Trace Metals
15.1 Introduction
Industrialized society is built upon the use of metals. Unlike many of the synthetic organic compounds used in industry, medicine, and agriculture, metals are part of natural biogeochemical cycles. Human activity influences their cycling in two interrelated ways: by altering the rate at which metals are transported among different reservoirs and by altering the form of the metals from that in which they were originally deposited.
Metals and other elements of economic interest are deposited when geochemical conditions reduce their mobility. Deposits range in quality from nearly pure elements, such as native copper, to highly disseminated deposits of marginal economic value. In addition, there are natural background levels of nearly all of the elements in what constitutes the average crustal rocks – the shales, sandstones, and igneous and metamorphic rocks that make up the continents and ocean floors. In the absence of human activities, elements are released to terrestrial and aquatic environments at rates corresponding to natural chemical and mechanical erosion times. Mining, construction, and large-scale changes to the natural environment alter the rate of release of elements to that part of the biogeochemical environment we call the ecosphere. These alterations, in turn, have a cascading effect on the rate at which metals are exchanged among various reservoirs in the ecosphere. In this way, the release of metals from, for example, the crustal reservoir, can affect biota in aquatic systems far from the original deposit site.
One of the characteristics of the cycle of metal mobilization and deposition is that the form of the metal is changed. This change in speciation of a metal has a profound effect on its fate. The link between metal speciation and fate is the central theme of this chapter.
This chapter differs from previous ones in that it describes the cycling of several chemical elements. These elements have many similar properties and can be considered as a group, but each also has properties that make it unique. One of the most important properties that distinguishes them from other elements is their tendency to bond reversibly with a very large number of compounds. The availability and nature of such compounds in a system can control the transport and fate of metals. In view of the importance of these reactions, this chapter starts with a broad overview of global metal cycling, which is then interpreted in the context of a discussion of the nature of metals and their chemical reactions. Finally, these concepts are applied in some detail to the natural and perturbed biogeochemical cycling of two specific metals.
15.2 Metals and Geochemistry
15.2.1 Metal Abundance and Availability
The average composition of the Earth’s crust is essentially the composition of igneous rocks, since metamorphic and sedimentary rocks constitute a relatively insignificant portion of the total crustal mass. Eight elements – O, Si, Al, Fe, Ca, Na, K, and Mg – make up nearly 99% of the total elemental mass; the remaining elements are differentiated throughout the crust according to their particular chemical properties. Geochemical differentiation based upon chemical affinities was first described by Goldschmidt (1954) in his proposal for a general geochemical classification scheme. In his framework, elements are considered to be siderophiles, chalcophiles, lithophiles, or atmophiles depending on their relative affinities for minerals containing iron, sulfide, or silicate, or for the atmosphere, respectively. Such a classification scheme was a significant advance in our understanding of the distribution of elements, making it possible to relate the general geochemical character of elements to their position in the Periodic Table and to fundamental chemical properties such as electronegativity and ion size.
With regard to geochemical cycling (as well as for economic considerations) it is important to distinguish between the abundance of an element and its availability. The availability of an element is related not only to its relative abundance on Earth but also to the stability of minerals of which it is a major constituent. Thus, a number of elements (e.g., copper, mercury, tin, and arsenic) which are scarce in terms of their average crustal abundance are easily isolated due to their ability to form mineral deposits. The most unavailable elements are those that form no major minerals of their own. Many of the rarer elements are available for economic use only to the extent that they are obtained as byproducts of the extraction of more abundant elements. Tellurium, for example, is produced during the electrolytic refining of copper.
15.2.2 Metal Mobilization
The availability of a metal describes one aspect of its potential to cycle among biogeochemical reservoirs. The initiation of the cycling process is called mobilization. Metals may be mobilized, that is, made available for transport away from their region of deposition, when the geochemical character of the depositional environment changes. These changes may be due to either natural or anthropogenic causes.
Natural mobilization includes chemical, mechanical, and biological weathering and volcanic activity. In chemical weathering, the elements are altered to forms that are more easily transported. For example, when basic rocks are neutralized by acidic fluids (such as rainwater acidified by absorption of CO2), the minerals contained in the rocks can dissolve, releasing metals to aqueous solution. Several examples are listed below of chemical reactions that involve atmospheric gases and that lead to the mobilization of metals:
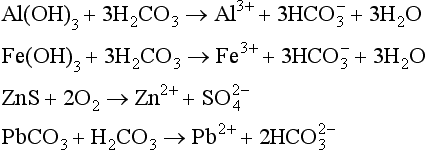
Biological and volcanic activities also have roles in the natural mobilization of elements. Plants can play multiple roles in this process. Root growth breaks down rocks mechanically to expose new surfaces to chemical weathering, while chemical interactions between plants and the soil solution affect solution pH and the concentration of salts, in turn affecting the solution-mineral interactions. Plants also aid in decreasing the rate of mechanical erosion by increasing land stability. These factors are discussed more fully in Chapter 6 and 7.
Volcanic activity has a significant effect on the mobilization of metals, particularly the more volatile ones, e.g., Pb, Cd, As, and Hg. Effects of volcanism are qualitatively different from those of the weathering and other near-surface mobilization processes mentioned above, in that volcanism transports materials from much deeper in the crust and may inject elements into the atmospheric reservoir.
15.2.3 Human Activities as Geochemical Processes
That humans have significantly altered the biogeochemical cycles of many metals is no longer an arguable point. What is uncertain is the magnitude of the effects from these alterations, particularly in the long term. Given that the flux of energy and materials through the biosphere is self-regulating, at least within certain limits, the issue becomes one of evaluating the ability of the biosphere to assimilate anthropogenic metal inputs and predicting the rate and types of changes that will occur as a new steady-state condition is approached. These sorts of predictions are hampered not only by the state of our understanding of the complex interactions that occur between metals and the environment, but also because the determination of background metal concentrations in uncontaminated environments is so difficult. In view of the worldwide dissemination of anthropogenic materials that has already occurred, locating an uncontaminated site is extremely difficult. For example, concentrations of mercury, selenium, and sulfur above background levels have been found in arctic ice sheets. These elemental “signals” correlate with the beginning of worldwide industrialization (Weiss et al., 1971a, b). In addition, there are problems associated with preparing and analyzing samples containing extremely low concentrations of metals without contaminating them.
Despite the difficulties, there have been many efforts in recent years to evaluate trace metal concentrations in natural systems and to compare trace metal release and transport rates from natural and anthropogenic sources. There is no single parameter that can summarize such comparisons. Frequently, a comparison is made between the composition of atmospheric particles and that of average crustal material to indicate whether certain elements are enriched in the atmospheric particulates. If so, some explanation is sought for the enrichment. Usually, the contribution of seaspray to the enrichment is estimated, and any enrichment unaccounted for is attributed to other natural inputs (volcanoes, low-temperature volatilization processes, etc.) or anthropogenic sources.
A second approach is to compare total mining production of a metal to an estimate of its total natural flux, making the implicit assumption that all mined materials will be released to the environment in the near future (a reasonable assumption when comparing with geologic processes).
Finally, some authors have computed metal loading to the environment from specific human activities, such as discharges of wastewater, and compared these with natural release rates. While the details of the computations and conclusions vary, the general observation for many metals is that anthropogenic contributions to metal ion transport rates and environmental burdens are approaching and in many cases have already exceeded natural contributions. A few such comparisons are provided in Tables 15-1–15-4.
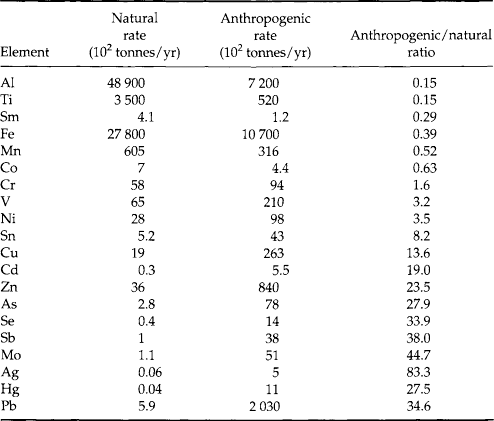
Table 15-2
Comparison between artificial and natural rates (103 tonnes/yr) of global metal injection into the oceans and atmospherea

Table 15-3
Calculated present-day fluxes (µg/(cm2 yr)) of heavy metals into the sediments of Lake Eriea
| Element | Anthropogenic | Natural |
| Cd | ||
| Stn 1 | 0.36 | 0.16 |
| Stn 3 | 0.02 | 0.02 |
| Stn 7 | 0.54 | 0.09 |
| Cu | ||
| Stn 1 | 12.0 | 7.8 |
| Stn 3 | 0.15 | 0.47 |
| Stn 7 | 8.8 | 4.8 |
| Pb | ||
| Stn 1 | 11.8 | 4.3 |
| Stn 3 | 0.33 | 0.44 |
| Stn 7 | 10.9 | 2.4 |
| Zn | ||
| Stn 1 | 36.2 | 14.8 |
| Stn 3 | 1.0 | 0.68 |
| Stn 7 | 30.6 | 5.9 |
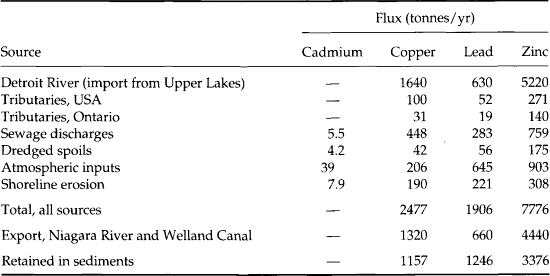
The amount of metals released as the byproduct of a single activity, the burning of coal, illustrates the potential importance of anthropogenic sources. Inorganic, non-combustible materials present in coal constitute the ash that remains after combustion. Fly ash (the ash that leaves the furnace and is collected by flue-scrubbing devices) is particularly enriched in metals, and in 1975 approximately 33 million tonnes of fly ash were produced in the United States (Theis and Wirth, 1977). This represents an average of 270 000 tonnes of fly ash per year for a typical 1000 MW power plant. A trace element with a concentration of 1 mg/kg (ppm) in the fly ash would be produced as waste at an average rate of 270 kg per 1000 MW plant per year. The composition of a few fly ash samples is shown in Table 15-5. As, Cd, Co, Cr, Cu, Hg, Pb, Se, V and Zn are present in concentrations ranging from 1 to 1000 ppm(mass). Furthermore, several trace metals including As, Pb, Cd, Se, Cr, and Zn are concentrated on the surfaces of the smallest particles, which have the greatest likelihood to escape the plant and be transported significant distances in the atmosphere. When these ashes are ponded or exposed to rain, the pH of the water can decrease to less than 4 or increase to greater than 13, depending on the chemistry of the fly-ash matrix. The fraction of the total metal solubilized under these conditions is a sensitive function of pH and, although this fraction is usually 10% or less, it can exceed 50%.
Table 15-5
Comparison of elemental concentrations in size-classified fly-ash fractiona
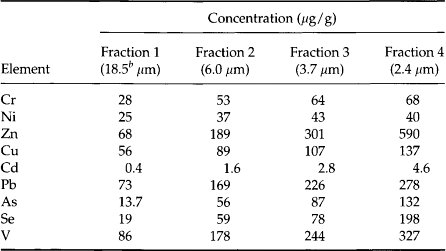
bMass median diameters determined by centrifugal sedimentation.
Once the anthropogenic release rates of metals are established, the next critical step is to evaluate their fate upon discharge to receiving waters. Sediment analyses are often useful in this regard because changes in metal concentration as a function of depth in the sediment can indicate historical trends. Also, the concentrations of metals are typically much greater in sediments than in the water column and are therefore easier to analyze and evaluate. Bruland et al. (1974) used metal/aluminum ratios in sediment to determine the magnitude of the anthropogenic component of the heavy metal transport rate to a Southern California basin. Assuming that aluminum has had a uniform rate of transport to the sediments over the past century from crustal rock sources, they concluded that Pb, Cr, Cd, Zn, Cu, Ag, V, and Mo are now accumulating at higher rates than a century or more ago. For all of these metals, the anthropogenic component represented at least one-third of the natural emission rate, and for Pb, Ag, and Mo, the anthropogenic rate exceeded the natural rate. A qualitatively similar conclusion applies to trace metal transport into the northern portion of Chesapeake Bay (Helz, 1976).
In a subsequent section, trace metal movement through some other aquatic systems will be reviewed and analyzed in the context of specific chemical reactions. First, though, the most important chemical reactions of metal ions are reviewed.
15.3 An Overview of Metal Ion Chemistry
15.3.1 Introduction
Metals, like many of the elements discussed in previous chapters, can exist in nature in several different oxidation states. When bonded to other elements, metal ions are almost always assigned a positive oxidation number and are somewhat electrophilic. Because of this they are stabilized by association with electron-rich atoms. In particular, atoms that have a free electron pair can “donate” some of their electron density to the metal to form a bond. Such atoms are Lewis bases. The most common and environmentally important donor atoms are oxygen, nitrogen, and sulfur. The bonds they form with metal ions range in strength from relatively weak associations such as those between a dissolved metal ion and water to very strong covalent bonds. These types of bonds are significant in both aqueous phase reactions and in the formation of insoluble compounds.
The characteristic affinity of metals for electron-rich donor atoms leads to an important distinction between the geochemical behavior of metal ions and that of the elements discussed earlier in this text. Specifically, metal ions in a single oxidation state can bind to donor atoms that are part of a variety of cationic, anionic, or neutral molecules. Thus, the metals can be found in and can move through the environment as parts of molecules spanning the complete range of charge, molecular weight, bioavailability, and other chemical characteristics. In addition, since metals constitute an extremely small fraction of biological organisms, the connection between metal transport through the environment and biological activity is generally less direct and of less quantitative importance than for, say, oxygen or phosphorus. (However, recent evidence is pointing to a more significant role for biota in controlling metal concentrations and transport in the ocean.) In a relative sense, then, purely chemical reactions are of more importance for metals than for the elements discussed earlier. The strength of the chemical bonding between the donor atoms O, N, and S and metals is the overriding factor controlling the geochemical cycling of metals. Once these reactions are understood, the behavior of metals, with respect to both transport and biological impacts, can be placed in a more logical framework.
15.3.2 Oxidation-Reduction Reactions
Metals exist in nature primarily in positive oxidation states, and many form stable compounds in more than one oxidation state. The formal oxidation number of the most common form can range from +1 to +6. The stable form in a given environment depends on the oxidation potential and chemical composition of that environment. Often the stable form at the Earth’s surface in the presence of molecular oxygen is different from that which is stable in anoxic sediments or waters.
Thermodynamically, virtually all metals in the elemental form are unstable with respect to redox reactions in environments where they are exposed to air and water, i.e., virtually all environments where they are used. Those metals least likely to oxidize (corrode) were long ago given the distinguished title “noble metals.” Efforts to prevent metals from corroding, and the cost of repairing and replacing metal structures that have done so, runs into the billions of dollars annually. Thus, one characteristic feature of the society’s use of metals is that the metals are continuously, albeit slowly, “degrading” to a less useful form from the moment they are put into use.
