Chapter 40
The Neurobiology of Sleep and Dreaming
Sleep is a behavioral state that alternates with waking and occupies one-third of our lives. It is characterized by a recumbent posture, a raised threshold to sensory stimulation, a low level of motor output, and a unique behavior: dreaming.
In contrast to sleep, the conscious behavior of waking is characterized by an active and deliberate sensorimotor discourse with the environment. For maintenance of waking behavior, neural gates must remain open for sensory input and motor output, the brain must be tuned and activated, and the chemical microclimate must be appropriate for processing and recording of information.
The Two States of Sleep: Rapid Eye Movement (REM) and Non–Rapid Eye Movement (NREM) Sleep
Sleep Architecture
The electrical activity of the brain during sleep reveals two distinct substates—rapid eye movement (REM) and non-REM (NREM) sleep—that alternate with a cycle of about 90 minutes (Fig. 40.1B), indicating their control by an endogenous neuronal oscillator. NREM phases are deeper and longer early in the night, whereas the proportion of each cycle occupied by REM is much greater during cycles later in the night (Fig. 40.1A).
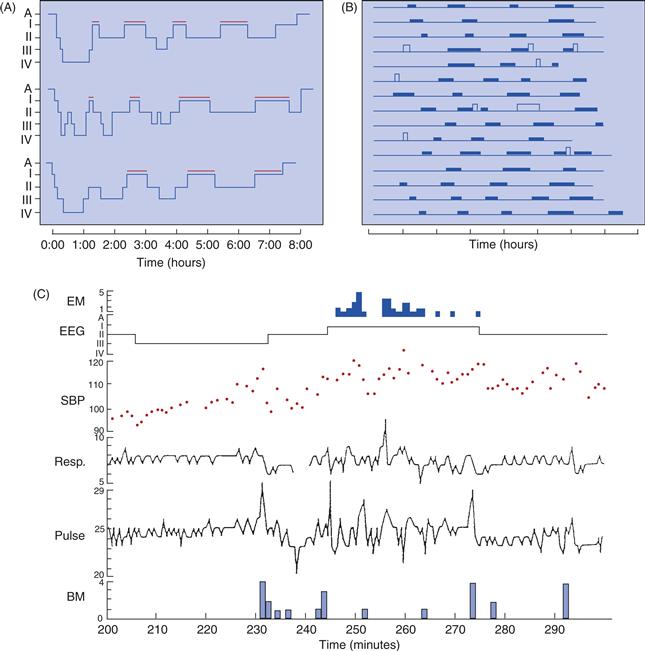
Figure 40.1 Periodic activation in sleep cycles. (A) The sleep stages of three people are graphed. The first two or three cycles of the night are dominated by deep stages (3 and 4) of NREM sleep, and REM sleep (indicated by red bars) is brief or nonexistent. During the last two cycles of the night, NREM sleep is lighter (stage 2), and REM episodes are longer, sometimes more than an hour. (B) Fifteen nights of sleep. Each line represents one night of sleep, with REM periods shown as solid bars and periods of wake as taller, open bars. Each record begins at the onset of sleep. The amount of time before the first episode of REM varies, but once REM has begun, the interval between episodes is fairly constant. (C) Eye movements (EM) in numbers per minute, EEG (as scored sleep stage), systolic blood pressure (SBP) in mmHg, respiration (Resp.) in breaths per minute, pulse in beats per quarter minute, and body movement (BM) in numbers per minute over 100 minutes of uninterrupted sleep. The interval from 242 to 273 minutes is considered the REM period, although eye movements are not continuous during that interval.
NREM and REM sleep are characterized and distinguished using polysomnography (PSG), an electrophysiological recording technique that employs electroencephalography (EEG), electrooculography (EOG), and electromyography (EMG) (Fig. 40.2, middle panel). Combinations of tonic (enduring) and phasic (transient) events in these three types of channels traditionally define sleep stages and substages using the Rechtschaffen and Kales (1968) manual, recently revised with new nomenclature (Iber, Ancoli-Israel, Chesson, & Quan, 2007). The EEG channel tracings of each of these stages are shown in Figure 40.3.
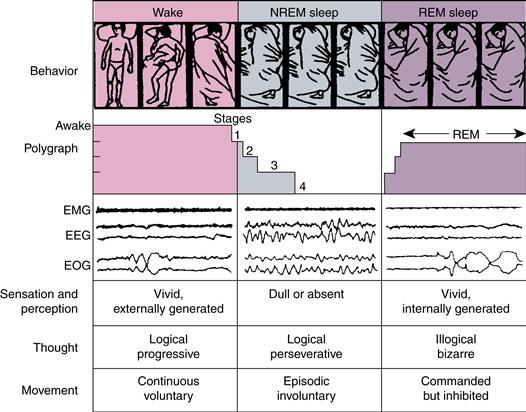
Figure 40.2 Behavioral states in humans. Body position changes during waking and at the time of phase changes in the sleep cycle. Removal of facilitation (during stages 1–4 of NREM sleep) and addition of inhibition (during REM sleep) account for immobility during sleep. In dreams, we imagine that we move, but no movement occurs. Tracings of electrical activity are shown in ~20-s sample records. The amplitude of the electromyogram (EMG) is highest in waking, intermediate in NREM sleep, and lowest in REM sleep. The electroencephalogram (EEG) and electrooculogram (EOG) are activated in waking and REM sleep and inactivated in NREM sleep.
Reprinted with permission from Hobson and Steriade (1986).
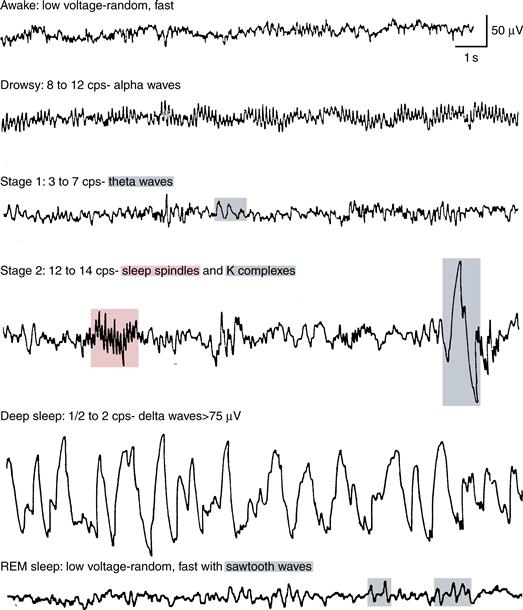
Figure 40.3 Electroencephalograms showing electrical activity of the human brain during different stages of sleep.
Sleep onset—that is, the onset of Stage 1 NREM sleep (N1 in new nomenclature)—is gradual and characterized by slowing in brain wave frequency from the waking EEG that consists predominantly of beta (14–25 Hz) and gamma (30–80 Hz) frequencies when the eyes are open (Fig. 40.3, first trace) and alpha (8–13 Hz) when the eyes are closed (Fig. 40.3, second trace), to a predominance of theta rhythm (4–7 Hz) in N1 (Fig. 40.3, third trace). During N1, characteristic slow eye movements are recorded in the EOG, EMG muscle tone is diminished relative to waking, awareness of the outside world is lost, and “hypnogogic” dreamlike imagery may occur. As NREM sleep deepens through NREM Stages 2, 3, and 4 (now termed N2 and N3, in which N3 encompasses prior Stages 3 and 4), muscle tone further diminishes, and, in the EEG, changes consist of a progressive decrease in the predominant EEG frequency that is accompanied by a progressive increase in average wave amplitude.
As N1 transitions to N2 (Fig. 40.3, fourth trace), the EEG further slows in the predominantly theta range, and two distinctive phasic wave forms, sleep spindles (De Gennaro & Ferrara, 2003) and K-complexes (Colrain, 2005) occur. Sleep spindles are intermittent clusters of spikes at sigma frequency (12–16 Hz) with characteristic waxing and waning morphology. Spindles occur in two varieties: slow spindles with frequencies approximately 12–14 Hz that appear most strongly in frontal scalp EEG leads and fast spindles (approximately 14–16 Hz) that appear most strongly at centro-parietal EEG leads (Merica, 2000). Sleep spindles are generated by a distinct thalamo-cortical circuitry detailed in a later section. K-complexes consist of a large negative (upward) deflection in the EEG followed by a positive (downward) deflection and are also generated by thalamo-cortical interactions.
In N3, also known as slow wave (SWS) or delta sleep (Fig. 40.3, fifth trace), many fewer sleep spindles occur, and EEG frequency further slows and displays increasing predominance of high amplitude delta (0.5–4.0 Hz) waves (Dijk, 2009). In the early 1990s, a yet slower oscillatory rhythm of less than 1 Hz, the “slow oscillation,” was described in cats and, shortly afterward, in humans (Steriade, 2006). The slow oscillation shows maximal spectral power in SWS and exerts an organizing or “grouping effect” on delta oscillations, spindles, and K-complexes (Steriade, 2006). As NREM deepens from N1 to N3, the threshold for arousal rises in proportion to the degree of EEG slowing, and during N3, awakenings are difficult, incomplete and brief. Those awakened from this state are confused, have difficulty reporting conscious experience, and return to sleep rapidly, especially early in the night. While dreaming also occurs during NREM sleep (Fig. 40.2, bottom panel), it is much more strongly associated with the activated brain state of REM.
REM sleep, discovered in 1953 by Eugene Aserinsky and Nathaniel Kleitman (Aserinsky & Kleitman, 1953), has also been termed “paradoxical” sleep because, whereas its fast mixed-frequency EEG resembles waking (Fig. 40.3, sixth trace), muscle tone reaches its minimum level (see Mallick et al., 2011, for extensive reviews). This EMG minimum in REM results from central, brainstem-mediated inhibition of spinal alpha motor neurons (“REM atonia”; Chase, 2008). Postural shifts precede and follow REM sleep (Fig. 40.2, upper panel). REM has also been termed “active” sleep because of its wakelike low-amplitude, high-mixed-frequency EEG, as well as “desynchronized” sleep, in contrast with “synchronized” NREM, especially SWS, in which high-amplitude waves reflect synchronous activity of thalamic and cortical neurons (described below). A characteristic EEG feature of REM sleep in humans is the “sawtooth wave” (Fig. 40.3, sixth trace). During REM, unlike waking, there is inhibition of sensory input and motor output. Subjects aroused from REM sleep, especially during periods with frequent eye movements, often give detailed reports of dreams characterized by vivid hallucinations, bizarre thinking, and intense emotion (Fig. 40.2 bottom panel).
As its name suggests, saccadic (rapid) eye movements (REMs) occur during REM (Fig. 40.1C), singly and in clusters, reflecting the fact that, unlike skeletal muscles, oculomotor muscles maintain tonus in REM. REMs are believed to reflect underlying, central phasic neuronal events also appearing as peripheral muscle twitches (that briefly overcome atonia), middle-ear muscle movements (MEMAs), and peri-orbital potentials (PIPs). In humans, phasic REM events are likely related to ponto-geniculo-occipital (PGO) waves. PGO waves are phasic field potentials that, in the REM sleep of cats, are recorded sequentially in the pons (P), lateral geniculate body of the thalamus (G), and the occipital cortex (O) (Datta & Maclean, 2007), and which also have recently been demonstrated in humans (Lim et al., 2007). In contrast to such phasic events, the tonic wakelike REM EEG reflects cortical activation by the cholinergic ascending arousal systems described below (Saper, Fuller, Pederson, Lu, & Scammell, 2010).
Central Autonomic Control Systems Are State Dependent
Sympathetic outflow diminishes in the transition from waking to NREM, and during SWS, parasympathetic outflow is predominant. Many autonomic and regulatory functions, such as heart rate, blood pressure, and respiration rate, diminish in NREM sleep (Fig. 40.1C), but some neuroendocrine activity, such as release of growth hormone from the pituitary, increases (Pannain & Van Cauter, 2007). With the onset of REM, sympathetic influence increases, although not to the level of waking, and cardiovascular and respiratory measures become more variable (Trinder et al., 2001; Fig. 40.1C). Active temperature and respiratory control are greatly diminished in REM sleep (Hobson, 1989). In mammals, sleep and thermoregulation can exert reciprocal influence on one another through shared circuitry in the preoptic anterior hypothalamus (Parmeggiani, 2003).
