Fig. 16-0: An outcrop of of banded iron formation (BIF) in Australia. These great accumulations of iron, the source of most iron ore today, were deposited over a rather brief period of Earth’s history as Earth gradually became more oxidized and converted the soluble reduced iron in seawater to the insoluble oxidized iron, which precipitated to make these spectacular rocks. See also color plate 25. (Courtesy of Simon Poulton and the Nordic Center for Earth Evoluton)
As we saw in the previous chapter, planetary evolution involved the progressive oxidation of surface reservoirs and the eventual rise of O2 in the atmosphere. The ancient reduced Earth surface contained H2, Fe as Fe2+ (FeO), S as sulphide (S–), and an atmosphere with no O2. Today H2 is minimal, Fe at the surface is Fe3+, S is S6+, and the atmosphere has 21% O2. These vast changes occurred by the gradual transformation of planet and life, the exterior renovation of the planet, a story that closely links Earth, life, and the sun. When and how did this happen during Earth’s history?
As we learned in the previous chapter, every molecule of CO2 converted to reduced carbon and stored in Earth creates an equivalent molecule of the highly reactive O2. The oxidizing power released by the stored molecules of life reacted with other planetary materials and gradually oxidized the ocean, soils, and atmosphere, converting Fe and S to their oxidized forms. Oxidized Fe and S contain almost all the O2 produced by burial of organic carbon over Earth’s history.
The rock record provides the history of this process, in large part because the solubility of iron and sulphur depends on their oxidation state. Under reducing conditions, iron is soluble and mobile in water and sulphur is immobile. For the early Earth, this led to an Fe-rich, S-poor ocean. Under oxidizing conditions, iron is insoluble and sulphur soluble. Today this leads to high S and vanishingly small Fe in the modern ocean, where seawater has an S/Fe ratio millions of times higher than the continental crust. The transition between these two states of the planetary surface caused the precipitation of massive Fe deposits known as banded iron formations (BIFs). The demise of BIFs near 2.0 Ga marks the initial rise of O2 in the atmosphere. From 2.0 to 0.8 Ga, O2 was likely a percent or two of present levels. Then in the Neoproterozoic, a second rise in O2 appears to have occurred, leading to the possibility of the Phanerozoic explosion of macroscopic life. Since ~600 Ma, O2 has been within 50% of modern levels. Both the 2.0 Ga and ~0.8 Ga oxidation events mysteriously also occur near times of global glaciations.
While this process is the surface renovation of Earth, the growth of oxygen and its steady-state disequilibrium is intimately connected to the operation of the whole earth system. The source of oxidizing power by burial of organic carbon requires steady addition of CO2 to the surface by volcanism. Addition of reduced materials to the surface from the mantle provides a steady sink for the oxygen. Subduction of carbon and other oxidized and reduced materials has had important long-term effects on the oxygen balance. Today the excess oxygen produced by life is compensated by subduction of oxidized iron produced by alteration of the ocean crust in the deep sea, largely at ocean ridges. While the broad outlines of the rise of O2 are clear, the complete story of the rise of oxygen is like a partially completed jigsaw puzzle where many of the pieces are well defined but do not yet come together into a unified and complete picture.
The development of chemical mechanisms that transitioned from a reduced early Earth with no free oxygen, to an Earth where oxygen was a pollutant, to an Earth where O2 could be a major atmospheric constituent, to a modern Earth where high levels of oxygen are essential for animal life, was the exterior renovation of the planet. It is closely tied to the development of evolutionary mechanisms in living organisms, and also to the geochemical cycles that control the abundances of elements in Earth’s various reservoirs. In the previous chapter we saw how life was the process that oxidized Earth’s surface, changing it from its reduced beginnings to the highly oxygenated environment that has been produced and is maintained by an inhabited planet. In this chapter our task is to try to discover the major events and mechanisms involving the inorganic Earth by which this exterior renovation took place. Were the changes steady or punctuated? When did they occur? What were the mechanisms and how were they recorded on the planetary surface?
To understand the history of O2, we need to delve a bit more deeply into Earth’s oxygen balance, because we can’t measure O2 in past atmospheres directly. Instead, this aspect of Earth’s history is a bit like a play where the protagonist has a major role but is never seen on stage. Fortunately, while O2 is the missing protagonist of this story, there are other starring characters that do make regular appearances.
These characters are the alpha-particle nuclides that occupy multiple oxidation states—C, Fe, and S—as well as the most abundant element in the universe, H. Of course, many other elements, such as manganese (Mn), arsenic (As), and molybdenum (Mo) have variable oxidation states and we will turn to these for additional evidence, but they are not of great importance to the overall oxygen budget (see Fig. 15-1).
Reactions among these elements reflect and control the oxidation state of Earth’s layers. The photosynthetic reaction—Equation (1) of Chapter 15—liberates oxygen. As we all know, organic matter is broken down by bacteria, is eaten by heterotrophs, and burns. These processes are simply the photosynthetic reaction running in reverse (Reaction [16-1]) as free O2 is consumed and recycled to CO2 and H2O. The oxygen that is not recycled in this way is available to react with other reduced species, as illustrated in Reactions (16-2)–(16-4).
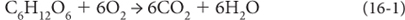
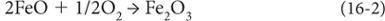
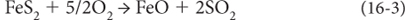
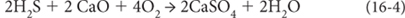
Note that all of these reactions are oxidation/reduction reactions where the valence states of Fe, S, and O are changed. And depending on the oxidation state, different solid materials are formed—reduced carbon is found in organic matter (e.g., coal). Reduced iron is in many silicates, while oxidized iron is found in hematite (Fe2O3) and magnetite (Fe3O4). Reduced sulfur is in pyrite (FeS2) and oxidized sulfur in gypsum (CaSO4). The materials found in rocks record the history of oxygen, even though oxygen itself is not measured.
Earth’s interior is relatively reduced. The core is reduced Fe metal, and the mantle contains about 93% of its Fe as FeO and most of its S as iron sulfide. This reduced oxidation state is inherited by magmas and gases expelled to the surface, so the operation of the plate tectonic geochemical cycle provides a steady diet of reduced species that would react with O2. Reduced species are also provided by exposure of the crystalline rocks of the continents. “Weathering” is the oxidation of these rocks as they are broken down by reactions with O2 and H2O. Without life’s continual re-supply of O2, all free O2 would be consumed, and Earth’s surface would revert to the same reduced state as the interior.
Because of this steady supply of reduced molecules from Earth’s interior, the presence of O2 in the atmosphere is a disequilibrium state, a “steady-state disequilibrium” reflecting the checks and balances of positive and negative feedbacks in the context of a very active geochemical cycle.
Today if O2 were to increase, the oxygen-consuming reactions would take place more quickly. If O2 were to reach something like 27% of the atmosphere instead of 21%, then fires would be rampant, putting Reaction (16-1) into overdrive, consuming organic carbon and reducing the mass of the biosphere and hence limiting O2 production. Or if oxygen were to drop, organic matter would be oxidized more slowly and the deep oceans would become reduced, causing greater burial of organic carbon and less oxidation of the rocks of the seafloor. Both would reduce the sinks for O2, and O2 would rise.
These processes apply to the present-day Earth, where, despite rampant O2 production by the biosphere through photosynthesis, atmospheric O2 stays constant at 21% because sources are exactly balanced by sinks (consumption of O2 by aerobic respiration, sulphide mineral oxidation, oxidation of iron, and oxidation of reduced volcanic gases). Feedbacks operate to preserve this level. Earlier in Earth’s history we know that O2 was nowhere near these levels. In fact this balance could theoretically occur at many other levels of O2 in the atmosphere, all depending on the relative rates of O2 production by photosynthesis and consumption by reactions such as (16-1)–(16-4).