The different oxidation states of a metal can have dramatically different chemical properties, which in turn affect their biogeochemical forms and significance. For example, almost 4 g/L ferrous iron, Fe(II), can dissolve in distilled water maintained at pH 7.0. However, if the water is exposed to air and the iron is oxidized to Fe(III) essentially all the iron will precipitate, reducing the soluble Fe concentration by more than eight orders of magnitude. Oxidation state can also affect a metal ion’s toxicity. For instance, the toxicity of As(III) results from its ability to inactivate enzymes, while As(V) interferes with ATP synthesis. The former is considerably more toxic to both aquatic organisms and humans.
The thermodynamically stable oxidation state of a metal in a given environment is a function of the prevailing oxidation potential. The value of the potential is given by the Nernst equation, which is described in Chapter 5 for the generic reduction half-cell:
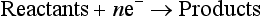
The electrochemical potential that would cause the reactants and products to be equilibrated with each other is
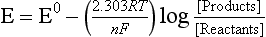
If the equilibrium half-cell potentials for two redox reactions are different, electrons will be transferred from the reduced species in the reaction with the less positive potential to the oxidized form with the more positive potential. The process is repeated until all exchangeable electrons have the same equilibrium potential. Water chemistry texts describe rapid graphical or computerized approaches to solve for the concentrations of all species once this equilibrium condition has been attained.
While these calculations provide information about the ultimate equilibrium conditions, redox reactions are often slow on human time scales, and sometimes even on geological time scales. Furthermore, the reactions in natural systems are complex and may be catalyzed or inhibited by the solids or trace constituents present. There is a dearth of information on the kinetics of redox reactions in such systems, but it is clear that many chemical species commonly found in environmental samples would not be present if equilibrium were attained. Furthermore, the conditions at equilibrium depend on the concentration of other species in the system, many of which are difficult or impossible to determine analytically. Morgan and Stone (1985) reviewed the kinetics of many environmentally important reactions and pointed out that determination of whether an equilibrium model is appropriate in a given situation depends on the relative time constants of the chemical reactions of interest and the physical processes governing the movement of material through the system. This point is discussed in some detail in Section 15.3.8. In the absence of detailed information with which to evaluate these time constants, chemical analysis for metals in each of their oxidation states, rather than equilibrium calculations, must be conducted to evaluate the current state of a system and the biological or geochemical importance of the metals it contains.
To summarize, an evaluation of the oxidation state of metals in an environment is central to determining their probable fate and biological significance. Redox reactions can lead to orders of magnitude changes in the concentration of metals in various phases, and hence in their mode and rate of transport. While equilibrium calculations are a valuable tool for understanding the direction in which changes are likely to occur, field measurements of the concentrations of metals in their various oxidation states are always needed to evaluate metal speciation, since equilibrium has often not yet been attained.
15.3.3 Volatilization
15.3.3.1 Volatilization from the solid state
The extent to which any chemical species is volatilized is governed by its vapor pressure, which is sensitive to temperature. Most metals and their compounds have very low vapor pressures at normal temperatures, low enough that their tendency to vaporize can be ignored. The major exceptions are metallic Hg and organometallic compounds. Nevertheless, in some environments significant quantities of metals can be volatilized either as elements or inorganic compounds such as oxides or carbonates. The most obvious such environments are high-temperature furnaces such as in smelters or fossil-fuel-burning power plants and in regions of geothermal activity or vulcanism. While the oxides, sulfates, carbonates, and sulfides of a metal all have somewhat different volatilities, the most volatile metals, regardless of the anion with which they are associated, are Hg, As, Cd, Pb, and Zn. Metallic Hg and organometallic compounds may be transported significant distances as gases and eventually be removed from the atmosphere by dissolution in rain droplets. However, most volatilized metals condense rapidly as they cool and fall to the surface associated with particulate matter, either as dry deposition or scavenged by precipitation. Some of the condensed particles are light enough to carry long distances, and those that fall to Earth may be resuspended by wind action or washed into a water body by surface runoff. Particles produced by high-temperature combustion processes are mostly in the < 2 µm size range and typically have atmospheric residence times of 7 to 14 days, while those generated by soil erosion are larger (>5 µm) (Hardy et al., 1985). Anthropogenic particles are typically enriched in trace metals (normalized to the concentration of Al) by a factor of 100 to 10 000 compared with atmospheric particulates generated by natural erosion and wind action. As an indication of the importance of volatilization of metals to their overall biogeochemical budgets, Galloway (1979) estimated that volatilization of As, Hg, and Se overwhelms total dust and volcanic emanation rates by factors of 7.5, 625, and 7.3, respectively.
15.3.3.2 Volatilization from solution
The equilibrium volatility of a species dissolved in water is characterized by its equilibrium constant for the reaction X(g) ↔ X(aq), i.e., its Henry’s Law constant. These equilibrium constants are related, but are not directly comparable to vapor pressures of the corresponding dry solids because of the effect of the solvent, water. (The most appropriate direct comparison involves the calculation of fugacities. The fugacity, or escaping tendency of a chemical species from the environment in which it exists, depends upon both the concentration of the chemical species of interest and the strength of its interactions with the surrounding (solvent) molecules. Details of the calculation are provided in chemical thermodynamics textbooks and in a number of articles by Mackay and co-workers (e.g. Mackay, 1979).) Suffice it to say that when volatile metal species, such as methylmercury, are present in water, there virtually always exists a driving force to strip them out of the aqueous phase, since their partial pressures in air are essentially zero. Thus, equilibrium between gaseous and aqueous phases is rarely a limiting factor. Rather, the factor limiting volatilization of these species is usually the rate at which they move through the water column and across the water-air interface.
15.3.3.3 Volatilization of mercury and lead
The ratio of anthropogenic emissions to total natural emissions is highest for the atmophilic elements Sn, Cu, Cd, Zn, As, Se, Mo, Hg, and Pb (Lantzy and Mackenzie, 1979). In the case of lead, atmospheric concentrations are primarily the consequence of the combustion of leaded gasoline. For many years, lead was used as a gasoline additive, in the form of an organometal compound, tetraethyl lead. When the fuel was burned, most of the lead was converted to inorganic forms and released in the exhaust. The widespread use of this compound as an anti-knock agent in gasoline engines led to its dispersion everywhere automobiles travel, and from there to very remote locations. As with metals near smelters, lead concentrations in the soil near major roadways decrease rapidly with distance from the source, but significant amounts of the metal are transported by wind, either directly after being emitted or by resuspension after a period of deposition.
Atmospheric fluxes of lead in the United States rose steadily from the first decades of this century, reaching a maximum in the early 1970s (see Eisenrich et al., 1986 and references therein). Passage of the Clean Air Act of 1972 and its subsequent amendments resulted in dramatic reductions in atmospheric lead concentrations, although lead fluxes worldwide still remain 10–1000 times background levels (Settle et al., 1982; Settle and Patterson, 1982).
Volatilization is also a dominant transport mode for mercury, which is the most volatile metal in its elemental state. As with lead, a key reaction that can increase the volatility of mercury is formation of an organometallic compound. In this case, the reactions take place in water and are primarily biological, being mediated by bacteria commonly found in the upper levels of sediments. These reactions and their importance in the global mercury cycle are discussed in some detail later in the chapter.
To summarize, metals can be transferred into the gas phase in high-temperature processes either in their elemental form or as inorganic compounds, and these compounds can then be transported long distances as gases or in other physical-chemical forms. Many organometallic compounds are also volatile. In a few cases, natural organometallic compounds may be formed that are volatile. These compounds are formed by microorganisms in mildly reducing aquatic environments and are then transported to the surface and across the air-water interface to enter the gas phase. Methylmercury compounds are probably the most important and certainly have been the most widely studied of these because of their central role in the bioaccumulation of mercury; others, such as methylarsine and organotin compounds, are environmentally important as well. Anthropogenic release to the atmosphere overwhelms natural sources for Hg, Cd, Cu, Ag, Zn, Pb, As, and Se, and possibly other metals.
15.3.4 Complexation Reactions
The stability of liquid water is due in large part to the ability of water molecules to form hydrogen bonds with one another. Such bonds tend to stabilize the molecules in a pattern where the hydrogens of one water molecule are adjacent to oxygens of other water molecules. When chemical species dissolve, they must insert themselves into this matrix, and in the process break some of the bonds that exist between the water molecules. If a substance can form strong bonds with water, its dissolution will be thermodynamically favored, i.e., it will be highly soluble. Similarly, dissolution of a molecule that breaks water-to-water bonds and replaces these with weaker water-to-solute bonds will be energetically unfavorable, i.e., it will be relatively insoluble. These principles are presented schematically in Fig. 15-1.
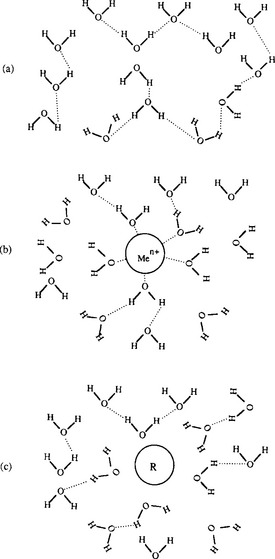
Fig. 15-1 Schematic representation of the change in water structure (water molecule orientation) due to the presence of a charged (hydrophilic) solute. (a) Pure water. (b) A solute forming strong bonds with water (dissolution favorable). (c) a solute forming weak bonds with water (dissolution unfavorable).
Cationic metal ions form strong bonds with water molecules. By orienting themselves in such a way that the metal “faces” an oxygen atom, the negative charge on the oxygen is partially distributed onto the metal, forming the analog of a strong hydrogen bond. Similarly, some of the charge on the metal ion is neutralized. (Recall the electrophilic nature of metal ions.) These bonds are strong enough that, in most aqueous environments, most metal ions are surrounded by an “inner hydration sphere” of four to eight strongly bound water molecules, as well as a loosely attached “outer hydration sphere” of variable size. Although chemical convention represents dissolved metal ions as Men+, a more accurate designation is Me(H2O)xn+. The strength of the metal-to-water bond increases with decreasing size and increasing charge of the metal ion, and also depends on the distribution of electrons around it.
The water molecules in the inner hydration sphere can undergo dissociation reactions just as water molecules far from a dissolved metal ion do, but the presence of the metal ion changes the equilibrium constant for this reaction. The magnitude of this alteration is directly related to the strength of the metal-to-water bond. If the metal attracts a large portion of the electron density from the oxygen, it weakens the oxygen-to-hydrogen bonds and enhances the tendency for the water molecule to dissociate. This leads to dissolved species that are typically designated hydrolyzed metal ions, Me(OH)yn – y, but which are more accurately portrayed as Me(H2O)x(OH)yn–y. As an extreme example, we can think of a metal like molybdenum bonding so strongly to four oxygen atoms that they lose their ability to bond to any hydrogen at all, forming MoO43−. In such a situation, the oxygens are not considered part of the hydration sphere at all, but as covalently bonded to the molybdenum. Nevertheless, in a qualitative sense this reaction is no different from the hydrolysis reactions described above. Comparison of a metal hydrolysis reaction with hydrolysis of pure water indicates that if the equilibrium constant for metal hydrolysis is larger than 10−14 (the dissociation constant of pure water) the metal acts as an acid, i.e., it has a greater tendency to release hydrogen ions to solution than pure water (Fig. 15-2). Furthermore, since metal ions are surrounded by several waters of hydration, they can act as multi-protic acids, releasing four, five, or even six hydrogen ions to solution and acquiring a net negative charge in the process.
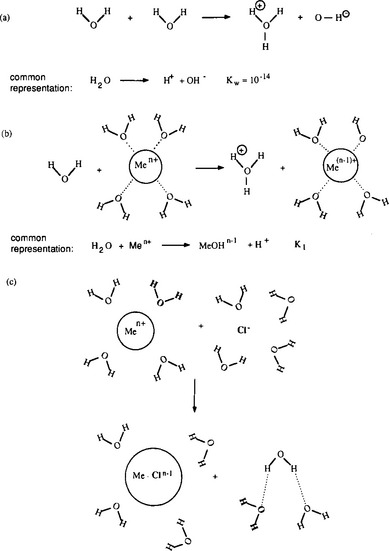
Fig. 15-2 Comparison of water dissociation in bulk solution (a) and in the hydration sphere of a metal ion (b). Exchange of water of hydration for a chloride ion (c) forms the Me–Cl complex. (from Manahan, 1979)
The comparisons between pure water and a solution containing dissolved metal ions can be extended by considering the behavior of other dissolved ions such as Cl−, SO42−, HCO3, S2−, and dissolved organic molecules. Just as metal ions do, these ions tend to orient themselves in a way that maximizes the bond strength between them and other constituents of the solution. For instance, the water molecules surrounding a chloride ion are somewhat structured by the negative charge of the chloride, so that the positive (hydrogen) ends of the water molecule are closer to the ion than is the oxygen. Typically, this tendency to attract and orient a hydration sphere is much weaker for anions than for metals. If a metal ion is dissolved in the same solution as the chloride, then an additional possibility presents itself. The chloride ion could replace one of the waters of hydration surrounding the metal, thereby exchanging a water-metal bond and a water-chloride bond for a metal-chloride bond and a water-water bond (Fig. 15-2c). Equilibrium constants for such reactions are called stability constants, and are tabulated in a number of sources. Values for these constants can range considerably. Very large values imply that metals and ligands are virtually certain to form complexes, even if both ions are present in very low concentrations.
The concept of exchange of water molecules in the hydration sphere of a metal ion for other dissolved species can be extended to include “mixed ligand complexes,” i.e., those in which water molecules have been replaced by two or more different types of ligands, and “multi-dentate” complexes, those in which a single molecule can bind to the metal through more than one atom, and therefore cause more than one water of hydration to be released for each ligand molecule binding (Fig. 15-3). As might be expected, the binding strength typically decreases as ligands are added, and the bond strength of a multi-dentate ligand is typically greater than that of mono-dentate ligands, as a result of the multiple bonds formed. When a ligand forms a strong multi-dentate complex it is referred to as a chelating agent, and the complex is called a chelate.
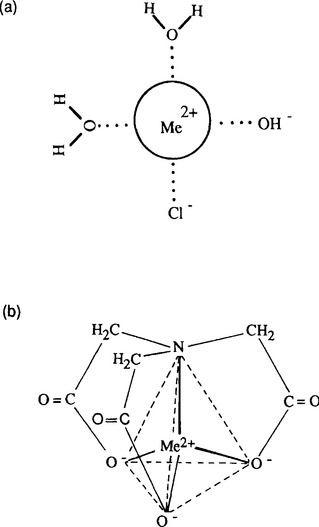
Fig. 15-3 Mixed ligand and multi-dentate complexes: (a) a hypothetical Me(OH)Cl0 complex; (b) nitrilotriacetate chelate of a divalent metal ion in a tetrahedral configuration.
It is important to recognize that although many ligands are anions and hence have an electrostatic attraction for the metal ion, this is not a requirement. For instance, the neutral ammonia molecule (NH3) is a strong complexing agent for many metals. Furthermore, if electrostatic attraction were critical to formation of these types of bonds, then complexation would cease once the positive charge on the metal was neutralized. Complexation can continue until several of the molecules in the hydration sphere are replaced, yielding a chemical species consisting of a positively charged central metal ion surrounded by as many as six negatively charged ligands, and carrying a net negative charge of –3 or – 4.
