).
Alternative Tryptophan Metabolic Pathways
Although serotonin is usually the endpoint of tryptophan metabolism in brain, serotonin can be metabolized further to yield active products. In the pineal gland, 5-HT is metabolized to form 5-methoxy-N-acetyltryptamine (melatonin), a hormone thought to play an important role in sleep.
In peripheral tissues, most tryptophan is not used to synthesize 5-HT but instead is metabolized by the kynurenine pathway. This kynurenine “shunt” is also present in the brain and leads to the accumulation of several interesting active substances (see Schwarcz, 2004). The two major tryptophan metabolites generated by the kynurenine shunt are quinolinic and kynurenic acids. Quinolinic acid is a potent activator of certain glutamate receptors and upon administration to animals causes cell loss and convulsions. In contrast, kynurenic acid is an agonist that activates one type of acetylcholine receptor in brain and decreases extracellular levels of the transmitter glutamate; levels of kynurenic acid have been reported to be increased in schizophrenia.
Inactivation of Released Serotonin
Serotonin is inactivated primarily by reuptake through SERT, the serotonin transporter that belongs to the same family of transporters as the catecholamine transporters. Selective serotonin reuptake blockers (SSRIs), which block SERT, are widely used as antidepressants; the prototypic SSRI is fluoxetine (Prozac). Because different antidepressant drugs increase levels of serotonin or NE, or both, current theories of depression posit critical modulatory roles for NE and 5-HT.
The enzymatic degradation of 5-HT is catalyzed by monoamine oxidase (MAO). The product of this reaction, 5-hydroxyindole acid aldehyde, is oxidized further to 5-hydroxyindoleacetic acid. MAO inhibitors prevent the metabolism of serotonin and thus increase 5-HT levels, and have been widely used as antidepressants (Box 6.1).
Acetylcholine
Our basic ideas about chemical synaptic transmission are based on early studies of acetylcholine (ACh). Pharmacological and electrophysiological studies at the neuromuscular junction led to an understanding that ACh is a chemical messenger between excitable cells. These ideas about cholinergic signaling between cells are the cornerstone of today’s understanding of chemical neurotransmission. For example, electrophysiological studies revealed fast excitatory responses of muscle fibers to the stimulation of nerves innervating the muscle. The presence of miniature end plate potentials (mEPPs) in muscle fiber was noted, and in the early 1950s, Fatt and Katz demonstrated that these mEPPs resulted from the slow “leakage” of ACh, with each mEPP representing the release of transmitter in one vesicle (termed a quantum). Overt depolarization generated an increase in the number of quanta released over a given period of time (see Chapter 7). Over the past half century many of the rules that govern ACh neurotransmission have been found to be general principles that apply to many transmitters.
The synthesis of ACh has a single step: the acetyl group from acetyl coenzyme A is transferred to choline by the enzyme choline acetyltransferase (ChAT), forming Ach. The acetyl-CoA that serves as the donor is derived from pyruvate generated by glucose metabolism. There is one interesting twist on ACh synthesis: acetyl-CoA is found in mitochondria, but the synthetic enzyme ChAT is cytoplasmic. This means that acetyl-CoA must exit the mitochondria to gain access to ChAT. Despite the fact that ChAT is the only enzyme involved in ACh synthesis, it is not the rate-limiting step in synthesis of the transmitter. The full enzymatic activity of ChAT is not expressed in vivo: ChAT activity measured in vitro is much greater than would be expected on the basis of measurements of ACh synthesis in vivo. The reason for this discrepancy has been suggested to be related to the requirement that acetyl-CoA be transported from the mitochondria to the cytoplasm. Alternatively, intracellular choline concentrations may determine the rate of ACh synthesis. This latter idea led to the use of choline precursors in attempts to increase ACh synthesis in Alzheimer’s disease, in which there is a marked decrease of cortical ACh levels. Unfortunately, these attempts have not proven successful.
ACh is taken up into storage vesicles by the vesicular cholinergic transporter (VAChT). Cloning of the human VAChT revealed that the gene is localized to chromosome 10, near the gene encoding ChAT. Subsequent studies demonstrated that the entire VAChT coding region is contained in the first intron of the ChAT gene. These observations suggested that both genes are coordinately regulated, a suspicion that subsequently was confirmed.
A major way of inactivating ACh is enzymatic, in which ACh is hydrolyzed to choline. There are two types of cholinesterases that accomplish this step: acetylcholinesterases (AChEs) and butyrylcholinesterases (Taylor & Brown, 2006). The former are relatively specific for ACh and are found in high concentration in the brain, whereas butyrylcholinesterases are enriched in the liver.
Acetylcholinesterase is present in high concentrations in cholinergic neurons. However, AChE is also found in moderately high concentrations in some noncholinergic neurons that receive cholinergic inputs. This observation is consistent with the fact that AChE is a secreted enzyme that is associated with the cell membrane. Thus, ACh hydrolysis takes place extracellularly, and the choline generated is conserved by the high-affinity reuptake process. In addition to its role in inactivating acetylcholine, AChE has been proposed to be a chemical messenger in the CNS (Greenfield, Zimmermann, & Bond, 2008).
γ-Aminobutyric Acid: The Major Inhibitory Neurotransmitter
Several amino acids fulfill most of the criteria for consideration as neurotransmitters. The best studied of these are γ-aminobutyric acid (GABA), the major inhibitory transmitter in brain, and glutamate, which is the brain’s major excitatory transmitter. Many of the principles concerning transmitter synthesis and inactivation that we have previously discussed also apply to amino acid transmitters. However, there are also some key differences. The most obvious difference is that GABA is derived from glucose metabolism (Fig. 6.3). Thus, mechanisms must exist to segregate the transmitter and general metabolic pools of the amino acid transmitters (Olsen & Betz, 2006). A second difference between amino acid and catecholamine transmitters is that inactivation of the former involves several types of transporters that are found on glial cells as well as neurons.
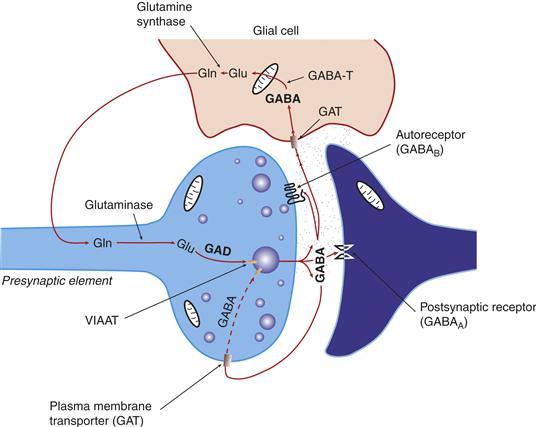
Figure 6.3 Schematic depiction of the life cycle of the transmitter pool of GABA. a-Ketoglutarate formed in the Krebs cycle is transaminated to glutamate (Glu) by GABA transaminase (GABA-T). The transmitter GABA is formed from the Glu by glutamic acid decarboxylase (GAD). GABA that is released is taken by high-affinity GABA transporters (GAT) present on neurons and glia.