Over the long term, for O2 to rise, the balance has to be perturbed so that O2 production is greater than consumption. Over Earth’s history, Reaction (16-1) has moved from right to left and Reactions (16-2) to (16-4) have on balance moved from left to right. Oxygenic photosynthesis reduced carbon to create organic matter and the highly reactive O2. But it does not matter how much O2 plants make if all of the organic matter is destroyed by oxidation. The O2 available to oxidize the surface environment requires the storage of an atom of organic carbon somewhere in the Earth system. Net O2 production is the excess of organic matter production over destruction, and this organic matter has to end up somewhere, unoxidized. The complementary other half of the story is that the net O2 produced ends up somewhere, either as O2 in the atmosphere or in the oxidized species of Fe and S. Today Fe in surface reservoirs is largely Fe3+ and S is sulfate (SO42–) with a valence of +6. Therefore, it is evident that the rise in atmospheric oxygen is only one aspect of the overall oxidation of Earth’s crust and ocean (Fig. 16-1). Indeed, oxygen could not persist in the atmosphere if the other surface reservoirs were not already oxidized, because in that case the oxygen sinks would be greater than they are currently. Over Earth’s history, progressive oxidation of the surface diminished the available sinks for O2, permitting it to rise in the atmosphere.
It is an imperfect analogy, but we can imagine Earth as if it were a person with a large amount of debt (reduced molecules). Earth is very fortunate because thanks to the sun, money keeps coming in (O2 generated by photosynthesis). As long as all of the income is spent (on reoxidation of organic molecules) the debt cannot be paid off. The saved portion (from sequestered organic matter) is first used to pay off the debt (oxidize the Fe and S), and then once the debt is paid off, the saved portion can go into building capital (raising the O2 of the atmosphere). When sufficient capital is built up, entirely new enterprises (multicellular life and advanced ecosystems) become possible.
So, if we have described the system accurately, we have a simple mass balance—the O2 production by organic matter burial should equal the O2 incorporated in oxidized species. This mass balance also provides us with two avenues to explore the history of oxygen, one through study of organic matter through time (the carbon cycle), which is the story of oxygen generation, and the other through study of the oxidized products (largely the Fe and S cycles), which is the story of oxygen consumption.
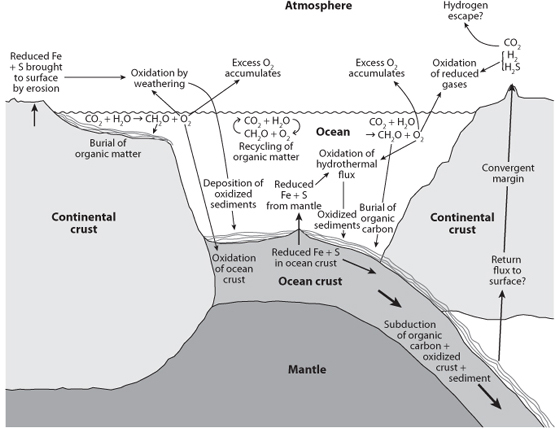
Fig. 16-1: Diagrammatic illustration of various components of the oxygen cycle. Oxygen is produced by photosynthesis. Most of the organic matter that is produced is then re oxidized. Burial of organic matter leads to excess oxygen that is consumed by various reactions. Reduced Fe and S coming from hydrothermal vents and exposure of the continental crust are oxidized, as are the reduced gases coming out of volcanoes. Subduction recycles various molecules to Earth’s interior. The oxidation state of sediments contains the record of these reactions through Earth history.
Reduced carbon resides in black shales, soils, coal, oil, and natural gas, resulting from billions of years of photosynthesis. This storage is what permits the high oxidation state of the planetary surface and the existence of animal life that depends on it. If we were to somehow recover and burn all this organic matter, we would reverse 3 billion years of planetary evolution and return Earth to its reduced state where only primitive life is possible. The current distribution of organic and inorganic carbon in Earth reservoirs is shown in Table 16-1. Notice that the biosphere, living or recently living organic matter, is only a very small portion of total organic carbon, that total organic carbon is about 17% of total crustal carbon, and that the mantle is by far the largest carbon reservoir.
Table 16-1
Reservoirs of carbon on Earth (in units of 1018 moles)
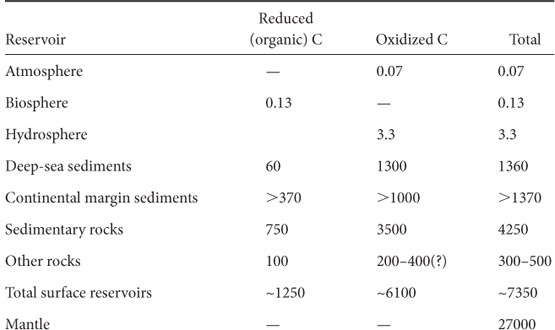
Reduced carbon estimates are from Des Marais, Rev. Mineral. Geochem. 43 (2005):555–78.
Tracking sources of O2 through time directly would require knowledge of how the mass of organic carbon has changed through time. This is an intractable problem because organic carbon resides in sediments, and these are rapidly recycled through Earth’s history. So scientists have had to turn to an indirect method, where the rate of formation of organic carbon is inferred from measurements of carbon isotopes in carbonates formed from seawater and in ancient organic matter.
The reasoning behind this approach makes use of the contrast in carbon isotope ratios between organic carbon and carbonate carbon. The starting point is that we can measure the average carbon isotope ratio of surface carbon reservoirs, and it turns out that this value is the same as the input to the system, which is carbon coming from the mantle. The mantle-derived carbon then becomes distributed by geological and biological processes to (inorganic) carbonate and organic carbon. As we learned in Chapter 13, biological processes that make the organic carbon preferentially incorporate 12C, the lighter of the two carbon isotopes, and the 13C/12C ratio of organic carbon is about 2.5% lower than the ratio in inorganic carbon. The variations are reported as per mil differences relative to an arbitrary standard (per mil is parts per thousand, as compared to percent, which is parts per hundred). These differences are conventionally assigned the symbol δ13C (“del C 13”). For example, a value of δ13C of –25 means the 13C/12C ratio is 25 per mil lower than the standard value.
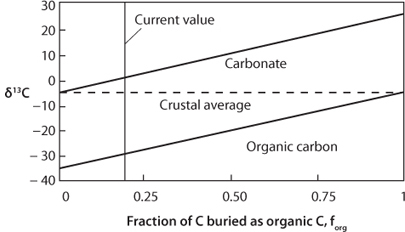
Mantle carbon has δ13C of ~–5, which must always be the value of the total carbon in the system. At the same time, there will also always be the offset between organic and inorganic carbon, which is about 30 per mil. If all the carbon were in the form of inorganic carbon, then the inorganic carbon would have to have the mean value of –5. The first organic molecule to form would be offset 30 per mil lighter to –35. Or if all the carbon were only organic carbon, then the organic carbon value would be –5 and the first inorganic carbon molecule would be offset to +25. The average is always –5 and the offset is always 30, but the carbon isotope values of organic and inorganic carbon depend on the relative amounts of each (Fig. 16-2). Mathematically, the mass balance can be written as:
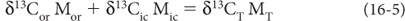
where MT is the total mass of carbon, Mor is the mass of organic carbon and Mic is the mass of inorganic carbong. Then MT = Mor + Mic. It is then possible to define a parameter, conventionally called f, which is the fraction of organic carbon to total carbon f = (Mor/MT). Substituting terms, we obtain:
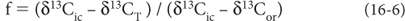
By measuring the δ13C values of organic and inorganic carbon in rocks of different ages, f can be estimated, permitting estimates of the proportion of organic to inorganic carbon through time (Fig. 16-2).
