Chapter 36
Food Intake and Metabolism
Eating is a familiar behavior. In humans, it is strongly influenced by cultural, social, and experiential factors. Consequently, eating has been a subject of considerable interest to social and behavioral scientists. In mammals, including humans, food intake is also a regulatory behavior with the primary function of supporting the continuous energy demands of body tissues. When considered from this biological perspective, food intake is influenced by hunger, satiety, and the physiological mechanisms that couple eating with internal caloric supplies and a stable body weight. This chapter discusses the signals important for the central nervous system’s control of food intake and glucose homeostasis, and also describes mechanisms thought to integrate those signals.
Despite decades of investigation, considerable differences of opinion still exist about how food intake is controlled. The multiple views follow one of two principles. The first considers eating to be a consequence of depleted energy stores in adipose tissue, reduced use of energy-rich metabolic fuel (glucose or lipid) in some critical tissue, or both. In this schema, the purpose of eating is to restore energy reserves in adipose tissue or to increase fuel utilization to some desired level, thereby eliminating the signal to eat. This traditional negative feedback model is not the approach taken in this chapter. Rather, we subscribe to a second principle, in which hunger is thought to be initiated in part by the gradual disappearance of inhibitory satiety stimuli that were generated by the previous meal. In addition, meal initiation may occur based on learning, habits, and various social and other situations, which allow animals to take maximal advantage of their environment. According to this view, caloric homeostasis influences meals, albeit indirectly, and eating contributes to caloric homeostasis by providing nutrients. The storage and use of those nutrients are regulated independently by physiological mechanisms described later. We describe the properties of caloric homeostasis before we consider the control of food intake.
Caloric Homeostasis
Homeostatic Mechanisms Provide a Continuous Supply of Metabolic Fuels to Cells
The purpose of caloric homeostasis is to preserve cellular metabolism. Cells oxidize metabolic fuels to drive all cellular processes; thus, the higher the cellular activity, the greater the demand for energy. Because most cells have limited amounts of stored energy, they rely on a steady supply of calories and oxygen from the bloodstream. Oxygen is dependably and instantaneously available via the respiratory system and is not stored in the body. In contrast, food calories can be scarce and require time after ingestion to become available to cells in significant quantities. One consequence of this functional organization is the capacity of animals to store sufficient energy to bridge long intervals during which no food is eaten.
Three categories of macronutrients—carbohydrates, lipids, and proteins—provide usable energy, but the use of specific macronutrients by the body varies depending on the tissue. Most tissues can oxidize carbohydrates in the form of glucose or lipids in the form of free fatty acids, depending on the availability of these nutrients and the levels of certain hormones in the blood. Notable exceptions are the liver, which requires lipids for proper functioning, and the brain, which has a large, continuous need for glucose despite the ability to oxidize lipids in the form of ketone bodies. When the supply of glucose to the brain is compromised, neurons cease to function and in a few minutes consciousness is lost. Death will ensue unless glucose delivery to the brain is restored. Therefore, maintenance of circulating glucose in sufficient amounts to support normal brain function is a critical goal of caloric homeostasis.
Two distinct metabolic states are defined by the availability of recently consumed food to cells. The prandial, or fed, state is characterized by an abundance of newly ingested and absorbed nutrients in the blood. These molecules are sequestered rapidly in tissues to prevent them from being excreted wastefully in urine. The postabsorptive, or fasted, state is characterized by the absence of calories entering the circulation from the gastrointestinal tract and a consequent reliance on energy from metabolic fuels less recently consumed and stored. These stores are released gradually into the blood during a fast. In both states, tissues take nutrients from the blood as needed for cellular metabolism.
Many tissues store carbohydrates in the form of glycogen, a polymer of glucose; the liver and skeletal muscles have the largest depots. Energy is stored more efficiently, mainly in adipose tissue, as triglyceride, each molecule of which consists of glycerol with three attached fatty acids. During the prandial period, newly ingested food is used immediately by the body or is stored as glycogen or triglyceride. Excess carbohydrate is largely converted to lipid (lipogenesis) because glycogen storage capacity is limited. During the fasting period, liver glycogen is converted back to glucose (glycogenolysis), which enters the blood and is available to all tissues. Similarly, stored triglycerides are mobilized from adipose tissue (lipolysis) and enter the circulation as fatty acids and glycerol. The fatty acids are used by tissues as needed or are converted to ketone bodies (ketogenesis), whereas glycerol is converted to glucose (gluconeogenesis) (Fig. 36.1).
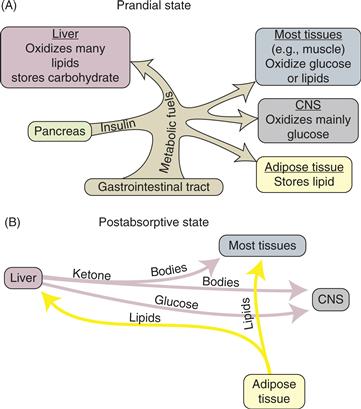
Figure 36.1 Schematic diagram of the fluxes of metabolic fuels in the (A) prandial and (B) postabsorptive states. Note the absence of insulin in the postabsorptive state, which greatly facilitates the mobilization of energy stores from the liver and adipose tissue. The adult human liver stores sufficient fuel to support metabolism during fasting for about 7 h, whereas the adipose mass has a far greater capacity.
The liver is the key organ of energy traffic. Lipogenesis (which also occurs in adipose tissue) and glycogen formation (which also occurs in skeletal muscle) occur in the liver during the prandial period, and glycogenolysis, ketogenesis, and gluconeogenesis occur during the fasting period. These processes are regulated (discussed later), as are delivery of metabolic fuels (from the small intestine into the circulation), storage of excess fuels, and mobilization of stored energy. The control system involves interplay among several hormones and the sympathetic and parasympathetic divisions of the autonomic nervous system.
Insulin is the Key Hormone Affecting Caloric Homeostasis
Secretion of the peptide hormone insulin from B cells of the pancreatic islets is influenced by several factors, among which interstitial glucose is critical. Insulin secretion increases in direct proportion to the concentration of glucose in the blood. Other substrates, such as amino acids and ketone bodies, also stimulate insulin secretion. In addition, autonomic nerves innervate the pancreatic islets: cholinergic parasympathetic activity stimulates secretion of insulin, and α-adrenergic sympathetic activity inhibits it.
When a hungry person anticipates a meal, the aroma and, subsequently, the taste of food initiate the secretion of insulin and many other pancreatic and gastrointestinal signals via neural activity. Neural signals descend from the forebrain (where the smell and taste of food are recognized) through the hypothalamus to the dorsal motor nucleus of the vagus in the caudal brainstem and then to the pancreas and gastrointestinal tract by way of cholinergic fibers of the vagus nerve. This cephalic phase of secretion of insulin and gastrointestinal hormones such as glucagon-like peptide-1 (GLP-1) helps reverse the mobilization of fuels that occurs during fasting and prepares the body for the entry of fuels from the gut. As ingested food enters the stomach and duodenum, several gastrointestinal hormones, which also are important in digestion, continue to stimulate pancreatic B cells to secrete more insulin. This gastrointestinal phase of insulin secretion ensures that the level of insulin in the circulation is high by the time digested nutrients first appear in the bloodstream. Finally, nutrients absorbed from the small intestine cause even greater stimulation of insulin secretion by their direct effect on the pancreas. This substrate phase of insulin secretion increases insulin levels further, and that effect lasts well beyond the cessation of eating. As a result of these coordinated meal-related events, prandial insulin secretion is rapid and appropriate to the caloric load, and ingested fuels are efficiently used and stored (Fig. 36.2).