Neural Control of the Sleep–Wake and REM/NREM Cycles
ARAS Sensorimotor and Neuromodulatory Neurons
Most of the classic reticular core neurons have very specific afferent inputs and highly organized outputs that underlie the multiple functions of the brainstem, such as sensory input, motor output, and autonomic control. These reticular neurons receive inputs from skin, muscle, bone, and joint receptors in the periphery, which they integrate and link to the vestibular and cerebellar circuits that determine posture and movement (see Chapter 29). Performing an entirely different function are small groups of neurons that send widely branching axons to distant parts of the brain where their neurotransmitters modulate brain function and play an essential role in behavioral state regulation including control of sleep/wake and REM/NREM cycles. These ARAS modulatory neurons differ from neighboring sensorimotor neurons in both morphology and characteristic firing patterns.
Sensorimotor neurons, approximately 50 to 75 microns in diameter, can fire continuously at high rates of up to 50 Hz and can generate bursts of up to 500 Hz. Their larger axons, especially those projecting to the spinal cord, have conduction velocities in excess of 100 m/s (Hobson & Steriade, 1986), making these neurons well suited to rapid posture adjustment and motor control. In contrast, modulatory neurons are smaller (10–25 microns in diameter), and even those with long axons conduct their signals very slowly (1 m/s). Modulatory neurons fire wider spikes (2 ms in duration) at much slower tonic frequencies (1–10 Hz) than sensorimotor neurons. In addition, they often show a very regular, metronome-like firing pattern, a reflection of their pacemaker properties. As their leaky membranes spontaneously and slowly depolarize, they reach threshold, fire, and then self-inhibit, becoming refractory even to exogenous excitatory inputs (Ramirez, Tryba, & Peña, 2004). Because of their vast postsynaptic domain, they can help set behavioral and mental states.
ARAS Neuronal Groups Involved in Behavioral State Regulation Produce Specific Neuromodulators
Modulatory ARAS neuronal groups produce specific types of neuromodulators (Dahlstrom & Fuxe, 1964). Norepinephrine (NE) neurons are located in the pons and medulla in two major groups. The lateral tegmental group consists of scattered neurons in the ventral and lateral reticular formation and appear to be involved in hypothalamic regulation and motor control (Moore & Card, 1984). Neurons of the second and major NE cell group, the locus coeruleus (LC), are located in the rostral pontine reticular formation and central gray matter. One group of LC neurons projects caudally to sensory regions of the brainstem and spinal cord, and another projects widely to the cerebellar cortex, dorsal thalamus, and cerebral cortex (Moore & Card, 1984). Thus, LC neurons are likely to be involved in regulating sensory input and cortical activation. Serotonin (5-HT) neurons are located in the raphe nuclei that extend, in the ARAS midline, from caudal medulla to midbrain in the dorsal (DR) and median raphe nuclei. These neurons project rostrally and innervate nearly the entire forebrain, again suggesting a role in regulation of behavioral state. The other important set of brainstem reticular formation nuclei involved in behavioral state control produce acetylcholine (ACh) and include two pontine nuclei: the laterodorsal tegmental (LDT) and the pedunculopontine (PPT) nuclei. Cholinergic neurons in these nuclei project to the brainstem reticular formation, hypothalamus, thalamus, and basal forebrain. A second set of cholinergic neurons in the basal forebrain (described below) projects to the limbic forebrain and neocortex. In contrast to these modulatory (5-HT, NE, and ACh) neurons, neurons in brainstem reticular formation nuclei involved in sensorimotor integration typically produce the excitatory transmitter glutamate or the inhibitory amino acids gamma-aminobutyric acid (GABA) and/or glycine.
Waking Requires Active Maintenance
The reticular formation is necessary to maintain the waking conscious state (Moruzzi & Magoun, 1949). Lesions in the midbrain reticular formation result in a state that resembles NREM sleep. Maintenance of the waking state requires brain mechanisms that compete with other active mechanisms that promote sleep. Multiple neurochemical systems maintain cortical arousal, and this redundancy allows the waking state to persist in the face of deficiency or destruction of specific arousal-related neurons (Saper et al., 2010).
These nuclei and their respective neurotransmitters and targets (Fig. 40.4) include the basal forebrain (BF) nuclei (medial septum, nucleus of the diagonal band, substantia innominata–nucleus basalis) that send ACh projections to topographically specific cortical and subcortical limbic areas (Jones, 2008); hypothalamic neurons (i) producing the neuropeptide orexin (also called hypocretin) in the lateral (LH) and perifornical (PFH) hypothalamus that project to other arousal systems as well as the entire cerebral cortex (Ohno & Sakurai, 2008), and (ii) histamine (HA)-producing neurons from the tuberomammilary nucleus (TMN) of the posterior hypothalamus (PH), which send widespread projections to diverse forebrain sites (Szymusiak & McGinty, 2008); midbrain neurons producing dopamine (DA) from specifically wake-active centers in the midbrain periaqueductal gray (PAG; Saper et al., 2010) that are distinct from midbrain substantia nigra pars compacta (SNpc) and ventral tegmental (VTA) area dopaminergic neurons (that, respectively, promote movement via projection to the basal ganglia, and appetitive behaviors via projection to the ventral striatum and prefrontal cortex); and mesopontine (junction of pons and midbrain) nuclei that include (i) ACh-producing PPT and LDT projecting to the diencephalon, basal forebrain, and restricted portions of the cortex; (ii) NE-producing LC neurons that send widespread projections to the forebrain; and (ii) 5-HT-producing DR neurons that also widely project to the forebrain (for reviews, see Pace-Schott & Hobson, 2002; Saper et al., 2010).
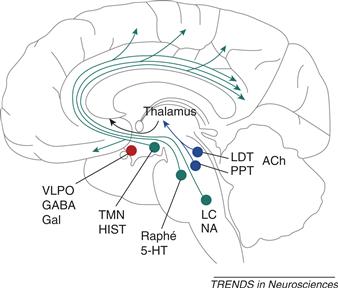
Figure 40.4 Ascending arousal systems of the brainstem and posterior hypothalamus and sleep-promoting areas of the anterior hypothalamus. Nuclei producing wake-promoting monoaminergic neuromodulators are shown in green. These include noradrenaline (NA)-producing neurons of the locus coeruleus (LC) and serotonin (5-HT)-producing neurons of the dorsal raphe (Raphe), both located in the mesopontine brainstem, as well as histamine (HIST)-producing neurons of the tuberomammilary nucleus (TMN), located in the posterior hypothalamus. These nuclei project to and activate widely distributed terminal fields throughout the forebrain. Nuclei shown in blue produce acetylcholine (ACh), which promotes forebrain activation in both waking and REM sleep. Cholinergic nuclei include the pedunculopontine (PPT) and laterodorsal (LDT) tegmental nuclei of the mesopontine brainstem as well as cholinergic nuclei of the basal forebrain (not shown). Cholinergic neurons of the PPT and LDT project to the thalamus and basal forebrain that then, in turn, send arousal-promoting projections to both distributed and regionally specific terminal fields in the forebrain. Shown in red are sleep-promoting neurons of the ventrolateral preoptic (VLPO) area of the anterior hypothalamus. VLPO neurons produce the inhibitory neuromodulator gamma-amino butyric acid (GABA) and the inhibitory neuropeptide galanin (Gal). VLPO neurons project to and inhibit the above wake-promoting neurons, thereby promoting and maintaining sleep. In turn, the wake-promoting neurons of the LC, DRN, and TMN reciprocally innervate and inhibit the neurons of the VLPO.
From Saper et al. (2001).
Certain neurochemical systems also promote cortical arousal indirectly. For example, cholinergic neurons of the LDT and PPT promote cortical activation and desynchronization of the EEG by exciting thalamo-cortical projection and, especially, inter-laminar neurons of the thalamus, as well as by inhibiting the inhibitory thalamic reticular neurons and exciting cholinergic neurons of the basal forebrain (Steriade, 2000). Similarly, orexinergic neurons stabilize the waking state by exciting other arousal systems in the hypothalamus, basal forebrain, and brainstem (Jones, 2008). Additionally, GABAergic neurons of the basal forebrain promote cortical activation by inhibiting inhibitory GABAergic cortical interneurons (Saper et al., 2010). Notably, at localized sites in the mesopontine brainstem, higher levels of GABA may actually promote wakefulness at the expense of REM and NREM sleep due to similar complex circuits of inhibition and disinhibition (Watson, Baghdoyan, & Lydic, 2011).
The Hypothalamus Controls the Sleep–Wake Transition
Lesions to anterior portions of the hypothalamus cause insomnia, whereas lesions of the posterior hypothalamus cause hypersomnolence (Saper, Chou, & Scammell, 2001). Neurons in the anterior hypothalamus increase firing during sleep, and their electrical stimulation promotes sleep, whereas neurons of posterior portions of the hypothalamus decrease firing during sleep, and their electrical stimulation promotes waking (Szymusiak & McGinty, 2008). Sherin, Shiromani, McCarley, and Saper (1996) demonstrated that neurons in the ventrolateral preoptic area (VLPO) of the anterior hypothalamus increase metabolic activity during NREM sleep in the rat. VLPO neurons produce both GABA and the inhibitory neuropeptide galanin and project to wake-promoting neurons in the TMN, LC, DR, LDT, and PPT (Saper et al., 2001). Activity in an adjacent region, the median preoptic area (MnPO), may respond to increased homeostatic sleep pressure (see next section) and contribute to subsequent activation of the VLPO that then persists to maintain sleep itself (Saper et al., 2010). Additionally, distinct subregions of the VLPO may be specialized for the maintenance of REM and NREM sleep (Saper et al., 2010). The LC, DRN, and TMN, in turn, innervate and inhibit the neurons of the VLPO (Saper et al., 2001).
Saper et al. (2001) suggested that this mutually inhibitory arrangement constitutes the biological analog of a bistable electrical “flip-flop” switch in which either the sleep (VLPO dominant) or wake (LC, DRN, TMN dominant) conditions are self-reinforcing stable states, whereas intermediate states are highly transient (Fig. 40.5). Orexin plays a key modulatory role in this flip-flop mechanism by providing excitatory drive on aminergic neurons, thereby further stabilizing the waking pole of the bipolar sleep–wake switch. In 1999, two research teams independently showed that deficiencies in orexinergic neurotransmission underlie the sleep disorder narcolepsy. Orexin-producing neurons are lost in human narcolepsy (Nishino, Ripley, Overeem, Lammers, & Mignot, 2000), likely due to an autoimmune mechanism, and, as a result, wake to sleep transitions can occur at inappropriate times (Saper et al., 2001). Orexin also opposes the atonia of REM, and, therefore, in narcolepsy, REM atonia can intrude into waking in the form of cataplexy (Alam, Szymusiak, & McGinty, 2011).
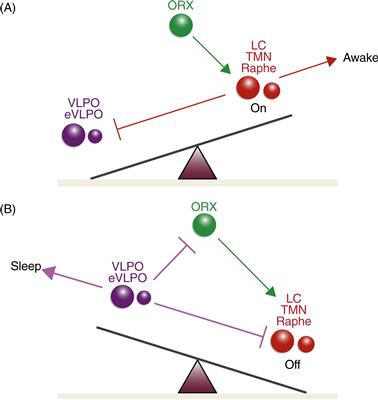
Figure 40.5 The flip-flop switch model of sleep–wake control proposed by Saper et al. (2001). (A) During waking, monoaminergic neurons (shown in red) of the locus coeruleus (LC), tuberomammillary (TMN), and dorsal raphe (Raphe) nuclei promote waking and inhibit the sleep-producing neurons of the ventrolateral preoptic area (VLPO) that are shown in purple. Orexinergic neurons (ORX) from the lateral hypothalamus (shown in green) excite and reinforce the wake-promoting activity of the monoaminergic neurons. (B) During sleep, neurons of the VLPO inhibit the wake-promoting monoaminergic neurons as well as the orexinergic neurons that help maintain their activity. By inhibiting the VLPO, monoaminergic neurons disinhibit their own wake-promoting activity, whereas, by inhibiting the monoaminergic nuclei, the VLPO disinhibits its own sleep-promoting activity. This mutually inhibitory interaction produces conditions analogous to an electrical flip-flop switch whereby the alternate states of sleep and waking are self-reinforcing but intermediate states are unstable and transient. This results in rapid transitions from waking to sleep and vice versa. Orexin serves to stabilize the waking state and prevent inappropriate transitions to sleep should the monoaminergic drive transiently weaken. In narcolepsy, orexinergic neurons are lost, thereby allowing such abrupt transitions from waking to sleep during the characteristic sleep attacks of this disorder.
(Modified from Saper, Scammel & Lu, 2005).