Fig. 16-2: Relationship between isotopic composition of carbonate carbon (δ13Cic) and organic carbon (δ13Corg) as a function of the fraction of carbon buried as organic matter. The offset between the two is always 30 per mil. The total carbon must always average –5 per mil. The vertical line represents the current value for the global carbon cycle, and this value has remained remarkably constant over Earth’s history. (Modified from Des Marais, Reviews in Minerology and Geochemistry v. 43, 555–78 (2001))
The principle behind a carbon isotope record is straightforward—to measure carbonate and organic matter of various ages that reflect seawater composition. In practice, there are many difficulties. Samples distributed over the various periods of Earth’s history with preserved carbonates and preserved organic matter are necessary. While carbonates are fairly abundant in the geological record and sometimes well preserved, organic matter is rarer and more easily modified by metamorphic processes. There are also the difficulties that the two types of material are often in different rocks from different locations, so the record requires correlation of ages across different continents. These aspects make it difficult to obtain a reliable record and to know whether measurements at particular time intervals in the Precambrian are global in extent or reflect local conditions. With such questions about the data, interpretations have “wiggle room.”
Despite the difficulties in detail, the long-term history of carbon isotopes tells a compelling story. The data for both organic and inorganic carbon are shown in Figure 16-3. The horizontal line provides a reference for the mantle value of δ13C. If no organic carbon were produced, we would expect carbonate carbon to have the same value as the mantle. As is apparent from the figure, throughout Earth’s history carbonate carbon has had higher δ13C than the mantle, and the offset on average varies within a small range. Since mantle carbon is –5, and carbonate carbon is about 0, this implies from Figure 16-2 that the carbonate carbon has made up about 80% of the active carbon reservoir, and inorganic carbon about 20%. The δ13C of organic carbon should then be –30. This value inferred from carbon isotopes is essentially the same as total carbon balance seen in Table 16-1.
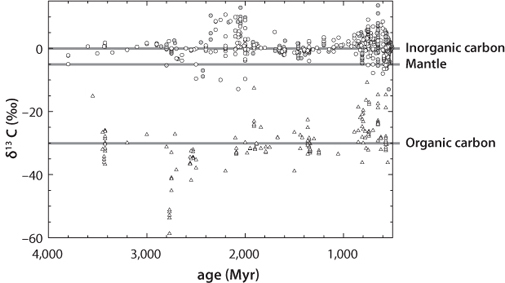
Fig. 16-3: Data for carbon isotopes from sediments over geological time. Open circles are carbonate (oxidized) carbon. Triangles are organic (reduced) carbon. Note the two periods of large variation in the carbon isotope composition of inorganic carbon near 2200 Ma and 800 to 600 Ma. (Adapted from Hayes and Waldbauer, Phil. Trans. R. Soc. B. 361 (2006):931–50)
The contrast between carbonate carbon and organic carbon is not a simple constant, however, and while modern organic carbon varies around –30 per mil, the sparse measurements of ancient organic carbon are lower. Simplifying and smoothing out the data by averaging leads to the variations in fraction of organic carbon through time shown in Figure 16-4. The scatter in the data preclude a definitive conclusion, but the data suggest a possible increase in proportion of organic matter from about 15% in the Archean to 20–25% today.
The carbonate isotope data also show two periods of increased variability, including excursions to much higher values of carbonate δ13C. These occur between 2.4 and 2.0 Ga and 0.8 to 0.55 Ga. Taken at face value, these would suggest higher proportions of organic matter, and greater production of oxidizing power, during these two periods. If this were true in a simple way, however, we would expect to see in Figure 16-3 a smooth and globally uniform increase in carbonate δ13C values over these time periods and a corresponding increase in the δ13C of organic matter during the same time periods, and neither of these is evident. The “noise” and lack of correspondence of the organic and inorganic carbon records is problematic. Furthermore, these two time periods also show some negative δ13C excursions. Many workers have interpreted these data to indicate when oxygen rose in the atmosphere, with a first-step increase in the 2.4–2.0 period and a second in the Neoproterozoic just prior to the development of multicellular organisms. What is clear is that these were times of rapid change and great variability, and that in contrast, the period from 2.0 to 1.0 Ga appears on present data to have been a remarkably long and stable period, sometimes referred to as the “boring billion.”
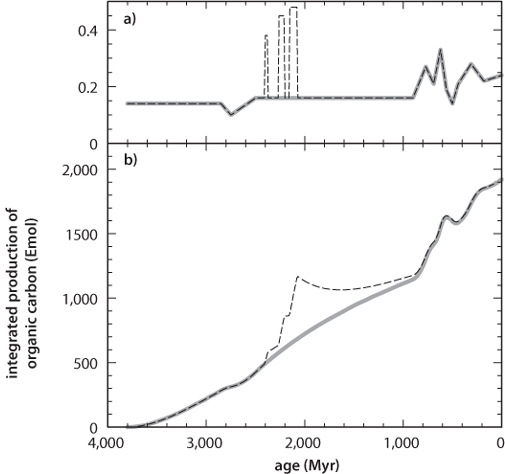
Fig. 16-4: Possible interpretation of smoothed data from figure 16-3. If CO2 is steadily outgassed from Earth’s interior, then the total amount of carbon at the surface increases with time. The constant fraction of organic carbon (top panel) then means that total buried organic carbon also increases with time (lower panel). There may have been short time periods of increased fraction of organic carbon burial (indicated by the dashed lines). Since O2 production has a one-to-one relationship with organic carbon burial, the lower panel could also be viewed as the increase in oxidized molecules at the surface (largely oxidized Fe and S). (Modified after Hayes and Waldbauer (2006))
What the geological record does show is an increasingly oxygenated surface over Earth’s history. This requires steady oxygen production in excess of the sinks. If oxygen were not steadily in excess, the progressive oxidation of crust, ocean, and atmosphere could not occur. Therefore, a constant organic fraction of carbon, and the fact that the integrated organic fraction in all rocks surviving today is similar to the long-term average, requires an increase in the total mass of carbon in external reservoirs, unless there is a phantom source of oxidizing power with no surviving rock record, a possibility we will come back to at the end of the chapter. If CO2 were steadily outgassed from Earth’s interior, thereby increasing over time at Earth’s surface, then a steady proportion of 20% organic matter removal would be a continuing flux to the surface, leading to a gradual increase in oxidizing power. It is not necessary to have periods of greater burial of organic matter to raise oxygen—oxygen is being produced all the time. A necessary consequence of this way of viewing the problem is that the total size of the organic matter reservoir would increase with time (as would the size of the carbonate reservoir). Determining the total carbon budget through Earth’s history is a difficult problem, however, because the rates of recycling and removal through subduction are poorly known, even for modern periods.
The data in Figure 16-3 also show a great deal of scatter and no clear correlation between variations in organic and inorganic carbon. The concept is clear. The data are noisy. Therefore the carbon isotopes provide important constraints and much food for thought about the rise of O2, but do not provide definitive answers.
The most important elements that track oxygen consumption are oxidized Fe and S. The rocks and seawater of Earth’s surface today contain abundant oxidized Fe (Fe3+) and S (S6+), in contrast to the reduced state of the mantle that delivers these elements to the surface through volcanism. The amount of oxygen tied up in these oxidized molecules vastly exceeds the amount of oxygen in the atmosphere (see Table 16-2). Therefore, almost all of the oxygen that has been produced in the formation of organic matter does not exist as atmospheric O2. It is largely stored in the oxidized molecules of Fe and S.
