Soils, Watershed Processes, and Marine Sediments
8.1 Introduction
The surface of the Earth is a dynamic place. Over geologic time, rocks uplifted above sea level break down and are converted into soils by weathering processes. Soils release soluble components into rivers and can be eroded and transported across landmasses until both soluble and particulate components are eventually deposited in marine sedimentary basins. Once buried, high pressures and temperatures gradually convert the sediment back to rock. Tectonic processes can then uplift the new rock and expose it again at the Earth’s surface, resulting in a cycle of uplift, erosion, deposition, burial, and renewed uplift called the rock cycle. The rock cycle continually modifies the Earth’s surface (Fig. 8-1). Soils are an especially reactive component of the Earth’s surface. They not only provide nutrients and water for terrestrial ecosystems, but they also store and exchange gases with the atmosphere, and affect the movement of surface and groundwater. Thus, soils affect and are affected by the biosphere, atmosphere, and hydrosphere.
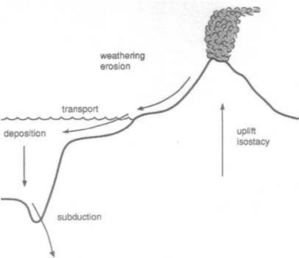
Soil is a key component of the rock cycle because weathering and soil formation processes transform rock into more readily erodible material. Rates of soil formation may even limit the overall erosion rate of a landscape. Erosion processes are also a key linkage in the rock cycle between soil production and the filling of sedimentary basins. When water falls onto the Earth’s surface it can seep into the ground and percolate down to the water table or it can run off downslope to collect into streams that ultimately combine to form rivers. Flowing water transports eroded material until it is either deposited in local depositional environments or delivered to the oceans. If there was no sink for these sediments, the oceans would fill up in less than 100 Myr, and if there was no source for uplift of rocks on the continents they would be degraded to ocean level in less than 50 Myr (Holland, 1978, p. 146). Global tectonic activity prevents these scenarios through rock uplift and the return of sediments accumulated on the ocean floor back to the continents either by accretion or subduction at plate margins.
The material transformations and interactions that occur between soil, sediment, rock, water and the atmosphere during geological cycles of uplift and erosion are important in global biogeochemical cycles. Over geologic time, for example, material incorporated into sedimentary rocks can be sequestered in long-term storage in marine basins. In contrast, weathering products are vented to the atmosphere over much shorter time scales. The development of soils, erosional processes, and deposition in marine basins all play key roles in global biogeochemical cycles.
8.2 Weathering
Weathering occurs because rocks and minerals become exposed to physical and chemical conditions that differ from conditions under which they formed. Rocks form at higher temperatures and pressures than that of the surficial environment so they are unstable at the temperatures and pressure of the Earth’s surface and are thus susceptible to weathering. The inorganic solid phase of any soil consists of a number of minerals displaying different degrees of weathering susceptibility. The extent of weathering of these minerals depends on the stabilities of the minerals and the physical and chemical environment to which the minerals are exposed in the soil or at surface conditions, including the supply of water and the removal or transport of weathering products (Garrels and Christ, 1965; Rai and Kittrick, 1989; Colman and Dethier, 1986).
Weathering can be separated into two types: physical and chemical. Physical weathering involves changes in the degree of consolidation with little or no chemical and mineralogical changes of rocks and minerals. Chemical weathering involves changes in chemical and mineralogical composition that generally act on the surfaces of rocks or minerals. Physical weathering increases the surface area of rocks and minerals such that chemical weathering can proceed at a faster rate. In nature these two processes occur concurrently and are difficult to separate (Jackson and Sherman, 1953; Birkeland, 1999).
8.2.1 Physical Weathering
Rocks and minerals break when stressed above their tensile strength. Commonly, rocks fracture along joints, fissures, or planes that have developed during cooling, tectonism, and sedimentary processes or along lines of weakness at the boundaries between mineral grains. When previously buried rock masses are exposed at the Earth’s surface, the lowering of the overburden pressure, or unloading, allows the rocks to expand. This expansion induces fracturing that aids in the conversion of rock to soil. Physical weathering processes expand these fractures or cause the development of new ones.
Frost wedging is the prying apart of materials by expansion of water when it freezes. The pressure produced by freezing water is well above the tensile strength of many rocks; however, this pressure may not be commonly attained in nature because rocks are not completely saturated but contain air gaps. Hydration shattering, the ordering and disordering of water molecules adsorbed at the surface of rocks, may be responsible for processes ascribed to frost wedging (Dunn and Hudec, 1972; Hudec, 1974). Nonetheless, the presence of shattered bedrock, and generally angular rock debris in cold environments provides sufficient evidence that frost wedging is at work. Laboratory experiments suggest that repeated freeze–thaw cycles can even produce clay-sized particles (Lautridou and Ozouf, 1982).
In arid environments, where the soluble products of weathering are not completely removed from the soil, saline solutions may circulate in the soil as well as in rock fractures. If upon evaporation the salt concentration increases above its saturation point, salt crystals form and grow (Goudie et al., 1970). The growth of salt crystals in crevices can force open fractures. Salt weathering occurs in cold or hot deserts or areas where salts accumulate. Boulders, blocks, and cliffs affected by salt weathering display cavities and holes and sometimes acquire grotesque forms, as observed in the cold desert of Antarctica (Ugolini, 1986). Frost and salt weathering combined have a synergistic effect that could be more effective at breaking down rocks than salt or frost alone (Williams and Robinson, 1981).
Thermal expansion induced by insolation may be important in desert areas where rocky outcrops and soil surfaces are barren. In a desert, daily temperature excursions are wide and rocks are heated and cooled rapidly. Each type of mineral in a rock has a different coefficient of thermal expansion. Consequently, when a rock is heated or cooled, its minerals differentially expand and contract, thereby inducing stresses and strains in the rock and causing fractures. Ollier (1969) discussed examples of rock weathering due to insolation. Fire can develop temperatures far in excess of insolation and be quite effective in fracturing rocks (Blackwelder, 1927).
Plants and animals disrupt and disaggregate rocks and fracture or abrade individual grains or minerals. Endolithic algae growing in deserts may be capable of disintegrating rocks through shrinking and swelling (Friedman, 1971). Lichens are effective agents in physical weathering by extending fungal hyphae into rocks and by expansion and contraction of the thalli (Syers and Iskandar, 1973). Higher plants grow roots in rock crevices and eventually the increased pressure breaks and disrupts the substratum. In addition, the physical mixing of rock and soil that occurs from tree throw is a primary process in the conversion of bedrock into soil in forested regions. Earthworms, as discussed by Darwin (1896), digest and abrade a considerable amount of soil. Mammals, such as moles, gophers, and ground squirrels tunnel and excavate a substantial amount of soil when they build dens (Black and Montgomery, 1991; Butler, 1995). Similarly, rodents break down rocks and create fine particles (Ugolini and Edmonds, 1983).
8.2.2 Chemical Weathering
Chemical weathering involves chemical changes of rocks and minerals under near-surface conditions. Mineral grains in soils (see Table 8-1) are bathed in a film of water and the dissolution of these minerals depends on a number of factors. First, the solubility of the mineral affects the potential of a mineral to be weathered; this is determined largely by the number and strength of chemical bonds within the crystal lattice. Second, temperature affects the rate of weathering reactions. Third, the composition of the soil solution surrounding the mineral grains will determine weathering rates; solution pH, organic acids, carbonic acid, concentration of other ions already in solution, redox, and complexing ligands can all affect how readily the ions released by weathering can go into solution. And last, water; water is not only the universal solvent in the weathering environment, but it is also the vehicle for the redistribution of products of weathering. The amount of contact between the soil solution and the mineral surface in conjunction with the frequency of removal of soil solution containing ions released by weathering (and its replacement with new soil solution) will all determine how readily a mineral weathers. Taking these factors into consideration it is possible to determine the thermodynamic stability of minerals, and predict the weathering sequence of minerals in an environment (Garrels and Christ, 1965). There are six fundamental processes that chemically weather minerals. These are dissolution, hydration, hydrolysis, acidolysis, chelation, and oxidation/reduction.
Table 8-1
Primary and secondary minerals commonly found in soils
| Primary minerals | Approximate composition | Weatherability |
| Quartz | SiO2 | — |
| K-Feldspar | KAlSi3O8 | + |
| Ca, Na-plagioclase | CaAl2Si2O8 to NaAlSi3O8 | + to (+) |
| Muscovite | KAl3Si3O10(OH)2 | +(+) |
| Amphibole | Ca2Al2Mg2Fe3Si5O22(OH)2 | +(+) |
| Biotite | KAl(Mg,Fe)3Si3O10(OH)2 | ++ |
| Pyroxene | Ca2(Al,Fe)4(Mg,Fe)4Si6O24 | ++ |
| Apatite | [3Ca3(PO4)2]·CaO | ++ |
| Volcanic glass | Variable | ++ |
| Calcite | CaCO3 | +++ |
| Dolomite | (Ca,Mg)CO3 | +++ |
| Gypsum | CaSO4·2H2O | +++ |
| Primary minerals | Approximate composition | Type |
| Kaolinite | Al2Si2O5(OH)4 | 1:1 layer-silicate |
| Vermiculite | (Al1.7Mg0.3)Si3.6Al0.4O10(OH)2 | 2:1 layer-silicate |
| Montmorillonite | (Al1.7Mg0.3)Si3.9Al0.1O10(OH)2 | 2:1 layer-silicate |
| Chlorite | (Mg2.6Fe0.4)Si2.5(Al,Fe)1.5O10(OH)2 | 2:1:1 layer-silicate |
| Allophane | (SiO2)1–2Al2O5·2.5–3(H2O) | Pseudocrystalline, spherical |
| Imogolite | SiO2Al2O3·2.5H2O | Pseudocrystalline, strands |
| Hallyosite | Al2Si2O5(OH)4·2H2O | Pseudocrystalline, tubular |
| Gibbsite | Al(OH)3 | Hydroxide |
| Goethite | FeOOH | Oxyhydroxide |
| Hematite | Fe2O3 | Oxide |
| Ferrihydrite | 5Fe2O5·9H2O | Oxide |
8.2.2.1 Dissolution
Dissolution of a mineral occurs when the crystal lattice breaks down and it separates into its component ions in water. Minerals most affected are salts, sulfates, and carbonates. For example, calcite dissolution is described by
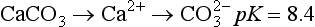
In this case the two ions, Ca2+ and CO2−3, are released into the soil solution and are able to react with water (to form bicarbonate or carbonic acid) or other solution components, or be removed from the soil by leaching. The
dissolution of CaCO3 is regulated by the following reactions:
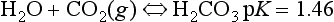
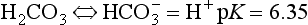

Overall:
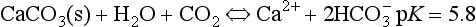
Dissolution of CaCO3 is a congruent reaction; the entire mineral is weathered and results completely in soluble products. The above reaction is driven to the right by an increase of CO2 partial pressure and by the removal of the Ca and/or bicarbonate. Any impurities present in the calcareous rock, such as silicates, oxides, organic compounds, and others, are left as residue. As the calcium and bicarbonate leach out over time, this residue becomes the substratum upon which soils develop in karst terrain found in areas of readily dissolved limestone. This terrain is characterized predominantly by underground drainage and marked by numerous abrupt ridges, fissures, sinkholes, and caverns.
8.2.2.2 Hydration and hydrolysis
Hydration is the incorporation of water molecule(s) into a mineral, which results in a structural as well as chemical change. This can drastically weaken the stability of a mineral, and make it very susceptible to other forms of chemical weathering. For example, hydration of anhydrite results in the formation of gypsum:
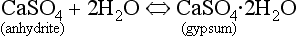
Gypsum is a relatively soluble mineral and can undergo dissolution whereas anhydrite is less soluble.
Hydrolysis is the incorporation of either H+ or OH−, the components of water, into a mineral. Although water has a low dissociation constant, it is abundant in most environments. Even though little H+ or OH− may be provided by dissociation of water, the sheer volume of water moving through a soil over time makes hydrolysis an extremely important reaction.
As in dissolution, a chemical and structural change can occur from hydrolysis as the ions replaced by H+ or OH− may be of a different size so that the crystal structure is stressed and weakened. An example of this is the weathering of feldspar or goethite by H+:
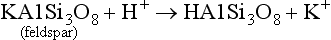
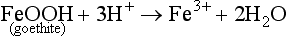
In Equation (7), an altered solid phase is produced by the weathering of feldspar with a K+ ion released – an example of incongruent weathering (not everything is weathered into solution). In Equation (8), the goethite goes completely into solution—another example of congruent weathering. Both of these examples demonstrate a critical property of weathering, namely that almost all weathering reactions consume H+. Thus, as long as weatherable minerals are present, weathering reactions can help counteract the natural tendency of soils to become acidic or neutralize the effects of acid rain.
8.2.2.3 Acidolysis
Acidolysis is a similar weathering reaction to hydrolysis in that H+ is used to weather minerals, but in this case the source of H+ is not water but organic or inorganic acids. Humic and fulvic acids (discussed in Section 8.3.2), carbonic acid, nitric or sulfuric acid, and low-molecular-weight organic acids such as oxalic acid can all provide H+ to weather minerals. All of these acids occur naturally in soils; in addition nitric and sulfuric acid can be added to soil by acid pollution. The organic acids are prevalent in the upper soil where they cause intense weathering. Carbonic acid and bicarbonate are more important to weathering in young soils, or deep in the soil profile where organic acids are not prevalent.
8.2.2.4 Chelation
Besides attacking minerals by providing H+, organic acids can also cause weathering by chelation. A chelator is a ligand capable of forming multiple bonds with a metal ion such as Fe, Al, or Ca, resulting in a ring-type structure with the metal incorporated into the complex. The large, complex organic acids formed in soils can act as chelators, and are capable of stripping metal ions from some primary minerals (Huang, 1989). Artificial chelators such as EDTA (ethylene diamine tetraacetic acid) are often used to test soils for the availability of micronutrients. Some low-molecular-weight organic acids are also capable of chelating metals.