Complexation reactions are of crucial importance in biogeochemical cycling of metals because a large fraction of the total dissolved metal may be complexed. Biological effects can be sensitive to the types of complexes present. In many cases free metal ions (fully hydrated metal ions) are more toxic than complexed ones, especially those complexed by multi-dentate organic ligands. There is also evidence that some organisms secrete chelating agents in response to high metal concentrations in their environment, presumably as a defense against metal ion toxicity. On the other hand, organisms can also produce chelating agents to acquire metals that are necessary for certain metabolic functions. These chelating agents are often extremely specific for a given metal and are used to “collect” metals from solution or maintain a desired concentration of metals inside the cell.
In addition to those complexing agents produced by organisms to regulate their environment, numerous complexing and chelating agents are present in aquatic systems as a result of the normal exchange of metabolites between organisms and their environment, cell death and decay or as a result of anthropogenic activities. The high-molecular-weight compounds known as fulvic and humic acids are particularly important. These acidic polymers are primarily the residue from organic decay processes and do not have a well-defined chemical structure. However, like synthetic chelating agents, they contain high concentrations of electron-rich donor atoms, especially oxygen in carboxylic and phenolic groups, which can complex or chelate metal ions. Sulfur- and nitrogen-containing portions of the molecules may also be important in complexing such metals. These molecules are especially effective chelators since they are large and flexible, and can have several complexing sites on each molecule. At least in theory, they may be able to arrange themselves in such a way that several donor groups are at the optimal positions to form very strong bonds.
The speciation (ignoring organic complexes) of selected metals in various aquatic environments is described in Table 15-6. The relative strengths of the complexes of a given metal, and differences among metals in the strength of binding to a given ligand, are apparent. Also apparent is the fact that the dominant form of a given metal often changes when the local environment changes pH or ionic strength (as in an estuary). Important changes also occur when the environment changes from aerobic to anaerobic (for instance in the bottom of a seasonally anoxic lake), or when the concentration of dissolved organic matter changes, such as near a sewage outfall.
Table 15-6
Model results for metal speciation in natural watersa,b
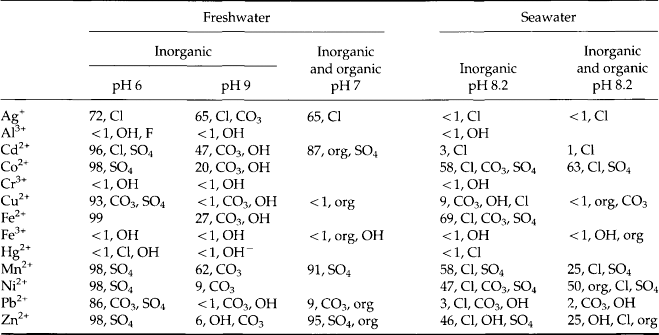
aData for inorganic freshwater and inorganic seawater from Turner et al. (1981). Data for systems with inorganics and organics from Stumm and Morgan (1981). Six organic ligands included corresponding to 2.3 mg/L total soluble organic carbon. Stability constants and inorganic composition of model water were not identical in the two studies, so comparisons are qualitatively valid but may have minor quantitative inconsistencies.
bEach entry has the % of total metal present as the free hydrated ion, then the ligands forming complexes, in decreasing order of expected concentration. For instance, in inorganic freshwater at pH 9, Ag is present as the free aquo ion (65%), chloro-complexes (25%), and carbonato-complexes (9%).
Summarizing this section, metal ions may be present in aqueous solution surrounded by and bonded to water molecules or a wide variety of inorganic and organic complexing agents. The bonds formed by complexation are often reversible, and their strength varies widely depending on the chemical and physical properties of the metal and ligand. As a result of complexation reactions, concentrations of total dissolved metal in an aquatic system can be orders of magnitude higher than those of free metal ions. Thus, if the concentration of free metal ion in a system is limited by virtue of low solubility, complexes can still cause large amounts of that metal to dissolve and be transported among the various geochemical reservoirs.
15.3.5 Precipitation Reactions
If metal concentrations in solution become large, soluble complexes that contain more than one metal ion can form. Eventually these can grow so large that a three-dimensional network is created in which most of the ions in the interior are not in contact with the bulk solution, and a separate phase is formed. In systems where large soluble polymeric metal complexes are stable, the exact point at which the new phase forms is open to question and is somewhat a matter of definition. In most practical situations, however, these large polymers have very limited stability and the system undergoes a dramatic change from a completely soluble state containing monomeric and relatively small polymeric species to one containing an identifiable second phase. The activation energy required to form this new phase is large, and solutions must be highly supersaturated for precipitation to be initiated in particle-free systems. On the other hand, the activation energy for growth of solids once a solid-liquid boundary has been established is much smaller, so once precipitation has started it can often proceed rapidly. In natural aquatic systems, the problem of high activation energies for formation of new phases is usually circumvented by new solids forming on the surfaces of existing solids with a compatible crystal structure.
Solubility equilibria are described quantitatively by the equilibrium constant for solid dissolution, KSP (the solubility product). Formally, this equilibrium constant should be written as the activity of the products divided by that of the reactants, including the solid. However, since the activity of any pure solid is defined as 1.0, the solid is commonly left out of the equilibrium constant expression. The activity of the solid is important in natural systems where the solids are frequently not pure, but are mixtures. In such a case, the activity of a solid component that forms part of an “ideal” solid solution is defined as its mole fraction in the solid phase. Empirically, it appears that most solid solutions are far from ideal, with the dilute component having an activity considerably greater than its mole fraction. Nevertheless, the point remains that not all solid components found in an aquatic system have unit activity, and thus their solubility will be less than that defined by the solubility constant in its conventional form.
Metal precipitates of biogeochemical significance are primarily oxides, hydroxides, carbonates, and sulfides. Not surprisingly, these are also the ligands with which many metals form strong complexes. However, the correspondence between the strength of bonding in a solid phase and that in a soluble complex is far from exact, because of the effects of the bonds with water in the latter cases. All of these anions have concentrations that are strongly dependent on pH. Because of this, a change in pH is one of the most important driving forces for precipitation or dissolution reactions. The other dominant factor is the oxidation-reduction potential of the solution. Metal sulfides tend to be extremely insoluble, but they can only exist in environments where the redox potential is sufficiently reducing for sulfur to exist in the –II oxidation state (H2S, HS− or S2−). Thus, some metals are soluble in the oxidized layers of sediments but are precipitated in lower anoxic layers, where sulfate (SO42−) is reduced to sulfide.
Model calculations that illustrate these points have been performed by Morel et al. (1975) for the speciation of a sewage-seawater mixture. They concluded that sulfides of Cu, Cd, Pb, Zn, Ag, Hg, and Co would be the predominant form of those metals in the sewage, along with oxidized precipitates Cr(OH)3 and Fe2O3, cyanocomplexes of Ni2+, and free Mn2+. However, as dilution and oxidation of the sewage by seawater proceed and the S(– II) concentration decreases, the sulfides dissolve and all the metals except Fe eventually go into solution. Their calculations show that CoCO3(s), ZnCO3(s), and CuO(s) may form as intermediates between the initial (sulfidic solid) and final (soluble) endstates.
In addition to effects on the concentration of anions, the redox potential can affect the oxidation state and solubility of the metal ion directly. The most important examples of this are the dissolution of iron and manganese under reducing conditions. The oxidized forms of these elements (Fe(III) and Mn(IV)) form very insoluble oxides and hydroxides, while the reduced forms (Fe(II) and Mn(II)) are orders of magnitude more soluble (in the absence of S(– II)). The oxidation or reduction of the metals, which can occur fairly rapidly at oxic–anoxic interfaces, has an important “domino” effect on the distribution of many other metals in the system due to the importance of iron and manganese oxides in adsorption reactions. In an interesting example of this, it has been suggested that arsenate accumulates in the upper, oxidized layers of some sediments by diffusion of As(III), Fe(II), and Mn(II) from the deeper, reduced zones. In the aerobic zone, the cations are oxidized by oxygen, and precipitate. The solids can then oxidize, as As(III) to As(V), which is subsequently immobilized by sorption onto other Fe or Mn oxyhydroxide particles (Takamatsu et al., 1985).
While the solubility constants for various potential solids can indicate which solid is thermodynamically stable under a given set of conditions, reactions involving precipitation or dissolution of a solid are typically more subject to kinetic limitations than are reactions that take place strictly in solution. As a result, a thermodynamically unstable solid (often referred to as “metastable”) may form and remain in place for geological time periods. Such is frequently the case when an insoluble but relatively less stable solid-phase precipitates, and the thermodynamically stable phase has a different crystal structure. Often the only way for the more stable solid to form is by dissolution of ions from the first solid and reprecipitation of the second. Since the first solid is quite insoluble itself, dissolution occurs very slowly. Since this causes the solution to be only very slightly supersaturated with respect to the second solid, the driving force for its formation is also small. Thus, for example, goethite (α-FeOOH) is more stable than lepidocrocite (γ-FeOOH), but lepidocrocite forms more rapidly than goethite when iron sulfide is oxidized by oxygen, and lepidocrocite can be found in geologic formations long after the oxidation has taken place.
An important result of the concepts discussed in this section and the preceding one is that precipitation and complexation reactions exert joint control over metal ion solubility and transport. Whereas precipitation can limit the dissolved concentration of a specific species (Men+), complexation reactions can allow the total dissolved concentration of that metal to be much higher. The balance between these two competing processes, taking into account kinetic and equilibrium effects, often determines how much metal is transported in solution between two sites.
15.3.6 Adsorption
The importance of one other type of reaction that metal ions undergo has been recognized and studied extensively in the past 40 years. This reaction is adsorption, in which metal ions bind to the surface of particulate matter and are thereby transported as part of a solid phase even though they do not form an identifiable precipitate. Conceptually, these reactions can be thought of as hybrids between complexation and precipitation reactions. Most studies of these reactions have used metal oxides or hydroxides as the solid (adsorbent) phase, and the reactions will be discussed in that context here, although sorption onto other solids may be important as well.
At a solid-solution interface, atoms are fixed in place by their attachment to the solid, but are freer to move and react with solution components than atoms in the solid. Put another way, these atoms are half in solution and half out. To the extent that their bonding requirements are not completely met by the bond to the bulk solid, they are energetically driven to react with solution components. In the case of a ferric oxide, for instance, an oxygen atom may be attached to an iron atom on the solid side, and may bind to a hydrogen ion on the solution side, to form the analog of an FeOH complex. If we treat the FeO− surface group as a reactant, we can write an equilibrium reaction describing this interaction. Furthermore, as shown in Fig. 15-4a we can postulate that the surface group might react with a second hydrogen ion to form the analog of a hydrated Fe ion. The result is that each surface Fe atom can behave as a diprotic acid, binding or releasing hydrogen ions in response to changes in solution pH. A second result is that the surface acquires a net charge, which may be positive or negative depending on the pH and the affinity of the surface for hydrogen ions. This charge is neutralized by ions of opposite charge accumulating in solution near the surface. This structure of the oxide–solution interface – a net surface charge balanced by a swarm of ions in the solution near the surface – is called the electrical double layer. This effect is also discussed in Chapter 8.
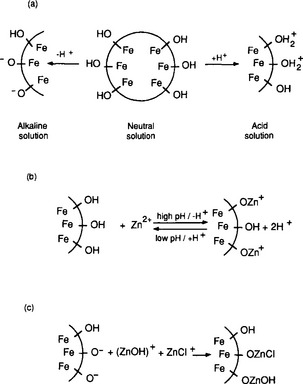
Fig. 15-4 Analogy between dissolved ligands and adsorbents (surface-bound ligands): (a) surface acid-base reactions; (b) surface complexation of free metals; (c) formation of “mixed-ligand” surface complexes.
Consider the situation of a metal ion, say Zn2+, dissolved in a solution containing FeOOH. Just as in the cases described earlier, the zinc ion might be present as a free aquo ion, or as a hydroxo- or other complex. However, if an iron oxide surface is available, another possibility is that the Zn might bind to a surface oxide site. The species that forms in such a case is analogous to a metal–ligand complex, with the surface serving as the ligand. The empirical evidence is that in many cases the most stable arrangement is for the Zn or other dissolved metal ion to form such complexes (Fig. 15-4b). As would be expected, since the reaction is effectively a competition between the metal and hydrogen ions to bind to the oxide, metal ion adsorption is enhanced at high pH and diminished at low pH (Fig. 15-5a). This conceptual model for adsorption is also consistent with the observation that the adsorptive bond strength of most metal ions onto oxide surfaces is strongly correlated with their ability to form hydroxo complexes in solution (Fig. 15-5b). That is, those metals that bind strongly to the oxygen atom of water molecules in solution also bond strongly to surface bound oxygen atoms.
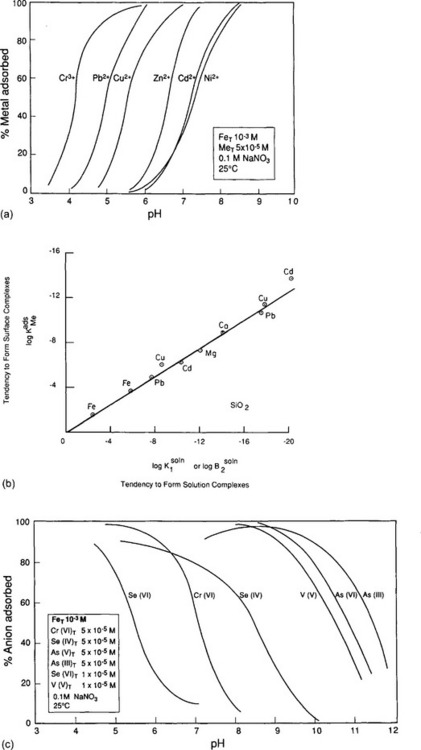
Fig. 15-5 Comparative adsorption of several metals onto amorphous iron oxyhydroxide systems containing 10−3 M Fet and 0.1 M NaNO3. (a) Effect of solution pH on sorption of uncomplexed metals. (b) Comparison of binding constants for formation of soluble Me-OH complexes and formation of surface Me-O-Si complexes; i.e. sorption onto SiO2 particles. (c) Effect of solution pH on sorption of oxyanionic metals. (Figures (a), (c) reprinted with permission from Manzione, M. A. and Merrill, D. T. (1989). “Trace Metal Removal by Iron Coprecipitation: Field Evaluation,” EPRI report GS-6438, Electric Power Research Institute, California. Figure (b) reprinted with permission from Balistrieri, L. et al. (1981). Scavenging residence times of trace metals and surface chemistry of sinking particles in the deep ocean, Deep-Sea Res. 28A: 101–121, Pergamon Press.)
As with the complexation reactions described earlier involving only dissolved species, the electrostatic attraction between a negatively charged surface and a positively charged metal ion can enhance the attraction of these reactants for one another, but much of the driving force for the reaction is provided by the formation of a chemical bond between the surface and the metal. Because of this, many metals can bind even to a positively charged surface, and in other cases can cause the surface charge to change sign from negative to positive as a result of their adsorption.