Table 16-2
Where the oxygen produced by organic matter resides (in units of 1018 moles of O2 equivalents)
O2 in atmosphere |
37.2 |
Fe3+ |
1375 |
SO42– |
410 |
Total O2 in oxidized compounds |
1847 |
Total O2 production by organic carbon from table 16.1 |
700–1280 |
Mass balance problem |
500–1000 *1018 moles of reduced carbon equivalent |
Modified after Hayes and Waldbauer, Phil. Trans. Roy. Soc. B 361 (2006):931–50.
There is a special characteristic of Fe and S that makes them particularly useful for tracking the history of oxygen and that also had fundamental impact on Earth’s history, and that is how their solubility varies as a function of their oxidation state.
Ferric iron is highly insoluble in water—this leads today to an oceanic concentration of Fe that is generally less than 1 part per billion, even though iron in the continental crust is about 5% (50,000,000 ppb). Ferrous iron, on the other hand, is quite soluble in water. The reduced fluids of hydrothermal vents, for example, can contain 100 ppm of Fe2+. When the fluid exits the vent and encounters oxidized seawater, the Fe2+ is converted to Fe3+ and precipitates to contribute to the “black smoke” of the deep sea hydrothermal vents (recall Fig. 11-4).
Sulfur has the opposite behavior. Reduced sulfur (S–1) is relatively insoluble, while oxidized sulfur (S6+) forms sulfate that can have very high concentrations in water. Sulfate in seawater today, for example, leads to concentrations of about 900 ppm sulfur, even higher than the continental crustal composition of 400 ppm. Sulfide concentrations in a reduced ocean would have been only a few ppm.
This opposite behavior of Fe and S is particularly useful when we examine the geological record. A reduced Earth has mobile Fe and immobile S, leading to reduced soils with low Fe and high S and oceans with very high Fe/S. As the surface becomes oxidized, Fe becomes immobile in sediments and insoluble in seawater, while S becomes mobile from weathering and soluble in seawater. This leads to seawater composition with very low Fe/S. The differences are not small. The S/Fe ratio in seawater today is 40 billion times greater than the ratio in the continental crust, whose weathering is contributing to the ocean. In the early Earth, Fe may have been more abundant than sulfur in the reduced ocean. Oxidation state can be a powerful element separator!
These changes in oxidation state influence the distribution of Fe and S in rivers and oceans and the mineralogy of soils and sediments. Reduced iron minerals are Fe silicates, pyrite, and the Fe carbonate, siderite (FeCO3). Reduced sulfur minerals are sulfides. Oxidized iron minerals are red and include silicates, oxides, and hydroxides. Oxidized sulfur minerals are sulfates, the most common of which is gypsum, CaSO4(H2O)2. The scientific detective work is then to identify where, when, and why these minerals occur in the sediments of the geological record, and how the change in transport and deposition could be preserved in the rock record. By examining the history of Fe and S recorded in the rocks formed at the surface, we can derive constraints on the history of O2.
The characteristics of many Fe- and S-bearing sediments have varied considerably over Earth’s history. Two distinctive rock types that have very particular time distributions are banded iron formations and red beds. While both these formations are red in color, their mineralogy and composition differ strikingly, and they reveal very different stages of the evolution of oxygen at the surface.
Banded iron formations (BIFs) are distinctive, colorful rocks that derive their name from the fine laminations of the rocks, where layers of very Fe-rich sediments alternate with layers of cherts, which are almost pure SiO2 (Fig. 16-5). They formed beneath shallow water and may have covered large portions of the continental shelves at the time of their maximum occurrence, at about 2,500 Ma. These unique rocks can contain up to 50% iron oxides, and are the chief source of iron ore for modern civilization.

Fig. 16-5: Sample from a banded iron formation (BIF) showing the prominent banding of alternating Fe-rich layers with chert layers. Such rocks are the dominant form of iron ore for modern civilization. See also color plate 26. (Courtesy of Harvard Museum of Natural History, Dick Holland Collection)
The detailed characteristics of the BIFs change with time. The formations that occur in the oldest rocks such as the 3.8 billion-year-old Isua sediments in Greenland are generally thin (tens of meters thick) and occur interlayered with volcanic rocks. When BIFs reach their peak some billion years later near 2.5 Ga, they can be a thousand meters thick and extend over hundreds of square km. They decline in abundance in younger rocks, except for a brief reappearance at 1.8 Ga, after which they disappear from the geological record (Fig. 16-6), with one minor exception in the Neoproterozoic during the proposed “snowball Earth” episodes (see Chapter 12). The Neoproterozoic BIFs are chemically distinguished by having higher ratios of ferric to ferrous iron, with 95% of their total iron as Fe3+, in contrast to the ~50% Fe3+ in the older BIFs.
Notable aspects of BIFs are their restriction to the Archean and early Proterozoic, and the vast concentrations of multivalent Fe that they represent. The challenges posed by these rocks are to understand how so much Fe could be concentrated in sediments in this period of Earth’s history, and never again, and what information they convey about the oxygen history of the surface.
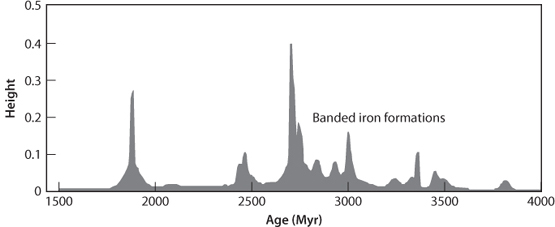
Fig. 16-6: Relative abundance of banded iron formations through time. Note the pronounced peak between 3000 and 2500 Ma, then there is a decline near 2400 Ma, and absence in rocks younger than 1800 Ma. (From Isley and Abbot, J. Geophys. Res. 104 (1999):15,461–77)
The influence of oxidation state on solubility must be central to BIF formation. Because the BIFs are rocks that precipitate from seawater, central to any model are two conditions: a global source of reduced Fe, which leads to high seawater concentrations, and a local oxidizing environment, which causes the Fe to precipitate on the continental shelf. Within this overall framework, different hypotheses are possible.
One scenario, pioneered by Preston Cloud in the 1970s, related BIFs to the rise of oxygenic photosynthesis. Prior to the invention of oxygenic photosynthesis, the atmosphere and the entire ocean would have been reduced. Rivers would have carried reduced iron from the continents to the oceans, and reduced iron coming out of hydrothermal vents would have remained dissolved in seawater. The ocean would have been relatively rich in Fe2+ and contain very little S. The early fully reduced ocean would not have had the oxidizing power to convert Fe2+ + to Fe3+, and Fe3+-bearing sediments would not form in abundance. After cyanobacteria appear and produce O2 in the shallow, sun-filled top of the ocean, the shallow ocean would have oxygen but the deep ocean would be reduced. Very little oxygen would exist in the atmosphere, because any oxygen that was present there would be reduced by reaction with volcanic gases and chemical interactions with the Fe- and S-bearing rocks of the continents.
At this stage, then, there could have been an oxygen source in the shallow ocean, particularly near continents where there would be sources of other nutrients, a reduced deep ocean, and a reduced atmosphere. When Fe-rich deep water reached shallow levels where oxygen was present, it would be susceptible to oxidation. Furthermore, the oxidation of Fe can produce energy, and bacteria likely evolved to take advantage of this reaction. Oxidation of Fe might even have made an advantageous environment for early ecosystems because it would remove the poisonous oxygen from the environment. The oxidized iron that formed would be highly insoluble and precipitate. If that occurred above the deep ocean, the Fe particles would sink to great depth, where they would be reduced and go back into solution. In shallow seas and continental shelves where the ocean depth was shallower than the depth of anoxia, the Fe could form very Fe-rich sediments. The change in oxidation state therefore would allow a kind of Fe pump to operate, where Fe is delivered to the ocean from deep hydrothermal vents and continental weathering and deposited beneath shallow, partially oxidized seas (Fig. 16-7). This chemical machine could operate only in a partially oxygenated Earth, where reduced settings mobilized Fe2+ and locally oxidized settings caused its precipitation as Fe3+. Then BIFs would be rare and thin in the oldest Archean rocks, prior to an initial rise of oxygen, and abundant in a transitional period with small amounts of oxygen present. Today Fe-rich precipitates are rare. They form in small quantities near the ocean ridges, where hydrothermal vents continue to pump Fe2+ into the local environment and particles form immediately and settle to the seafloor. They cannot form in abundance today because atmosphere and the entire ocean are oxidized, the Fe content of seawater is vanishingly small, and oxidized Fe of the continental crust is not transportable. In this scenario, BIFs occur after the development of oxygenic photosynthesis and reflect a transitional stage of oxidation of Earth’s surface reservoirs.