8.2.2.5 Oxidation and reduction
Oxidation and reduction reactions weather minerals by the transfer of electrons. Minerals containing elements that can have multiple valence states such as Fe, Mn, S, or even N, are susceptible to redox reactions. A common reaction that occurs in soil involves both the oxidation and reduction of iron, which when present in a mineral is usually in the Fe(II) form. Fe(II) in the parent material oxidizes slowly in well-drained and aerated soils (Bohn et al., 1985; Birkeland, 1999). In this oxidizing environment an electron may be removed from Fe2+ at mineral edges causing disruption in the crystal due to charge imbalance, triggering disintegration of mineral edges or making the mineral more susceptible to other forms of weathering. The Fe3+ released by weathering is very insoluble and readily combines with oxygen and water to form goethite
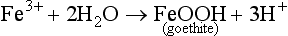
This occurs in well-drained temperate soils, and is the reverse of Equation (8). In very well-drained warm soils, hematite can form. For example, a primary Fe-bearing mineral such as a pyroxene or amphibole weathers through oxidation to release Fe3+ into solution, which then precipitates out as goethite or hematite, depending on the environment. An important aspect of this process is that ultimately H+ is released and able to weather other minerals. Minerals containing oxidized elements can undergo reduction reactions in anaerobic soils causing them to weather. Weathering of goethite in an anaerobic soil will release Fe2+ into solution.
Overall, weathering controls the chemistry of material that is transported into the sediment and that which stays behind in the soil. As an example, consider a general weathering reaction for an aluminosilicate (Stumm and Morgan, 1995):
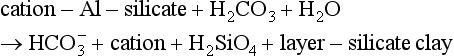
A specific example of this would be the weathering of K-feldspar and the formation of kaolinite (see Table 8-1 for mineral definitions), a layer-silicate clay:
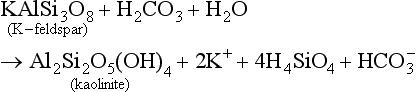
Minerals formed in the soil can weather further with increasing quantities of H2CO3 and H2O, or other sources of H+ (Pedro, 1982). The cations released into solution (in this case K+) can be moved out of the soil into groundwater, rivers, and ultimately to the oceans and marine sediments. Al and Fe, however, tend to persist in the soil. Since they are not readily lost from soil and other cations are, soils ultimately become richer in Fe and Al oxides such as goethite, hematite, or gibbsite. See Section 8.3.2 for more information about clay formation in soil.
The weathering reactions given above show the key effects of weathering: the breakdown of the original rock minerals, the consumption of H+, and the release of cations and silica into solution which can then be used to make new minerals or be lost from the soil into the groundwater and rivers.
8.3 Soils
Soil is a multi-phase system consisting of solids, liquids, and gases. In a typical soil, solids, liquids and gases compose about 50%, 20–30% and 20–30% respectively of the total soil volume (Brady and Weil, 1999). The solid phase can be broken down into two components: inorganic and organic matter, with organic matter ranging from 1 to 5% of the soil.
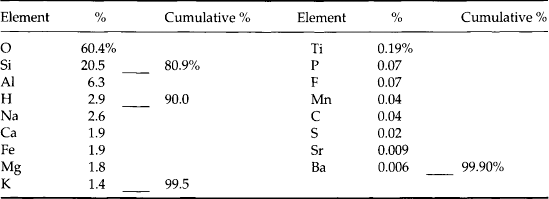
The inorganic component of soil is dominated by four elements: O, Si, Al, and Fe (Jackson, 1964). Together with Mg, Ca, Na, and K they constitute 99% of the soil mineral matter (see Table 8-2). Minerals in soil are divided into primary and secondary minerals. Primary minerals, which occur in igneous, metamorphic, and sedimentary rocks, are inherited by soilfrom the parent material. Secondary minerals form in soils and include layer-silicate clays, amorphous (or non-crystalline) minerals, carbonates, phosphates, sulfides and sulfates, oxides, hydroxides, and oxyhydroxides. The most common primary and secondary minerals in soils are given in Table 8-1. Primary minerals are typically larger than secondary minerals. Primary minerals are seldom clay-sized (≤ 2 µm), whereas secondary minerals are rarely larger than clay-sized. The small size of secondary minerals gives them a very high specific surface area, making them extremely reactive in soils.
Organic matter is incorporated into the soil from the detritus of organisms living on and within the soil. Plant litter is composed largely of C (44%), H (8%), and O (40%), with N, S, P, and other nutrients making up the remaining 8%. Soil organic matter consists of three fractions: recognizable litter, humus and colloidal organics. Humus forms from litter that has undergone decomposition and synthesis into a new amorphous organic compound with a brown to black color. Colloidal organics are soluble organic acids and other organic compounds that can stay in suspension or coat particle surfaces. Soils store substantial amounts of carbon – approximately 1500 Pg C is stored in the upper 1 m of soil in the world (Schlesinger, 1997). Living organisms are also a vital part of the soil as they facilitate many soil processes.
Soil air differs in composition from the atmosphere due to the activities of organisms and their interactions with the soil. Soil air is generally higher in CO2 (frequently about 1%, but may be as high as 10%) and lower in oxygen (5 to 20%). The higher CO2 and lower O2 of soil air results from decomposition and root and microbial respiration that releases CO2 and consumes oxygen. The exact composition of the soil air depends on the porosity of the soil. A soil with a high total porosity and large pores will allow faster diffusion of soil air to the atmosphere and have lower concentrations of CO2 and higher concentrations of O2. A soil with a low porosity or a soil that has its pores largely filled with water may have very high CO2 and little O2.
The liquid phase of the soil system is the soil water, or the soil solution as it is more appropriately called. Water can enter the soil from at the surface by rainfall or snowmelt, by upward movement of groundwater, or by lateral flow through soil. When water is in intimate contact with soil minerals, organics, organisms, and air, it acts as an interface between these different soil phases, providing a transport mechanism for elements from one location in the soil to another. Water is an excellent solvent; it dissolves many ions and contains organic and inorganic colloids in suspension. It also serves to remove elements from the soil by leaching, and can physically remove soil by erosion.
8.3.1 Soil Formation
Soil genesis is the result of four fundamental types of processes simultaneously operating at any part of the Earth’s surface. As a soil develops, matter and energy enter the soil, can be transformed or translocated, and can leave the soil. The nature and magnitude of inputs, outputs, transformations, and translocations can vary widely from one site to another and result in numerous different types of soils.
8.3.1.1 Inputs and outputs
One reason soils form is because of the endless migration of ions, molecules, and particles into the soil from meteoric inputs. Examples of meteoric inputs include H2O, CO2, O2, nitrogenous compounds, pollutants, salts, and dust. These molecules and compounds come from space, from the atmosphere and the oceans, and from other terrestrial systems.
Litter inputs come from vegetation and animals. These may be above ground litter as in tree leaves falling in autumn, or below ground as a plant dies and its roots become part of the soil organic matter. The composition of the meteoric inputs can be dramatically changed as inputs pass through the vegetative canopy (see Cronan (1984) and Johnson and Lindberg (1992) for a discussion of canopy processes and development of throughfall). Solar energy is another critical input to soils. Most soil reactions are driven by the energy released during decomposition of organic matter and by solar energy.
The release of ions through weathering is also considered an input to soils. Elements that were bound in mineral crystals are released into the soil solution. These ions can be involved in soil processes and the formation of new organic or inorganic materials, or leached from the soil into the groundwater.
Leaching is an important output from soils through which dissolved elements or suspended materials are carried downward by the soil solution and enter the groundwater. Most soils act as very good filters, retaining nutrients and organic matter, and even most metals and pesticides added as pollutants. Thus leaching losses to groundwater are normally minimal. However, the capacity of the soil to sorb ions or filter particulates can be exceeded in highly polluted, overfertilized, or eroded soils allowing passage of materials which then pollute the groundwater.
Erosion and gases released from the soil can also remove material from soils. Erosive outputs are usually from the upper portion of the soil, and may adversely impact soil processes and soil fertility by removing large quantities of the soil organic matter. Such losses of organic matter may remove substantial amounts of nutrients needed for plant growth. Gaseous losses are a normal efflux from soils to the atmosphere. Carbon is usually in the form of CO2 when it effluxes from the soil to the atmosphere, but can be released as methane (CH4) in wetlands where anaerobic processes are dominant. Nitrogen can evolve from the soil to the atmosphere as N2, N2O, or NH3 depending on whether the soil is aerobic or anaerobic. Under anaerobic conditions,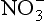 can be converted to N2 or N2O. Gaseous losses of S are typically as H2S from wetlands. Volatile losses of C, N, and S through wildfire can be important in some environments.
can be converted to N2 or N2O. Gaseous losses of S are typically as H2S from wetlands. Volatile losses of C, N, and S through wildfire can be important in some environments.
8.3.1.2 Transformations
Organic matter and rocks are the building materials of soils, which both undergo extensive transformations within soil. These transformations include changes in physical as well as chemical properties and result in unique new soil characteristics. Weathering is one type of transformation of inorganic matter that occurs in soil (see discussion above). The formation of secondary minerals and the development of cation exchange capacity are others. The development of humus from fresh litter is another transformation that affects soil organic matter.
8.3.1.2.1 Secondary minerals.
As weathering of primary minerals proceeds, ions are released into solution, and new minerals are formed. These new minerals, called secondary minerals, include layer silicate clay minerals, carbonates, phosphates, sulfates and sulfides, different hydroxides and oxyhydroxides of Al, Fe, Mn, Ti, and Si, and non-crystalline minerals such as allophane and imogolite. Secondary minerals, such as the clay minerals, may have a specific surface area in the range of 20–800 m2/g and up to 1000 m2/g in the case of imogolite (Wada, 1985). Surface area is very important because most chemical reactions in soil are surface reactions occurring at the interface of solids and the soil solution. Layer-silicate clays, oxides, and carbonates are the most widespread secondary minerals.
Layer-silicate structure, as in other silicate minerals, is dominated by the strong Si–O bond, which accounts for the relative insolubility of these minerals. Other elements involved in the building of layer silicates are Al, Mg, or Fe coordinated with O and OH. The spatial arrangement of Si and these metals with O and OH results in the formation of tetrahedral and octahedral sheets (see Fig. 8-2). The combination of the tetrahedral and octahedral sheets in different groupings, and in conjunction with different metal oxide sheets, generates a number of different layer silicate clays (see Table 8-1).
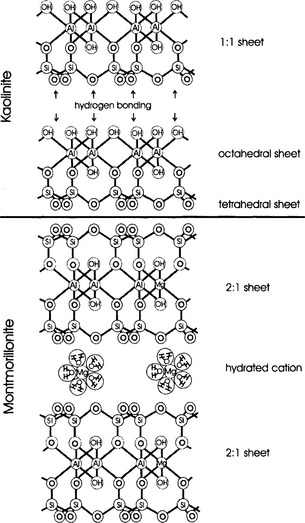
Once a layer-silicate clay forms, it does not necessarily remain in the soil forever. As conditions change it too may weather and a new mineral may form that is more in equilibrium with the new conditions. For example, it is common in young soils for the concentrations of cations such as K, Ca, or Mg in the soil solution to be high, but as primary minerals are weathered and disappear, cation concentrations will decrease. With a decrease in solution cations, a layer-silicate such as vermiculite will no longer be stable and can weather. In its place, montmorillonite or another clay can form. Ultimately, kaolinite or Fe and Al oxides are most stable and, therefore most common in the oldest, most highly developed and leached soils.
8.3.1.2.2 Ion exchange capacity.
A characteristic common to all the layer silicates is the electric charge present at their surfaces. This charge can develop when a clay is forming. With an abundance of Al3+ in solution when the clay is forming, some of the sites where Si4+ normally occurs in the crystal can be filled by Al3+ in place of Si4+ due to its similar ionic radius. This process is called isomorphic substitution. While the crystalline structure can persist with this substitution, the leftover negative charge from the O2− and OH− making up the rest of the clay molecule must be satisfied. This is achieved by attracting cations to the surface of the clay or in between the layers of the clay mineral. Clays such as the smectites, which have little substitution and low charge (∼0.6 to 0.25 charge per unit cell) can expand and allow water and cations to move in between the clay plates using the cations to balance the negative charge. The cations are electrostatically held; this allows one cation to easily take the place of another cation so that the cations are exchangeable. Thus, if a plant root releases H+ into the soil solution, the H+ can replace an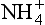 ion exchangeably held by a clay so that it goes into solution and can be taken up by the root. The quantity of cations that soils can exchangeably store is called its cation exchange capacity (CEC). It is fortunate for humans that the phenomenon of cation exchange capacity exists. Without this property, the nutrients released by weathering and decomposition would be easily lost to rivers and the ocean, leaving an infertile land. According to Jackson (1969), life that had originated in the seas was able to move onto the land because the clay produced by weathering had the capacity to hold cations and make them available to plants.
ion exchangeably held by a clay so that it goes into solution and can be taken up by the root. The quantity of cations that soils can exchangeably store is called its cation exchange capacity (CEC). It is fortunate for humans that the phenomenon of cation exchange capacity exists. Without this property, the nutrients released by weathering and decomposition would be easily lost to rivers and the ocean, leaving an infertile land. According to Jackson (1969), life that had originated in the seas was able to move onto the land because the clay produced by weathering had the capacity to hold cations and make them available to plants.
In a fertile agricultural soil, the exchangeable cations include most of the macronutrients needed by plants (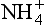 , K+, Ca2+, and Mg2+) along with Na+. In an acid soil, many of the cation exchange sites are filled by Al3+ or H+ ions, neither of which is essential for plants. When a soil has many of its exchange sites (≥ 50%) filled by nutrient cations or Na+, it has a high base saturation. Conversely, when a soil has mostly Al3+ and H+ on its exchange sites, it has a low base saturation. Other layer silicates such as mica or chlorite (∼0.9 to 1.0 charge per unit cell) have high negative charges and tend to fix cations (K and Mg, in particular) between their plates so that they do not expand and have little CEC. On the other hand, kaolinite has a charge of zero and thus has a CEC of almost zero, but kaolinite does not expand because hydrogen bonds hold the sheets together.
, K+, Ca2+, and Mg2+) along with Na+. In an acid soil, many of the cation exchange sites are filled by Al3+ or H+ ions, neither of which is essential for plants. When a soil has many of its exchange sites (≥ 50%) filled by nutrient cations or Na+, it has a high base saturation. Conversely, when a soil has mostly Al3+ and H+ on its exchange sites, it has a low base saturation. Other layer silicates such as mica or chlorite (∼0.9 to 1.0 charge per unit cell) have high negative charges and tend to fix cations (K and Mg, in particular) between their plates so that they do not expand and have little CEC. On the other hand, kaolinite has a charge of zero and thus has a CEC of almost zero, but kaolinite does not expand because hydrogen bonds hold the sheets together.