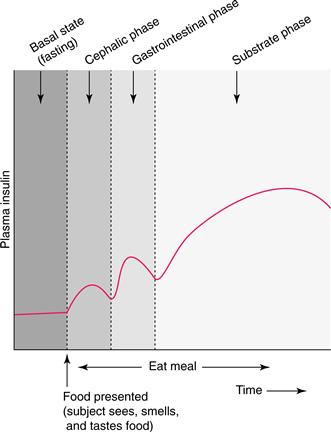
Figure 36.2 The three phases of meal-related insulin secretion. The cephalic phase, during which parasympathetic vagal activity stimulates the pancreas, is initiated when food is seen, smelled, and tasted. The gastrointestinal phase is mediated by the direct action of digestive hormones on the insulin-secreting B cells. The prolonged substrate phase is caused by metabolic fuels (mainly glucose) directly stimulating pancreatic B cells. When meals are prolonged, the three stimuli operate simultaneously and have additive effects.
Neural and endocrine factors control the ebb and flow of metabolic fuels from body stores. Circulating insulin is the most important factor that promotes storage. Insulin enables most tissues to take up glucose from the blood for immediate oxidation or for storage during the prandial period. Conversely, the most important factor for fuel mobilization is the near disappearance of insulin from the circulation during the postprandial, postabsorptive period, when fuel delivery from the small intestine has ended and parasympathetic activity to the pancreas is no longer elevated. Insulin secretion at this time is reduced greatly but is not inhibited and therefore can be stimulated immediately if necessary. For example, when more than the needed amount of stored fuels is mobilized, substrates directly stimulate the pancreas to secrete insulin, thereby slowing substrate mobilization to a rate appropriate to need. Thus, insulin is pivotal for storing calories in the fed state and for allowing stored calories to be mobilized in measured amounts during the postabsorptive state.
The amount of body fat (adiposity) is superimposed on other factors that influence insulin secretion. People with low adiposity have a relatively large number of active insulin receptors on adipose tissue, whereas obese individuals have fewer active insulin receptors. Consequently, in response to a given stimulus, secretion of insulin is lower in people who are lean than in those who are obese, but its net effects on adipose and other tissues are comparable. In healthy, nondiabetic people, this reciprocal relationship between the secretion of insulin and the sensitivity of most tissues to insulin ensures efficient storage and use of fuels independent of body fat. In these individuals, plasma insulin levels in the prandial and the postprandial periods are reliable correlates of adiposity.
Summary
All cells require continuous supplies of metabolic fuels to support ongoing activity. The fuels enter the circulation from the small intestine during the prandial period and from storage depots during fasting. The availability of fuels to tissues is controlled primarily by the liver and by the hormone insulin. Hepatic function and insulin secretion are in turn controlled in large part by the autonomic nervous system.
Role of Caloric Homeostasis in Control of Food Intake
Meals Generate Biological Satiation Signals
The traditional view of caloric homeostasis is that animals eat when they need calories. However, as discussed earlier, the delivery of metabolic fuels to cells is continuous, and animals may experience urgent needs for calories to support cellular metabolism only infrequently. Instead, caloric homeostasis can be related to the control of food intake in ways that are unrelated to acute cellular needs.
Animals consume food in distinct bouts (i.e., meals); therefore, daily food intake reflects the cumulative intake of multiple meals. Control factors influence the time when each meal is initiated and the amount of food consumed before the meal is terminated. Satiation signals terminate an ongoing meal whereas satiety signals prolong the interval until a subsequent meal is initiated. The first comprehensive study of meals was reported in 1966 by Le Magnen and Tallon. They maintained laboratory rats in cages and allowed them to eat ad libitum for weeks, during which time intake was recorded by photosensors and electronic relays. They consequently obtained a full record of when meals were initiated, how much was eaten during each meal, how much time passed before the next meal, and so on. No relationship was found between how much a rat ate in a meal and how much time had passed since it had last eaten; in other words, meal sizes were unpredictable. However, the larger the meal, the longer the interval before the next meal, and the relationship was strongest during the night, when rats ate the largest meals. Thus, eating appeared to be inhibited by a satiety signal generated in proportion to the size of a meal, and eating resumed when that signal disappeared. Similar observations have since been made in many laboratories.
Gastric Distension, Cholecystokinin, and Other Satiation Signals
Several meal-related variables might plausibly provide satiation signals. These include signals based on the smell, taste, and texture of food. Signals arising from the stomach could also play a role, as could intestinal and postabsorptive signals that arise after ingested food has left the stomach. In studies of rats, pregastric signals have been ruled out because little satiation is achieved when ingested food is immediately drained out through an esophageal or gastric fistula; these animals eat continuously, as if they had no satiation despite the passage of very large amounts of food through the oropharynx (Fig. 36.3). Because meals end long before significant digestion and absorption occur, gastric signals have been considered a likely source of satiation. Gastric volume is an obvious possibility, and, in fact, meals are known to end when substantial gastric distension occurs. The stomach wall is richly endowed with stretch receptors whose activity increases in proportion to the volume of the stomach. Those signals are communicated by way of the vagus nerve to the nucleus of the solitary tract (NST) and adjacent area postrema in the brainstem. The signals travel to the hypothalamus and, ultimately, to the cortex, where gastric distension is perceived.
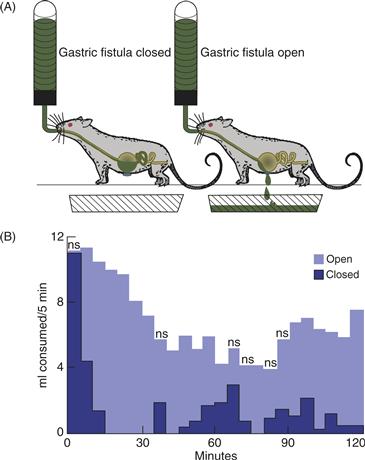
Figure 36.3 (A) Rats consuming liquid diet, with gastric fistula closed (real eating) or open (“sham eating”). When the gastric fistula is closed, food passes through the stomach and into the small intestine normally. When the gastric fistula is open, ingested food drains out the open fistula instead of accumulating in the stomach to distend it, and no food enters the small intestine. (B) Mean consumption of liquid diet (ml/5 min) by rats with a gastric fistula after 17 h of food deprivation. The rats consumed food in discrete meals when the fistula was closed. When the fistula was open, they ate continuously throughout the test period and never displayed satiety. Thus, rats consumed significantly more food during the 2-h test period when their fistulas were open than when they were closed. ns, intervals in which the amount of food consumed did not differ significantly between open and closed fistulas.
Gastric stretch that accompanies meals presumably interacts with other signals to produce satiation. For example, aside from directly affecting digestion, the intestinal peptide cholecystokinin (CCK), which is secreted during meals, acts on receptors located on vagal afferent fibers that carry gastric stretch signals from the pyloric region of the stomach to the brainstem. Thus, relatively small amounts of CCK can inhibit feeding in rats by acting synergistically with gastric distension. When given in larger doses, CCK can inhibit food intake even when the stomach is empty (Gibbs, Young, & Smith, 1973). As might be expected, total or selective gastric vagotomy (or ablation of brainstem areas to which the gastric vagus projects) decreases the ability of gastric volume and peripherally administered CCK to reduce meal size. Conversely, systemic administration of CCK receptor antagonists increases the size of meals (Box 36.1).
Box 36.1 Validating Satiation Factors
Several metabolically important peptides are secreted during meals, including CCK, insulin, leptin, glucagon-like peptide-1 (GLP-1), Peptide YY (PYY) and others. The specific individual nutrients being consumed are thought to determine the specific mix of peptides secreted, and their combination is considered to elicit satiation. Consistent with this perspective, each of these peptides has been found to reduce food intake when administered systemically to laboratory animals. By themselves, these findings do not prove that the peptides normally function as endogenous satiation agents; blood levels of an administered peptide must be within the physiological range. In addition, the behavioral effects of the peptide must be specific to the inhibition of food intake and cannot merely reflect a secondary consequence of illness, behavioral depression, or motor incapacitation. Because investigators cannot be certain what animals sense, they must infer whether animals experience satiation in association with an observed reduction in food intake.