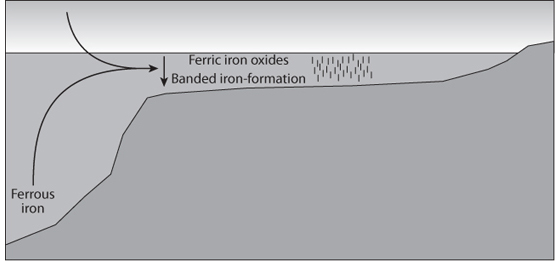
Fig. 16-7: Illustration of how banded iron formations (BIFs) might have formed. Soluble Fe2+ is released from hydrothermal vents and from continental weathering and remains in dissolved form in the reduced ocean. Continental shelves are sites of photosynthesis that convert Fe2+ to Fe3+, leading to its precipitation. As long as the oceans at depth are largely reduced, then Fe can be pumped continually from the mantle to allow formation of BIFs on the continental shelves.
While this scenario has great appeal, it does not account easily for several aspects of BIFs. The first is the alternating layers of chert and Fe minerals. A successful model needs to account for both the Si and the Fe deposition. The second is that while BIFs do contain oxidized iron, they also contain a large amount of reduced Fe. Where does the reduced iron come from? And finally, we do not know for certain from independent evidence when oxygenic photosynthesis became important.
An alternative model, recently elaborated by Woody Fischer and Andy Knoll, forms BIFs by anoxygenic photosynthesis. Without oxygenic photosynthesis there must be another source of electrons to produce organic matter. A source that does not involve oxygen and could lead to deposition of Fe3+ could be:
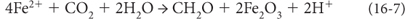
In this scenario, Fe-oxidizing photosynthetic bacteria, which are known to exist today, would take advantage of the abundant Fe2+ in seawater to create Fe3+ minerals by photosynthesis, using the energy from the sun. The oxidized iron creates very reactive particles that scavenge Si from seawater. Reaction of the oxidized Fe and the reduced carbon in the organic matter in the sediment would create reduced Fe and oxidized carbon, leading to the precipitation of the reduced iron mineral siderite (FeCO3). This mechanism carries both Si and Fe to the sediment, permits the reduction of some of the Fe, and leads to the formation of the siderite found in BIFs.
Whatever the detailed mechanism, the large mass of BIFs requires a high Fe content of seawater in the shallow ocean where sunlight is available, hence a much more reduced Earth than exists presently. And the BIFs also represent a major oxidation event of Earth’s surface, since a great deal of Fe3+ was generated from Fe2+. Every four moles of oxidized iron would reflect one mole of stored organic matter produced from CO2.
When we turn to land, another piece of evidence from Fe emerges from the rare preserved ancient soils, called paleosols. Dick Holland studied such soils through geological time and showed that prior to 2.2 billion years soils were deficient in Fe, formed reduced Fe minerals, and did not contain oxidized iron minerals. Soils became more oxygenated in younger rocks. He used this and other evidence to propose a “Great Oxidation Event” around this time period.
The Fe evidence from these ancient sediments thus indicates a world with little free O2. Even if oxygenic photosynthesis were underway, there were huge sinks for O2 in the reduced Fe and S species at Earth’s surface. The vast quantities of reduced species would require large amounts of formation and burial of organic matter before free oxygen was able to exist. These conditions were sufficient to keep atmospheric O2 at such low levels that continental weathering took place under a reduced atmosphere, mobilizing Fe and immobilizing S.
Much more precision on the timing of the early rise of oxygen came from the tools of sulfur isotope geochemistry. We have made use several times already of stable isotope variations to reveal earth processes, but all of the variations that we have discussed thus far have been mass dependent fractionations that make use of slight differences in chemical behavior of two isotopes of the same element with different masses. Because this behavior is mass dependent, when there is double the difference in mass, the change in behavior is twice as great. For example, if some process changes the 18O/16O ratio by 2%, then the 17O/16O ratio changes by about 1%, because it has half the mass difference.
There are also processes that can cause mass independent fractionation of stable isotopes (MIF), and these can be studied in elements that have more than two isotopes. Apart from the nucleosynthetic variations produced in stars, mass independent fractionations are much less common and smaller in magnitude than mass dependent fractionations. One of the ways they can be produced is by photochemical reactions that are activated by light from the sun. For example, mass independent fractionation of oxygen occurs in the stratosphere today during formation of ozone.
Sulfur has isotopes of mass 32, 33, 34, and 36 and therefore is subject to both mass dependent and mass independent fractionation. Indeed, MIF of sulfur isotopes (SMIF) is observed during experiments on photochemical reactions and takes place to a very minor extent in today’s upper atmosphere. All of Earth’s major surface reservoirs today, however, show less than 0.02% mass independent fractionation. This turns out not to be the case in the ancient past. Rocks older than 2.0 Ga show larger MIF variations in sulfur, and rocks older than 2.45 Ga show variations of up to 0.4%, twenty times greater than anything seen in the last 2 billion years (Fig. 16-8).
There are two requirements for such large MIF to be preserved in ancient rocks. First, there must have been atmospheric processes that created large variations. And second, the sulfur cycle has to be in a state where multiple sulfur species with diverse isotopic composition can coexist in the atmosphere and the variations are not destroyed by mixing in the ocean. Today, for example, the ocean is a huge reservoir of sulfate, with a sulfate concentration of ~0.3% and a residence time of 9 million years. This contrasts with the ocean mixing time of thousands of years, so there is a huge mass of well-mixed sulfur in the oceans, and it takes a very large mass of distinctive sulfur to modify its isotopic composition.
Laboratory experiments and the details of SMIF suggest it is created by SO2 and SO photolysis driven by deep ultraviolet radiation. Ozone is the present atmospheric constituent that absorbs ultraviolet radiation, and prevents both the photolysis and the coexistence of multiple sulfur species that would preserve the isotope variations. Therefore, there is no large SMIF today. The large SMIF in the Archean appears to require very low atmospheric ozone, consistent with low O2 levels prior to 2 Ga. More detailed modelling has led to the conclusion that SMIF can be preserved only for oxygen levels 100,000 times lower than the present atmosphere. Furthermore, the experimental results suggest that the atmospheric effects lead to SMIF variations of 6.5%, so the preservation of variations of .1–.2% requires 1–2% of sedimentary sulfur derived from the atmosphere, which would require a smaller oceanic sulfur reservoir.