GABA Synthesis
The GABA Shunt and GABA Transaminase
GABA ultimately is derived from glucose metabolism. α-Ketoglutarate formed by the Krebs cycle is transaminated to the amino acid glutamate by GABA-transaminase (GABA-T). In those cells in which GABA is used as a transmitter, the presence of another enzyme, glutamic acid decarboxylase (GAD), generates GABA from glutamate (Fig. 6.3). The presence of GAD is therefore an unambiguous marker of neurons that use GABA as a transmitter.
An unusual feature of GABA synthesis is that intraneuronal GABA is inactivated by the actions of GABA-T, which appears to be associated with mitochondria. Thus, GABA-T is both a synthetic and a degradative enzyme! GABA-T metabolizes GABA to succinic semialdehyde but only if α-ketoglutarate is present to receive the amino group that is removed from GABA. This unusual GABA shunt serves to maintain supplies of GABA.
How Are Transmitter and Metabolic Pools Separate?
A major question concerning amino acid transmitters is how the transmitter pool is separately maintained from the general metabolic roles of amino acids. The GABA synthetic enzyme GAD is a cytosolic enzyme, but GABA-T, which converts α-ketoglutarate into the GAD substrate glutamate, is present in mitochondria. Thus, the metabolic pool is present in mitochondria, and glutamate destined for the transmitter pool must be exported from mitochondria to the cytosol.
Glutamate is not only a precursor to GABA, but it is also the major excitatory neurotransmitter found in the brains of mammals. Why GABA neurons fail to use the precursor glutamate as a transmitter probably involves different vesicular transporters that accumulate glutamate and GABA into vesicles, as well as different biosynthetic enzymes for the transmitter and metabolic pools of glutamate. In the case of glutamate, three different vesicular glutamate transporters are responsible for determining if glutamate is used as a transmitter.
Storage and Release of GABA
Vesicular Inhibitory Amino Acid Transporter
A vesicular GABA transporter was cloned on the basis of homology to a protein found in the worm Caenorhabditis elegans. The strategy of moving from invertebrate to mammalian species has been very useful in identifying a variety of mammalian transmitter-related genes. The vesicular GABA transporter shares with the vesicular monoamine transporters a lack of substrate specificity, and will transport the inhibitory transmitter glycine as well as GABA.
Autoreceptor Regulation of GABA Release
There are two major classes of GABA receptors. One type of GABA receptor (the GABAA receptor) forms ion channels and is mainly on cells postsynaptic to GABA terminals (see Chapter 8). A second type of receptor, the GABAB receptor, is coupled to intracellular G proteins. Pharmacological studies indicate that the autoreceptor-mediated regulation of GABA neurons involves GABAB receptors located on GABAergic axon terminals. Anatomical studies have revealed that GABAB receptors also are found on postsynaptic non-GABAergic neurons. It is possible that these postsynaptic GABAB sites respond to GABA released from a neuron that is presynaptic to another GABA neuron. Interestingly, when one GABA neuron terminates on another GABA cell, the inhibition of the second (postsynaptic cell) will have the same functional consequence as an autoreceptor (decreasing subsequent transmitter release) on the third cell in the chain.
Inactivation of GABA
Reuptake is the primary mode of inactivation of the transmitter GABA. There are four GABA transporters (GATs), providing a diverse means of regulating GABA neurons. Early studies defined the different GABA transporters as neuronal or glial based on pharmacological criteria. However, anatomical studies found that one GAT that was defined on pharmacological grounds as a glial transporter is present in both neurons and glia. The reason for multiple GABA transporters is not clear. GATs are expressed in both GABAergic and non-GABAergic cells (presumably cells that receive a GABA innervation). Consistent with the promiscuous uptake of transmitters by other transporters, amino acids other than GABA are also substrates for GATs.
Glutamate and Aspartate: Excitatory Amino Acid Transmitters
Excitatory amino acid transmitters account for most of the fast synaptic transmission that occurs in the mammalian brain. Glutamate and aspartate are the major excitatory amino acid neurotransmitters, and several related amino acids, such as N-acetylaspartylglutamate, are also thought to have neurotransmitter roles.
Neither glutamate nor aspartate crosses the blood–brain barrier, and in the brain these transmitters are derived by local synthesis from glucose. Two processes lead to glutamate synthesis in the nerve terminal. As discussed earlier, glutamate is formed from glucose through the Krebs cycle and transamination of α-ketoglutarate. Glutamate can also be formed directly from glutamine. Because glutamine is synthesized in glial cells, both neurons and glia are important in determining the transmitter pool of glutamate. Glutamine is exported from glia and incorporated by nerve terminals before being converted into glutamate by a glutaminase enzyme (Hassel & Dingledine, 2006).
Because of the intermingling of glial and neuronal contributions to glutamate synthesis, and the lack of specific enzymes or other proteins to distinguish the metabolic pool of glutamate from the transmitter glutamate, the identification of glutamatergic neurons was initially made on the basis of physiological studies. More recently the identification of three vesicular glutamate transporters, VGluT1-3, has provided a relatively simple means for identifying glutamatergic neurons. However, in neuroscience facts are rarely unambiguous, and anatomical studies over the past several decades have revealed that many glutamate neurons also contain one or more other transmitters (Fremeau, Voglmaier, Seal, & Edwards, 2004), with some even containing GABA. Because GABA hyperpolarizes cells in adult neurons, while glutamate depolarizes neurons, one would not expect to find transmitter pools of GABA and glutamate in the same neuron. The functional significance of such an arrangement is not clear.
Just as there are several vesicular glutamate receptors, there are several glutamate transporters that terminate the action of glutamate. An unusual aspect is that the major cell type taking up glutamate is the astrocyte, which expresses very high levels of one glutamate transporter. This is one of several findings that point to glia as releasing glutamate in a signaling capacity (Halassa & Haydon, 2010). These findings have led to an increasing awareness of glia as being far more than structural support cells and argue for a much broader contribution of glia to neuronal communication.