One common approach is to determine whether the peptide causes a learned flavor aversion in animals. People readily learn to avoid food or drink that, when ingested, produces nausea. Rats and other animals seem to respond in the same way; moreover, when a toxin that elicits nausea is administered systemically soon after consumption of an uncontaminated drink with a novel flavor, the animals subsequently avoid fluids of that flavor and behave as if the drink had contained the toxic agent that had made them sick. In addition, electrophysiological recording from the first gustatory relay nucleus in the brainstem, the nucleus of the solitary tract, shows that the response elicited by a taste that has been associated with a toxin is similar to the pattern of activity typical of a naturally aversive flavor. Nausea is critical to the process; damage to the “emetic center” in the area postrema eliminates the sensation of nausea and prevents the formation of the learned taste aversion.
Another approach to distinguishing nausea from satiation is to monitor biological variables that occur in association with nausea but not satiation, or vice versa. One such variable is neurohypophyseal hormone secretion. For example, administration of lithium chloride (LiCl) causes nausea in humans and vomiting in monkeys, and elicits a learned flavor aversion in rats. LiCl stimulates vasopressin secretion in humans and monkeys. In this example, elevated plasma vasopressin can be considered a biological marker of nausea because it does not itself cause nausea or vomiting. Curiously, rats secrete the other neurohypophyseal peptide, oxytocin (and not vasopressin), in response to large doses of LiCl and other nauseants. Control experiments have shown that following an ordinary meal, vasopressin is not secreted in primates nor is oxytocin secreted in rats. Thus, when rats are administered a chemical agent that stimulates pituitary oxytocin secretion, nausea, rather than satiation, is the suspected basis of the reduction in food intake.
Such experiments are helpful for interpreting the anorexia that occurs when a peptide is administered exogenously, but insight into the normal effects of endogenously secreted chemical signals requires an alternative experimental approach. For example, a drug that blocks the effect of an endogenous peptide on its receptors could be administered. If the hormone normally functions to reduce food intake, then the blocking agent should increase the size of meals. Such drugs are not yet available for all peptides suspected of mediating endogenous satiation, but increases in meal size were found in experiments using drugs to reduce the activity of CCK, GLP-1, and several other peptides at their respective receptors. These observations strongly suggest that these peptides normally function as endogenous satiation factors.
Gastric distension is one of the factors that control suckling in neonatal rats. The dam controls the timing of meals, and at each meal pups consume as much milk as their stomachs allow; gastric volume, not caloric content, provides the signal to stop. Consistent with the importance of gastric distension to suckling, pups are particularly sensitive to the inhibitory effects of CCK on food intake, suggesting that a combination of gastric distension and CCK may be the principal signals that inhibit the initiation of spontaneous meals in suckling rats.
As rat pups mature, signals related to the caloric content of food begin to participate in the control of meal size and become integrated with CCK, GLP-1, gastric distension, and other factors as satiation signals. This developmental change allows adults to ingest, assess, and respond to foods whose caloric density is not as constant as that of milk. For example, when liquid food is diluted with water, adult rats compensate by increasing the volume consumed in each meal, allowing daily caloric intake to remain stable. This observation reveals that gastric distension is but one of several possible satiation signals in adult rats and that greater distension can be accommodated in circumstances in which the calorie-related satiation signals have diminished.
The mechanism by which caloric signals are monitored is an area of intense investigation. Although early reports suggested that the stomach monitored caloric nutrients, later findings did not support this hypothesis. For example, when hungry rats were equipped with closed pyloric cuffs to prevent ingested food from entering their small intestine, the rats decreased their food intake in proportion to the volume, and not the caloric content, of intragastric infusions (Phillips & Powley, 1996). In contrast, when the cuff was open so that gastric contents could empty into the duodenum, caloric nutrients were much more effective in reducing food intake than calorie-free loads of equal volume. These observations suggest that the stomach does not detect caloric nutrients but instead contributes primarily inhibitory signals related to gastric distension.
Postgastric Effects of Meals Provide Additional Satiation Signals
During the course of a meal, some of the ingested food enters the small intestine and is absorbed, thus allowing postgastric signals to contribute to satiation. In this regard, small amounts of specific nutrients infused directly into the duodenum produce a robust suppression of eating that cannot be attributed to gastric factors. It is likely that both the upper small intestine as well as postabsorptive sites are responsible for detecting the administered calories and generating the signals that limit meal size. The luminal surface of the intestine contains mainly enterocytes that absorb digested nutrients and pass them into the blood. Interspersed among the enterocytes are scattered enteroendocrine cells that have tentacle-like projections which extend into the intestinal lumen and express many of the same taste receptors as occur on taste buds on the tongue. As a result, these enteroendocrine cells “taste” specific nutrients (e.g., glucose) that are in the intestinal lumen, and they respond by secreting peptide hormones that contribute to the ongoing process of digestion. There are a dozen or so different types of enteroendocrine cells, each secreting a different peptide or peptides. As an example, I-cells secrete CCK when they “taste” fatty acids in the lumen, and the CCK in turn enters the blood and causes the liver/gall bladder to secrete bile into the intestine to facilitate the digestion of fats. Analogously, L-cells secrete GLP-1 and PYY in response to “tasting” carbohydrates, and GLP-1 increases insulin secretion during the meal. Importantly, many of the peptide secretions of the enteroendocrine cells also double as satiation signals, acting either locally on afferent nerves in the wall of the intestine or else circulating to a distant organ such as the liver or brain to elicit satiation. These peptides include CCK, GLP-1, and PYY.
As discussed earlier, the liver plays a critical role in caloric homeostasis, and the delivery of nutrients absorbed from the small intestine to the liver may be monitored to provide an important postgastric signal for the inhibition of food intake. Consistent with this hypothesis are findings that infusion of glucose or lipids into the hepatic portal vein reduces food intake. Importantly, infusion of fructose (a sugar utilized by the liver but not by the brain) into the general circulation no longer reduces food intake in rats after hepatic vagotomy. Collectively, these findings suggest that like the intestines themselves, the liver provides a satiation signal. All of these signals would diminish as absorption slows and satiation occurs.
Intravenously administered glucose reduces food intake but not by as much as the caloric content of the glucose. In contrast, when glucose solution is consumed orally or fed through a tube directly into the stomach, the compensatory reduction in food intake is equivalent to the caloric load. In this case, gastric and/or intestinal signals such as GLP-1 contribute to the increased satiation and augment insulin secretion above that elicited by the glucose per se; that is, more insulin is secreted in response to infusion of glucose into the stomach or intestine than into a vein, which might contribute to the observed reduction in appetite. In fact, intravenously infused glucose reduces food intake by an equivalent caloric amount when insulin is added to the infusate. Together, these observations suggest that insulin contributes to the satiating effect of glucose whether given by gavage or ingested during a meal. Consistent with this possibility, rats made diabetic by destruction of their pancreatic B cells eat discrete meals more frequently as if they experienced less postprandial satiety (Box 36.2).
Box 36.2 The Glucostatic Hypothesis
Almost 60 years ago, Mayer (1955) proposed that the onset and termination of meals are determined by changes in the amounts of glucose used by certain areas of the brain. More specifically, Mayer hypothesized that food intake was controlled by a region of the ventral hypothalamus whose glucose utilization, unlike the rest of the brain, was sensitive to insulin. In diabetic rats, the presence of hyperphagia despite elevated blood glucose emphasized the point that diminished glucose utilization in these insulin-dependent cells (as opposed to a reduction of blood glucose levels per se) was the critical factor in removing satiety and stimulating food intake.