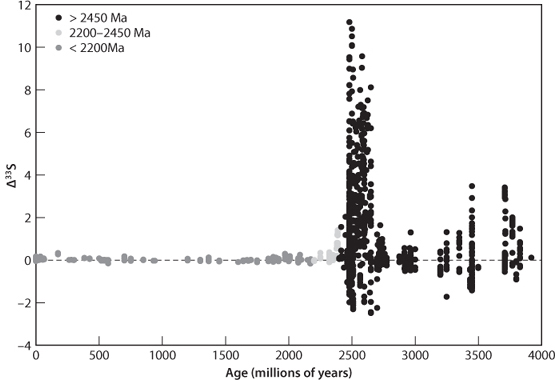
Fig. 16-8: Mass independent fractionation of sulfur (SMIF) through time. Prior to 2.4 Ga, SMIF was present on Earth, requiring anoxic conditions. The decline at 2.4 Ga, and absence of any SMIF in samples younger than 2.0 Ga, requires a rise in O2 in the atmosphere at that time. (Courtesy of David Johnston, Harvard University)
How much O2 is needed to turn off the SMIF signal? The experimental data suggest that levels of ~10–2 the present atmospheric level of O2 (PAL) are sufficient to prevent the necessary UV radiation to create the SMIF variations in the atmosphere. The simplest interpretation of the SMIF data, therefore, is that oxygen levels were <10–5 PAL prior to 2.45 Ga, gradually increased from 2.45 Ga to 2.0 Ga, and were at or above 1% PAL starting at 2.0 Ga.
Note that BIFs, SMIF, and carbon isotopes all indicate a major change occurring at about the same time near 2 billion years ago, suggesting a fundamental change at that time in the oxygen cycle. Might this change have had something to do with life itself?
A biogenic factor limiting oxygen may have been the intolerance of ancient organisms for high oxygen levels. Since oxygen breaks down organic molecules, it would have been a poison for early photosynthesizers, and too much oxygen is even a kind of poison for us today, hence, as said earlier, the popularity of “antioxidants.” Anaerobic bacteria today, the likely descendants of earliest life, are killed by oxygen. The development of metabolic protection against high oxygen would be necessary for oxygenic photosynthesis to flourish. Otherwise there would be a negative feedback where high O2 killed off O2 production. We do not know when this evolutionary adaptation occurred, but it is conceivable that it relates to the geological rise of oxygen.
At some point bacteria did develop protection against oxygen, which would permit thriving oxygenic ecosystems. With photosynthesis providing a steady stream of oxygen to the atmosphere, we can imagine that eventually the oxygen overcame the reduced species available to consume it. At this point the atmosphere would have significant oxygen, though far less than modern values, leading to oxidizing conditions during weathering of the continents. This would impede the continental supply of Fe2+ by weathering and cut off one source of Fe for BIF formation. It would also lead to enhanced weathering of sulfur and a likely rise in seawater sulfate. If the oceans remained reduced at depth, the sulfate at depth would reduce to sulfide, causing precipitation of pyrite and removal of Fe from the deep sea. No longer able to be transported and unable to persist in the ocean, Fe would have dropped to low concentrations in seawater, cutting off the lifeblood of BIF formation.
The disappearance of BIFs and SMIF near 2 billion years ago appears to require only a slight rise in oxygen level, to less than 1% of the present atmospheric concentration. Of course there are important remaining details, such as the fact that SMIF declines abruptly at 2.4 billion years and stops at 2.0 billion years, and BIFs show a renewed pulse at 1.8 billion. Given the diversity of geological environments and conditions, however, the overall coherence of the two signals is quite conclusive. This change also occurs at the same time as the change in the mineralogy of soils and the great variability of carbonate δ13C. This time period clearly involved a major exterior modification of the surface from an anoxic to partially oxygenated world.
The other distinctive red rocks in the geological record are the red beds, sandstones such as the rocks that can be seen in the glorious landscapes of the American Southwest. These rocks are mostly quartz grains with a coating of fully oxidized Fe in the form of hematite. Virtually all of the iron in these rocks is Fe3+ and the total iron content is low, usually less than 2%. The red color does not reflect high iron content but an oxic coating on mineral surfaces. Like BIFs, these rocks begin to appear only in rocks younger than 2.0 Ga, suggesting a more oxic environment commencing at that time (Fig. 16-9). The consensus is that red beds reflect a fully oxidized surface environment that can convert all Fe2+ to Fe3+.
This existence of an oxic Phanerozoic atmosphere is supported by other lines of evidence. We know that modern multicellular life requires high oxygen levels, and the advent of preserved fossils suggests quite high O2 during the Cambrian and Ordovician. Large animals later in the fossil record, along with charcoal showing that organic matter burned, require high oxygen levels. Levels even higher than today during some time intervals are suggested by the existence of very large insects (e.g., dragonflies with the wingspan of eagles) and geochemical modeling. High model values are inferred from the large deposition of organic carbon (e.g., vast coal deposits) during the aptly named Carboniferous period ~300 Ma, when carbon isotopes also showed an increase supporting a higher proportion of organic carbon deposition.
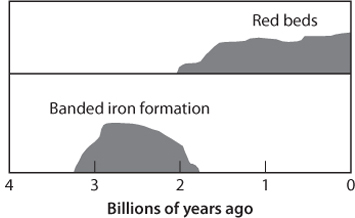
Fig. 16-9: Schematic illustration of the decline in BIFs and beginning of red beds in the geological record. Red beds are thought to occur only in an at least partially oxidized surface environment.
High Phanerozoic O2 values can also be inferred from the compositions of evaporites. Evaporites form when water in closed basins evaporates, leaving its dissolved solids as precipitated sediments. We can see evaporites forming today, for example, around the Caspian Sea and the Dead Sea in the Middle East. Huge evaporite deposits formed in the Mediterranean when it dried up during the Miocene period ~5 Ma. Modern seawater composition has so much sulfate in it that modern evaporites precipitate the sulfate mineral gypsum even before halite. Phanerozoic evaporates also have an abundance of gypsum, suggesting similar oxidation conditions and a high sulfate concentration in seawater. It thus appears that Earth’s surface layers have had similar O2 levels to the present for the last several hundred million years. An Earth inhabited by plants and animals appears to have been possible only with the presence of an oxygenated atmosphere.
The rock record has allowed us to work forward from the Archean to see an initial rise of oxygen near 2.0 Ga, and to work backward from the present to suggest high O2 for the last ~600 Ma. In between is the 2.0 Ga to 0.6 Ga interval of the Proterozoic, during which oxygen levels likely rose from perhaps 1% at 2.0 Ga to the 10–20% levels required for animal life at the dawn of the Phanerozoic. Did this change occur abruptly following a prolonged state at low levels, or was it a slow and gradual increase, or perhaps a series of step changes? If we look again at the carbon isotope record of Figure 16-3, it is apparent that much of this time interval appears remarkably stable, and then at the end, in the Neoproterozoic, the carbon isotopes fluctuated wildly with large positive and negative excursions, particularly during the proposed “snowball Earth” episodes (Fig. 16-10). The long stability followed by great instability and a change from unicellular to multicellular biota that occurs at the same time has naturally led to the suggestion that the next big change in oxygen content of surface reservoirs took place during the Neoproterozoic.
There is emerging evidence from the chemical compositions of black shales that supports this possibility. The evidence comes from an unlikely source, the trace element molybdenum (Mo), which is another element with multiple oxidation states and changes in solubility with oxidation. Despite its low abundance in the total Earth, Mo concentrations in seawater today are higher than any other transition metal! This strange result occurs because oxidized Mo is easily weathered from continents and relatively soluble in oxidized seawater. Under more reducing conditions when the ocean would have had significant sulphide, Mo would have been efficiently removed from seawater. The record of Mo concentrations in black shales, while sparse, suggests a marked change in Mo concentration in seawater in the Neoproterozoic between 800 and 500 Ma. This result would be consistent with a fully oxygenated ocean appearing at this time, preventing Mo removal (Fig. 16-11). A popular current point of view, therefore, is an oxidized shallow ocean and reduced deep ocean during much of the Proterozoic, and then oxidation of the deep ocean associated with a second rise of oxygen in the Neoproterozoic. The increasingly oxygenated environment then permitted the development of multicellular organisms and ultimately the Cambrian explosion of life.