Dissolved metal complexes such as MeClxn-x or Me(HCO3)yn-yare also potential adsorbing species and can form the analog of mixed-ligand complexes at the surface (Fig. 15-4c). In most cases these simple inorganic complexes tend to adsorb somewhat less strongly than aquo or hydroxo complexes. This may be partially accounted for simply by statistical factors: when one of the metal’s waters of hydration is replaced by another ligand, there are fewer waters of hydration that can be exchanged for the surface oxide ligand. Also, the bond strength of the complex-to-surface bond may be less than the aquo metal-to-surface bond because the ligand satisfies some of the bonding requirements of the metal ion. In any case, complexation by simple inorganic ligands generally acts to retain metal ions in solution at the expense of metal ion adsorption. Formation of Cd–Cl complexes, for instance, has been cited to explain the desorption of Cd from suspended matter as it passes through an estuary (Paulson et al., 1984).
The effects on metal ion adsorption of ligands that can themselves adsorb strongly can be quite different from that described above. Many multi-atomic ligands can bond to oxide surfaces through atoms different from those they use to bind to metal ions. For example, the sulfidic sulfur of thiosulfate (—S—S—O32−) is thought to bind to polarizable metal ions such as Ag+ or Cd2+, while the sulfate group is more likely to bond to an oxide surface site. In such a case, the ligand may simultaneously bind to both the surface and the metal, thereby acting as a bridge between them. To explain the interactions in such a system, it is necessary to discuss ligand adsorption briefly.
The adsorption of anions, including metalloids such as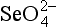 ,
,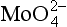 , and
, and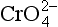 , anionic ligands such as
, anionic ligands such as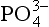 and
and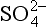 , and fulvic acids, is similar to that of cationic metals except that it usually has the inverse dependence on solution pH. That is, the sorption reaction involves competition with OH− ions for the surface, and ligand adsorption is therefore strong at low pH, where competition is weak (Fig. 15-5c). The complete set of equilibria describing the interactions in a system containing dissolved metal and ligand molecules and a solid that can adsorb either of these is complicated, but conceptually it involves a balance among all the relatively simple individual reactions discussed thus far. These interactions are summarized schematically in Fig. 15-5c. Low pH favors adsorption of the free ligand and ligand-bridged metals. High pH favors adsorption of free metal ions and metal-bridged ligands. The complexation of the metal and ligand in solution may or may not be a function of pH. The net interaction of all these factors can lead to solutions where the adsorption of metal ions is enhanced or diminished compared to that in ligand-free systems. Interesting examples of some of these interactions have been provided by Davis and co-workers, using both synthetic ligands and natural organic matter as the complexing agents (Fig. 15-6).
, and fulvic acids, is similar to that of cationic metals except that it usually has the inverse dependence on solution pH. That is, the sorption reaction involves competition with OH− ions for the surface, and ligand adsorption is therefore strong at low pH, where competition is weak (Fig. 15-5c). The complete set of equilibria describing the interactions in a system containing dissolved metal and ligand molecules and a solid that can adsorb either of these is complicated, but conceptually it involves a balance among all the relatively simple individual reactions discussed thus far. These interactions are summarized schematically in Fig. 15-5c. Low pH favors adsorption of the free ligand and ligand-bridged metals. High pH favors adsorption of free metal ions and metal-bridged ligands. The complexation of the metal and ligand in solution may or may not be a function of pH. The net interaction of all these factors can lead to solutions where the adsorption of metal ions is enhanced or diminished compared to that in ligand-free systems. Interesting examples of some of these interactions have been provided by Davis and co-workers, using both synthetic ligands and natural organic matter as the complexing agents (Fig. 15-6).
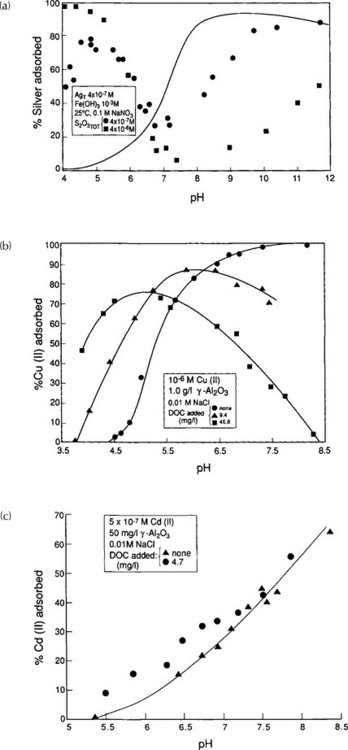
Fig. 15-6 Adsorbing, complexing ligands can enhance sorption of metals at low pH and interfere with it at high pH. This is shown (a) in a strictly inorganic solution (line shows adsorption curve with ligands absent) and (b) in a solution with natural organic matter as the complexing agent. (c) Metals that do not form strong complexes with the ligand are unaffected by its presence. (Figure (a) modified with permission from Davis, J. A. and Leckie, J. O. (1978). Effect of adsorbed complexing ligands on trace metal uptake by hydrous oxides, Environ. Sci. Technol. 12, 1309–1315, American Chemical Society. Figures (b), (c) reprinted with permission from Davis, J. A. (1984). Complexation of trace metals by adsorbed natural organic matter, Geochim. Cosmochim. Acta 48, 605–615, Pergamon Press.)
In addition to the interactions discussed above, which all depend in part on the ionizability, or at least polarizability, of the surface and the adsorbates, hydrophobic parts of ligands may bind to corresponding parts of surfaces. Thus, if a metal ion is complexed or irreversibly bonded to a hydrophobic molecule, the metal may be incorporated into the bulk or surface of a particle via hydrophobic interaction between the molecule and the solid phase. Such interactions may be quantitatively significant in systems with high concentrations of dissolved and particulate organic matter.
The fraction of the total metal bound to particulate matter in an aquatic system can be large, at times dominating the fraction in bulk solution. Davies-Colley et al. (1984) investigated the distribution of Cd among various model sediment components and concluded that for “typical” oxidized estuarine sediments, most of the trace metal will be bound to hydrous iron oxides and organic matter, with the organic-bound fraction of Cu being greater than that of Cd. Manganese oxides and clays contribute negligibly to the total binding capacity for both metals, according to their work.
Another approach to assess the partitioning of metals among the phases comprising natural particulate matter is to sequentially and selectively extract or dissolve portions of natural particulate matter. Based on the release of trace metals accompanying each step, associations between the trace metal and the extracted phase are inferred. Both of the above approaches have drawbacks, and at this time it is impossible to predict in advance how and to what extent metals and particulate matter will bond to one another in a natural system. Despite the uncertainties, empirical results can often be interpreted using the framework provided here, offering insights into the behavior of metals in complex systems and ultimately leading to improved predictive capability. Experimental measurements of the overall fractionation of metals between particulate and soluble phases in a few aquatic systems are presented in Fig. 15-7.
Fig. 15-7 Relationship between dissolved and total heavy metal concentrations in several rivers. Cross-hatched bands represent range of values from the Ruhr (Imhoff et al., 1980); W and F represent winter and fall values at a selected station in the Mississippi (Eisenreich et al., 1980); Triangles represent values from the Rhine river (Davis, 1984).
A related phenomenon is the adsorption of organic matter, metals, and particulate matter at another interface – that between the bulk water and air. At the air-water boundary, a microlayer of approximately 50 µm thickness is established with physical-chemical properties different from those of the bulk water. These properties cause metals, particles, microorganisms, and dissolved organic matter to accumulate in concentrations 5 to 1000 times greater than in bulk solution (Hardy et al., 1985). While this layer does not contain a significant fraction of the total metal in the aquatic system, it comprises much of the material that is transferred to the atmosphere as spray and represents an important region through which substances must pass to enter the bulk water from the atmosphere. During the several hours that particles are thought to spend in this enriched environment, important dissolution, complexation, photochemical, and biological reactions may take place.
15.3.7 Reactions Involving Organisms
Metals play an essential role in many enzyme systems, and virtually all metals can be toxic at concentrations that exceed the levels at which they are required or are normally found in the environment. Microorganisms play a central role in converting inorganically and organically bound metals to other chemical forms and transporting metals among various compartments of aquatic ecosystems as adsorbed or absorbed species. The effects of metals on biological systems is an established field of study in itself, while the importance of biological reactions in local or global metal cycling has only recently begun to become clear. An overview of the current understanding in these areas is provided in this section, focusing once again on the importance of metal speciation to the understanding of the system.
15.3.7.1 Effects of organisms on metal speciation and cycling
The biological contribution to metal cycling has been mostly studied in metal systems for which biological reactions contribute significantly to the total global flux, or for which biological transformations have significant implications in humans. Mercury and arsenic fall in this category. The biological cycles of these metals were first described clearly by Wood (1974). He emphasized that they, and perhaps many other metals, have natural biological cycles even though the metals themselves are not biologically essential. Biological conversions of metals may be evolutionary responses that microorganisms have developed as detoxification mechanisms. However, at times these “detoxification” reactions in one group of organisms actually intensify the toxicity of the metal to higher organisms. This is the case with of mercury and arsenic, which are both methylated by lower organisms.
In the case of mercury (Fig. 15-8), Wood suggested that reduction of Hg2+ to Hg0 and alkylation to form methyl- or dimethylmercury can both be viewed as detoxification reactions, because all the products are volatile and can be lost from the aqueous phase. Organisms can also convert the methylated forms to Hg¯, which is more volatile and less toxic. However, both the methylated and the reduced forms are more toxic to humans and other mammals than is Hg2+.
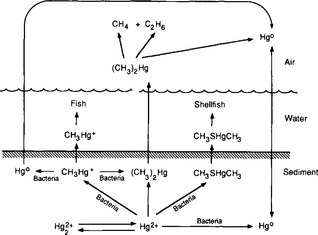
Fig. 15-8 The mercury cycle, demonstrating the bioaccumulation of mercury in fish and shellfish. Reprinted with permission from “An Assessment of Mercury in the Environment” (1978) by the National Academy of Sciences, National Academy Press, Washington, DC.
The arsenic cycle in ocean waters and sediments also has important biological steps (Andreae, 1979). Arsenate (As(V)) can be biologically converted into arsenite (As(III)) and at least eight different organo-arsenic compounds, all presumably representing detoxification processes mediated by bacteria in reduced sediments or by algae in the water column. On the other hand, biologically catalyzed demethylation and perhaps oxidation of arsenite serve to return arsenate to the water. In the bulk water below 400 m, one study found the ratio of arsenite to arsenate to be at least 12 orders of magnitude greater than that expected from equilibrium calculations, indicating the importance of biological reactions, since the kinetics of the abiological reactions controlling this equilibrium must be slow.
Even if microorganisms do not mediate a reaction with the metal directly, e.g., by changing its oxidation state or forming organometallic compounds, they still may play an important role in the cycling of many metals. For instance, Wood pointed out that bacteria can facilitate the mobilization of mercury from mineral deposits by oxidizing sulfide and thereby allowing mercury which had been sequestered in the extremely insoluble solid cinnabar (HgS) to dissolve. The same mechanism can be important in release of many other metal sulfides. Additionally, recent evidence has shown that the cycling of several metals in the open ocean is closely tied to the cycling of particulate organic matter, i.e., microorganisms (Jones and Murray, 1984). Figure 15-9 shows correlations of cadmium and phosphate in the ocean off the coast of California. Like phosphate, Cd is depleted in the surface waters due to biological uptake and released to the aqueous phase in deeper water as the sinking organic solids decay. It is not yet known whether the metal uptake is active and carried out only by living organisms, or is passive and involves adsorption on dead cells as well. Similar correlations have been reported for Ni, Zn, and Cu.
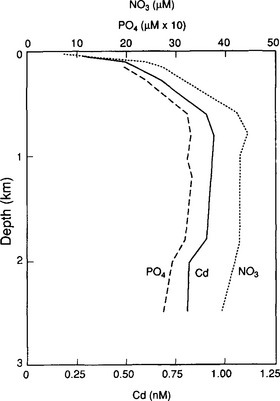
Fig. 15-9 Depth profiles for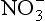 , PO43−, and Cd observed at station 64 off the coast of California in April 1977. (Reprinted with permission from Bruland, K. W. et al. (1978). Cadmium in Northeast Pacific waters, Limnol. Oceanogr. 23, 618–625, Society for Limnology and Oceanography.)
, PO43−, and Cd observed at station 64 off the coast of California in April 1977. (Reprinted with permission from Bruland, K. W. et al. (1978). Cadmium in Northeast Pacific waters, Limnol. Oceanogr. 23, 618–625, Society for Limnology and Oceanography.)
The range of processes that must be considered in the cycle of metals is described in Fig. 15-10 (Nelson et al., 1977). Both the complexity of metal cycle analysis in a real system and the importance of speciation are well-stated by Andreae (1979) in his overview of the arsenic cycle in seawater:
Fig. 15-10 Summary of reactions and processes important in metal biogeochemical cycling. (after Nelson et al., 1977).
The biological cycle of arsenic in the surface ocean involves the uptake of arsenate by plankton, the conversion of arsenate to a number of as yet unidentified organic compounds, and the release of arsenite and methylated species into the seawater. Biological demethylation of the methylarsenicals and the oxidation of arsenite by as yet unknown mechanisms serve to regenerate arsenate. The concentrations of the arsenic species are then controlled primarily by the relative rates of biologically mediated reactions, superimposed on processes of physical transport and mixing. The presence of arsenite in the deep ocean at concentrations far from thermodynamic equilibrium further emphasizes the importance of kinetic restraints on redox equilibration in the ocean.
15.3.7.2 Effects of metals on organisms: Toxicity and bioavailability
Recognition of the importance of metal speciation has had an enormous impact in the area of toxicity studies. Before this recognition, data were often difficult to interpret even from a single study, let alone from a range of investigations each with slightly different conditions. Recent analysis of speciation has led to a remarkably consistent conclusion: for all metals that have been studied (Cu, Cd, and Zn have been investigated the most) the toxicity of metals to algae, various aquatic invertebrates, and fish is strongly related to the activity of the free metal ion, regardless of the concentration of total metal in solution. This means, for example, that a solution containing 1.0 mg/L total Cu and enough organic matter so that 99% of the copper is strongly complexed, will be less toxic than an otherwise comparable solution with only 0.05 mg/L total dissolved Cu and little or no complexing organic matter. Other investigations have reached the same conclusion with respect to metal ion availability as an essential nutrient.
Strongly complexed metals, especially those complexed by chelating agents such as EDTA or by natural humic material, appear to be completely unavailable and non-toxic. A typical experiment showing such a result is summarized in Fig. 15-11. Weaker complexes formed with monodentate ligands such as chloride or carbonate also provide some protection against toxicity, although it is not yet clear whether these complexes are as innocuous as the chelates. Of course, the overall effect of potentially toxic metals depends not only on metal speciation but on all aspects of solution chemistry and on the identity and previous exposure history of the test organism. For example, one of the unresolved issues in this field has to do with the effects of water hardness (conferred primarily by dissolved Ca and Mg ions) and alkalinity on metal toxicity. In some studies these variables are negatively correlated with toxicity, and in others they seem to have no effect. Undoubtedly the results depend partially on the ability of the hardness ions to compete with the toxic ones for specific binding sites and on other responses of the organism to changes in the concentrations of these ions, quite apart from the response to the toxin. Regardless of the ultimate resolution of the remaining issues, it is clear that consideration of speciation has been the key step in rationalizing studies of toxicity and bioavailability in recent years and will be a crucial part of any such future studies.