Homeostatic and Circadian Factors Interact to Control Sleep Onset and Offset
The timing of transitions between sleep and waking that are effected in the hypothalamus, however, depend upon input from extra-hypothalamic sources. Alexander Borbely’s two-process model of sleep propensity (Fig. 40.6) suggests that homeostatic mechanisms (Process S) interact with circadian factors (Process C) to regulate behaviorally expressed sleep and waking (Borbely, 1982). Process C (Fig. 40.6A) constitutes a circadian rhythm (see Chapter 39) in the propensity to fall asleep that is controlled by an endogenous circadian clock in the suprachiasmatic nucleus (SCN) of the anterior hypothalamus (Dibner, Schibler, & Albrecht, 2010). Reliable output from the SCN, possessing a near–exact 24-hour periodicity in humans (Czeisler et al., 1999), results in a daily rhythm in sleep propensity that is maximal in the early morning, corresponding to the circadian nadir in body temperature and is minimal in the evening, corresponding to a “forbidden zone” for sleep (Lavie, 1986). Another smaller increase in circadian sleep propensity, the “post-prandial dip,” may occur in the early afternoon around “siesta time” (Bes, Jobert, & Schulz, 2009). If a research subject is deprived of sleep, sleepiness will continue to follow a circadian rhythm, indicating that sleepiness cycles can be independent of actual sleeping and waking.
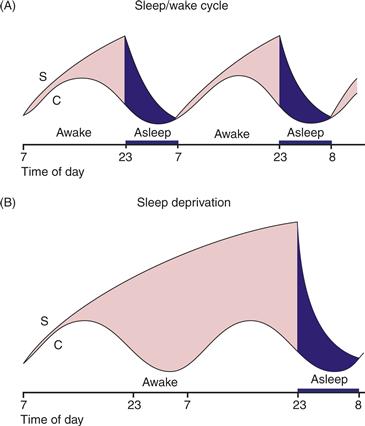
Figure 40.6 The Borbely and Daan model of sleep regulation. Sleep is assumed to result from the actions of process C and process S. Process C follows a circadian rhythm and is independent of sleeping and waking. Process S, on the other hand, depends on sleep–wake behavior; S declines during sleep and rises continuously during sleep deprivation. The period of recovery sleep that follows sleep deprivation is more intensive but only slightly longer than normal. If curve C represents the threshold for waking up, then at any time, “sleep pressure” is the (vertical) distance between the S and C curves. The greater the distance, the greater the pressure to fall asleep.
Reprinted with permission from Daan et al. (1984).
Briefly, the molecular and cellular basis of Process C involves synchronized molecular clocks in each cell of the SCN that maintain 24-hour periodicity via interlocking positive and negative feedback control in the transcription and translation of circadian genes (see Chapter 39 and Dibner et al., 2010). This endogenous clock is entrained to the ambient photoperiod via nonvisual, photoreceptive pigments such as melanopsin in retinal ganglion cells (Peirson & Foster, 2006) that send glutamatergic signals to the SCN via the retinohypothalamic tract. The SCN conveys circadian information via the subparaventricular and dorsomedial nuclei of the hypothalamus to the VLPO to influence sleep–wake transitions (Saper, Chou, & Scammel, 2005). Secretion of the sleep hormone melatonin from the pineal gland is also indirectly controlled by the SCN, increases just prior to normal sleep onset, and provides an internal indicator of circadian time as well as feedback influence on the SCN, serving to further stabilize circadian rhythms via action at melatonin receptors on the SCN itself (Arendt, 2006).
Sleep homeostasis, or Process S, is a mechanism that increases sleep pressure as a function of the amount of time since the last sleep episode (Fig. 40.6B). Process S generates the powerfully increased drive to sleep that accompanies prolonged sleep deprivation and is believed to result from sleep-promoting substances, “endogenous somnogens,” that accumulate during waking and dissipate during sleep. The purine nucleoside adenosine, which accumulates in extracellular space as a product of metabolism, is currently the most likely candidate (Porkka-Heiskanen & Kalinchuk, 2011). Evidence also exists for endogenous somnogenic actions of interleukin-1B, prostaglandin D2, and growth hormone–releasing hormone (Szymusiak & McGinty, 2008). A primary mechanism for the sleep-promoting effects of adenosine is believed to be activation of inhibitory adenosine A1 receptors on wake-promoting cholinergic cells in the basal forebrain that project to and activate the cortex and desynchronize the EEG. However, recent findings suggest that adenosine may also produce somnogenic effects elsewhere in the brain, such as at excitatory A2A receptors in the hypothalamus and ventral striatal areas. Interestingly, the most commonly employed drug to counteract high homeostatic sleep pressure, the nonspecific adenosine receptor agonist caffeine, acts not at the basal forebrain but at postsynaptic A2A receptors in the basal ganglia (Lazarus et al., 2011). Adenosine may also mediate localized sleeplike electrophysiology in restricted areas of the cortex reflecting a prior-use dependent, restorative function (Krueger et al., 2008). Astrocytic release of adenosine, which inhibits local neurons by binding to A1 receptors, may allow localized emergence of sleeplike activity such as the slow (<1 Hz) oscillation (Halassa, 2011).
By acting in concert, processes C and S promote a single, daily consolidated nocturnal sleep bout in healthy adult humans (Dijk & Czeisler, 1994; Fig. 40.6). Specifically, in the evening, when sleep homeostatic pressure is maximal following a day’s waking, the circadian “forbidden zone” maintains wakefulness until the normal time of sleep onset. In the early morning at the circadian nadir, when homeostatic sleep pressure has largely dissipated, Process C maintains the drive to sleep until the normal time of awakening.
NREM Sleep Rhythms Require Thalamo-Cortical Interaction
Following the transition from waking to NREM, neuronal firing rates in all wake-active nuclei decline with deepening NREM. This decline allows the emergence of intrinsic oscillations in reciprocally interconnected thalamic and cortical circuits that appear in the human EEG as the characteristic wave forms of N2 and N3 sleep: spindles, K-complexes, delta waves, and the (<1 Hz) slow oscillation (Steriade, 2006; Fig. 40.7). During waking, ascending arousal systems modulate thalamo-cortical circuits with the arousal-promoting neuromodulators (ACh, NE, 5-HT, and HA) and prevent the emergence of these oscillations. Similarly, during REM sleep, thalamo-cortical rhythms are prevented by cholinergic activation of the thalamus from the LDT and PPT.
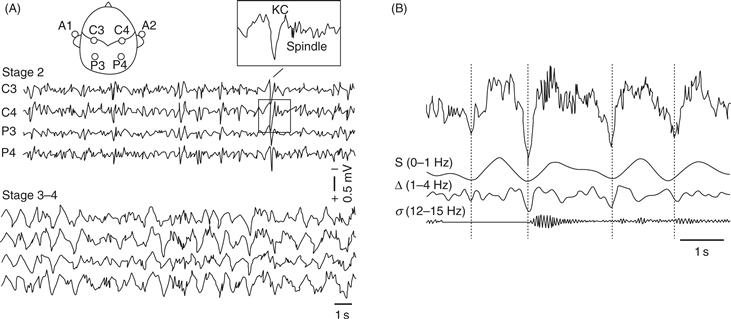
Figure 40.7 Characteristic NREM oscillations in the human EEG. (A) Recordings from central (C3, C4) and parietal (P3, P4) scalp sites referenced to the contralateral ears (A1, A2) during stage 2 NREM sleep (upper traces) and stages 3 and 4 NREM or slow wave sleep (lower traces). The insert shows the characteristic Stage 2 K-complex and sleep spindle wave forms. (B) Frequency composition of the NREM EEG showing the slow (0–1 Hz), delta (1–4 Hz), and 12–15 Hz sleep spindle (sigma frequency) oscillations. Note the organizing or grouping effect of the slow oscillation on the faster NREM oscillations.
The sleep spindles, K-complexes, and delta waves of NREM reflect the influence of the cortical slow oscillation on the thalamic reticular nucleus, a thin sheet of GABAergic neurons surrounding the thalamic periphery that inhibit other thalamic neurons (Steriade, 2000). The slow oscillation results from an alternation between a prolonged hyperpolarized (“down-state”) phase and a shorter, rapidly spiking, depolarized (“up-state”) phase that is synchronized among large assemblages of cortical neurons (Menicucci et al., 2009). The slow oscillation can be generated entirely within the cortex and may be an intrinsic property of cortical networks (Halassa, 2011), although the thalamus may also be crucial to its expression in vivo (Crunelli & Hughes, 2010). In human sleep, slow oscillations originate in frontal areas and travel posteriorly within the cortex (Massimini, Huber, Ferrarelli, Hill, & Tononi, 2004) but can also occur locally in the cortex (Nir et al., 2011). The slow oscillation exerts an organizing or grouping influence on other intrinsic NREM sleep rhythms (Fig. 40.6).
The cortical slow oscillation initiates sleep spindles when, during its depolarized up-phase, neurons projecting from the cortex to the thalamus excite neurons of the thalamic reticular nucleus that, in turn, inhibit thalamo-cortical neurons projecting from the thalamus to the cortex (Steriade, 2000). This periodic inhibition of thalamo-cortical neurons changes their firing pattern from a “transmission mode,” in which firing frequency is proportional to the strength of a sensory input, to a “bursting mode,” in which transmission of sensory information to the cortex is blocked (McCormick & Bal, 1997; Fig 40.8). This bursting mode is initiated when thalamo-cortical neurons are sufficiently hyperpolarized to activate a unique ion channel that opens only when their membrane potential falls well below its normal resting potential (McCormick & Bal, 1997). Opening of this channel begins a series of membrane events (“H” currents followed by “T” or “low-threshold calcium spike” currents) that culminates in thalamo-cortical neurons emitting a series of spikes that impinge on the cortex at spindle frequency.
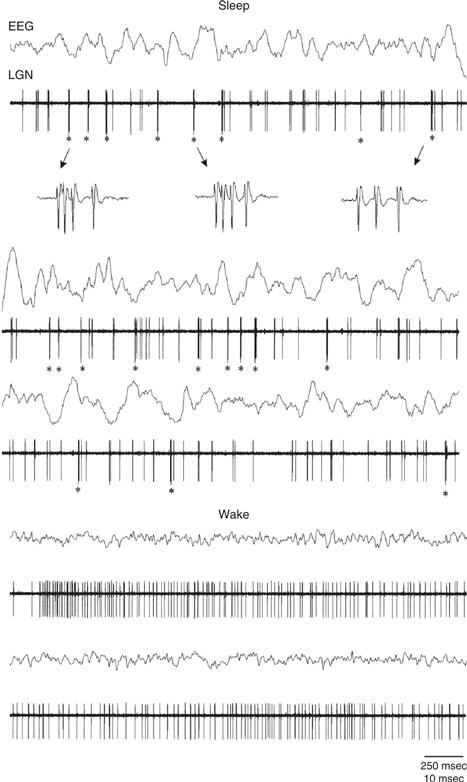
Figure 40.8 Two alternate firing modes of thalamic relay neurons: the bursting mode that is typical of slow wave sleep and the tonic (transmission) mode typical of waking. Traces show recordings in the thalamic lateral geniculate nucleus (LGN) of the cat along with simultaneous EEG from the occipital cortex. The top 7 traces show burst firing during sleep and the resultant synchronized low-frequency, high amplitude occipital EEG. The bottom 4 traces show recordings from these same sites during waking along with the resultant desynchronized, high-frequency EEG. Asterisks indicate bursting periods and the third trace shows an enlargement of the LGN channel during bursts. In the tonic mode during waking, firing frequency is proportional to the strength of a sensory input whereas during the bursting mode in sleep, transmission of sensory information to the cortex via the thalamus is blocked. For additional details on these modes of firing in thalamic relay neurons, as well as the membrane currents that underlie the rhythmic bursting mode, please see Chapter 5, Figure 5.14.
From Weyand et al. (2001), J. Neurophysiology 85:1107–18).
Delta frequency EEG oscillations may have both thalamic and cortico-thalamic origins (Steriade, 2000). Delta-frequency oscillations can arise via thalamic neurons’ intrinsic delta-frequency pacemaker property or, alternatively, when cortico-thalamic neurons excite thalamic reticular neurons that, in turn, hyperpolarize the thalamo-cortical neurons, causing them to stimulate the cortex at delta frequency. Both of these mechanisms also result from H and T currents. Because spectral power in the delta (Ferrara et al., 2002) as well as the slow oscillation (Bersagliere & Achermann, 2010) frequencies increase in response to the duration of prior waking, both are indicative of increased sleep pressure.