At first widely accepted, the glucostatic theory fell into disfavor for several reasons. For one, although eating can be induced by low availability of glucose to the brain, no evidence supports suppression of eating by high glucose availability. In addition, concentrations of blood glucose after administration of large doses of insulin are now known to be far lower than what occurs during a fast or at the normal onset of a meal. Thus, eating in response to insulin-induced hypoglycemia is now considered an “emergency response” that is not relevant to normal feeding. (A similar emergency response occurs after a systemic injection of 2-deoxyglucose induces an acute decrease in cellular glycolysis.) Finally, the prevailing view of the ventromedial hypothalamus as a satiety center has changed considerably since the original formulation of the glucostatic hypothesis. This region of the brain is better regarded as a regulatory control center that influences many aspects of hormonal and autonomic function. Diabetic hyperphagia may therefore represent the ineffectiveness of insulin or leptin to activate neural systems that inhibit food intake. In contrast, the earlier glucostatic hypothesis would have explained diabetic hyperphagia as a consequence of the decreased uptake of glucose into special hypothalamic neurons that mediate satiety.
In recent years, evidence has emerged that neurons in the hypothalamic arcuate nucleus are sensitive to fluctuations of the adiposity signals leptin and insulin. Neurons in the same area also are sensitive to fluctuations of glucose as well as to certain fatty acids (especially oleic acid) and amino acids (especially leucine), and local manipulations of either adiposity signals or nutrients influence food intake, pancreatic insulin secretion, and the release of glucose from the liver. Hence, the view is emerging that the arcuate nucleus integrates long-term energy stores (principally body fat) with acute availability of nutrients to control several aspects of energy homeostasis.
Stephen C. Woods and Edward M. Stricker
Reference
1. Mayer J. Regulation of energy intake and body weight: The glucostatic theory and the lipostatic hypothesis. Annals of the New York Academy of Science. 1955;411:221–235.
In addition to providing useful nutrients, consumption of food increases plasma osmolality, yet another factor influencing the size of a meal. Increased plasma osmolality stimulates thirst, and for this reason water intake usually accompanies eating. The important point, however, is that less food is consumed during meals when drinking water is not available, a phenomenon known as dehydration anorexia. Similarly, increased plasma osmolality caused by systemic injection of hypertonic saline reduces food intake in proportion to the administered osmotic load. The well-described forebrain osmoreceptors that cause thirst and influence vasopressin secretion (Chapter 39) apparently do not mediate this inhibitory effect on food intake, which persists after those osmoreceptors have been destroyed surgically. Instead, osmoreceptors in the viscera or in the caudal brainstem might mediate the inhibitory effect of increased plasma osmolality on food intake.
Body Weight Influences Food Intake
The control of food intake is also associated with the maintenance of body weight (adiposity, actually). For example, after a period of food deprivation and forced loss of body weight, animals (including humans) eat relatively large meals until adiposity returns to pretreatment levels. Conversely, after a period of forced feeding, during which the increased daily intake of calories causes weight gain, animals eat relatively small meals (or no meals at all) until normal adiposity is restored. Long-term stability of adiposity in adult animals is attributed in part to these compensatory responses to weight fluctuations that are caused by acute changes in food intake or energy expenditure.
Adiposity may indirectly influence food intake by modulating how quickly food passes through the gastrointestinal tract, how long nutrients remain in the circulation, or how nutrients interact with the liver. For example, after fasting, less prolonged gastric distension or more rapid absorption and storage of ingested calories would diminish the duration of satiety signals, thereby increasing the frequency of meals. These effects would promote the hyperphagia (overeating) associated with the restoration of body weight in animals that have been food restrained and lost weight. Similarly, the satiating properties of ingested food would be reduced by diversions of ingested food calories to the fetus during pregnancy, to milk during lactation, or to skeletal muscle during exposure to cold temperatures, thereby contributing to the hyperphagia associated with each of these conditions.
Alternatively, adipose tissue may directly signal the brain to modulate food intake. Although afferent nerves from adipose tissue to brain have been described, communication from adipose tissue is thought to be mediated primarily by a humoral (circulating) factor. Parabionts, created by surgically joining two animals at the flank muscles and skin, support this hypothesis. Vascular interconnection within a parabiont is demonstrated when a dye injected into one animal appears in the blood of its partner. Because the nervous systems of the two animals remain independent, any physiologic communication between the animals must occur through the vascular system. When two lean rats were joined parabiotically, each consumed its normal amount of food and maintained its usual body weight. Similar results were obtained when two obese rats with lesions in the ventromedial hypothalamus (see later) were joined together. However, when a lean animal was joined to an obese animal, the result was striking: The lean animal reduced its food intake markedly and consequently lost a considerable amount of weight. These experiments have been interpreted to mean that a circulating factor, secreted by the obese animal in large amounts proportional to adiposity, enters the blood of the lean animal and reduces food intake, as if the lean animal was receiving a signal that it was too fat (Coleman & Hummel, 1969).
Investigations have identified leptin, a hormone synthesized by adipose tissue, as a key circulating adiposity signal that regulates food intake and body weight. Leptin concentration in plasma is directly proportional to adiposity, and a receptor-mediated transport process passes it into the brain. Animals that lack the gene for synthesizing leptin (ob/ob mice) are hyperphagic and obese, and administering leptin to ob/ob mice causes them to eat less and lose weight. Because lower doses of leptin have this action when administered into the ventricles of the brain than when administered systemically, leptin is believed to reduce food intake and body weight via a central site of action. Consistent with this hypothesis, the receptor for leptin has been identified and found to be located within the hypothalamus. Like ob/ob mice, animals with mutated leptin receptors (db/db mice and fatty Zucker rats) are obese, but unlike ob/ob mice, they do not respond to exogenous leptin. Hence, leptin appears to function as a negative feedback signal to the brain. When fat stores increase in adipose tissue, more leptin is secreted and enters the brain, causing greater inhibition of food intake and loss of body fat. When circulating leptin levels are low, feeding and other anabolic responses are disinhibited. In this way, leptin acts to promote the maintenance of relatively stable adiposity over long intervals. The neural circuitry through which leptin inhibits food intake and influences caloric homeostasis is discussed later.
Insulin provides a second circulating signal that informs the brain of body fat levels. Plasma levels of insulin are directly correlated with adiposity, and, like leptin, insulin is transported through the blood–brain barrier where it can act on insulin receptors in the hypothalamus and other sites. Insulin administered into the brain reduces food intake and body weight, whereas animals that do not secrete insulin are hyperphagic. Of course, severe diabetics do not become obese because their adipocytes cannot store fat in the absence of insulin. Mice lacking insulin receptors on brain cells are both hyperphagic and obese. In short, both leptin and insulin provide blood-borne signals that enter the brain and act on their respective receptors to reduce food intake and body fat.
Obesity, the accumulation of greater than normal fat in the body, is increasing throughout the world and creating a major public health problem, in part because it increases the risk for developing cardiovascular disorders, several cancers, type-2 diabetes mellitus, and other metabolic problems, and increases mortality. Obese individuals have both leptin resistance and insulin resistance; that is, although they secrete large amounts of both leptin and insulin in proportion to their adiposity, these signals are relatively ineffective in controlling metabolic parameters. Consequently, considerably more insulin is required to maintain plasma glucose within normal limits, and the brain is insensitive to the catabolic action of both leptin and insulin. The latter is thought to be due to a combination of reduced passage of these hormones through the blood–brain barrier as well as fewer leptin and insulin receptors within the brain itself. See Box 36.3 for more on obesity.
Box 36.3 Treatment of Obesity
Obesity, the accumulation of greater than normal fat in the body, results when energy intake exceeds energy expenditure over long intervals. Obesity often is defined in terms of the body mass index (BMI), which is weight in kilograms divided by height in meters squared. A BMI of 18.5 to 24.9 is considered normal, with <18.5 being underweight, 25 to 29.9 being overweight, and >30 being obese. A BMI over 40 is morbid obesity. However, the calculation of BMI does not consider the distribution of fat within the body, yet this is recognized as a major determinant of health risk. Abdominal fat (mainly around the viscera) is more metabolically active, more highly vascularized and innervated, and more likely to be a site of inflammation in the body than subcutaneous fat; abdominal fat (typified by the “potbelly” or “apple” stereotype) therefore carries a much higher health risk than subcutaneous fat (characterized as a “pear” shape). Because of this, assessment of the circumference of the waist (abdomen) at the top of the iliac crest is thought to be a better predictor of health risk than the BMI. By this determination, obesity is defined as a waist circumference of >102 cm in men and >88 cm in women.