Nonclassical Neurotransmitters
Classical transmitters account for relatively few of all neurotransmitters. For example, many more transmitters are peptides, which share many but not all key attributes of classical transmitters. Both classical and peptide transmitters usually are well conserved across species, and indeed many of the peptide transmitters were initially isolated from amphibians. In addition, both classical and peptide transmitters are synthesized in neurons, where they are stored in vesicles and released in a Ca2+-dependent manner. However, the synthetic mechanisms and the modes of inactivation of the two types of transmitters are quite different. We will first consider the question of the significance of multiple neurotransmitters, and then turn to general principles of peptide transmitter life cycle.
Why Do Neurons Have So Many Transmitters?
About a dozen molecules are generally accepted as “classical” transmitters. Many more neuropeptides serve as transmitters. And there are “unconventional” transmitters that fail to meet most of the criteria for classical transmitters yet have clearly been shown to function as chemical messengers over restricted spatial and temporal domains. If transmitters simply serve as a chemical bridge that conveys information between two spatially distinct cells, why have so many chemical messengers?
Convergence of Different Transmitter-containing Axons on a Common Neuron
Perhaps the simplest explanation for multiple transmitters is that many nerve terminals synapse onto a single neuron. How can a neuron distinguish between multiple inputs that carry different information? One way is to segregate the place on the neuron at which an input arrives, such as the soma, axon, or dendrite. However, because many afferents terminate in close proximity, another means of distinguishing inputs and their information is necessary. Chemical coding of inputs from various neurons can provide a high degree of both spatial and temporal resolution.
Colocalization of Neurotransmitters
The idea that a neuron is limited to one transmitter can be traced to Henry Dale, or more accurately, to an informal restatement of what is termed Dale’s principle. In the 1940s, Dale posited that a metabolic process that takes place in the cell body can reach or influence events in all processes of the neuron. Sir John Eccles restated Dale’s view to suggest that a neuron releases the same transmitter from all of its processes. Illustrating the dangers of scientific sound bites, this “principle” was soon misinterpreted to indicate that only a single transmitter can be present in a given neuron, a view that held sway for about 40 years. We now know that neurons can contain multiple transmitters, including both a classical transmitter (such as DA) and a peptide transmitter (such as neurotensin). Indeed, it appears that few, if any, neurons contain only one transmitter, and in many if not most cases three or more transmitters are found in a single neuron.
The presence of multiple transmitters in single neurons suggests that different transmitters are used by a neuron to signal different functional states to its target cell. The firing rates of the neurons differ considerably, and it may be useful to encode fast firing by one transmitter and slower firing by a different transmitter. The firing pattern of neurons also conveys information. For example, a neuron may discharge five times every second on average; this may mean that the cell regularly discharges every 200 ms, but can also represent a cell that has a burst of five discharges during an initial 100-ms period followed by 900 ms of silence. Peptide transmitters often are released at higher firing rates and particularly under burst-firing patterns.
The different synthetic steps in peptides and classical transmitters lead to differential release. Classical transmitters can be replaced rapidly because their synthesis occurs in nerve terminals, with rapid uptake of the released transmitter and energy-efficient recycling of the transmitter. In contrast, peptide transmitters must be synthesized in the cell body and transported to the terminal. Thus, it is useful to conserve peptide transmitters for situations of high demand because they would otherwise be depleted rapidly.
Transmitter Release from Different Processes
The restatement of Dale’s principle by Eccles held that a transmitter is found in all processes of a neuron. In invertebrate species a transmitter can be localized to different parts of a neuron, and it is now clear that most proteins in mammalian neurons are not randomly distributed but present in distinct “compartments” of a neuron, often being reversibly tethered there by anchoring proteins. For example, if a transmitter were restricted to a particular part of a neuron, the neuron would need multiple transmitters to account for different release sites. Receptors are very specific in their locations on neurons, and certain receptors are recruited to “hot spots” on neuronal processes by the activity patterns of presynaptic inputs.
Synaptic Specializations versus Nonjunctional Appositions between Neurons
The anatomical relations between one cell and its follower may contribute to the need for different transmitters. We usually think of synaptic specializations as the physical substrate of communication between two neurons. However, nonsynaptic communication between two neurons can also take place, including over longer distances than that of conventional synapses. In such a situation the requirements for transmitter action would differ from those discussed previously because the distance traversed by the transmitter molecule would be farther than at a synaptic apposition. Thus, transmitters that lack an efficient reuptake system, such as peptide transmitters, might be favored at nonsynaptic sites. Because a single neuron can form both synaptic and nonsynaptic specializations, a single neuron may require more than one neurotransmitter.
Fast versus Slow Responses of Target Neurons to Neurotransmitters
Different firing rates or patterns of firing may be accompanied by changes in the type or relative amounts of a transmitter being released from a neuron. For example, stimulation of receptors that form ion channels leads to very rapid changes, whereas actions at metabotropic receptors that are coupled to intracellular events through specific transduction molecules have slower response characteristics. Differences in temporal response characteristics allow the receptive neuron to respond differently to a stimulus depending on the antecedent activity in the cell. A transmitter can change the response characteristics of a particular cell to subsequent stimuli by seconds or even minutes, and thus short-term changes can occur independent of changes in gene expression.
Peptide Transmitters
Synthesis and Storage of Peptide Transmitters
Classical transmitters are synthesized in the axon from which they are released. In contrast, genes encoding most peptide transmitters give rise to a prohormone, which is incorporated into secretory granules, after which the prohormone is acted on by peptidases to form the peptide transmitter (Fig. 6.4). This process typically occurs in the cell body, and the peptide-containing vesicles are then transported to the axon; a small number of peptide transmitters are synthesized enzymatically.
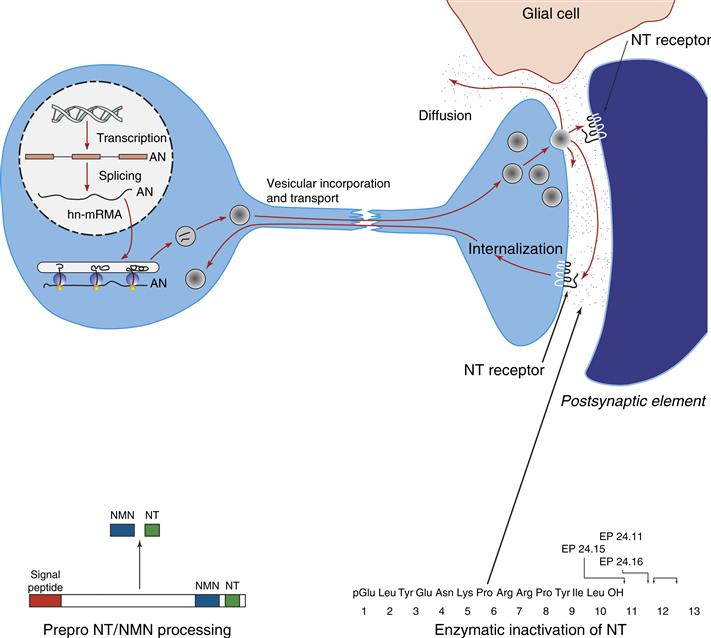
Figure 6.4 Schematic illustration of the synthesis, release, and termination of action of the peptide transmitter neurotensin (NT). The illustrative panels on the bottom show processing of NT from the prohormone (left) and enzymatic inactivation of NT (right).