REM Sleep Is Initiated in the Brainstem
In the transition from NREM to REM sleep, firing rates of LC, DR, and TMN neurons and respective levels of NE, 5-HT, and HA at their terminal fields decline to their minimum levels. In contrast, firing rates of PPT/LDT neurons return to levels comparable to waking (reviewed in Pace-Schott & Hobson, 2002). Like aminergic neurons, orexin neurons of the LH/PFH decline in activity at sleep onset and remain quiescent in REM (Ohno & Sakurai, 2008). In the transition from NREM to REM, activation of the thalamus by brainstem cholinergic (LDT and PPT) neurons blocks the intrinsic oscillations of NREM and results in cortical EEG desynchronization (Steriade, 2000). The PPT and LDT also activate cortically projecting BF cholinergic neurons, resulting in further desynchronization of the cortex (Jones, 2008).
The cellular and molecular events that produce the regular alternation of REM and NREM sleep emerge under brainstem control once sleep has been achieved (reviewed in Steriade & McCarley, 2005 and Datta & Maclean, 2007). The cardinal neural features of REM are initiated by specific brainstem cell groups, illustrated in Figure 40.9, and include: (i) EEG desynchronization resulting from ARAS activation of the thalamus and basal forebrain; (ii) PGO waves in the cat, or P-waves in the rat, originating from different distinct pontine generators. PGO waves result from tonic disinhibition and phasic excitation of bursting cells in the peribrachial region of the mesopontine tegmentum and, like REM sleep, they can be triggered and augmented by cholinergic stimulation see below; (iii) rapid eye movements brought about by pre-motor neurons in proximity to oculomotor (abducens) nuclei; (iv) a change in hippocampal EEG from irregular rhythms to a regular theta rhythm (Vertes & Kocsis, 1997); and (v) skeletal muscle atonia resulting when pontine REM-active glutamatergic neurons transmit signals from pontine REM-control regions to inhibitory glycinergic and GABAergic cells of the medulla that, in turn, suppress somatic motorneurons to produce REM atonia (Chase, 2008). Other peripheral signs triggered by activity in specific brainstem nuclei include penile and clitoral tumescence, fluctuations of autonomic functions such as heart rate and blood pressure, and skeletal muscle twitches.
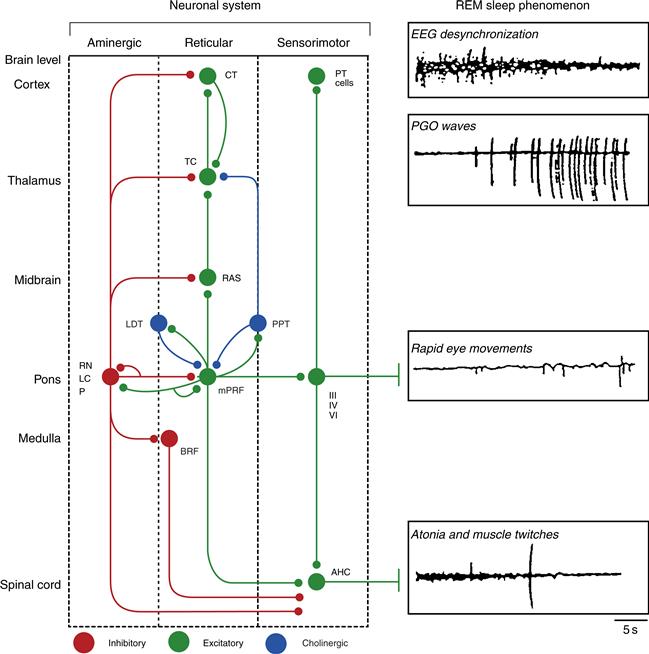
Figure 40.9 Schematic representation of the REM sleep generation process. A distributed network involves cells at brain levels from the spinal cord to the cortex (listed on the left). The network is represented as a diagram of three neuronal systems (aminergic, reticular, and sensorimotor) that mediate REM sleep phenomena (depicted in tracings on the right). Postulated inhibitory connections are shown as red circles; postulated excitatory connections as green circles; and cholinergic pontine nuclei are shown as blue circles. It should be noted that the actual synaptic signs of many of the aminergic and reticular pathways remain to be demonstrated, and, in many cases, the neuronal architecture is known to be far more complex than indicated here (e.g., the thalamus and cortex). During REM, additive facilitatory effects on pontine REM-on cells are postulated to occur via disinhibition (resulting from the marked reduction in firing rate by aminergic neurons at REM sleep onset) and through excitation (resulting from mutually excitatory cholinergic–noncholinergic cell interactions within the pontine tegmentum). The net result is strong tonic and phasic activation of reticular and sensorimotor neurons in REM sleep. REM sleep phenomena are postulated to be mediated as follows: EEG desynchronization results from a net tonic increase in reticular, thalamocortical, and cortical neuronal firing rates. PGO waves are the result of tonic disinhibition and phasic excitation of burst cells in the lateral pontomesencephalic tegmentum. Rapid eye movements are the consequence of phasic firing by reticular and vestibular cells; the latter (not shown) excite oculomotor neurons directly (see Chapter 32). Muscular atonia is the consequence of tonic postsynaptic inhibition of spinal anterior horn cells by the pontomedullary reticular formation. Muscle twitches occur when excitation by reticular and pyramidal tract motorneurons phasically overcomes the tonic inhibition of the anterior horn cells. RN, raphe nuclei; LC, locus coeruleus; P, peribrachial region; PPT, pedunculopontine tegmental nucleus; LDT, laterodorsal tegmental nucleus; mPRF, meso- and mediopontine tegmentum (e.g., gigantocellular tegmental field, parvocellular tegmental field); RAS, midbrain reticular activating system; BIRF, bulbospinal inhibitory reticular formation (e.g., gigantocellular tegmental field, parvocellular tegmental field, magnocellular tegmental field); TC, thalamocortical; CT, cortical; PT cell, pyramidal cell; III, oculomotor; IV, trochlear; V, trigmenial motor nuclei; AHC, anterior horn cell.
From Hobson et al. (2000).
The earliest cellular model of the brainstem REM sleep generator, the Reciprocal Interaction (RI) Model (Hobson, McCarley, & Wyzinski, 1975), suggested that regular alternation of REM and NREM sleep results from the interaction of aminergic REM-off and cholinergic REM-on neuronal populations in the pontine brainstem as illustrated in Figures 40.10A and B. This hypothesis was supported by extensive experimental and clinical findings showing, for example, that micro-injection of cholinergic agonists (or drugs like neostigmine that block the enzymatic degradation of naturally produced ACh) into the pontine ARAS of cats produced a state indistinguishable from naturally occurring REM sleep. In addition, REM sleep was reduced or blocked by micro-injection of noradrenergic or serotonergic agonists into similar sites. Moreover, in humans, REM is suppressed by systemic administration of drugs that block the synaptic re-uptake or enzymatic degradation of naturally produced monoamines, such as many antidepressants.
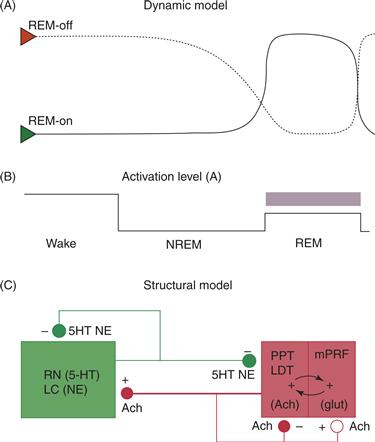
Figure 40.10 The Reciprocal Interaction (RI) Model (A) In the original RI model (Hobson et al., 1975; McCarley & Hobson, 1975), REM-on cholinergic neurons (Green triangle, solid line) both self-excite and excite aminergic REM-off neurons (Red triangle, dashed line). Aminergic REM-off neurons both inhibit cholinergic REM-on neurons and self-inhibit. This interaction leads to the alternation of behavioral states, depicted in (B), as follows: During waking, the REM-off aminergic system is active continuously and inhibits the pontine cholinergic system. During NREM sleep, aminergic activity decreases, allowing cholinergic activity to rise. The number of activated cholinergic REM-on neurons then exponentially increases due to self-excitation until sufficient numbers of aminergic REM-off neurons are also excited and begin to suppress the cholinergic REM-on population. However, these aminergic REM-off neurons then self-suppress, allowing the emergence of the next REM period. (C) In a subsequent revision of the RI hypothesis, mutually excitatory interactions between cholinergic and glutamatergic neurons underlie the rapidly escalating firing of pontine reticular REM-on neurons during REM sleep and cholinergic REM-on cells both self-excite and self-inhibit via cholinergic autoreceptors.
From Hobson et al. (2000).
Based upon subsequent reports, revision of the RI model added intermediate synaptic steps for both the activation of REM-on neurons and the suppression of REM-off aminergic nuclei (reviewed in Hobson et al., 2000). For example, mutually excitatory interactions between cholinergic and glutamatergic neurons may underlie the rapidly escalating firing of pontine reticular REM-on neurons during REM sleep (Fig. 40.10C). Similarly, GABAergic inhibition by midbrain or local mesopontine interneurons may be responsible for inhibiting REM-off serotonergic and noradrenergic cells thereby allowing REM (Luppi et al., 2006). GABAergic inhibition of cholinergic cells in the PPT may prevent REM during SWS, while glutamatergic activation of a certain percentage of these cholinergic cells may trigger REM with further increase in percentage activated leading to waking (Datta & Maclean, 2007).
Other models on the control of REM onset and offset have emphasized interactions between brainstem GABAergic cell populations in which GABAergic inhibition plays both REM facilitatory and REM inhibitory roles. For example, Luppi et al. (2006) have suggested that the onset of REM in the rat is triggered by the release of noncholinergic, possibly glutamatergic, neurons of the pontine sublaterodorsal (SLD) nucleus from tonic GABAergic inhibition by midbrain reticular neurons that is present during NREM and wake. Most recently, Saper et al. (2010) have proposed that a GABAergic flip-flop switch, analogous to the above hypothalamic flip-flop switch, controls the alternation of REM and NREM sleep states in the rat. GABAergic REM-off regions in the ventrolateral periaqueductal gray and lateral pontine tegmentum project to and are mutually inhibitory with REM-on regions in the mesopontine SLD, precoeruleus, and periventricular gray areas. In this model, aminergic and cholinergic influences are modulatory, but a mutually inhibitory GABAergic flip-flop circuit is the final common pathway of REM-NREM switching. Aminergic inputs from the LC and DR excite the REM-off GABAergic regions and inhibit the REM-on ones, whereas cholinergic input from the LDT/PPT displays the opposite pattern. Similarly, orexinergic neurons excite the REM-off GABAergic regions, thereby inhibiting REM, whereas GABAergic projections from REM-promoting regions of the VLPO inhibit this same region. Discrepancies between models may result from species differences. For example, Datta and Maclean (2007) hypothesize different anatomical origins of pontine (PGO and P) waves in predator versus prey species.
Other Neurotransmitter Systems Participating in Behavioral State Control
Many additional neurotransmitter systems participate in the modulation of sleep states. Such modulatory neurochemicals may exert their effects on behavioral state by interacting with aminergic, cholinergic, GABAergic, and glutaminergic neurons that exert a more direct “executive” control over sleep and waking states, or they may mediate only specific physiological signs of REM and NREM sleep. Little attention was initially paid to dopamine (DA) in behavioral state control because, unlike the other monoamines, firing rates of dopaminergic cells in the SNpc and VTA did not appear to vary with behavioral state. However, synaptic levels of DA increase in REM when VTA neurons shift from tonic to burst firing modes (Dahan et al., 2007) and recently discovered wake-active dopaminergic neurons in the ventral PAG may constitute a dopaminergic component of ascending arousal analogous to serotonergic, noradrenergic, and histaminergic systems (Saper et al., 2010). In addition to the roles of galanin, orexin, and the cytokine peptides described above, other neuropeptides implicated in the REM–NREM and sleep–wake cycles include vasoactive intestinal polypeptide, as well as numerous hormones, such as corticotropin releasing hormone (reviewed in Prospéro-García, Méndez-Díaz, Ruiz-Contreras, & Pérez-Morales, 2011). Nitric oxide (NO), an intercellularly diffusible gaseous neurotransmitter, is co-produced by cholinergic mesopontine neurons and may play a role in maintaining the cholinergically mediated REM sleep state in both the pons and the thalamus (Leonard & Lydic, 1999).
Molecular Biology of Behavioral State Control
Research on behavioral state control now extends inquiry beyond neurotransmitters and their receptors to intracellular second messenger cascades and gene transcription. The cyclic AMP-protein kinase A (cAMP-PKA) intracellular signaling pathway has been identified as the target of glutamatergic activation as well as GABAergic inhibition of PPT cholinergic cells generating physiological signs of REM (Datta & Maclean, 2007). For example, Bandyopadhya, Datta, and Saha (2006) have shown that intracellular concentrations of the catalytic subunit of PKA that enter the nucleus to phosphorylate CRE-binding protein vary with the prior amount of REM sleep. The specific profile of genes selectively activated in different sleep–wake states is also a current area of intense investigation (Cirelli, 2009), as is the genetic basis for variability in normal and disordered human sleep patterns (Landolt & Dijk, 2011). For example, genetic variation in adenosine deaminase, the degratory enzyme of adenosine, affects inter-individual variation in response to increased sleep pressure (Landolt & Dijk, 2011).