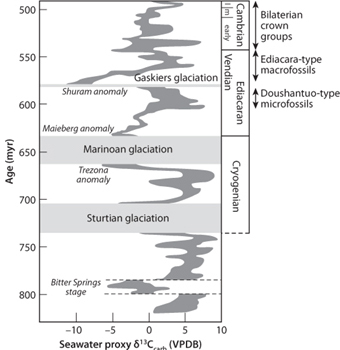
Fig. 16-10: The wildly varying carbon isotopes during the end of the Neoproterozoic. The first multicellular life (without hard body parts) occurred in the Ediacaran, just after the two major “snowball Earth” episodes. Since a snowball episode also occurred before the initial rise of oxygen near 2.3 Ga, and multicellular life likely requires high O2, it is possible that the Proterozoic snowball episodes were also associated with a second rise in oxygen. (Modified from Halverson et al., GSA Bulletin 117 (2005), no. 9–10: 1181–207)
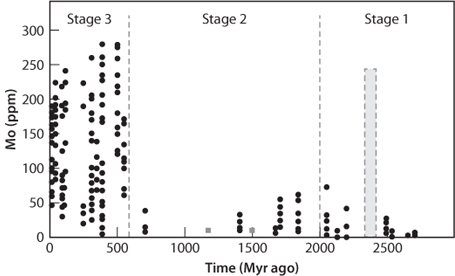
Fig, 16-11: Change of molybdenum concentrations in sediments through time. A reduced oxidation state makes Mo insoluble, hence low in concentration in seawater and oceanic sediments. A high oxidation state of Mo leads to solubility in seawater, making it one of the most abundant elements in seawater today despite its low abundance in surface rocks. The large change in its behavior near 600 Ma is consistent with a second rise in surface O2 at that time. The shaded area shows the interval where sulfur isotopes indicate O2 levels first rose (see Fig. 16-8). (Modified after Scott et al., Nature 452:456–59)
These observations do not explain why oxygen rose to close to modern levels at this time. Various models have been proposed relating to increasing sources and decreasing sinks. Sources of oxidizing power are caused by removal of electrons from the system, normally brought about by organic carbon burial. What could precipitate excess burial of organic carbon? Once oxygenic photosynthesis became dominant and there was an essentially infinite source of electrons and hydrogen from breaking down H2O, the limiting factor on organic matter production became supply of nutrients to the ocean. This is the case today, and very productive areas of the ocean are all areas where there is a supply of nutrients. Some tectonic factor that would increase nutrient supply could then lead to more organic matter production. One possibility is the breakup of large continents. Smaller land masses weather more efficiently because there is a greater proportion of coastline to land area. Another possibility is continental collision, since mountains weather more than flat plains. Periodic plate tectonic events could thus lead to pulses of organic carbon burial and a series of stepwise increases in O2. Another impact on carbon burial could be sea level and the extent of inland seas and extended continental shelves, where there could be high productivity and efficient burial. All these pulses of organic carbon burial, however, should reveal themselves in the carbon isotope record, and there such signals are not clear.
There are two other ways to remove electrons from the system and cause the surface to become more oxygenated. One is the subduction of organic carbon. If the 1:5 organic/inorganic carbon ratio of subducted sediments is the same as sediments that remain at the surface, then a large amount of organic carbon could find its way to the mantle. Variations in the budget of subducted carbon would then have consequences for the oxygen budget. Variations in the ratio of subducted sulfate to sulfide might also have an effect.
Alternative models would diminish the sinks for O2. One possibility is a locally more oxidized mantle through time at convergent margins, owing to subduction of oxidized iron. Then gases coming from volcanoes would be less reducing, diminishing the atmospheric sink for oxygen. If organic matter production were kept constant, O2 would rise. Alternatively, metamorphic gases, which depend on the oxidation state of the continental crust, could have become more oxidizing, lessening a sink. Another diminished sink from continents would be the gradual oxidation of continental rocks so that the weathering flux contained less reduced Fe and S and had less capacity to absorb oxygen. Or the deep ocean could become more oxidizing and lessen the amount of organic carbon burial as well as sulfate reduction and pyrite burial. As sulfate concentration increases in the ocean, the flux of Fe from hydrothermal vents is also lessened. All of these are positive feedbacks—a smaller sink leads to more oxidized materials, which further diminish the sink.
These various models are not definitive because hard evidence from the rocks to support them is difficult to come by. We know the answer to the question of change in the oxidation state of atmosphere and ocean—oxygen rose. Much remains to be understood concerning the detailed mechanisms.
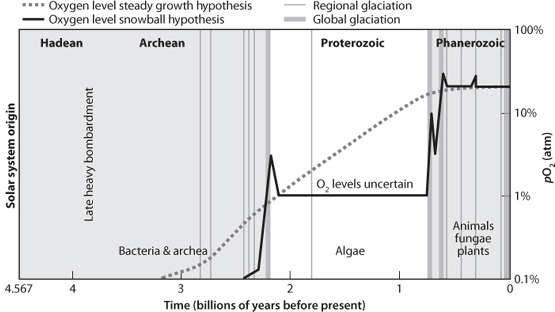
Fig. 16-12: One possible path for the evolution of O2 in the atmosphere. This figure uses a hypothetical link between “snowball Earth” episodes (marked by vertical bands at 2.2 Ga and 0.6–0.7 Ga) and abrupt rises in O2. It is also possible that rises were more progressive through time (the dotted gray line). Clear bounds are the absence of SMIF and decline of BIF near 2.0 Ga, and the advent of multicellular life that requires modern levels of O2 about 0.6 Ga. (Figure modified from Paul Hofmann; http://www.snowballearth.org)
The end result of all this information is an attempt to graph the change of oxygen in the atmosphere through time (Fig. 16-12). Such a diagram has some clear fixed points, such as a zero O2 early Earth, a significant change in atmospheric O2 near 2.0 Ga, and a rise to modern levels at the end of the Proterozoic. To what extent the changes are gradual or abrupt, and the exact levels at the different periods of Earth’s history, are less clear.
Whatever the details of the oxygen history, in the end there should be a one-to-one relationship between the electron addition to form organic matter and electron subtraction to create oxidized compounds, including O2. To approach this question in the most general way, we can simply add up the oxidized and reduced reservoirs and see if the one-to-one relationship between organic carbon and O2 is preserved.
Reduced carbon exists in myriad forms in rocks—for example, as fossil fuels—but most of it occurs in sediments, particularly in black shales that are abundant in the geological record. Total organic carbon abundance is estimated to be about 700–1,300 × 1018 moles (recall that one mole is 6.022 × 1023 molecules). The observable oxidized reservoirs are the atmosphere, ocean, and crustal rocks. As can be gathered from Table 16-2, O2 in the atmosphere is a trivial reservoir, and oxidized iron is the most important reservoir. Constraints on oxidized iron are quite good, because we have estimates for the Fe content and Fe3+/Fe2+ ratio of the continental crust and sediments. We can use the oxidized Fe of the continental crust to estimate about 1,250 × 1018 moles of O2 equivalents residing in Fe3+. An additional contribution of 175 × 1018 moles equivalent comes from current oxidation of the ocean crust. As shown by comparison of tables 16-1 and 16-2, the oxidized reservoirs are then about double the reduced carbon estimates. This is the mass balance problem concerning the rise of oxygen—the oxygen sinks tell us that there has been a greater source of oxygen through Earth’s history than what is preserved in the organic carbon remaining at the surface today.