In neurons that use classical transmitters, demands for increased amounts of transmitter are met by increasing local transmitter synthesis or activity at the axon terminal. However, increasing the amount of a peptide transmitter requires an increase in gene expression to yield a prohormone, with a subsequent delay in delivery to the axon terminal of the peptide. Thus, classical transmitters respond to increased demand rapidly, but peptide transmitters cannot.
The storage of peptides and classical transmitters also differs. Classical neurotransmitters are usually stored in small (~50 nm) synaptic vesicles, while neuropeptide transmitters are stored in large (~100 nm) dense-core vesicles. Because peptide transmitters are usually released at a high neuronal firing frequency or when neurons show a burst-firing pattern, different mechanisms may be involved in the release of peptide- and classical transmitter-containing vesicles. Although the release of both classical and peptide transmitters is calcium-dependent, distinct but related molecular mechanisms subserve the release of small and large dense-core vesicles (Sieburth, Madison, & Kaplan, 2007).
Inactivation of Peptide Transmitters
There are major differences in how peptide and classical transmitters are inactivated. Classical transmitter actions primarily depend on a high-affinity reuptake process to remove the transmitter from the extracellular space. There are no such transporters for peptide transmitters, which are inactivated enzymatically or by diffusion.
Enzymatic inactivation of peptide transmitters differs from that of classical transmitters. Peptide transmitters are short chains of amino acids, but the inactivating enzymes are specific for certain dipeptide sequences and are not specific to any single peptide. For example, an enzyme that inactivates small opioid-like peptide transmitters (enkephalins) is often referred to as enkephalinase, but also cleaves other peptide transmitters.
A final difference between the inactivation of peptide and classical transmitters is the nature of the final product. When classical transmitters are broken down enzymatically, the metabolites are inactive at the transmitter’s receptor. However, certain peptide fragments derived from the enzymatic “inactivation” of peptide transmitters are biologically active. An example is angiotensin, in which angiotensin I is metabolized to yield angiotensin II and III, each successively more active than the parent angiotensin I. It can therefore sometimes be difficult to distinguish between transmitter synthesis and transmitter inactivation. The peptide that is stored in vesicles and then released is therefore considered the transmitter, although the actions of certain peptidases may lead to other biologically active fragments. As a means of exploring aspects of the life cycle of peptide transmitters, we will now examine as a representative example the neuropeptide neurotensin.
Neurotensin Synthesis
Neurotensin (NT) is widely expressed in the central nervous system and in certain peripheral tissues, such as the small intestine. A related peptide, neuromedin N (NMN), is also present. A 170 amino acid prohormone precursor of NT is the product of a single gene that is transcribed to yield two mRNAs. The smaller transcript is the major form in the intestine; the two mRNA species are equally abundant in most brain areas. The precursor contains one copy each of NT and NMN. The molar ratios of NT/NMN vary across different tissues, suggesting different enzymatic processing of the prohormone or, alternatively, the generation of different transcripts. Because NT and NMN are contained in the same exon of the NT-NMN gene, differences in relative abundance of the two are due to differential processing of the precursor.
NT and other peptide transmitters are stored in large dense-core vesicles. Although there are some differences in the mechanisms controlling release of the contents of small and large dense-core vesicles, the general characteristics of NT release are similar to those of classical transmitters. Thus, depolarization evokes the Ca2+-dependent release of both NT and NMN (Kitabgi, De Nadal, Rovere, & Bidard, 1992). The impulse-dependent release of NT varies as a function of frequency and pattern of impulses, with higher frequencies of firing or burst firing patterns eliciting greater peptide release (Bean & Roth, 1992).
Inactivation of Neurotensin
Peptides are inactivated by enzymatic actions or diffusion, or both. NT is degraded by three endopeptidases, grandly named 24.11, 24.15, and 24.16. Endopeptidase 24.11 cleaves NT at two specific sites to yield a decapeptide, as does endopeptidase 24.16 (Fig. 6.4); the other endopeptidase acts at a different set of amino acid residues. Because these enzymes act at dipeptides that are found in many peptides and proteins, and two of the three act at the same site, it is obvious that enzymes that inactivate peptides are not very specific.
Although there are no known membrane transporters for peptide transmitters, peptides can be accumulated by neurons. This occurs by internalization of a neuropeptide bound to its receptor. G protein-coupled receptors undergo internalization via an endocytotic mechanism, where they are either recycled to the membrane after various steps or shipped to lysosomes for degradation (Hanyaloglu & von Zastrow, 2008). However, once inside the cells the peptide and its receptor can dissociate, leaving the free neuropeptide to exert some actions.
Coexistence of Neurotensin and Classical Transmitters
Neurotensin is colocalized with dopamine in certain hypothalamic and midbrain neurons. The colocalization of NT and DA has provided a useful system in which to explore the interrelationships between two colocalized transmitters. In the prefrontal cortex of the rat, NT is found only in DA axons, although there are some DA axons that do not contain NT. Neurotensin release in the prefrontal cortex is increased when neuronal firing is increased or when DA neurons display a burst-like firing pattern (Bean & Roth, 1992). In addition, the release-modulating dopamine autoreceptor on DA axons regulates NT release, but in a very different manner than it regulates DA release: DA agonists (which decrease DA release) enhance NT release from colocalized DA-NT axons. Conversely, antagonists at the DA autoreceptor decrease NT release but enhance DA release. Thus, the release of NT and DA are reciprocally regulated by actions at release-modulating DA autoreceptors.
Unconventional Transmitters
The differences between peptide and classical transmitters substantially delayed the general acceptance of peptides as transmitters. However, once peptides were recognized as neurotransmitters, a gate opened for the consideration of radically different molecules as transmitters.
Remarkable technical advances have allowed us to measure substances in the brain that are present in minute quantities or are very unstable. The early “classical” neurotransmitters were present in relatively high concentrations, at least in some brain sites, permitting the measurement of these compounds by neurons after electrical or chemical stimulation. Peptide transmitters are usually found in smaller amounts, and sensitive biochemical assays for measuring neuropeptides were not widely used until the 1970s. When it became clear that there were many compounds, including certain peptides, that might potentially serve as chemical messengers between neurons, the basic criteria for designation of a molecule as a neurotransmitter came into question.