Systems Neuroscience of Sleep and Dreaming
Electromagnetic Physiology
As described above, slow oscillatory rhythms produced in reentrant thalamo-cortical and cortico-cortico circuits predominate in human slow wave sleep (SWS). High-density EEG shows that the slow oscillation can originate in frontal regions of the brain and then travel posteriorly across the cortex (Massimini et al., 2004), as well as occur as a local cortical phenomenon (Nir et al., 2011). Frontal areas are also the first regions of the brain to show EEG slowing at sleep onset, as well as those that most reflect the increase in slow wave activity following sleep deprivation (Ferrara et al., 2002).
In contrast to NREM, wakelike electromagnetic brain activity in REM contains fast, gamma range (low gamma = 30–80 Hz, high gamma = 80–150 Hz) oscillations (Ferri et al., 2001). Gamma oscillations are associated with effortful waking cognitive activity such as working memory or attention to perceptual stimuli. Long-range synchronization of gamma oscillations across different brain regions has been proposed as one way in which the brain generates the coherent experience of a percept or memory across different sensory modalities (“binding”), and such binding may also create coherent hallucinatory percepts during REM-sleep dreams (Kahn, Pace-Schott, & Hobson, 1997). Interestingly, another difference in REM compared to waking, the loss of coherence (a quantitative measure of EEG synchrony) at gamma frequencies between frontal executive-control areas and posterior perceptual areas of the cortex, may be one mechanism by which dream bizarreness could arise (Corsi-Cabrera, Guevara, & Del Rio-Portilla, 2008).
Functional Neuroimaging of Sleep
Early positron emission tomography (PET) studies showed decreasing brain activation with sleep onset. As NREM deepens to SWS, global brain activity can decline by as much as 40% below waking (Maquet, 2000). Deactivation of the thalamus may occur at an early stage in this process and contribute to subsequent deactivation of widespread cortical, limbic, and striatal areas (Dang-Vu et al., 2010). fMRI has further demonstrated decreases in perfusion initially of frontal cortical areas and, as NREM deepens, also in the hypothalamus, striatum, thalamus, hippocampal formation, and posterior cortex (Kaufmann et al., 2006).
Recent studies have examined the effect of NREM sleep on activity in the brain’s default network, a group of (mostly) midline brain areas that characteristically show a decrease in regional cerebral blood flow signal in response to a wide variety of goal-directed tasks, especially those requiring attention to exteroceptive (i.e., outside the body), perceptual stimuli (Buckner, Andrews-Hanna, & Schacter, 2008). In the absence of exteroceptive attention, these regions are believed to carry out adaptive, homeostatic, interoceptive (directed toward the internal milieu), and self-related functions, including imaginal simulation of future needs and desires, retrieval of autobiographical episodic memory, and self-regulation. In the blood oxygen level dependent (BOLD) signal of fMRI, the default network manifests as temporal synchrony of low-frequency (0.01–0.1 Hz) oscillations among its different regions (Buckner et al., 2008). This signature synchrony of BOLD fluctuations that defines the default mode during task-free waking persists into early stages of NREM sleep (N1, N2) but is lost during SWS (Samann et al., 2011).
With transition from NREM to REM, most lateral portions of the cortex remain less active than in waking, whereas activation, sometimes to levels exceeding that of waking, occurs in the pons, midbrain, thalamus, amygdala, hypothalamus, and basal ganglia, as well as limbic-related cortices (reviewed in Pace-Schott, 2011b). These regions comprise what Nofzinger et al. (2004) call the “anterior paralimbic REM activation area,” and these findings have led to an important expansion of theories on the neural origin of REM dreams, as described in the next section.
A Neurobiological Model of Dreaming
From evidence such as the above, we can suggest a neurobiological model of REM-sleep dreaming—the major conscious state that humans experience during sleep. Although REM dreams—being the most vivid, motoric, emotional, and bizarre—are modeled, dreaming also occurs at lower intensity and rate of recall during NREM (Hobson et al., 2000). (More detailed presentations of this model, shown in Fig. 40.11, can be found in Pace-Schott, 2011b.) Table 40.1 lists the phenomenological features of dreams that this neurobiological model seeks to explain.
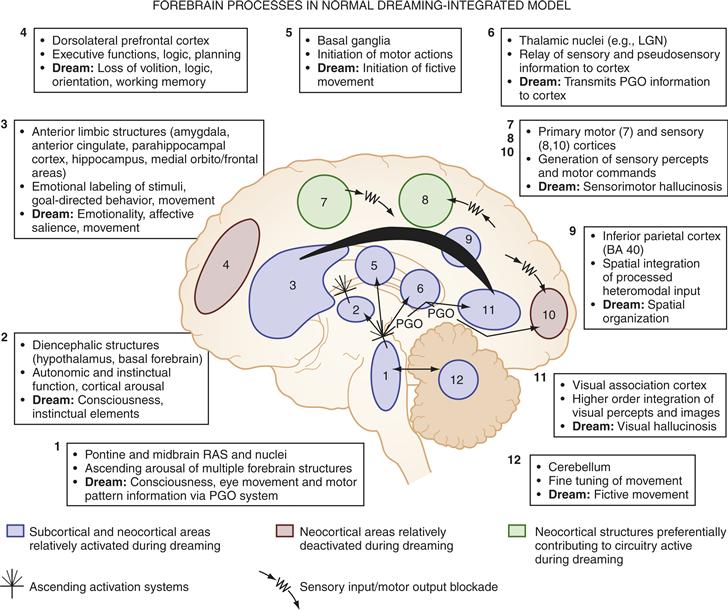
Figure 40.11 Forebrain processes in normal dreaming—an integration of neurophysiological, neuropsychological, and neuroimaging data. Regions 1 and 2: ascending arousal systems; 3: subcortical and cortical limbic and paralimbic structures; 4: dorsolateral prefrontal executive association cortex; 5: motor initiation and control centers; 6: thalamocortical relay centers and thalamic subcortical circuitry; 7: primary motor cortex; 8: primary sensory cortex; 9: inferior parietal lobe; 10: primary visual cortex; 11: visual association cortex; 12: cerebellum.
Table 40.1 Phylogeny of Rest and Sleep
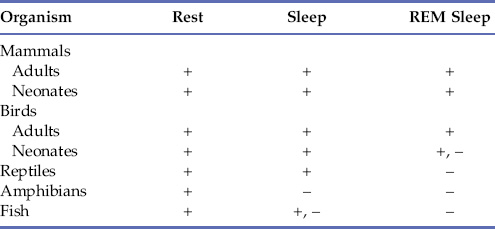
Note: REM, rapid eye movement; +, present; +, –, ambiguously or inconsistently present; –, absent.
In REM, the forebrain is activated, as in waking, by ARAS cholinergic nuclei and their brainstem targets that, by activating the thalamus and basal forebrain, allow these forebrain structures to, in turn, activate the limbic system and cortex via glutamatergic and cholinergic mechanisms, respectively. As originally hypothesized in the Activation-Synthesis Hypothesis (Hobson & McCarley, 1977), such forebrain activation results in a markedly different sort of conscious experience owing to the almost complete lack of noradrenergic, serotonergic, and histaminergic neuromodulation of the cortex. Notably, as detailed in Chapter 46 and 50, aminergic modulation facilitates higher cognitive functions such as attention and working memory.
Activity in the “anterior paralimbic REM activation area” may generate the emotionally salient features of dreaming. For example, activity in the amygdala, related basal forebrain areas (e.g., bed nucleus of the stria terminalis) and medial prefrontal (e.g., subgenual) areas, which are components of fear-processing circuits, may result in anxious or fearful dreams (Levin & Nielsen, 2007). On the other hand, activity in the midbrain, ventral striatum, and pallidum (components of reward circuits) may produce appetitive dreams (Solms, 1997). Similarly, activity in the hypothalamus and brainstem (e.g., periaqueductal gray) may trigger instinctual programs resulting in sexual, panic, or rage experiences (Jouvet, 1999). In addition, the richly social and interpersonally salient nature of dreams may result from activation of medial prefrontal areas involved in social cognition, such as the capacity for forming a “theory of mind,” empathizing, or moralizing.
Veridical replay of actual episodic experiences is quite rare in dreams (Stickgold, Hobson, Fosse, & Fosse, 2001), but recognition memory can sometimes be bizarrely enhanced, such as when we “just know” who a dream character is despite marked differences from the real-life counterpart (Kahn, Stickgold, Pace-Schott, & Hobson, 2000). Such credulity in dreams may be one manifestation of how low activity in lateral prefrontal areas results in dream bizarreness. Ventromedial frontal areas active in REM are involved in rapid, feelings-based appraisal of memory accuracy and may accept dream events without the verification provided by lateral and polar prefrontal areas that are needed for critical thinking and strategic search of memory. In a similar manner, other forms of dream bizarreness, such as incongruous juxtapositions, sudden discontinuities of plot, or bizarre uncertainty (see Hobson, 1988), may be uncritically accepted due to deactivation of lateral and polar prefrontal cortex. Interestingly, damage to lateral frontal areas has little effect on dreaming (Solms, 1997).
Although dreams abound with hallucinatory motion, REM atonia prevents contraction of skeletal muscles for the enactment of motor commands except in the case of REM behavior disorder (see below). However, REM-related activity in basal ganglia may produce the sense of initiated motion, while activity in the cerebellum may produce vestibular sensations such as flying, falling, or moving.
The Activation-Synthesis Hypothesis suggested that the striking visual hallucinations of dreams arise when phasic signals from the brainstem during REM, such as PGO waves, arrive at the cortex through a dedicated visual pathway and are interpreted by the visual cortex as true sensory input (Hobson & McCarley, 1977). Activation of visual association areas during REM by such ascending signals may allow emergence of complex hallucinations of actual objects and persons (Hobson et al., 2000). For example, regions of the ventral stream of visual processing (see Chapter 44), such as the fusiform face area or the parahippocampal place area, increase their activity during REM (Braun et al., 1998). Interestingly, damage to such ventral stream areas can result in nonvisual dreams (Solms, 1997).
Much, however, remains unexplained. For example, why are dream hallucinations integrated into elaborate plots in the absence of higher-order functions that, in waking, would be needed to create a coherent story? Similarly, why should damage to the inferior parietal cortex result in dream cessation when this area is among those that remain deactivated during REM? And, of course, how are dreams generated in NREM sleep?
Development and Phylogeny of Sleep
Changes in Sleep over the Human Lifespan
Human sleep shows striking developmental changes across the lifespan (Fig. 40.12). Newborns spend 17 to 18 hours per day sleeping in 3- to 4-hour bouts that, by approximately age 6, have consolidated into a single 10- to 12-hour nocturnal sleep period (Scher, 2008). An important transition during this period involves consolidation of diurnal sleep, first into a single daytime nap, that is then abandoned for solely nocturnal sleep (Davis, Parker, & Montgomery, 2004). SWS is absent at birth and develops during the first year of life, as do sleep spindles. In contrast, REM makes up around 50% of sleep in newborns, who also display incomplete atonia with a poorly differentiated EEG. By age 4, however, REM has declined to approximately adult levels of 20 to 25%. As the preadolescent enters adolescence, there is an endogenous circadian phase delay resulting in later sleep onset and rising time (Carskadon, 2011) as well as a decline in SWS. In young adults, on average, total sleep time is around 6 to 8 hours, of which about 5% is N1, 50 to 60% N2, 15 to 20% SWS, and 20 to 25% REM (Ohayon, Carskadon, Guilleminault, & Vitiello, 2004). Healthy aging is characterized by a diminished quality and duration of sleep, with increased time awake after sleep onset (Pace-Schott & Spencer, 2011). Older adults awaken more frequently and show a dramatic decline in SWS along with less marked decreases in numbers of sleep spindles and K-complexes. Opposite to changes entering adolescence, circadian rhythms of temperature, melatonin, and cortisol become phase advanced (earlier sleep and rising) in older adults.