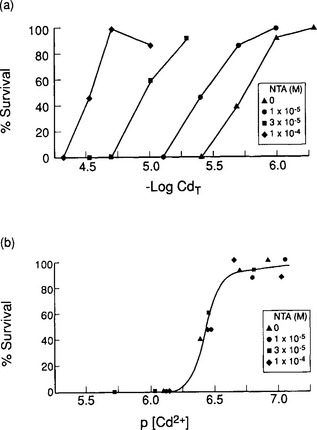
Fig. 15-11 Effects of strong complexation on metal ion toxicity. (a) Increasing concentration of NTA, a strong multi-dentate complexing agent, decreases the toxicity of Cd to grass shrimp. All systems have equal concentrations of total Cd. (b) When the results are replotted showing survival as a function of Cd2+ concentration, the data for all concentrations of NTA collapse to a single curve. (Reprinted with permission from W. G. Sunda et al. (1978). Effect of chemical speciation on toxicity of cadmium to grass shrimp, Palaemonetes pugio: importance of free cadmium ions, Environ. Sci. Technol. 12, 409–413, American Chemical Society.)
15.3.7.3 Toxic sediments
The widespread use of many metals such as silver, cadmium, copper, mercury, nickel, lead, and zinc has resulted in their accumulation in the environment. Sediments are often the repositories of toxic metals (e.g., Table 15-2). For example, copper is used as an anti-biofouling agent in marine paints and many harbor sediments contain markedly elevated levels of copper.
A considerable number of studies has demonstrated that the adverse biological effects of dissolved metals is largely regulated by the activity (i.e., “effective” concentration) of free metal ions (see Section 15.3.7.2), although some reports indicate that other metal species or variables may, at times, be important (e.g., Erickson et al., 1996). One challenge facing environmental scientists is the development of methodologies for assessing the toxicity of sediments contaminated with toxic metals.
As is the case with assessments of the toxicity of dissolved trace metals, the development of sediment quality criteria (SQC) must be based on the fraction of sediment-associated metal that is bioavailable. Bulk sediments consist of a variety of phases including sediment solids in the silt and clay size fractions, and sediment pore water. Swartz et al. (1985) demonstrated that the bioavailable fraction of cadmium in sediments is correlated with interstitial water cadmium concentrations. More recent work (e.g., Di Toro et al., 1990; Allen et al., 1993; Hansen et al., 1996; Ankley et al., 1996, and references therein) has demonstrated that the interstitial water concentrations of a suite of trace metals is regulated by an extractable fraction of iron sulfides.
The role of iron sulfides in regulating sediment interstitial water concentrations is shown by the following reactions (Di Toro et al., 1990):
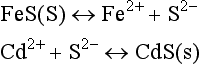
where ’(s)’ indicates that the substance is a solid. Anoxic sediments contain a large reservoir of (amorphous) iron sulfides. The first reaction represents the dissociation of iron sulfide; cadmium (the second reaction) associates with sulfide to form the insoluble cadmium sulfide. The overall reaction is
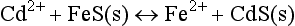
which is strongly distributed to the products’ side (the right-hand side) of the equation. The molar ratio of Cd2+ to FeS is 1:1; consequently, Cd will be essentially unavailable in the free aquo form (i.e., Cd2+) until the FeS has been exhausted. Only if the ratio of Cd to FeS (the acid volatile sulfide) is greater than 1 will the Cd2+ be potentially bioavailable in substantial amounts.
Figure 15-12 is a schematic illustration of a technique known as acid volatile sulfides/simultaneously extracted metals analysis (AVS/SEM). Briefly, a strong acid is added to a sediment sample to release the sediment-associated sulfides, acid volatile sulfides, which are analyzed by a cold-acid purge-and-trap technique (e.g., Allen et al., 1993). The assumption shown in Fig. 15-12 is that the sulfides are present in the sediments in the form of either FeS or MeS (a metal sulfide). In a parallel analysis, metals simultaneously released with the sulfides (the simultaneously extracted metals) are also quantified, for example, by graphite furnace atomic absorption spectrometry. Metals released during the acid attack are considered to be associated with the phases operationally defined as “exchangeable,” “carbonate,” “Fe and Mn oxides,” “FeS,” and “MeS.”
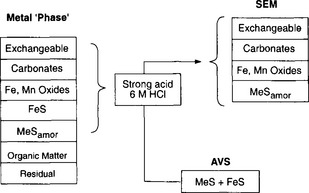
Fig. 15-12 Schematic illustration of the acid volatile sulfides (AVS)/simultaneously extractable metal (SEM) analysis. Refer to the text for details.
Sediment toxicity is evaluated by equilibrating 200 ml of the target sediment with 600 ml of water (e.g., saltwater for estuarine sites). Lethality tests are run for a specific period of time (e.g., 10 days) after which the indicator organism is counted for mortality. Figure 15-13 shows generalized results for a hypothetical suite of sediments. Percent mortality of the indicator organism is plotted as a function of the SEM/AVS ratio. Significant organism mortality (≥ 24%) appears at SEM/AVS molar ratios greater than 1 as anticipated from the discussion on metal sulfides given above.
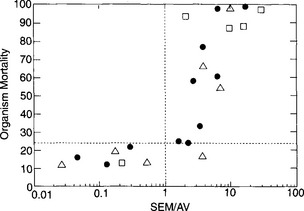
Fig. 15-13 Organism mortality as a function of SEM/AVS ratio for a hypothetical sediment. This figure is generalized from results typical of sediment toxicity tests (e.g., Hansen et al., 1996). Organisms evaluated in such tests include amphipods and polychaetes. The symbols represent different sediments. The vertical line at 100 is positioned at an SEM/AVS ratio of 1.0; the horizontal line at 24% represents the limit of toxicity, that is, mortality ≤24% is defined as not the consequence of toxicity.
15.3.8 Geochemical Kinetics
To this point, we have emphasized that the cycle of mobilization, transport, and redeposition involves changes in the physical state and chemical form of the elements, and that the ultimate distribution of an element among different chemical species can be described by thermochemical equilibrium data. Equilibrium calculations describe the potential for change between two end states, and only in certain cases can they provide information about rates (Hoffman, 1981). In analyzing and modeling a geochemical system, a decision must be made as to whether an equilibrium or non-equilibrium model is appropriate. The choice depends on the time scales involved, and specifically on the ratio of the rate of the relevant chemical transition to the rate of the dominant physical process within the physical-chemical system.
Comparisons of time scales for various physical processes have been discussed by Lerman (1979) and Schwartzenbach and Imboden (1983). In general, physical processes in lakes have characteristic times in the range of 10−4 to 102 years. Oceanic processes span times from days to thousands of years (Broecker, 1974) and geologic events may occur catastrophically (e.g., earthquakes or landslides exposing new weathering surfaces) or very slowly, as in the case of continental subduction. If the characteristic chemical reaction times (τchem) are short compared to the characteristic times of the dominant physical processes (τphys) then it is probably appropriate to consider the chemical transition in terms of equilibrium concepts. If τchem is large compared to τphys then reactions will proceed only slightly, if at all, from their initial conditions. When τchem is of the same order as τphys then quasi-equilibrium or kinetic descriptions may be employed (Morgan and Stone, 1985; Morel, 1983; Keck, 1978). Thus, a description of the chemical transitions occurring in each reservoir of a biogeochemical system must consider the relative physical and chemical time scales characteristic of that reservoir. For example, the hydrolysis of Fe(III) has a characteristic time (τchem) of 3.2 × 10−7 seconds (Hemmes et al., 1971). Since most natural physical processes are much slower than this rate, the Fe(III) hydrolysis reaction can be considered to be at equilibrium in those systems. In contrast, the dissolution of silica proceeds at a much slower rate, with a characteristic time of approximately 8 years. The oxygenation of Mn(II) is more ambiguous. It has characteristic times ranging from weeks to tens of years depending on whether the reaction is homogeneous, mediated by bacteria, or catalyzed by the presence of metal-oxide surfaces (Morgan and Stone, 1985).
The suite of chemical reactions taking place in a geochemical compartment, such as the atmosphere or a lake, are often quite complex, involving higher-order reaction kinetics and multiple, linked reactions (e.g., Pankow and Morgan, 1981). Nevertheless, the principle of comparing the relative time scales of physical versus chemical process is still valid. What is needed is an understanding of the rate controlling reaction(s) and the dominant physical processes. A variety of chemical reactions and their characteristic times are shown in Fig. 15-14. Also shown on the figure are the major physical processes extant in lakes and the range of their characteristic times. As described in Chapter 4, the characteristic time is the time required for the transformation to proceed to within the fraction 1/e of completion. This is also known as the system response time (Lasaga, 1980). The characteristic times of the transformations shown in Fig. 15-14 range from approximately 1 h to half a year. Of the chemical reactions shown, a significant number have characteristic times of the order of the predominant physical processes, suggesting that non-equilibrium approaches to the geochemical modeling need to be considered.
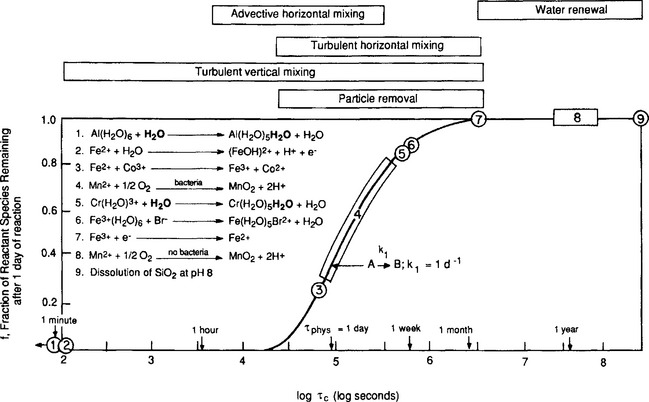
15.4 Observations on Metals in Natural Systems
15.4.1 Combining Physical and Chemical Information
All the factors mentioned in the previous sections play a role in the movement of metals through their overall biogeochemical cycle: injection into the atmosphere, deposition onto land or water surfaces, transport via rivers and estuaries to the oceans, and sedimentation and ultimate burial in the sediments. The physical-chemical form in which each metal is transported in the aquatic phases of the cycle will depend on the specific metal and its interactions with other dissolved and suspended constituents in accord with the principles discussed above. However, it is important to keep in mind that the physical processes of fluid flow and sediment transport must be combined with the chemical reactions to gain insight into the functioning of the complete system. This point has been well-stated by Turekian (1977) in a discussion of metal concentrations in the ocean:
Why are the oceans so depleted in these trace metals? Certainly it is not for the lack of availability from rock weathering or because of constraints imposed by the solubility of any unique compound of these elements. The reason must lie in the dynamics of the system of delivery of the metals to the oceans and their subsequent behavior in an ocean that cannot be simulated by simple in vitro experiments involving homogeneous reaction kinetics.
In this section, an overview of the net transport of metals through rivers, estuaries, and the oceans provides an example of how these chemical and physical forces interact.
Recent studies have reported that from 10% to more than 90% of the trace metal load of streams is carried in the particulate fraction, with the fraction of particle-bound Fe, Pb, Cu, As, and Zn typically being greater than that of Cd, Co, and Ni. As noted earlier, the exact balance will depend on the concentration of inorganic and organic complexing ligands, the type and quantity of particulate matter available, and the concentrations of other ions competing for the binding sites. In interpreting these data one must bear in mind that the fraction defined as being “particulate” does not represent an absolute measure; rather, it is defined by the separation technique employed. Any material that will not pass a 0.45 µm filter is often defined as particulate. However, a significant fraction of the “soluble” trace metals, Fe and Mn, and organic material that does pass the filter can subsequently be filtered out using filters with smaller pores. Furthermore, much of the 0.45 µm filterable material is converted to non-filterable matter (still using 0.45 µm filters) when the concentrations of Ca2+, Mg2+, and Na+ increase to levels they attain in estuaries. This process and its implications merit some special attention.
15.4.2 Particle and Metal Interactions in Estuaries
Estuaries exhibit physical and chemical characteristics that are distinct from oceans or lakes. In estuaries, water renewal times are rapid (10−3 to 10−4 years compared to 1 to 10 years for lakes and 104 years for oceans), redox and salinity gradients are often transient, and diurnal variations in nutrient concentrations can be significant. The biological productivity of estuaries is high and this, coupled with accumulation of organic debris within estuary boundaries, often produces anoxic conditions at the sediment-water interface. Thus, in contrast to the relatively constant chemical composition of the oceans, the chemical environment in an estuary varies dramatically in time and space.
An estuary can be defined as a system in which ocean waters have been diluted by freshwater from land drainage. Many ions are more abundant in coastal or oceanic waters than river water, e.g., Na, K, Mg, Ca, Cl,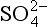 , and
, and . However, nutrients (N, P, and Si) and many transition series metals, including Fe, are more concentrated in river water. Dissolved and particulate organic matter (DOM and POM) are one to two orders of magnitude more concentrated in rivers than in ocean water. As end-member waters mix, dissolved constituents originally in each of the end-member waters may remain in solution during mixing (i.e., behave conservatively), interact with sinking particulate matter, or precipitate out of solution. The wide variety of mixing regimes and end-member waters makes it difficult to generalize about the estuarine behavior of many metals of interest to environmental geochemists. Generally, however, the major components of seawater – Na, Ca, K, and SO4 – behave conservatively. Some elements, such as zinc, may behave conservatively in some estuaries but be rapidly removed in others. Iron, by contrast, is removed rapidly and efficiently in nearly all estuarine environments, i.e., its behavior is highly non-conservative.
. However, nutrients (N, P, and Si) and many transition series metals, including Fe, are more concentrated in river water. Dissolved and particulate organic matter (DOM and POM) are one to two orders of magnitude more concentrated in rivers than in ocean water. As end-member waters mix, dissolved constituents originally in each of the end-member waters may remain in solution during mixing (i.e., behave conservatively), interact with sinking particulate matter, or precipitate out of solution. The wide variety of mixing regimes and end-member waters makes it difficult to generalize about the estuarine behavior of many metals of interest to environmental geochemists. Generally, however, the major components of seawater – Na, Ca, K, and SO4 – behave conservatively. Some elements, such as zinc, may behave conservatively in some estuaries but be rapidly removed in others. Iron, by contrast, is removed rapidly and efficiently in nearly all estuarine environments, i.e., its behavior is highly non-conservative.
As river water (Fig. 15-15a) high in dissolved and particulate iron and other inorganics (PIM) and organic matter enters an estuarine mixing zone (Fig. 15-15b), rapid flocculation of the DOM and coagulation of particulate matter occurs (Fig. 15-15c). The extent to which this occurs is a function of the salinity of the water mixture: flocculation and coagulation generally increase with increasing salinity up to a salinity of about 15‰, with greater salinities having little added effect. While systems containing only colloidal iron oxides have been shown to coagulate as salinity increases, the presence of organic matter increases the coagulation rate and leads to the formation of Fe oxide-organic aggregates. The efficiency of metal removal by the flocculation process generally follows the relative order of trace metal-DOM complex stabilities: Fe3+ > Al3+ > Cu2+ ∼ Ni2+, etc. Reactions that may initiate the iron-organic removal process include coagulation of pre-existing colloidal particles or precipitation of previously soluble humic acids, and adsorption of trace metals onto particles before or after either of the above processes.