What should be the key attribute(s) of a neurotransmitter? One simple approach would be to designate as a neurotransmitter any compound that permits information flow from one neuron to another. However, this definition could encompass hormone signaling and does not address the temporal characteristics of transmitter action. Moreover, it does not address the possibility of a neurotransmitter being a molecule that conveys information between neurons and non-neuronal brain cells, such as glia. Finally, this definition does not accommodate unconventional roles for transmitters, such as the regulation of neuronal development or intracellular trafficking of proteins. We will now discuss what we call, for lack of a better term, “unconventional” transmitters.
Endocannabinoids
The psychoactive properties of marijuana have been known for thousands of years. Marijuana contains dozens of structurally related cannabinoids. Cannabinol was the first of the cannabinoids to be structurally characterized. The major psychoactive cannabinoid, Δ-9-tetrahydrocannabinol (THC), was subsequently identified. The fact that marijuana and THC had psychoactive properties in humans led to search for a receptor for THC, the first of which, CB1, was cloned a full half century after the structure of cannabinol was first elucidated. As has been the case for other “orphan” receptors, the search for an endogenous ligand that binds to the CB1 receptor culminated two years after the cloning of CB1 with the identification of a compound that bound with high affinity to the receptor. Over the past 15 years, the identification of multiple endocannabinoids (ECs), the enzymes involved in synthesizing and degrading endocannabinoids, and the characterization of two cannabinoid receptors have led to the realization that ECs serve as chemical messengers between neurons.
Synthesis, Inactivation, and Release of Endocannabinoids
Endocannabinoids are hydrophobic lipids that pass easily through plasma membranes. This free diffusion across membranes means that ECs cannot be stored in vesicles. Instead, they are synthesized and released when needed from lipid precursors located in neuronal membranes. The basic mechanisms involved in the synthesis of ECs are reasonably well characterized, although some issues regarding regulation of EC synthesis have not been resolved (Alger & Kim, 2011).
There are two major ECs in brain, N-arachidonoyl-ethanolamide (anadamide) and 2-arachidonoylglycerol (2-AG). The synthesis of the latter has been reasonably well characterized. An increase in intracellular calcium stimulates phospholipase C (PLC), which metabolizes membrane phosphoinositides to diacylglycerol (DAG). In turn, DAG is metabolized by DAG lipase to 2-AG. There is also a second minor route through which DAG can be produced from phosphatidic acid.
Anandamide is a fatty acid amide and is the best characterized of the ECs. However, the synthesis of anadamide can be accomplished through alternative pathways, and it is unclear which of these multiple synthetic routes predominates in vivo.
The membrane localization of the EC precursors for anandamide and 2-AG, coupled with the highly permeable nature of the two ECs, initially led to controversy concerning the ability of ECs to be synthesized only when the (“on-line”) need for their release occurs. However, newer data favor the idea that ECs are indeed synthesized on demand.
Three proteins play prominent roles in EC inactivation. Two are enzymes (fatty acid amide hydrolyase [FAAH] and monoacylglycerol lipase [MGL]); the third protein is an anadamide transporter. 2-AG is metabolized primarily by MGL, but it can also be acted on by another enzyme (β hydrolase 6). In contrast, anandamide is metabolized by fatty acid hydrolase. The availability of mice with genetic deletions of EC synthetic or degradative enzymes has been critical in establishing the functional roles of anandamide and 2-AG. The anandamide transporter is a sodium- and energy-dependent carrier protein that facilitates diffusion.
Recent studies have demonstrated that ECs act as retrograde messengers at synapses. Thus, ECs are produced on demand in an activity-dependent manner by a neuron through the cleavage of membrane lipids. The resultant ECs travel backward (retrogradely) across the synapse to activate presynaptic cannabinoid receptors on axon terminals, resulting in a suppression of transmitter release. Because the presynaptic CB1 cannabinoid receptor is found on both GABAergic and glutamatergic nerve endings, EC signaling can modulate both excitatory and inhibitory transmission. EC-mediated retrograde signaling appears to be crucial for certain types of short- and long-term synaptic plasticity that underlie learning and memory.
Although still early, studies of ECs have inspired the development of novel treatment strategies for neuropsychiatric disorders. Consistent with subjective reports that marijuana increases eating (the “munchies”), early clinical trials reported some success of CB1 receptor antagonists in the treatment of obesity. However, other potential behavioral side effects of CB1 antagonists led to a halt in the development of these drugs, at least for the time being. Similarly, CB1 antagonists have been advanced as an aide to smoking cessation.
Gases as Unconventional Transmitters
Nitrates such as nitroglycerine have long been used to treat angina pectoris, in which pain arises because of insufficient blood delivery to the heart. Nitrates dilate cardiac blood vessels, thereby increasing blood flow and diminishing or relieving the pain. However, the mechanism by which nitrates dilate coronary arteries was not known until relatively recently. In 1980, a “relaxing” factor contained in the cells lining blood vessels was found to potently and rapidly dilate blood vessels. This factor was soon shown to be the gas nitric oxide (NO). Other studies found that the transmitter glutamate released a factor that caused blood vessels in the brain to dilate. It then became apparent that endothelial-derived relaxing factor and the glutamate-induced factor that caused vasodilation were the same compound, NO. Thus, NO was present in neurons as well as cells forming blood vessels. These data led to the proposal that NO may take part in intercellular communication (see Box 6.2).
Box 6.2 Going for Gases as Neurotransmitter
One of the most rewarding experiences for a scientist is to find that long-held prejudices are altogether wrong and that a new, correct insight reveals a novel scientific principle. In the late 1950s, only acetylcholine and the biogenic amines were known to be neurotransmitters. The next decade saw amino acids acknowledged as neurotransmitters. The discovery of enkephalins and endorphins in the mid-1970s reinforced gradually accumulating evidence that peptides are transmitters, and now we find that there are over 100 different bioactive brain peptides.
In the 1970s and early 1980s, nitric oxide (NO) was found to mediate the ability of macrophages to kill tumor cells and bacteria and to regulate blood vessel relaxation. A short report suggested that NO can be formed in brain tissue. Progress in the NO field at that time was slow because assaying NO synthase (NOS), the enzyme that oxidizes the amino acid arginine to NO, was quite tedious, based on the accumulation of nitrite formed from the NO. A much simpler approach was to monitor the conversion of [3H]arginine into [3H]citrulline, which is formed simultaneously with NO; this assay could process 100 or more samples in an hour. Research on blood vessels revealed that NO acts by stimulating cyclic GMP formation. In the brain, the excitatory neurotransmitter glutamate was known to augment cyclic GMP levels. Glutamate, acting through its NMDA receptor, triples NO synthase activity in a matter of seconds, and arginine derivatives that inhibit NOS activity block the elevation of cyclic GMP. This finding causally linked the actions of so prominent a neurotransmitter as glutamate to NO.