The incidence of obesity is increasing throughout the world and creating a major public health problem, in part because it increases the risk for developing cardiovascular disorders, several cancers, type-2 diabetes mellitus, and other metabolic problems, and increases mortality. Obese individuals have both leptin resistance and insulin resistance; that is, although they secrete large amounts of both leptin and insulin in proportion to their adiposity, those signals are relatively ineffective at controlling metabolic parameters. Consequently, considerably more insulin is required to maintain plasma glucose within normal limits, and the brain is insensitive to the catabolic action of both leptin and insulin. The latter is thought to be due to a combination of reduced passage of these hormones through the blood–brain barrier as well as fewer leptin and insulin receptors within the brain itself.
Hypotheses as to why the incidence of obesity has been increasing so substantially in recent years abound, and often presume some combination of increased availability of energy-dense palatable foods and/or decreased average energy expenditure. It is axiomatic to state that obesity can be prevented or cured by reversing the trend—that is, by eating less and exercising more. Unfortunately, many humans in modern society cannot realistically modify these behaviors for long intervals, and they have consequently turned to other methods to aid in weight loss. Historically, several different neuropharmacologic approaches have been used, most of them targeting brain neurotransmitter systems thought to be important in appetite control. Specifically, pharmaceuticals developed to treat obesity have targeted the serotonin, catecholamine, or endocannabinoid systems, either with ligands for their respective receptors or else with chemical agents that influence their secretion, degradation, or reuptake. However, because each of these transmitter systems influences multiple brain circuits, and because none of the compounds available to date have been appetite-specific and often have undesirable side effects on mood and other behaviors, none are currently approved by the FDA in the United States to treat obesity. Further, actual weight loss achieved any by these pharmacological approaches was rarely greater than 10%.
During the last decade, bariatric surgery has become the treatment of choice, especially for morbidly obese individuals, and these surgeries often result in far greater weight loss than that achieved pharmacologically (i.e., up to 70% of excess fat or more). In the conceptually simplest procedure, a band is placed around the stomach near the esophagus. The result of this gastric banding procedure is that food is swallowed into a very small gastric pouch, severely limiting the amount of food that can be consumed at once due to gastric restriction. Distension of the remaining gastric pouch is thought to result in smaller meals. Even greater loss of weight may occur with more invasive procedures that involve rearrangement of the normal routing of ingested food through the gastrointestinal system. In a roux-en-Y gastric bypass (RYGB), swallowed food enters a small gastric segment and then is immediately shunted into the distal small intestine, effectively bypassing half or more of the small intestine and theoretically reducing the absorptive area for food. In a vertical sleeve gastrectomy (VSG), most of the stomach is surgically removed and consequently swallowed food enters a small cylinder of remaining stomach leading directly into the small intestine, thereby eliminating most of the stomach’s normal ability to store ingested food. These commonly performed procedures result in rapid, substantial, and comparable weight loss; for example, many patients have their diabetes greatly improved, often within days after the surgery.
The mechanisms for the weight loss and reduction of diabetic symptoms after RYGB or VSG are not well understood. Although increased gastric stretch may be a factor in some procedures, it is not common to all, and malabsorption seems to have a minimal effect at best. Instead, a major contributing factor may be an alteration of the normal neural and endocrine signaling between the GI tract and the brain. For example, one feature common to the successful procedures is greatly increased meal-induced secretion of peptides such as GLP-1 and PYY from enteroendocrine cells, perhaps thereby creating a greater satiation signal as well as a greater stimulation of insulin secretion during meals. In this regard, many diabetic patients lose substantial amounts of body weight when their pharmacotherapy includes GLP-1 agonists.
Summary
Three effects of eating limit the size of an ongoing meal: gastric distension (potentiated by CCK), postgastric detection of calories (via satiation signals such as CCK and GLP-1, perhaps potentiated in the liver by insulin), and increased plasma osmolality. These signals reach the brain through visceral afferent fibers (especially those traveling in the vagus nerve) and the circulatory system. Meal size invariably increases after the inhibitory effects are experimentally removed by blocking the detection of gastric stretch or of satiating peptides, diluting the food with noncaloric material, or rehydrating the animal. Inhibition appears to be integrated so that as one effect diminishes (e.g., the signal associated with gastric distension), another increases (e.g., the signal associated with the postabsorptive delivery of calories and insulin to the liver), thereby maintaining satiety and prolonging the interval between meals. When the satiety signals disappear, hunger emerges and stimulates initiation of another meal.
Food intake is also normally linked closely with body weight. In experimental animals, periods of starvation or forced feeding are followed by self-regulated compensatory changes in eating patterns until prior body size is regained. Links between body weight and food intake can be mediated indirectly by altered gastrointestinal motility as well as by the blood-borne, centrally active hormones leptin and insulin.
Central Control of Food Intake
Early theories of the control of food intake focused on signals derived from stomach and blood. Peripheral factors, such as the concentration of glucose in the blood, were plausibly associated with food intake and were accessible to experimentation. Theories involving the brain emerged after development of a stereotaxic instrument that enabled discrete lesions of the cerebrum to be made in laboratory animals, permitting evaluation of ideas about brain function (Fig. 36.4). The first findings from such experiments were striking: Bilateral electrolytic lesions of the ventromedial hypothalamus (VMH) caused marked hyperphagia and obesity in rats. This observation was interpreted to mean that the animals had become less sensitive to incoming signals of satiation and that they therefore overate and became fat. Bilateral lesions in the adjacent ventrolateral hypothalamus (VLH) were later found to cause aphagia (absence of eating), which led to death by starvation. This finding was interpreted to mean that these rats no longer detected hunger signals, and thus they starved to death while unaware of their internal state. Collectively, these results provided the foundation of a dual-center hypothesis, in which a satiety center in the VMH was thought to suppress activity in a hunger center in the VLH. Both the syndrome of hyperphagia and obesity and the syndrome of aphagia and weight loss that result from focal hypothalamic lesions have been reproduced in multiple laboratories and in multiple species. However, additional observations have caused reinterpretation of these familiar and reliable findings, and they now have a very different meaning.
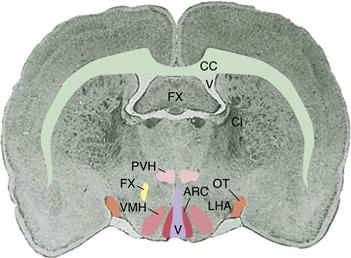
Figure 36.4 Coronal section through an adult rat brain at the level where the optic tract enters the two hemispheres. The lateral ventricle and third cerebral ventricle (V) are shaded, and selected fiber tracts are outlined for reference (OT, optic tract; CC, corpus callosum; FX, fornix). Key hypothalamic sites important in caloric homeostasis, present in each hemisphere, are paraventricular nuclei (PVN), ventromedial nuclei (VMN), arcuate nuclei (ARC), and the lateral hypothalamic area (LHA).
Food Intake is Not Controlled by Hypothalamic Hunger and Satiety Centers
The strength of the traditional dual-center model, in which one center mediates hunger and the other satiety, was that it provided a simple answer to the question of how the brain controls intake of food. The principal limitation of the model, however, was that it could not answer the next level of questions: What signals control individual meals? How are these signals integrated with physiological aspects of caloric homeostasis, including long-term maintenance of body weight? What is the influence of other neural sites and systems in the brain on the control of food intake? How are excitatory and inhibitory influences on eating integrated? Investigators are addressing these and related questions, but they now usually take a perspective that does not presume the existence of brain centers mediating hunger and satiety.