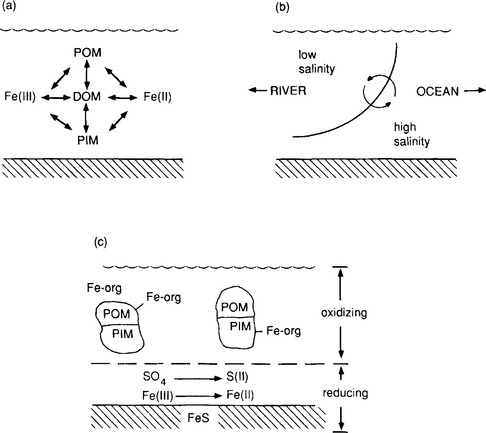
Fig. 15-15 Schematic representation of Fe and organic matter interactions in an estuary. POM = particulate organic matter; DOM = dissolved organic matter; PIM = particulate inorganic matter.
The efficient removal of iron in estuaries allows very little of the initial river-borne iron to escape the estuary to the coastal waters. “Soluble” iron concentrations are extremely low in river water and in estuaries. Nearly all of the iron entering the estuary is transported in particulate form to sediments on the estuary floor where, due to the high accumulation of organic matter, reducing conditions are often met. Fe(III) is reduced in anaerobic sediments and, since S is often also accumulating in the sediments, Fe(II) may be precipitated as iron sulfides. Resuspension of sediments may reintroduce iron to the oxygenated layer of the estuary where it can be rapidly oxidized, forming new colloidal particles, which can sorb other metals, associate with suspended organic matter and return to the sediments in repeated episodes of internal cycling.
The ultimate result is that not only Fe, but most metals that interact strongly with organic matter or oxide adsorbents, are likely to settle out of the water column in estuaries. Not only does this process reduce the metal flux reaching the ocean, but 50% of the time the current at the bottom of an estuary actually facilitates the movement of metals upstream.
While Fe- and Mn-oxides certainly are important in the binding and transport of the trace metals in the estuarine system, organic matter appears to play a dominant role in the coagulation step. For instance, adjustment of the solution pH to values where humics normally precipitate (pH = 1 to 3) causes all the components (Fe and Mn, organics, and trace metals) to coagulate into filterable particles. By contrast, adjustment of the pH upward, which would normally favor precipitation of Fe- and Mn-oxides, has no effect. In view of this it is not surprising that the tendency of trace metals to be removed from estuarine waters corresponds roughly with their tendency to bind to organics, with Cu being the most strongly affected. In one study, for instance, approximately 40% of the dissolved Cu, 15% of the dissolved Cd and Ni, and less than 5% of the dissolved Co and Mn were filterable after coagulation was induced by addition of Ca and Mg to river water (Sholkovitz and Copland, 1981). As noted earlier, desorption of metals owing to the formation of soluble Me–Cl complexes may partially or completely counteract this process for some metals.
To summarize, even though some Fe- and Mn-oxides may dissolve in the anoxic sediments of estuaries and thereby release trace metals, re-oxidation of the Fe and Mn in the upper layers of the sediments, coagulation of Fe-Mn-humictrace metal mixtures in the water column, and circulation patterns within the estuary all combine to return trace metals to the upstream particulate fraction. Turekian has suggested that while these processes allow significant internal cycling of trace metals in the estuary, they prevent any more than a small fraction of the total trace metal load from reaching the ocean. In describing the role that soils and rocks play in attenuating the movement of metals from the land to water bodies and the scavenging of metals from the water column by particles in the ocean in combination with the estuarine processes, Turekian dubbed the overall process “the great particle conspiracy,” working to maintain soluble trace metal concentrations at extremely low and remarkably consistent levels throughout the world’s open oceans.
15.4.3 Application of Chemical Principles to Evaluate Field Data
Examples were given earlier of a few systems for which the theoretical speciation of metals was calculated, and a few others in which it was analyzed experimentally, at least into some subgroups if not specific species. These approaches have rarely been combined to critically evaluate data or hypotheses regarding metal transport through a real system. One study, already noted earlier in this chapter, can serve as a model for combining theoretical considerations with field data. In that study, Morel et al. (1975) used data for the partitioning of metals between the soluble and particulate fractions to estimate the redox potential of primary-treated sewage. Knowing the composition of the Southern California coastal waters into which the sewage was discharged, they were then able to interpret the relative changes in the concentrations of several metals as a function of dilution and oxidation. Combining chemical analyses with data for sedimentation rates of metals and organic carbon, they suggested that most metals are not mobilized in the nearfield deposition zone, and they offered plausible explanations for the deviation of Pb and Cd from this pattern. As the authors noted, extension of their results to other systems is not warranted, but extension of the methodology, i.e., combination of experimental data with chemical speciation models to test hypotheses and suggest profitable research directions, certainly is.
15.4.4 Acid Mine Drainage (AMD)
From the beginnings of ecology as a discipline, the mining industry has been at the center of the battle over preservation versus exploitation. As discussed in the introduction to this chapter, human activities such as mining, power production from fossil fuels and discharges of industrial and municipal wastes not only increase the rate at which metals enter the biosphere but may also drastically alter the speciation of metals from what it would be in the undisturbed geologic cycle.
A major consequence of the activities associated with the exploitation of mineral deposits (i.e., exploration, the development of mines and processing facilities, the extraction and concentration, which is also called beneficiation, of ores containing the desired minerals, and the decommissioning or abandonment of mine facilities) is the production of extremely large volumes of unwanted materials. Waste volumes vary from ca. 30% of the mass of the ore in the case of gypsum and other non-metals, to about 50% for base metals to more than 80% for strip-mined coal (see Ripley et al., 1996, and references therein).
In a number of milling and mining operations (e.g., metal mines containing sulfide minerals either in the ore or surrounding rock, or coal or lignite deposits) the oxidation of sulfides, particularly iron pyrite, is of primary concern for the contamination of water. The oxidation process, which generates acid drainage from the deposit, occurs under natural conditions but is greatly accelerated as the consequence of mining activities. In this case it is then referred to as acid mine drainage (AMD).
The existence of acid drainage has been recognized for some time:
The spring water issuing through fissures in the hills, which are only masses of coal, is so impregnated with bituminous and sulphurous particles as to be frequently nauseous to the taste and prejudicial to the health. (T. M. Morris, 1803, in MacKenthum, 1969)
However, the seriousness of the problem, and its underlying causes, have only relatively recently been addressed by the mining industry (Ripley et al., 1996). As a consequence, many mining operations conducted before the 1980s were not managed to control the deleterious effects of acid drainage.
Some of the basic processes that contribute to the generation of acid drainage are discussed in earlier portions of this section (e.g., the oxidation of Fe(II) to Fe(III)). Although, as Langmuir (1997) points out, the presence of iron pyrite in coal or metal deposits does not “predetermine” that mining operations will generate significant amounts of AMD, the amount and nature of the pyrite present is an important factor in the generation of AMD.
Basically, the oxidation of iron pyrite, FeS2, results in the production of iron(III) sulfate and sulfuric acid, H2SO4. However, two overall reaction stoichiometries are possible and each will yield a different acid generation capacity (e.g., Langmuir, 1997; Baird, 1995):
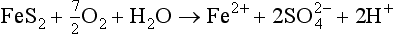
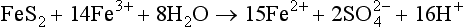
In both reactions, Fe2+ is produced as a consequence of the oxidation of sulfur from the – 1 oxidation state of pyritic sulfur to the +6 state of sulfate.
A second oxidation step is the oxidation of Fe2+ to Fe3:
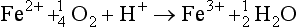
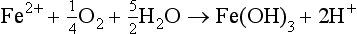
The abiotic rate of the first oxidation reaction is slow; the rate of the second reaction increases with increasing pH. The second iron oxidation reaction produces Fe(OH)3(s), ferric hydroxide. “Yellow boy,” a limonitic precipitate, is produced when the ferric hydroxide mixes with ferric sulfates; when formed, “Yellow boy” gives receiving waters an unappealing yellow tint.
The rates of oxidation are significantly increased in the presence iron- or sulfur-oxidizing bacteria. Iron-oxidizing bacteria, which are aerobic, include Gallionella and Sphaerotilus, which are autotrophs (i.e., obtaining carbon for synthesis of new biomolecules from carbonate species) and heterotrophs (require organic carbon), respectively. Sulfur-oxidizing bacteria include Thiobacillus thiooxidans and Ferrobacillus ferrooxidans; all such bacteria are aerobic and autotrophic.
15.5 Examples of Global Metal Cycling
In this final section, the global cycles of two metals, mercury and copper, are reviewed. These metals were chosen because their geochemical cycles have been studied extensively, and their chemical reactions exemplify the full gamut of reactions described earlier. In addition, the chemical forms of the two metals are sufficiently different from one another that they behave differently with respect to dominant transport modes and pose different risks to organisms.
15.5.1 Mercury
Mercury occupies a unique (and infamous) place in environmental history because it was the first chemical for which a direct connection was proven between relatively low concentrations in a natural water system, bioaccumulation up the food chain, and a serious health impact on a human population at the top of the food chain. Within a relatively few years, such epidemics were documented at two sites in Japan, and somewhat less definitive evidence suggesting mercury poisoning was reported in Canada. In addition, consumption of animals that had been fed grain seed contaminated by methylmercury fungicide led to acute poisoning of a family in the United States, and, in one of the worst cases of acute human exposure to an environmental hazard, over 6000 Iraqis were hospitalized with symptoms of mercury poisoning after consuming home-made bread made from seed wheat treated with this same type of fungicide. In the latter case, over 500 deaths were reported by hospitals, and it is likely that many other affected individuals did not report to the hospital.
15.5.1.1 Global mercury cycling
Unlike most heavy metals, the natural and anthropogenic cycles of mercury are dominated by atmospheric transport. Metallic mercury has the highest vapor pressure of any heavy metal, and it is released in geochemically significant quantities by volcanic activity, volatilization from land and ocean surfaces, and in high-temperature industrial processes such as smelting of minerals and burning of fossil fuels. Figure 15-16a,b provides an estimate of the natural and anthropogenic mercury cycles (Mason et al., 1994). The two parts demonstrate many salient points, among which are the following:
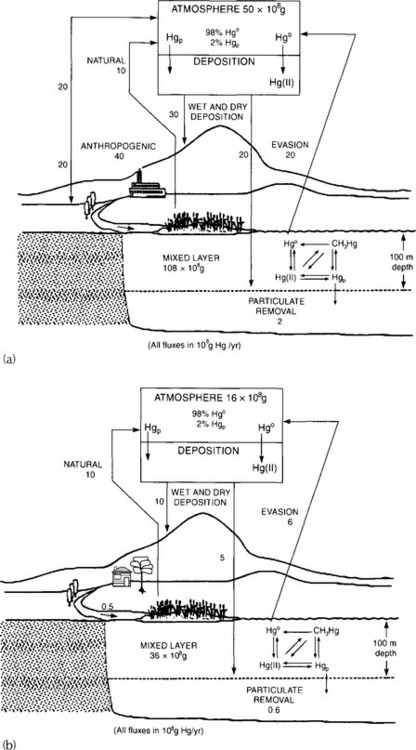
Fig. 15-16 The (a) present day and (b) pre-industrial global cycles for mercury. Units are 108 g Hg (burdens) and 108 g Hg/yr (fluxes). Hgp refers to mercury in particles. Redrawn from Mason et al., 1994.
1. Exchange rates of mercury between the atmosphere and the land and between the atmosphere and the ocean are much greater than transport from the land directly into the ocean via riverine discharge.
2. While the natural exchange of mercury between the land and atmosphere and the atmosphere and oceans is balanced, human activity has tipped this balance. There is now about three times more mercury in the atmosphere and fluxes of more than four times to and from the atmosphere.
3. The transport rate of mercury flowing from the land to the oceans in rivers has been increased by a factor of about three by human activity. While the increased rate is still relatively less important than the total transport of Hg through the atmosphere, it can represent a significant stress on the exposed organisms, particularly since the increased flux is unevenly distributed. That is, human activity has created local environments where the transport of mercury or its concentration in a river or estuary is many tens of times higher than background levels.
4. The average residence times for mercury in the atmosphere, terrestrial soils, oceans, and oceanic sediments are approximately 1 yr, 1000 yr, 3200 yr, and 2.5 × 108 yr, respectively. (See Bergan et al. (1999) for more details on atmospheric residence times.)
15.5.1.2 Environmentally important forms of mercury
There are several environmentally significant mercury species. In the lithosphere, mercury is present primarily in the +II oxidation state as the very insoluble mineral cinnabar (HgS), as a minor constituent in other sulfide ores, bound to the surfaces of other minerals such as oxides, or bound to organic matter. In soil, biological reduction apparently is primarily responsible for the formation of mercury metal, which can then be volatilized. Metallic mercury is also thought to be the primary form emitted in high-temperature industrial processes. The insolubility of cinnabar probably limits the direct mobilization of mercury where this mineral occurs, but oxidation of the sulfide in oxygenated water can allow mercury to become available and participate in other reactions, including bacterial transformations.
In aqueous solution, mercury forms strong complexes with both organic and inorganic ligands. In particular, mercury is strongly complexed by compounds containing reduced sulfur atoms or carboxyl groups, by hydroxide, and by chloride. As a result, much of the dissolved mercury in freshwater is associated with dissolved organic matter, most likely humic and fulvic acids. Even in extremely “pure” river water, containing few dissolved minerals or organic compounds, mercury exists not as the divalent free aquo metal ion, but as a neutral or anionic hydroxo complex, depending on the solution pH. In seawater, dissolved mercury exists primarily as chloro and possibly organic complexes and mixed ligand complexes of chloride, bromide, and hydroxide.
Dissolved mercury also reacts to form strong complexes with inorganic and probably organic particulate matter. Figure 15-17 shows the distribution of mercury between dissolved and adsorbed phases in an idealized system consisting of water, an artificially prepared oxide and various concentrations of chloride. The mercury is strongly sorbed in the absence of the chloride, but as more of this ligand is added to the system, the mercury remains in solution at pH values where it had previously sorbed. Chemically, the effect of the chloride is to convert the mercury from Hg–OH complexes to Hg–Cl or Hg–Cl–OH complexes. These complexes apparently do not sorb as strongly as the Cl-free mercury species. While the conditions in any natural water are more complex, it is clear that this type of interaction may have a significant effect on the form and fate of mercury under changing environmental conditions, for instance in water moving through an estuary.
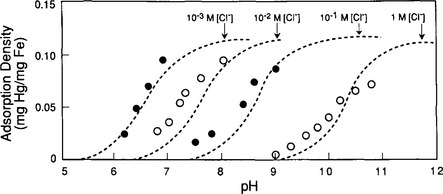
Fig. 15-17 The effect of chloride on adsorption of mercury by hydrous iron oxide at constant total mercury concentration of 3.4 × 10−5 M. The lines represent the predicted adsorption assuming that Hg–Cl complexes do not sorb at all. (Reprinted with permission from P. V. Avotins, “Adsorption and coprecipitation studies of mercury on hydrous iron oxides,” 1975, Ph.D. dissertation, Stanford University, Stanford, CA.)