To determine if NO was a neurotransmitter, it was necessary to ascertain whether NOS was localized in neurons. The most straightforward approach would be to generate an antibody to use in anatomical studies. However, purifying NOS protein to generate an antibody proved very difficult because the enzyme lost its activity in attempts to purify it. The addition of calmodulin was found to stabilize the enzyme. Because calmodulin is a calcium-binding protein, this finding immediately explained how NO formation can be triggered rapidly by synaptic activation through glutamate. When glutamate activates its NMDA receptor, Ca2+ rushes into the cell, binds to calmodulin, and activates NOS.
The ability of purified NOS led to antibodies being developed and NOS being localized by immunohistochemistry. The neuronal form of NOS (nNOS) is present in only about 1% of the neurons in the brain. However, these cells give rise to processes that ramify so extensively that probably every neuron in the brain is exposed to NO. The purified NOS protein also allowed an amino acid sequence to be obtained and the gene for the enzyme cloned. The structure of NOS revealed that it is regulated by many more factors than virtually any other enzyme in biology, including at least five oxidative-reductive cofactors, four phosphorylating enzymes, and three binding proteins. This makes sense because of the unique properties of NO as a gaseous neurotransmitter. Most neurotransmitters are stored in vesicles with large storage pools so that only a small amount of the transmitter is released with each nerve impulse. In contrast, every time a neuron wishes to release a molecule of NO, it must activate NOS—hence, a requirement for exquisitely subtle regulation of the enzyme.
Neurotransmitters come in chemical classes such as biogenic amines, amino acids, and peptides. Might there be at least one other gaseous neurotransmitter? Carbon monoxide (CO) is normally formed in the body by the enzyme heme oxigenase, which is primarily responsible for degrading heme in aging red blood cells. It cleaves the heme ring to form biliverdin, which is reduced rapidly to bilirubin, the pigment that accounts for jaundice in patients with a degradation of red blood cells. When the enzyme cleaves the heme ring, CO is released as a single carbon fragment.
The biosynthetic enzyme for a transmitter should be localized to selected neuronal populations. Heme oxigenise-2, the neuronal form of the enzyme, was shown to be localized to discrete neuronal populations throughout the brain. To seek a neurotransmitter function, the peripheral nervous system (in which synaptic transmission is characterized more readily than in the brain) was used. The myenteric plexus of nerves regulates intestinal peristalsis. A previously unidentified neurotransmitter of myenteric plexus neurons accounts for the relaxation phase of peristalsis. nNOS had already been localized to neurons of the myenteric plexus, and some functional evidence showed that NO might be a neurotransmitter of this pathway. Heme oxigenise-2 was found to be localized to the same myenteric plexus neurons as NOS. Mice with targeted deletions of the genes for nNOS or heme oxigenise-2 were used to elucidate function. In both types of gene knockout mice, intestinal relaxation evoked by neuronal depolarization was reduced about 50%, implying that NO and CO each contribute half of the relaxation. This finding, along with other evidence, established transmitter functions for both NO and CO and suggested that they are functioning as cotransmitters, although exactly how they interact remains a mystery. Such a cotransmitter role reminds us of the fact that most neurons in the brain contain at least two and sometimes more neurotransmitters. Thus, in addition to overturning a number of dogmas about neurotransmission, NO and CO may help resolve the riddle of cotransmission.
Solomon H. Snyder
Nitric oxide is a well-known air pollutant. The idea that an unstable toxic gas could serve as a transmitter led to several incredulous but pointed questions, including “how can a gas be stored for release in an impulse-dependent manner?” The answer is simple: it is not stored. If one accepts the argument that NO is a transmitter, the classical definition of a neurotransmitter becomes untenable. Many theories can accommodate one exception. However, NO is not the only gaseous neurotransmitter. Other gases, including carbon monoxide and hydrogen sulfide, also play transmitter-like roles (Mustafa, Gadalla, & Snyder, 2009), leading to the introduction of the term gasotransmitter!
Synthesis of Nitric Oxide
Nitric oxide synthesis involves a single step, the conversion of L-arginine to NO and citrulline. The enzyme responsible for NO synthesis is aptly named nitric oxide synthase (NOS). There are three isoforms of NOS. Endothelial is found in the cells lining blood vessels, where the NO story began. Macrophage-inducible NOS is present in microglia, while the third isoform is localized to neurons (nNOS). All three isoforms require tetrahydrobiopterin as a cofactor and NADPH as a coenzyme. The presence of NADPH explains why the enzyme NADPH diaphorases, which had been described well before NOS was known, is found in high concentration in nNOS-positive neurons.
Storage and Release of Nitric Oxide
The subheading “Storage and Release of Nitric Oxide” promises a discussion of the storage of NO. As previously discussed, storage of a compound (usually in a place poised for release) was originally considered to be necessary for designation of a molecule as a transmitter. However, there is no storage of NO. Instead of NO being synthesized in a neuron and released in a calcium-dependent exocytosis from vesicles, NO is an uncharged molecule that diffuses freely across cellular membranes, including the membranes of vesicles.
The ability of NO to diffuse across cell membranes opens the possibility that NO serves as a chemical messenger to convey information regarding the activity of one cell to a number of other cells, which can be pre- or postsynaptic to the neuron from which NO is liberated, or even be non-neuronal cells (in which case it is not synaptic transmission in the original sense of the phrase). Nitric oxide release from neurons occurs in response to glutamate release and subsequent activation of N-methyl-D-aspartate (NMDA) receptors (and voltage-dependent calcium channels) on the postsynaptic cell that allows calcium to enter the neuron. The increase in intracellular calcium in turn activates the enzyme nNOS, resulting in diffusion of NO to reach nearby cells.
Termination of Action of Nitric Oxide
The formation and diffusion of NO mean that it can affect many nearby cells. Although no receptors for NO have been identified, cell surface receptors are not necessary for a molecule that freely diffuses; once inside of nearby neurons and other cells NO can act on second messenger cascades (notably cGMP-mediated events; see Chapter 8).