Oxides, non-crystalline minerals, and humified organic matter can also develop charges at their surfaces by reactions with the soil solution. In this case, the surface can have positive (CEC) or negative charges (anion exchange capacity, AEC) depending on the pH of the solution surrounding the particles. Figure 8-3 shows how this occurs. A pH-dependent charge can also occur at the incomplete bonds at the ends of layer silicate clay minerals. Ions held by pH-dependent charges are also completely exchangeable. The pH-dependent charges can aid in keeping either cations or anions (such as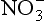 ) in the soil and prevent them from leaching. An acidic soil has a much lower CEC than a basic soil, but may have AEC.
) in the soil and prevent them from leaching. An acidic soil has a much lower CEC than a basic soil, but may have AEC.
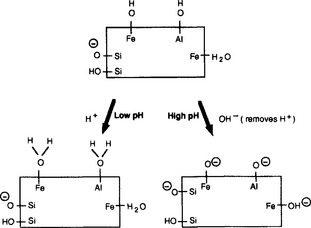
Fig. 8-3 Diagram illustrating the development of positively and negatively charged sites on surfaces of soil constituents, at low and high pH. (Reproduced with permission from R. L. Parfitt (1980). Chemical properties of variable charge soils. In “Soils with Variable Charge” (B. K. G. Theng, ed.), p. 168. New Zealand Society of Soil Science Offset Publications.)
8.3.1.2.3 Humus fractionation.
Another extremely important transformation that takes place in soils is the conversion of fresh organic litter into humus. Organic matter in soils is made up of partially decomposed plant and animal remains, substances synthesized during decomposition, and microbial bodies. Organic matter is derived directly or indirectly from photosynthesis. Decomposition processes incompletely break down the complex organic molecules, so that organic matter tends to accumulate in soils. Decomposition is an extremely important process in terms of releasing energy and nutrients to the soil system.
Plant litter consists mainly of sugars, cellulose, hemicellulose, lignin, waxes, and polyphenols, and to a lesser extent proteins and cations (Paul and Clark, 1996). The rate of litter decomposition is a function of climate and the composition of the litter. The nitrogen content is often a limiting factor in decomposition, but lignin content may also control decomposition rates (Edmonds, 1979). In general, decomposition is most rapid in well-aerated, moist, mesic, and nearly-neutral pH soils. Cold, humid environments with high water tables and acidic conditions favor the accumulation of undecomposed organic matter and carbon storage in soils. When C is converted from CO2 to an organic molecule during photosynthesis, it is reduced (gains electrons). Under aerobic conditions, decomposition oxidizes the C back to the 4+ valence state in CO2 by using O as an electron acceptor. Anaerobic decomposition requires that a different electron acceptor be used, as little or no O is available. Nitrogen, Mn, Fe, S, and C can act as electron acceptors when O is not present. If C is used as an electron acceptor, methane is produced. Using C as an electron acceptor gives decomposers less energy than using any other element, thus decomposition will proceed extremely slowly under such a strongly reducing anaerobic environment.
Complete decomposition of organic matter releases CO2, water, nutrients, and energy. Partial decomposition resynthesizes fresh organic matter into new organic compounds that are collectively referred to as humus or humic substances. Humic substances are most usefully described in terms of their solubility. Humic substances are “amorphous, dark-colored, hydrophilic, acidic, partly aromatic, chemically complex organic substances that range in molecular weight from a few hundred to several thousand” (Oades, 1989). Humic substances are fractionated into three main groups based on their solubility in acidic and basic extracts. Humic acids are soluble in base, but precipitate with acidification of the extract to pH 2. Fulvic acids are soluble in both alkaline and acidic water. Humin, the last humic group, is insoluble in either acid or base. Whereas these three humic fractions are structurally similar, their molecular weight and functional groups differ. Humic acid and humin contain more H, N, and S, but less O than fulvic acid. The total acidity and the number of carboxylic functional groups of fulvic acid are greater than those of humic acid and humin (Oades, 1989).
Humic substances are involved in many soil reactions, largely due to their high surface area (800–900 m2/g) which can give humic material a high exchange capacity and a high absorptive capacity. This makes humic substances important for water retention, aggregation of soil particles, nutrient supply, and buffering of soil pH (Bohn et al., 1985). Specific humic groups are also essential to certain soil processes. For example, fulvic acids with their high acidity and solubility, and high number of complexing functional groups, can weather Fe3+ and Al3+ from minerals, chelate them, and keep them in solution, thereby allowing their movement down the soil profile where they may deposit Fe and Al in lower horizons. On the other hand, humic acids, with their high molecular weight and lower solubility, help to create organic-rich surface soils; humic acids can complex with Ca, become very insoluble and create a deep, highly fertile, surface soil.
8.3.1.3 Translocations
Movement of raw and transformed materials can take place within the soil and results in zones of accumulation, depletion, or mixing. Formation, migration, and accumulation of different elements, clays, oxides, and organic matter can occur in different parts of the soil. These different zones or layers in soil that are approximately parallel to the surface are called soil horizons. Depleted or enriched soil horizons result in different depths in the soil having different chemical and physical properties. Translocations are caused by a combination of physical, chemical, and biological processes.
As an example, the migration of clay from the surface of a soil to a lower horizon results from several processes occurring when certain soil and environmental properties exist. First, clay-sized minerals must form, usually requiring weathering to have occurred. Clay minerals formed in the surface soil can then go into suspension when salt concentrations in solution are low. Seasonal rains can move the clay down as water percolates through the soil and deposit the clay when the wetting front stops or soil pores become plugged. This process is called lessivage and results in a clay-enriched horizon (illuviated) beneath a coarser textured horizon (Duchaufour, 1982).
Movement of carbonates and salts can also occur in a similar fashion. As these minerals are weathered in the upper soil profile, their component ions go into solution and are moved down through the soil by rainfall entering the soil. As the water moves down the soil there may not be enough water to move the ions out of the soil, so they precipitate in a lower horizon where they accumulate. Such accumulations are common in arid environments with limited rainfall. In high rainfall areas, carbonates and salts are usually completely removed from the soil through leaching.
The kind of vegetation and environment in which a soil develops also can influence the mobility of organic and inorganic substances. One particular example occurs commonly in areas that have Ericaceous plants or coniferous forests, and cold, wet environments in which Fe, Al, and humic substances are translocated. This process, called podzolization, is particularly favored by a coarse soil texture so that water can move easily through the upper soil. In podzolization, the vegetation drops litter that is low in nutrients and favors the production of organic acids by incomplete decomposition. The soluble organic acids (fulvic acids in particular) migrate into the mineral soil where they can weather minerals by release of H+ or chelation. The Fe and Al can then migrate down the soil profile along with the organic acids creating a zone of soil depleted in sesquioxides and organics (eluviated). Subsequently, the Fe, Al, and organic matter can precipitate out at a lower depth, creating a zone enriched in these materials. Further decomposition of this enriched horizon (illuviated) may release Al and Fe which may move further down the soil to create another zone enriched only in Fe and Al and where amorphous minerals can form (Lundstrom, 1994).
Some translocations can disrupt the concentration of materials and thereby provide mixing of different horizons rather than segregating them. Worms and other soil organisms do this by carrying down litter and organic rich soil into lower horizons and then tunneling up to the surface again carrying soil with them – thereby mixing horizon material in the process. This process is called bioturbation. Worms also contribute to the formation of humic substances with the passage of fresh litter through their gut and the release of partially decomposed humus. Some soil organisms such as worms are important for the creation of a deep, highly fertile, organic-rich surface soil because they aid in moving organic matter deeper into the soil. Mixing that can disrupt different accumulation zones in soils can also occur due to shrinking and swelling of clays in a soil with highly seasonal rainfall. Shrinking and cracking of the soil can allow surface soil to slough into the cracks and be moved lower in the soil, while lower soil swells upward in the wet season. In this case, organisms are not responsible for this type of soil mixing, called pedoturbation. Frost action and windthrow are other examples of pedoturbation.
8.3.1.4 Soil horizons and profile development
Soil horizons differ from one another in composition (e.g. clay or organic matter content), physical properties (e.g. color or particle size), or chemical properties such as pH or CEC. Five different soil horizons can form. An all-organic horizon (O horizon), typically occurs in wetlands or at the surface of forest soils. A mineral horizon (A horizon) that is rich in humified organic matter, may be found in prairies. A loss of Fe, Al, organic matter, or clay will create an E horizon (eluviated horizon). B horizons are zones of soil that have accumulated material from above or well-weathered soil that shows evidence of pedogenic processes through changes in color or development of soil aggregates. The lowest soil horizon is typically a C horizon, which is the least weathered zone of soil and is most similar to the original material the soil is forming from.
To provide even more information about a soil, subordinate horizon designations are used. These are modifiers of the master horizons that indicate specific properties of a horizon. Table 8-3 gives a summary of master horizon characteristics and some subordinate horizon designations used to modify them.
Table 8-3
Master soil horizons and some common subordinate horizon designationsa

aaSee “Keys to Soil Taxonomy” (Soil Survey Staff, 1998) for a complete listing.
A soil profile is a vertical cross-section of the soil showing all of its constituent horizons. Different soils will have different soil profiles. Soil profiles may differ in the types of horizons they contain, the location of the horizons, or the depth of the horizons. Figure 8-4 gives examples of two different types of soil profiles. The dominant soil-forming processes at a site will determine what type of profile forms. In Fig. 8-4, the presence of a coniferous forest with a cold climate and high rainfall results in O, E, Bhs, Bs, and C horizons (podzolization). Fulvic acids are important in the creation of the E, Bhs, and Bs horizons as described above. Within a prairie, grasses input large quantities of organic matter directly into the soil from root turnover. In conjunction with deep mixing by organisms such as worms and prairie dogs, and with migration of carbonates to the lower profile due to seasonal rains, a deep A horizon develops which is rich in humic acids and humin, often with a Bk horizon below. Prairie soils may also have a Bw or Bt horizon below the A horizon. Other environments and organisms result in many other types of soil profiles that combine to create the pedosphere.
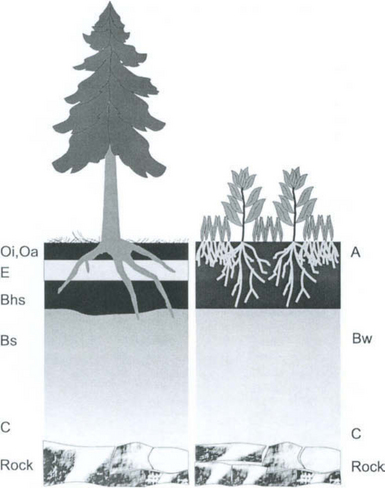
8.3.2 The Pedosphere
The pedosphere is the envelope of the Earth where soils occur and where soil-forming processes are active. Although the biosphere overlaps the pedosphere, the two are not coincident. Soil can develop in areas that are virtually abiotic such as in the high Arctic or in the ice-free areas of Antarctica (Ugolini and Jackson, 1982; Claridge and Campbell, 1984; Campbell and Claridge, 1987). Nevertheless, most soil formation does occur in the presence of life, and soil formation proceeds at a faster rate when biota are abundant (Ugolini and Edmonds, 1983). The rate of development of soils and the type of soil that forms are directly related to the soil-forming factors.
8.3.2.1 Soil-forming factors
In the late 19th century, five factors were found to determine the rate and dominant processes that lead to the development of an individual soil. These are: climate, organisms, topography, parent materials, and time. The effects of climate on soils are numerous. For example, climate determines leaching rates by the quantity of water moving through the soil and determines reaction rates by temperature. It can also affect translocation in a soil by rainfall occurring constantly throughout the year (resulting in increased leaching) or rains occurring seasonally, which may be insufficient to leach ions, and only move material from one horizon to another. Organisms can influence soil by altering the quality and quantity of organic matter inputs and how rapidly they are decomposed or converted to humic substances. Topographic position can alter soil by changing whether a soil is well-aerated or waterlogged. Parent materials, the geologic materials or organic materials that the soil develops from, influence soil texture, acidity, weathering rates, and clay formation. Time determines how well developed a soil is relative to its environment. By continuing soil development for thousands to hundreds of thousands of years, soils become deeper, more highly weathered, contain more clay and, ultimately, under a strong leaching regime become rich in oxides.
8.3.2.2 Soil orders
Soils that have developed in different environments, or with different parent materials or ages result in different combinations of soil horizons and different soil properties. Twelve major types of soil are recognized by the USDA Soil Taxonomy. These 12 soil types are called soil orders, the highest level of soil classification used in the US. Other classification systems use different but similar designations for their highest level of classification (e.g., FAO, 1971–81, Soil Survey Staff, 1998). Soil orders are distinguished by unique diagnostic horizons, unique climatic factors that control soil formation, or unique processes that create specific horizons within a soil. Table 8-4 shows the different soil orders and some unique characteristics and environmental factors that distinguish one from another. The table is arranged so that the soils with the least pedogenic development are at the top and those that are most highly developed are at the bottom. For example, Entisols are soils that have little horizon development, and usually do not even have a B horizon; they may have only a C horizon, or an A–C profile. In contrast, Oxisols are extremely old soils that have lost all their weatherable minerals, most cations, and residual Fe and Al oxides persist in the soil; normally these soils only develop in tropical areas where both temperatures and rainfall are high. Many other soils develop in other environments throughout the world and will be the “climax” soil for that environment.
Table 8-4
USDA soil orders with common ecosystems or environments in which they occur and shown in approximate order from low to high pedogenic developmenta
| Soil order | Unique features | Ecosystem/environment |
| Entisols | Young, normally no B horizon | Variable |
| Gelisols | Cryoturbation; permafrost | Cold soils; polar areas |
| Inceptisols | Young, but have a Bw horizon | Variable |
| Andisols | Andic soil properties (Al humates, low pH, darkly colored A) | Volcanic parent materials |
| Aridisols | Variable | Dry soils; deserts |
| Histosols | All organic soil | Wetlands, riparian areas |
| Mollisols | Thick, dark A horizon with high base saturation | Temperate grasslands |
| Vertisols | > 30% shrink-swell clays, cracking in dry season, deep A | Temperate to subtropical grasslands |
| Alfisols | Bt horizon with moderate base saturation | Temperate deciduous forests |
| Spodosols | Bhs and/or Bs horizon | Cool, wet, coniferous forests |
| Ultisols | Low base saturation and Bt horizon | Tropical to temperate, often forested |
| Oxisols | Highly weathered and leached, residual Fe and Al oxides | Tropical areas with a stable landscape |
aaSee “Keys to Soil Taxonomy” (Soil Survey Staff, 1998) for complete criteria for each soil order.