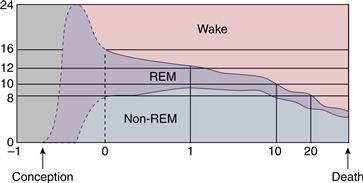
Figure 40.12 Portions of a 24-hour day that are devoted to waking, REM sleep, and non-REM (NREM) sleep change over a lifetime. Although the timing of these changes in utero is not known with certainty (dotted lines), data from premature infants are consistent with REM sleep occupying most of life at a gestational age of 26 weeks. After 26 weeks, the time spent in waking increases until death.
Phylogeny of Sleep
All mammals display REM and NREM sleep, but the duration and architecture of sleep vary widely. In general terms, smaller mammal species sleep more than larger ones, brain weight positively correlates with length of the REM-NREM cycle, and those born less mature have more REM (Hobson, 1989). In some marine mammals and birds, sleep occurs in one hemisphere at a time which, in the case of cetaceans, allows continued breathing (Rattenborg, Amlaner, & Lima, 2000). Birds have very short sleep and REM/NREM cycles, with minimal REM lacking atonia. Reptiles, amphibia, fish, and invertebrates have periods of quiescence with both circadian and seasonal periodicities, but the equivalence of these states to the sleep of homeotherms is just now being elucidated (Allada & Siegel, 2008). As noted in Chapter 39, the genetic basis of circadian rhythms is remarkably conserved from the most primitive to the most advanced phyla. Table 40.2 shows what is currently believed true about sleep states in the various phyla.
Table 40.2 Physiological Basis of Changes That Occur during Dreaming
| Function | Change (Compared with Waking) | Hypothesized Cause |
| Sensory input | Blocked | Presynaptic inhibition |
| Perception (external) | Diminished | Blockade of sensory input |
| Perception (internal) | Enhanced | Removal of inhibition from networks that store sensory representations |
| Attention | Lost | Aminergic modulation decreases, causing a decrease in the ratio of signal to noise |
| Memory (recent) | Diminished | Because of a decrease in aminergic activity, activated representations are not stored in memory |
| Memory (remote) | Enhanced | Removal of inhibition from networks that store memory representations |
| Orientation | Unstable | Internally inconsistent signals are generated by cholinergic systems |
| Thought | Poor reasoning, processing hyper-associative | Loss of attention, memory, and volition leads to failure of sequencing and rule inconstancy; analogy replaces analysis |
| Insight | Self-reflection lost | Failures of attention, logic, and memory weaken second- (and third-) order representations |
| Language (internal) | Confabulatory | Aminergic demodulation frees the use of language from the restraint of logic |
| Emotion | Episodically strong | Cholinergic hyperstimulation of the amygdala and related structures of the temporal lobe triggers emotional storms, which are not modulated by aminergic activity |
| Instinct | Episodically strong | Cholinergic hyperstimulation of the hypothalamus and limbic forebrain triggers fixed motor programs, which are experienced fictively but not enacted |
| Volition | Weak | Cortical motor control cannot compete with disinhibited subcortical networks |
| Output | Blocked | Postsynaptic inhibition |
Sleep Disorders
Disrupted Control of Behavioral State Can Lead to Insomnia and Hypersomnia
When brain aminergic activity is excessive due to stress, disease, or genetic predisposition, behavioral state is shifted in the direction of hyperarousal, and insomnia can result. Behavioral and pharmacologic interventions can be used to reduce this hyperarousal (Ellenbogen & Pace-Schott, 2012). Among pharmacological treatments, the most common are drugs such as zolpidem that target the benzodiazepine binding site on GABAA receptors. Binding to the benzodiazepine receptor site increases the ability of GABA to trigger chloride influx through the ionotropic GABAA receptor. The resulting hyperpolarization of the membrane then leads to neuronal inhibition and behavioral sedation. As described in Chapter 8, the GABAA receptor is a pentameric structure whose subunits exist in various isoforms. By targeting specific isoforms, hypnotic drugs have been developed that promote sleep with a lesser tendency to be addictive or to cause cognitive problems compared to “traditional benzodiazepines” like valium.
Abnormal decreases in CNS aminergic activity have the opposite effect on cognitive and behavioral systems. In the case of narcolepsy, the reduced orexinergic drive on aminergic systems allows sleep attacks. In such conditions, the REM generator is disinhibited, as can be seen in REM sleep attacks of narcoleptics. These symptoms can be decreased by aminergic agonists, such as antidepressants and psychostimulants, that block amine reuptake. The drug now most commonly used to treat hypersomnolence, modafinil (and its R-enantiomer armodafinil), shows weak reuptake blockade of norepinephrine and dopamine but may also augment electrical coupling among cells of the ARAS (Ellenbogen & Pace-Schott, 2012).
Respiratory Dysfunction Is Common in Sleep
One of the more dramatic problems associated with sleep is an exaggeration of the normal decline in drive from brainstem respiratory centers that occurs during sleep, a condition known as central sleep apnea (Fig. 40.13). A much more common condition, obstructive or peripheral sleep apnea (OSA), results from mechanical collapse or compression of the airway, a condition especially common in obese people (see Kryger et al., 2011, for extensive reviews; Fig. 40.13). In OSA, central respiratory drive causes continued respiratory effort. Eventually, the increase of CO2 in the brain leads to an arousal that in turn produces increased muscle tone in the oropharynx, and the airway opens. As the person falls into deeper sleep again, the oropharynx relaxes and the airway can become obstructed again until arousal recurs. In severe cases, arousals every one to two minutes all night are common.
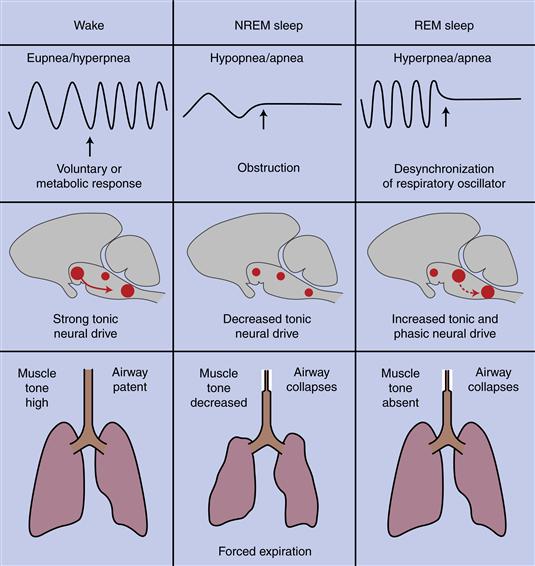
Figure 40.13 Central and obstructive sleep apnea. During waking, the respiratory oscillator of the medulla receives tonic drive from other neural structures and can respond to voluntary and metabolic signals to change breathing pattern. Muscle tone keeps the oropharynx open to the flow of air. In NREM sleep, central drive decreases, and the rate and depth of ventilation fall. If the decrease is excessive, central sleep apnea results, including a complete, albeit temporary, cessation of breathing. If the airway collapses, obstructive sleep apnea may result, with a similar cessation of breathing. During REM sleep, activation of pontine generator neurons drives the respiratory oscillator, and desynchronization may lead to breathing efforts that are too frequent or strong (hyperpnea) or that stop. During REM sleep the oscillator also becomes unresponsive to metabolic signals.
People with sleep apnea usually feel excessively sleepy during the daytime because frequent arousal prevents deep, sustained sleep. They are often unaware of their sleep-dependent breathing pattern because they are never fully aroused from sleep. Instead, observant friends or family often describe seeing these signs of respiratory dysfunction. In a sleep laboratory, EEG, respiratory effort, and cardiovascular parameters (e.g., heart rate, blood pressure, and oxygen saturation) are measured to quantify the problem. OSA can be treated by having the person wear a continuous positive airway (CPAP) mask through which air is forced gently, helping to keep the airways open. The pressure of the air fluctuates to allow regular exhalation. People with OSA are treated not only to alleviate their constant drowsiness but to prevent long-term effects of sleep apnea, such as cardiopulmonary complications and cognitive impairment.
Motor Disturbances Can Occur during Sleep
Parasomnias that affect the skeletal motor system can occur in either REM or NREM sleep. In NREM sleep, these parasomnias (e.g., sleep walking, sleep talking, tooth grinding, and night terrors) are mainly seen in young people, and reflect motor responses to the outputs of central motor pattern generators. Normally, responses to these generators are disinhibited at sleep onset and inhibited during REM sleep (by the REM atonia previously described). Excessive motor output during REM, termed REM sleep behavior disorder (RBD), can be an early sign of degenerative brain disease in the elderly when the brainstem neural circuits controlling REM atonia have become damaged. Because of a failure to inhibit the expression of motor acts in REM, people with this disorder may literally enact their dream scenarios in their bedrooms. A syndrome strikingly similar to RBD can be induced experimentally by bilateral lesions of the pontine tegmentum in cats. When in REM sleep, these animals perform stereotyped behavior sequences, such as hissing, piloerection, pouncing, and jumping. Jouvet called these “hallucinatory behaviors,” and Morrison called the phenomenon “REM sleep without atonia” (see Jouvet, 1999). Because the ability of a cat to inhibit motor pattern commands during REM sleep is impaired by the lesions, they act out commands that are normally initiated during REM sleep. The instinctual aspect of these released behaviors is relevant to the psychophysiology of dreaming as described above.
Other Sensorimotor Disturbances
Among the most interesting of sleep disorders is restless leg syndrome (RLS), a disorder that is often accompanied by another sleep disorder, periodic limb movement disorder (PLM). In RLS, while trying to go to sleep, the patient experiences a highly unpleasant burning or crawling sensation in the legs that can be relieved only by movement, often by arising and pacing. RLS is treated with dopaminergic agents that compensate for deficient output from dopaminergic neurons of the A11 region of the posterior hypothalamus that is the sole source of spinal cord dopamine (Paulus & Schomburg, 2006). Dopamine regulates sensory input to the dorsal horn of the spinal cord as well as the transmission of this input to the brain. In RLS, dopamine deficiency may allow excessive transmission of the normal low-level activity entering the spinal cord from peripheral receptors that, when transmitted to the brain, then produces abnormal sensations. Similarly, dopaminergic medications treat PLM by boosting dopaminergic inhibition of spinal early flexor reflexes that, when such inhibition is deficient, can be abnormally triggered by such baseline sensory inputs.
Disorders of Circadian Misalignment
Misalignment of waking activities with endogenous circadian rhythms is a common source of disordered sleep. As has been discussed, abnormal delay of sleep onset and offset (delayed sleep phase syndrome) is more common in adolescents and young adults, whereas abnormally early sleep onset and offset (advanced sleep phase syndrome) is more common in the elderly. In addition, overnight shift work or travel across time zones can lead to a desired sleep bout becoming misaligned with one’s endogenous circadian propensity for sleep and waking. Because light can entrain the circadian oscillator in the SCN, precisely timed treatment with bright light is the most commonly employed strategy for treating those circadian disorders. Melatonin and its agonists are also used to promote sleep onset in disorders of circadian misalignment.
The Purpose of Sleep
Sleep Appears to Have Multiple Functions
As a quiescent and ecologically protected behavior, sleep fosters conservation of energy, defense from predation, and an opportunity for repair of injury. The anabolic character of NREM sleep (e.g., brain and body inactivity and hormone release) suggests a rest and restoration function. For example, Benington and Heller (1995) propose that an important function of NREM sleep is to restore brain glycogen stores. In contrast, the high proportion of REM sleep in the developing brain (Fig. 40.12) suggests a role of REM sleep in brain development and plasticity (Marks, Shaffery, Oksenberg, Speciale, & Roffwarg, 1995).
The crucial anabolic and conservative role of sleep is illustrated by studies showing that prolonged sleep deprivation results in the disruption of metabolic and caloric homeostasis and eventually death (Rechtschaffen & Bergmann, 2002). In rats, sleep deprivation was fatal when it persisted for 4 to 6 weeks. Early in the deprivation period, the rats began to eat more but could not maintain their body weight. Later, they lost their ability to maintain body temperature, developed strong heat-seeking behavior, and finally died. It is suspected their death resulted from overwhelming sepsis because of immunodeficiency. The linkage between sleep and immune function is further emphasized by their numerous interactions (Imeri & Opp, 2009). For example, NREM sleep is enhanced by the cytokines interleukin-1 and interleukin-2, both of which are released during NREM sleep (Krueger, Rector, & Churchill, 2007).
Sleep also plays a key role in hormonal regulation. Growth and development are enhanced by the release of growth hormone and gonadotropins during SWS (Leproult & Van Cauter, 2010). Such hormone release declines, along with SWS, after growth and sexual maturation are complete (Van Cauter, Leproult, & Plat, 2000). In addition, sleep loss has been linked to dysregulation of glucose metabolism and increased risk of the metabolic syndrome diabetes and obesity (Van Cauter et al., 2007).