In addition to causing hyperphagia, VMH lesions profoundly reduce sympathetic tone and increase parasympathetic tone and vagal reflexes. For example, the high concentration of insulin in animals with VMH lesions is not simply a secondary consequence of hyperphagia but occurs even when food intake is limited. Because of the change in autonomic tone, caloric equilibrium in adipose tissue shifts away from lipolysis and toward lipogenesis, thereby allowing more rapid storage of ingested calories after a meal. Increased parasympathetic activity also allows faster gastric emptying. Importantly, VMH lesions do not cause loss of satiety. Meal sizes continue to correlate well with postmeal intervals, but the interval after a meal of any size is shorter in animals with VMH lesions than in control animals. Because the duration of their postprandial satiation signals is shorter than normal, rats with VMH lesions eat more frequent meals. They remain hyperphagic until the equilibrium between lipogenesis and lipolysis is reestablished in adipocytes. For this to happen, triglyceride stores must accumulate in the cells to levels sufficient to create insulin resistance, whereupon lipogenesis subsides, gastrointestinal motility diminishes, meals are eaten less frequently, and daily food intake is reduced. The animal maintains its obese state as if it had a new “set point” for body weight. Thus, after obese rats with VMH lesions have been deprived of food and forced to lose weight, hyperphagia reoccurs until the prior level of high adiposity is reattained. Similarly, when neurologically normal rats are overfed and become obese prior to receiving a lesion of the VMH, they are not hyperphagic after the lesion but rather eat to maintain their elevated weight.
In sum, rats gain body fat after VMH lesions, and as they are becoming obese (indeed, in part because they are becoming obese), the duration of satiety after meals is relatively low. However, as fat accumulates the duration of satiety after meals increases back toward normal. This explanation contrasts with early hypotheses that body weight increased after VMH lesions because animals became obligatorily hyperphagic. The primary phenomenon is now recognized as a change in autonomic tone to promote vagal reflexes and fat storage. Because that change is observed even when increased food intake is prevented, hyperphagia cannot be the primary phenomenon. The key element appears to be the chronic, neurally stimulated increase in insulin secretion. When the increased insulin secretion is prevented by denervating the pancreas, obesity does not develop and hyperphagia is greatly attenuated.
In contrast to VMH lesions, rats with large VLH lesions do not eat and may starve to death. However, they also do not drink water when dehydrated and they do not move about in their cages or respond to diverse stimulation. In other words, the most prominent abnormalities in these rats are akinesia and sensory neglect. In this respect, rats with VLH lesions resemble human patients with Parkinson’s disease, a neurological disorder that has been attributed to the degeneration of dopamine-containing neurons of the nigrostriatal bundle (see Chapters 30 and 31). Dopaminergic fibers course through the internal capsule just lateral to the VLH as they ascend from the ventral mesencephalon to the striatum along the medial forebrain bundle (Chapter 43). Large electrolytic lesions of the VLH area interrupt these dopaminergic fibers. More selective damage to the dopaminergic neurons near the VLH by intracerebral administration of the neurotoxin 6-hydroxydopamine also produces akinesia and sensory neglect in association with loss of food intake, and it does so without disturbing parasympathetic reflexes. Thus, the aphagia induced by large VLH lesions does not result from damage to a putative hunger center in the brain but instead reflects a more general disruption of movement and sensorimotor integration (Ungerstedt, 1971), at least in part (see later).
Summary
The ability of brain circuitry to control food intake initially was based on the consequences of stereotaxic lesions of ventromedial and ventrolateral hypothalamic nuclei leading to hyperphagia or aphagia, respectively. Subsequent studies revealed that each lesion also induces changes in autonomic function, in gastrointestinal activity, and in the equilibrium between lipolysis and lipogenesis in adipocytes. In addition, these lesions affected eating behavior through influences on spontaneous mobility and sensory responsivity to food-derived stimuli.
Neuropeptides and the Control of Food Intake
Although the details of the central control of food intake are incompletely understood, many neuropeptides have been implicated. To simplify the discussion, we focus on two broad categories of neuropeptides: those that are mainly anabolic and those that are mainly catabolic. Both of these subgroups in turn are modified by multiple influences, including satiation signals, adiposity signals, experience, habit, emotional state, the social situation, and so on. Anabolic peptides cause increased eating, decreased energy expenditure, and, when chronically elevated, increased body fat accumulation. Catabolic peptides reduce food intake, increase energy expenditure, and lead to loss of body fat. Figure 36.5 presents a simplified model of some of the better-known components of this control system. An interesting feature of the model is that major efferent outputs from the hypothalamus to autonomic nuclei and to motor nuclei controlling ingestion per se emanate from the VMH and VLH, the central neural sites that were the main focus of attention over a half-century ago.
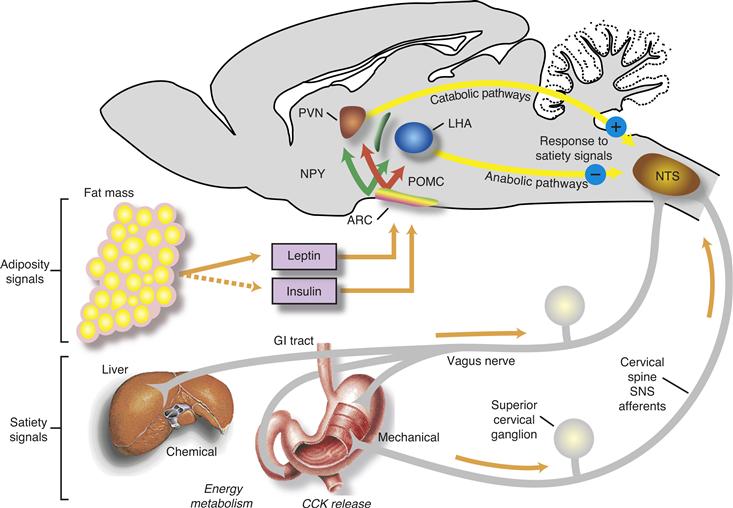
Figure 36.5 Schematic diagram of the signals that control caloric homeostasis. Satiety signals arising in the periphery such as gastric distension and CCK are relayed to the nucleus of the solitary tract (NTS) in the brainstem. Leptin and insulin, the two circulating adiposity signals, enter the brain and interact with receptors in the arcuate nucleus (ARC) and other brain areas. These adiposity signals inhibit ARC neurons that synthesize NPY and AgRP (NPY cells in the diagram) and stimulate neurons that synthesize proopiomelanocortin (POMC), the precursor of α-MSH, and CART. These ARC neurons in turn project to other hypothalamic areas, including paraventricular nuclei (PVN) and the lateral hypothalamic area (LHA). Catabolic signals from the PVN and anabolic signals from the LHA are thought to interact with the satiety signals in the brainstem to determine when meals will end.
The Hypothalamic Arcuate Nucleus (ARC) Mediates the Effect of Adiposity on Food Intake and Body Weight
The amount of body fat is signaled to the brain by leptin and insulin. Receptors for both peptides are located (among other sites) in the ARC on two distinct groups of neurons. The first group of ARC neurons synthesizes the neuropeptides, α-melanocyte-stimulating hormone (α-MSH) and cocaine–amphetamine-related transcript (CART); these neurons are activated by leptin and insulin. α-MSH and CART are potent catabolic peptides, and when either is administered locally into the third cerebral ventricle animals eat less food, have increased energy expenditure, and lose weight (Cone, 1999). α-MSH is in the melanocortin family of peptides, a group that also includes adrenocorticotrophic hormone (ACTH). These peptides act on melanocortin (MC) receptors, and two of these receptors, termed MC3 and MC4, are expressed in several nuclei of the hypothalamus, including the paraventricular nucleus (PVN), VMH, and VLH, where α-MSH-containing axons project from the ARC and where α-MSH acts as an agonist. Animals that lack either MC3 or MC4 receptors become obese; animals with no MC3 receptors gain weight gradually over their lifetime without overeating, whereas animals without MC4 receptors are hyperphagic and become much fatter. The administration of selective MC4 antagonists causes neurologically intact animals to overeat and, if prolonged, to become obese. Hence, hypothalamic melanocortins, exemplified by α-MSH, exert a tonic catabolic effect to keep body weight from increasing (Cone, 1999). Importantly, the ability of exogenous leptin to reduce food intake and body weight is completely attenuated in the presence of an antagonist to MC3 and MC4 receptors, suggesting that the “downstream” pathway by which adiposity influences caloric homeostasis is via melanocortin signaling (Seeley et al., 1997).