8.3.2.3 Global soil patterns
Jenny (1941) attempted to quantitatively relate the factors of soil formation to soil properties such as N, C, or clay content, depth of leaching of carbonates, and others. Determining the role of each environmental factor in influencing the development of any particular soil is difficult. Many soils have developed on the landscape over a long time, such soils are therefore polygenetic as they have acquired some of their properties under a constellation of soil-forming factors different from those currently in operation. The soil-forming factors are seldom independent. This is particularly true for organisms, especially vegetation, which is both influenced by climate and can alter microclimate. Many other interactions between soil-forming factors occur, such as between relief or topography and parent materials or time. Nevertheless, knowledge of the soil-forming factors can allow prediction of a general global soil pattern.
Figure 8-5 shows soil orders that form in relation to climate and vegetation along a transect from the poles to the equator along with processes occurring in the soil. In this conceptual diagram, it is assumed that there is a uniform parent material, similar topography and equivalent time for soil formation. The effects of a cold climate and little vegetation result in slow, limited weathering and little organic inputs to soil. In slightly warmer climates, there is more vegetation and more organic inputs to soils. However, due to the cold climate, partial decomposition of litter results in formation of organic acids and intense leaching in the upper part of the soil, with accumulation of Fe and Al in the lower part of the soil – podzolization is dominant here, producing Spodosols. Under deciduous temperate forests, soils can undergo lessivage, resulting in clay accumulations in the lower profile. In such environments deeper soil profiles are common. These soils belong to the Alfisols and Ulfisols. In grasslands and deserts low rainfall may prevent carbonates from leaching out of the soil and keep cations in the soil so that base saturation is high. Grasses can help develop deep A horizons and humic acids can aid in weathering and clay formation (Mollisols). Under a tropical rainforest, old soils can be extremely weathered, cations will have been leached, primary minerals are mostly gone, and only Fe and Al oxides remain in the soil along with kaolinite (Oxisols).
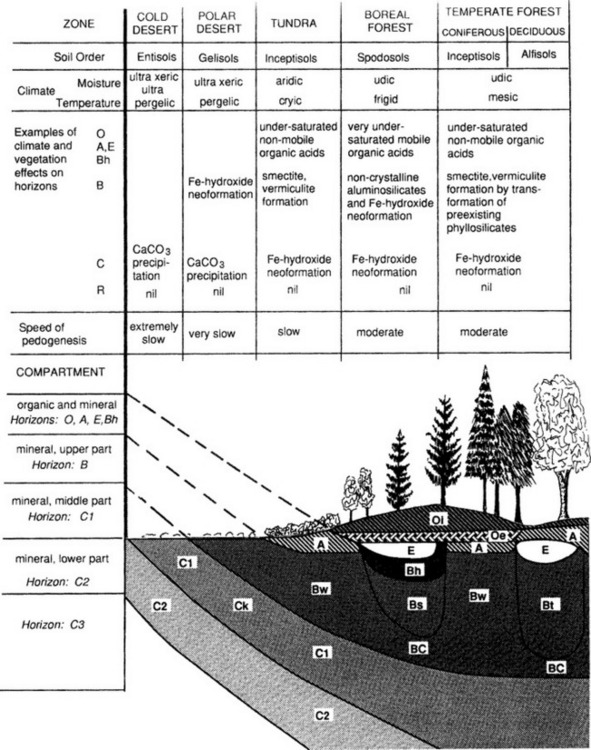
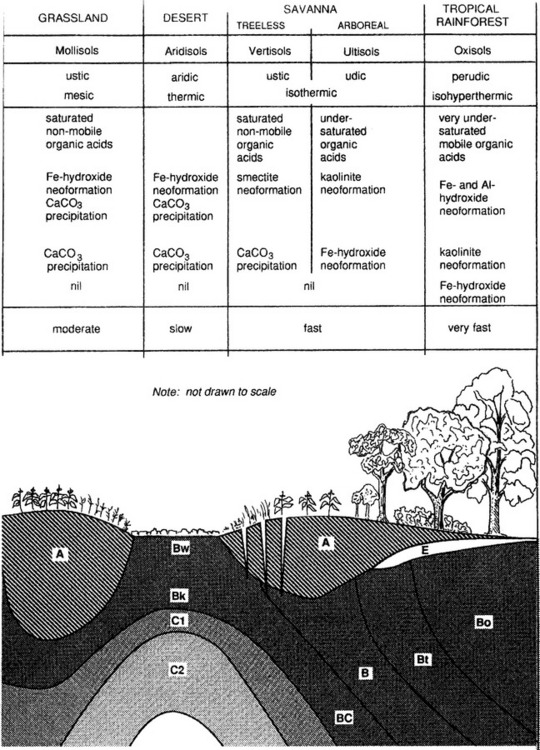
Fig. 8-5 Soil and soil forming processes – a global view. The moisture and temperature regimes are generalized and intended only to show major pedoclimatic environments. Spodosols, for example, can also occur in a cryic regime and even in equatorial regions. Other orders could also occur in more than one moisture and temperature environment. (overleaf)
Plate 3 shows a map of dominant soil orders for the entire world. Although this map necessarily lacks detail due to its scale, the relationship between soils and the biosphere is evident. Different terrestrial ecosystems are correlated with climatic conditions and different soils are correlated with both. For example, Mollisols are common in areas where there are prairies or steppes; a result of grasses as the dominant vegetation and low, seasonal rainfall. Spodosols occur where coniferous forests dominate and the climate is cold and wet. Comparing Fig. 8-5 and Plate 3 carefully will show how strong this correlation is for the entire Earth.
8.3.2.4 Biogeochemical cycling in soils
One of the most important functions of the pedosphere is the cycling of elements that occurs within soils and the transfers that occur between the atmosphere, lithosphere, biosphere, and hydrosphere through soils. Soil is an interface between the atmosphere and lithosphere, between the biosphere and lithosphere, and between roots and soil organisms and the atmosphere. In many ways, soil acts as a “membrane” covering the continents and regulating the flow of elements between these other systems of the Earth.
This soil “membrane” has inputs and outputs, and can transform the elements entering it before these elements leave. Consider the simple cycle of potassium shown in Fig. 8-6. Inputs to the surface of the soil come from atmospheric deposition of particulates, fertilizer applications, and litterfall. Potassium that is released from a crystalline matrix by weathering is also considered an input to soils from rocks even though the rocks are contained within the pedosphere (Zabowski, 1990). Likewise, roots that die and begin to decompose provide inputs of K to the soil – the conversion of organically bound elements to an inorganic form is called mineralization. Within the soil, K can be used to form new clays, attached or released by cation exchange sites, released by dissolution of clays, or taken up by soil organisms (immobilized) where eventually they may be mineralized again when these organisms die. Note how the soil solution functions as the transfer mechanism. Potassium may also be removed from the soil by uptake into the biosphere (in which case it may eventually return to the soil through litterfall), erosion of soil, or leaching to groundwater which flows away from the soil to rivers. By knowing inputs and the quantity of an element in the soil, mean residence times (τr) and turnover times (τ0) can be calculated. The basis for these calculations is presented in Chapter 4. Biogeochemical cycling in soils is further complicated by the different soil processes occurring in the different soil orders (see Fig. 8-6 and Table 8-4) which all cycle K and other elements at different rates.
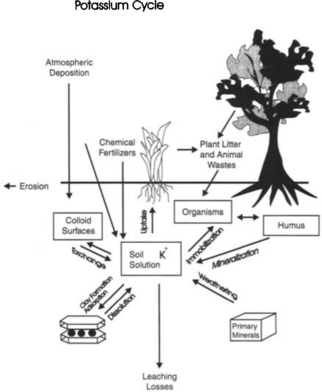
The cycle of potassium is quite simple, as K does not change valence states or have a gaseous phase. In contrast, elements such as carbon and nitrogen both change phase and undergo redox reactions and undergo much more complicated cycling. For example, carbon captured by plants from the atmosphere is reduced to form organic matter, which is then oxidized by either organisms in the soil, or in roots to provide energy with a release of CO2 if the soil is aerobic. Methane may be released if the soil is anaerobic. Other conversions of carbon to humic substances can make it very resistant to further decomposition. The τr of C in various soil fractions can range from a few months to thousands of years (Schlesinger, 1997). Nitrogen cycling is also very intricate, because N can exist as N2, N2O,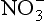 ,
,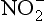 ,
,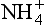 , and NH3 in soils. Although nitrogen is rarely input to soils by weathering, nitrogen-fixing organisms capture atmospheric N2 and thereby act as a source of the element to soils. The rates at which nitrogen converts from one form to another in soil affect the rate of transfer of N from one “sphere” to another, owing to the fact that soils are at the interface of the atmosphere, biosphere, and hydrosphere, where nitrogen compounds reside. A complicating factor is that nitrogen is the nutrient most commonly limiting to plant growth. Carbon and nitrogen cycling are discussed in more detail in Chapter 11 and 12, respectively.
, and NH3 in soils. Although nitrogen is rarely input to soils by weathering, nitrogen-fixing organisms capture atmospheric N2 and thereby act as a source of the element to soils. The rates at which nitrogen converts from one form to another in soil affect the rate of transfer of N from one “sphere” to another, owing to the fact that soils are at the interface of the atmosphere, biosphere, and hydrosphere, where nitrogen compounds reside. A complicating factor is that nitrogen is the nutrient most commonly limiting to plant growth. Carbon and nitrogen cycling are discussed in more detail in Chapter 11 and 12, respectively.
8.4 Watershed Processes
Watersheds, also known as drainage basins, define a natural context for the study of relationships among soils, geology, terrestrial ecosystems, and the hydrologic system because water and sediment travel downslope under the influence of gravity. This material is a continuation of some of what was presented in Chapter 6.
The concept of a drainage basin has no inherent scale, since watersheds range from small headwater valleys to the catchments of huge rivers that drain continents. The physical and chemical load carried in a river is produced by watershed processes, including weathering and exchange processes in the soil, how the water that runs off of a landscape as streamflow (often simply called runoff) is generated, and the relative importance of different geomorphological processes, climate, and the lithology of the bedrock. Climate and topography are two of the strongest influences on the sediment load of rivers, although sediment routing processes and long-term storage of sediment in floodplains and structural valleys can create substantial time lags and differences in net sediment delivery to the oceans.
8.4.1 Runoff Processes
The processes through which rainfall is turned into runoff, together with the nature of the material through which water moves, control the chemical characteristics of streamflow. Specific runoff mechanisms operating in a landscape control the flowpaths by which water moves through the landscape. Flowpath-dependent differences, such as the total time that water spends in contact with different soil horizons or bedrock (residence time), can strongly influence runoff amounts and timing, the relative contribution of event (new) versus stored (old) water, and runoff chemistry.
8.4.1.1 Runoff mechanisms
The translation of rainfall into runoff occurs by a variety of mechanisms associated with different environments (refer back to Fig. 6-7). Horton overland flow (HOF) occurs primarily in arid or disturbed landscapes where rainfall intensity exceeds the infiltration rate of the ground surface long enough for ponding to occur (Horton, 1933). Observations that most rainfall infiltrates into the soil in humid, soil-mantled landscapes, and therefore that HOF is rare in such environments, led to the recognition of subsurface flow as a major mechanism of storm runoff (Loudermilk, 1934; Hursh, 1936). Subsurface stormflow (SSSF) dominates runoff generation in steep soil-mantled terrain where precipitation infiltrates and flows laterally either through macropores, or over a lower conductivity zone, such as at the base of a root mat or at the soil-bedrock boundary. Saturation overland flow (SOF) occurs in soil-mantled landscapes when an initially shallow water table rises enough to intersect the ground surface over a portion of the catchment (Hewlett and Hibbert, 1967), which then causes runoff by either return flow or direct precipitation onto saturated areas (Dunne and Black, 1970). Topographically driven patterns of soil moisture favor development of SOF in low-gradient, convergent topography where flow is concentrated. Water that infiltrates through the soil to the regional groundwater table contributes to groundwater flow that maintains low flows between storm events. The relative importance of these runoff generation mechanisms depends on several key ratios: rainfall intensity to infiltration rate; drainage area to local slope; and bedrock to soil conductivity (Fig. 8-7).
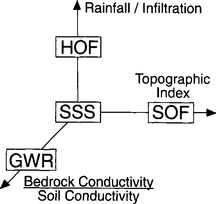
Fig. 8-7 Three principal ratios control the style of runoff generation prevalent in a landscape: (1) ratio of rainfall intensity to the infiltration capacity of the soil; (2) ratio of bedrock conductivity to soil conductivity; and (3) the topographic index defined by the ratio of the upslope drainage area to the ground slope. HOF = Horton overland flow; SOF = saturation overland flow; SSS = subsurface stormflow; GWR = groundwater flow.
8.4.1.2 Runoff chemistry
The soil is the connecting link between rainfall and river flows, and the interaction of runoff processes and soils regulates the dissolved load in the hydrologic system. The soil plays a complex role in biogeochemical cycles, and the elements found in rivers as soluble salts or as particles are derived from weathering of rocks (Si, Al, Fe, Mg, Ca, K, P) or atmospheric fallout (S, N). One could predict the quality and quantity of this load to depend strongly upon the soils surrounding the river, but this is not always the case. In fact, the chemistry of rivers flowing on granitic terrains in western Europe and tropical Africa appear rather similar, although the soils in these two regions are dramatically different (Tardy, 1969). Such counter-intuitive results indicate that the dissolved load of rivers reflect not only soil properties, but also runoff pathways, lithological influences, and other environmental factors.
Most soil orders can be divided into two major compartments: an organo-mineral compartment on top, referred to as the biogeochemical compartment, and an underlying mineral compartment, referred to as the geochemical compartment. The character of runoff from the biogeochemical compartment depends strongly on the complex interactions among the climate, biota, parent material, topography and time, whereas in the geochemical compartment these interactions are less important. The chemistry of a river depends mostly on whether runoff flows overland, and thereby has little interaction with the soil, or on the level at which the water leaves the soil, as the soil solution can change dramatically as water moves through the different soil horizons.