Several recent theories extend the role of sleep to the cellular and molecular levels. The synaptic homeostasis theory (Tononi & Cirelli, 2006) suggests that sleep, especially SWS, may serve to reduce or “downscale” excessive synaptic potentiation produced during prior waking, eliminating the more weakly potentiated synaptic connections and leaving only those associated with the most salient waking events. A particularly intriguing idea is that sleep is necessary to optimize correct folding of proteins in the endoplasmic reticulum (ER), as evidenced by the fact that sleep deprivation increases production of chaperone proteins involved in the ER stress (or “unfolded protein”) response (Naidoo, 2009). As further detailed in the next section, sleep and memory consolidation are closely interconnected, and the cAMP-PKA-CREB, second messenger cascade, important to memory formation in wake, also appears to be important in sleep-related memory consolidation (Hernandez & Abel, 2011).
Sleep and Memory Are Interdependent
Perhaps the clearest evidence of a function for sleep is its role in consolidating recently formed memories (for recent reviews, see Diekelmann & Born, 2010; Walker & Stickgold, 2006). One of the most notable examples is the discovery, in rats, by Carlyle Smith, of “REM windows” (see Smith, 1995), which are time periods after an animal is trained on specific tasks when the animal shows enhanced quantities of REM sleep and when retention of learning could be decreased by selective REM sleep deprivation. Thus, not only does REM sleep appear to be critical for the maintenance of this learning, but animals also increase their REM sleep after training, presumably by a homeostatic mechanism that regulates the amounts of different sleep stages based on the needs of the organism. Subsequent studies in humans have led to the theory that REM sleep is especially important for the consolidation of procedural memories, which are defined as memories of how to do things in the absence of explicit rules, such as how to ride a bicycle or read a map (Walker & Stickgold, 2006). In contrast, SWS has been implicated in the consolidation of explicit or declarative memory, which includes autobiographical memories as well as general knowledge of facts (Diekelmann & Born, 2010). However, studies on a procedural visual discrimination task have shown a requirement for both REM and SWS on the night following training in order for next-day improvement on the task to be observed (Stickgold, Whidbee, Schirmer, Patel, & Hobson, 2000b). This finding supports an earlier theory based on animal studies in which a two-step process involving both REM and SWS was hypothesized. In humans, the requirement for sleep immediately following learning was shown when subjects who were sleep deprived the night following training, but who then were allowed two nights of unrestricted recovery sleep before testing, showed no significant task improvement (Stickgold, James, & Hobson, 2000a). In addition to procedural learning tasks, REM effects have been reported in humans on learning of complex logic games, foreign language acquisition, intensive studying in general, and both REM and SWS effects have been reported for the consolidation and processing of emotional memories. In contrast, sleep-dependent consolidation of simple motor skill learning, such as for a sequential finger tapping task, correlates with Stage II NREM sleep (Walker, Brakefield, Morgan, Hobson, & Stickgold, 2002).
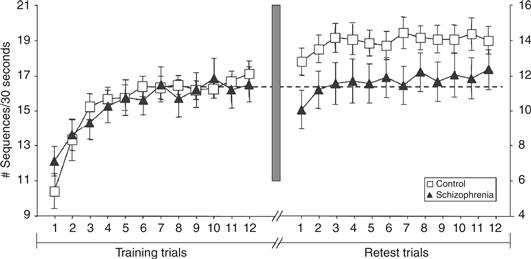
Figure 40.14 Sleep-dependent memory consolidation. Subjects were asked to type the sequence “41324” as quickly and accurately as they could for 30 seconds and then rest for 30 seconds before doing it again. Twelve trials were performed on each of two successive days. While the control subjects showed a 16% increase in speed overnight, the schizophrenia patients showed none.
The Functions of Dreaming, If Any, Are Poorly Understood
Does dreaming also serve a function? Answering this question is not yet possible, because we do not know how to separate possible functions of the experience of dreaming from functions of the neurobiological processes that produce it. But REM sleep dream content can predict who will become clinically depressed following marital divorce or separation (Cartwright, Young, Mercer, & Bears, 1998), suggesting that dreaming may serve to modulate emotional states during waking.
Dreaming may also play a role in modifying associative memory networks. Subjects awakened from REM sleep show preferential activation of more distantly related associated than seen either during waking or following NREM sleep (Stickgold et al., 1999). Such preferential activation of weak associations might explain the bizarre content of dreams during REM sleep and may permit the brain to discover creative solutions to problems during sleep. Indeed, sleep has been shown to increase the likelihood of such discoveries (Fig. 40.15; Wagner, Gais, Haider, Verleger, & Born, 2004). Most recently, it has been demonstrated that individuals who dream about a cognitive task following practice perform better on that task when they awaken (Wamsley, Tucker, Payne, Benavides, & Stickgold, 2010). But whether the conscious experience of dreaming is crucial for any of these sleep-dependent processes remains unknown.
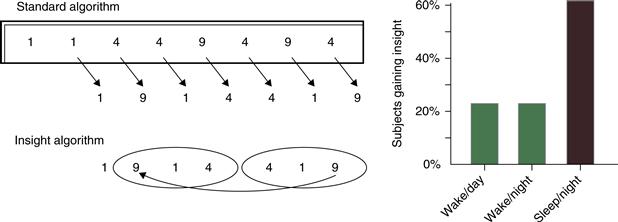
Figure 40.15 Sleep facilitates mathematical insight. Subjects are taught to reduce an 8-digit sequence to a single digit (9 at the right), through six intermediate calculations (in italics) with a standard algorithm. Unknown to the subjects, the task is designed so that the last three calculations form a mirror image of the preceding three, and thus the second intermediate calculation matches the final answer. Right: Subjects who slept between training and testing were more than twice as likely to discover the insight algorithm than those not allowed to sleep.
Dreaming may be viewed as evidence for a state of primary consciousness whose function is to provide the brain with an updatable virtual reality model of the brain that it uses in waking consciousness (Hobson, 2009). This view is surprisingly compatible with thermodynamic hypotheses about the brain (Friston, 2010).
References
1. Alam MN, Szymusiak R, Mcginty D. Hypocretinergic system: Role in REM-sleep regulation. In: Mallick BN, Pandi-Permual SR, Mccarley RW, Morrison AR, eds. Rapid eye movement sleep, regulation and function. Cambridge: Cambridge University Press; 2011.
2. Allada R, Siegel JM. Unearthing the phylogenetic roots of sleep. Current Biology. 2008;18:R670–R679.
3. Amzica F, Steriade M. Progressive cortical synchronization of ponto-geniculo-occipital potentials during rapid eye movement sleep. Neuroscience. 1996;72:309–314.
4. Arendt J. Melatonin and human rhythms. Chronobiology International. 2006;23:21–37.
5. Aserinsky E, Kleitman N. Regularly occurring periods of eye motility, and concomitant phenomena, during sleep. Science. 1953;118:273–274.
6. Bandyopadhya RS, Datta S, Saha S. Activation of pedunculopontine tegmental protein kinase A: A mechanism for rapid eye movement sleep generation in the freely moving rat. Journal of Neuroscience. 2006;26(35):8931–8942.
7. Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Progress in Neurobiology. 1995;45:347–360.
8. Bersagliere A, Achermann P. Slow oscillations in human non-rapid eye movement sleep electroencephalogram: effects of increased sleep pressure. Journal of Sleep Research. 2010;19:228–237.
9. Bes F, Jobert M, Schulz H. Modeling napping, post-lunch dip, and other variations in human sleep propensity. Sleep. 2009;32:392–398.
10. Borbely AA. A two process model of sleep regulation. Human Neurobiology. 1982;1:195–204.
11. Braun AR, Balkin TJ, Wesensten NJ, et al. Dissociated pattern of activity in visual cortices and their projections during human rapid eye movement sleep. Science. 1998;279:91–95.
12. Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Annals of the New York Academy of Science. 2008;1124:1–38.
13. Carskadon MA. Sleep in adolescents: The perfect storm. Pediatric Clinics of North America. 2011;58:637–647.
14. Cartwright R, Young MA, Mercer P, Bears M. Role of REM sleep and dream variables in the prediction of remission from depression. Psychiatry Research. 1998;80:249–255.
15. Chase MH. Confirmation of the consensus that glycinergic postsynaptic inhibition is responsible for the atonia of REM sleep. Sleep. 2008;31:1487–1491.
16. Cirelli C. The genetic and molecular regulation of sleep: From fruit flies to humans. Nature Reviews. 2009;10:549–560.
17. Colrain IM. The K-complex: A 7-decade history. Sleep. 2005;28:255–273.
18. Corsi-Cabrera M, Guevara MA, Del Rio-Portilla Y. Brain activity and temporal coupling related to eye movements during REM sleep: EEG and MEG results. Brain Research. 2008;1235:82–91.
19. Crunelli V, Hughes SW. The slow (<1 Hz) rhythm of non-REM sleep: A dialogue between three cardinal oscillators. Nature Neuroscience. 2010;13:9–17.
20. Czeisler CA, Duffy JF, Shanahan TL, et al. Stability, precision, and near-24-hour period of the human circadian pacemaker. Science. 1999;284:2177–2181.
21. Daan S, Beersma DGM, Borbely AA. Timing of human sleep: Recovery process gated by a circadian pacemaker. American Journal of Physiology. 1984;246:R161–R178.
22. Dahan L, Astier B, Vautrelle N, Urbain N, Kocsis B, Chouvet G. Prominent burst firing of dopaminergic neurons in the ventral tegmental area during paradoxical sleep. Neuropsychopharmacology. 2007;32:1232–1241.
23. Dahlstrom A, Fuxe K. Localization of monoamines in the lower brain stem. Experientia. 1964;20:398–399.
24. Dang-Vu TT, Schabus M, Desseilles M, et al. Functional neuroimaging insights into the physiology of human sleep. Sleep. 2010;33:1589–1603.
25. Datta S, Maclean RR. Neurobiological mechanisms for the regulation of mammalian sleep-wake behavior: Reinterpretation of historical evidence and inclusion of contemporary cellular and molecular evidence. Neuroscience and Biobehavioral Reviews. 2007;31:775–824.
26. Davis KF, Parker KP, Montgomery GL. Sleep in infants and young children: Part one: normal sleep. Journal of Pediatric Health Care. 2004;18:65–71.
27. De Gennaro L, Ferrara M. Sleep spindles: An overview. Sleep Medicine Reviews. 2003;7:423–440.
28. Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: Organization and coordination of central and peripheral clocks. Annual Review of Physiology. 2010;72 517–49.
29. Diekelmann S, Born J. The memory function of sleep. Nature Reviews Neuroscience. 2010;11:114–126.
30. Dijk DJ. Regulation and functional correlates of slow wave sleep. Journal of Clinical Sleep Medicine. 2009;5:S6–S15.
31. Dijk DJ, Czeisler CA. Paradoxical timing of the circadian rhythm of sleep propensity serves to consolidate sleep and wakefulness in humans. Neuroscience Letters. 1994;166:63–68.
32. Ellenbogen JM, Pace-Schott E. Drug-induced sleep: Theoretical and practical considerations. Pfluegers Archive European Journal of Physiology. 2011;463:177–186.
33. Ferrara M, De Gennaro L, Curcio G, Cristiani R, Corvasce C, Bertini M. Regional differences of the human sleep electroencephalogram in response to selective slow-wave sleep deprivation. Cerebral Cortex. 2002;12:737–748.
34. Ferri R, Cosentino FI, Elia M, et al. Relationship between Delta, Sigma, Beta, and Gamma EEG bands at REM sleep onset and REM sleep end. Clinical Neurophysiology. 2001;112:2046–2052.
35. Friston K. The free-energy principle: A unified brain theory?. Nature Reviews. 2010;11:127–138.
36. Halassa MM. Thalamocortical dynamics of sleep: Roles of purinergic neuromodulation. Seminars in Cell and Developmental Biology. 2011;22:245–251.
37. Hernandez PJ, Abel T. A molecular basis for interactions between sleep and memory. Sleep Medicine Clinics. 2011;6:71–84.
38. Hobson JA. Sleep New York, NY: Scientific American Library; 1989.
39. Hobson JA. REM sleep and dreaming: Towards a theory of protoconsciousness. Nature Reviews. 2009;10:803–813.
40. Hobson JA, McCarley RW. The brain as a dream state generator: An activation-synthesis hypothesis of the dream process. American Journal of Psychiatry. 1977;134:1335–1348.
41. Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: Reciprocal discharge by two brainstem neuronal groups. Science. 1975;189:55–58.
42. Hobson JA, Steriade M. The neuronal basis of behavioral state control. In: Bloom FE, ed. Bethesda, MD: American Physiological Society; 1986; Handbook of physiology - the nervous system. Vol. IV.