The other type of ARC neuron influenced by adiposity signals synthesizes the neuropeptides neuropeptide Y (NPY) and agouti-related protein (AgRP). Both peptides are potent anabolic compounds in that the administration of either into the third ventricle results in hyperphagia, reduced energy expenditure, and weight gain. Although NPY is synthesized in many areas of the brain including the dorsomedial nucleus of the hypothalamus (DMN), AgRP synthesis is limited to the ARC. Neurons containing NPY and AgRP express receptors for both leptin and insulin, and the local administration of either insulin or leptin near the ARC reduces the synthesis of both NPY and AgRP. Hence, adiposity signals exert net catabolic effects through the joint mechanisms of stimulating α-MSH/CART neurons and simultaneously inhibiting NPY/AgRP neurons. For all these reasons, the ARC can be considered to be the brain’s sense organ that detects body adiposity by monitoring the levels of leptin and insulin. In turn, axons from these two groups of ARC neurons innervate many other hypothalamic nuclei as they modulate aspects of caloric homeostasis.
Two important tracts convey NPY-containing axons to the PVN, one from the ARC that co-expresses AgRP and one from the DMN that has no AgRP. The PVN expresses receptors for NPY, and careful mapping studies have identified the PVN as the most sensitive site to the orexigenic effect of exogenously administered NPY. Consistent with this finding, when antisense oligonucleotides to NPY receptors are given locally within the PVN, rats that have been deprived of food eat less than they usually would. Among the numerous sites that synthesize NPY within the brain, only those in the arcuate nucleus are sensitive to changes of food intake as signaled by leptin and insulin (Schwartz et al., 2000). NPY mRNA is increased in the ARC and DMN when animals are fasted and returns to baseline upon refeeding. Fasting also increases the levels of NPY in axon terminals in the PVN and increases secretion of NPY within the PVN. Hence, the fasted animal is primed to eat more food due in part to the action of elevated NPY in the area of the PVN. Circulating insulin and leptin are decreased during fasting, and local administration of either into the brain of a fasted rat lowers the elevation of NPY mRNA in the ARC and lowers the amount of food eaten by food-deprived rats (Schwartz et al., 2000). Hence, the metabolic state, as reflected by leptin and insulin, has a marked influence on arcuate NPY-containing neurons and consequently on neural pathways that influence energy intake and expenditure. Endogenous levels of NPY in the ARC–PVN system normally peak when daylight ends and nocturnal activity begins (Leibowitz, 1990), which also is the time when rats typically eat their largest meal of the day. Hence, NPY activity in the PVN appears to be a key mediator of anabolic activity.
The other neuropeptide synthesized in NPY neurons in the arcuate nucleus, AgRP, is an endogenous antagonist of MC3 and MC4 receptors (Cone, 1999). AgRP therefore promotes food intake by acting at MC receptors in the PVN and other areas to block the action of α-MSH. When exogenous AgRP is administered into the third cerebral ventricle, the resultant hyperphagia lasts for up to six days (Hagan et al., 2000). In contrast, the hyperphagia elicited by NPY lasts only a few hours. Hence, activation of the arcuate NPY/AgRP neurons results in an acute but robust increase of food intake caused by the actions of NPY and a steady and more prolonged increase caused by the actions of AgRP. Furthermore, the two peptides act in different ways. NPY acts through its receptors to stimulate anabolic pathways directly, whereas AgRP acts through a different receptor by antagonizing tonically active catabolic peptides. It is not known whether NPY and AgRP from ARC neurons act on the same or different target cells in the PVN, VLH, and elsewhere. An important distinction is that whereas NPY originating in both the ARC and the DMN project to the PVN and VLH, only DMN NPY neurons project to areas of the brainstem where satiation signals are relayed from the vagus (Moran, 2010).
In sum, the ARC appears to be a chemosensor in the brain that detects body adiposity by means of insulin and leptin receptors. One type of ARC neuron synthesizes peptide transmitters (α-MSH and CART) having a net catabolic effect, whereas another type synthesizes peptide transmitters (NPY and AgRP) with a net anabolic effect. Both types of neuron project to other hypothalamic areas where they influence specific aspects of caloric homeostasis.
Other Hypothalamic Peptides also Influence Food Intake
Several other neuropeptides important in the control of caloric homeostasis are synthesized in the VLH, including the orexins and melanin-concentrating hormone (MCH) (Shimada, Tritos, Lowell, Flier, & Maratos-Flier, 1998). Administration of either of these peptides into the brain stimulates food intake, whereas mice that lack the gene for MCH are lean and obesity resistant. Administering orexins into the brain also elicits wakefulness, and as orexin levels decline after a meal, wakefulness wanes and drowsiness takes over. It appears likely that the aphagia exhibited by animals with VLH lesions is due in part to reduced levels of these peptides and that the hyperphagia caused by electrical stimulation of the VLH is due in part to release of orexins and MCH.
Oxytocin is a peptide synthesized in the PVN and supraoptic nuclei of the hypothalamus and is secreted from the posterior lobe of the pituitary. In the systemic circulation of female mammals, this hormone is well known for stimulating uterine contractions for parturition and stimulating milk letdown in mammary glands during lactation. However, the hypothalamus and pituitary of males contain as much oxytocin as those of females; therefore, the peptide has long been suspected of being secreted in other circumstances and having additional functions. In fact, research has revealed that gastric distension, CCK administration, and plasma hyperosmolality, treatments that decrease food intake in rats, each elicit pituitary oxytocin secretion in male and female rats. In addition, increases in plasma concentrations of oxytocin correlate highly with observed decreases in food intake.
Although observations were consistent with oxytocin being an appetite suppressant, later experiments showed that this was not the case: Systemic administration of oxytocin does not affect food intake. Instead, the reduced food intake seen after treatment with CCK or hypertonic saline appears to be mediated by oxytocin secreted from neurons whose cell bodies are located in the hypothalamic PVN but whose axons project within the central nervous system rather than to the pituitary gland. Consistent with this hypothesis, central injection of oxytocin into the cerebral ventricles (icv) decreases food intake in rats, an effect that is blocked by icv pretreatment of the animals with an oxytocin receptor antagonist. More to the point, the effects of exogenous CCK and hypertonic saline on food intake are blunted by icv pretreatment with an oxytocin receptor antagonist. A summary of some of the hypothalamic peptides that modulate food intake is depicted in Figure 36.6.
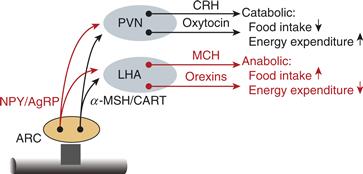
Figure 36.6 Hypothalamic neuropeptides that influence caloric homeostasis. The adiposity hormones, leptin and insulin, are transported through the blood–brain barrier and influence neurons in the arcuate nucleus (ARC). ARC neurons that synthesize and release NPY and AgRP are inhibited by adiposity signals, whereas ARC neurons that synthesize and release α-MSH and CART are stimulated by adiposity signals. NPY/AgRP neurons are inhibitory to the PVN and stimulatory to the LHA, whereas α-MSH/CART neurons are stimulatory of the PVN and inhibitory of the LHA. The PVN in turn has a net catabolic action, whereas the LHA has a net anabolic action.