8.4.1.2.1 Overland flow.
Runoff by overland flow carries material to the river as particles of sand, silt, clay, and organic matter. This material consists of plant debris, more or less decomposed, and aggregates of humified organic material and mineral particles. Runoff from different soil orders developed on similar bedrock will also reflect soil properties; Spodosols developed on granite, for example, produce overland flow runoff that consists of very acidic organic matter, whereas overland flow from a Mollisol developed on a similar lithology yields aggregates of neutral humic acids and clay. In most environments, however, the load carried to the rivers by overland flow generally has a high nutrient content.
8.4.1.2.2 Biogeochemical compartment.
The biogeochemical compartment consists of the O, A, E, and B horizons of soils, in which substantial organic matter can be an integral component of the soil. The load of the water leaving the soil from the biogeochemical compartment consists mainly of soluble compounds because the matrix of the soil acts as a filter that retains particulate matter. In some cases, however, clay and particulate humic substances are also carried away. The total dissolved load strongly reflects whether the soil experiences intense or mild weathering. Intense weathering is due to very acidic conditions or a very weatherable parent material which results in a soil solution that contains relatively high amounts of silica as H4SiO4. This silica cannot react with Al or Fe, because the latter elements are effectively complexed by humic substances. In addition, different soil orders yield different types of solution from the biogeochemical compartment. For example, Spodosols yield a very acidic and yellow solution containing fulvic acids partially saturated with Al and Fe, and containing some N and P, whereas Mollisols yield a neutral solution that contains relatively small amounts of organic matter. In general, the water exiting the biogeochemical compartment is likely to contain low concentrations of nutrients such as N and P, and higher concentrations of Ca, Mg, K, and Na in solution, accompanied by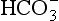 and
and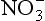 , small amounts of phosphate, and very little Fe and Al.
, small amounts of phosphate, and very little Fe and Al.
8.4.1.2.3 Geochemical compartment.
The geochemical compartment consists of some lower B horizons, C horizons, and bedrock flowpaths. The solution entering streamflow from the geochemical compartment generally contains very little organic matter. For this reason, organic C and N, as well as Al and Fe, are present only in very small amounts, if at all. In this mineral environment, pH is a critical factor affecting the mobility of P because it is often too alkaline or too acid for P to exist as a soluble anion. The load carried to rivers from the geochemical compartment consists mostly of alkaline cations (Mg2+, Ca2+, Na+, K+), and the anion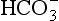 . Silicic acid (H4SiO4) losses are strongly attenuated under mild weathering conditions, and therefore the Si concentration of the water reaching a river from the geochemical compartment is relatively low.
. Silicic acid (H4SiO4) losses are strongly attenuated under mild weathering conditions, and therefore the Si concentration of the water reaching a river from the geochemical compartment is relatively low.
The combined influences of runoff generation mechanisms, runoff flowpaths, and soil properties together control runoff chemistry. In spite of the wide range of interactions that characterize terrestrial environments, a few broad generalities can be offered, as the chemical composition of streamflow typically contains little H4SiO4, variable amounts of alkaline cations (Ca, Mg, Na, K), and the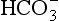 anion. There is little organic C or N, and a virtual absence of Al, Fe, and P. Prediction of runoff chemistry is complicated because the specific runoff pathways operating in a particular environment can have a greater impact on the composition of the river water than the composition of the soil itself.
anion. There is little organic C or N, and a virtual absence of Al, Fe, and P. Prediction of runoff chemistry is complicated because the specific runoff pathways operating in a particular environment can have a greater impact on the composition of the river water than the composition of the soil itself.
8.4.2 Sediment Load of Rivers
The material transported by rivers consists of dissolved ions (dissolved load), sediment suspended in the flow (suspended load), and sediment transported along the bed of the river (bedload). The total load and the proportion of the load represented by these phases varies widely among rivers in different environments. In particular, climate, topography, and erosion influence the amount and composition of riverine sediment loads.
A typical watershed can be considered to be composed of a source area in its uplands, where sediment is produced from rock and introduced into the fluvial system, transport zones in which there is little storage of material, and downstream depositional areas where substantial sediment storage may occur (Schumm, 1977). The specific characteristics of a watershed control the connections and time lags between initial erosion of a sediment and its ultimate delivery to the marine environment. Sediment storage in floodplains can substantially delay sediment delivery as material may be stored there as floodplain deposits for decades to millenia before it is reintroduced to the river. The time required to route sediment out of a drainage basin is important for interpreting the cause and timing of erosional events that are recorded in marine sediments. Church and Slaymaker (1989), for example, showed that reworking of late Quaternary deposits dominates the contemporary sediment yield of the Fraser River in British Columbia, Canada.
The sediment load of a channel and the sediment yield of its drainage basin are expressed in different ways. The sediment load is the total mass of material moved by the river in a specified period of time, whereas the sediment yield of a drainage basin is the sediment delivered from the basin divided by the basin area and typically expressed in tonnes/km2 per year. Hence, the sediment yield for a basin may decrease downstream, due to sediment storage, even though the total sediment load of the river draining it increases downstream. Comparison of sediment yields allows assessment of relative erosion rates among drainage basins on an equal area basis, and thereby normalizes for the effect of basin size on sediment load.
8.4.3 Dissolved Load
The dissolved load of rivers consists of material leached from the soil. In most natural ecosystems, the water leaving the soil and entering rivers contains mainly the bicarbonate anion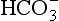 , a primary product of mineral weathering, and alkaline cations (mostly Ca2+ and Na+). Nutrients such as N and P are added to the rivers in small amounts by other processes – soil erosion at the edge of the river, runoff, and from direct inputs such as leaves and other organic debris. Therefore, the nutrient content of rivers and lakes is generally low. In a heavily forested and still pristine watershed of southeast Alaska, Stednick (1981) measured the output of N and P to be about 4.5 and 0.8 kg/ha per year, (1 ha = 103 m3) while it reaches 185 kg/ha per year for
, a primary product of mineral weathering, and alkaline cations (mostly Ca2+ and Na+). Nutrients such as N and P are added to the rivers in small amounts by other processes – soil erosion at the edge of the river, runoff, and from direct inputs such as leaves and other organic debris. Therefore, the nutrient content of rivers and lakes is generally low. In a heavily forested and still pristine watershed of southeast Alaska, Stednick (1981) measured the output of N and P to be about 4.5 and 0.8 kg/ha per year, (1 ha = 103 m3) while it reaches 185 kg/ha per year for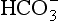 , 275 kg/ha per year for Ca, 40 kg/ha per year for Na and 80 kg/ha per year for Si (measured as H4SiO4). One of the most important anthropogenic impacts for temperate ecosystems is the huge input of nutrients such as N and P to rivers that can occur from clear cutting of forests on steep slopes, drainage of wetlands, excessive fertilizer application, and agricultural practices that induce topsoil erosion.
, 275 kg/ha per year for Ca, 40 kg/ha per year for Na and 80 kg/ha per year for Si (measured as H4SiO4). One of the most important anthropogenic impacts for temperate ecosystems is the huge input of nutrients such as N and P to rivers that can occur from clear cutting of forests on steep slopes, drainage of wetlands, excessive fertilizer application, and agricultural practices that induce topsoil erosion.
In arid areas, runoff is often the main source of water reaching the valley bottom. The rivers carry a high nutrient load consisting mainly of N and P. Throughout the world, estuaries of rivers draining arid lands, or the lakes they empty into, are incredibly rich in aquatic life. Flood plains located downstream of arid areas are also known for their rich soils. The Yellow River in China, the Colorado in the United States, and the Nile in Africa, are only some of the most famous examples. The construction of dams along these rivers allows flood control, and water for irrigation and power, but the retention of sediment behind the dams severely impacts some of the richest and most productive ecosystems in the world.
In humid areas, the concentration of soil-derived elements in river water is generally low. With humid and tropical climates, the concentration of H4SiO4 in soil solution is dictated by the equilibrium with kaolinite and tends to be low (Tardy, 1969; Kittrick, 1977; Velbel, 1985; Clayton, 1986; Pavich, 1986). In parent materials that are well drained and coarse textured, the H4SiO4 concentration in the soil solution going through the lower mineral compartment can be low enough to lead to gibbsite formation, even in temperate climates (Green and Eden, 1971; Dejou et al., 1972; Macias-Vaquez et al., 1987). In wetlands, river loads originate within the organo-mineral compartment and the rivers are rich in nutrients; however, the acidity of the water inhibits development of aquatic biomass and wildlife, especially in cold climates.
8.4.4 Bedload and Suspended Load
The physical transport of particles in a river occurs by two primary modes: bedload and suspended load. Bedload consists of material moved along the bed of the river by the tractive force exerted by flowing water. Bedload may roll or hop along the bottom, and individual particles may remain stationary for long periods of time between episodes of movement. Suspended load consists of material suspended within the flow and that is consequently advected by flowing water. Rivers and streams are naturally turbulent, and if the upward component of turbulence is sufficient to overcome the settling velocity of a particle, then it will tend to remain in suspension because the particles become resuspended before they can settle to the bottom of the flow. Suspended load consists of the finest particles transported by a river, and in general is composed of clay- and silt-sized particles, but larger material can be suspended in especially fast water. The finest component of the suspended load is transported as washload that never settles and is therefore rapidly transported from a drainage basin. Suspended load constitutes the majority of sediment transported by most rivers, usually more than 80%, but there is substantial variability in the ratio of these two types of transport.
The travel time for suspended load is controlled by the flow velocity and the distance to the basin outlet. Flow velocities do not change much downstream in a typical river system (Leopold, 1953) and typically range from 0.1 to several m/s. Hence, suspended load should be able to travel at least 10 to 100 km per day and the travel time for suspended sediment to traverse even the longest rivers in the world should be less than a season. Although some of the suspended load will be deposited in floodplains, the component of the suspended load that does not get sequestered in terrestrial depositional environments is delivered almost as fast as the water that it flows in. Bedload travels much more slowly. In mountain drainage basins, the velocity of individual bedload clasts is on the order of kilometers per year in steep mountain channels and may be on the order of only from one gravel bar to the next (tens to hundreds of meters) per year in lower-gradient pool-riffle type channels. Hence, we can think of a decoupling of transport rates, with transport of fine sediment involving relatively rapid response and coarser sediment potentially having much greater time lags between erosion and ultimate deposition in the ocean. At present, the combined global total for suspended load discharge into the oceans is ca. 20 Pg/yr (Milliman and Syvitski, 1992). A map showing the distribution of this discharge for rivers around the world is given in Fig. 8-8.
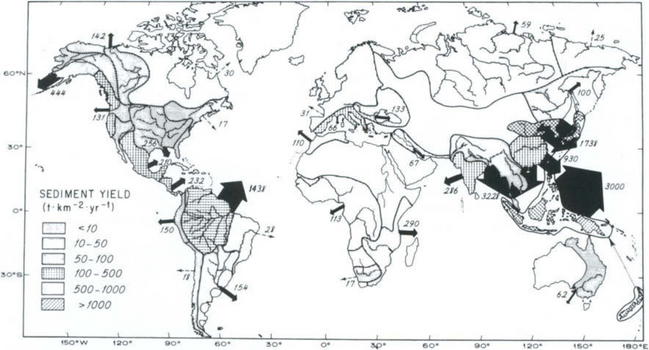
Fig. 8-8 Annual suspended sediment discharge. Relative discharge is indicated by the width of the arrows. (From Milliman and Meade, 1983, reproduced with permission from University of Chicago Press.)
8.4.5 Climate, Topography, and Erosion
A fundamental distinction among landscapes is whether the net sediment flux (the total load carried by the river) is limited by the ability of erosional processes to carry sediment (transport-limited environments) or the availability of erodible material (weathering-limited environments). In general, soil-mantled landscapes can be considered transport limited, whereas landscapes with bedrock hillslopes are weathering limited. While the thickness of the soil mantle on hillslopes is dictated by the ratio of soil production to soil erosion rates (bedrock hillslopes result where erosion rates chronically exceed soil production rates), climate and vegetation are primary controls on both soil production and erosion. Wet climates with abundant vegetation tend to have soil-mantled landscapes, whereas dry climates with little vegetation tend to have relatively greater exposure of bedrock. Soil production increases with both greater precipitation and more extensive vegetation, whereas soil erosion rates also increase with greater precipitation but are inversely related to the extent of vegetation cover. The combined influence of vegetation acting to protect the ground surface and the tendency for higher rainfall to cause greater erosion leads us to expect a relation between mean annual precipitation and erosion rates in which erosion rates increase with increasing precipitation in arid areas to a maximum in semi-arid lands and then decrease in temperate and tropical areas due to the binding effect of vegetation counteracting the erosive potential of rainfall in areas with a complete ground cover of vegetation (Fig. 8-9).
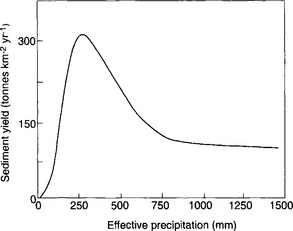
Fig. 8-9 Generalized variation of sediment yield with precipitation. (Modified from Langbein and Schumm, 1958.)
The proportion of the total load of a river that is composed of dissolved load varies globally from close to 90% in the St. Lawrence river that drains low-gradient portions of Canada where numerous lakes act as effective traps for suspended and bedload material, to less than 10% for the Ganges river which receives a huge load of mechanical debris from the very high erosion rates in the Himalaya (Table 8-5). There are also general climatic controls on the relative importance of chemical and mechanical erosion on the composition of the load of rivers. The weathering history of a landscape is an important control on sediment yield (McLennan, 1993). Tropical areas with high rainfall and high temperatures tend to have high chemical loads, whereas cold environments tend to have relatively high mechanical loads due to relatively slow chemical weathering in such environments. However, watershed-specific geologic factors superimpose substantial local variability onto such general global patterns.
Table 8-5
Proportion of total load represented by dissolved load for large rivers.
| River | Load(%) |
| Ganges | 8 |
| Amazon | 18 |
| Mississippi | 20 |
| Yukon | 28 |
| Zaire | 42 |
| Volga | 64 |
| St. Lawrence | 89 |
(Data from Summerfield, 1991)
Climate also exerts a primary control on the rates of mechanical erosion processes. While there has long been controversy over whether rivers or glaciers produce higher erosion rates, the consistently highest contemporary sediment yields come from glaciated catchments (Hallet et al., 1996). Whether a glacier is frozen to its bed is also a very important control on the erosion rates, since such “cold bed” glaciers cause far less abrasion of their beds than do “warm bed” sliding glaciers. Hence, mountain drainage basins with valley glaciers in temperate environments typically have higher sediment yields than ice caps in polar landscapes.