These approximate calculations permit some comments on models for the rise of oxygen. The first is the great importance of oxidized iron. Consideration of the iron cycle also allows a fresh approach to the possible change in the oxidation state of the mantle, sometimes called upon to cause a diminished supply of reduced gases from volcanoes. It seems reasonable qualitatively to call upon subduction of oxidized iron to oxidize the mantle. But the upper mantle alone contains so much Fe that to raise Fe3+ by just 1%, less than can be resolved by measurement, would require the equivalent of 6 billion years of subduction of ocean crust as oxidized as it is today, which is unreasonable. This amount of subducted Fe3+ would also quintuple the shortfall of organic carbon, creating a far worse mass balance problem. And based on other evidence discussed above, the deep ocean may have been oxidized for <1 billion years, and prior to that time oxic alteration of the ocean crust would not occur. Oxidation of the mantle by organic matter production thus is very unlikely.
Instead, for the mantle to solve the mass balance problem, reduced atoms need to have been subducted. One possibility is ancient subduction of organic carbon. The reduced oceans of the ancient past could have led to some organic matter in deep sediments. If this material were subducted and not returned to the surface, there could be a large storehouse of electrons in the mantle. There would have to have been a mass of some 750 × 1018 moles of C subducted in excess of oxidized Fe. The amount of iron in the upper mantle is so great that this reduced carbon would have negligible impact on the iron oxidation state. It is also a small amount of carbon relative to the ~30,000 × 1018 moles of carbon in the mantle. Net subduction of organic carbon is thus a potential solution to the bookkeeping problem.
An alternative solution to the mass balance problem is to postulate a source of electron removal in addition to reduced carbon. The one likely candidate is loss of H2 to outer space. Earth’s early reducing atmosphere may have contained hydrogen. This creates a possible energy source for life, through the energy-producing reaction:
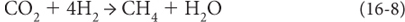
Some bacteria, called methanogens, use this reaction to provide the energy needed to produce organic matter from CO2 and H2. In ancient Earth environments with H2 available, a waste product of organic matter production would be methane, and methane is a product of anaerobic environments on Earth even today.
In today’s atmosphere, methane is not a stable gas, since it reacts rapidly with the O2 molecules that are present. Molecules of methane produced by life (e.g., cows produce an enormous amount of methane) survive in the atmosphere for an average only twelve years. In the absence of oxygen, however, methane can be stable in the atmosphere. The moon Titan, for example, has more than 1% methane in its atmosphere. In the low oxygen environment of the early Earth, methane might have been an important atmospheric constituent. Methane is also a very effective greenhouse gas and may have contributed to the early greenhouse effect that was able to keep Earth warm when the Sun was less luminous than it is today.
If methane were abundant in the atmosphere, then the ionizing radiation from the sun might have broken it up in carbon and hydrogen molecules. The H2 formed in this process is able to escape from the upper atmosphere. Since H2 is neutrally valent, it has two more electrons than hydrogen atoms combined in more complex molecules such as H2O. Loss of H2 is thus a way to have a flux of electrons from Earth’s surface to space, creating a reduced reservoir of unknown size that is no longer measurable. Calculations by David Catling and colleagues suggest this flux could lead to an irreversible oxidation of the early Earth and solve the electron mass balance problem. An additional advantage of the methane formation/hydrogen loss model is that it may account for the fact that the rises in atmospheric oxygen appear to have occurred during times when Earth underwent severe glaciations. If methane were present in the atmosphere, it would keep the climate warm. A rise of oxygen would dramatically reduce the methane, causing a sudden ice age.
This model has the appeal that methanogenic bacteria are likely important parts of the early biosphere, an early reducing atmosphere would be consistent with presence of methane, and lowering methane could cause the heavy glaciations that seem to have occurred at the same times as oxygen rose rapidly. This type of model is very difficult to test, however, since the hydrogen lost to space can never be measured, and early methanogenic ecosystems likely have no exact parallels on the modern Earth.
The mass balance issues also relate to the current steady-state O2 in the atmosphere. The carbon isotope data suggest that f has increased in the Phanerozoic, and CO2 outgassing followed by organic matter burial should continually supply more O2 to the surface reservoirs. Why is O2 not then increasing? The traditional answer to this question would relate to variations in the fraction of burial of organic matter, as discussed earlier in this chapter. That fraction, however, appears to have been relatively high throughout the Phanerozoic. Furthermore, subduction of carbon in the 1:5 organic/carbonate proportions would contribute further to an increasingly oxidized surface. What is needed is a flux of oxidized material out of the system. One solution would be a modern oxidized flux to the mantle. The current outgassing flux of CO2 is 3.4 × 1012 moles per year. If 20% of that becomes stored organic carbon, that is a flux of 0.68 × 1012 moles per year of reduced carbon. The current flux of oxidized iron in subducting ocean crust is ~2.0 × 1012 moles per year of O2 equivalents, which more than accounts for the incremental oxidizing power produced by modern life. Plate-tectonic geochemical cycles appear to have played a very significant role in the oxygen balance in both the ancient and modern Earth.
Earth’s atmosphere, soils and ocean today are oxidized environments consistent with a high level of O2 in the atmosphere. This contrasts with the early Earth, where Earth’s external reservoirs would have been in a reduced state. The journey from a reduced to oxidized surface is long, multifaceted, and complex. The broad picture of the change is apparent from the geological record, but many important details as to timing, mechanisms, and the actual levels of O2 in the atmosphere through time remain to be fully elucidated. What is clear is that the ultimate driver of this process is life, which has made use of solar and chemical energy sources to generate a current of electrons to reduce carbon and form organic molecules. Storage or loss of these electrons has left behind the oxidized counterparts in the surface reservoirs, creating the oxic conditions that enable life to make use of aerobic respiration, which is the metabolic process that generates the most energy. Over time, through evolutionary change, life has created conditions that enhance habitability and maximize access to energy.
The exterior modification of the surface is closely related to geochemical cycles that involve Earth’s interior. Because the fraction of organic/carbonate carbon stays roughly constant through Earth’s history, continued generation of organic carbon and increase of oxidized molecules at the surface requires a continual flux of CO2 from Earth’s interior through volcanism. The ultimate source of surface oxidation is thus the plate-tectonic geochemical cycle. The mantle also supplied the reduced material to the surface, principally reduced Fe and S, with some H2, which were the ultimate source of electrons to form reduced carbon. Oxygen is the intermediary of this process, donating electrons to form organic matter during oxygenic photosynthesis, and then receiving them back by oxidizing Fe and S, the long-term reservoirs of oxidized material. The steady-state O2 in the atmosphere and ocean is the reactive reservoir involved in this transfer, and in terms of volume it is a small by-product of this overall process.
Important questions about the rise of O2 remain unanswered. It appears that O2 had a series of steady-state values at different levels through Earth’s history, beginning at essentially zero in the Archean, rising to about 1% between 2.4 and 2.0 Ga after the rise of oxygenic photosynthesis, and then rising to close to modern values in the Neoproterozoic. The existence of long periods of steady state followed by step changes is a hypothesis that remains to be tested. Rigorous evidence of the causes of change and the detailed feedbacks that preserve different O2 levels in each era also remain to be discovered.
James Callen Gray Walker. 1977. Evolution of the Atmosphere. New York: Macmillan.
H. D. Holland. 2006. Oxygenation of the atmosphere and oceans. Phil. Trans. R. Soc. B. 361:903–15.
J. Hayes and J. Waldbauer. 2006. The carbon cycle and associated redox processes through time. Phil. Trans. R. Soc. B. 361:931–50.
D. C. Catling and M. W. Claire. 2005. How Earth’s atmosphere evolved to an oxic state: A status report. Earth Planet. Sci. Lett. 237:1–20.