We previously noted that unrestrained release of a neurotransmitter does not convey to the receptive follower cells information regarding the activity of a presynaptic neuron and is often deleterious. Because NO is a gas, one might envision a molecule of NO careening through neurons like a crazed arrow. There are no degradative enzymes that inactivate NO, nor are there transporters that accumulate NO that is in the extracellular space for a fleeting time. Instead, the inactivation of NO is passive, with NO spontaneously decaying to a nitrate, with a half life of about 30 seconds.
Actions of NO on Neurons
Once in the cytosol of neurons and other cells in the brain, NO can exert function effects by stimulating guanylyl cyclase to produce cGMP levels and initiating a cascade of intracellular effectors. Because NO is produced in response to activation of nNOS by a glutamate-mediated increase in intracellular calcium levels, NO is mainly synthesized in the dendrites of neurons. The diffusion of NO starts from the dendrites and then moves unpredictably to nearby cells, many of which are presynaptic to the released glutamate that started NO’s brief wandering. We have previously discussed a number of mechanisms that inform the presynaptic neuron of its activity, allowing for feedback modulation; these include processes ranging from release-modulating autoreceptors to end-product inhibition of biosynthetic enzymes. Because NO diffuses freely and can influence neurons both pre- and postsynaptic to the cell from which it originates, NO (like endocannabinoids) can provide a “report” to the presynaptic neuron about the physiological effect NO is having on the postsynaptic cell. In this way, “synaptic transmission” becomes bidirectional.
Nitric Oxide as a Neurotransmitter
The list of exceptions posed by NO to the dogma of traditional neurotransmitters is long. NO is not stored in cells, is not released in an exocytotic manner, lacks an active process that terminates its action, does not interact with specific membrane receptors on target cells, and often acts as a retrograde transmitter to regulate axon terminals presynaptic to the neuron in which NO is synthesized. It is not difficult to understand the skepticism that first met the hypothesis that NO is a neurotransmitter, or to have some sympathy for those who expressed the view that NO is not a transmitter but an alien event, benign or otherwise, intent on making neuroscientists question their most cherished beliefs (Fig. 6.5).
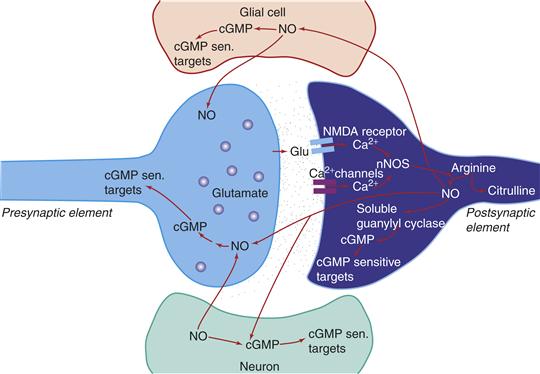
Figure 6.5 Schematic representation of a nitric oxide (NO)-containing neuron. NO is formed from arginine by the actions of different nitric oxide synthases (NOS). NO diffuses freely across cell membranes and can thereby influence both presynaptic neurons (such as the glutamatergic presynaptic neuron in the figure) or other cells that are not apposed directly to the NOS-containing neuron; these other cells can be neurons or glia.
Summary
We have discussed two “unconventional” transmitters, one a gas and the other a lipid. This growing category of transmitters contains many other similarly unconventional molecules, including growth factors, neuroactive steroids, and others still to be uncovered. All offer a significant challenge to our definitions of what constitutes a neurotransmitter, but with the advantage of expanding our knowledge of how the nervous system works in health and disease.
The Expanding Synapse and Gliotransmission
Our discussion of neurotransmission has been focused on the molecular signaling between pre- and postsynaptic neurons necessitated by the presence of a synaptic cleft that separates the two neurons. However, there are cells in the brain other than neurons, including different types of glial cells. The processes of one of these glial cells, the astrocyte, have long been known to be closely positioned near synapses and to be closely apposed to blood vessels. The latter structural arrangement was thought to contribute to, in part, the blood–brain barrier. The astrocyte was also known to play important roles in potassium homeostasis in the brain, as well as in scavenging cellular debris after brain injury.
However, as noted in earlier parts of this chapter, various molecules involved in the life cycle of classical transmitters, particularly glutamate and GABA, are expressed not only by neurons but also by astrocytes. For example, the major glutamate transporter involved in termination of action of glutamate is expressed by astrocytes rather than neurons. A metabotropic glutamate receptor that functions as a glutamate autoreceptor is also expressed by astrocytes, as is the enzyme responsible for the synthesis of kynurenic acid. These and similar observations raised the possibility that astrocytes play an important role in synaptic transmission, and coupled with structural data led to the formulation of the “tripartite synapse” (Araque, Parpura, Sanzgiri, & Haydon, 1999) involving classic pre- and postsynaptic neuronal elements and astrocytes.
The past decade has seen a dramatic rise in our understanding of the multiple roles played by astrocytes (see Allen & Barres, 2009; Halassa & Haydon, 2010; Panatier et al., 2011). Astrocytes repond to neuronal activity by increases in intracellular calcium, which can lead to the release of several transmitters from astrocytes; the precise mechanisms of the release of these gliotransmitters are not completely understood. Among these transmitters are glutamate, D-serine (which binds to a site on the glutamatergic NMDA receptor complex), kynurenic acid, and ATP. Because astrocytic processes of a single astrocyte can envelope thousands of synapses, gliotransmitters can coordinately regulate a very large number of neurons. In addition, the astrocytic “end feet” apposed to capillaries, which were thought to be a structural compartment of the blood–brain barrier, are now thought to be involved in glucose transport from the microvasculature to the brain and thus be critical to the metabolism and activity of neurons. Finally, glia have recently been shown to be critically involved in synaptogenesis and the pruning of synapses seen in the brain during development (Eroglu & Barres, 2010).
These transmitter-like roles played by astrocytes have recently been explored in the context of neuropsychiatric disorders, including substance abuse, schizophrenia, depression, and sleep disorders. Thus, in a very short period of time, glia have been shown to be critical partners with neurons and be involved in the pathophysiology of a large (and growing) number of disorders.
Concluding Remarks
Several of the key proteins involved in different aspects of chemical neurotransmission in mammals were originally identified on the basis of homologies to proteins found in invertebrates, such as the worm C. elegans or the fly Drosophila melanogaster. Some of the same molecules used as neurotransmitters in animals can be found in plants, along with their receptors. Although we tend to think of neurotransmitters, their synthesizing and degradative enzymes, and their transporters as having a single or very restricted roles, as nervous systems have evolved, many transmitter-related proteins have maintained roles that are not directly related to transmitter function or, alternatively are involved in less discrete and more spatially elaborate signaling.