Erosion rates by fluvial, hillslope, or glacial processes generally increase with increasing slope, and bedrock outcrops are found on the steepest slopes in most landscapes. Local relief (the difference between the highest and lowest points in an area of interest) is also correlated with erosion rates (Fig. 8-10) (Ruxton and McDougall, 1967; Ahnert, 1970), as areas with steep slopes tend to characterize areas of high relief and result in higher erosion. Deeply incised topography with steep slopes tends to produce the highest erosion rates.
Fig. 8-10 Erosion rate as a function of relief for (a) mid-latitude medium-sized drainage basins, and (b) for Hydrographer’s Volcano, Papua New Guinea. (modified from Ahnert, 1970), (data source: Ruxton and McDougall, 1967)
Human or natural actions that significantly alter the erosion resistance of the ground surface can lead to dramatic increases in erosion rates. Channel networks dissect natural landscapes down to a fine-scale limit controlled by a threshold of channel initiation (Montgomery and Dietrich, 1992). In effect, channels begin where sufficient discharge collects to overcome the erosion resistance of the ground surface (Horton, 1945; Montgomery and Dietrich, 1988). Consequently, human or natural actions that alter the critical shear stress of the ground surface can trigger expansion of the channel network to generate a network of rills, small channels and gullies that can rapidly dissect formerly undissected hillslopes, leading to very high sediment yields. Different styles of land use can result in very different erosion rates. For example, Dunne (1979) studied erosion rates from undisturbed forest, cut forest, and agricultural lands in Kenya and showed that sediment yields were substantially higher in managed landscapes where ground disturbance was common (as shown in Fig. 8-11). Human-induced acceleration of erosion can result in sediment yields that greatly exceed the range of natural variability in all but the most catastrophically disturbed environments. Accelerated erosion from human activity is thought to have doubled the sediment yield to oceans over the past several thousand years, although recent dam construction has reduced this effect (Hay, 1998).
Fig. 8-11 Annual sediment yield for various land uses in northern Mississippi vs. annual precipitation. (After Ursic and Dendy, 1965.)
8.4.5.1 Sediment delivery
Sediment transport rates by fluvial processes are proportional to the product of flow depth and the energy gradient, or water surface slope (Richards, 1982). Hence, sediment transport increases with either deeper flow or steeper slopes, and conversely sediment transport rates decrease in shallower flow or on gentler slopes. Consequently, the transport capacity of a river decreases where flow spreads out across wide valley bottoms (such as in floodplains), or where channel gradients decrease abruptly (such as in alluvial fans at the foot of mountain ranges). The downstream decrease in transport capacity in turn leads to deposition of sediment in these locations. The proportion of the sediment eroded from a drainage basin that actually makes it to the mouth of the basin defines the sediment delivery ratio for the basin. This ratio tends to decrease with increasing basin size due to storage effects along floodplains or in structural valleys, and ranges from virtually complete delivery for short, steep catchments to less than half of the sediment load for large, continent-spanning rivers. In large floodplain river systems, such as the Amazon and the Ganges-Brahmaputra, roughly a third of the sediment load can be sequestered into long-term storage in floodplain sediments (Dunne et al., 1998; Goodbred and Kuehl, 1998).
8.5 Marine Sediments
Sediments record aspects of the environments from which they were eroded and into which they were deposited. Consequently, a sequence of sedimentary layers contains information about environmental changes over time. Sediments are therefore a library of Earth history that can be read through an understanding of the processes behind their formation and post-depositional evolution. The recent sedimentary record reveals cultural impacts on the environment during the industrial era, while ancient sedimentary rocks provide insight into the evolution of free oxygen in the atmosphere during the Precambrian Era. But sediments are not simply inert records of past conditions; during formation and diagenesis they take an active part in biogeochemical cycles.
8.5.1 Formation of Sediments
Disintegration of rocks by weathering processes produces three types of weathering products: detrital material from the rock and vegetation; solutes from the dissolution of minerals and organic matter; and new minerals from chemical reactions between solutes and minerals. Marine sediments are formed by the deposition of these particles once they are transported to the ocean by rivers, glaciers and wind. Episodic delivery processes result in marine sediments that consist of depositional layers that record discrete depositional events, whereas a soil exhibits weathering horizons that record developmental processes, as discussed in Section 8.3.2. The mineral composition of detrital material depends on the type of source rock, and also on the duration and intensity of weathering and transport.
8.5.2 Composition of Sediments
Like soil, sediment contains three phases, and interactions among these phases can alter the nature of sediments through time.
8.5.2.1 The solid phase
The solid components of sediments are classified in a number of ways. Grain size conveys some basic information for exchange processes and correlates to a certain extent with the minerogenic distribution. Clay minerals, for example, occur in the < 2 µm size fraction. For biogeochemical purposes, however, a granulometric description is far from sufficient. A more satisfactory classification is based on the material and genetic differences of marine sediments (Table 8-6). The arrangement of the particles is of even greater importance as it governs the permeability and porosity of the sediments. Marine sediments (with few exceptions) are composed of relatively porous clay aggregates and randomly distributed coarser grains of detrital material (Fig. 8-12).
Table 8-6
Material-genetic classification of marine sedimentsa
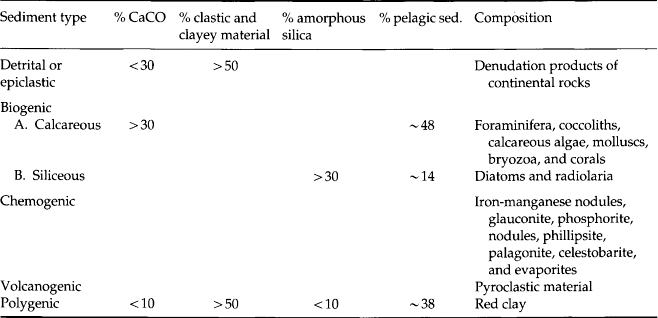
aaModified from Lisitzin (1972) and Sverdrup et al. (1942).
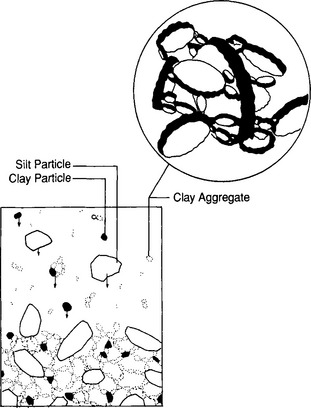
Fig. 8-12 Hypothetical particle arrangement in a sediment and a clay aggregate. (Adapted from Casagrande, 1940.)
The aggregates of sedimentary particles are usually arranged before deposition. Within these aggregates, forces are set up between atoms, molecules, and ions that depend on the ionic strength of the electrolyte. As a result, the arrangements of the particles are different in brackish and marine environments. Clays deposited in fresh water form relatively porous aggregates with small voids, while marine clays form large dense aggregates separated by large voids. Salinity variations in interstitial water are most significant in coastal areas and different degrees of permeability and porosity therefore may be expected in sediments of the continental margins as compared to deep sea sediments of the same grain size.
8.5.2.2 The liquid phase
The liquid phase of sediment consists of three different types of aqueous electrolytic solutions: (1) “normal” water of random ionic ordering at some distance from a solid surface; (2) adsorbed water, essentially free from ions, on a solid surface; and (3) “structured” water with a layering of ionic ordering of types (1) and (2) near a solid surface. Because of the close relationship to the mineral particles in the sediment, interlamellar water is usually of types (2) and (3). The various types of water in sediments strongly influence other processes taking place in the aqueous phase.
8.5.2.3 The gaseous phase
The gaseous phase of the sediment-water interface includes oxygen, nitrogen, hydrogen sulfide, methane, carbon dioxide, and ammonia. The last-named occurs principally as the ammonium ion at pH values in the range 5 to 8. Oxygen and hydrogen sulfide react with each other spontaneously, and consequently these two gases are usually regarded as non-coexistent. In a sediment, however, heterogeneity of organic matter and permeability give rise to microenvironments of different chemical composition that can exist close to one another as more or less isolated chemical and biological systems. The presence of hydrogen sulfide is not therefore a reliable indicator of entirely anoxic conditions within the sediment. Fecal pellets may serve as such microniches at the uppermost part of the sediment, while burrowing organisms may distribute oxygen to the reduced part of the sediment.
Methane is produced by bacteria in the strongly reduced part of the sediment. Methane-producing bacteria are obligatory anaerobes and are most active below the zone of hydrogen sulfide production. Methane is used as a carbon source by heterotrophic microorganisms in the overlaying sediments. However, methane concentrations decrease rapidly below the sediment-water interface, and its importance for the biogeochemical cycles of carbon in the marine system is negligible.
Carbon dioxide is produced as a result of metabolism of all heterotrophic organisms. The concentrations of CO2 in pore water of reduced sediments are therefore high. Autotrophic microorganisms consume CO2 in the oxidized part of the sediment, which can vary in depth from a meter in deep sea sediments to a few mm in organic-rich sediments of the continental margins. The buffer capacity of ocean water is very low and an addition of 0.5 mmol/kg of CO2 will reduce the pH of seawater by more than one pH unit. It is therefore surprising that the concentration of CO2 in pore water of marine sediments may reach values of 60 mmol/kg (Presley, 1969), and yet show pH shifts less than one unit. The increase in CO2 therefore must be counterbalanced by other processes that tend to increase the pH (such as production of ammonia and increase in total alkalinity) so that the result is a fairly constant pH.
8.5.3 Diagenesis
Physico-chemical reactions within the sea-sediment interface tend to reach equilibrium. Those reactions that are so rapid that they occur prior to burial are referred to as halmyrolysis (e.g. formation of clay aggregates) while those that take place in the sediment are termed diagenesis. The diagenetic processes include cementation, compaction, diffusion, redox reactions, transformation of organic and inorganic material, and ion exchange phenomena. Only a short survey of some diagenetic processes is presented here; for a more detailed presentation of diagenetic processes the reader is referred to Berner (1980).
8.5.3.1 Cementation
Cementation is the precipitation of a binding material around grains, thereby filling the pores of a sediment. Berner (1971, p. 97) states that “cementation by silica must be predominantly a phenomenon of later diagenesis because almost no examples are found in recent marine sediments.” In contrast, cementation by calcium carbonate may occur rapidly after deposition. A good example is beachrock, a mix of beach and intertidal sand (usually carbonate and skeletal fragments) cemented by CaCO3 in subtropical to tropical climates. The cement forms so rapidly that human artifacts only a few decades old are commonly found cemented into the beachrock. Other examples of early diagenetic CaCO3 cementation are provided by subtidal lithified calcarenites, reefs, and pelagic oozes (Mackenzie et al., 1969). In all cases the cements are aragonite or high magnesium calcite indicating formation from seawater or other magnesium-rich solutions.
8.5.3.2 Compaction
Compaction is the decrease in volume of a sediment resulting primarily from an expulsion of water due to compression from deposition of overlying sediment. Rosenqvist (1962) stated on the basis of a study of approximately 100 stereoscopic micrographs of undisturbed marine clays that the particle arrangements within the clay aggregates were of the “corner/plane cardhouse” type (Fig. 8-12) suggested by Tan (1957) and Lambe (1958). From microstructural investigations of these “cardhouse” aggregates (Pusch, 1970) it can be concluded that during the compaction process, the aggregates approach one another and assume new, more stable orientations.
Thus the initial state of compaction results in a collapse of the more unstable original structures and a rearrangement of the particles into a tighter packing. More efficient packing will expel interstitial water from the sediment and decrease the porosity. As different clay minerals exhibit different packing characteristics, which in turn depend upon the chemical composition of the interstitial water, relationships become too complex for a simple generalization (Meade, 1966). The total compaction that occurs during accumulation of a sediment layer can be calculated from the following formula with the assumption that the rate of burial of sediment and the diagenetic processes, including bioturbation, are constant:
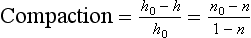
where h0 = thickness of the layer at deposition; h = observed thickness of the layer; n0 = porosity of the top-most sedimentary layer; n = porosity of the observed layer.
For the upper part of the sediment, where the sea-sediment interaction is most predominant, steady-state compaction is a reasonable assumption, as porosity and compaction undergo a linear change during burial.
8.5.4 Diffusion
Diffusion as referred to here is molecular diffusion in interstitial water. During early diagenesis the chemical transformation in a sediment depends on the reactivity and concentration of the components taking part in the reaction. Chemical transformations deplete the original concentration of these compounds, thereby setting up a gradient in the interstitial water. This gradient drives molecular diffusion. Diffusional transport and the kinetics of the transformation reactions determine the net effectiveness of the chemical reaction.
The presence of particles in the fluid medium complicates diffusion in a sediment due to the effects of porosity, represented by n, and tortuosity. Since tortuosity of natural sediments is seldom known it is more convenient to use the term “formation factor” or “lithological factor,” denoted L, which takes into account everything but porosity. Fick’s diffusion constant D is replaced by the whole sediment diffusion constant Ds, where Ds < D.
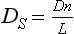
Manheim (1970) presents a critical review of the confusing variety of Ds data found in the literature, but according to Horne et al. (1969), pressures up to several thousand atmospheres seem to have no significant effect on Ds. Values for Ds are sometimes not available or are difficult to estimate but can be obtained indirectly by means of electrical conductivity measurements (Klinkenberg, 1951). Table 8-7 provides a list of Ds values found in the literature for the most common dissolved chemical species in sediment.
Table 8-7
Diffusion constants of sediments (Ds) for some common chemical speciesa
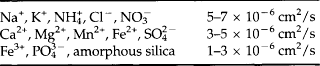
aa The data given should serve only as reference values following the rule, the higher the ionic potential, the thicker the hydration layer of the water molecules around the ion, and the slower the ionic diffusion. Cations generally diffuse more rapidly than anions.
Interactions between diffusion and chemical transformation determine the performance of a transformation process. Weisz (1973) described an approach to the mathematical description of the diffusion-transformation interaction for catalytic reactions, and a similar approach can be applied to sediments. The Weisz dimensionless factor compares the time scales of diffusion and chemical reaction:
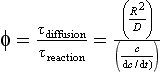
where R is a characteristic size of sediment particles. Using this approach, we can identify the following cases: (1) when ϕ < 1 the time scale for diffusion is short compared with that for chemical reaction and reactivity will determine the rate of change of concentration; and (2) for ϕ > 1 the time scale for diffusion is long and diffusion will play an important role in determining the reaction rate.