43. Iber C, Ancoli-Israel E, Chesson A, Quan SF. The AASM manual for the scoring of sleep and associated events: Rules, terminology and technical specification Westchester, IL: American Academy of Sleep Medicine; 2007.
44. Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nature Reviews. 2009;10:199–210.
45. Jones BE. Modulation of cortical activation and behavioral arousal by cholinergic and orexinergic systems. Annals of the New York Academy of Sciences. 2008;1129:26–34.
46. Kahn D, Pace-Schott EF, Hobson JA. Consciousness in waking and dreaming: The roles of neuronal oscillation and neuromodulation in determining similarities and differences. Neuroscience. 1997;78:13–38.
47. Kahn D, Stickgold R, Pace-Schott EF, Hobson JA. Dreaming and waking consciousness: A character recognition study. Journal of Sleep Research. 2000;9:317–325.
48. Kaufmann C, Wehrle R, Wetter TC, et al. Brain activation and hypothalamic functional connectivity during human non-rapid eye movement sleep: An EEG/fMRI study. Brain. 2006;129:655–667.
49. Krueger JM, Rector DM, Churchill L. Sleep and Cytokines. Sleep Medicine Clinics. 2007;2:161–169.
50. Krueger JM, Rector DM, Roy S, et al. Sleep as a fundamental property of neuronal assemblies. Nature Reviews. 2008;9:910–919.
51. Landolt H, Dijk D. Genetic basis of sleep in healthy humans. In: Kryger MH, Roth T, Dement WC, eds. Principles and practice of sleep medicine 5th edition. Philadelphia, PA: Elsevier; 2011.
52. Lavie P. Ultrashort sleep-waking schedule III “Gates” and “forbidden zones” for sleep. Electroencephalography and Clinical Neurophysiology. 1986;63:414–425.
53. Lazarus M, Shen HY, Cherasse Y, et al. Arousal effect of caffeine depends on adenosine A2A receptors in the shell of the nucleus accumbens. Journal of Neurosciences. 2011;31:10067–10075.
54. Leonard TO, Lydic R. Nitric oxide, a diffusible modulator of physiological traits and behavioral states. In: Mallick BN, Inoue S, eds. Rapid eye movement sleep. New York, NY: Marcel Dekker; 1999;167–193.
55. Leproult R, Van Cauter E. Role of sleep and sleep loss in hormonal release and metabolism. Endocrine Development. 2010;17:11–21.
56. Levin R, Nielsen TA. Disturbed dreaming, posttraumatic stress disorder, and affect distress: A review and neurocognitive model. Psychological Bulletin. 2007;133:482–528.
57. Lim AS, Lozano AM, Moro E, et al. Characterization of REM-sleep associated ponto-geniculo-occipital waves in the human pons. Sleep. 2007;30:823–827.
58. Luppi PH, Gervasoni D, Verret L, et al. Paradoxical (REM) sleep genesis: the switch from an aminergic-cholinergic to a GABAergic-glutamatergic hypothesis. Journal of Physiology, Paris. 2006;100:271–283.
59. Maquet P. Functional neuroimaging of normal human sleep by positron emission tomography. Journal of Sleep Research. 2000;9:207–231.
60. Marks GA, Shaffery JP, Oksenberg A, Speciale SG, Roffwarg HP. A functional role for REM sleep in brain maturation. Behavioural Brain Research. 1995;69:1–11.
61. Massimini M, Huber R, Ferrarelli F, Hill S, Tononi G. The sleep slow oscillation as a traveling wave. Journal of Neurosciences. 2004;24:6862–6870.
62. McCarley RW, Hobson JA. Neuronal excitability modulation over the sleep cycle: A structural and mathematical model. Science. 1975;189:58–60.
63. McCormick DA, Bal T. Sleep and arousal: Thalamocortical mechanisms. Annual Review of Neuroscience. 1997;20:185–215.
64. Menicucci D, Piarulli A, Debarnot U, et al. Functional structure of spontaneous sleep slow oscillation activity in humans. PLoS ONE. 2009;4:e7601.
65. Merica H. Fast and slow frequency spindles in sleep: Two generators?. Clinical Neurophysiology. 2000;111:1704–1706.
66. Moore RY, Card JP. Noradrenaline-containing Neuron System Amsterdam: Elsevier; 1984.
67. Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electroencephalography and Clinical Neurophysiology. 1949;1:455–473.
68. Naidoo N. Cellular stress/the unfolded protein response: Relevance to sleep and sleep disorders. Sleep Medicine Reviews. 2009;13:195–204.
69. Nir Y, Staba RJ, Andrillon T, et al. Regional slow waves and spindles in human sleep. Neuron. 2011;70:153–169.
70. Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355:39–40.
71. Nofzinger EA, Buysse DJ, Germain A, et al. Increased activation of anterior paralimbic and executive cortex from waking to rapid eye movement sleep in depression. Archives of General Psychiatry. 2004;61:695–702.
72. Ohayon MM, Carskadon MA, Guilleminault C, Vitiello MV. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: Developing normative sleep values across the human lifespan. Sleep. 2004;27:1255–1273.
73. Ohno K, Sakurai T. Orexin neuronal circuitry: Role in the regulation of sleep and wakefulness. Frontiers in Neuroendocrinology. 2008;29:70–87.
74. Pace-Schott EF. REM sleep and dreaming. In: Mallick BN, Pandi-Permual SR, McCarley RW, Morrison AR, eds. Rapid eye movement sleep—regulation and function. Cambridge: Cambridge University Press; 2011a.
75. Pace-Schott EF. The neurobiology of dreaming. In: Kryger MH, Roth T, Dement WC, eds. Principles and practice of sleep medicine 5th edition. Philadelphia, PA: Elsevier; 2011b.
76. Pace-Schott EF, Hobson JA. The neurobiology of sleep: Genetics, cellular physiology and subcortical networks. Nature Reviews Neuroscience. 2002;3(8):591–605.
77. Pace-Schott EF, Spencer RMC. Age-related changes in the cognitive function of sleep. Progress in Brain Research. 2011;191:75–89.
78. Pannain S, Van Cauter E. Modulation of endocrine function by sleep-wake homeostasis and circadian rhythmicity. Sleep Medicine Clinics. 2007;2:147–159.
79. Parmeggiani PL. Thermoregulation and sleep. Frontiers in Bioscience. 2003;8:S557–S567.
80. Paulus W, Schomburg ED. Dopamine and the spinal cord in restless legs syndrome: Does spinal cord physiology reveal a basis for augmentation?. Sleep Medicine Reviews. 2006;10:185–196.
81. Peirson S, Foster RG. Melanopsin: Another way of signaling light. Neuron. 2006;49:331–339.
82. Porkka-Heiskanen T, Kalinchuk AV. Adenosine, energy metabolism and sleep homeostasis. Sleep Medicine Reviews. 2011;15:123–135.
83. Prospéro-García O, Méndez-Díaz M, Ruiz-Contreras AEA, et al, Pérez-Morales M. Neuropeptides and REM sleep. In: Mallick BN, Pandi-Permual SR, McCarley RW, Morrison AR, eds. Rapid eye movement sleep—regulation and function. Cambridge: Cambridge University Press; 2011.
84. Ramirez JM, Tryba AK, Peña F. Pacemaker neurons and neuronal networks: An integrative view. Current Opinion in Neurobiology. 2004;14(6):665–674.
85. Rattenborg NC, Amlaner CJ, Lima SL. Behavioral, neurophysiological and evolutionary perspectives on unihemispheric sleep. Neuroscience and Biobehavioral Reviews. 2000;24:817–842.
86. Rechtschaffen A, Bergmann BM. Sleep deprivation in the rat: An update of the 1989 paper. Sleep. 2002;25:18–24.
87. Rechtschaffen A, Kales A. A manual of standardized terminology techniques and scoring system for sleep stages of human subjects Los Angeles, CA: University of California, Brain Information Service/Brain Research Institute; 1968.
88. Samann PG, Wehrle R, Hoehn D, et al. Development of the brain’s default mode network from wakefulness to slow wave sleep. Cerebral Cortex. 2011;21:2082–2093.
89. Saper CB, Lu J, Chou TC, Gooley J. The hypothalamic integrator for circadian rhythms. Trends in Neurosciences. 2005a;28:152–157.
90. Saper CB, Chou TC, Scammell TE. The sleep switch: Hypothalamic control of sleep and wakefulness. Trends in Neuroscience. 2001;24:726–731.
91. Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–1042.
92. Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263.
93. Scher MS. Ontogeny of EEG-sleep from neonatal through infancy periods. Sleep Medicine. 2008;9:615–636.
94. Sherin JE, Shiromani PJ, Mccarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219.
95. Smith C. Sleep states and memory processes. Behavioural Brain Research. 1995;69 137–45.
96. Solms M. The neuropsychology of dreams: A clinico-anatomical study Mahwah, NJ: Lawrence Erlbaum Associates; 1997.
97. Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience. 2000;101:243–276.
98. Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–1106.
99. Stickgold R, Hobson JA, Fosse R, Fosse M. Sleep, learning, and dreams: Off-line memory reprocessing. Science. 2001;294:1052–1057.
100. Stickgold R, James L, Hobson JA. Visual discrimination learning requires sleep after training. Nature Neuroscience. 2000a;3:1237–1238.
101. Stickgold R, Scott L, Rittenhouse C, Hobson JA. Sleep-induced changes in associative memory. Journal of Cognitive Neuroscience. 1999;11:182–193.
102. Stickgold R, Whidbee D, Schirmer B, Patel V, Hobson JA. Visual discrimination task improvement: A multi-step process occurring during sleep. Journal of Cognitive Neuroscience. 2000b;12:246–254.
103. Szymusiak R, McGinty D. Hypothalamic regulation of sleep and arousal. Annals of New York Academy of Science. 2008;1129:275–286.
104. Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Medicine Reviews. 2006;10:49–62.
105. Trinder, J., Kleiman, J., Carrington, M., Smith, S., Breen, S., Tan, N., … Kim, Y. Autonomic activity during human sleep as a function of time and sleep stage. Journal of Sleep Research, 10, 253–264.
106. Van Cauter E, Holmback U, Knutson K, et al. Impact of sleep and sleep loss on neuroendocrine and metabolic function. Hormone Research. 2007;67(Suppl 1):2–9.
107. Van Cauter E, Leproult R, Plat L. Age-related changes in slow wave sleep and REM sleep and relationship with growth hormone and cortisol levels in healthy men. JAMA. 2000;284:861–868.
108. Vertes RP, Kocsis B. Brainstem-diencephalo-septohippocampal systems controlling the theta rhythm of the hippocampus. Neuroscience. 1997;81:893–926.
109. Wagner U, Gais S, Haider H, Verleger R, Born J. Sleep inspires insight. Nature. 2004;427:352–355.
110. Walker M, Brakefield T, Morgan A, Hobson JA, Stickgold R. Practice with sleep makes perfect: Sleep dependent motor skill learning. Neuron. 2002;35:205–211.
111. Walker MP, Stickgold R. Sleep, memory, and plasticity. Annual Reviews in Psychology. 2006;57:139–166.
112. Wamsley EJ, Tucker M, Payne JD, Benavides JA, Stickgold R. Dreaming of a learning task is associated with enhanced sleep-dependent memory consolidation. Current Biology. 2010;20:850–855.
113. Watson CJ, Baghdoyan HA, Lydic R. Neuropharmacology of sleep and wakefulness. Sleep Medicine Clinics. 2011;5:513–528.
114. Weyand TG, Boudreaux M, Guido W. Burst and tonic response modes in thalamic neurons during sleep and wakefulness. Journal of Neurophysiology. 2001;85:1107–1118.
Suggested Readings
1. Hobson JA. The dreaming brain New York, NY: Basic Books; 1988.
2. Hobson JA, Pace-Schott EF, Stickgold R. Dreaming and the brain: Toward a cognitive neuroscience of conscious states. Behavioral and Brain Sciences. 2000;23:793–842 discussion 904–1121.
3. Jouvet M. The paradox of sleep: The story of dreaming Cambridge: MIT Press; 1999.
4. Kryger MH, Roth T, Dement WC, eds. Principles and practice of sleep medicine 5th edition. Philadelphia, PA: Elsevier; 2011.
5. Mallick BN, Pandi-Permual SR, Mccarley RW, Morrison AR. Rapid eye movement sleep–regulation and function Cambridge: Cambridge University Press; 2011.
6. Steriade M, McCarley RW. Brain control of wakefulness and sleep New York, NY: Kluwer Academic/Plenum; 2005.