Stress is also well known to decrease food intake in animals, and various stressors activate the hypothalamic–pituitary–adrenal (HPA) axis. A key mediator of this effect is corticotropin-releasing hormone (CRH), which is synthesized and secreted from the PVN. CRH stimulates the secretion of ACTH from the anterior pituitary, and ACTH in turn elicits steroid hormone secretion from the adrenal cortex (see Chapter 40). In rats, icv injection of CRH decreases food intake in the absence of stress and in association with oxytocin secretion from the posterior pituitary. In addition, icv pretreatment with an oxytocin receptor antagonist eliminates CRH-induced inhibition of food intake. These findings are consistent with central oxytocinergic neurons mediating the inhibition of food intake that occurs during stress. Such inhibitory effects on eating would complement the known inhibitory effects of central oxytocin on gastric motility and emptying. CRH neurons also stimulate activity of the sympathetic nervous system, and the PVN is considered to be the major integrative site where stressors activate both the HPA axis and the sympathetic nervous system. Some individuals when stressed increase their consumption of “comfort foods,” which are generally palatable and calorically dense. Recent evidence suggests that consumption of these foods is a buffer against the magnitude of the physiological responses to both physical and psychological stressors (Ulrich-Lai et al., 2010).
The Brainstem Plays an Important Role in the Control of Food Intake
The NST and the adjacent area postrema in the brainstem receive sensory fibers from gustatory receptors in the mouth and throat (see Chapter 24), as well as afferent information from the stomach, small intestine, pancreas, and liver. In the central control of food intake, these linked brainstem sites likely are where sensory input from the viscera is first integrated with input from taste buds. This sensory information is relayed along prominent neural projections from the brainstem to the hypothalamus, amygdala, and other portions of the limbic system, as well as to the thalamus and gustatory cortex. Reciprocal neural connections to the brainstem from these rostral sites allow emotion and cognitive function to influence the control of eating. Complementary and linked efferent elements participate in the central control of the gastrointestinal tract and liver, and of autonomic function generally.
Evidence of the brainstem’s control over food intake comes from investigations of the chronic decerebrate rat in which all axonal connections between the caudal brainstem and the forebrain are severed at the midcollicular level of the midbrain (Grigson, Kaplan, Roitman, Norgen, & Grill, 1997). This animal does not seek food or initiate spontaneous meals, but, like an intact animal, it will reflexively swallow liquid food put directly into its mouth via an oral fistula. The decerebrate rat will swallow a sucrose solution when its stomach is empty, such as after a period of food deprivation; however, it will not swallow water or saline when food deprived, nor will it swallow sucrose solution when its stomach is full or when it has received a systemic injection of CCK or hypertonic saline (but instead lets the administered fluid passively drip from its mouth). These experiments suggest that considerable control of food intake exists entirely within the caudal brainstem, which receives signals that arise when ingested food contacts peripheral sensory organs (i.e., taste buds, gastric stretch receptors, intestinal CCK-secreting cells). Nonetheless, decerebrate animals are incapable of receiving input from forebrain sites that could modify intake based on learning or other experience, on body adiposity, or on time of day.
Other observations provide additional support for an important role of the brainstem in the control of meal size. When the area postrema is ablated, rats eat larger meals than normal, although total daily food intake is normal; that is, they compensate for their large meals by eating less frequently, indicating that long-term controls of food intake are not impaired. A similar effect is seen in rats pretreated with capsaicin, a neurotoxin that destroys most gastric afferent vagal axons, eliminating the food intake-reducing effects of CCK and presumably damaging the signal of gastric distension.
Summary
We began this chapter with a description of the physiology of caloric homeostasis in order to provide a biological context within which to consider the purpose of eating and the fate of ingested food. Two principles were emphasized. First, meal size is determined by the integrated effects of several acute stimuli. Delivery of calories to the stomach and small intestine upon food consumption and postabsorptive delivery of nutrients to the liver elicit sensory neural signals from the stomach and the liver, respectively, to the brainstem. Second, food intake is also influenced by chronic signals associated with body adiposity. Adiposity indirectly affects gastric and hepatic signals of satiety. In addition, humoral signals proportional to adiposity enter the brain and affect food intake. One signal, leptin, emanates from adipose tissue itself, and the other, insulin, comes from the pancreas. These two peptides appear to be important in the central control of food intake; they modulate the response of the brain to the acute neural signals that affect food intake.
These neural and humoral signals are related to caloric homeostasis and are inhibitory in nature. During meals, they combine to stop ongoing ingestion; later, when these signals disappear in the postabsorptive state, new meals begin. Other inhibitory signals are unrelated to caloric homeostasis yet can have a pronounced effect on the cessation of eating. These signals include gastric distension, toxins (in the food and consumed inadvertently, or else injected systemically), and dehydration.
The central nervous system exerts control over eating at many levels. The spinal cord and brainstem influence all aspects of caloric homeostasis via the autonomic nervous system. The hypothalamus and limbic forebrain receive signals about ingested food and body adiposity and integrate them with information about the taste of the food, the memory of that food, the experience of previous meals, the competition of other desires, and aspects of the environment. Thus, the central control of eating involves many areas of the central nervous system in the collective maintenance of caloric homeostasis.
References
1. Coleman DL, Hummel KP. Effects of parabiosis of normal with genetically diabetic mice. American Journal of Physiological. 1969;217:1298–1304.
2. Cone RD. The central melanocortin system and energy homeostasis. Trends in Endocrinology Metabolism. 1999;10:211–216.
3. Gibbs J, Young RC, Smith GP. Cholecystokinin decreases food intake in rats. Journal of Comparative and Physiological Psychology. 1973;84:488–495.
4. Grigson PS, Kaplan JM, Roitman MF, Norgen R, Grill HJ. Reward comparison in chronic decerebrate rats. American Journal of Physiological. 1997;273:R479–R486.
5. Hagan MM, Rushing PA, Pritchard LM, et al. Long-term orexigenic effects of AgRP-(83–132) involve mechanisms other than melanocortin receptor blockade. American Journal of Physiological. 2000;279:R47–R52.
6. Le Magnen J, Tallon S. La periodicite spontanee de la prise d’aliments ad libitum du rat blanc. Journal of Physiological, (Paris). 1966;58:323–349.
7. Leibowitz SF. Hypothalamic neuropeptide Y, galanin, and amines: Concepts of coexistence in relation to feeding behavior. Annals of the New York Academy of Science. 1990;611:221–235.
8. Moran TH. Hypothalamic nutrient sensing and energy balance. Forum of Nutrition. 2010;63:94–101.
9. Phillips RJ, Powley TL. Gastric volume rather than nutrient content inhibits food intake. American Journal of Physiological. 1996;271:R766–R779.
10. Schwartz MW, Woods SC, Porte Jr D, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671.
11. Seeley RJ, Yagaloff KA, Fisher SL, et al. Melanocortin receptors in leptin effects. Nature. 1997;390:349.
12. Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674.
13. Smith GP, Gibbs J, Young RC. Cholecystokinin and intestinal satiety in the rat. Federation Proceedings. 1974;33:1146–1150.
14. Ulrich-Lai YM, Christiansen AM, Ostrander MM, et al. Pleasurable behaviours reduce stress via brain reward pathways. Proceedings of the National Academy of Science of United States of America. 2010;107:20529–20534.
15. Ungerstedt U. Adipsia and aphagia after 6-hydroxydopamine induced degeneration of the nigro-striatal dopamine system. Acta Physiologic Scandinavica Supplementum. 1971;367:95–122.
Suggested Readings
1. Barsh GS, Farooqi IS, O’Rahilly S. Genetics of body-weight regulation. Nature. 2000;404:644–651.
2. Elmquist JK, Elias CF, Saper CB. From lesions to leptin: Hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232.
3. Langhans W. Metabolic and glucostatic control of feeding. The Proceedings of the Nutrition Society. 1996;55:497–515.
4. Levin BE, Magnan C, Dunn-Meynell A, Le Foll C. Metabolic sensing and the brain: Who, what, where, and how?. Endocrinology. 2011;152:2552–2557.
5. Woods SC. Insulin and the brain: A mutual dependency. Progress in Psychobiology and Physiological Psychology. 1995;16:53–81.
6. Woods SC, Schwartz MW, Baskin DG, Seeley RS. Food intake and the regulation of body weight. Annal Review in Psychology. 2000;51:255–277.