Finally, it must be stressed that diffusion of dissolved species in solutions is a key physico-chemical process for the sea/sediment interaction and energy exchange at the sediment-water interface. The reader is referred to Cussler (1984) for a more comprehensive presentation of diffusion in fluid systems.
8.5.5 Redox Conditions
Reducing and oxidizing conditions in a sediment determine the chemical stability of the solid compounds and the direction of spontaneous reactions. The redox state can be recognized as a voltage potential measured with a platinum electrode. This voltage potential is usually referred to as E or Eh defined by the Nernst equation, which was introduced in Chapter 5, Section 5.3.1:
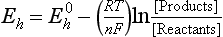
The Nernst equation is applicable only if the redox reaction is reversible. Not all reactions are completely reversible in natural systems; activities of reacting components may be too low or equilibrium may be reached very slowly. In a sediment, the biotic microenvironment may create a redox potential that is different from the surrounding macroenvironment. For this reason, measurements of Eh in natural systems must be cautiously evaluated and not used strictly for calculations of chemical equilibria. Calculations of redox equilibria are in some cases valuable, in the sense that they will give information about the direction of chemical reactions.
If a system is not at equilibrium, which is common for natural systems, each reaction has its own Eh value and the observed electrode potential is a mixed potential depending on the kinetics of several reactions. A redox pair with relatively high ion activity and whose electron exchange process is fast tends to dominate the registered Eh. Thus, measurements in a natural environment may not reveal information about all redox reactions but only from those reactions that are active enough to create a measurable potential difference on the electrode surface.
In marine sediments, usually only the uppermost layer of the sediment exhibits oxidizing conditions while the rest is reduced. The thickness of the oxidized layer and the reducing capacity of the sediment below depend on:
1. concentration of oxygen in the ambient bottom water;
2. the rate of oxygen penetration into the sediment;
3. the accessibility of utilizable organic matter for the bacterial activity.
The depositional rate of organic matter is higher in the coastal areas than in the open sea. Even though the exchange of water is higher in shallow areas of the ocean the deep water is not deficient in oxygen. We should thus expect to find a general trend of thicker oxidized sediments with increasing distance from the shoreline. Lynn and Bonatti (1965), for example, found that the thickness of the oxidized upper sediment in the Pacific Ocean between 15° S and 20° N increased with distance from the continent. Thicknesses were < 1 cm near the shore and 8–15 cm at distances of 800 km offshore.
The inflection point of the redox gradient, constituting the boundary between oxidizing and reducing environments in sediments lies at around +250 mV. This boundary, the redoxcline (Hallberg, 1972), is recognized as comparable to other natural boundaries, such as the halocline and thermocline. The redoxcline is usually situated close to the sediment-water interface resulting in a redox turnover from oxidizing to reducing conditions during early diagenesis. The redox turnover will, in turn, produce disequilibrium within the sediment, causing dissolution of certain minerals and compounds and increased ion exchange across the redoxcline. The change of redox potential is a useful tool for describing the sedimentary environment.
8.5.6 Bioturbation
In oceanic sediments macro- (> 1 mm) and microorganisms play an important role in the mixing of surface sediment layers. Burrowing by organisms in marine sediments is so common that it is the preservation of depositional structures that requires explanation, not their destruction (Arrhenius, 1952, p. 86). Bioturbation is the mixing of sediments by in-situ fauna, and is usually attributed to macro- and mesofauna (0.1–1 mm of sieved sediments). Very little is known about the role of microfauna. The mixing of sediments by the former can easily be observed in coastal areas where their abundance is high and the resuspension and cycling of the annual sediment influx can be as high as 99% (Young, 1971). In anaerobic sediments, however, where macro- and mesofauna are absent due to lack of oxygen, there is still a large community of microorganisms; 107 individuals/cm3 of sediments is not an uncommon value. Several types are motile and have been observed to swim 3 mm/day (Oppenheimer, 1960). If all bacteria in a cubic centimeter of sediment moved 1 mm/day they would cover a total distance of 10 km/day. These organisms cannot move particles but may have a significant effect on the mixing of the interstitial water, and thus also on the exchange between water and sediments.
8.6 Soils, Weathering, and Global Biogeochemical Cycles
The soil may represent a thin film on the surface of the Earth, but the importance of soils in global biogeochemical cycles arises from their role as the interface between the Earth, its atmosphere, and the biosphere. All terrestrial biological activity is founded upon soil productivity, and the weathering of rocks that helps to maintain atmospheric equilibrium occurs within soils. Soils provide the foundation for key aspects of global biogeochemical cycles.
The soil, an open system in the context of biogeochemical cycles, receives inputs and outputs of C and N, and mineral elements and is the foundation of the primary production of terrestrial ecosystems. The amount of carbon present in the soil is closely connected to the CO2 concentration of the atmosphere, but atmospheric CO2 is regulated mainly by the ocean rather than by the soil (see Chapter 10 and 11). The amount of N in the soil also does not influence the N in the atmosphere because the atmosphere is a huge reservoir regulated mainly by the ocean (see Chapter 12). Nevertheless, the soil has a tremendous influence on the nitrate load of rivers. Biogeochemical processes that occur in soils and the processes controlling the delivery of the products of these reactions to the oceans exert profound influences on global biogeochemical cycles.
Weathering processes take an active part in the cycling of oxygen and carbon, but does chemical weathering affect these cycles to a significant extent? Consider the following examples.
Oxygen is formed by photosynthesis according to the reaction
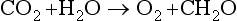
The annual primary production of organic carbon through photosynthesis is on the order of 70 Pg/yr. The major part of this carbon is decomposed or respired in a process that also involves the biogeochemical transformation of nitrogen, sulfur, and many other elements. Only a small part of the annual primary production of organic carbon escapes decomposition and is buried in marine sediments. On average, sediments and sedimentary rocks contain 3% carbon, which corresponds to a net production of oxygen of about 300 Tg/yr. This production has the potential to change atmospheric oxygen over a time scale of 4 Myr. In contrast, the fossil record indicates that the partial pressure of oxygen in the atmosphere has fluctuated very little, at least during the last 600 Myr (Conway, 1943). Obviously there must be a sink that can accommodate the net annual production of oxygen. This sink is the annual weathering of rocks, which can be estimated from the approximately 18 × 1023 g of sediments formed during the last 600 Myr (Gregor, 1968). During this period, soil formation and sediment formation have fluctuated greatly, but the average weathering rate is 30 × 1014 g/yr. The average continental rock contains ferrous iron and sulfide sulfur (mainly as pyrite) and organic carbon. During the weathering processes these constituents are oxidized and consume oxygen. The amounts of oxygen necessary for each of these reactions are given in Table 8-8.

The total annual consumption of oxygen by weathering can be estimated (Holland, 1978) as follows:
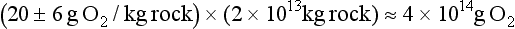
Note that this estimate of the annual O2 loss to weathering processes is approximately equal to the estimated annual production of oxygen estimated above. Hence, the weathering of rocks and burial of organic carbon in sediments during their formation are important processes for the oxygen content of the atmosphere.
Weathering of rocks is also a sink for CO2. Garrels and Mackenzie (1971) have estimated that the formation of 1500 g of sedimentary rocks requires 100 g of CO2. If we use the same number as above for the annual formation of sedimentary rocks, we have a CO2 sink of 200 Tg/yr. About half of that comes from the oxidation of organic carbon in the weathered rock. The rest is from the atmosphere. The burning of fossil fuel increases the partial pressure of CO2 in the atmosphere, but increased CO2 levels increase the weathering rate. Thus, over geologic time, the weathering rate and sediment formation ultimately control the carbon dioxide content of the atmosphere.
Questions
8-1. Discuss how physical weathering operates in each of the following environments: (1) sea shore, (2) hot desert, (3) temperate forest.
8-2. Rewrite the weathering reactions shown in Section 8.3.2.2 using HNO3 in place of H2CO3.
8-3. Why do we speak in terms of soil horizons and sediment layers?
8-4. Discuss the significance of clay minerals in a description of the solid phase of a sediment.
8-5. Give examples of early diagenetic processes.
8-6. Why do continental margins play a dominant role on the biogeochemical cycling of elements?
8-7. Give examples of bacterial transformations in a sediment that are of special importance for biogeochemical cycles.
8-8. Write a balanced reaction for formation of hematite from Fe2+.
8-9. Explain the difference between a Mollisol and a Spodosol. How would cycling of Ca differ in each? N?
8-10. Track the possible fate of a K+ ion from the moment it is released by weathering in a soil to its burial at sea.
Plate 1. Overview of the cyclic processes on Earth. The placements of the geological and geographic features are not meant to represent their true placement on Earth, but rather are included to provide a view of many of the processes discussed in this book in one pictorial figure.
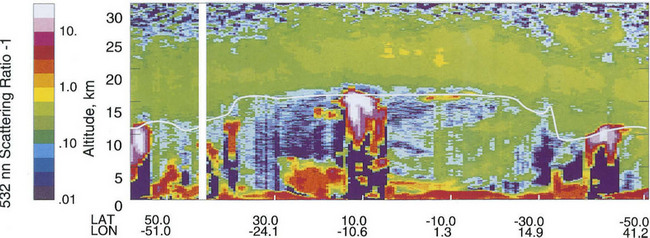
Plate 2. Altitude-distance image of the atmosphere obtained by LIDAR aboard the NASA Space Shuttle on 17th September 1994, across the Atlantic Ocean from approximately Newfoundland to South Africa. (Lidar stands for Light Detection And Ranging the instrumental analog of RADAR with 532 nm radiation.) Approximately 30 minutes elapsed in going from the northern to southern hemisphere. The white and red tones are bright objects-clouds and thick dust, while the green and yellow tones are thin aerosols. Blue is clear air and black is no return. The horizonal white line is the tropopause obtained from metrological data. The vertical white stripe is due to momentary data loss. Visible are: the stratospheric aerosol left over from the emissions of Mt. Pinatubo (June 1991); the clouds of the ITCZ; Sahara dust over the Atlantic being carried by the trade winds; and the mid-latitude clouds associated with ordinary weather systems. Compare to Fig. 7-4 to see the consequences of the general circulation. (Figure courtesy of NASA, and is explained in detail Kent et al., 1997.)
Plate 3. Global soil regions. (Courtesy of US Department Agriculture, Natural Resources Conservation Service, Soil survey Division, World Soil Resources.)
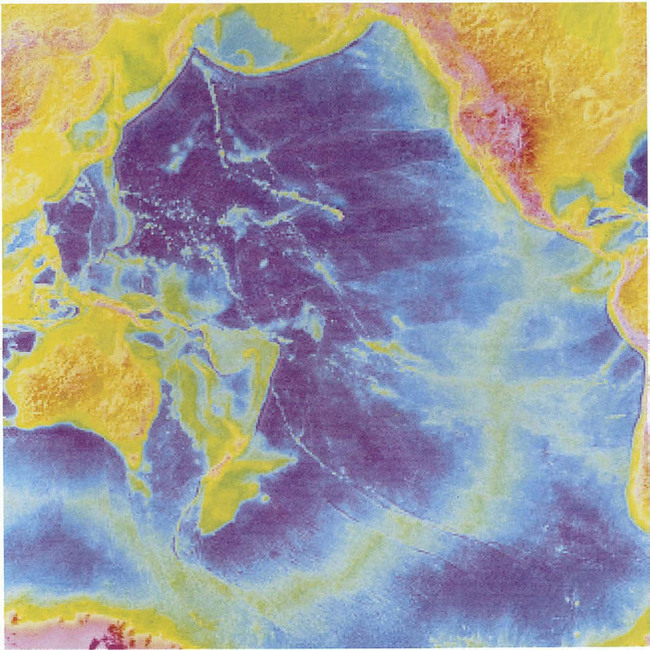
Plate 4. The global sea floor topography from satellite altimetry and ship depth soundings, as described in Smith and Sandwell, 1997. (Kindly provided by Dr Walter Smith of NOAA.)
Ahnert, F. Functional relationships between denudation, relief, and uplift in large mid-latitude drainage basins. Am. J. Sci. 1970; 268:243–263.
Arrhenius, G. Rep. Swedish Deep-Sea Expedition (1947–1948). 1952; 5:227.
Berner, R. A. Principles of Chemical Sedimentology. New York: McGraw-Hill; 1971.
Berner, R. A. Early diagenesis — a Theoretical Approach. New York: Princeton University Press; 1980.
Birkeland, P. W. Soils and Geomorphology, 2nd edn. Princeton, NJ: Oxford University Press; 1999.
Black, T. A., Montgomery, D. R. Sediment transport by burrowing mammals, Marin County, California. Earth Surf. Process. Landforms. 1991; 16:163–172.
Blackwelder, E. B. Fire as an agent in rock weathering. J. Geol. 1927; 35:134–140.
Bohn, H. L., McNeal, B. L., O’Conner, G. A. Soil Chemistry, 2nd Edn. New York: John Wiley; 1985.
Brady, N. C., Weil, R. The Nature and Properties of Soils, 12th edn. New York: Prentice Hall; 1999.
Butler, D. R. Zoogeomorphology: Animals as Geomorphic Agents. Cambridge University Press; 1995.
Campbell, I. B., Claridge, G. G. C., Antarctica: Soils, Weathering Processes and Environment. Developments in Soil Science; Vol 16. Elsevier, Cambridge, 1987.
Casagrande, A. The structure of clay and its importance in foundation engineering. J. Boston Soc. Civ. Engrs. 1940; 19(4):168–209.
Church, M., Slaymaker, O. Disequilibrium of Holocene sediment yield in glaciated British Columbia. Nature. 1989; 337:452–454.
Claridge, G. G. C., Campbell, I. B. Mineral transformations during weathering of dolerite under cold arid conditions. N. Z. J Geol. Geophys. 1984; 25:537–545.
Chap. 19 Clayton, J. L. An estimate of plagioclase weathering rate in the Idaho batholith based upon geochemical transport rates. In: Coleman S. M., Dethier D. P., eds. Rates of Chemical Weathering of Rocks and Minerals. Amsterdam, The Netherlands: Academic Press; 1986:453–466.
Colman S. M., Dethier D. P., eds. Rates of Chemical Weathering of Rocks and Minerals. New York: Academic Press, 1986.
Conway, E. J., The chemical evolution of the ocean. Proc. R. I. A. ; 48B. 1943:161–212.