A good example of this is acetylcholine, the synthesis of which depends on the enzyme choline acetyltransferase. The mRNA for ChAT is not only found in the brain, but is also present in the testes, where it is functional and contributes to the appearance of acetylcholine in spermatozoa. ChAT mRNA has also been reported to be present in lymphocytes, as have certain muscarinic cholinergic receptors. Also found in these white blood cells is the mRNA encoding AChE. Decreases in both acetylcholinesterase and butyrylcholinesterase enzyme activity have been reported in lymphocytes in Alzheimer’s disease. Still another example is the presence of AChE in bone marrow and peripheral blood cells in certain leukemias; recent data indicate that inhibition of AChE gene expression in bone marrow suppresses programmed cell death (apoptosis; see Chapter 17). These examples clearly indicate that although a neurotransmitter may have one role in the brain, the same molecule may play a totally different role in other tissues. Such multifunctional roles of transmitters and related proteins should at the very least encourage us to attend to developments in diverse fields.
We have discussed chemically coded synaptic transmission. However, one cannot discuss the biochemistry and pharmacology of synaptic transmission without referring to and appreciating critical information about the structure (anatomy) and function (physiology and behavior) of neurons. Neuroscience is multidisciplinary, requiring an understanding of different aspects of cellular function to come to grips with the basic principles of synaptic communication.
Our concepts of synaptic transmission are in a greater degree of flux than ever before, and require frequent reevaluation and revision. Although some aspects of neurotransmission have been known for about a 100 years, others have become apparent only within the past year. Classical transmitters are but one branch of the family of transmitters, with other relatives including peptides, gases, growth factors, and lipid-derived transmitters such as endocannabinoids. The synapse has historically been viewed as the close apposition of two neurons. It is now clear that astrocytes, once viewed as structural support elements of the brain, play critical roles in transmitter function, ranging from terminating the actions of glutamate to promoting organized multineuronal activation patterns. The use of the terms “conventional” and “unconventional” in discussing transmitters is indicative of our unease with the expanding definition of neurotransmitters. Because our knowledge of neuroscience is constantly changing we must be open and receptive to challenges to long-taught dogma.
References
1. Agnati LF, Guidolin D, Guescini M, Genedani S, Fuxe K. Understanding wiring and volume transmission. Brain Research Reviews. 2010;64:137–159.
2. Alger BE, Kim J. Supply and demand for endocannabinoids. Trends in Neurosciences. 2011;34:304–315.
3. Allen JA, Barres BA. Glia–more than just brain glue. Nature. 2009;457:675–677.
4. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: Glia, the unacknowledged partner. Trends in Neurosciences. 1999;22:208–215.
5. Bean AJ, Roth RH. Dopamine-neurotensin interactions in mesocortical neurons: Evidence from microdialysis studies. Annals of the New York Academy of Sciences. 1992;668:43–53.
6. Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. Journal of Neurochemistry. 2000;75:889–907.
7. Chaudhry FA, Lehre KP, van Lookeren Campagne M, Otterson OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1996;15:711–720.
8. El Mestikawy S, Wallén-Mackenzie A, Fortin GM, Descarries L, Trudeau LE. From glutamate co-release to vesicular synergy: Vesicular glutamate transporters. Nature Reviews Neuroscience. 2011;12:204–216.
9. Erlander MG, Tobin AJ. The structural and functional heterogeneity of glutamic acid decarboxylase: A review. Neurochemical Research. 1991;16:215–226.
10. Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231.
11. Fremeau RT, Voglmaier S, Seal RP, Edwards RH. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. TINS. 2004;27:98–102.
12. Gainetdinov RR, Jones SR, Fumagalli E, Wightman RM, Caron MG. Re-evaluation of the role of the dopamine transporter in dopamine system homeostasis. Brain Research Reviews. 1998;26:148–153.
13. Greenfield SA, Zimmermann M, Bond CE. Non-hydrolytic functions of acetylcholinesterase: The significance of C-terminal peptides. FEBS Journal. 2008;275:604–611.
14. Halassa MM, Haydon PG. Integrated brain circuits: Astrocytic networks modulate neuronal activity and behavior. Annual Review of Physiology. 2010;72:335–355.
15. Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annual Review of Pharmacology and Toxicology. 2008;48:537–568.
16. Hassel B, Dingledine R. Glutamate. In: Siegel GJ, Albers RW, Brady S, Price DL, eds. Basic neurochemistry. 7th ed. San Diego, CA: Elsevier–Academic Press; 2006;267–290.
17. Iversen L, Iversen S, Bloom FE, Roth RH. Introduction to neuropsychopharmacology New York, NY: Oxford University Press; 2008.
18. Jiang J, Amara SG. New views of glutamate transporter structure and function: Advances and challenges. Neuropharmacology. 2011;60:172–181.
19. Kitabgi P, De Nadal F, Rovere C, Bidard JN. Biosynthesis, maturation, release, and degradation of neurotensin and neuromedin N. Annals of the New York Academy of Sciences. 1992;668:30–42.
20. Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291:2419–2423.
21. Mustafa AK, Gadalla MM, Snyder SH. Signaling by gasotransmitters. Science Signaling. 2009;2 re2.
22. Olsen RW, Betz H. GABA and glycine. In: Siegel GJ, Albers RW, Brady S, Price DL, eds. Basic neurochemistry. 7th ed. San Diego, CA: Elsevier–Academic Press; 2006;291–302.
23. Panatier A, Vallée J, Haber M, Murai KK, Lacaille JC, Robitaille R. Astrocytes are endogenous regulators of basal transmission at central synapses. Cell. 2011;146:785–798.
24. Schwarcz R. The kynurenine pathway of tryptophan degradation as a drug target. Current Opinion in Pharmacology. 2004;4:12–17.
25. Shepherd GM. Foundations of the neuron doctrine New York, NY: Oxford University Press; 1991.
26. Sieburth D, Madison JM, Kaplan JM. PKC-1 regulates secretion of neuropeptides. Nature Neuroscience. 2007;10:49–57.
27. Taylor P, Brown JH. Acetylcholine. In: Siegel GJ, Albers RW, Brady S, Price DL, eds. Basic neurochemistry. 7th ed. San Diego: Elsevier-Academic Press; 2006.
28. Valenstein ES. The war of the soups and the sparks New York, NY: Columbia University Press; 2005.
29. Xu E, Gainetdinov RR, Wetsel WC, et al. Mice lacking the norepinephrine transporter are supersensitive to psycho-stimulants. Nature Neuroscience. 2000;3:465–471.
30. Zucchi R, Chiellini G, Scanlan TS, Grandy DK. Trace amine-associated receptors and their ligands. British Journal of Pharmacology. 2006;149:967–978.