Finally, in addition to being an unusual metal because of its high volatility, mercury is unusual because of the importance of biological reactions in altering its form. It has been pointed out that biological activity is critical in converting oxidized (+2) mercury to metallic (0) mercury in soils, allowing subsequent volatilization. Mercury also undergoes biomethylation, which is of overriding significance with respect to its toxicity. In this reaction, which takes place in the upper layers of fresh- and saltwater sediments, divalent mercury forms covalent bonds with organic methyl groups. The organo-mercury compounds are more volatile and more easily bioaccumulated than their inorganic precursors. They may sorb to organic matter in the sediment, but once released they are rapidly taken up by organisms and are concentrated up the food chain, eventually becoming available for human consumption. This is the path that led to the first recognized epidemic of mercury poisoning affecting humans, at Minamata Bay, Japan. The rate of biomethylation of mercury is proportional to the concentration of inorganic mercury in the system and the concentration of organisms capable of carrying out the reaction. Thus, it has been suggested that limiting the nutrient supply discharged to receiving waters may be an effective method of limiting biomethylation in contaminated areas. Biodemethylation has also been observed, and may be a microorganismal response to mercury intoxication.
15.5.1.3 Mercury movement in aquatic systems: two case studies
Although there have been numerous efforts to describe the global mercury flux, studies in which fluxes of the individual species in a well-defined natural system were evaluated are rare. Two studies that have been reported – one for a highly contaminated saltwater system, the other for a relatively less contaminated stretch of a river – provide an interesting synthesis of the above information. In the former case, the movement of mercury out of Minamata Bay over a period of about 25 years was estimated (Kudo and Miyahara, 1983). The results support a remarkably strong affinity between the discharged mercury and bottom sediments, so strong in fact that the majority of the mercury discharged to the Bay remained there, attached to bottom sediments, 25 years after all Hg discharges had ceased. This result is all the more surprising considering the rapid flushing rate of the Bay: the residence time of water in the Bay is only 2.5 days due to vigorous tidal action. Even so, it is estimated that between 1960 and 1975 the rate of mercury elimination from the Bay to the sea was only 0.4% per year, which could easily be accounted for by sediment transport. The rate of movement of mercury from the Bay to the sea increased by approximately a factor of 10 in the period 1975–78, which the authors of the report hypothesize was caused by increased sediment movement generated by an increase in the traffic of large ships in the Bay during this period. As supporting evidence for the speed and tenacity with which mercury binds to sediments in the Bay, the investigators point out that the sea sediments just outside of Minamata Bay were not contaminated by the initial mercury discharges, indicating that sorption or other processes immobilizing mercury in the Bay must have occurred very quickly, on a time scale of hours. Thus, whatever the immobilization process is, it appears to be rapid, efficient, and long-lasting.
A detailed study of mercury in a 3-mile (4.9 km) section of the Ottawa River (Kudo, 1983), represents one of the few comprehensive studies in which the transport of mercury in several physical and chemical forms was evaluated simultaneously. This stretch of river had received mercury as an industrial discharge for many years, a practice that was stopped 3 years prior to the start of the study. In addition some mercury is derived from natural weathering reactions in the river’s headwaters region. The vast majority of the total mercury in the test section (>96%) was associated with the bed sediments, and in conformity with the study of Minamata Bay described above, this mercury was not easily released to the solution phase. Thus, despite the relatively large reservoir of mercury in the sediments, slow bed sediment velocities limited the significance of sediment-mediated mercury transport. Although the water contained only 13 ng/L total dissolved Hg, it accounted for 58% of the total mercury flux through the system, while 41% could be attributed to suspended soil transport. Of the total mercury in the system, less than 6% was methylmercury. This mercury species was apparently less strongly sorbed to suspended sediments than was inorganic mercury, and as a result the aqueous phase accounted for 80% of its transport, with the remaining 20% moving with the suspended sediments. While the exact percentages would undoubtedly vary with such parameters as suspended sediment load, average water velocity, and sediment composition, these results are useful as a reference point with which other systems can be compared. This study is also representative of the type of analysis necessary for an evaluation of heavy metal movements in water systems and the risks they pose to aquatic and human populations.
Mercury provides an excellent example of the importance of metal speciation in understanding biogeochemical cycling and the impact of human activities on these cycles. Mercury exists in solid, aqueous, and gaseous phases, and is transported among reservoirs in all these forms. It undergoes precipitation-dissolution, volatilization, complexation, sorption, and biological reactions, all of which alter its mobility and its effect on exposed populations. The effect of all these reactions on the environmental behavior of mercury has been indicated in this section. The importance of analyzing speciation is perhaps most evident from the fact that the most toxic form of mercury and the only form that has been identified as having affected large populations of humans, methylmercury, is relatively insignificant in the global mercury balance. In the next section, a similar summary will be presented for copper. Despite significant differences from mercury with respect to certain chemical properties and environmental transport modes, it will once again be shown that an understanding of chemical speciation is critical to an appreciation of the metal’s environmental behavior.
15.5.2 Copper
The biogeochemical cycling of copper has been extensively studied. While some questions remain regarding the gross inventories of copper in various environmental reservoirs, studies dating from as early as the turn of the century provide considerable insight into the flux rates among the reservoirs. The speciation of copper in natural waters has also been well-studied, and in recent years a rough consensus seems to have been reached regarding the dominant copper species in “typical” environments, although the exact values of the stability constants of some important copper complexes are still somewhat uncertain.
Unlike mercury, copper is known to be a metabolically essential element for virtually all organisms. It displays a well-established property of many of the heavy metals, being essential to growth at low concentrations and toxic at high concentrations. The requirement for Cu stems from its inclusion in several proteins, in which it is always coordinated to N, S, or O ligands (Moore and Ramamoorthy, 1984). At the other extreme, copper’s toxic properties have been exploited to reduce growth of unwanted organisms such as algae and fungus in water bodies and in soils. Copper biogeochemistry has been discussed and reviewed frequently in recent years. In this section, only an outline of the literature will be provided, with emphasis on how the speciation of copper controls its behavior.
15.5.2.1 Global cycling
The global cycling of copper has been reviewed by Nriagu (1979) and is described schematically in Fig. 15-18. Like most heavy metals and in contrast to mercury, the flux of metal from terrestrial to oceanic reservoirs is dominated by transport in rivers. Copper reaching the oceans by atmospheric transport is of the same order of magnitude as that by three strictly anthropogenic sources: direct discharge of wastes and sludge into the oceans, discharge of domestic and industrial wastes into rivers which eventually discharge into the ocean, and the use of copper-containing anti-fouling paints. Each of these sources is several hundred times less than the copper burden carried to the oceans by riverine runoff as part of the natural biogeochemical cycle. The transport that does occur through the atmosphere is essentially all via particulate matter, not transport of gaseous copper species, and roughly 90% of this particulate matter is injected into the atmosphere as a result of smelting, fossil fuel burning and other human activities. Despite the relative insignificance of human inputs to the global cycle of copper, atmospheric transport is thought to be responsible for most of the transport to some inland water systems, e.g. the Great Lakes, and there is no question that in localized ecosystems anthropogenic inputs can dominate over natural ones.
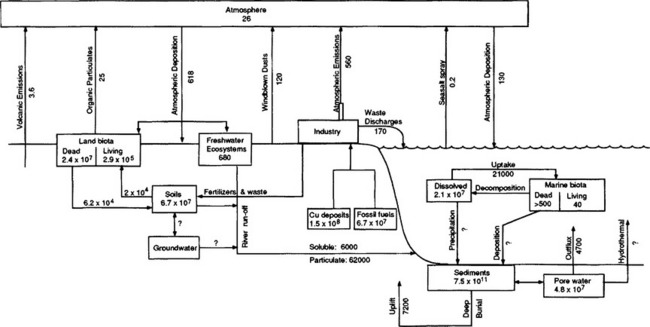
15.5.2.2 Copper in rocks, minerals, and atmospheric dust
Copper exists in crustal rocks at concentrations ranging from about 10 to a few hundred ppm, with 70 ppm being about average. In addition, at least 20 copper minerals have been identified, containing copper in the 0, +I, or +II oxidation state. These are primarily sulfides, hydroxides, and carbonates, of which chalcopyrite (CuFeS2), is most common. Copper is also found in relatively high concentrations in deep-sea ferromanganese nodules, in many cases at concentrations greater than 0.5% and occasionally greater than 1.0%.
Solids containing oxidized anions (carbonates, sulfates, hydroxides, and oxides) are the dominant forms of Cu in airborne particulate matter. In the few studies that have addressed the reactions of these particles in atmospheric washout, about 50% of the copper has been found to be soluble. Since the solubility is strongly dependent on pH, acid precipitation and acidification of receiving waters may have a significant effect on the form and fate of airborne copper.
15.5.2.3 Reactions of copper in aquatic systems
15.5.2.3.1 Redox reactions and complexation.
Copper in solution, as in solids, can carry either a +1 or +2 charge. The divalent form is the stable one in oxygenated water and is the prevalent form in natural waters. The distinction between the two forms is particularly significant to copper transport because they tend to form complexes with different ligands. Cuprous (Cu+) ions form strong bonds with Cl, which probably controls its speciation in marine systems, but these have been little studied since Cu(I) is rarely detected in such systems other than in anoxic sediments. By contrast, cupric ions (Cu2+) form strong complexes with hydroxide, carbonate, phosphate, and ammonia, and its complexes with organic molecules are typically the second strongest (after mercury) of any of the heavy metals. Even in waters with low concentrations of dissolved organic carbon, dissolved copper is associated primarily with organic matter, i.e., humic or fulvic acids. Turner et al. (1981) computed that in seawater with a total humic acid concentration of 10−6 M as carbon, organic complexes could account for 47% of the total dissolved copper, 2% of the lead, and less than 0.1% of the Mn, Co, Ni, Zn, Cd, or Hg. Metals other than Cu are prevented from complexing with the organics by a combination of competition for the ligand by Ca2+ and Mg2+ and complexation of the metals themselves by inorganic ligands.
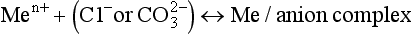
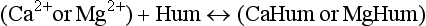
In seawater most of the non-organically bound Cu2+ is complexed with inorganic carbon. In freshwater, because both types of competition shown above are less significant than in seawater, and because humic and fulvic acid concentrations are greater than in seawater, all metals, including Cu2+, are more likely to exist as organic complexes. Thus, frequently gt;90% of the dissolved copper in rivers is reported to be organically bound. (It should be remembered that this may include significant quantities of suspended Cu–Fe–humate colloids.) The tendency to form such complexes would be even greater in waters contaminated by organic discharges.
Equilibrium complexation constants for Cu reactions with natural organic matter and the details of Cu speciation are bound to remain somewhat uncertain, since the composition of the complexing molecules varies from site to site. What is not in dispute is that the fraction of dissolved copper present as free aquo Cu2+ is probably very small in any natural water. In extremely pristine waters, hydroxide and carbonate complexes may dominate, but organic complexes usually dominate in waters containing more than a few tenths of a mg/L organic carbon.
15.5.2.3.2 Adsorption.
Considering the similarities between adsorption and complexation reactions, it is not surprising that Cu2+ is among the most strongly sorbing of the heavy metals, and that it sorbs onto both inorganic and organic solids. Sorption of copper onto oxides and clays has been investigated extensively, both because of its importance to biogeochemical cycling and as part of studies of the availability of soil-bound copper to plants. Typical results showing the relative adsorptive strength of copper and several other metals in inorganic systems were presented in Fig. 15-5.
In a completely inorganic system, binding of Cu2+ to suspended and bottom sediments would remove much of the Cu from the dissolved phase. The formation of Cu–organic complexes and the presence of organic solids complicates the analysis, however. In essence, free Cu2+ has a strong tendency to react with many components of the system, and when these components are tested one at a time, each is able to sequester most of the Cu in the system. In a real system these reactants compete and interact with one another, leading to complex speciation patterns that depend on the relative concentrations of each component and solution pH. The general picture that emerges from studies of the speciation of Cu in real and simulated natural waters is that natural organic complexing agents can bind Cu2+ so strongly that direct copper-to-surface bonding is effectively inhibited. Rather, the copper remains bonded to the organic matter under almost all conditions realistically expected in natural waters, and is adsorbed only under conditions where the organics sorb. Copper bound to organic particulates, or to organic matter adsorbed onto inorganic particulates, is thus the most likely form of Cu to reach bottom sediments, and speciation studies have consistently found Cu to be primarily associated with organics in aerobic sediments. The role of coagulation of colloidal matter in facilitating transport of Cu to estuarine sediments has already been discussed. In anaerobic sediments, reduction of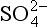 to
to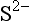 leads to precipitation of extremely insoluble CuS or mixed metal sulfides containing Cu.
leads to precipitation of extremely insoluble CuS or mixed metal sulfides containing Cu.
15.5.2.4 Biological effects
The strength of the copper-organic bond and the well-established dual potential of copper as a biological stimulant or inhibitor has led to many studies of the effect of copper speciation on growth. A few of these studies were mentioned earlier, showing that strong organic and inorganic complexes of most metals, including Cu, are non-toxic. The general conclusion that hydrated Cu2+ appears to be more biologically active than chelated or certain organically or inorganically complexed forms, bears repeating. This fact becomes more significant when put in the context of the very small ratio of Cu2+ to total Cu in most natural waters. More than for any other metal, it has been demonstrated for copper that toxicity does not correlate well with total copper or even total soluble copper in a system. Some uncertainty remains regarding the toxicity of certain inorganic complexes (particularly the hydroxo- and carbonato-complexes) and the roles of alkalinity, hardness, and other heavy metals in modifying copper toxicity. Nevertheless, studies of Cu toxicity interpreted in terms of speciation have been extremely successful and have helped establish the value of such an approach. Analysis of speciation has shifted from the state-of-the-art to the mainstream in toxicity studies in the past decade and will undoubtedly continue to shed light on the biological effects of all heavy metals.
15.6 Summary
This chapter provides an overview of the important processes and reactions controlling biogeochemical cycling of metals. While each metal has some unique properties, several generalizations apply. The most important of these have to do with metal speciation, that is, the physical-chemical form in which the metal exists in a given environment. Because metals tend to undergo a greater number and variety of relatively rapid, reversible reactions than the elements discussed in previous chapters, they are found in the environment in solid, aqueous, or gaseous phases, associated with literally thousands of different compounds. These reactions often reflect the affinity of metal ions for other atoms with free electron pairs, in particular O, N, or S.
The critical processes controlling global metal cycling are volatilization (exchange between aqueous and gaseous phases), adsorption-precipitation-dissolution (exchange between aqueous and solid phases), and complexation (conversion among various dissolved forms of the metal). For most metals, transport as a gaseous species is of little quantitative importance except in very high-temperature environments. A few metals, most notably mercury, can exist as gases at ambient temperatures, and several metals form volatile organometallic compounds that may dominate transport of the metal in local environments. Transport of particles suspended in the air is an important process for distributing many metals to regions far from their sources.
Transport in solution or aqueous suspension is the major mechanism for metal movement from the land to the oceans and ultimately to burial in ocean sediments. In solution, the hydrated metal ion and inorganic and organic complexes can all account for major portions of the total metal load. Relatively pure metal ores exist in many places, and metals from these ores may enter an aquatic system as a result of weathering. For most metals a more common sequence is for a small amount of the ore to dissolve, for the metal ions to adsorb onto other particulate matter suspended in flowing water, and for the metal to be carried as part of the particulate load of a stream in this fashion. The very insoluble oxides of Fe, Si, and Al (including clays), and particulate organic matter, are the most important solid adsorbents on which metals are “carried.”