Cronan, C. S. Biogeochemical responses of forest canopies to acid precipitation. In: Linthurst R. A., ed. Direct and Indirect Effects of Acidic Deposition on Vegetation. New York: Butterworth; 1984:65–79.
Cussler, E. L. Diffusion-Mass Transfer in Fluid Systems. Boston, MA: Cambridge University Press; 1984.
Darwin, C., The Formation of Vegetable Mould Through the Action of Worms with Observations of their Habitats. D. Appleton, Cambridge, 1896. 305–313.
Section 10 Dejou, J., Guyot, J., Chaumont, C., La gibbsite, minéral banal d’altération des formations superficielles et des sols dévelopés sur roches cristallines dans les zones tempérées humides. Int. Geol. Cong., 24th. 1972:417–425.
(translated by T. R. Patan) Dochaufour, P. Pedology. New York: George Allen and Unwin; 1982.
Highway Research Board Dunn, J. R., Hudec, P. P., Frost and sorbtion effects in argillaceous rocks. Frost action in Soils. Natl. Acad. Sci. -Natl. Acad. Eng.; 393. 1972:65–78.
Dunne, T. Sediment yield and land use in tropical catchments. J. Hydrol. 1979; 42:281–300.
Dunne, T., Black, R. D. Partial area contributions to storm runoff in a small New England watershed. Water Resour. Res. 1970; 6:1296–1311.
Dunne, T., Leopold, L. B. Water in Environmental Planning. London: W. H. Freeman; 1978.
Dunne, T., Mertes, L. A. K., Meade, R. H., Richey, J. E., Forsberg, B. R. Exchanges of sediment between the flood plain and channel of the Amazon River in Brazil. Geol. Soc. Am. Bull. 1998; 110:450–467.
Edmonds, R. L. Decomposition and nutrient release in Douglas-fir needle litter in relation to stand development. Can. J. For. Res. 1979; 9:132–140.
Legend and 9 volumes FAO. The FAO-UNESCO Soil Map of the World. San Francisco: UNESCO; 1981.
Friedman, E. I. Light and scanning electron microscopy of the endolithic desert algal habitat. Phycologia. 1971; 10:411–428.
Garrels, R. M., Christ, C. L. Solutions, Minerals, and Equilibria. Paris: Harper and Row; 1965.
Garrels, R. M., Mackenzie, F. T. Evolution of Sedimentary Rocks. New York: W. W. Norton; 1971.
Goodbred, S. L., Jr., Kuehl, S. A. Floodplain process in the Bengal Basin and the storage of Ganges-Brahmaputra River sediment: an accretion study using 137Cs and 210Pb geochronology. Sediment. Geol. 1998; 121:239–258.
Goudie, A., Cooke, R., Evans, I. Experimental investigation of rock weathering by salts. Area. 1970; 4:42–48.
Green, C. P., Eden, M. J. Gibbsite in weathered Dartmoor granite. Geoderma. 1971; 6:315–317.
Gregor, C. B. The rate of denudation in Post-Algonkian time. Koninkl. Ned. Akad. Wetenschap. Proc. 1968; 71:22.
Hallberg, R. O. Sedimentary sulfide mineral formation — an energy circuit system approach. Mineral. Deposit. 1972; 7:189–201.
Hallet, B., Hunter, L., Bogen, J. Rates of erosion and sediment yield by glaciers: A review of field data and their implications. Glob. Planet. Change. 1996; 12:213–235.
Hay, W. W. Detrital sediment fluxes from continents to oceans. Chem. Geol. 1998; 145:287–323.
Hewlett, J. D., Hibbert, A. R. Factors affecting the response of small watersheds to precipitation in humid areas. In: Sopper W. E., Lull W. H., eds. International Symposium on Forest Hydrology. New York: Pergamon; 1967:275–290.
Holland, H. D. The Chemistry of the Atmosphere and Oceans. New York: Wiley-Interscience; 1978.
Horne, R. A., Day, A. F., Young, R. P. Ionic diffusion under high pressure in porous solid materials permeated with aqueous, electrolytic solution. J. Phys. Chem. 1969; 73:2782–2783.
Horton, R. H. The role of infiltration in the hydrologic cycle, EOS. Trans. Am. Geoph. U. 1933; 14:446–460.
Horton, R. E. Erosional development of streams and their drainage basins: Hydrophysical approach to quantitative morphology. Bull. Geol. Soc. Am. 1945; 56:275–370.
Hudec, P. P. Weathering of rocks in arctic and subarctic environments. In: Aitken J. D., Glass D. J., eds. Canadian Arctic Geology: Symposium on the Geology of the Canadian Arctic, Saskatoon 1973. New York: Geological Association of Canada/Canadian Society for Petrology and Geology; 1974:313–335.
Huang, P. M. Feldspars, olivines, pyroxenes, and amphiboles. In: Dixon J. B., Weed S. B., eds. Minerals in Soil Environments. 2nd edn. Calgary, Alberta: Soil Sci. Soc. Am. ; 1989:975–1050.
Hursh, C. R. Storm-water and adsorption, EOS. Trans. Am. Geoph. U. 1936; 17:301–302.
Jackson, M. L. Chemical composition of soils. In: Bear F. E., ed. Chemistry of Soil. Madison, WI: Reinholt; 1964:71–151.
Jackson, M. L., Soil Chemical Analysis. Advanced Course. 2nd edn. 1969. Dep. of Soil Sci., Univ. of Wisconsin, New York, 1973.
Jackson, M. L., Sherman, G. D. Chemical weathering of minerals in soils. Adv. Agron. 1953; 5:219–318.
Jenny, H. Factors of Soil Formation: A System of Quantitative Pedology. Madison, WI: McGraw-Hill; 1941.
Johnson, D. W., Lindberg, S. E. Atmospheric deposition and forest nutrient cycling: a synthesis of the integrated forest study. New York: Springer-Verlag; 1992.
Kittrick, J. A. Mineral equilibria and the soil system. In: Dixon J. B., Weed S. B., eds. Minerals in Soil Environments. New York: Soil Sci. Soc. Am. ; 1977:1–25.
Klinkenberg, L. J. Analogy between diffusion and electrical conductivity in porous rocks. Bull. Geol. Soc. Am. 1951; 62:559–563.
paper 1654 Lambe, T. W. The structure of compacted clay. J. Soil Mech. Found. Div., Proc. ASCE, SM. 1958; 2:84.
Langbein, W. B., Schumm, S. A. Yield of sediment in relation to mean annual precipitation. Trans. Am. Geoph. U. 1958; 39:1076–1084.
Lautridou, J. P., Ozouf, J. C. Experimental frost shattering: 15 years of research at the Center de Geomorphologie du CNRS. Prog. Phys. Georg. 1982; 6:215–232.
Leopold, L. B. Downstream change of velocity in rivers. Am. J. Sci. 1953; 251:606–624.
Lisitzin, A. P. Sedimentation in the world ocean. Soc. Econ. Paleontol. Mineral. Spec. Publ. 1972; 17:1–218.
Loudermilk, W. C. The role of vegetation in erosion control and water conservation. J. Forest. 1934; 32:529–536.
Lundstrom, U. S. Significance of organic acids for weathering and the podzolization process. Environ. Int. 1994; 20:21–30.
Lynn, D. C., Bonatti, E. Mobility of manganese in diagenesis of deep-sea sediments. Marine Geol. 1965; 3:457–474.
Macias-Vasquez, F. M., Fernandez-Marcos, L., Chesworth, W. Transformations mineralogique dans les podzols et les sols podzolique de Galice (NW Espagna). In: Righi D., Chauvel A., eds. Podzols et Podzolisation. Madison, WI: Inst. Nat. de la Recher. Agronomique; 1987:163–177.
Mackenzie, F. T., Ginsburg, R. N., Land, L. S., Bricker, O. P. Carbonate cements. Bermuda Biological Station for Research Special Publication No. 3. 1969; * 325.
Manheim, F. T. The diffusion of ions in unconsolidated sediments. Earth Planet. Sci. Lett. 1970; 9:307–309.
Mason, B., Moore, C. B. Principles of Geochemistry, 4th edn. Plaisir et Paris: John Wiley; 1982.
McLennan, S. M. Weathering and global denudation. J. Geol. 1993; 101:295–303.
Meade, R. H. Factors influencing the early stages of the compaction of clays and sands — review. J. Sediment. Petrol. 1966; 36:1085–1101.
Milliman, J. D., Meade, R. H. World-wide delivery of river sediment to the oceans. J. Geol. 1983; 91:1–21.
Milliman, J. D., Syvitski, J. P. M. Geomorphic/tectonic control of sediment to the ocean: The importance of small mountainous rivers. J. Geol. 1992; 100:525–544.
Montgomery, D. R., Dietrich, W. E. Where do channels begin? Nature. 1988; 336:232–234.
Montgomery, D. R., Dietrich, W. E. Channel initiation and the problem of landscape scale. Science. 1992; 255:826–830.
Oades, J. M. An introduction to organic matter in mineral soils. In: Dixon J. B., Weed S. B., eds. Minerals in Soil Environments. 2nd edn. New York: Soil Sci. Soc. Am. ; 1989:89–159.
Ollier, C. D. Weathering. Madison, WI: Oliver and Boyd; 1969.
Oppenheimer, C. H. Bacterial activity in sediments of shallow marine bays. Geochim. Cosmochim. Acta. 1960; 19:244–260.
Parfitt, R. L. Chemical properties of variable charge soils. In: Theng B. K. G., ed. Soils with Variable Charge. Edinburgh: New Zealand Society of Soil Science Offset Publications; 1980:167–194.
Paul, E. A., Clark, F. E. Soil Microbiology and Biochemistry. Palmerston North, New Zealand: Academic Press; 1996.
Pavich, M. J. Processes and rates of saprolite production and erosion on a foliated granitic rock of the Virginia Piedmont. In: Coleman S. M., Dethier D. P., eds. Rates of Chemical Weathering of Rocks and Minerals. San Diego: Academic Press; 1986:551–590.
Pedro, G. The conditions of formation of secondary constituents. In: Bonneau M., Souchier B., eds. Constituents and Properties of Soils. New York: Academic Press; 1982:63–81.
Presley, B. J. Chemistry of interstitial water from marine sediments. Ph. D. thesis, Univ. Calif., Los Angeles. 1969.
Document D8 Pusch, R. Clay microstructure. New York: National Swedish Building Research; 1970.
Rai, D., Kittrick, J. A. Mineral equilibria and the soil system. In: Dixon J. B., Weed S. B., eds. Minerals in Soil Environments. 2nd edn. Soil Sci. Soc. Am. ; 1989:161–198.
Richards, K. Rivers: Form and Process in Alluvial Channels. Madison, WI: Methuen & Co. ; 1982.
Rosenqvist, I. T., The influence of physicochemical factors upon the mechanical properties of clays. Proc. 9th Nat. Conf. on Clays and Clay Minerals. 1962:12–27.
Ruxton, B. P., McDougall, I. Denudation rates in northeast Papua from potassium-argon dating of lavas. Am. J. Sci. 1967; 265:545–561.
Schlesinger, W. H. Biogeochemistry: an Analysis of Global Change. London: Academic Press; 1997.
Schumm, S. A. The Fluvial System. San Diego: Wiley & Sons; 1977.
Soil Survey Staff. Keys to Soil Taxonomy, 8th edn. New York: USDA Natural Resource Conservation Service; 1998.
Stednick, J. D., Precipitation and streamwater chemistry in an undisturbed watershed in southeast Alaska. Res. Pap. PNW-291. US Department of Agriculture, Forest Service, Pacific Northwest and Range Experiment Station, 1981.
Stumm, W., Morgan, J. J. Aquatic Chemistry, 3rd edn. Portland, Oregon: John Wiley and Sons; 1995.
Summerfield, M. A. Global Geomorphology. New York: Longman Scientific & Technical; 1991.
Sverdrup, H. U., Johnson, M. W., Fleming, R. H. The Oceans. Essex: Prentice-Hall; 1942.
Syers, J. K., Iskandar, I. K. Pedogenic significance of lichens. In: Ahmadjian V., Hare M., eds. The Lichens. Englewood Cliffs, New Jersey: Academic Press; 1973:225–248.
Tan, T. K., Discussion on: Soil properties and their measurement. Proc. 4th Int. Soil Mech. and Found. Engng. ; 3. 1957:87–89.
Tardy, Y., Geochimie des alterations. Etude des arenes et des eaux de quelques massifs cristallins d’Europe et d’Afrique. These Doc. Etat, Univ. Strasbourg. 1969.
Ugolini, F. C. Processes and rates of weathering in cold and polar desert environments. In: Colman S. M., Dethier D. P., eds. Rates of Chemical Weathering of Rocks and Minerals. New York: Academic Press; 1986:193–235.
Ugolini, F. C., Edmonds, R. L. Soil biology. In: Walding L. P., Smeck N. E., Hall G. F., eds. Pedogenesis and Soil Taxonomy 1. Concepts and Interactions. New York: Elsevier; 1983:193–231.
Ugolini, F. C., Jackson, M. L. Weathering and mineral synthesis in Antarctic soils. In: Craddock C., ed. Antarctic Geoscience. Amsterdam, The Netherlands: University of Wisconsin Press; 1982:1101–1108.
Ursic, S. J., Dendy, F. E. Sediment yields from small watersheds under various land uses and forest covers. Proceedings, Federal Inter-Agency Sedimentation Conference 1963. US Dept. of Agriculture Misc. Publ. 1965; 970:47–52.
Velbel, M. A. Hydrogeochemical constraints on mass balances in forested watersheds of the southern Appalachians. In: Drever J. I., ed. The Chemistry of Weathering. Madison, WI: G. Reidel; 1985:231–247.
Wada, K. The distinctive properties of Andosols. Adv. Soil Sci. 1985; 2:173–229.
Weisz, P. B. Diffusion and chemical transformation. Science. 1973; 179:433–440.
Williams, R. B. G., Robinson, D. A. Weathering of sandstone by the combined action of frost and salt. Earth Surf. Process. Landforms. 1981; 6:1–9.
Suppl. 22 Young, D. K. Vie et Milieu. 1971; * 557–571.
Proceedings, IEA/BE A3 Workshop, South Island, NZ, March 1989. IEA/BE T6/A6 Report No. 2. Forest Research Inst. Bulletin No. 159. Zabowski, D., The role of mineral weathering in long-term site productivityDyck W. J., Mees C. A., eds. Impact of Intensive Harvesting on Forest Site Productivity. 1990:55–71.