The distribution of metals between dissolved and particulate phases in aquatic systems is governed by a competition between precipitation and adsorption (and transport as particles) versus dissolution and formation of soluble complexes (and transport in the solution phase). A great deal is known about the thermodynamics of these reactions, and in many cases it is possible to explain or predict semi-quantitatively the equilibrium speciation of a metal in an environmental system. Predictions of complete speciation of the metal are often limited by inadequate information on chemical composition, equilibrium constants, and reaction rates.
Metals act upon and are acted upon by biota in important ways. Through their effect on the chemical environment in soils, sediments, or open bodies of water, organisms can help dissolve, complex, or precipitate metals. Additionally, as noted above, suspended organic particulates can be important adsorbents for metals, carrying them through an aquatic system to its outlet or through a water column to the bottom sediments. Organisms can also directly mediate reactions involving metals, such as by formation of organometal compounds.
Metals, in turn, can either stimulate or inhibit biological activity. In this regard, the different effects of dissolved metal species are especially important. For most metals, the hydrated metal ion and simple inorganic complexes have a much greater effect on organisms (in both the stimulatory and inhibitory ranges) than large organic complexes or adsorbed metal ions.
The full appreciation of the overriding importance of metal speciation in evaluating the transport and effects of metals in an environment is a relatively recent event. As more information is gathered on the forms in which metals exist and are transported through various environmental compartments, it will become possible to predict more accurately the response of the biological communities exposed to the metals and hopefully avert or mitigate the adverse effects.
Questions
15-1. Imagine that an industrial spill allows an acidic solution containing chromate ion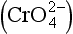 to enter a turbid, slow-moving stream containing organic-rich runoff. The streamwater pH is lowered to about 5 by the spill. As the water moves downstream, some of the particles settle, eventually being buried in the sediment. Other particles remain suspended, and as additional tributaries mix in, the pH of the water returns to a normal level of around 7.5. Describe the various fates that chromate ions might experience in this system.
to enter a turbid, slow-moving stream containing organic-rich runoff. The streamwater pH is lowered to about 5 by the spill. As the water moves downstream, some of the particles settle, eventually being buried in the sediment. Other particles remain suspended, and as additional tributaries mix in, the pH of the water returns to a normal level of around 7.5. Describe the various fates that chromate ions might experience in this system.
15-2. In the treatment of water for public consumption, iron chloride is frequently added to the water. After pH adjustment, the iron forms a precipitate of Fe(OH)3, which is thought to enhance coagulation and subsequent settling of suspended colloids. Discuss what you think would happen to dissolved copper in the water when this iron solid precipitates. Consider two “types” of water supply: a relatively pristine supply generated by snowmelt from a remote area, and a supply high in natural organic matter (humic matter) generated by percolation of rainwater through organic-rich vegetated zones.
15-3. Using the equilibrium constants below, calculate the concentrations of free (uncomplexed) cadmium ion in a freshwater with a chloride concentration of 15 mg/L, and in sea water containing 17 000 mg/L chloride. Ignore complexation with other ions.
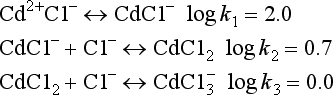
15-4. Calculate the specific surface site concentration (moles of surface sites per gram) for quartz (SiO2) particles 1 µm in diameter. Assume a site density, ns, of 5 sites/nm2 (1 nm = 10−9 m). What will be the concentration of silica surface sites if the suspended quartz is present at 2 ppm (mg/L)?
Allen, H. E., Fu, G., Deng, B. Analysis of acid-volatile sulfide (AVS) and simultaneously extracted metals (SEM) for the estimation of potential toxicity in aquatic sediments. Environ. Toxicol. Chem. 1993; 12:1441–1453.
Andreae, M. O. Arsenic speciation in seawater and interstitial waters: the role of biological-chemical interactions on the chemistry of a trace element. Limnol. Oceanog. 1979; 24:440–452.
Ankley, G. T., Di Toro, D. M., Hansen, D. M., Berry, W. J. Technical basis and proposal for deriving sediment quality criteria for metals. Environ. Toxicol. Chem. 1996; 15:2056–2066.
Avotins, P. V., Adsorption and coprecipitation studies of mercury on hydrous iron oxides. Ph. D. Dissertation, Stanford University, Stanford, CA. 1975.
Baird, C. Environmental Chemistry. New York: W. H. Freeman and Co. ; 1995.
Balistrieri, L., Brewer, P. G., Murray, J. W. Scavenging residence times of trace metals and surface chemistry of sinking particles in the deep ocean. Deep-Sea Res. 1981; 28A:101–121.
Bergan, T., Gallardo, L., Rodhe, H. Mercury in the global troposphere: A three-dimensional study. Atmos. Environ. 1999; 33:1575–1585.
Bertine, K. K., Goldberg, E. D. Fossil fuel combustion and the major sedimentary cycle. Science. 1971; 173:233–235.
Broecker, W. S. Chemical Oceanography. New York: Harcourt Brace Jovanovich; 1974.
Bruland, K. W., Bertine, K., Koide, M., Goldberg, E. D. History of metal pollution in Southern California coastal zone. Environ. Sci. Technol. 1974; 8:425–432.
Bruland, K. W., Knauer, G. A., Martin, J. H. Cadmium in Northeast Pacific waters. Limnol. Oceanog. 1978; 23:618–625.
Coles, D. G., Ragaini, R. C., Ondor, J. M., Fisher, G. L., Silberman, D., Prentice, B. A. Chemical studies of stack fly ash from a coal-fired power plant. Environ. Sci. Technol. 1979; 13:455–459.
Davies-Colley, R. J., Nelson, P. O., Williamson, K. J. Copper and cadmium uptake by estuarine sedimentary phases. Environ. Sci. Technol. 1984; 18:491–499.
Davis, J. A. Complexation of trace metals by adsorbed natural organic matter. Geochim. Acta. 1984; 48:679–691.
Davis, J. S., Leckie, J. O. Effect of adsorbed complexing ligands on trace metal uptake by hydrous oxides. Environ. Sci. Technol. 1978; 12:1309–1315.
Di Toro, D. M., Mahony, J. D., Hansen, D. J., Scott, K. J., Hinks, M. B., Mayhr, S. M., Redmond, M. S. Toxicity of cadmium in sediments: the role of acid volatile sulfide. Environ. Toxicol. Chem. 1990; 9:1487–1502.
Adv. in Chem. Series #189 Eisenreich, S. J., Hoffman, M. R., Rastetter, D., Yost, E., Maier, W. J. Metal transport phases in the upper Mississippi River. In: Kavanaugh M., Leckie J. O., eds. Particulates in Water. New York: American Chem. Soc., 1980.
Eisenrich, S. J., Metzer, N. A., Urban, N. R. Response of atmospheric lead to decreased use of lead in gasoline. Environ. Sci. Technol. 1986; 20:171–174.
Erickson, R. J., Benoit, D. A., Mattson, V. R., Nelson, H. P., Leonard, E. N. The effects of water chemistry on the toxicity of copper to flathead minnows. Environ. Toxicol. Chem. 1996; 15:181–193.
Galloway, J. N. Alteration of trace metal geochemical cycles due to the marine discharge of wastewater. Geochim. Cosmochim. Acta. 1979; 43:207–218.
Goldschmidt, V. M. Geochemistry. Washington, DC: Oxford University Press; 1954.
Hansen, D. J., Berry, W. J., Mahony, J. D., Boothman, W. S., Di Toro, D. M., Robson, D. L., Ankley, G. T., Ma, D., Tan, Q., Pesch, C. E. Predicting the toxicity of metal contaminated field sediments using interstitial concentration of metals and acidvolatile sulfide normalizations. Environ. Toxicol. Chem. 1996; 12:2080–2094.
Hardy, J. T., Apts, C. W., Crecelius, E. A., Fellingham, G. W. The sea-surface microlayer: fate and residence times of atmospheric metals. Limnol. Oceanog. 1985; 30:93–101.
Helz, G. R. Trace element inventory for the Northern Chesapeake Bay with emphasis on the influence of man. Geochim. Cosmochim. Acta. 1976; 40:573–580.
Hemmes, P., Rich, L. D., Cole, D. L., Eyring, E. M. Kinetics of hydrolysis of ferric ion in dilute aqueous solution. J. Phys. Chem. 1971; 75:929–932.
Hoffman, M. R. Thermodynamic, kinetic and extra-thermodynamic considerations in the development of equilibrium models for aquatic systems. Environ. Sci. Technol. 1981; 15:345–353.
Imhoff, K. R., Koppe, P., Dietz, F. Heavy metals in the Ruhr River and their budget in the catchment area. Progr. Water Technol. 1980; 12:735–749.
Jones, C. J., Murray, J. W. Nickel, cadmium and copper in the northeast Pacific off the coast of Washington. Limnol. Oceanog. 1984; 29:711–720.
Keck, J. C. Rate-controlled constrained equilibrium method for treating reactions in complex systems. In: Levine R. D., Tribus M., eds. Maximum Entropy Formalism. Fairlawn, NJ: M. I. T. Press, 1978.
Kudo, A. Physical/chemical/biological removal mechanisms of mercury in a receiving stream. In: Armstrong N. E., Kudo A., eds. Toxic Materials—Methods for Control. Cambridge, MA: The Center for Research in Water Resources, University of Texas at Austin, 1983.
Kudo, A., Miyahara, S. Migration of mercury from Minamata Bay. In: Armstrong N. E., Kudo A., eds. Toxic Materials — Methods for Control. Austin, TX: The Center for Research in Water Resources, University of Texas at Austin, 1983.
Langmuir, D. Aqueous Environmental Geochemistry. Austin, TX: Prentice-Hall; 1997.
Lantzy, R. J., Mackenzie, F. T. Atmospheric trace metals: global cycles and assessment of man’s impact. Geochim. Cosmochim. Acta. 1979; 43:511–525.
Lasaga, A. The kinetic treatment of geochemical cycles. Geochim. Cosmochim. Acta. 1980; 44:815–828.
Final Report EPRI-RP-910-1 Leckie, J. O., Appleton, A. R., Ball, N. B., Hayes, K. F., Honeyman, B. D. Adsorptive removal of trace elements from fly-ash pond effluents onto iron oxyhydroxide. New York: Electric Power Research Institute; 1986.
Lerman, A. Geochemical Processes: Water and Sediment Environments. Palo Alto, CA: Wiley-Interscience; 1979.
Mackay, D. Finding fugacity feasible. Environ. Sci. Technol. 1979; 13:1218–1223.
US Dept of Interior, Federal Water Pollution Control Administration MacKenthum, K. M., The Practice of Water Pollution Biology, 1969.
Manahan, S. E. Environmental Chemistry, 3rd edn. New York: Willard Grant Press; 1979.
Mason, R. P., Fitzgerald, W. F., Morel, F. M. M. The biogeochemical cycling of elemental mercury: Anthropogenic influences. Geochim. Cosmochim. Acta. 1994; 58:3191–3198.
Moore, J. W., Ramamoorthy, W. Heavy Metals in Natural Waters: Applied Monitoring and Impact Assessment. Boston, MA: Springer-Verlag; 1984.
Morel, F. M. M. Principles of Aquatic Chemistry. New York: Wiley-Interscience; 1983.
Morel, F. M. M., Westall, J. C., O’Melia, C. R., Morgan, J. J. Fate of trace metals in Los Angeles County wastewater discharge. Environ. Sci. Technol. 1975; 9:756–761.
Morgan, J. J., Stone, A. T. Kinetics of chemical processes of importance in lacustrine environments. In: Stumm W., ed. Chemical Processes in Lakes. New York: Wiley, 1985.
NAS. An Assessment of Mercury in the Environment. New York: National Academy of Sciences; 1978.
Nelson, M. B., Davis, J. A., Benjamin, M. M., Leckie, J. O. The Role of Iron Sulfides in Controlling Trace Heavy Metals in Anaerobic Sediments: Oxidative Dissolution of Ferrous Monosulfides and the Behavior of Associated Trace Metals. Air Force Weapons Laboratory, Technical Report 425. 1977.
Niragu, J. O. Copper in the Environment, Part I: Ecological Cycling. Washington, DC: Wiley-Interscience; 1979.
Pankow, J. F., Morgan, J. J. Kinetics for the aquatic environment. Environ. Sci. Technol. 1981; 15:1155–1164.
Paulson, A. J., Feely, R. A., Curl, H. C., Gendron, J. F. Behavior of Fe, Mn, Cu, and Cd in the Duwamish River estuary downstream of a sewage treatment plant. Water Res. 1984; 18:633–641.
Ripley, E. A., Redman, R. E., Crowder, A. A. Environmental Effects of Mining. New York: St. Lucie Press; 1996.
Schwartzenbach, R. P., Imboden, P. M. Modelling concepts for hydrophobic pollutants in lakes. Ecol. Modelling. 1983; 22:171.
Settle, D. M., Patterson, C. C. Magnetites and sources of precipitation and dry deposition fluxes of industrial and natural leads to the North Pacific at Enewetak. J. Geophys. Res. 1982; 87:8857–8869.
Settle, D. M., Patterson, C. C., Turekian, K. K., Cochran, J. K. Lead precipitation fluxes at tropical ocean sites determined from 210Pb measurements. J. Geophys. Res. 1982; 87:1239–1245.
Sholkovitz, E. R., Copland, D. The coagulation, solubility, and adsorption properties of Fe, Mn, Cu, Ni, Cd, Co, and humic acids in a river water. Geochim. Cosmochim. Acta. 1981; 45:181–189.
Stumm, W., Morgan, J. J. Aquatic Chemistry, 2nd edn. Delray Beach, FL: Wiley-Interscience; 1981.
Sunda, W. G., Engel, D. W., Thuotte, R. M. Effect of chemical speciation on toxicity of cadmium to grass shrimp, Palaemonetes pugio: importance of free cadmium ion. Environ. Sci. Technol. 1978; 12:409–413.
Swartz, R. C., Ditzworth, G. R., Schultz, D. W., Lamberson, J. O. Sediment toxicity to a marine infaunal amphipod: cadmium and its interaction with sewage sludge. Mar. Environ. Res. 1985; 18:133–153.
Takamatsu, T., Kawashima, M., Koyama, M. The role of Mn-rich hydrous manganese oxide in the accumulation of arsenic in lake sediments. Water Res. 1985; 19:1029–1032.
Theis, T. L., Wirth, J. L. Sorptive behavior of trace metals on fly ash in aqueous systems. Environ. Sci. Technol. 1977; 11:1096–1100.
Turekian, K. K. Rivers, tributaries, and estuaries. In: Wood D. W., ed. Impingement of Man on the Ocean. New York: Wiley, 1971.
Turekian, K. K. The fate of metals in the oceans. Geochim. Cosmochim. Acta. 1977; 41:1139–1144.
Turner, D. R., Whitfield, M., Dickson, A. G. The equilibrium speciation of dissolved components in freshwater and seawater at 25°C and 1 atm pressure. Geochim. Cosmochim. Acta. 1981; 45:855–881.
Weiss, H. V., Koide, M., Goldberg, E. D. Selenium and sulfur in a Greenland ice sheet: relation to fossil fuel combustion. Science. 1971; 172:261–263.
Weiss, H. V., Koide, M., Goldberg, E. D. Mercury in a Greenland ice sheet: evidence of recent input by man. Science. 1971; 174:692–694.
Wood, J. Biological cycles for toxic metals in the environment. Science. 1974; 183:1049–1052.